

Abstract
The success of CAR T cell therapy in solid tumors, unlike in hematologic malignancies, is limited by inadequate tumor infiltration and T cell dysfunction and exhaustion. Regional delivery of CAR T cells in patients with solid tumors is safe and feasible, promotes infiltration, proliferation, and trafficking, and ignites functionally persisting systemic immunity.
Keywords: Chimeric antigen receptor (CAR) T cells, Regional therapies, Adoptive cell therapy, Solid tumors cell therapy
The goal of any solid tumor-targeted immune therapy should not only generate an effective immune response against visible primary tumor but also establish circulating, long-lasting immunity to eradicate micrometastatic disease. Data from preclinical and clinical studies suggest that regional chimeric antigen receptor (CAR) T cell therapy counters the spatiotemporal obstacles and immune inhibition of solid tumors, generating the T cell memory required to eradicate disease and prevent recurrence.
Understanding T cell dysfunction informs development of solid tumor cell therapy
The complete clinical responses attained with CD19 antigen-targeted T cell therapy modified to include CD28 or 4–1BB costimulatory signaling in the majority of patients with treatment-refractory hematologic cancers highlights the potential of engineered T cells (June and Sadelain, 2018). This potential has yet to be fully realized for solid tumors due to several obstacles; T cell dysfunction resulting from anergy, ignorance, tolerance, or exhaustion is one such obstacle. It was the understanding that anergy is a hyporesponsive state of T cells activated in the absence of costimulatory signaling and that tumor cells lack the costimulatory ligand expression needed for optimal T cell activation that led to genetically engineered costimulation (Maher et al., 2002), the breakthrough leading to clinical success in treating hematologic malignancies.
Unlike hematologic malignancies—which reside within the peripheral compartment, into which intravenously administered costimulated CAR T cells are delivered—solid tumors are sequestered within an immunosuppressive niche that also impedes efficient T cell infiltration due to poor tumor vascularity and stromal barriers. Furthermore, intravenously administered T cells do not routinely infiltrate noninflamed tumor tissues and instead are sequestered within nontarget organs (Figure. 1A) (Adusumilli et al., 2014), which is an example of T cell ignorance. In this context, tumors are an immune-privileged site shielded from recognition unless an inciting inflammatory event triggers T cell priming and infiltration.
Figure 1. Regional versus systemic delivery of CAR T cells.
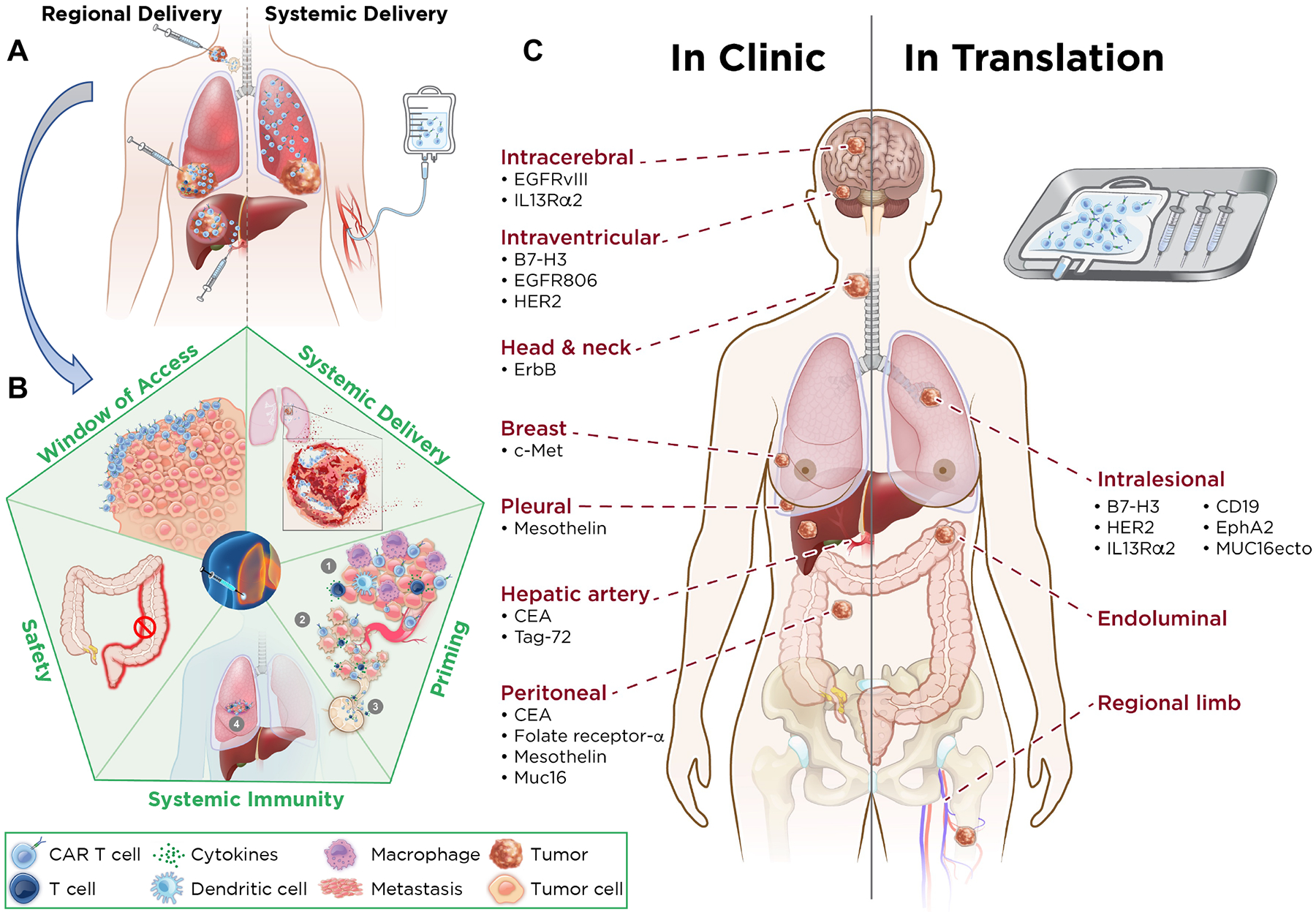
A. Characteristics of CAR T cells delivered regionally versus systemically. Regional delivery allows for increased intratumoral proliferation of CAR T cells, increased depth of penetration of CAR T cells, augmentation of CD4 help, immune balance, greater trafficking to site(s) of metastasis, and drainage to lymph nodes. Systemic delivery results in sequestration of CAR T cells in the lung, with shallow penetration into the tumor, no CD4 help, and little effect on metastatic site(s). B. Advantages of regional administration of CAR T cells. Regional delivery acts as a window of access for CAR T cells to generate systemic immunity and enter metastatic site(s) better, results in tumor lysis, and primes the environment for systemic immunity via the induction of tumor immune microenvironment changes, including decreased immune suppression via effector response in the tumor microenvironment, increased tumor cell death leading to strong immune response (via release of neoantigens that are drained to regional lymph nodes), increased susceptibility of tumor blood vessels to immune cell traffic, and increased T cell circulation and entry into metastatic sites. Regional delivery generates a local immune response that results in a long-lasting circulating T-cell immunity to eliminate systemic disease and prevent tumor recurrence and avoids systemic toxicity—that is, there is no colitis when CAR T cells are delivered intrahepatically. C. Comparing regional therapy modalities in clinic and in translation. Regional therapy is currently being administered for intracerebral, intraventricular, head and neck, breast, pleural, hepatic (via the hepatic artery), and peritoneal tumors. For each tumor type, the relative CAR target is highlighted (Left). CAR T cell targets in translation by intralesional administration are listed. Potential other routes of administration are endoluminal and regional limb infusion (Right).
One of the goals of immunotherapy is to promote tumor infiltration of T cells, modulating a “cold” tumor environment to “hot.” To effectively eliminate tumor, tumor-infiltrating T cells must maintain function in spite of repeated antigen activation within an environment rich in inhibitory signaling. The large antigen burdens of well-established solid tumors promote T cell exhaustion, a hyporesponsive state defined transcriptionally, functionally, and phenotypically. Attempts to reinvigorate exhausted CAR T cell states have led to rational combinations with checkpoint blockade therapy.
Regional delivery provides a window of access for T cell invasion
To overcome ignorance and exhaustion, as well as to generate the circulating immunity required for a durable clinical response, regional therapy has been applied. In studies using clinically relevant animal models, CAR T cells are administered into a locoregional body cavity that can serve as a window of access to the tumor, allowing the generation of a robust local immune response and functioning as a distribution center for long-lasting circulating T cell immunity (Figure. 1B) (Adusumilli et al., 2014; Adusumilli et al., 2021; Cherkassky et al., 2016). Whereas the lack of a tumor chemokine gradient can limit tumor infiltration of systemically administered CAR T cells, regionally delivered CAR T cells undergo antigen activation and proliferation (dose expansion) and upregulate chemokine receptors, which facilitates systemic circulation and effective distant-site tumor infiltration (Mulazzani et. al. 2019). Furthermore, CAR T cell–secreted cytokines and chemokines can modulate the regional tumor immune microenvironment to facilitate activation of endogenous immunity (Brown et. al. 2016). This cascade of antitumor immunity can result in a treatment response in patients with solid tumors, including complete metabolic responses in patients with heterogenous antigen expression, as demonstrated in a recently published report on mesothelin-targeted regional CAR T cell therapy in patients with pleural malignancies (Adusumilli et al., 2021).
Modeling an in vivo T cell stress test
Adequate testing of T cell function in a preclinical model of regional delivery requires an “in vivo CAR T cell stress test” in an orthotopic model with an established large solid tumor, recapitulating the high antigen burden, locoregional invasion, necrotic and hypoxic zones, stromal barriers, and inhibitory environment characteristic of human solid tumors. In one such clinically representative model of orthotopic pleural mesothelioma with established large tumor burden (control mice die in 2 weeks), CAR T cells were administered at a low effector/target ratio, creating a condition whereby T cells must persist in function upon repeated antigen stimulation to achieve tumor elimination. To further investigate systemic persistence of functional immunity, tumor rechallenge months after initial tumor eradication can be conducted (Cherkassky et al., 2016).
It is with these models the benefits of regional therapy were demonstrated. Whereas intravenously administered CAR T cells are sequestered within the lungs, limiting tumor infiltration and activation, intrapleurally administered CAR T cells are able to deeply penetrate pleural tumor, followed by rapid kinetics of expansion and function to counteract the inhibitory tumor environment, thereby achieving tumor regression and eradication at a lower dose (Adusumilli et al., 2014). Similar observations of enhanced tumor infiltration and efficacy have been made in brain tumor and head and neck cancer models (Mulazzani et. al. 2019, Theruvath et. al. 2020, Pituch et al., 2018, Vitanza et al., 2021). CD8+ CAR T cells rapidly and efficiently lyse tumor cells but may be particularly predisposed to dysfunction and activation-induced cell death, requiring CD4+ CAR T cells as a source of IL-2. CD4+ T cells endowed with high-avidity CAR-target ligation are able to lyse tumor cells without losing their helper function. Thus, regional therapy maximizes these contributions from both CD8+ and CD4+ CAR T cell subsets to help realize the therapeutic potential.
Translational regional CAR therapies
Regional CAR therapy approaches for several solid tumors are in translation to clinical trials (Figure. 1C). These include intracerebroventricular administration for central nervous system (CNS) tumors, intraperitoneal delivery for peritoneal surface disease, and hepatic arterial infusion for colorectal cancer liver metastases, all of which pose unique obstacles for CAR therapy. In CNS tumors, the blood-brain barrier may constitute an especially immune-privileged compartment. In peritoneal disease, tumors establish multiple sites of disease and produce ascitic fluid containing immunosuppressive cells and signals. In cases of liver metastases, treatment must overcome the decidedly immunosuppressive environment of the liver.
Using these models, investigators have expanded and reinforced the therapeutic promise of regional therapy. Theruvath et al. used intraventricular-administered CAR T cells in brain tumor models to highlight efficient intratumoral infiltration, early T cell expansion, and effective T cell distribution throughout the local environment to eliminate multifocal sites of disease (Theruvath et al., 2020). CAR T cells administered intracerebroventricularly or intratumorally in mice with cerebral tumors exhibited faster kinetics, greater potency, and reduced systemic levels of inflammatory cytokines, compared with CAR T cells administered intravenously (Theruvath et. al. 2020). Mulazzani et al. performed intravital microscopy in a model of the primary CNS to demonstrate that intracerebral-administered CD19-targeted T cells infiltrate deep into the tumor core with single-cell resolution (Mulazzani et al., 2019). They showed that tumor cell–antigen encounter and killing by CAR T cells do not proceed in an orderly fashion, one cell layer at a time. Instead, CAR T cells infiltrate deep into the tumor substance for antigen activation and effector function, contrary to the behavior of other locally administered biologic and gene therapies, including oncolytic viruses, in which agent distribution is confined to the first few cell layers of tumor and systemic distribution is limited.
Regional CAR therapy could be a push-key ignition for systemic immunity
Early CAR T cell activation and dose-expansion following regional therapy facilitates CAR T cell biodistribution to remote sites of disease, and the rapid acquisition of an effector phenotype may account for the efficient trafficking. The resulting immunity is able to control metastasis of disease and tumor rechallenge at orthoptic and heterotopic sites (Figure. 1B). Theruvath et al. rechallenged cured mice at distant sites and documented circulating, functional immunity (Theruvath et al., 2020). Mulazzani et al. observed T cells within the periphery 6 months after a single intracerebral administration of T cells (Mulazzani et al., 2019). Pituch et al. noted a memory response to rechallenge with an antigen-negative variant in an immunocompetent model (Pituch et al., 2018), which suggests that endogenous T cells specific for antigens other than those targeted by the CAR can contribute to long-term immunity following regional tumor immunomodulation. Regional therapy resulted in migration of intrapleurally administered CAR T cells out of the pleural cavity; these T cells were measurable at extrapleural tumor sites within 1 week after administration in a preclinical model (Adusumilli et al., 2014) and in patients who received mesothelin-targeted CAR T cell therapy in a phase I clinical trial (Adusumilli et al., 2021). The possibility that CAR T cell therapy can recruit endogenous immunity is particularly attractive since solid tumors are frequently antigen heterogeneous and recruitment of polyclonal immunity may limit outgrowth of antigen-negative variants, thereby preventing antigen escape.
Early-stage clinical trials of regional CAR T cell therapies
Regional CAR T cell therapies are under investigation in the clinic (Table 1). A group in City of Hope treated patients with recurrent glioblastoma with IL13Rα2-targeted CAR T cells. In an initial report of 12 patients, recurrent tumors were resected followed by infusion of first-generation (CD3ζ signaling only) CAR T cells into the resection cavity via indwelling catheter. The investigators observed necrosis of recurrent glioblastoma after regional CAR T cell retreatment and regrowth of antigen-negative variants, demonstrating both a signal of efficacy and resistance to antigen immune escape, overcoming a key obstacle to the successful treatment of solid tumors. To enhance distribution and persistence, they switched to intraventricular infusion of central memory 4–1BB–costimulated CAR T cells. A durable complete clinical response was achieved in a patient with multifocal recurrent glioblastoma with intracranial and spinal metastases (Brown et al., 2016). The absence of cytokines and circulating CAR T cells despite ten intraventricular administrations suggests either a lack of extravasation beyond the blood-brain barrier, inadequate T cell persistence (which might explain the eventual recurrence), or regrowth of antigen-negative variants.
Table 1.
Clinical studies of regional delivery of CAR T Cells
| Cancer | Target (Costimulation) | Delivery | Sample Size | NCT # | Phase/Site First Author, Publication, Year Study Features/Notable Findings |
|---|---|---|---|---|---|
| Breast | c-Met (4–1BB) | Intratumoral | 6 | NCT01837602 | Phase 0/University of Pennsylvania |
| CNS (glioblastoma, pediatric diffuse pontine glioma, diffuse midline glioma) | B7-H3 | Intracranial Intraventricular |
— | NCT04185038 | Phase I/Seattle Children’s Hospital |
| EGFR806 | Intracranial Intraventricular |
— | NCT03638167 | Phase I/Seattle Children’s Hospital | |
| EGFRvIII | Intracerebral | — | NCT03283631 | Phase I/National Cancer Institute, Duke University | |
| HER2 | Intracranial Intraventricular |
3 | NCT03500991 | Phase I/Seattle Children’s Hospital | |
| HER2 | Intracranial | — | NCT02442297 | Phase I/Baylor College | |
| HER2 | Intraventricular | — | NCT03696030 | Phase I/National Cancer Institute | |
| IL13Rα2 | Intracranial | 3 | NCT00730613 | Phase I/City of Hope | |
| IL13Rα2 (4–1BB) | Intracavitary Intratumoral Intraventricular |
1 | NCT02208362 | Phase I/City of Hope | |
| IL13Rα2 (CD28) | Intratumoral | 6 | NCT01082926 | Phase I/City of Hope | |
| Cavitary (ovarian, fallopian tube, primary peritoneal, peritoneal mesothelioma, peritoneal metastases) | CEA | Intraperitoneal | — | NCT03682744 | Phase I/Rutgers, Roger Williams Medical Center |
| Folate receptor–alpha | Intraperitoneal | — | NCT03585764 | Phase I/University of Pennsylvania | |
| Mesothelin | Intraperitoneal | 11 | NCT03608618 | Phase I/National Cancer Institute | |
| Muc16 (CD28/IL-12 secreting) | Intraperitoneal Intravenous |
18 | NCT02498912 | Phase I/Memorial Sloan Kettering | |
| Head and neck Squamous | ErbB | Intratumoral | NCT01818323 | Phase I/King’s College London Guy’s and St Thomas |
|
| Liver metastases (from colorectal, pancreatic) | CEA (CD28) | Intrahepatic | 6 | NCT01373047 | Phase I/Roger Williams Medical Center |
| CEA (CD28) | Intrahepatic | 8 | NCT02416466 | Phase I/Roger Williams Medical Center | |
| CEA (CD28) | Intrahepatic | 1 | NCT02850536 | Phase I/Roger Williams Medical Center | |
| Tag-72 | Intrahepatic Intravenous |
6 | — | Phase I/University of California, San Francisco, Stanford Mary Crowley | |
| Mesothelin-expressing cancers (lung adenocarcinoma, ovarian, fallopian tube, pleural and peritoneum mesothelioma) |
Mesothelin | Intrapleural Intravenous |
— | NCT03054298 | Phase I/University of Pennsylvania |
| Thoracic (lung, pleural disease, mesothelioma) | FAP | Intrapleural | 3 | NCT01722149 | Phase I/University of Zurich |
| Mesothelin (CD28) | Intrapleural | 41 | NCT02414269 | Phase I/II/Memorial Sloan Kettering | |
| Mesothelin (1XX/PD1DNR) | Intrapleural | — | NCT04577326 | Phase I/Memorial Sloan Kettering Cancer |
We searched PubMed and the National Clinical Trials website for clinical trials related to the following search terms: “CAR T cells regional delivery,” “CAR T cells locoregional delivery,” “intracerebral,” “intrahepatic,” “intrapleural,” “intratumoral,” and “intraventricular.”
Katz et al. reported their phase I investigations of regional intrahepatic CAR T cells for malignancies metastatic to the liver, including in a patient with carcinoembryonic antigen–expressing pancreatic adenocarcinoma who had a complete metabolic response after intra-arterial infusions of CAR T cells (Katz et al., 2015). Maher et al. developed a gene-modified cellular therapy called T4 immunotherapy in which autologous T cells are engineered to undergo selective ex vivo expansion using IL-4. An ongoing phase I study investigating intratumoral administration of these cells in patients with head and neck cancer has demonstrated disease control (stable disease) in a majority of patients who had rapidly progressing tumors before treatment (Papa et al., 2018).
Supporting potential T cell persistence, circulating CAR T cells longer than 100 days were observed after a single infusion intrapleural mesothelin-targeted CAR T cells (Adusumilli et al., 2014),up to 12 weeks after Muc16-targeted intraperitoneal delivery for ovarian cancers and CAR T cells in the blood 48 weeks after hepatic intra-arterial infusion of Tag-72-targeted CAR T cells for colorectal cancer liver metastases (Hege et al., 2017).
Safety considerations
Preclinical models of regional administration have demonstrated an absence or lower levels of cytokines in the peripheral blood, suggesting that regional administration may limit the systemic release of cytokines and reduce the incidence of cytokine-mediated organ damage. Brown et al. also observed an absence of cytokines in the peripheral blood in a patient with glioblastoma treated with intraventricular regional therapy (Brown et al., 2016). In an intrapleural trial, the number of CAR T cells in the pleural fluid was associated with peak levels of cytokines in the pleural fluid at week 2, whereas concentrations of cytokines in the peripheral blood were many fold lower (Adusumilli et al., 2021). Locoregional delivery of HER2-specific CAR T cells exhibited clinical as well as correlative laboratory evidence of local CNS immune activation, including high concentrations of CXCL10 and CCL2 in the cerebrospinal fluid (Vitanza et al., 2021). Regional administration can be combined effectively with systemic administration. A second dose of GD2-targeted CAR T cells was administered by regional intracerebroventricular delivery following initial systemic administration and showed radiographic or clinical benefit. Regional administration was associated with less systemic toxicity, such as cytokine release syndrome, and with enhanced proinflammatory cytokines and reduced immunosuppressive cell populations in the cerebrospinal fluid (Majzner et al., 2022).
Regional administration offers a way to circumvent normal tissue, on-target, off-tumor toxicity. Although regional CAR T cells are eventually exposed to normal tissues, they may be present at lower numbers in normal tissues than intravenously administered T cells. The absence of toxicity to normal tissues which express a much lower level of antigen, compared with tumor—suggests that targeting shared cancer-associated antigens in solid tumors is safe as long as a single-chain fragment variable with an affinity below the threshold for normal levels of antigen expression is used. Since cytokine-related toxicities and on-target, off-tumor adverse events result from or are tightly linked to the antitumor function of CAR T cells, conclusions of safety without efficacy may be misleading, indicating ineffective therapeutic targeting and insufficient T cell persistence.
Combining regional therapy with immune checkpoint inhibitors
Whereas regional therapy–induced immediate antigen activation, dose expansion, and trafficking of CAR T cells are beneficial, both CAR and endogenous T cells can become exhausted in the presence of the overwhelming tumor burden with predominant immunosuppression that is typical in patients with advanced solid tumors. The administration of immune checkpoint inhibitors after regional CAR T cell therapy has the potential to rescue exhausted CAR, endogenous, and neoantigen-specific T cells. Results from preclinical studies supported the translation of a combination of regional CAR therapy and PD-1 checkpoint blockade in a phase I clinical trial, which achieved partial responses by mRECIST criteria, including two complete metabolic responses on PET/CT (Adusumilli et al., 2021). That close to 40% of patients harbored detectable CAR T cells in the peripheral circulation at longer than 100 days and up to 6 months after a single dose underscores the potential to achieve long-lasting systemic antitumor immunity. Gene-engineered PD-1 checkpoint blockade—such as the PD-1 dominant negative receptor being tested in an ongoing clinical trial (Cherkassky et al., 2016)—may decrease systemic adverse events and reliance on repeated antibody administration. Preclinical studies with CRISPR-Cas9 disruption of PD-1 in universal EGFRvIII CAR T cells prolonged the survival of mice bearing intracranial tumors after intracerebral administration but not after intravenous administration (Choi et al., 2019).
Limitations for regional CAR T cell therapy
While image-guided intervention radiological approaches facilitate CAR T cell therapy to pleural, peritoneal or intracranial cavities, regional delivery to solid organ metastases is restricted by safe access and the volume of infusion required, typically 30–120 ml. In patients with multiple sites of metastases, selection of target site of administration solely based on ease of access may not be appropriate; repeat administration is not practical unlike in systemic delivery. Intraperitoneal administration of biological agents in patients with advanced peritoneal carcinomatosis can result in bowel perforation or obstruction, the risk of which may be exaggerated by CAR T cell-induced regional cytokine storm. Patient and target site selection limits the potential of regional therapy as a universal approach.
Concluding remarks
The clinical utility of regional CAR therapy is being increasingly realized, as accessible sites for administration, made possible by improvements in interventional radiologic delivery methods, are paired with appropriate antigen targeting. The site of regional administration serves as a charging and distribution center for CAR T cells so that they can overcome solid tumor obstacles for effective control of primary tumor and establish circulating immunity. The establishment of circulating T cell memory is the key to realizing the full potential of CAR T cell therapy: durable clinical responses and long-term survival.
Acknowledgments
We thank Summer Koop and David Sewell for editorial assistance and Waseem Cheema and Amy Zhu for assistance in compiling figure and table. P.S.A.’s laboratory work is supported by grants from the National Institutes of Health (P30 CA008748, R01 CA236615-01, and R01 CA235667), the U.S. Department of Defense (BC132124, LC160212, CA170630, CA180889, and CA200437), the Batishwa Fellowship, the Comedy vs Cancer Award, the DallePezze Foundation, the Derfner Foundation, the Esophageal Cancer Education Fund, the Geoffrey Beene Foundation, the Memorial Sloan Kettering Technology Development Fund, the Miner Fund for Mesothelioma Research, the Mr. William H. Goodwin and Alice Goodwin, the Commonwealth Foundation for Cancer Research, and the Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center. P.S.A.’s laboratory receives research support from ATARA Biotherapeutics.
Declaration of Interests
P.S.A. declares research funding from ATARA Biotherapeutics; Scientific Advisory Board Member and Consultant for ATARA Biotherapeutics, Bayer, Carisma Therapeutics, Imugene, ImmPactBio, Takeda Therapeutics; Patents, royalties and intellectual property on mesothelin-targeted CAR and other T cell therapies, which have been licensed to ATARA Biotherapeutics, issued patent method for detection of cancer cells using virus, and pending patent applications on PD-1 dominant negative receptor, wireless pulse-oximetry device, and on an ex vivo malignant pleural effusion culture system. All other authors do not have competing interests to disclose.
Memorial Sloan Kettering Cancer Center (MSK) has licensed intellectual property related to mesothelin-targeted CARs and T cell therapies to ATARA Biotherapeutics, and has associated financial interests.
References
- Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR, and Sadelain M (2014). Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med 6, 261ra151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adusumilli PS, Zauderer MG, Riviere I, Solomon SB, Rusch VW, O’Cearbhaill RE, Zhu A, Cheema W, Chintala NK, Halton E, et al. (2021). A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with malignant pleural disease, in combination with the anti-PD-1 agent pembrolizumab. Cancer Discov 11, 2748–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, et al. (2016). Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 375, 2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, and Adusumilli PS (2016). Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 126, 3130–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BD, Yu X, Castano AP, Darr H, Henderson DB, Bouffard AA, Larson RC, Scarfo I, Bailey SR, Gerhard GM, et al. (2019). CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J Immunother Cancer 7, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, Lin AA, Schlom J, June CH, and Sherwin SA (2017). Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, and Sadelain M (2018). Chimeric antigen receptor therapy. N Engl J Med 379, 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, Khare PD, Thorn M, Ma Q, Stainken BF, et al. (2015). Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res 21, 3149–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher J, Brentjens RJ, Gunset G, Riviere I, and Sadelain M (2002). Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. NatBiotechnol 20, 70–75. [DOI] [PubMed] [Google Scholar]
- Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, Richards RM, Jiang L, Barsan V, Mancusi R, et al. (2022). GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature [Online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulazzani M, Fräßle SP, von Mücke-Heim I, Langer S, Zhou X, Ishikawa-Ankerhold H, Leube J, Zhang W, Dötsch S, Svec M, et al. (2019). Long-term in vivo microscopy of CAR T cell dynamics during eradication of CNS lymphoma in mice. Proc Natl Acad Sci U S A 116, 24275–24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa S, Adami A, Metoudi M, Achkova D, van Schalkwyk M, Pereira AP, Bosshard-Carter L, Whilding L, van der Stegen S, Davies D, et al. (2018). A phase I trial of T4 CAR T-cell immunotherapy in head and neck squamous cancer (HNSCC). J Clin Oncol 36, 3046. [Google Scholar]
- Pituch KC, Miska J, Krenciute G, Panek WK, Li G, Rodriguez-Cruz T, Wu M, Han Y, Lesniak MS, Gottschalk S, and Balyasnikova IV (2018). Adoptive transfer of IL13Ralpha2-specific chimeric antigen receptor T cells creates a pro-inflammatory environment in glioblastoma. Mol Ther 26, 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, Labanieh L, Dhingra S, Leruste A, Majzner RG, et al. (2020). Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med 26, 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitanza NA, Johnson AJ, Wilson AL, Brown C, Yokoyama JK, Kunkele A, Chang CA, Rawlings-Rhea S, Huang W, Seidel K, et al. (2021). Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: an interim analysis. Nat Med 27, 1544–1552. [DOI] [PubMed] [Google Scholar]