

Abstract
VP64 is the smallest transactivation domain that can be packaged together with the sgRNA into a single adeno-associated virus (AAV) vector. However, VP64-based CRISPRa often exerts modest activation to the target gene when only one sgRNA is used. Herein, we used PAM-flexible dual base editor-mediated mutagenesis and self-activation strategies to derive VP64 variants with gain-of-function mutations. First, we generated an HEK293FT transgenic clone to stably expressing pTK-CRISPRa-GFP. The sgRNA of CRISPRa was designed to target the TK promoter, thereby allowing self-activation of CRISPRa-GFP. Base editors were then used to randomly mutagenesis VP64 in this transgenic cell. VP64 with enhanced potency would translate into increment of GFP fluorescence intensity, thereby allowing positive selection of the desired VP64 mutants. This strategy has enabled us to identify several VP64 variants that are more potent than the wild-type VP64. ΔCRISPRa derived from these VP64 variants also efficiently activated the endogenous promoter of anti-aging and longevity genes (KLOTHO, SIRT6, and NFE2L2) in human cells. Since the overall size of these ΔCRISPRa transgenes is not increased, it remains feasible for all-in-one AAV applications. The strategies described here can facilitate high-throughput screening of the desired protein variants and adapted to evolve any other effector domains.
Keywords: adeno-associated virus, base editing, dual base editor, nCas9-NG, positive feedback, SpRY, VP16, VP64
Graphical abstract
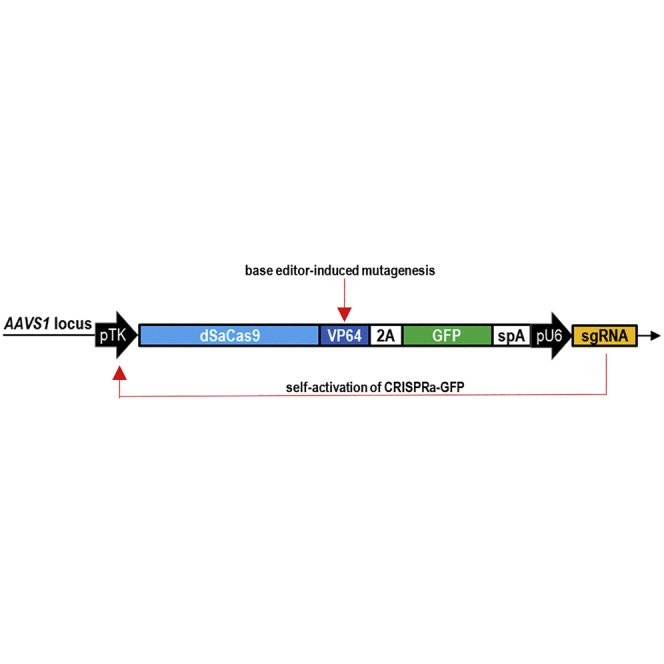
To enable the use of an all-in-one AAV vector platform, we enhanced VP64 potency and CRISPRa function through the use of PAM-flexible dual base editor-mediated random mutagenesis and self-activation strategies. These strategies can be potentially adapted to evolve any other effector domains and CRISPR components.
Introduction
CRISPR activator (CRISPRa) offers several advantages over nuclease-based genome editing, such as restoring normal gene expression and correct gene misregulation without altering the underlying DNA sequence and inducing DNA damage.1,2 Moreover, CRISPRa allows physiological or near-physiological expression of target endogenous genes by modulating gene regulatory elements (e.g., promoter and enhancer).3 Importantly, CRISPRa can upregulate the most relevant splice variants of the targeted gene even when the function of this splice variant is unknown. In contrast, the physiologically relevant splice variants could be missed if cloning expression cassette from the cDNA for ectopic overexpression.3 Similarly, overexpressing a certain splice variant from the open reading frame may result in missing other physiologically relevant splice variants. Protein overexpression can also be harmful to the cell since the production of the protein amount is uncontrollable and can upset the balance in protein complexes. Ectopic overexpression often uses strong exogenous promoters that can result in supraphysiological expression levels. It is also a great challenge to clone multiple genes, a long gene, a highly repetitive sequence, or GC-rich genes into the viral vector for overexpression.
Adeno-associated virus (AAV) has become an increasing popular viral vector for therapeutic gene transfer in vivo. Besides being non-pathogenic to humans, AAV elicits a very mild immune response in vivo, and its genetic material is rarely integrated into the host genome.4 Also, AAV is attractive for in vivo applications owing to its high efficacy of viral delivery and transduction.5 Apart from dividing cells, AAV is capable of infecting non-dividing cells, such as postmitotic neurons in the central nervous system and somatic tissues in the adult animals. AAV provides long-term transgene expression in these quiescent cells due to its ability to replicate epichromosomally by forming stable concatemers. However, owing to the limited packaging capacity of AAV, careful design is required to package an all-in-one CRISPRa for in vivo target gene activation.
We have recently developed an all-in-one CRISPRa that can be packaged into a single AAV vector for targeted gene activation in vitro and in vivo.6 In addition, this CRISPRa could be efficiently delivered by our newly engineered AAV1-PHP.B variant for targeted transgene activation in the mouse brain via systemic administration. In our design, CRISPRa is a fusion of catalytically inactive SaCas9 (dSaCas9)7 and VP64 transcriptional activator domain,8, 9, 10 and linked to truncated regulatory elements. Owing to the limited packaging capacity of AAV, only one single guide RNA (sgRNA) can be packaged together with the dSaCas9-VP64 into an AAV vector. Despite this CRISPRa vector being minimal in size, its transgene expression and functionality were not compromised. On the other hand, AAV1-PHP.B variant was engineered by inserting a 7-mer PHP.B peptide on an AAV1 capsid.6 PHP.B peptide was previously identified through an in vivo AAV9 capsid library screen in adult mice.11 PHP.B peptide insertion to the AAV9 capsid significantly enhanced the blood-brain-barrier crossing ability and broadened biodistribution and transduction of AAV9 in the adult mouse central nervous system. Thereafter, these enhancement properties of AAV9-PHP.B were similarly reported in many other studies.12, 13, 14 As seen for AAV9, we demonstrated that this enhanced blood-brain-barrier crossing of PHP.B modification could be extended to AAV1. Our AAV1-PHP.B can efficiently deliver this CRISPRa to cross the blood-brain barrier and be taken up by the brain tissue upon lateral tail vein injection in mice.6,11
Despite the attractiveness of these engineered CRISPRa and AAV tools, our existing AAV-CRISPRa system requires co-delivery of several different AAV vectors to simultaneous express multiple sgRNAs for effective endogenous gene activation. This is because only one sgRNA can be packaged together with the dSaCas9-VP64 into this all-in-one CRISPRa construct. A single AAV vector platform is desired because it ensures co-delivery of both dSaCas9-VP64 and sgRNA into each cell. However, VP64-based CRISPRa often exerts modest activation to the target gene when only one sgRNA is employed. Although a number of recently reported activator domains (e.g., VPR,15 TET1,16 P300,17 and PRDM918) are more potent than the VP64 activator, their large size has ruled out the possibility of co-packaging them with dSaCas9 and its sgRNA into a single AAV vector. Since VP64 is the only transactivation domain that can be selected to construct CRISPRa for application of a single AAV vector platform, here, we attempted to use PAM-flexible dual mutations base editor-mediated mutagenesis and self-activation strategies for enhancing VP64 potency.
To derive VP64 variants with gain-of-function mutations, we first generated a HEK293FT clone to stably express pTK-CRISPRa-GFP via CRISPR/Cas9-mediated homologous recombination. The sgRNA of CRISPRa was designed to target the TK promoter, thereby allowing self-activation of CRISPRa-GFP. Diverse VP64 mutants were then generated by random mutagenesis of the VP64 in this transgenic cell using different PAM-flexible dual base editors with multiple sgRNAs. Next, we screened and identified the VP64 variants with gain-of-function mutations by constructing various pCMV-ΔCRISPRa to perform luciferase reporter assay and targeting endogenous genes in vitro. Finally, ΔCRISPRa with the most potent VP64 mutant was packaged into the AAV1-PHP.B vector for activating target endogenous genes in vitro. The strategies described here can minimize complexity and facilitate high-throughput screening of the desired protein variants. Thus, it can be potentially adapted to evolve any other effector domains and CRISPR components.
Results
Generation of an HEK293FT transgenic clone to stably express pTK-CRISPRa-GFP
To generate a transgenic clone stably expressing pTK-CRISPRa-GFP, we co-transfected CRISPR-Cas9 targeting the AAVS1 site and donor pTK-dSaCas9-VP64-2A-GFP-pU6-sgRNA harboring two flanking AAVS1 homology arms into the HEK293FT cells (Figure 1A). Upon CRISPR-Cas9-induced DNA double-strand breaks, the donor vector was integrated into the AAVS1 locus via homologous recombination. After 1 week of co-transfection, the GFP-expressing cells were sorted and expanded on a 96-well plate. Genomic DNA from each single-cell derived clone was then extracted for PCR genotyping. Three pairs of primer were used for PCR genotyping (Figure 1B). Primer pair 3 (P3) was used to confirm site-specific integration of a donor pTK-CRISPRa-GFP at the AAVS1 locus, while the other two primer pairs (P1 and P2) were used to ensure insertion of full-length pTK-CRISPRa-GFP. PCR genotyping with these three pairs of primer confirmed targeted integration of the full-length pTK-CRISPRa-GFP sequences at the genomic AAVS1 locus in HEK293FT C2.8 clone.
Figure 1.

Generation of a HEK293FT transgenic clone stably expressing pTK-CRISPRa-GFP
(A) Targeted integration of a donor pTK-CRISPRa-GFP into the genomic AAVS1 locus via CRISPR-Cas9-mediated homologous recombination. (B) Confirming site-specific integration of the full-length pTK-CRISPRa-GFP sequences at the genomic AAVS1 locus by PCR genotyping. AAVS1, adeno-associated virus integration site 1; GFP, green fluorescent protein; pTK; thymidine kinase promoter.
Enhanced CRISPRa function by VP64 mutagenesis
We performed VP64 mutagenesis in pTK-CRISPRa-GFP expressing HEK293FT C2.8 clone using PAM-flexible (SpRY19 and nCas9-NG20) and dual base editors (A&C-BEmax-nCas921 and TadA-nCas9-NG-CDA120), as well as dCas9-based base editors (ABE7.10,22 BE3,22 ABEmax,23 AncBE4max,23 BE4max,23 and AID24). Base editor was used because it enables clean mutational knockin by direct conversion of nucleotides in genomic DNA without inducing double-strand DNA breaks and generating indel mutations.22,25 Adenine base editor (e.g., ABE7.1022 and ABEmax23) mediates the direct conversion of adenine to inosine, which eventually leads to an A-to-G (or T-to-C) substitution. Meanwhile, cytidine base editor (e.g., BE3,22 AncBE4max,23 and BE4max23) mediates the direct conversion of cytidine to uridine, which eventually leads to a C-to-T (or G-to-A) substitution. AID directs somatic hypermutation to a given genomic locus by deamination of cytosine to uracil, which eventually converts cytidines or guanines to any other three bases at the desired loci.24
However, if using dCas9-based base editors, only four target sites with adjacent PAM sequences can be designed on VP64 transgene due to the PAM sequence constraints. To enable exhaustive discovery of VP64 with gain-of-function mutations, we further employed PAM-flexible dual base editors for maximizing the diversity of VP64 mutants generated. SpRY19 and nCas9-NG20 have more relaxed PAM requirements than the SpCas9. The PAM requirement of nCas9-NG is NG, while SpRY is a near-PAMless SpCas9 variant that can target NRN and to a lesser extent NYN PAMs. Using these PAM-flexible base editors, we were able to design 22 sgRNAs to target every sequence on the VP64 transgene (Figure 2A). To expand the range of DNA alterations, dual base editors were used to allow concurrent adenine and cytosine editing.20,21
Figure 2.

Enhanced CRISPRa function by VP64 mutagenesis
(A) sgRNA target sites of base editor on VP64 transgene. Arrows indicate the sense or antisense orientation of sgRNAs designed to recognize target DNA sequences. (B) Sorting of VP64 mutagenesis cells. Multiple sgRNAs and various base editors were used to target and for mutagenesis of the VP64 transgene in the HEK293FT C2.8 clone. The brightest GFP-positive cells in the gate region are sorted for single-cell cloning, followed by PCR genotyping and DNA sequencing. Density plots are shown, with the highest density cells in the red areas. GFP, green fluorescent protein; SSC, side scatter.
One week after the transfection of base editors into this HEK293FT C2.8 clone, the brightest GFP-positive cells were sorted for single-cell cloning (Figure 2B). We speculate that the introduction of gain-of-function mutations to the VP64 transgene can improve the activation potency of VP64, thereby improving CRISPRa function and increasing GFP expression. As expected, only a very small portion of cells displayed higher GFP expression than the original HEK293FT C2.8 clone upon random mutagenesis of the VP64 transgene. These brightest GFP-expressing cells were then isolated for culturing and expanding. A large portion of cells has reduced GFP expression owing to the introduction of deleterious mutations to the VP64 transgene. In this case, CRISPRa no longer can self-activate its promoter to express the GFP. To support this notion, we added additional control by designing the CRISPR-Cas9 to knock out the sgRNA of this CRISPRa in the HEK293FT C2.8 original clone. Upon knockout, CRISPRa no longer can activate the TK promoter in this clone, thereby decreasing the GFP fluorescent intensity. Thus, a fluorescent density plot similar to VP64 mutagenesis is observed. This knockout experiment strongly proved that this single sgRNA was enough to enable CRISPRa to activate its TK promoter, thereby confirming our self-activation strategy is functional.
After single-cell sorting and expansion, we successfully derived 86 clones with GFP expression higher than the original HEK293FT C2.8 clone. We then performed PCR to amplify the VP64 sequence from the genomic DNA of each clone for sequencing the full-length VP64 transgene. From these 86 clones, we identified 19 variants of VP64 (Figures 3 and S1). Apart from substitution mutations, designing guide sequences to target multiple sites on VP64 transgene would lead to the deletion of the intervening segment between two sgRNA target sites. To minimize this deletion issue, we also designed multiple sgRNAs of SpRY or nCas9-NG that are targeted only on one of the DNA strands (sense or antisense).
Figure 3.

VP64 variants identification. Amino acid alignments of wild-type and mutant VP64
Blue highlighted red letters indicate the substitution mutations on VP64 sequence, while red dashed lines indicate the deleted sequences on VP64. Green highlighted amino acids indicate four copies of VP16 in wild-type VP64, while yellow highlighted green letters represent the most critical amino acid (Phe442) in VP16. The full length of VP64 transgene is 165 base pairs (55 amino acids).
Functional validation of the VP64 variants
All the newly identified VP64 mutations were functionally validated by performing luciferase reporter assays. In this episomal reporter assay, AAV-compatible CRISPRa (CMV-dSaCas9-ΔVP64) derived from different VP64 variants was constructed. The size of the CRISPRa is within the AAV packaging capacity (<4.7 kb). In this assay, each of this ΔCRISPRa plasmid was co-transfected with the pTK-Ren luciferase plasmid into the wild-type HEK293FT cells. The guide sequence of ΔCRISPRa was designed to target the TK promoter that drives the expression of the Renilla luciferase gene. To identify the most efficient sgRNA for CRISPRa, we designed eight sgRNAs to target the TK promoter (Figure 4A). From these eight sgRNAs, sgRNA-7 exerted the strongest Renilla luciferase activity. Therefore, sgRNA-7 was selected for constructing ΔCRISPRa plasmids and testing in subsequent luciferase assay (Figure 4B).
Figure 4.

Functionality screening of VP64 variants
(A) Identification of the most potent sgRNA target site on TK promoter for constructing CMV-dSaCas9-ΔVP64. A 750-base pair TK promoter was cloned at the upstream of the chimeric intron and Renilla luciferase gene (Rluc). Arrows indicate the sense or antisense orientation of sgRNAs designed to recognize target DNA sequences. (B) Renilla luciferase activity with CRISPRa bearing different VP64 mutants. pAAV-CMV-dSaCas9-ΔVP64 (CRISPRa) transgene targeting the TK promoter was co-transfected with pTK-Rluc vector into HEK293FT cells. Two independent experiments with four replicates were carried out. The statistical significance levels from VP64-WT are indicated as ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. All data are presented as mean ± SD. ITR, inverted terminal repeats; NLS, nuclear localization signal; pTK; thymidine kinase promoter; Rluc, Renilla luciferase.
Two days after the plasmid transfections, the luciferase activities induced by the ΔCRISPRa were quantified and compared. Compared with CRISPRa with the wild-type VP64, our luciferase reporter assays revealed a higher luciferase activity induced by CRISPRa bearing VP64 variants such as A2, S4, M10, CA8.15, and CA10.11 (Figure 4B). Among these VP64 variants, CA10.11, A2, and S4 consistently showed the highest luciferase activity, therefore, these three VP64 variants were selected for activating endogenous genes and packaging into the AAV vector.
In the subsequent validation, pCMV-ΔCRISPRa derived from CA10.11, A2, and S4 were used to target and activate the promoter of target endogenous genes (KLOTHO, SIRT6, and NFE2L2) in human HEK293FT cells. We also tested ΔCRISPRa bearing both A2 and CA10.11 mutations. These endogenous genes were targeted for several reasons. Previous studies have shown that the promoter of KLOTHO gene could be robustly activated by CRISPRa (dSpCas9-VP64) in HEK293T cells.26 On the other hand, SIRT6 and NFE2L2 genes were targeted because our qPCR data confirmed that these two genes are expressed in HEK293FT cells. KLOTHO is a well-known longevity gene and it encodes membrane-bound and secreted forms of Klotho protein. The secreted form (circulating hormone) displays an important role in anti-aging and longevity.27,28 Overexpression of Klotho gene could extend life span in mice by inhibiting insulin and insulin-like growth factor 1 signaling, while a defect in Klotho gene expression in mice would lead to aging and age-related phenotypes.27 On the other hand, SIRT6 encodes Sirtuin 6 that functions in DNA repair and telomere maintenance.29 Knockout of Sirt6 shortens lifespan and causes premature aging,30,31 while overexpression of Sirt6 could prolong the life span in male mice.32 Meanwhile, NFE2L2 encodes the Nrf2 transcription factor that can regulate the expression of a large number of cytoprotective genes to defend against oxidative stress.33 In fact, many pharmacological drugs in pre-clinical testing have been developed to target and activate NFE2L2 for the treatment of various age-related human diseases.34,35 By designing sgRNAs to target three different sites on each endogenous promoter, we found that ΔCRISPRa particularly CA10.11 (120% Klotho and 38% Sirt6 mRNA levels higher than VP64-WT) and A2+CA10.11 (20% Nrf2 mRNA levels higher than VP64-WT) exerted stronger activation effects than the original CRISPRa (Figure 5). However, the degree of activation is gene-dependent.
Figure 5.

Activation of endogenous genes in HEK293FT cells by evolved CRISPRa
Targeted activation of endogenous human (A) KLOTHO, (B) SIRT6, and (C) NFE2L2 genes. Red arrows indicate the sense or antisense orientation of sgRNAs designed to recognize target DNA sequences on the promoter. Bottom panel shows the relative expression level of endogenous genes in HEK293FT cells. The statistical significance levels from VP64-WT are indicated as ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. All data are presented as mean ± SD (n = 3).
AAV delivery of ΔCRISPRa for targeted endogenous gene activation in vitro
Given the promising activation effects on two endogenous genes observed for ΔCRISPRa bearing CA10.11-, A2-, or S4-VP64, we packaged these ΔCRISPRa transgenes into the AAV1-PHP.B vector (Figure 6A). AAV1-PHP.B is a capsid-modified AAV serotype 1 (AAV1) that provided higher transduction efficiency than the wild-type AAV1 (Figure 6B). Similar to the results of plasmid transfection, through AAV delivery, these three ΔCRISPRa particularly the one bearing CA10.11-VP64 exerted stronger activation effects (20% Sirt6 mRNA level higher than the VP64-WT) to SIRT6 promoter than the original CRISPRa (Figure 6C).
Figure 6.

AAV delivery of an evolved CRISPRa for targeted gene activation in HEK293FT cells
(A) Packaging of CRISPRa (CMV-dSaCas9-ΔVP64) by AAV1-WT and AAV1-PHP.B. AAV1-PHP.B was generated by inserting a 7-mer PHP.B peptide sequence in between S588 and T589 of the AAV1 capsid. (B) Transduction efficiency of AAV1-WT and AAV1-PHP.B in HEK293FT cells. Flow cytometry analysis and GFP fluorescent images were taken 3 days after the AAV transduction. (C) qPCR of SIRT6 expressions upon targeted activation of this endogenous gene by AAV delivery of CRISPRa. Red arrows indicate the antisense orientation of sgRNAs designed to recognize target DNA sequences on the promoter. The statistical significance levels from VP64-WT are indicated as ∗p < 0.05. All data are presented as mean ± SD (n = 3).
Discussion
Here, we have successfully enhanced VP64 activation potency and improved overall CRISPRa function through PAM-flexible dual base editor-mediated genetic diversification of the VP64 at a genomic safe harbor, followed by self-activation of modified CRISPRa in the living cells. This integrated approach used CRISPR-SpCas9-mediated homologous recombination and CRISPR-dSpCas9-mediated base editing to derive CRISPR-dSaCas9 activator with an enhanced function. The PAM sequence requirements for dSpCas9 of base editor and dSaCas9 of CRISPRa are different, thereby avoiding competition between base editor and CRISPRa for sgRNA in the cell. The PAM-flexible dual base editor-mediated mutagenesis and self-activation strategies described here allow positive selection of GFP-expressing cells with the desired VP64 mutants for culturing and expanding.
We generated diverse VP64 mutants in the living cells by mutagenesis of the VP64 transgene using different CRISPR base editors with multiple sgRNAs. Compared with the structure-based rational design, our base editor-mediated mutagenesis and self-activation strategies allow selection of novel variants having greater enhancement effect without relying on protein structural and evolutionary features. Notably, our integrated approach enables feasible selection of the most promising traits from a large number of variants upon mutagenesis, thereby minimizing complexity and improving feasibility in high-throughput screening. Base editor is preferable to nuclease-based editing tools because it enables direct nucleotide substitutions without damaging the target DNA or requiring donor DNA.22,23,25,36 Therefore, it will not cause reading frameshift that can lead to the generation of truncated, non-functional VP64. Base editor also is more efficient than homology-directed oligonucleotide integration approaches.22,25 Base editor also has a broad editing window (4–5 nucleotides), thereby enabling introduction of diverse substitution mutations to the VP64.22,25 Therefore, base editor can create a diverse repertoire of point mutations in situ without the need to use error-prone PCR to create mutations in vitro or design a large number of DNA oligos for incorporating into a genomic locus.22,25
We have identified several VP64 variants, particularly CA10.11 and A2 that are more potent than the wild-type VP64. VP64 bears four VP16 core transactivation domains.37 Virion protein 16 (VP16) of herpes simplex virus type 1 was identified as a potent acidic transcriptional activator of the viral immediate-early genes.38 Both ionic and structure-specific hydrophobic interactions contribute to VP16 function. However, bulky hydrophobic residues, not acidic residues, are the most critical for VP16 activation activity.39 Multimerized VP16 activation domains could induce transcription synergistically in mammalian cells by increasing the stability of the transcription complex formation.37
The structural elements critical for VP16 function have been extensively studied.40, 41, 42, 43 A phenylalanine residue at position 442 of VP16 was exquisitely sensitive to substitution mutation, thus Phe442 is very critical for VP16 activation activity.40,42 Phe442 of VP16 makes a critical hydrophobic contact with transcription factor II D (TFIID) that makes up the RNA polymerase II preinitiation complex. It is also important for the structure of the VP16 activation domain. The substitution with the nonaromatic residues at Phe442 could completely abolish transcriptional activation by VP16. In fact, in our luciferase screening, none of the VP64 variant with mutation at this residue displayed higher activation activity than the wild-type VP64.
In our study, the substitution mutations of CA10.11 (Asp443Asn and Asp437His of VP16) and A2 (Asp445Asn of VP16) of VP64 were located in the vicinity of Phe442. For Asp443Asn and Asp445Asn, the acidic charge of aspartic acid (Asp) was replaced with the uncharged -NH2 group of asparagine (Asn). A previous study has shown that asparagine substitution to the aspartate residues proximal to the Phe442 conserved most of the VP16 activation activity, while glutamate and alanine were poor substitutes for aspartate.40 Although a subsequent study revealed aromatic amino acids such as tyrosine and tryptophan are the best substitutes for Phe442, the effects of Asp443Asn and Asp445Asn substitutions on VP16 activation activity have not been explored.43 In the case of VP64, asparagine substitution to the aspartate residues close to Phe442 of VP16 enhanced VP64 transcription activation potency, probably by affecting VP64 folding, the physical contact of the molecular target of VP64, or transcriptional synergism in multimerized VP16 activation domains. To support this notion, mutated at Phe442 alone was shown insufficient to affect transcriptional synergism of VP16, and other residues in the vicinity of Phe442 are more important for synergistic effects of VP16.44
Compared with Asp443Asn and Asp445Asn, Asp437His is located slightly farther away from the Phe442 residue. Therefore, substitution of basic charged histidine to the aspartate residue in CA10.11 VP64 may not affect VP64 function as much as Asp443Asn. Moreover, net negative charge is not the sole determinant of VP16 activation activity, but also the domain structure and surface interactions with target proteins.43
Although our ΔCRISPRa is more potent than the original CRISPRa, it exerted a modest activation to the target endogenous genes in the presence of one sgRNA. To derive more potent AAV-compatible CRISPRa, smaller Cas proteins (e.g., CRISPR-CasPhi45) or truncated small dCas9 orthologues (e.g., mini-dSaCas946) can be used alongside with other larger effector domains. Apart from small VP64 transactivation domain, mutagenesis of a large dSaCas9 can give more possibility to greatly improve CRISPRa potency and minimize the overall size of CRISPRa for efficient AAV packaging. To further reduce the payload size, truncated effector domains such as truncated VPR transactivation domain can be used.46,47 For example, a recent study has successfully packaged a compact and robust miniCas9 activator (dCjCas9-VPR) into an all-in-one AAV vector for in vivo target gene activation.48 Alternatively, to enable synergistic transcription activation with a single AAV vector platform, self-cleaving ribozyme or tRNA can be used to replace U6 promoter for driving multiple sgRNA expressions.49
Instead of using AAV1-PHP.B vector, the use of AAV-PHP.eB50 or AAV.CAP-B1051 could further improve ΔCRISPRa performance, particularly in neuronal cells by maximizing the cellular transduction efficiency. AAV-PHP.eB is an enhanced variant of AAV-PHP.B, which provides higher blood-brain-barrier crossing ability and transduction efficiency of neurons throughout the adult mouse brain.50 Therefore, it can lower the systemic dose and viral load required to transduce the central nervous system. AAV-PHP.eB (DGTLAVPFK) was evolved from the AAV-PHP.B (TLAVPFK) by screening an AAV9-based 7-mer library in GFAP-Cre mice. On the other hand, AAV.CAP-B10 was further evolved from AAV-PHP.eB to provide higher specificity for neurons in the central nervous system, while decreasing liver targeting.51
Materials and methods
Cell line cultures
Human HEK293FT cells were cultured in Dulbecco’s modified Eagle’s medium (high glucose) (Gibco 11,965,092) with 10% fetal bovine serum (Gibco 10,270,106). The cells were cultured under a standard cell culture condition (37°C, 5% CO2) in a humidified incubator.
Donor vector construction (pTK-dSaCas9-VP64-2A-GFP-pU6-sgRNA)
First, we inserted a 22-bp guide sequence targeting TK promoter to the sgRNA of our previously constructed pMecp2-dSaCas9-VP64-pU6-sgRNA6 using BsaI. Next, we constructed pMecp2-dSaCas9-VP64-2A-GFP-pU6-sgRNA by replacing VP64 of pMecp2-dSaCas9-VP64-pU6-sgRNA with VP64-2A-GFP using BamHI and EcoRI. The VP64-2A-GFP was amplified from dCAS9-VP64-GFP vector (Addgene 61,422). Then, we replaced pMecp2 with pTK to construct pTK-dSaCas9-VP64-2A-GFP-pU6-sgRNA using XbaI and AgeI. The pTK was amplified from pRL-TK vector (Promega E2241). Then, we inserted a left AAVS1 homology arm to this pTK-dSaCas9-VP64-2A-GFP-pU6-sgRNA using XhoI and XbaI. Finally, the right AAVS1 homology arm was inserted using NotI. The left and right AAVS1 homology arms were amplified from genomic DNA of HEK293FT cells. This donor transgene pTK-dSaCas9-VP64-2A-GFP-pU6-sgRNA harboring two flanking AAVS1 homology arms is abbreviated as pTK-CRISPRa-GFP. Primers used for vector construction are listed in Table S1.
Generation of a HEK293FT transgenic clone stably expressing pTK-CRISPRa-GFP
At 1 day prior to plasmid transfection, 1 × 105 HEK293FT cells were seeded into each well of a 6-well plate. Briefly, 1,250 ng of donor pTK-CRISPRa-GFP and 1,250 ng of CRISPR-Cas9 (Addgene 42,230) targeting the AAVS1 site were mixed with 5 μL of P3000 Reagent and 7.5 μL of Lipofectamine 3000 (Invitrogen) in 1 mL of Opti-MEM I Reduced Serum Medium (Gibco). The transfection mixture was added to the cells and incubated overnight. After 7 days, GFP-positive cells were single-cell sorted and expanded on a 96-well plate. The genomic DNA of these clones was then extracted for PCR genotyping. Primers used for AAVS1-directed knockin are listed in Table S2.
VP64 mutagenesis in the HEK293FT clone expressing pTK-CRISPRa-GFP
The dCas9-based base editors, including ABE7.1022 (Addgene 102,919), BE322 (Addgene 73,021), ABEmax23 (Addgene 112,095), AncBE4max23 (Addgene 112,094), BE4max23 (Addgene 112,093), and AID(P182X)24 (Addgene 83,260) were used for mutagenesis of VP64 transgene in the HEK293FT C2.8 clone expressing pTK-CRISPRa-GFP. Using these dCas9-based base editors, only four sgRNAs (Addgene 53,188) of base editor could be designed to target four different sites on the VP64 transgene. To maximize the number of VP64 variants generated, we also used PAM-flexible (SpRY19 and nCas9-NG20) (Addgene 139,999, 140,003) and dual base editors (A&C-BEmax-nCas921 and TadA-nCas9-NG-CDA120) (Addgene 140,244, 157,947) for VP64 mutagenesis. At 1 day prior to plasmid transfection, 1 × 105 of HEK293FT cells expressing pTK-CRISPRa-GFP were seeded into each well of a 24-well plate. Briefly, 250 ng of base editors and 250 ng of sgRNAs were mixed with 1 μL of P3000 Reagent and 1.5 μL of Lipofectamine 3000 (Invitrogen) in 200 μL of Opti-MEM I Reduced Serum Medium (Gibco). For co-transfection of multiple base editors or multiple sgRNAs, an equal amount of each plasmid vector was added. After 7 days, the brightest GFP-expressing cells were single-cell sorted and expanded on a 96-well plate. The clones with the GFP fluorescence intensity stronger than the original transgenic cell (before VP64 mutagenesis) were selected for genomic DNA extraction. PCR was then carried out to amplify the full-length VP64 sequence for DNA sequencing. Primers used for VP64 mutagenesis experiments are listed in Table S3.
Construction of CRISPRa variants (pAAV-CMV-dSaCas9-ΔVP64)
First, we replaced pMecp2 with pCMV to our previously constructed pAAV-pMecp2-dSaCas9-VP64(WT)-pU6-sgRNA using XbaI and AgeI. The pCMV was amplified from SP-dCas9-VPR vector (Addgene 63,798). Next, we replaced wild-type VP64 with mutated VP64 (ΔVP64) using BamHI and EcoRI. The ΔVP64 sequences were amplified from the genomic DNA of HEK293FT clones expressing pTK-CRISPRa-GFP-pU6-sgRNA. For construction of ΔCRISPRa bearing both ΔVP64 A2 and CA10.11, duplexed ultramer DNA oligonucleotides (IDT) consisting of the full-length VP64 sequence bearing these two mutations were custom synthesized. The successful ΔVP64 A2+CA10.11 replacement to ΔCRISPRa was verified using SphI and HindIII. Primers used for construction of CRISPRa variants and their guide sequences are listed in Tables S4 and S5.
Design of CRISPR guide sequences
To construct the guide sequence, a pair of annealed oligos was cloned into the sgRNA of CRISPR-Cas9 and base editors using BbsI. A guide sequence was cloned into the sgRNA of CRISPRa using BsaI. The CRISPR RGEN Tool, Cas-Designer (http://www.rgenome.net/cas-designer/), was used to identify the target sequence of sgRNAs. The first nucleotide of the transcribed gRNA was a guanine nucleotide (G) to maximize U6 promoter activity. For CRISPRa, BsaI and HindIII were used to screen the clones with target sequence insertion. Then the pU6-seq primer was used for DNA sequencing to confirm successful guide sequence insertion in the sgRNA.
Luciferase bioluminescence reporter assays
At 1 day prior to plasmid transfection, 1 × 104 HEK293FT cells were seeded into each well of a 96-well plate. Using Lipofectamine 3000 Transfection Reagent (Invitrogen), the pRL-TK (Promega E2241) (50 ng/well) and pAAV-CMV-dSaCas9-ΔVP64 (50 ng/well) plasmid vectors were co-transfected at a ratio of 1:1 into the 96-well plate. After 48 h of transfection, cells were lysed and quantified for the luciferase activity using the Renilla Luciferase Assay System (Promega E2810). The Renilla luminescence on a 96-well white polystyrene plate was measured with the SpectraMax M5e Microplate Reader (Molecular Devices). Two independent experiments with four replicates for each experimental group were performed.
Plasmid transfection of CRISPRa
At 1 day prior to plasmid transfection, 1 × 105 HEK293FT cells were seeded into each well of a 24-well plate. Briefly, 500 ng of pAAV-CMV-dSaCas9-ΔVP64 targeting KLOTHO or SIRT6 or NFE2L2 were mixed with 1 μL of P3000 Reagent and 1.5 μL of Lipofectamine 3000 (Invitrogen) in 200 μL of Opti-MEM I Reduced Serum Medium (Gibco). The transfection mixture was added to the cells and incubated overnight. After 2 days of plasmid transfection, cells were harvested for total RNA extraction.
Recombinant AAV vector production and tittering
At 1 day before plasmid transfection, ∼7 × 106 HEK293FT cells were seeded on each 100-mm dish. To generate recombinant AAV vectors, HEK293FT cells were transfected with transgene plasmid (pAAV-CMV-dSaCas9-ΔVP64 or pAAV-CMV-GFP), pAAV1 serotype vector (AAV1-WT or AAV1-PHP.B), and pHelper vector at a 1:1:1 ratio using Calcium Phosphate Transfection Kit (Invitrogen K278001), with some modifications. Briefly, 4 h prior to transfection, the culture media in each 100-mm dish were replaced with 12 mL of fresh media consisting of 1% heat-inactivated fetal bovine serum (Gibco 10,082,139). Then, 600 μL of transfection mixture that consisted of 36 μL of 2M CaCl2, 20 μg of DNA, 300 μL of 2x HEPES-buffered saline, and H2O was added to the cells. After 72 h of plasmid transfection, culture media from each 100-mm dish were collected into a 15-mL tube for AAV particle extraction. AAV particles from media were extracted using AAVanced Concentration Reagent (SBI AAV100A-1). Briefly, one volume of cold AAVanced Concentration Reagent was added to four volumes of AAV particle-containing media. The mixture was then incubated at 4°C in the refrigerator overnight. The next day, the mixture was spun at 1500 x g for 30 min, followed by pellet washing once and spinning again for 3 min. Finally, the pellet was resuspended in 50 μL using cold PBS. The concentrated virus was aliquoted for storing in −80°C freezer. Viral genomes were extracted and titered using TaqMan qPCR according to our recent published paper (Table S6).6
AAV transduction of CRISPRa
At 1 day prior to AAV transduction, 1 × 105 HEK293FT cells were seeded into each well of a 24-well plate. On the next day, the culture media in each well were replaced with 250 μL of 1% heat-inactivated fetal bovine serum (FBS) media. After 4 h of media replacement, AAV particles were added directly to the cells. After 24 h of AAV infection, the transduction mixture was removed and the cells were maintained in 1 mL of fresh culture media containing 1% heat-inactivated FBS (Gibco). For transductions of AAV-CMV-dSaCas9-ΔVP64, AAV particles were added at an MOI of 30,000 viral genomes/cell. After 3 days of AAV infection, cells transduced with AAV-CMV-dSaCas9-ΔVP64 were harvested for total RNA extraction. For cells transduced with AAV-CMV-GFP, GFP-expressing individual cells were analyzed with flow cytometry and imaged with fluorescent microscopy.
qPCR and melt curve analysis
Total RNA was extracted from the HEK293FT cells with the RNeasy Mini Kit (QIAGEN 74104) and treated with DNase I to digest genomic DNA (QIAGEN 79254). The concentration and purity of total RNAs were then determined with a Biochrom spectrophotometer. The total harvested RNA was reverse transcribed using SuperScript III First-Strand Synthesis System (Invitrogen 18,080,051), according to the manufacturer’s protocol.
For qPCR, the SsoAdvanced Universal SYBR Green Supermix (Bio-Rad 1,725,271) was used to amplify the synthesized cDNA. The mixture for each qPCR reaction was 10 μL of 2X SsoAdvanced Universal SYBR Green Supermix, 0.2 μL of 10 μM forward primer, 0.2 μL of 10 μM reverse primer, 2 μL of 2.5 ng/μL cDNA sample, and 7.6 μL of nuclease-free water. qPCR was done by using Thermal Cycling (Bio-Rad CFX96 Connect System). qPCR was carried out as follows: an initial denaturation step at 95°C for 3 min followed by 40 cycles at 95°C for 15 s and 55°C for 45 s (for Klotho mRNA), 58°C for 45 s (for Sirt6 mRNA), or 60°C for 45 s (for Nrf2 mRNA). The housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (Gapdh) was used for internal normalization. Relative quantification of gene expression was calculated using the 2-ΔΔCt method. Technical triplicates for each sample were performed. Melt curve analysis was done at 65°C–95°C with 0.5°C increments and 2 to 5 s/step to check specificity of the primers and to ensure no primer-dimer formation during the qPCR. An exon-exon junction primer was designed to avoid genomic DNA amplification during the qPCR. Primers used for qRT-PCR are listed in Table S7.
Cell sorting and flow cytometry analysis
BD FACSMelody cell sorter (BD Biosciences) was used to sort GFP-expressing cells, while BD FACSVerse flow cytometer (BD Biosciences) was used to analyze GFP-expressing cells. The fluorescent channel fluorescein isothiocyanate (FITC) was used to detect the GFP-expressing cells; 100,000 cells were analyzed for each sample.
Statistical analysis
Using the Minitab Express Program, significant differences in continuous variables among subject groups were confirmed by ANOVA, followed by Dunnett post hoc multiple comparisons. All p values were two tailed. The statistical significance levels are indicated as ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. All data are presented as mean ± SD.
Conclusions
We have successfully utilized PAM-flexible dual base editor-mediated mutagenesis and self-activation strategies to derive VP64 variants with gain-of-function mutations. Importantly, these VP64 variants could improve the potency of an all-in-one CRISPRa for application of a single AAV vector platform. However, multiple sgRNAs are still needed for robust target gene activation due to the modest improvement of the function of our current ΔCRISPRa. This mutagenesis study not only provided insights into the structural elements critical for VP64 function, it also represents a proof of concept for evolving CRISPR activators through integration use of different CRISPR variants and approaches.
Data availability statement
All data and supporting materials are available within the article and supplemental information.
Acknowledgments
This work was supported by the Research Grants Council of the Hong Kong Special Administrative Region [grant number CityU11213717, CityU11104220 and CityU11215619] and City University of Hong Kong [grant number CityU7005088].
Author contributions
C.T. conceived and supervised the project. C.H.L. and S.H. designed and performed the experiments. C.H.L. and S.H. collected and analyzed the data. C.H.L., S.H., R.H.W.L, and C.T. discussed the results, and wrote and revised the manuscript. All authors read, corrected, and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.04.014.
Supplemental information
References
- 1.Lau C.H. Applications of CRISPR-Cas in bioengineering, biotechnology, and translational research. CRISPR J. 2018;1:379–404. doi: 10.1089/crispr.2018.0026. [DOI] [PubMed] [Google Scholar]
- 2.Kampmann M. CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine. ACS Chem. Biol. 2018;13:406–416. doi: 10.1021/acschembio.7b00657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.La Russa M.F., Qi L.S. The new state of the art: Cas9 for gene activation and repression. Mol. Cell. Biol. 2015;35:3800–3809. doi: 10.1128/mcb.00512-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grimm D., Büning H. Small but increasingly mighty: latest advances in AAV vector research, design, and evolution. Hum. Gene. Ther. 2017;28:1075–1086. doi: 10.1089/hum.2017.172. [DOI] [PubMed] [Google Scholar]
- 5.Lau C.H., Suh Y. In vivo genome editing in animals using AAV-CRISPR system: applications to translational research of human disease. F1000Res. 2017;6:2153. doi: 10.12688/f1000research.11243.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lau C.H., Ho J.W.T., Lo P.K., Tin C. Targeted transgene activation in the brain tissue by systemic delivery of engineered AAV1 expressing CRISPRa. Mol. Ther. Nucleic Acids. 2019;16:637–649. doi: 10.1016/j.omtn.2019.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S., et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maeder M.L., Linder S.J., Cascio V.M., Fu Y., Ho Q.H., Joung J.K. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods. 2013;10:977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H., Guimaraes C., Panning B., Ploegh H.L., Bassik M.C., et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perez-Pinera P., Kocak D.D., Vockley C.M., Adler A.F., Kabadi A.M., Polstein L.R., Thakore P.I., Glass K.A., Ousterout D.G., Leong K.W., et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods. 2013;10:973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deverman B.E., Pravdo P.L., Simpson B.P., Kumar S.R., Chan K.Y., Banerjee A., Wu W.L., Yang B., Huber N., Pasca S.P., Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galvan A., Petkau T.L., Hill A.M., Korecki A.J., Lu G., Choi D., Rahman K., Simpson E.M., Leavitt B.R., Smith Y. Intracerebroventricular administration of AAV9-PHP.B SYN1-EmGFP induces widespread transgene expression in the mouse and monkey central nervous system. Hum. Gene Ther. 2021;32:599–615. doi: 10.1089/hum.2020.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silva-Pinheiro P., Cerutti R., Luna-Sanchez M., Zeviani M., Viscomi C. A single intravenous injection of AAV-PHP.B-hNDUFS4 ameliorates the phenotype of Ndufs4 (-/-) Mice. Mol Ther Methods Clin Dev. 2020;17:1071–1078. doi: 10.1016/j.omtm.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim J.A., Yi H., Gao F., Raben N., Kishnani P.S., Sun B. Intravenous injection of an AAV-PHP.B vector encoding human acid alpha-glucosidase rescues both muscle and CNS defects in murine pompe disease. Mol Ther Methods Clin Dev. 2019;12:233–245. doi: 10.1016/j.omtm.2019.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chavez A., Scheiman J., Vora S., Pruitt B.W., Tuttle M., P R Iyer E., Lin S., Kiani S., Guzman C.D., Wiegand D.J., et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods. 2015;12:326–328. doi: 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X.S., Wu H., Ji X., Stelzer Y., Wu X., Czauderna S., Shu J., Dadon D., Young R.A., Jaenisch R. Editing DNA methylation in the mammalian genome. Cell. 2016;167:233–247.e17. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilton I.B., D'Ippolito A.M., Vockley C.M., Thakore P.I., Crawford G.E., Reddy T.E., Gersbach C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cano-Rodriguez D., Gjaltema R.A.F., Jilderda L.J., Jellema P., Dokter-Fokkens J., Ruiters M.H.J., Rots M.G. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 2016;7:12284. doi: 10.1038/ncomms12284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walton R.T., Christie K.A., Whittaker M.N., Kleinstiver B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020;368:290–296. doi: 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grünewald J., Zhou R., Lareau C.A., Garcia S.P., Iyer S., Miller B.R., Langner L.M., Hsu J.Y., Aryee M.J., Joung J.K. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat. Biotechnol. 2020;38:861–864. doi: 10.1038/s41587-020-0535-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X., Zhu B., Chen L., Xie L., Yu W., Wang Y., Li L., Yin S., Yang L., Hu H., et al. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat. Biotechnol. 2020;38:856–860. doi: 10.1038/s41587-020-0527-y. [DOI] [PubMed] [Google Scholar]
- 22.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018;36:843–846. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma Y., Zhang J., Yin W., Zhang Z., Song Y., Chang X. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat. Methods. 2016;13:1029–1035. doi: 10.1038/nmeth.4027. [DOI] [PubMed] [Google Scholar]
- 25.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A∗T to G∗C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen C.D., Zeldich E., Li Y., Yuste A., Abraham C.R. Activation of the anti-aging and cognition-enhancing gene Klotho by CRISPR-dCas9 transcriptional effector complex. J. Mol. Neurosci. 2018;64:175–184. doi: 10.1007/s12031-017-1011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurosu H., Yamamoto M., Clark J.D., Pastor J.V., Nandi A., Gurnani P., McGuinness O.P., Chikuda H., Yamaguchi M., Kawaguchi H., et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto M., Clark J.D., Pastor J.V., Gurnani P., Nandi A., Kurosu H., Miyoshi M., Ogawa Y., Castrillon D.H., Rosenblatt K.P., Kuro-o M. Regulation of oxidative stress by the anti-aging hormone klotho. J. Biol. Chem. 2005;280:38029–38034. doi: 10.1074/jbc.m509039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mostoslavsky R., Chua K.F., Lombard D.B., Pang W.W., Fischer M.R., Gellon L., Liu P., Mostoslavsky G., Franco S., Murphy M.M., et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 30.Michishita E., McCord R.A., Berber E., Kioi M., Padilla-Nash H., Damian M., Cheung P., Kusumoto R., Kawahara T.L.A., Barrett J.C., et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peshti V., Obolensky A., Nahum L., Kanfi Y., Rathaus M., Avraham M., Tinman S., Alt F.W., Banin E., Cohen H.Y. Characterization of physiological defects in adult SIRT6-/- mice. PLoS. One. 2017;12:e0176371. doi: 10.1371/journal.pone.0176371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanfi Y., Naiman S., Amir G., Peshti V., Zinman G., Nahum L., Bar-Joseph Z., Cohen H.Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–221. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- 33.Nguyen T., Nioi P., Pickett C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009;284:13291–13295. doi: 10.1074/jbc.r900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gazaryan I.G., Thomas B. The status of Nrf2-based therapeutics: current perspectives and future prospects. Neural. Regen. Res. 2016;11:1708. doi: 10.4103/1673-5374.194706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Sawaf O., Clarner T., Fragoulis A., Kan Y.W., Pufe T., Streetz K., Wruck C.J. Nrf2 in health and disease: current and future clinical implications. Clin. Sci. (Lond.) 2015;129:989–999. doi: 10.1042/cs20150436. [DOI] [PubMed] [Google Scholar]
- 36.Liu X., Li G., Zhou X., Qiao Y., Wang R., Tang S., Liu J., Wang L., Huang X. Improving editing efficiency for the sequences with NGH PAM using xCas9-derived base editors. Mol. Ther. Nucleic Acids. 2019;17:626–635. doi: 10.1016/j.omtn.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emami K.H., Carey M. A synergistic increase in potency of a multimerized VP16 transcriptional activation domain. EMBO. J. 1992;11:5005–5012. doi: 10.1002/j.1460-2075.1992.tb05607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sadowski I., Ma J., Triezenberg S., Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 39.Sullivan S.M., Horn P.J., Olson V.A., Koop A.H., Niu W., Ebright R.H., Triezenberg S.J. Mutational analysis of a transcriptional activation region of the VP16 protein of herpes simplex virus [published erratum appears in Nucleic Acids Res 1998 Dec 1;26(23):537-8] Nucleic. Acids. Res. 1998;26:4487–4496. doi: 10.1093/nar/26.19.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cress W.D., Triezenberg S.J. Critical structural elements of the VP16 transcriptional activation domain. Science. 1991;251:87–90. doi: 10.1126/science.1846049. [DOI] [PubMed] [Google Scholar]
- 41.Berger S.L., Cress W.D., Cress A., Triezenberg S.J., Guarente L. Selective inhibition of activated but not basal transcription by the acidic activation domain of VP16: evidence for transcriptional adaptors. Cell. 1990;61:1199–1208. doi: 10.1016/0092-8674(90)90684-7. [DOI] [PubMed] [Google Scholar]
- 42.Ingles C.J., Shales M., Cress W.D., Triezenberg S.J., Greenblatt J. Reduced binding of TFIID to transcriptionally compromised mutants of VP16. Nature. 1991;351:588–590. doi: 10.1038/351588a0. [DOI] [PubMed] [Google Scholar]
- 43.Regier J.L., Shen F., Triezenberg S.J. Pattern of aromatic and hydrophobic amino acids critical for one of two subdomains of the VP16 transcriptional activator. Proc. Natl. Acad. Sci. U S A. 1993;90:883–887. doi: 10.1073/pnas.90.3.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghosh S., Toth C., Peterlin B.M., Seto E. Synergistic activation of transcription by the mutant and wild-type minimal transcriptional activation domain of VP16. J. Biol. Chem. 1996;271:9911–9918. doi: 10.1074/jbc.271.17.9911. [DOI] [PubMed] [Google Scholar]
- 45.Pausch P., Al-Shayeb B., Bisom-Rapp E., Tsuchida C.A., Li Z., Cress B.F., Knott G.J., Jacobsen S.E., Banfield J.F., Doudna J.A. CRISPR-CasΦ from huge phages is a hypercompact genome editor. Science. 2020;369:333–337. doi: 10.1126/science.abb1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma D., Peng S., Huang W., Cai Z., Xie Z. Rational design of mini-Cas9 for transcriptional activation. ACS Synth. Biol. 2018;7:978–985. doi: 10.1021/acssynbio.7b00404. [DOI] [PubMed] [Google Scholar]
- 47.Vora S., Cheng J., Xiao R., VanDusen N., Quintino L., Pu W., Vandenberghe L., Chavez A., Church G. Rational design of a compact CRISPR-Cas9 activator for AAV-mediated delivery. bioRxiv. 2018 doi: 10.1101/298620. Preprint at. [DOI] [Google Scholar]
- 48.Zhang X., Lv S., Luo Z., Hu Y., Peng X., Lv J., Zhao S., Feng J., Huang G., Wan Q.L., et al. MiniCAFE, a CRISPR/Cas9-based compact and potent transcriptional activator, elicits gene expression in vivo. Nucleic Acids Res. 2021;49:4171–4185. doi: 10.1093/nar/gkab174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu L., Zhao L., Gao Y., Xu J., Han R. Empower multiplex cell and tissue-specific CRISPR-mediated gene manipulation with self-cleaving ribozymes and tRNA. Nucleic Acids Res. 2017;45:e28. doi: 10.1093/nar/gkw1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan K.Y., Jang M.J., Yoo B.B., Greenbaum A., Ravi N., Wu W.L., Sánchez-Guardado L., Lois C., Mazmanian S.K., Deverman B.E., Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goertsen D., Flytzanis N.C., Goeden N., Chuapoco M.R., Cummins A., Chen Y., Fan Y., Zhang Q., Sharma J., Duan Y., et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat. Neurosci. 2021;25:106–115. doi: 10.1038/s41593-021-00969-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and supporting materials are available within the article and supplemental information.