

Abstract
Pregnant women are often excluded from research without clear justification, even when the research poses minimal fetal risk. Little is known about institutional review board (IRB) decision-making practices when reviewing such research. We conducted a survey of IRB personnel in the US to elicit their interpretations of “minimal risk” – a formal regulatory category -- and to identify factors that may influence IRB decisions to approve or disapprove research involving pregnant participants. Study results revealed some consensus among IRB members about the risk level of individual research procedures and hypothetical study vignettes. However, we uncovered important variations not only in the assessment of risk but also in the willingness of IRB members to approve minimal risk research that includes pregnant women. Based on our findings, guidance is needed to assist IRB members in characterizing risk, applying federal regulations, and appropriately ensuring the inclusion or justified exclusion of pregnant women in research.
Keywords: Clinical trials, pregnancy, minimal risk, institutional review board (IRB), human subjects research, survey
Introduction:
In recent years, attention to profound evidence gaps on the safety and efficacy of medical interventions during pregnancy has highlighted the underrepresentation of pregnant women in clinical research1. Reluctance to conduct research involving pregnant women often stems from a well-intentioned desire to avoid fetal harm. Yet, evidence suggests that even when clinical trial participation poses little to no fetal or maternal risk, pregnant women2 are often excluded from trial participation3. One contributing factor to this may be confusion or disagreement over how to interpret the category of “minimal risk.” According to the U.S. Code of Federal Regulations, “minimal risk” means that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests4. The category of “minimal risk” has been the subject of much discussion, and empirical evidence in the context of pediatric research indicates that IRBs can vary widely in determining which research interventions constitute “minimal risk”5.
Uncertainty about how to apply the minimal risk standard may also complicate IRB assessments of research risk in the context of pregnancy. According to Subpart B of Code of Federal Regulations (45 CFR 46.204), when studies do not offer the prospect of direct benefit to the woman or fetus, they can only be approved if the risk to the fetus is “no greater than minimal” and if the “purpose of the research is the development of important biomedical knowledge which cannot be obtained by any other means.”6 IRBs may reasonably differ in their interpretation of this guidance, particularly in their evaluation of and tolerance for risk when there is no prospect of direct benefit – a category termed non-beneficial research. Non-beneficial research in the form of observational studies, surveys, and other studies with no study-related interventions offers no prospect of direct benefit to participants. Yet, such research generally poses less risk than clinical drug trials and other types of interventional studies which may offer the prospect of direct benefit but also expose participants to risks associated with the study interventions. Ambiguities in the regulatory language, uncertainty about characterizing research risks, or other factors may present challenges for IRBs deciding whether to allow pregnant women to enroll in non-beneficial research under review. Given that IRBs serve as the gateway for research approval, understanding the factors influencing IRB assessments of non-beneficial, low risk research may help to facilitate a paradigm shift from the presumed exclusion of pregnant women to their responsible inclusion in clinical research7.
In order to identify factors potentially influencing IRB member decision-making and identify areas for developing specific guidance around the enrollment of pregnant participants, we conducted a survey of IRB members in the United States. Our goal was to assess similarities and differences among IRB members in their regulatory interpretations, assessments of research risk, and approval determinations across hypothetical examples of minimal risk, non-beneficial research with pregnant women. With the exception of two small qualitative studies, one involving six IRB members sharing their perspectives on acceptable perinatal mental health research and another involving nine investigators and five administrators describing their experiences at a single institution, to our knowledge there have been no other empirical studies characterizing the range of IRB practices, interpretations, risk assessments, and decisions regarding the enrollment of pregnant women8.
Study Methods
The IRB at the University of North Carolina at Chapel Hill approved our study. No validated instrument exists for probing the attitudes and perceptions we wished to study, therefore the authors developed a de novo survey instrument through an iterative process that included a comprehensive review of the literature, draft survey development, cognitive pretesting and revisions with assistance from the University of North Carolina Odum Institute, which provided resources and expertise in survey methodology and design. Our survey underwent further external review by a team of bioethicists at the National Institutes of Health who reviewed it for content validity and by a senior IRB analyst at the University of North Carolina who assisted with wording and clarity. Pilot testing was completed with eight IRB members and administrators who were representative of our planned study population. In the pilot, each participant completed the entire survey using either a mobile device or computer, followed by a semi-structured interview in which we probed their understanding, perceptions, and responses to individual survey questions and hypothetical research vignettes. Based on the feedback of each participant, we revised the survey language and format accordingly.
The finalized survey was in the form of a web-based, self-administered questionnaire containing dichotomous and Likert-scale quantitative questions, as well as closed-ended and open-ended qualitative questions, and was divided into four sections. Section one asked respondents to describe their professional backgrounds, IRB experience, familiarity with Subpart B interpretation, and application of Subpart B by their respective IRBs. Section two, which was limited to IRB members with a background in medicine, nursing, or biomedical science, asked respondents to assign a risk level of “minimal risk” or “greater than minimal risk” to ten research procedures in the context of pregnancy. To avoid potential confusion about varying degrees of procedure risk depending on the trimester of pregnancy, we limited these ten research procedures in section two to the third trimester of pregnancy. Section three used vignette methodology to explore IRB members’ views on the general idea of enrolling pregnant women in minimal risk research involving no prospect of direct benefit, as well as their responses to three hypothetical research scenarios involving pregnant women. Three vignettes of different study types (Table 1) were presented in random order to survey respondents. These three vignettes were not limited to any particular trimester of pregnancy, as we intentionally designed each scenario to involve clear examples of minimal risk procedures regardless of trimester, with the purpose of probing IRB member reactions to the approval of studies involving these procedures. For each vignette, we asked respondents to categorize the hypothetical research study as “minimal risk” or “greater than minimal” risk and to indicate whether they believed Subpart B would allow the enrollment of pregnant women. We then used Likert scale questions to assess both how likely individual respondents as well as their collective IRBs would be to vote to approve the enrollment of pregnant women. To explore underlying reasons for voting decisions, our team consulted with experts in survey development and used feedback from pilot testing to generate a list of ten possible ethical considerations, attitudes, and beliefs involving risk, justice, and scientific validity that might impact IRB member decision-making. Based on this list, we included five-point Likert scale questions to probe the extent to which voting decisions for each vignette may have been influenced “a lot” to “not at all” by these possible considerations or other factors that respondents could specify in open-ended comments. Finally, section four of the survey explored administrators’ perspectives on the main issues they observed their IRBs deliberating when reviewing research involving pregnant women and recommendations for areas of needed IRB guidance.
Table 1:
Hypothetical Research Scenarios
| Vignette | Scenario Description (as presented in survey) |
|---|---|
| 1. Observational study | A researcher is studying the natural history of chronic dermatitis in a particular region of the United States that has high levels of pollen. The researcher is recruiting adult volunteers with eczema or other dermatitis who live in this region for a multi-site, observational study to better understand the causes and natural history of chronic dermatitis. Because the study is observational, the scientific review committee has determined that there are no safety or scientific concerns that would preclude pregnant women from entering the study. There is also no prospect of direct benefit for the participants. Study participants will have an initial visit that involves a physical examination, single small blood draw (10mL), and urine test. There are 6 additional visits over 6 months each expected to last one hour. Pictures of any dermatologic findings will be taken at each visit. A small volume of blood (10mL) will be drawn once per month, which is within the acceptable limit for the collection of blood samples according to the Office for Human Research Protections expedited review categories. Participants are asked to record any topical or systemic medications they use that are over the counter or are prescribed by their personal clinicians during the study period. |
| 2. Survey Study | A group of investigators studying the associations between obesity (defined by body mass index) and mental health are conducting a survey of women with obesity. The survey includes questions about nutrition, physical activity, work stress, relationship status, depression history, and past emotionally stressful experiences they attribute to their weight. A scientific review committee has already determined that the questions are unlikely to trigger emotional distress for participants, and appropriate safeguards are in place. |
| 3. PK Study |
A researcher proposes to enroll
pregnant women in a
pharmacokinetic (PK) study
of a once-daily anti-retroviral medication for treatment of HIV.
The drug is widely in use among pregnant women. However, the
therapeutic dose of this anti-retroviral treatment in pregnancy
remains unknown. Initial PK studies of this medication enrolled
primarily men, and the drug was FDA approved for men and
non-pregnant women. There are no birth defects known to be
associated with this anti-retroviral treatment in women who became
pregnant while taking this medication, preclinical animal data have
revealed no adverse fetal effects, and preliminary safety data from
Phase 1 trials of non-pregnant women have been favorable.
Researchers therefore plan to conduct a PK study with
pregnant women with HIV who are already taking the
standard dose of this drug in the course of clinical
care. The study would not
involve any changes to participants’ current medication
regimens as prescribed by their
physicians. The study will involve the following research procedures: a series of blood draws for PK analysis to be collected at 1,2,3,4,8, and 12 hours after dosing (total volume less than 50 mL, which is within the acceptable limit for the collection of blood samples according to the Office for Human Research Protections expedited review categories); and common safety parameters for a Phase 1 study, including electrocardiogram (EKG) monitoring, vital signs, physical exam, clinical laboratory panels to identify any laboratory abnormalities that arise during the PK study, urinalysis, and monitoring of adverse events. |
We distributed the survey to IRB personnel in the United States between 10/2017 and 9/2018. We recruited IRB personnel from a range of institutional types, including academic and non-academic medical centers, government agencies, and independent/commercial IRBs. In order to reach IRB members from diverse backgrounds, we collaborated with Public Responsibility in Medicine and Research (PRIM&R) to distribute a series of electronic invitations to over 1200 active members who had indicated a willingness to participate in research. To enhance recruitment, we contacted IRB directors of major academic centers affiliated with the Maternal Fetal Medicine Units (MFMU) Network and other pregnancy-related research networks to distribute the survey to their IRB personnel. Study participants were eligible if they were current or former IRB members who had served on an IRB within the past five years, had served for at least one year, and had reviewed at least one study protocol that included pregnant women in their prior or last year of IRB service. IRB administrators were eligible to participate in section four of the survey if they held a leadership role for an IRB that reviewed at least one study in the prior year that included pregnant women. We offered a $20 Amazon gift card incentive to all eligible participants who completed the survey.
We conducted descriptive analyses of the quantitative data using SAS version 9.3. Our approach to analyzing open-ended qualitative responses was informed by thematic analysis9. We reviewed responses for emergent themes and used data display matrices to identify patterns between and across respondents. Representative quotes for themes are presented.
Results
Initially 206 respondents agreed to participate. Of these, 53 were excluded because they reported less than one year IRB experience, were not current/former IRB members or senior administrators, did not recall reviewing at least one study protocol in the prior year that included pregnant women, or their IRB was located outside of the United States (Figure 1). An additional 21 respondents did not complete the survey and were therefore excluded. A total of 132 participants (93 IRB members and 39 administrators) met eligibility criteria and completed the survey.
Figure 1:
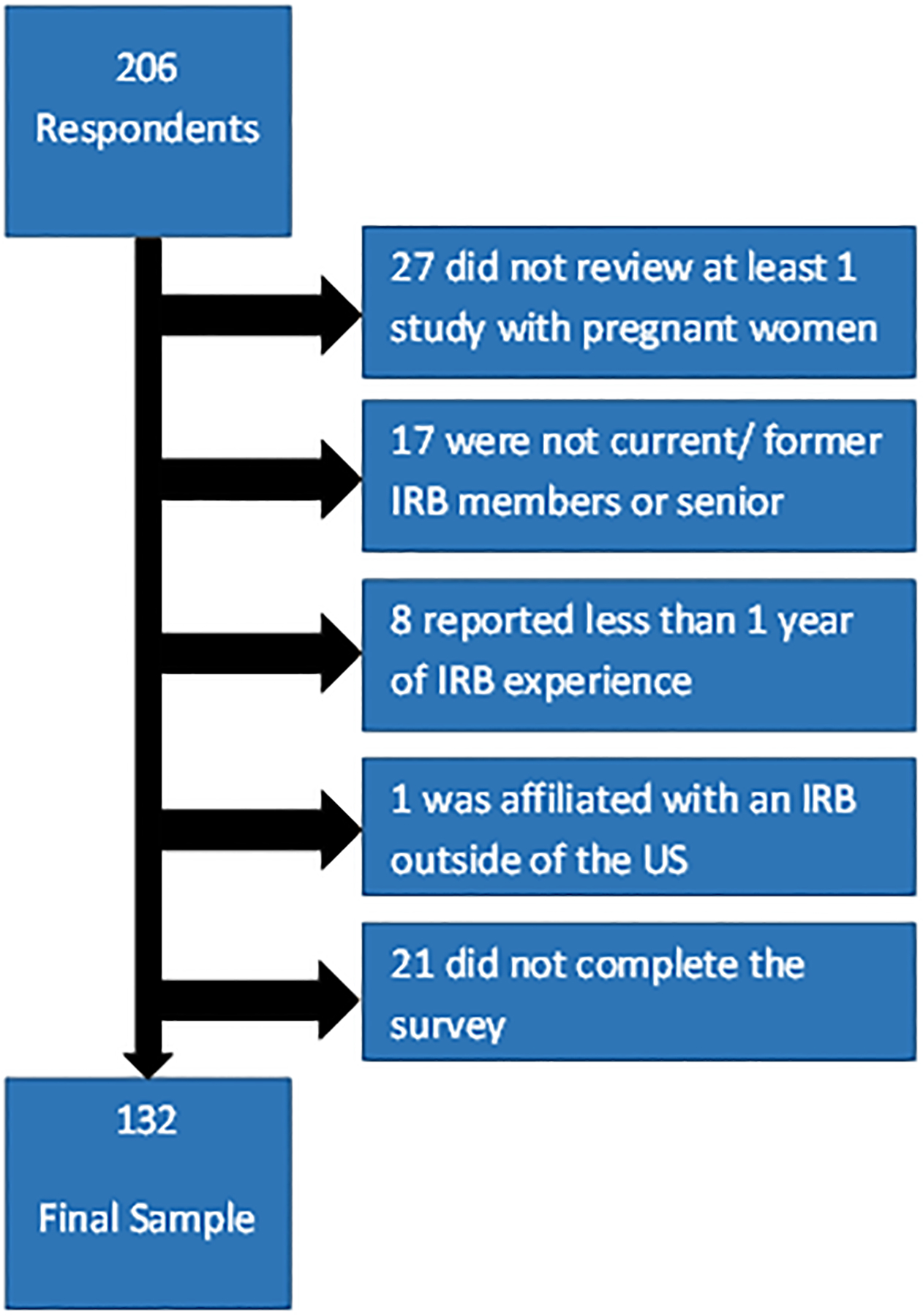
Eligible Participants
Section 1: Respondents’ backgrounds, experience, and familiarity with Subpart B interpretation and application
Of the 93 IRB members, 19 were IRB chairpersons (20.4%). The majority were female (76%), had greater than five years of IRB experience (69%), and served on an IRB affiliated with an academic medical center, university, or research institute (78%). Table 2 presents demographic information for IRB member survey respondents. Overall, 71.3% of IRB members in our cohort reported that they were mostly or very familiar with Subpart B. Roughly half described Subpart B as somewhat easy or very easy to understand, and 20.4% thought it was somewhat difficult or very difficult to understand. Additionally, 25.8% described the application of Subpart B as somewhat difficult or very difficult for them personally, and 22.5% as somewhat difficult for their IRB.
Table 2:
IRB Member Demographics (n=93)
| IRB Personnel Characteristics | ||
|---|---|---|
| Age | Range | 25–69, Median 44 |
| Gender | Male | 22 (23.6%) |
| Female | 71(76.3%) | |
| Professional background (more than 1 choice could be selected) | Medicine | 18 (19.6%) |
| Nursing | 7 (7.5%) | |
| Biomedical science | 19 (20.4%) | |
| Behavioral/ Social science | 34 (36.5%) | |
| Public Health | 7 (7.5%) | |
| Law | 10 (10.8%) | |
| Other | 21 (22.6%) | |
| IRB member role | IRB Chair | 19 (20.4%) |
| Scientific member | 34 (36.6%) | |
| Nonscientific member | 31 (33.3%) | |
| Community member | 1 (1.1%) | |
| Another role | 8 (8.6%) | |
| Total IRB experience | 1–5 years | 29 (31.2%) |
| 6–10 years | 28 (30.1%) | |
| >10 years | 36 (38.7%) | |
| IRB Affiliation | Academic Medical Center/ University / Research Institute | 73 (78.5%) |
| Non-academic Medical Center | 7 (7.5%) | |
| Government Agency | 16 (17.2%) | |
| Independent/Commercial IRB | 4 (4.3%) | |
| Other | 0 | |
| Types research protocols (more than 1 choice could be selected) | Biomedical research | 92 (98.9%) |
| Social Science | 63 (67.7%) | |
| Public Health | 46 (49.5%) | |
| Other | 4 (4.3%) | |
| IRB Experience With Subpart B | ||
| Frequency of reviewing IRB protocols involving pregnant women | Low frequency | 63 (67.7%) |
| High frequency (half or more of IRB meetings) | 30 (32.2%) | |
| Frequency of IRB referencing Subpart B | Never or unsure | 5 (5.4%) |
| Sometimes | 49 (52.7%) | |
| Always | 38 (40.9%) | |
| Familiarity with Subpart B | Very familiar (from memory) | 21 (22.6%) |
| Mostly familiar | 47 (50.5%) | |
| A little familiar | 22 (23.6%) | |
| Not familiar at all | 3 (3.2%) | |
| Ease of understanding Subpart B | Somewhat or very easy | 47 (50.5%) |
| Not easy or difficult | 27 (29.0%) | |
| Somewhat or very difficult | 19 (20.4%) | |
| Ease of applying Subpart B to protocol review | Somewhat or very easy | 44 (47.3%) |
| Not easy or difficult | 25 (26.9%) | |
| Somewhat or very difficult | 24 (25.8%) | |
Of the 39 administrators, the majority were female (97.4%) and had at least five years of experience on their current IRB (56.4%). Overall, 71.8% of administrators in our cohort reported that they were mostly or very familiar with Subpart B.
Section 2: IRB member characterization of research procedure risks
The subgroup of 40 IRB members who endorsed a background in medicine, nursing, and/or biomedical science (18, 7, and 19 respondents, respectively) assigned a risk level to ten hypothetical research procedures in the third trimester of pregnancy (Figure 2). More than 97% of this subgroup characterized a mouth swab and external fetal heart rate monitoring as minimal risk. Almost all characterized a chest/abdominal CT scan, amniocentesis, and vaginal microbicide for HIV prevention at delivery as greater than minimal risk. However, respondents disagreed on the risk level of other procedures. For example, while 82.5% characterized a potentially emotionally distressing interview as minimal risk, 15% assigned a greater than minimal risk level. Respondents were evenly divided over a single foot x-ray with abdominal shielding, for which 47.5% assigned minimal risk and 45.0% assigned greater than minimal risk. Opinions differed even among the three IRB members reporting a background in obstetrics and gynecology, as they disagreed on the risk level of the foot x-ray, CT scan, and emotionally distressing interview.
Figure 2:
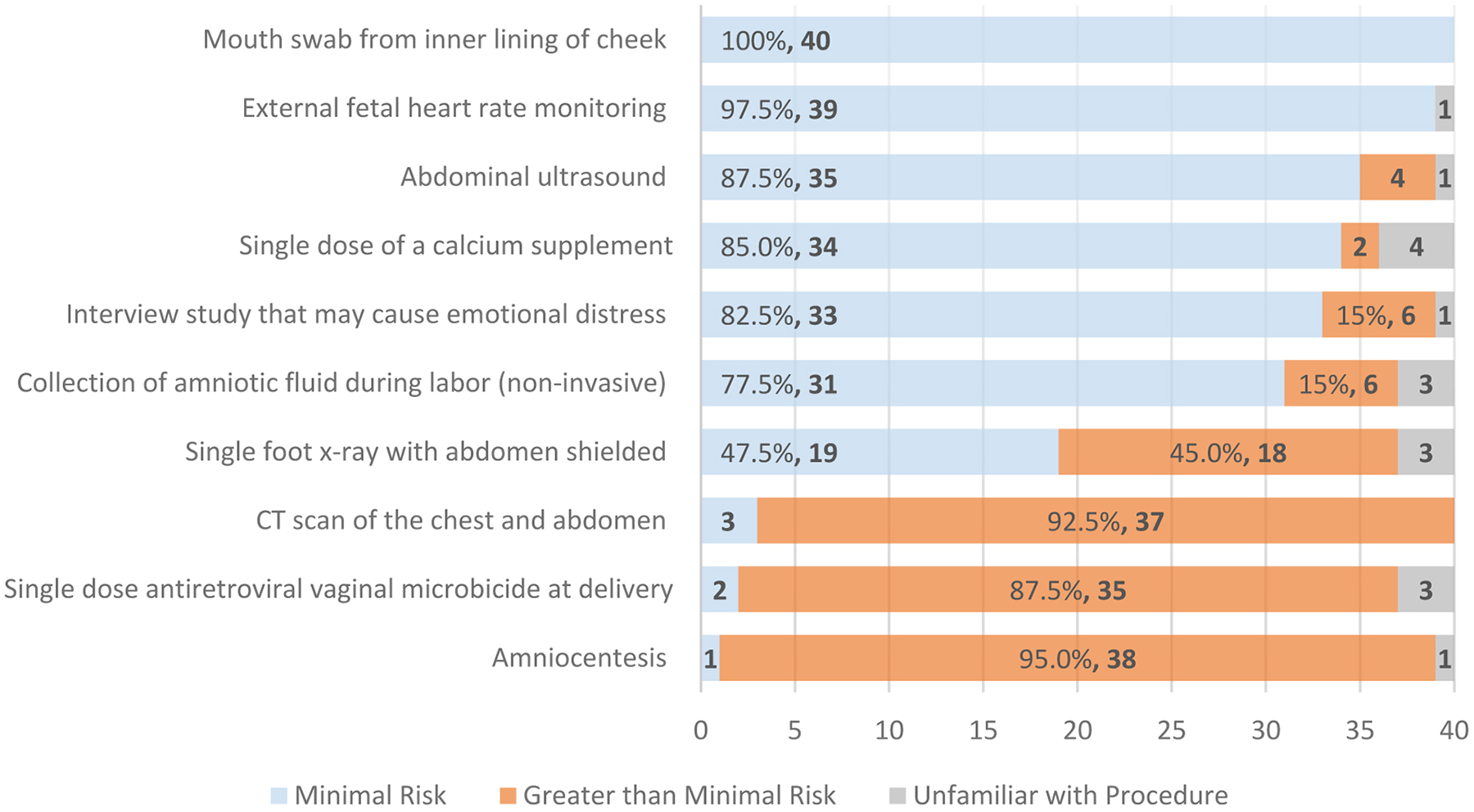
IRB Member Assessment of Risk Level for Research Procedures in the Third Trimester of Pregnancy (n=40)
Section 3: IRB member responses to hypothetical research scenarios
Attitudes toward the idea of minimal risk research during pregnancy
When asked to assess the appropriateness of enrolling pregnant women in a hypothetical, generic scenario about a minimal risk study that offered no prospect of direct benefit but was considered important biomedical research, 5.4% of the 93 respondents believed pregnant women should be excluded, and 21.5% thought it was ethically preferable to enroll only non-pregnant women if the study could be done without pregnant women. On the other hand, 49.5% of IRB members thought there was no reason to prefer non-pregnant or pregnant women, 4.3% believed pregnant women should be allowed to enroll, and 19.4% thought enough pregnant women should be encouraged to enroll to reach an adequate sample size for subgroup analysis.
Risk level assigned to vignettes
When asked to assign a risk level to each of the three vignettes, 96.8% and 97.8% of 93 respondents selected a minimal risk classification for the observational study and survey study, respectively. A smaller but sizeable majority (82.8%) assigned minimal risk to the PK study of an antiretroviral medication already prescribed in clinical care while a sizeable minority assigned a greater than minimal risk level (17.2%) (Table 3).
Table 3:
IRB Member Risk Assessment and Approval Decisions for Vignettes (n=93)
| Study Vignette | Assigned minimal risk | Subpart B allows enrollment | Would vote to approve enrollment | Anticipated IRB approval |
|---|---|---|---|---|
| Observational study | 90 (96.8%) | 85 (91.4%) | Very likely – 72
(77.4%) Somewhat likely – 17 (18.3%) |
Very likely – 61
(65.6%) Somewhat likely – 26 (27.9%) |
| Survey study | 91 (97.8%) | 85 (91.4%) | Very likely – 70
(75.3%) Somewhat likely – 14 (15%) |
Very likely – 68
(73.1%) Somewhat likely – 18 (19.4%) |
| PK study | 77 (82.8%) | 86 (92.5%) | Very likely – 59
(63.4%) Somewhat likely – 29 (31.2%) |
Very likely – 48
(51.6%) Somewhat likely – 37 (39.8%) |
Open ended responses from the minority who regarded a given vignette as greater than minimal risk revealed some of their reasons. For all three vignettes, this minority mentioned the perception of an increased burden on or discomfort of pregnant women from study participation. Another reason involved a perceived conflict between the criteria for minimal risk and criteria for using expedited review procedures, which regulatory guidelines permit for a subset of minimal risk research involving a limited set of research procedures (45 CFR 46.110)10. Even though designation as minimal risk research does not ensure qualification for expedited review, one IRB member indicated that the PK study should not be categorized as minimal risk because the number of required blood draws would preclude using expedited review. Additionally, two respondents described their perception that studying the PK of a drug that lacks FDA approval for use in that population automatically makes the study greater than minimal risk. Finally, one respondent conveyed their perception that research intended to support FDA drug relabeling for a new indication cannot be minimal risk. However, our PK study vignette did not involve FDA drug relabeling.
Propensity to approve the research
Despite high numbers of IRB members classifying the vignettes as minimal risk studies, fewer IRB members reported a high likelihood of approving the enrollment of pregnant women in those studies. For the observational study, 95.7% of IRB members thought pregnant women “should be allowed” to enroll in the study based on the vignette description and 91.4% believed that Subpart B would allow this. However, only 77.4% reported being very likely to vote for study approval, and even fewer believed their IRB would be very likely to approve the study. Similarly, although 89.2% of respondents believed pregnant women should be allowed to enroll in the survey study and 91.4% indicated Subpart B would allow their enrollment, just 75.3% were very likely to vote for study approval. The sharpest decline occurred for the PK study. Despite 92.5% of respondents indicating that Subpart B would allow the enrollment of pregnant women, only 63.4% were very likely to vote for study approval (Table 3).
Attitudes and beliefs impacting IRB member approval decisions
When asked to indicate the extent to which a list of competing factors might have influenced their voting decisions for each vignette on a five-point Likert scale ranging from “a lot” to “not at all,” at least 50% of IRB members considered several factors “a lot” in addition to research risk (Table 4). These factors included the following attitudes and beliefs: 1) a diverse study population is needed to best understand the condition being studied, 2) access to research should not discriminate against pregnant women, 3) certain conditions can affect pregnant women and should be studied, and 4) efforts should be made to fill research gaps on conditions that affect pregnant women. In the PK study, at least 50% of respondents considered these additional factors “a lot”: 1) whether a treatment was already in widespread use, 2) a perception that important biomedical knowledge cannot be obtained without pregnant women, and 3) the idea that all populations at risk for HIV need accurate PK data on available treatments.
Table 4:
Main Factors Impacting Decision-Making to Approve Research Vignettes for Majority of IRB Members
| Issues considered “a lot” | IRB members selecting this issue (n = 93) | ||
|---|---|---|---|
| Observational study | Survey study | PK study | |
| The risks of research participation for the fetus or pregnant woman are truly minimal | 80 (86.0 %) | 76 (81.7%) | 71 (76.3%) |
| All kinds of people with the condition should be included to best understand the condition | 71 (76.3%) | 67 (72.0%) | n/a* |
| Access to research should not discriminate against pregnant women | 64 (68.8%) | 62 (66.7%) | 62 (66.7%) |
| Certain conditions can affect pregnant women and should be studied | 62 (66.7%) | 65 (69.9%) | n/a* |
| Efforts should be made to fill research gaps on conditions that affect pregnant women | 48 (51.6%) | 53 (57.0%) | 70 (76.1%) |
| This treatment is already widely being used and should be studied | n/a* | n/a* | 83 (89.2%) |
| Important biomedical knowledge cannot be obtained without pregnant women | n/a* | n/a* | 80 (86.0%) |
| All populations at risk for HIV need accurate PK data on available treatments | n/a* | n/a* | 67 (72.0%) |
n/a = issue was not a selection option
Reasons for excluding pregnant participants
Of those who explained their rationale for indicating that pregnant women should not be enrolled in one or more of the study vignettes, interestingly, few cited that the studies or individual procedures were too risky. Instead, the most common explanation for exclusion from the observational and survey studies was concern about undermining scientific validity, either due to pregnancy confounding study results or an inadequate sample size to generate meaningful results. For all three studies, some respondents believed that federal regulations would not permit the enrollment of pregnant women. Others noted that the purpose of the study was not about a pregnancy-related condition, viewed pregnancy as a life stressor that could complicate the study, wanted more information about informed consent or safety monitoring, or simply acknowledged a habit of excluding pregnant women. As one participant explained, “We are generally conditioned to expect pregnant women to be excluded.”
Open-ended comments provided insights into misunderstandings or uncertainty interpreting Subpart B requirements. In response to whether Subpart B would allow the inclusion of pregnant women in the observational study, one respondent explained that “the criteria of ‘important knowledge which cannot be obtained by any other means’ is difficult to interpret.” Some interpreted this language to mean that the observational and survey studies could, and therefore should, be conducted without including pregnant women. Additionally, two respondents expressed the mistaken belief that Subpart B required the prospect of direct benefit for research approval. One respondent commenting on general confusion around the application of Subpart B explained, “Subpart B does not address…survey research. I believe most IRBs just ignore this oversight…” Indeed, federal regulations do specify criteria for the exemption of surveys, educational tests, interviews, and public behavior observation studies from Subpart B requirements if certain conditions are met regarding the nature and identifiability of the data11.
Responses to the PK study highlighted additional areas of concern, ambivalence, and deliberation. This vignette involved an opportunistic study design relying on clinical care as an opportunity for PK data collection. Open-ended comments revealed notable uncertainty for some respondents about whether to classify the HIV antiretroviral drug prescribed in clinical care as a research procedure and whether the drug’s risks should be considered research-related risks. Many IRB members concluded that research participation introduced no additional fetal risk compared to the woman’s clinical care and classified the overall study as minimal risk. These individuals explained their motivation to approve the study based on an understanding that the only research procedures were blood draws and safety monitoring. However, other IRB members reasoned that since drug administration at some time point was a necessary step in any PK study, they believed that the drug itself should be considered a research intervention. Since this would require classifying the overall study risk as greater than minimal, further uncertainty arose around whether federal regulations would permit the enrollment of pregnant women. Thus, responses to this vignette revealed key areas of disagreement related to defining research procedures in an opportunistic PK study, classifying research risks and, by extension, allowing pregnant women to enroll.
Moreover, concerns about the scientific validity of the observational and survey study vignettes emerged as a common theme. While some IRB members believed the inclusion of pregnant women could ensure validity by reducing bias in subject selection and data collection, others thought their inclusion could threaten scientific validity. One respondent was ambivalent and explained that “investigators could make a case for the care of pregnant women being different than non-pregnant women because their primary care physicians/dermatologists may treat them with different medications during pregnancy, so I would be ok if the investigators had a reason to exclude them and would also be ok if they were included.”
We asked IRB members how they would handle a non-pregnant participant becoming pregnant after enrollment in the observational and survey studies. Whereas roughly 75% of respondents were very likely to allow the initial enrollment of pregnant women in both studies, over 90% would allow a participant who later became pregnant to remain enrolled, with roughly one-third requiring a re-consent process in each case. The most common reason given by the fewer than 10% who would require study withdrawal was concern about scientific validity.
Likelihood of requiring justification for exclusion
In two of the vignettes, we asked IRB members how likely they would be to require investigators to provide a compelling reason for excluding pregnant women if their protocols did not specify any justification. For the observational study, nearly half (48.4%) reported being “very likely” to require justification. The remaining IRB members reported being “somewhat likely” (29%), “somewhat unlikely” (15.1%), or “very unlikely” (7.5%) to require any justification. A similar pattern emerged for the survey study. These findings suggest that requiring justification for the exclusion of pregnant women is a relatively inconsistent practice among IRB members, even for studies that most would classify as minimal risk.
Perspectives on inclusion of pregnant women if not clear in protocol
Respondents were also divided on whether investigators for these same two vignettes ought to specify in the study protocols whether pregnant women can enroll. While 65.6% thought the investigator should specify this in the observational study protocol, 34.3% disagreed. Similarly, 53.8% thought the survey study protocol ought to clearly state whether pregnant women may enroll, but the remaining 46.2% had the opposite view. As one IRB member explained, “I think pregnancy should only be listed as an inclusion/exclusion [criterion] when pregnancy is relevant to the research question and/or risks.” This implies that unless pregnancy is central to the study or a reason for exclusion, some believe it is better left unmentioned.
Section 4: Administrator perspectives and recommendations for IRB guidance
We asked the 39 IRB administrators who participated in the survey to select all applicable responses and/or write open-ended comments describing the main issues they observed their IRBs deliberating when reviewing research involving pregnant women. The most common issue selected was how to determine the overall risk level of the study (79.5%), followed by how to determine the risk to the fetus of research procedures/interventions (72.8%), whether/when to apply Subpart B (51.3%), and how to interpret the language of Subpart B (41%). Administrators offered various recommendations for guidance they believed would be useful for IRB members (Table 5). The single most common suggestion was for a decision aid in the form of a checklist, flowchart, examples, or targeted questions that could assist IRB members in deciding whether to apply Subpart B to a protocol and whether Subpart B applies to studies allowing participants who later become pregnant to remain enrolled. This type of decision aid could also clarify when exemption criteria should be considered. Many who witnessed IRB deliberation in assigning risk levels to procedures recommended additional guidance from the Office for Human Research Protections (OHRP) and FDA that includes a list of examples to help IRB members make comparative risk determinations and a list of FDA-approved drugs in pregnancy. Another recommendation called for guidance on interpreting Subpart B language on “important biomedical knowledge,” the appropriateness of enrolling pregnant women in studies that are not about pregnancy, and the requirement for paternal consent. Finally, some administrators suggested that IRB members would benefit from guidance on ensuring equitable subject selection, clarification of what investigators should specify in a protocol regarding enrollment of pregnant women, and general IRB and investigator education on the inclusion of pregnant women in research. As one administrator explained, “We continue to have an issue with PI’s excluding pregnant women from the studies and it is not because of risk. We are consistently educating our institution that pregnant women can be involved in certain types of research and do [not] need to always be excluded.”
Table 5:
IRB Administrator Observations and Recommendations for Guidance (n=39)
| Common Issues Observed in IRB Deliberation | Main Guidance Recommendations |
|---|---|
|
|
Discussion
In this first large empirical study of IRB members regarding research involving pregnant women, IRB member reluctance emerged as a potential barrier to approving research with pregnant women. This reluctance seemed partially related to discrepancies in risk assessment and different views on what Subpart B regulations allow, though we found notable consensus around these issues in IRB member responses to the vignettes. However, even when IRB members deemed the inclusion of pregnant women ethically and legally permissible, a general reluctance to approve enrollment or require justification for exclusion appeared to stem from other factors unrelated to risk, including concerns around scientific validity, ambivalence about inclusion, or habitual IRB practices.
Discrepancies in judgments about the risks of certain research procedures may have contributed to some of the reluctance we observed. Virtually equal numbers of IRB members evaluated the risk level of a foot x-ray as minimal and as greater than minimal risk. Participants also assessed different levels of risk related to the volume and number of blood draws involved in the PK study vignette. Such variations in risk assessment highlight the challenge of interpreting and applying the federal minimal risk standard in the context of pregnancy – a challenge which has been noted in other contexts as well12. Although the Secretary’s Advisory Committee on Human Research Protections (SACHRP) provides recommendations for assessing risk in pediatric research with examples of routine tests and activities of daily life, similar guidance is not available to help investigators and IRB members better characterize minimal risk procedures in research with pregnant women13.
Another discrepancy in characterizing risk arose in opinions about what counts as a research procedure. The PK study vignette involved an opportunistic study design that provides what many regulatory experts and bioethicists view as an ethically appropriate means of gathering much needed data on drugs already prescribed in clinical care without incurring additional research risks beyond a series of maternal blood draws14. This type of study design, when used to obtain pharmacokinetic data on understudied drugs in pediatrics, has been considered minimal risk research15. However, several respondents in our survey perceived that the drug under study should be considered a research procedure classified as greater than minimal risk, even though the drug was prescribed as a part of clinical care, leading a sizeable minority to be unwilling to approve our hypothetical PK study. Distinguishing clinical from research-related drug risks in opportunistic drug trials in pregnancy hinges on whether the decision to administer the drug was independent of study participation16. Of note, since launching our survey, the FDA has issued draft guidance clarifying that “when a study collects data about a drug treatment during pregnancy, but the drug was prescribed before study enrollment by the patient’s [health care provider], then the risks associated with the drug use are not research-related risks.”17 Our survey highlights a need for clear IRB guidance on research procedures and risk assessments in opportunistic PK studies, particularly given the vast number of drugs that are widely prescribed without appropriate pharmacokinetic and pharmacodynamic studies to guide proper dosing in pregnancy.
In addition to risk assessment as a source of reluctance, we identified differences in how IRB members interpreted regulatory language. Subpart B interpretations fell into two distinct patterns of understanding the meaning of “the purpose of the research is the development of important biomedical knowledge that cannot be obtained by any other means.” Some IRB members placed more emphasis on “important biomedical knowledge” in choosing to approve research that was not primarily about pregnancy but could yield valuable knowledge if pregnant women were included. On the other hand, those who emphasized “by any other means” understood the regulations to exclude pregnant women from research that could be conducted with non-pregnant participants. The observed divergence highlights an additional need for clarification regarding appropriate interpretation of this regulatory language. Qualitative responses also uncovered questions about the relationship between FDA approval of a drug and the designation of minimal risk. The FDA has recently clarified their position that approval of a drug for use in the adult population is inclusive of pregnant women absent specific contraindication, and that prescription of approved medications in that absence does not constitute “off-label” usage18. Further efforts to clarify and communicate this and other guidance may be useful in reducing misclassification of research interventions.
Despite variations in assessing risk and interpreting regulatory guidance, the majority of IRB members agreed that all three vignettes posed minimal risk and that federal regulations would permit the enrollment of pregnant women. Yet, even among those who acknowledged the low risk and regulatory acceptability of the vignettes, willingness to approve the research varied. This suggests that IRB members were weighing factors unrelated to research risk or federal regulations in the decision-making process.
One such factor involved opposing views on whether scientific validity of the observational and survey studies could be maintained with the enrollment of pregnant women. Some participants suggested that enrollment would strengthen scientific validity due to a reduction in selection bias while others reasoned that enrollment would undermine scientific validity by introducing confounding bias. Those who focused on reducing selection bias pointed out that by including pregnant women, the data collected would be more representative of the general population and less likely to suffer from errors in sampling a select group of participants falsely presumed to represent the general population. On the other hand, those most concerned about introducing confounding bias highlighted differences between non-pregnant and pregnant physiology that might influence the variables being studied. While physiologic differences between research subjects may complicate a study, it does not immediately follow that pregnancy is a confounding variable or that excluding pregnant women is necessary to avoid spurious study conclusions. Yet, for our observational and survey studies, some IRB members seemed willing to accept or even presume that this added complexity undermined the scientific validity of the research without a deeper explanation. Finally, several IRB members in our survey had concerns about enrolling pregnant women sporadically in insufficient numbers to generate scientifically valid results. These issues highlight the need for thoughtful consideration of how to understand and address biases related to subject selection, confounding, and sample size limitations that can weaken scientific validity.
Lessons from the historical exclusion of women from clinical trials may offer insights on ways to mitigate concerns about physiologic differences, small sample sizes, and scientific validity. Prior to the NIH Revitalization Act of 1993, women were often excluded from research based on investigator claims of physiologic sex differences between males and females that would complicate data collection and analysis19. Such androcentric claims were highly problematic, for they prioritized male bodies in research as the “norm” and inappropriately regarded non-male bodies as different, muddying the study population, and possibly skewing study results20. In fact, the exclusion of women on such grounds led to incomplete, inaccurate, and sometimes harmful study conclusions that affected half the population21. Legislation established that mere acknowledgement of physiologic difference no longer sufficed as grounds for exclusion. Investigators had to provide stronger justification based on the health of participants, purpose of the study, or other scientifically grounded rationale. In the absence of such justification, investigators were not only compelled to include women but also to analyze whether study variables had different effects in different subpopulations22.
Mandating this type of subgroup analysis was controversial, for simply enrolling a small number of participants from a subgroup could fail to show meaningful results and could lead to erroneous conclusions about that group23. Avoiding such errors prompted the use of various study designs, statistical approaches, and power analyses to calculate the necessary sample size for subgroup analysis without routinely excluding women from research participation24. Even if the ideal sample size could not be reached, collecting data from a subgroup of women would allow for the possibility of later analysis with pooled data from other studies25. A similar approach may be necessary and useful in the case of enrolling pregnant women when sample sizes are insufficient for separate data analysis. While much progress has been made, the NIH issued a policy in 2015 requiring the study of sex as a biological variable, including in non-human animal studies, with strong justification needed for NIH-funded studies proposing to study only one sex26. Although no mandate currently requires the inclusion of pregnant women, IRBs are in the position to demand more compelling justification for exclusion than some did in our survey.
Moreover, our survey revealed additional areas of disagreement or ambivalence that likely contributed to reluctance. IRB members in our survey disagreed on the level of detail and transparency on the enrollment of pregnant women that investigators should include in a protocol. A surprising number of IRB members were reluctant to require any justification for excluding pregnant women from study participation in vignettes that most categorized as minimal risk. Reasons for this were not entirely clear but may relate to ambivalence about inclusion or presumptions about scientific validity that investigators and IRB members are conditioned to accept as an implied justification for exclusion.
Respondents also held very different opinions about whether investigators should explicitly disclose that pregnant women are invited to participate in a study. Over one third of IRB members opposed such specification in the protocol. Which approach is ethically preferable is unclear and may best be determined on a case by case basis. On one hand, a protocol requirement to clarify whether pregnant women may enroll could prompt more intentional analysis of how to ensure scientific validity, whether a power analysis is needed for sample size projections, and the overall study impact in terms of generalizability and filling research gaps. Alternatively, extra clarification may be redundant since requiring adequate justification for exclusion would give the same information about whether pregnant women may enroll. We found widespread reluctance among IRB members to require justification for the exclusion of pregnant women from minimal risk research and disagreement about the need to explicitly invite pregnant women to participate. Further study is warranted to determine how to best foster the presumption of inclusion among investigators during protocol development. A recent qualitative study identified successful strategies that helped investigators conduct clinical drug, device, and other interventional trials with pregnant women at their academic institution. These strategies included formal institutional policies requiring investigators to provide justification for the exclusion of pregnant women, worksheets with checklists and structured questions to help investigators and IRBs apply Subpart B to individual protocols, and required training for IRB members in making risk assessments in the context of pregnancy27. Our study findings suggest that similar strategies may help to foster a presumption of inclusion for even minimal risk research.
Importantly, we were able to gain insights into certain attitudes and beliefs that appeared to shape decision-making in favor of approving research with pregnant women. While IRB members gave considerable weight to the assessment of research risks, they also considered other pro-enrollment factors including the potential value of a diverse study population for observing the condition being studied and a belief that pregnant women deserve fair access to research.
Proposed areas for IRB guidance
Based on our findings of diverse IRB member interpretations of minimal risk, Subpart B language, and other factors that impeded research approval, we propose the development of specific guidance to assist IRB members in key areas. First, development of a decision aid checklist or flowchart, as administrators suggested, would be valuable for IRB members attempting to decide whether Subpart B should be applied to protocol review. OHRP already provides decision charts on whether an activity is human subjects research that requires IRB review, whether research is eligible for exemption, and other topics28. Adding a decision chart for the application of Subpart B could help to clarify the appropriate level of protocol review for research involving pregnant women, including conditions in which exemption is appropriate. Second, guidance is needed on applying the minimal risk standard to procedures in the context of pregnancy. Since the permissibility of non-beneficial research hinges in large part on the risk classification of individual research procedures, IRB members could benefit from clear examples of minimal risk, routine procedures in prenatal care and activities of daily life that can be applied to research procedures such as a foot x-ray. Third, instructions for characterizing research procedures in opportunistic studies such as PK studies would help to distinguish research risks from those arising from clinical care in order to facilitate approval. Fourth, guidance on properly assessing confounding bias, selection bias, and scientifical validity in studies involving pregnant women would help to alleviate hesitation to approve research already deemed to be minimal risk and allowable under Subpart B requirements. Finally, IRB guidance should urge consistency in requiring clear and compelling justification for excluding pregnant women from enrollment or continuation in a trial if pregnancy occurs after enrollment.
Limitations
Our study has several limitations. Vignette design, although useful for probing aspects of decision-making in hypothetical scenarios, may not capture actual IRB member practices in the real world29. We did not use an externally validated survey instrument, although we followed a rigorous process of survey development. Additionally, recruiting IRB participants from academic centers affiliated with the MFMU network in addition to the PRIMR listserv may have introduced selection bias favoring the participation of academically affiliated IRB members with different experiences and perceptions than the broader population of IRB members. Our sample size precluded our ability to detect statistical differences between groups of IRB members who would vote to approve versus prohibit the research presented in the hypothetical vignettes or to identify statistically significant factors that may influence or predict IRB decision-making. Additionally, we limited administration of the questions about characterizing research procedure risk to respondents with backgrounds in medicine, nursing, or biomedical science, who are most likely to be familiar with medical procedures. However, IRB members with other backgrounds, who will have a voice and vote in IRB deliberations, may have had relevant perspectives on procedure risks that we did not capture. We also did not assess respondents’ familiarity with the medical language in the list of research procedures or in the vignettes, which might have limited their ability to give meaningful responses to the survey questions. Furthermore, while we intentionally avoided specifying the pregnancy trimester in the vignettes in order to allow respondents to consider study risks at any stage of pregnancy, some respondents may have assigned study risk levels differently if we had specified a trimester for the inclusion of pregnant women in the vignettes. Our survey also did not directly question respondents on their knowledge of and familiarity with regulatory exemption criteria for research involving pregnant women. Since only one respondent mentioned this, and our survey did not prompt participants to consider exemption criteria, it was not possible to assess whether other IRB members were aware of exemption eligibility criteria. Finally, our data are subject to the effects of recall bias, given that we asked former IRB members to recall their comfort level interpreting and applying Subpart B. Future studies should explore IRB decision-making prospectively, with a greater number and wider diversity of IRB members, and with attention to additional aspects of regulatory guidance that may influence decisions on approving research with pregnant women.
Conclusion
This study makes important contributions to our understanding of barriers to the responsible inclusion of pregnant women in clinical research. Our data provide valuable insights on IRB decision-making, reflecting the perspectives of IRB members and their reasons for hesitation to approve research with pregnant women. Building on conceptual literature describing a tendency to mischaracterize risk in the context of pregnancy, our study offers empirical evidence of risk assessment challenges among IRB members30. Yet, even beyond challenges in risk assessment, our study revealed additional barriers to the inclusion of pregnant women stemming from concerns around scientific validity and a general culture of reluctance to approve research even when deemed to be minimal risk. These data can help inform specific guidance for IRBs and others, particularly in reviewing minimal risk research that may not offer the prospect of direct benefit but is critical for gaining knowledge to guide the care of pregnant women and the children they bear.
Further development of guidance will likely require collaboration between the clinical, research, and regulatory communities to overcome barriers related to IRB member interpretation and application of federal regulations that govern research in pregnancy. In this way, IRB members can become equipped to facilitate a paradigm shift away from the presumption of exclusion toward the responsible inclusion of pregnant women in clinical research.
Acknowledgments:
Funding
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI108368 (with Lyerly as the PI) and a Diversity Supplement R01AI108368-02S1, supporting White. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Human subjects protection statement: The IRB at the University of North Carolina at Chapel Hill approved this study.
Contributor Information
Amina White, Department of Obstetrics & Gynecology, University of North Carolina at Chapel Hill.
Christine Grady, Clinical Center Department of Bioethics, National Institutes of Health.
Margaret Little, The Kennedy Institute of Ethics, Georgetown University.
Kristen Sullivan, Center for Bioethics, Department of Social Medicine, School of Medicine, University of North Carolina at Chapel Hill.
Katie Clark, University of North Carolina at Chapel Hill, Research Associate, Yale University.
Anne Drapkin Lyerly, Department of Social Medicine and Associate Director, Center for Bioethics, University of North Carolina at Chapel Hill.
References
- 1.Lyerly AD, Little MO, and Faden R, “The Second Wave: Toward Responsible Inclusion of Pregnant Women in Research,” International Journal of Feminist Approaches to Bioethics 1, no. 2 (2008): 5–22; [DOI] [PMC free article] [PubMed] [Google Scholar]; Blehar MC, et al. , “Enrolling Pregnant Women: Issues in Clinical Research,” Women’s Health Issues : Official Publication of the Jacobs Institute of Women’s Health 23, no. 1 (2013): e39–45; [DOI] [PMC free article] [PubMed] [Google Scholar]; Little MO, Wickremsinhe M, and Lyerly AD, “Research in Pregnancy: The Ethics of Risk-Benefit Tradeoffs Between Woman, Fetus, and Future Child [21G],” Obstetrics & Gynecology 129 (2017): 76S. [Google Scholar]
- 2.We recognize that pregnant individuals may have diverse gender identities. Please note that in this article, we will refer to pregnant “women” to reflect the language of our survey instrument.
- 3.Shields KE, and Lyerly AD, “Exclusion of Pregnant Women From Industry-Sponsored Clinical Trials:,” Obstetrics & Gynecology 122, no. 5 (2013): 1077–81. [DOI] [PubMed] [Google Scholar]
- 4.45 C.F.R. 46.102(j)
- 5.Shah S et al. , “How Do Institutional Review Boards Apply the Federal Risk and Benefit Standards for Pediatric Research?,” JAMA 291, no. 4 (2004): 476–82. [DOI] [PubMed] [Google Scholar]
- 6.45 C.F.R. 46.204b[0]
- 7.Blehar et al. , “Enrolling Pregnant Women”; White, A.,“Accelerating the Paradigm Shift toward Inclusion of Pregnant Women in Drug Research: Ethical and Regulatory Considerations,” Seminars in Perinatology, Medications in Pregnancy and Lactation, 39, no. 7 (2015): 537–40; P. [DOI] [PubMed] [Google Scholar]; Payne, “Including Pregnant Women in Clinical Research: Practical Guidance for Institutional Review Boards,” Ethics & Human Research 41, no. 6 (2019): 35–40; [DOI] [PubMed] [Google Scholar]; Farrell R, Michie M, and Pope R, “Pregnant Women in Trials of Covid-19: A Critical Time to Consider Ethical Frameworks of Inclusion in Clinical Trials,” Ethics & Human Research 42, no. 4 (2020): 17–23, [DOI] [PMC free article] [PubMed] [Google Scholar]; and; Mastroianni AC, et al. , “The Pathway Forward: Insights on Factors That Facilitate Research with Pregnant Women,” Ethics & Human Research 42, no. 4 (2020): 2–16. [DOI] [PubMed] [Google Scholar]
- 8.Brandon AR, et al. , “Ethical Challenges in Designing, Conducting, and Reporting Research to Improve the Mental Health of Pregnant Women: The Voices of Investigators and IRB Members,” AJOB Empirical Bioethics 5, no. 2 (2014): 25–43; [Google Scholar]; van der Zande ISE, et al. , “Fair Inclusion of Pregnant Women in Clinical Research: A Systematic Review of Reported Reasons for Exclusion,” in Clinical Research Involving Pregnant Women, ed. Baylis F and Ballantyne A, Research Ethics Forum (Cham: Springer International Publishing, 2016), 65–94.; [Google Scholar]; Mastroianni AC, et al. , “The Pathway Forward.” [Google Scholar]
- 9.Guest G, MacQueen KM, and Namey EE, Applied Thematic Analysis (Thousand Oaks: SAGE Publications, 2011). [Google Scholar]
- 10.45 C.F.R. 46.110.
- 11.Ibid.
- 12.Shah S, et al. , “How Do Institutional Review Boards Apply the Federal Risk and Benefit Standards for Pediatric Research?” [DOI] [PubMed]
- 13.“Appendix B: Recommendations Regarding Risk in Research Involving Children,” Text, HHS.gov, March 28, 2016, accessed February 20, 2020, at https://www.hhs.gov/ohrp/sachrp-committee/recommendations/2005-july-28-letter-appendix-b/index.html. [Google Scholar]
- 14.Blehar et al. , “Enrolling Pregnant Women”; White, “Accelerating the Paradigm Shift toward Inclusion of Pregnant Women in Drug Research”; Lyerly, Little, and Faden, “The Second Wave.” [Google Scholar]
- 15.Gonzalez D et al. , “Use of Opportunistic Clinical Data and a Population Pharmacokinetic Model to Support Dosing of Clindamycin for Premature Infants to Adolescents,” Clinical Pharmacology & Therapeutics 96, no. 4 (2014): 429–37; [DOI] [PMC free article] [PubMed] [Google Scholar]; Autmizguine J et al. , “Pharmacokinetic Studies in Infants Using Minimal-Risk Study Designs,” Current Clinical Pharmacology 9, no. 4 (2014): 350–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheffield JS, et al. , “Designing Drug Trials: Considerations for Pregnant Women,” Clinical Infectious Diseases: An Official Publication of the Infectious Diseases Society of America 59 Suppl 7 (2014): S437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.“Draft Guidance: Pregnant Women: Scientific and Ethical Considerations for Inclusion in Clinical Trials Guidance for Industry,” Food and Drug Administration, April 2018, accessed January 28, 2021, at https://www.fda.gov/media/112195/download. [Google Scholar]
- 18.Yao LP “Risk Communication Advisory Meeting: Communicating Information about Risks of Prescription Products and Vaccines Used During Pregnancy,” Food and Drug Administration, March 5, 2018, accessed January 29, 2021, at https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/RiskCommunicationAdvisoryCommittee/UCM599983.pdf. [Google Scholar]
- 19.Bennett JC, “Inclusion of Women in Clinical Trials--Policies for Population Subgroups,” The New England Journal of Medicine 329, no. 4 (1993): 288–92; [DOI] [PubMed] [Google Scholar]; Baird KL, “The New NIH and FDA Medical Research Policies: Targeting Gender, Promoting Justice,” Journal of Health Politics, Policy and Law; Durham 24, no. 3 (1999): 531–65. [DOI] [PubMed] [Google Scholar]
- 20.Merton V, “The Exclusion of Pregnant, Pregnable, and Once-Pregnable People (a.k.a. Women) from Biomedical Research,” SSRN Scholarly Paper (Rochester, NY: Social Science Research Network, 1993); [PubMed] [Google Scholar]; DeBruin DA, “Justice and the Inclusion of Women in Clinical Studies: An Argument for Further Reform,” Kennedy Institute of Ethics Journal 4, no. 2 (1994): 117–46. [DOI] [PubMed] [Google Scholar]
- 21.Merton, “The Exclusion of Pregnant, Pregnable, and Once-Pregnable People (a.k.a. Women) from Biomedical Research.” [PubMed]
- 22.“NIH Policy and Guidelines on The Inclusion of Women and Minorities as Subjects in Clinical Research, Grants.nih.gov,” accessed February 24, 2020, at https://grants.nih.gov/policy/inclusion/women-and-minorities/guidelines.htm.
- 23.Bennett, “Inclusion of Women in Clinical Trials--Policies for Population Subgroups.” [DOI] [PubMed]
- 24.Ibid.
- 25.Ibid.
- 26.“NOT-OD-15–102: Consideration of Sex as a Biological Variable in NIH-Funded Research,” accessed August 3, 2020, at https://grants.nih.gov/grants/guide/notice-files/NOT-OD-15-102.html.
- 27.Mastroianni et al. , “The Pathway Forward.”
- 28.“Human Subject Regulations Decision Charts,” Text, HHS.gov, February 25, 2016, accessed February 28, 2020, at https://www.hhs.gov/ohrp/regulations-and-policy/decision-charts/index.html. [Google Scholar]
- 29.Aguinis H and Bradley KJ, “Best Practice Recommendations for Designing and Implementing Experimental Vignette Methodology Studies,” Organizational Research Methods 17, no. 4 (2014): 351–71. [Google Scholar]
- 30.Lyerly AD, et al. , “Risk and the Pregnant Body,” Hastings Center Report 39, no. 6 (2009): 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]