

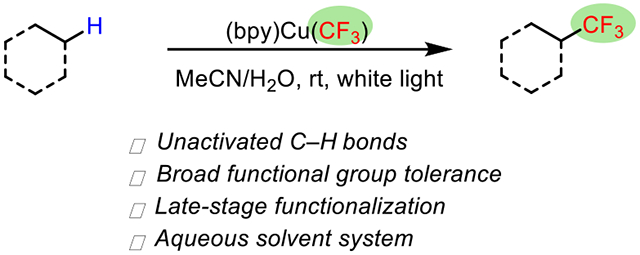
Abstract
A straightforward method for the undirected trifluoromethylation of unactivated methylene units was developed. The reaction proceeds in aqueous acetonitrile with Grushin’s reagent, bpyCu(CF3)3, under broad-spectrum white-light irradiation. The trifluoromethylation tolerates a wide range of functional groups including ketones, esters, nitriles, amides, alcohols, and carboxylic acids. The C–H cleavage step is performed via intermolecular H atom abstraction, and the selectivities across a range of methylene units are reported. Mechanistic studies offer a general reaction coordinate for the overall transformation.
Graphical Abstract
The introduction of fluorine atoms into small molecules selectively and safely has become of paramount importance across a range of industries. Numerous approved drugs and agrochemicals possess judiciously located fluorine atoms.1 In general, the two most common fluorinated motifs in biologically active small molecules include simple monofluorinated motifs with a single C–F bond or the presence of the more electron-withdrawing trifluoromethyl group—CF3.2 The installation of such motifs is driven by a variety of reasons that range from modulating the pKa of nearby functionalities, dramatically changing the overall dipole of the target molecule, and even interrupting biological oxidation of the molecule to tune pharmacokinetics.2 What is striking about a variety of these examples is the large percentage of aromatic C–F and C–CF3 groups. Far more methods exist to target their installation relative to aliphatic fluorination.3 This leads to a dearth of such compounds in typical screening libraries and underrepresentation in chemical campaigns.4 By expending the toolbox for selective installation of aliphatic C–F-containing functionality, we can expand libraries and simplify potential chemical routes to hypothetical molecules of interest.
These issues have driven academic and industrial groups alike to focus on the problem of trifluoromethylation.5 One strategy to generate aliphatic C(sp3)–CF3 bonds proceeds through carbon-based radicals generated from carboxylic acids6,7 or halides;8,9 however, the direct functionalization of carbon–hydrogen (C–H) bonds–the most abundant moiety in organic molecules—represents a more direct approach to trifluoromethylation.10 In this context, a small number of recent publications have sought different approaches to convert Csp3–H bonds into Csp3–CF3 systems. While somewhat limited at this time, the most common trifluoromethylation employs benzylic C–H substrates (Scheme 1a).5a,11 Remarkably, similar conditions were reported near simultaneously by our group5a and the Liu group in 2018.11a Catalytic copper in combination with bpyZn(CF3)2 was accomplished by Li the following year.11b Fluoroamide-directed C–H functionalization, pioneered by our group in 2016,12 has expanded beyond fluorination to include other functionalities, including trifluoromethylation (Scheme 1b).13 Recently, Macmillan and co-workers demonstrated the trifluoromethylation of pyrrolidine using the electrophilic trifluoromethyl source of Togni’s reagent II.14 While these studies offer unique solutions to these specific substrates, we sought a more general trifluoromethylation reaction of unactivated, aliphatic C–H bonds (Scheme 1c).
Scheme 1.

Methods for Csp3–H Trifluoromethylation
To begin, we surveyed the reaction conditions for the trifluoromethylation of cyclohexane with Grushin’s reagent, bpyCu(CF3)3 (Table 1). The trifluoromethylation proceeded well with or without persulfate present (Table 1, entries 1–3), but not without light irradiation (Table 1, entries 4–6). As Grushin’s reagent can be excited by both longwave UV and blue light,5a,b the reaction proceeded well under 365 nm LED irradiation or with broad-spectrum white-light irradiation (Table 1, entries 1 and 5). While the trifluoromethylation proceeded in low yield in water, the addition of acetonitrile cosolvent dramatically increased the yield—presumably by solubilizing bpyCu(CF3)3 (Table 1, entries 7–11). Supersilane inhibited the reaction,5a suggesting a critical role for the initial trifluoromethyl radical produced upon irradiation (Table 1, entry 13). With the optimal conditions, more than one trifluoromethyl from Grushin’s reagent can produce the new carbon–carbon bond (Table 1, entry 1).
Table 1.
Optimization of Pertinent Reaction Parametersa

| ||
|---|---|---|
|
| ||
| entry | catalyst | yieldb (%) |
| 1 | standard conditions | 110 |
| 2 | without K2S2O8 | 47 |
| 3 | under air | 66 |
| 4 | no white LEDs | nd |
| 5 | 365 nm | 85 |
| 6 | 50 °C instead of white LEDs | trace |
| 7 | 0.05 M MeCN/H2O (11:1) | 62 |
| 8 | MeCN/H2O (1:5) | trace |
| 9 | MeCN/H2O (1:1) | 82 |
| 10 | MeCN/H2O (5:1) | 98 |
| 11 | acetone instead of MeCN | trace |
| 12 | (CH3)3COH instead of MeCH | 23 |
| 13 | adding (TMS)3SiH | trace |
Unless otherwise noted, all the reactions were run with 1a (0.2 mmol) and 2a (0.04 mmol) in 0.4 mL of solvent for 12 h.
Yields were determined by 19F NMR spectroscopy with fluorobenzene as the internal standard.
With suitable conditions for the trifluoromethylation of cyclohexane obtained, a variety of unactivated, aliphatic substrates were evaluated (Scheme 2). The reaction worked reasonably well across a range of methylene substrates to produce products 3a–3q. Moreover, the exceptionally mild reaction conditions tolerated a number of commonly reactive functional groups such as ketones (3c–3e), ethers (3q), nitriles (3f), esters (3g and 3p), a range of amide derivatives (3h–3j, 3l), and even a free carboxylic acid (3n). The reaction could also provide Ruppert–Prakash derivative 3o.15 While the reaction performed reasonably well over a range of substrates, we were intrigued by the various selectivities observed in the reaction. While in some cases, trifluoromethylation α to an acidifying functional group was detected in small quantities (3c, 3f, 3j and 3n), other systems provided no detectable a trifluoromethylation (3d, 3e, 3m, and 3p). Moreover, the yield of certain substrates could be improved by changing the ratio of MeCN/H2O. For example, 3i was produced in only 42% yield in 11:1 MeCN/H2O, but trifluoromethylation improved to 56% in 1:1 MeCN/H2O. Consequently, these conditions represent an operationally simple and convenient method for the trifluoromethylation of a wide range of substrate classes..
Scheme 2. Substrate Scope for Trifluoromethylationa.

aAll reactions were run on a 0.2 mmol scale with 1 (1 mmol) and 2a (0.2 mmol) in 2 mL of solvent for 12 h unless otherwise noted.
bYields and regioselectivities determined by 19F NMR analysis of the crude reaction mixtures with fluorobenzene as the internal standard.
c90% in 1.0 mmol scale.
d24 h.
With access to this unique trifluoromethylation reaction, we sought to understand some of the fundamental steps involved in the reaction (Scheme 3). Based on previous work demonstrating the homolysis of Grushin’s reagent under both long-wave UV and visible-spectrum light,5a,b we postulated the formation of trifluoromethyl-based radicals as key, long-lived species in the reaction. Interestingly, we found the combination of TEMPO and Grushin’s reagent, lacking substrate 1, produced TEMPO–CF3 under the reaction conditions (Scheme 3a). Moreover, we found a positive correlation between the amount of organic solvent in the reaction with the amount of TEMPO–CF3 produced (entry 1 vs entry 2, Scheme 3a). We attribute this responsive photophysical behavior to the lack of aqueous solubility of bpyCu(CF3)3, but studies remain ongoing. Control reactions with deuterated substrate (entries 1 and 4, Scheme 3b) and deuterated reagents (entries 2 and 3, Scheme 3b) suggest that the C–H bond-cleaving step is performed by the trifluoromethyl-based radicals produced in the reaction. To examine whether C–H cleavage might be the slow step of the overall transformation, we conducted a parallel KIE study (Scheme 3c). Not surprisingly, a large, positive KIE of 5.4 was observed in this experiment. Taken together, these data enabled the formulation of a mechanistic hypothesis for the overall transformation (Figure 1).
Scheme 3.

Key Mechanistic Experiments
Figure 1.

Proposed mechanism.
The proposed roles for the reagents needed for the trifluoromethylation of unactivated methylene groups is delineated in Figure 1. The reaction likely proceeds through the homolysis of Grushin’s reagent to generate an active, relatively long-lived trifluoromethyl-based radical. The newly formed trifluoromethyl-based radical can proceed through path b to abstract a C–H bond of the substrate, thereby generating a new carbon-based, secondary radical. This radical can recombine with the newly formed Cu(II) species to form a secondary alkyl copper species that undergoes rapid reductive elimination to give the desired products 3 and inactive Cu(I)CF3. We cannot rule out a second path wherein homolyzed persulfate represents an alternative C–H abstracting entity for the reaction (path a). That said, previous work by our group has also demonstrated that the role of persulfate may be more important for providing an ancillary ligand on copper intermediates to lower the activation barrier to radial recombination and reductive elimination.5b
In summary, we have developed an efficient copper-based system for the trifluoromethylation of unactivated methylenes in a wide range of chemical environments with interesting selectivities. Using simple, air- and moisture-tolerant reagents, the efficient construction of C–CF3 and Si–CF3 bonds can be accomplished under aqueous conditions. The reaction proved tolerant of a range of common functional groups that should provide a valuable transformation for practicing organic chemists. Mechanistic studies and experiments on the relative rates of Grushin’s reagent homolysis in different solvents remain ongoing.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge funds from Indiana University in partial support of this work. We also gratefully acknowledge the NIH (R01GM121668). Eli Lilly & Co. and Amgen supported this work through the Lilly Grantee Award and the Amgen Young Investigator Award. We thank IU mass spectrometry for HRMS spectra (NSF Grant No. CHE1726633).
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.orglett.0c03891
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.0c03891.
Experimental details, compound characterization, and NMR data (PDF)
The authors declare no competing financial interest.
Contributor Information
Jiachen He, Department of Chemistry, Indiana University, Bloomington, Indiana 47405-7102, United States.
Truong N. Nguyen, Department of Chemistry, Indiana University, Bloomington, Indiana 47405-7102, United States
Shuo Guo, Department of Chemistry, Indiana University, Bloomington, Indiana 47405-7102, United States.
Silas P. Cook, Department of Chemistry, Indiana University, Bloomington, Indiana 47405-7102, United States.
REFERENCES
- (1).(a) Ma J-A; Cahard D J. Fluorine Chem 2007, 128, 975–996. [Google Scholar]; (b) Wang J; Sánchez-Roselló M; Aceña JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Chem. Rev 2014, 114, 2432–2506. [DOI] [PubMed] [Google Scholar]; (c) Nie J; Guo H-C; Cahard D; Ma J-A Chem. Rev 2011, 111, 455–529. [DOI] [PubMed] [Google Scholar]; (d) Ilardi EA; Vitaku E; Njardarson JT J. Med. Chem 2014, 57, 2832–2842. [DOI] [PubMed] [Google Scholar]; (e) Kirk KL Org. Process Res. Dev 2008, 12, 305–321. [Google Scholar]; (f) Zhou Y; Wang J; Gu Z; Wang S; Zhu W; Aceñ a JL; Soloshonok VA; Izawa K; Liu H Chem. Rev 2016, 116, 422–518. [DOI] [PubMed] [Google Scholar]
- (2).(a) Müller K; Faeh C; Diederich F Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]; (b) Studer A Angew. Chem. Int. Ed 2012, 51, 8950–8958. [DOI] [PubMed] [Google Scholar]; (c) Meanwell NA J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]; (d) Shimizu M; Hiyama T Angew. Chem. Int. Ed 2005, 44, 214–231. [DOI] [PubMed] [Google Scholar]; (e) Koike T; Akita M Acc. Chem. Res 2016, 49, 1937–1945. [DOI] [PubMed] [Google Scholar]; (f) Furet P; Guagnano V; Fairhurst RA; Imbach-Weese P; Bruce I; Knapp M; Fritsch C; Blasco F; Blanz J; Aichholz R; Hamon J; Fabbro D; Caravatti G Bioorg. Med. Chem. Lett 2013, 23, 3741–3748. [DOI] [PubMed] [Google Scholar]
- (3).Bume DD; Harry SA; Lectka T; Pitts CR J. Org. Chem 2018, 83, 8803–8814. [DOI] [PubMed] [Google Scholar]
- (4).(a) Alonso C; Martínez de Marigorta E; Rubiales G; Palacios F Chem. Rev 2015, 115, 1847–1935. [DOI] [PubMed] [Google Scholar]; (b) Inoue M; Sumii Y; Shibata N ACS Omega 2020, 5, 10633–10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Guo S; AbuSalim DI; Cook SP J. Am. Chem. Soc 2018, 140, 12378–12382. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guo S; AbuSalim DI; Cook SP Angew. Chem., Int. Ed 2019, 58, 11704–11708. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xiao H; Shen H; Zhu L; Li C J. Am. Chem. Soc 2019, 141, 11440–11445. [DOI] [PubMed] [Google Scholar]; (d) Le C; Chen TQ; Liang T; Zhang P; MacMillan DWC Science 2018, 360, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Tan X; Liu Z; Shen H; Zhang P; Zhang Z; Li C J. Am. Chem. Soc 2017, 139, 12430–12433. [DOI] [PubMed] [Google Scholar]
- (7).Kautzky JA; Wang T; Evans RW; MacMillan DWC J. Am. Chem. Soc 2018, 140, 6522–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kornfilt DJP; MacMillan DWC J. Am. Chem. Soc 2019, 140, 6853–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shen H; Liu Z; Zhang P; Tan X; Zhang Z; Li C J. Am. Chem. Soc 2017, 139, 9843–9846. [DOI] [PubMed] [Google Scholar]
- (10).Zhu L; Fang Y; Li C Chin. J. Chem 2020, 38, 787–789. [Google Scholar]
- (11).(a) Paeth M; Carson W; Luo J-H; Tierney D; Cao Z; Cheng M-J; Liu W Chem. - Eur. J 2018, 24, 11559–11563. [DOI] [PubMed] [Google Scholar]; (b) Xiao H; Shen H; Zhu L; Li C J. Am. Chem. Soc 2019, 141, 11440–11445. [DOI] [PubMed] [Google Scholar]
- (12).Groendyke BJ; AbuSalim DI; Cook SP J. Am. Chem. Soc 2016, 138, 12771–12774. [DOI] [PubMed] [Google Scholar]
- (13).Liu Z; Xiao H; Zhang B; Shen H; Zhu L; Li C Angew. Chem, Int. Ed 2019, 58, 2510–2513. [DOI] [PubMed] [Google Scholar]
- (14).Sarver PJ; Bacauanu V; Schultz DM; DiRocco DA; Lam Y.-h.; Sherer EC; MacMillan DWC Nat. Chem 2020, 12, 459–467. [DOI] [PubMed] [Google Scholar]
- (15).(a) Prakash GKS; Mandal M J. Fluorine Chem 2001, 112, 123–131. [Google Scholar]; (b) Prakash GKS; Jog PV; Batamack PTD; Olah GA Science 2012, 338, 1324–1327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.