

Abstract
Abstract
Coronavirus disease-19 (COVID-19) pandemic caused by severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) has infected approximately 26 million people and caused more than 6 million deaths globally. Spike (S)-protein on the outer surface of the virus uses human trans-membrane serine protease-2 (TMPRSS2) to gain entry into the cell. Recent reports indicate that human dipeptidyl peptidase-4 inhibitors (DPP4 or CD26) could also be utilized to check the S-protein mediated viral entry into COVID-19 patients. RNA dependent RNA polymerase (RdRp) is another key virulence protein of SARS-CoV-2 life cycle. The study aimed to identify the potential anti-SARS-CoV-2 inhibitors present in Withania somnifera (Solanaceae) using computer aided drug discovery approach. Molecular docking results showed that flavone glycoside, sugar alcohol, and flavonoid present in W. somnifera showed − 11.69, − 11.61, − 10.1, − 7.71 kcal/mole binding potential against S-protein, CD26, RdRp, and TMPRSS2 proteins. The major standard inhibitors of the targeted proteins (Sitagliptin, VE607, Camostat mesylate, and Remdesivir) showed the − 7.181, − 6.6, − 5.146, and − 7.56 kcal/mole binding potential. Furthermore, the lead phytochemicals and standard inhibitors bound and non-bound RdRp and TMPRSS2 proteins were subjected to molecular dynamics (MD) simulation to study the complex stability and change in protein conformation. The result showed energetically favorable and stable complex formation in terms of RMSD, RMSF, SASA, Rg, and hydrogen bond formation. Drug likeness and physiochemical properties of the test compounds exhibited satisfactory results. Taken together, the present study suggests the presence of potential anti-SARS-CoV-2 phytochemicals in W. somnifera that requires further validation in in vitro and in vivo studies.
Graphical Abstract

Supplementary information
The online version contains supplementary material available at 10.1007/s42535-022-00404-4.
Keywords: SARS-CoV-2; Withania somnifera; Phytochemical, TMPRSS2; RNA dependent RNA polymerase
Introduction
Coronavirus disease-19 (COVID-19) has become pandemic in humans by targeting the severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) entry and replication in the viral host. Numerous studies have reported the potential druggable targets to control the SARS-CoV-2 infection (Gupta et al. 2021; Wu et al. 2020; Kushwaha et al. 2021a; Maurya et al. 2020). SARS-CoV-2 interacts with host cellular membrane ACE2 receptors (angiotensin converting enzyme-2) and TMPRSS2 co-receptors (trans-membrane serine protease-2) with its spike glycoprotein and initiate host-pathogen interaction. One of the Spike-protein’s co-receptor viz. DPP4/CD26 (dipeptidyl peptidase-4) involved in COVID-19 pathophysiology has received much attention. Solerte et al. (2020) determined that CD26 inhibitors may helpful to reduce the viral entry, replication, cytokine storm and inflammation in COVID-19 patients. Recently, genetics-based study from our group revealed the coding and regulatory variations of TMPRSS2 and CD26 in prolonging SARS-CoV-2 infection in humans (Senapati et al. 2020). The spike glycoprotein S1 and S2 domains involved in the protein-protein interaction and membrane fusion establish viral entry into the cell. Binding of cellular receptor to the S1 domain induce conformational change in the S2 domain which create cleavage site for TMPRSS2, a human serine protease. TMPRSS2-induced cleavage generates S-fragments and releases them into the cellular supernatant. After processing in the early and late endosome, viral-genome is being replicated with the support of RNA dependent RNA polymerase-RdRp and transcribed into viral mRNA. Following translation, synthesized viral proteins undergoes maturation and generation of viral particle. Targeting entry of virus particle into the cell by inhibiting spike and/or host membrane receptor/co-receptors is a better strategy to develop SARS-CoV-2 anti-viral drug discovery. Moreover, targeting TMPRSS2 and RdRp proteins may decrease the SARS-CoV-2 mediated virulence and host pathogenicity.
Type II transmembrane serine proteases is an important protein family involved in the pathophysiology of cancer and viral disease. TMPRSS2 is a member of hepsin/transmembrane protease, serine (TMPRSS) subfamily. TMPRSS2 is known to prime S2 domain of SARS-CoV-2 which is a key step in viral infection and virulence. Various in vitro, and in vivo studies showed TMPRSS2 inhibition mediated decrease in coronavirus (SARS-CoV, SARS-CoV-2 and MERS-CoV) induced immunopathology, virulence and airway spread of the disease (Iwata-Yoshikawa et al. 2019; Hoffmann et al. 2020) reported that a clinically proven TMPRSS2 inhibitor has potential to inhibit viral infection in SARS-CoV-2 infected lung cells. Cluster of differentiation 26 (CD26), also known as dipeptidyl peptidase-4 (DPP4) is a human cell surface glycoprotein having dipeptidyl peptidase activity at extracellular domain. Besides, it is also present in soluble form in body fluids and possesses significant serine exopeptidase activity (Casrouge et al. 2018). DPP4 inhibitors (DPP4i) are clinically approved antidiabetic drug for type 2 diabetes mellitus. Pharmacologically active CD26 inhibitors are known to decrease pro-inflammatory cytokines and lower the incidence risk of autoimmune diseases (Kim et al. 2015). Acute respiratory failure and deregulated inflammation is the major cause related to SARS-CoV-2 death. Reports suggest that CD26 inhibitor sitagliptin could be utilized as a therapeutic candidate for acute respiratory distress syndrome (ARDS). Clinically approved CD26 inhibitors including vildagliptin and saxagliptin either act as pseudo-substrates or substrate competitive inhibitors that binds to the same catalytic site of CD26 receptor (Berger et al. 2017). Importantly, the active site of the receptor is different from the viral-human (Spike glycoprotein-CD26) protein-protein interaction interface (Lu et al. 2013; Vankadari and Wilce 2020), where CD26 interacts with S1 domain of SARS-CoV-2 spike glycoprotein (Vankadari and Wilce 2020). SARS-CoV-2 like other coronaviruses utilizes CD26 other than ACE2 as a functional receptor to enter into the host cell.
Natural products have been used overtime for the treatment of various diseases (Kushwaha et al. 2019, 2020a, b, 2021b; Kumar et al. 2019; Waseem et al. 2017). Phytochemicals present in medicinal plants are known to possess antiviral potential by targeting different proteins of Ebola, Herpes Simplex Virus 1 and 2, Human Immunodeficiency Virus, Adenovirus, Respiratory Syncytial virus, including SARS-CoV-2 virus (Li et al. 2005; Naveed et al. 2018; Kushwaha et al. 2021c). Withania somnifera (Solanaceae) is a well-known Indian medicinal plant. Phytochemicals present in W. somnifera possess DPP4 inhibitory and immunomodulatory potential (Kempegowda et al. 2018). Besides, the plant is well documented for its antiviral potential against several viruses (such as H1N1 influenza and HIV virus). Research demonstrates that W. somnifera phytochemicals inhibit viral protein, alters inflammation and immune system (Cai et al. 2015; Jain et al. 2018; Ganguly et al. 2018). Previously, our team identified W. somnifera phytochemicals having potential to bind SARS-CoV-2 main protease. In the present study, for the first time, we studied the SARS-CoV-2 (Spike-protein, RdRp) and human host protein (CD26 and TMPRSS2) binding efficacy of W. somnifera phytoconstituents. These phytochemicals were docked against test proteins to identify their binding potential in comparison to respective standard inhibitors. Further, the MM-GBSA and molecular dynamics (MD) simulation was performed to identify the binding free energy, stability and protein conformation of the lead phytochemical-protein complexes. Moreover, the druglikeness and physiochemical properties of the lead compounds were also studied.
Materials and methods
Withania somnifera phytochemical retrieval and preparation
.
Phytochemicals present in Withania somnifera were retrieved from previous published literature Kushwaha et al. 2021c.) The 3D or 2D structures of standard compounds against respective targeted proteins were retrieved from the NCBI PubChem in .sdf format. The .sdf structure files were converted into .mol to get the respective 3D conformation of the compound with the assistance of Open babel molecule format converter (O’Boyle et al. 2011). Energy of the test ligands were minimized by applying mmff94 force field and conjugate gradients optimization algorithm using PyRx-Python prescription 0.8 for 200 steps (Dallakyan 2008).
Protein retrieval and preparation
Crystal structure of target proteins viz., CD26 (PDB ID: 4PNZ), Spike glycoprotein (PDB ID: 6VSB), and RdRp (PDB ID: 6M71) were obtained from Protein Data Bank (https://www.rcsb.org/) with a resolution of 1.90 Å, 3.46 Å, and 2.90 Å (Berman et al. 2000). TMPRSS2 modeled protein (PDB ID: 5CEL was used as template) was adapted from our previous publication (Kushwaha et al. 2021c). Three dimensional structure of the targeted proteins were prepared for molecular docking using UCSF Chimera (Pettersen et al. 2004). These proteins were cleaned and optimized by removing the ligands and other heteroatoms including water. Following this step, the energy minimization of the proteins was performed using steepest descent method having 100 steps (step size 0.02 Å), and a conjugate gradient method with 10 steps (step size 0.02 Å) using UCSF Chimera.
Molecular docking studies
For docking analysis, receptors and ligands were loaded in Auto Dock Tools 1.5.6 (ADT) (Trott and Olson 2010; Singh et al. 2021; El Khatabi et al. 2021; Verma et al. 2020). Following the merging of non-polar hydrogens and torsions applied to the ligands by rotating all rotatable bonds, the Gestgeiger partial charges were assigned. Docking calculations were performed for the entire protein model. Auto Dock tools were used to assign polar hydrogen atoms, Kollman charges, and solvation parameters. For the exploration of the space of active binding with different efficacy, the Lamarckian Genetic Algorithm option of Auto Dock 4.2 was used. The grid boxes were prepared for the entire binding site (Spike glycoprotein) or the active site (CD26, RdRp and TMPRSS2) of the target proteins to provide sufficient space for the ligand’s translational and rotational walk. A maximum number of 27,000 GA operations were generated on a single population of 150 individuals for each of the 30 independent runs. UCSF Chimera (v.1.10.2) was used for visualization and LigPlot+ (v.1.4.5) used for the analysis of interface between receptors and ligands (Laskowski et al. 2011).
MM-GBSA analysis
The binding free energies of the targeted protein(s) complexed with the lead phytochemicals were calculated by using Prime MM-GBSA (Molecular Mechanics/Generalized Born Model and Solvent Accessibility) calculations. The calculation includes the VSGB solvent model, OPLS_2005 force field, and rotamer search algorithms. The docked PDB file was used to Prime MM-GBSA simulation and subsequent computation of the binding free energy.
Molecular dynamics simulation
All-atom MD simulations were performed for TMPRSS2 and RdRp protein in unbound and lead phytochemical/standard inhibitor bound complex. Simulations were performed using GROMOS 54A7 force field in the GROMACS 5.1.1 package (Van Der Spoel et al. 2005; Ezebuo et al. 2019; Kushwaha et al. 2021c). Ligand topologies and parameters generated using PRODRG server and were combined with the protein topology to make simulation complexes (Schüttelkopf and van Aalten 2004; Singh et al. 2020a, b). All systems were solvated using the SPC water model in a cubic box. Energy minimization was performed to get rid of steric clashes by using 1500 steps of the steepest descent approach. Following energy minimization, the temperature of all systems was increased from 0 to 300 K in equilibrium phase of 100 ps at a constant volume and stable pressure of 1 bar. The final MD run was set to 10,000 ps for all six systems. Trajectory analysis was performed using various GROMACS utilities such as gmx rms, gmx rmsf, gmx gyrate, gmx sasa, gmx hbond and gmx do_dssp for the analysis of rmsd, rmsf, radius of gyration, solvent accessible surface area, hydrogen bonds and secondary structure respectively.
Pharmacological and physiochemical property prediction
Swiss ADME (Swiss Institute of Bioinformatics, Switzerland), a widely used web tool was utilized for the prediction of physicochemical properties, pharmacokinetics, drug likeness and water solubility by bioavailability radar generation. Bioavailability radar confers six physicochemical properties i.e. flexibility, lipophilicity, saturation, size, polarity and solubility. The Egan BOILED-Egg method of SwissADME tool was applied to determine the absorption of the inhibitors in the gastrointestinal tract and brain (Daina et al. 2017). BOILED-Egg (Brain or Intestinal Estimated permeation predictive model), also called Egan egg, was used to predict a threshold and a clear graphical representation of how far a molecular structure is from the ideal one for good absorption. In 2D graphical representation, the molecules present in yolk sac will passively permeate through the blood–brain barrier (BBB), whereas the molecules present in the white region are expected to be passively absorbed by the gastrointestinal (GI) tract.
Result
In the present study, a total of 93 Withania sominfera compounds were searched from the literature and docked against SARS-CoV-2 (spike glycoprotein and RdRp) and human host proteins (CD26 and TMPRSS2). Binding energy of the lead compounds (cut-off value ≤− 4.5 kcal/mole) against targeted test proteins is shown in Table 1. Two dimensional structure of the lead compounds against all targeted protein with cut-off value ≤ binding energy of standard inhibitors are depicted in Fig. 1.
Table 1.
Binding energy of W. somnifera phytochemicals against SARS-CoV-2 and human host targeted proteins with cutoff ≤ − 4.5 kcal/mole
| S. No. | PMID | Phytochemical | Group of compound | Binding energy kcal/mole |
|---|---|---|---|---|
| Human CD26 | ||||
| 1 | 10,190,763 | Quercetin-3-rutinoside-7-glucoside | Flavonoid | − 11.69 |
| 2 | 5,280,805 | Rutin | Flavonoid | − 9.49 |
| 3 | 5988 | 5-phospho-D-ribosylamine | Pentose phosphate | − 8.62 |
| 4 | 5,315,599 | Caffeoyl quinic acid | Polyphenol | − 7.91 |
| 5 | 595 | Cystine | Glucosinolates | − 7.48 |
| 6 | 189,702 | Sitoindoside X | Saponin | − 7.15 |
| 7 | 439,341 | Maltose | Sugar | − 7.14 |
| 8 | 11,850 | Dulcitol | Sugar alcohol | − 6.61 |
| 9 | 12,306,778 | Anahygrine | Alkaloid | − 6.39 |
| 10 | 5,280,343 | Quercetin | Flavonoid | − 6.37 |
| 11 | 44,423,097 | 17-Alpha-hydroxywithanolide D | Steroidal lactones | − 6.22 |
| 12 | 3,802,778 | Isochlorogenic acid B | Phenolics | − 6.04 |
| 13 | 443,143 | Anaferine | Alkaloid | − 6.01 |
| 14 | 92,987 | Pelletierine | Alkaloid | − 6.01 |
| 15 | 12,444,363 | Tropigline | Alkaloid | − 5.87 |
| 16 | 5810 | Hydroxyproline | Amino acid | − 5.8 |
| 17 | 1,201,543 | Cuscohygrine | Alkaloid | − 5.62 |
| 18 | 5,280,460 | Scopoletin | Coumarin | − 5.6 |
| 19 | 89,594 | Nicotine | Alkaloid | − 5.54 |
| 20 | 189,586 | Sitoindoside IX | Saponin | − 5.37 |
| 21 | 5862 | Cysteine | Amino acid | − 5.36 |
| 22 | 101,687,980 | Somnifericin | Alkaloid | − 5.18 |
| 23 | 8424 | Tropine | Alkaloid | − 5.1 |
| 24 | 449,293 | Psuedotropine | Alkaloid | − 5.1 |
| 26 | 1,794,427 | Chlorogenic acid | Phenolics | − 5.05 |
| 27 | 3931 | 9, 12-Octadecadienoic acid | Fatty acid | − 5.02 |
| 28 | 21,679,022 | Withanolide J | Steroidal lactones | − 4.94 |
| 29 | 21,606,678 | (18R)-Withaphysalin F | Steroidal lactones | − 4.79 |
| 30 | 101,281,365 | Withanolide Q | Steroidal lactones | − 4.54 |
| 31 | 5,742,590 | Sitogluside | Phytosterol-like compound | − 4.53 |
| 32 | 23,266,146 | Withanolide O | Steroidal lactones | − 4.5 |
| SARS-CoV-2 Spike protein | ||||
| 1 | 11,850 | Galactitol | Sugar alcohol | − 10.1 |
| 2 | 1,794,427 | Chlorogenic acid | Phenolics | − 9.92 |
| 3 | 10,190,763 | Quercetin-3-rutinoside-7-glucoside | Flavonoid | − 9.66 |
| 4 | 3,802,778 | Isochlorogenic acid B | Phenolics | − 8.51 |
| 5 | 5,280,805 | Rutin | Flavonoid | − 8.38 |
| 6 | 5,280,343 | Quercetin | Flavonoid | − 8.25 |
| 7 | 5,315,599 | Caffeoyl quinic acid | Polyphenol | − 8.21 |
| 8 | 5988 | 5-phospho-D-ribosylamine | Pentose phosphate | − 7.67 |
| 9 | 439,341 | Maltose | Sugar | − 6.86 |
| 10 | 189,702 | Sitoindoside X | Saponin | − 6.33 |
| 11 | 70,684,083 | 2,3-Didehydrosomnifericin | Steroidal lactone | − 6.09 |
| 12 | 23,266,147 | Withanolide N | Steroidal lactone | − 5.63 |
| 13 | 13,743,195 | 4-Deoxyphysalolactone | Steroidal lactone | − 5.53 |
| 14 | 102,066,417 | Somniwithanolide | Steroidal lactone | − 5.52 |
| 15 | 101,687,980 | Somnifericin | Alkaloid | − 5.41 |
| 16 | 6508 | Quinic acid | Cyclitol | − 4.94 |
| 17 | 5,280,460 | Scopoletin | Coumarin | − 4.88 |
| 18 | 101,687,981 | Withaoxylactone | Steroidal lactone | − 4.85 |
| 19 | 189,586 | Sitoindoside IX | Saponin | − 4.83 |
| 20 | 44,423,097 | 17-Alpha-hydroxywithanolide D | Steroidal lactone | − 4.83 |
| 21 | 21,679,022 | Withanolide J | Steroidal lactone | − 4.8 |
| 22 | 10,767,792 | 2, 3- Dihydro-3beta-Methoxy-Wihaferin A | Steroidal lactone | − 4.72 |
| 23 | 131,751,517 | 2,3-Dihydrowithanolide E | Steroidal lactone | − 4.69 |
| 24 | 179,575 | Wihanolide L | Steroidal lactone | − 4.68 |
| 25 | 387,980 | Withafastuosin E | Steroidal lactone | − 4.67 |
| 26 | 101,710,595 | Withsomniferol A | Steroidal lactone | − 4.58 |
| 27 | 101,281,365 | Withanolide Q | Steroidal lactone | − 4.57 |
| 28 | 73,621 | 4-Beta-Hydroxywithanolide E | Steroidal lactone | − 4.52 |
| 29 | 21,606,678 | (18R)-Withaphysalin F | Steroidal lactone | − 4.5 |
| Human TMPRSS2 | ||||
| 1 | 5,280,805 | Rutin | Flavonoid | − 7.71 |
| 2 | 3,802,778 | Isochlorogenic acid B | Phenolics | − 6.68 |
| 3 | 5,315,599 | Caffeoyl quinic acid | Polyphenol | − 5.89 |
| 4 | 70,684,083 | 2,3-Didehydrosomnifericin | Steroidal lactone | − 5.58 |
| 5 | 1,794,427 | Chlorogenic acid | Phenolics | − 5.05 |
| 6 | 5,280,460 | Scopoletin | Coumarin | − 4.93 |
| 7 | 179,575 | Wihanolide L | Steroidal lactone | − 4.85 |
| 8 | 15,411,208 | 2, 3-Dihydrowithaferin A | Steroidal lactone | − 4.73 |
| 9 | 13,743,195 | 4-Deoxyphysalolactone | Steroidal lactone | − 4.67 |
| 10 | 54,606,507 | Withacnistin | Steroidal lactone | − 4.61 |
| 11 | 101,710,596 | Withasomniferol B | Steroidal lactone | − 4.6 |
| 12 | 189,586 | Sitoindoside IX | Saponin | − 4.55 |
| SARS-CoV-2 RdRp | ||||
| 1 | 10,190,763 | Quercetin-3-rutinoside-7-glucoside | Flavonoid | − 11.61 |
| 2 | 5,280,805 | Rutin | Flavonoid | − 9.07 |
| 3 | 439,341 | Maltose | Sugar | − 8.05 |
| 4 | 1,794,427 | Chlorogenic acid | Phenolics | − 7.57 |
| 5 | 5988 | 5-phospho-D-ribosylamine | Pentose phosphate | − 6.9 |
| 6 | 3,802,778 | Isochlorogenic acid B | Phenolics | − 6.63 |
| 7 | 11,850 | Dulcitol | Sugar alcohol | − 6.61 |
| 8 | 189,586 | Sitoindoside IX | Saponin | − 6.59 |
| 9 | 5,315,599 | Caffeoyl quinic acid | Polyphenol | − 6.53 |
| 10 | 5,280,343 | Quercetin | Flavonoid | − 6.5 |
| 11 | 1,201,543 | Cuscohygrine | Alkaloid | − 6.14 |
| 12 | 189,702 | Sitoindoside X | Saponin | − 6.08 |
| 13 | 102,066,417 | Somniwithanolide | Steroidal lactone | − 5.03 |
| 14 | 8424 | Tropine | Alkaloid | − 5 |
| 15 | 449,293 | Psuedotropine | Alkaloid | − 5 |
| 16 | 92,987 | Pelletierine | Alkaloid | − 4.9 |
| 17 | 12,306,778 | Anahygrine | Alkaloid | − 4.88 |
| 18 | 6508 | Quinic acid | Cyclitol | − 4.84 |
| 19 | 443,143 | Anaferine | Alkaloid | − 4.79 |
| 20 | 13,743,195 | 4-Deoxyphysalolactone | Steroidal lactone | − 4.79 |
| 21 | 5810 | Hydroxyproline | Amino acid | − 4.78 |
| 22 | 101,687,981 | Withaoxylactone | Steroidal lactone | − 4.68 |
| 23 | 101,687,980 | Somnifericin | Alkaloid | − 4.66 |
| 24 | 5,742,590 | Sitogluside | Phytosterol-like compound | − 4.54 |
| 25 | 12,444,363 | Tropigline | Alkaloid | − 4.52 |
| 26 | 5862 | Cysteine | Amino acid | − 4.5 |
| 27 | 11,049,407 | Withanolide S | Steroidal lactone | − 4.5 |
Fig. 1.

Structure of lead phytochemicals present in W. somnifera plant identified in the present study against targeted SARS-CoV-2 and Human host targeted proteins. (Duke 1992; Kumar and Patnaik 2016; Singh et al. 2017)
In the present study, galactitol, chlorogenic acid, quercetin-3-rutinoside-7-glucoside, isochlorogenic acid, rutin, quercetin, and caffeoyl-quinic acid present in W. somnifera showed potential binding (− 10.1, − 9.92, − 9.66, − 8.51, − 8.38, − 8.25, and − 8.21 kcal/mole respectively) against SARS-CoV-2 Spike glycoprotein S1 domain. Standard inhibitor VE607 showed binding energy − 7.181 kcal/mole. Docking poses all lead (binding energy cutoff value ≤ standard inhibitor) and standard compound at S1 domain is shown in Fig. 2 A. Surface structure of top two lead phytochemicals and standard inhibitor at S1 domain is depicted in Fig. 2B−D. Binding energies and type of interaction of standard inhibitor and lead W. somnifera phytochemicals with Spike glycoprotein S1 domain is provided in Tables 1 and 2. VE607, galactitol, chlorogenic acid, quercetin-3-rutinoside-7-glucoside, isochlorogenic acid, rutin, quercetin, and caffeoyl-quinic acid interact with the active site of the protein using 3, 4, 3, 3, 4, 7, 2 and 4 hydrogen bonds respectively. Notably VE607 and galactitol showed hydrogen bonding with similar amino acid residues and both ligands interact with Asn422 and Leu492 via hydrophobic interaction. Rutin interacted with minimum (01 residue), while Chlorogenic acid showed maximum (11 residue) hydrophobic interaction with the S1 domain of SARS-CoV-2 spike glycoprotein.
Fig. 2.

Withania somnifera lead phytochemical docking pose and interaction with SARS-CoV2 Spike glycoprotein (S-protein) protein. A Docking pose of standard inhibitor and lead phytochemicals at S1 domain of S-protein. B Surface structure of S-protein interacted with lead compound galactitol. C Surface structure of S-protein interacted with lead compound chlorogenic acid. D Surface structure of S-protein interacted with standard inhibitor VE607. Green and orange color represents the amino acid involved in hydrogen bonding and hydrophobic interaction respectively. The molecular docking was performed as per methodology discussed in material and method section
Table 2.
Amino acid residues and type of interaction involved in binding of standard ligands and W. somnifera lead phytochemicals with target SARS-CoV-2 and human host proteins
| S. No. | Compound name | Hydrogen binding site | Hydrophobic binding site |
|---|---|---|---|
| SARS-CoV-2 Spike Glycoprotein | |||
| 1 | VE607 | Gln493(3.00), Ser494(2.96; 3.21), Tyr495(3.06) | Asn422, Asp442, Leu492, Pro491, Phe497, Tyr351, Tyr421, Val350 |
| 2 | Galactitol | Gln493(2.95; 2.72; 3.05), Ser494(2.96), Tyr495(3.19), Val350(3.03) | Asn422, Leu492, Ser349 |
| 3 | Chlorogenic acid | Asn422(2.95), Gln493(2.90; 2.85, 2.97), Phe347(2.93; 3.01) | Ala348, Ile418, Lys417, Leu492, Phe497, Ser349, Ser494, Tyr351, Tyr495, Val350, Val401 |
| 4 | Quercetin-3-rutinoside-7-glucoside | Pro491(2.95), Ser494(2.78, 2.71), Tyr495(3.05) | Asn422, Gln493, Ile418, Lys417, Leu492, Ser349, Tyr351 |
| 5 | Isochlorogenic acid B | Asp422(2.73, 3.07), Gln493(2.80), Phe347(3.31), Tyr351(3.11) | Lys417, Phe497, Ser349, Ser494, Tyr495, Val350, Val401 |
| 6 | Rutin | Asp442(2.59), Arg509(3.27), Phe347(2.95, 2.68), Phe497(2.71), Ser494(2.93), Thr345(2.76, 2.81), Tyr495(3.09) | Arg346 |
| 7 | Quercetin | Asp442(2.74, 2.76), Val350(2.86) | Phe497, Ser349, Tyr495, Val401 |
| 8 | Caffeoyl-quinic acid | Gln493(2.94), Ser494(2.96), Tyr351 (2.34), Tyr495(3.08) | Asn422, Ser349, Val350 |
| Human CD26 | |||
| 1 | Sitagliptin | Arg669(3.05), Asp708, Glu206, His740, Ser630 | Arg125, Arg358, Asn710, Glu205, Phe357, Ser209, Tyr547, Tyr662, Tyr666, Val207 |
| 2 | Quercetin-3-rutinoside-7-glucoside | Arg358(2.72, 2.85), Asp302(2.64, 2.82, 3.08), Glu205(2.96), Glu206(2.74), Glu361(2.73), Ile405(2.94, 3.17), Tyr662(3.07), Val207(3.18) | Arg125, Arg356, Phe208, Phe357, Ser209, Trp215, Tyr585, Tyr666 |
| 3 | Rutin | Arg125(2.89, 3.33), Glu206(2.94, 3.03), Ser209(2.96), Ser630(3.20, 2.97), Tyr547(2.63), Tyr662(2.73, 2.83) | Glu205, Gln553, Lys554, Phe357, Ser552, Trp629, Tyr666, Val207 |
| 4 | Caffeoyl quinic acid | Arg125(3.26, 2.93), Asn710(3.03), Cys551(2.78), Glu206(2.85), Gln553(3.04), Tyr547(2.77) Tyr585(3.08) Tyr662(2.80) | His740, Glu205, Phe357, Ser552, Tyr666, Ser630 |
| 5 | Sitoindosite X | Arg356(2.94), Gln553(3.20), Lys554(3.0) | Arg125, Asp739, Glu361, Gly741, His740, Ile405, Phe357, Ser360, Tyr546, Tyr547, Tyr585, Trp629 |
| 6 | Dulcitol | Arg358(3.16), Arg669(3.08), Glu205(3.06), Glu206(2.85), Ser209(2.84, 3.01, 3.19), Val207(2.78) | Tyr666, Phe357 |
| Human TMPRSS2 | |||
| 1 | Camostat mesylate | Arg277(2.99), Tyr227(3.00) | Phe394, Ile279, Thr324, Pro325, Phe231, Ala280, phe193, Cys278, Gly190, Leu188 |
| 2 | Rutin | Arg219(2.76, 3.79), Asn229(2.29), Glu326(2.87, 2.81), Trp327(3.01) | Arg526, Ile279, Lys228, Phe231, Pro391, Tyr232, Thr324 |
| 3 | Isochlorogenic acid B | Asn229(2.76), Asp528(2.67), Met525(2.98), Lys387(3.03), Cys278(3.24) | Ala280, Lys228, Trp327, Arg277, Phe231, Ile279, Pro325, Thr324, Glu326 |
| 4 | Caffeoyl-quinic acid | Arg277(2.73), Asn229(3.05), Glu326(2.86) | Ala280, Cys278, Ile279, Met525, Phe231, Phe394, Thr324, Trp327 |
| 5 | 4-2-3-Didehydrosomnifericin | Asn229(3.30), Glu326(2.62), Leu392(3.24), Trp327(3.24) | Ile279, Met525, Phe231, Pro325, Pro391, Thr324, Thr393, |
| 6 | Chlorogenic acid | Asn229(2.97), Met525(3.08) | Asp528, Glu326, Ile279, Lys228, Phe231, Pro325, Thr324 |
| SARS-CoV-2 RdRp | |||
| 1 | Remdesivir | Arg553(3.08, 2.83), Thr556(3.25), Asp623(2.39), Cys622(2.85), Tyr619(2.77) | Asp452, Arg24, Arg555, Tyr455, Ala554, Lys621, Asp618, Asp760 |
| 2 | Quercetin-3-rutinoside-7-glucoside | Asp618(2.73), Asp623(2.88, 2.98), Asp760(2.88), Asp761(3.22), Glu811(2.96), Lys621(2.75), Lys798 (2.36), Ser814(3.31), Trp800(3.00) | Arg553, Pro620, Trp617, Tyr619 |
| 3 | Rutin | Asp760(2.85, 2.83), Lys551(3.33), Lys621(2.84, 2.85), Lys798(2.89), Tyr619(2.74, 3.29), Trp800(2.90) | Arg553, Asp618, Asp761, Cys662, Gly616, Ser814, Ser759, Trp617, |
| 4 | Chlorogenic acid | Asp618(3.21), Lys551(2.91), Lys621(2.81), Lys798(2.92), Tyr619(3.18), Trp800(3.09) | Asp760, Asp761, Glu811, Pro620, Trp617 |
Numerical values given in bracket represent hydrogen bond length in Å
Analysis of phytochemical groups and binding efficacy against targeted proteins showed that flavonoid/polyphenolics showed lowest binding followed by other groups, saponins, alkaloids and steroidal/benzenoid lactone (Fig. 6).The binding free energies of SARS-CoV-2 spike glycoprotein- Galactitol, RdRP- Quercetin-3-rutinoside-7-glucoside, TMPRSS2-rutin and CD26- Quercetin-3-rutinoside-7-glucoside were calculated by using MM-GBSA method and the results are shown in Fig. 7. Mostly the overall binding free energy of all the protein ligand complexes was more negative in comparison to unbound protein
Quercetin 3-rutinoside-7-glucoside, rutin, caffeoylquinic acid, sitoindoside X, and dulcitol present in W. somnifera plant showed potential binding (binding energy − 11.69, − 9.49, − 7.91, − 7.15, and − 6.61 kcal/mole) against human CD26 active site. Standard inhibitor sitagliptin showed binding energy of − 6.6 kcal/mole. Docking pose of the lead phytochemicals (binding energy cutoff value ≤ standard inhibitor) and the standard compound, at the human CD26 active site is depicted in Fig. 3A. Surface structure of the top two lead phytochemicals and standard inhibitor at human CD26 active site is directed in Fig. 3B−D. Binding energies and type of interaction of standard inhibitor and lead W. somnifera phytochemicals with CD26 active site is shown in Tables 1 and 2. Sitagliptin, quercetin 3-rutinoside-7-glucoside, rutin, caffeoylquinic acid, sitoindoside X, and dulcitol interact with the active site of the protein using 5, 8, 6, 8, 3 and 6 hydrogen bonds respectively. These six phytochemicals against human CD26 showed binding with mostly similar type of amino acid residues (Table 2). Furthermore, W. somnifera compounds were docked on protein-protein interaction interface of human CD26 to find the CD26-S1 domain protein-protein interaction inhibition potential of the compounds. Result showed tight binding of dulcitol, quercetin-3-rutinoside-7-glucoside, and rutin phytochemicals on CD26-Spike protein (S1 domain) protein-protein interaction interface with − 8.19, − 7.13, and − 7.08 kcal/mole binding energy. Dulcitol, quercetin-3-rutinoside-7-glucoside, and rutin formed hydrogen bond with Ser239 (two bond), Tyr241(two bond); Ala707, Asp192, Asp739, Gln123, Lys122, Lys250; and Arg253, Asp192, Glu237, Leu235, Pro249, Thr231, Thr251 amino acid residues at CD26-Spike protein-protein interaction interface.
Fig. 3.

Withania somnifera lead phytochemical docking pose and interaction with human host CD26 protein active site. A Docking pose of standard inhibitor and lead phytochemicals at CD26 active site. B Surface structure of CD26 active site with lead compound Quercetin-3-rutinoside-7-glucoside. C Surface structure of CD26 active site with lead compound rutin. D Surface structure of S-protein interacted with standard inhibitor sitagliptin. Green and orange color represents the amino acid involved in hydrogen bonding and hydrophobic interaction
In the present study, rutin, isochlorogenic acid, caffeoyl-quinic acid, 4-2-3-didehydrosomnifericin, and chlorogenic acid present in W. somnifera plant showed potential binding (− 7.71, − 6.68, − 5.89, − 5.58, and − 5.05 kcal/mole) against human TMPRSS2 active site. Standard inhibitor camostat mesylate showed binding energy − 5.146 kcal/mole. Docking poses for all lead (binding energy cutoff value ≤ standard inhibitor) and standard compound at human TMPRSS2 active site is depicted in Fig. 4A. Surface structure of top two lead phytochemicals and standard inhibitor at human TMPRSS2 active site is depicted in Fig. 4B−D. Binding energies and type of interaction of standard inhibitor and lead W. somnifera phytochemicals with TMPRSS2 is provided in Tables 1 and 2. Camostat mesylate, rutin, isochlorogenic acid, caffeoyl-quinic acid, 4-2-3-didehydrosomnifericin, and chlorogenic acid interacted with the active site of the protein using 2, 4, 5, 3, 4 and 2 hydrogen bonds. Camostat mesylate and top five W. somnifera compounds (rutin, isochlorogenic acid, caffeoyl-quinic acid, 4-2-3-didehydrosomnifericin, and chlorogenic acid) against human TMPRSS2 showed binding with three (Ile279, Phe231, Thr324) common amino acid residues (Table 2). Moreover, isochlorogenic acid and caffeoyl-quinic acid showed maximum binding similarity with standard inhibitor against host TMPRSS2 among top five lead molecules.
Fig. 4.

Withania somnifera lead phytochemical docking pose and interaction with human host TMPRSS2 protein. A Docking pose of standard inhibitor and lead phytochemicals at TMPRSS2 protein active site. B Surface structure of TMPRSS2 protein with lead compound rutin. C Surface structure of TMPRSS2 protein with lead compound Isochlorogenic acid B. D Surface structure of TMPRSS2 interacted with standard inhibitor camostst mesylate. Green and orange color represents the amino acid involved in hydrogen bonding and hydrophobic interaction respectively
In the present study, quercetin-3-rutinoside-7-glucoside, rutin, and chlorogenic acid present in W. somnifera plant showed potential binding (− 11.61, − 9.07, and − 8.05 kcal/mole) against SARS-CoV-2 RdRp active site. Standard inhibitor remdesivir showed binding energy − 7.56 kcal/mole. Docking poses all lead (binding energy cutoff value ≤ standard inhibitor) and standard compound at SARS-CoV-2 RdRp active site which is depicted in Fig. 5A. Surface structure of top two lead phytochemicals and standard inhibitor at SARS-CoV-2 RdRp active site is provided in Fig. 5B−D. Binding energies and type of interaction of standard inhibitor and lead W. somnifera phytochemicals with RdRp is shown in Tables 1 and 2. Remdesivir, quercetin-3-rutinoside-7-glucoside, rutin, and chlorogenic acid interacted with the active site of the protein using 5, 9, 6 and 6 hydrogen bonds. Remdesivir and quercetin-3-rutinoside-7-glucoside interacted with 6 similar amino acid residue (Arg553, Tyr619, Asp623, Asp618, Asp760 and, Lys621) of SARS-CoV-2 RdRp (Table 2).
Fig. 5.

Withania somnifera lead phytochemical docking pose and interaction with SARS-CoV-2 RdRp active site. A Docking pose of standard inhibitor and lead phytochemicals at SARS-CoV-2 RdRp active site. B Surface structure of RdRp active site with lead compound Quercetin-3-rutinoside-7-glucoside. C Surface structure of RdRp active site with lead compound rutin. D Surface structure of RdRp active site interacted with standard inhibitor remdesivir. Green and orange color represents the amino acid involved in hydrogen bonding and hydrophobic interaction respectively
Fig. 6.
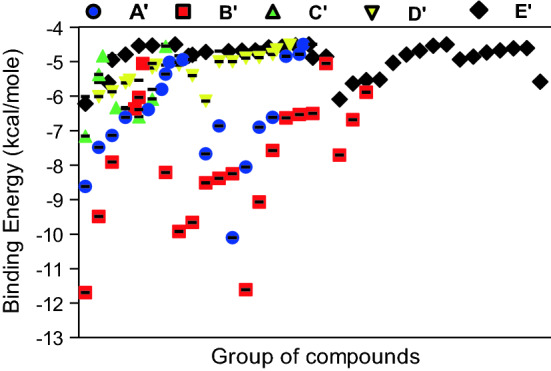
Binding energy of W. somnifera lead phytochemical groups against the targeted SARS-CoV-2 (Spike-glycoprotein and RdRp) and Human host (CD26 and TMPRSS2) test proteins. The lead (more potent binding in comparison to standard inhibitors of the targeted proteins) phytochemicals were groped as A’-Other phytochemicals; B’-Flavonoids/Polyphenoilcs; C’-Saponin; D’-Alkaloids; E’-Steroidal/Benzenoid lactones. Other phytochemicals included pentose phosphate, glucinolates, amino acids, fatty acids and cyclitol. The analysis was done on GraphPad Prism software using data provided in Table 1
On the basis of docking results, we selected proteins that are involved in the viral entry (TMPRSS2) and virulence (RdRP) to perform MD simulation study. The simulations were carried out on unbound state and lead phytochemical bound state of the respective proteins to elucidate the dynamic behavior of both proteins. Simultaneously, the protein bounded with their biologically proven inhibitors also ran to compare the results. MD simulation results for RdRP-Quercetin-3-rutinoside-7-glucoside (RQ) complex, TMPRSS2-Rutin (TR) complex and their respective unbound proteins and complex with standard inhibitors are shown in Figs. 8 and 9. Favipiravir and camostat mesylate were used as biologically approved RdRP and TMPRSS2 protein inhibitors in MD simulation study. The MD trajectory of both proteins was stable under most parts of 10 ns simulation period. Consequently, these trajectories were used for further analysis. RMSD analysis revealed that RQ and TR complexes did not shown any significant deviation from their respective unbound proteins (Figs. 8A and 9A). RMSD of both complexes were comparable to their respective biological known inhibitors complex. To understand the effect of inhibitor binding on the flexible areas of the protein structure, root-mean-square fluctuation (RMSF) was analyzed. Little fluctuation was observed in the RQ and TR complexes in comparison to unbound proteins (Figs. 8B and 9B). Binding of standard inhibitors to their respective proteins showed similar pattern of fluctuation during simulation period.
Fig. 8.

Simulation results of W. somnifera lead phytochemical targeting RdRP protein. Plot of molecular dynamic simulation trajectories of COVID-19 RdRP protein and protein-ligand complexes during 10 ns simulation. A The root mean square deviation (RMSD) of solvated SARS-CoV2 RdRP protein and RdRP -favipiravir and RdRP-Quercetin-3-rutinoside-7-glucoside complex during 10 ns molecular dynamics simulation. B The root mean square fluctuation (RMSF) values of solvated SARS-CoV2 RdRP protein and RdRP-favipiravir and RdRP-Quercetin-3-rutinoside-7-glucoside complex plotted against residue numbers. C Plot of solvent accessible surface area (SASA) during 10 ns molecular dynamics simulation of SARS-CoV2 RdRP protein and RdRP-favipiravir and RdRP-Quercetin-3-rutinoside-7-glucoside complex. D Plot of radius of gyration (Rg) during 10 ns molecular dynamics simulation of SARS-CoV2 RdRP protein and RdRP-favipiravir and RdRP-Quercetin-3-rutinoside-7-glucoside complex. E Plot of number of hydrogen bond in the SARS-CoV2 RdRP protein in unbound state and bound with favipiravir and Quercetin-3-rutinoside-7-glucoside. F Plot of number of hydrogen bonds formed between RdRP and favipiravir along with RdRP and Quercetin-3-rutinoside-7-glucoside. Unbound protein parameters are depicted in black color. Parameters for RdRP-favipirvavir complex and RdRP-Quercetin-3-rutinoside-7-glucoside complex are represented in red and green color respectively
Fig. 9.

Simulation results of W. somnifera lead phytochemical targeting TMPRSS2 protein. Plot of molecular dynamic simulation trajectories of Human TMPRSS2 protein and protein-ligand complexes during 10 ns simulation. A The root mean square deviation (RMSD) of solvated Human TMPRSS2 protein and TMPRSS2-Camostat mesylate and TMPRSS2-rutin complex during 10 ns molecular dynamics simulation. B The root mean square fluctuation (RMSF) values of solvated Human TMPRSS2 protein and TMPRSS2-Camostat mesylate and TMPRSS2- rutin complex plotted against residue numbers. C Plot of solvent accessible surface area (SASA) during 10 ns molecular dynamics simulation of Human TMPRSS2 protein and TMPRSS2- Camostat mesylate and TMPRSS2- rutin complex. D Plot of radius of gyration (Rg) during 10 ns molecular dynamics simulation of Human TMPRSS2 protein and TMPRSS2-Camostat mesylate and TMPRSS2- rutin complex. E Plot of number of hydrogen bond in the Human TMPRSS2 protein in unbound state and bound with Camostat mesylate and rutin. F Plot of number of hydrogen bonds formed between TMPRSS2 and Camostat mesylate along with TMPRSS2 and rutin. Unbound protein parameters are depicted in black color. Parameters for TMPRSS2- Camostat mesylate complex and TMPRSS2- rutin are represented in red and green color respectively
Solvent accessible surface area analysis revealed that there were no significant difference in the SASA value found for RQ and TR complexes, unbound proteins and their complex with respective standard inhibitors (Figs. 8C and 9C). The SASA value for RQ and RdRP- Favipiravir complexes as well as the unbound protein progressively decreased during the 10ns simulation period (Fig. 8C). Radius of gyration for RQ and TR complexes are compared with their respective unbound protein and standard inhibitor bound protein in Figs. 8D and 9D. TQ complex showed lesser Rg value during the 10 ns simulation period in comparison to unbound and camostat mesylate protein complex (Fig. 9D). Hydrogen bond analysis was performed for RQ and TQ complexes during simulation period and results are shown in Figs. 8E, F, 9E, F. Binding of Quercetin-3-rutinoside-7-glucoside to RdRP protein resulted in increase in the number of hydrogen bonds in the RdRP protein (Fig. 8E). In an event of TMPRSS2, binding of identified lead inhibitor had no overall effect on intra-molecular hydrogen bonding network of TMPRSS2 (Fig. 9E). In both cases (RQ and TQ complexes), number of hydrogen bonds between protein and respective identified lead inhibitor were significantly higher than their standard inhibitors (Fig. 8F and 9F).
We further performed secondary structure analysis for both RQ and TR complex, unbound RdRP and TMPRSS2 proteins and standard inhibitor bound complexes during the 10 ns simulation (Supplementary Fig. 1). Secondary structure analysis was conducted to assign the secondary structure content of the proteins as a function of time. The various secondary structures of protein such as, helix, sheet, bridge, and turn were divided for particular residues for every step in 10 ns simulation. The number of amino acid residues participating in formation of a particular secondary structure can be seen in secondary structure plots of both RdRP and TMPRSS2 protein simulations in unbound and lead/standard inhibitor bound complex (Supplementary Fig. 1). The average number of residues participating in the formation of secondary structure in RdRP-favipiravir complex and RdRP-Quercetin-3-rutinoside-7-glucoside complex seems to increase slightly in comparison to unbound RdRP protein. In case of TMPRSS2, there was a slight decrease in the number of residues involve in the secondary structure formation in TMPRSS2- Camostat mesylate due to the increase in the percentage of bend (Supplementary Fig. 1). However in TMPRSS2- rutin complex, there is a slight increase in the number of residues involved in the formation B-sheet along with a slight increase in the number of residues forming the structured region.
Drug-likeness and toxicity properties of all the lead phytochemicals (binding energy ≤ respective standard inhibitor) against targeted proteins were performed and results are shown in Fig. 10. Compound itoindoside IX ; caffeoyl quinic acid; dulcitol; sitoindoside X, chlorogenic acid and galactitol were predicted as not absorbed and not brain penetrant as they were located outside the egg (shown as white circle) whereas compound 2,3-didehydrosomnifericin and quercetin were predicted as well-absorbed (inside the egg) (Fig. 10). Moreover, these compounds (2, 3-didehydrosomnifericin and quercetin) showed blood brain barrier inaccessibility (out of egg yolk). It is noteworthy that compound 2, 3-didehydrosomnifericin and quercetin showed higher probability (white circle area of the egg) of passive absorption by the gastrointestinal tract. Anahygrine, cuscohygrine, (-)-anaferine molecules showed potential to cross blood brain barrier (yellow colour circle as egg yolk). Notably, the compound (-)-Anaferine and 2, 3-Didehydrosomnifericin were predicted to be actively effluxed by P-glycoprotein (blue dot) while compound Anahygrine, cuscohygrine, and quercetin were predicted as non-substrate of P-gp (red dot). Moreover, among the not absorbed compounds sitoindoside IX and sitoindoside X were predicted actively effluxed by P-gp while Caffeoyl quinic acid, dulcitol, 2, 3-didehydrosomnifericin and galactitol showed P-gp non-substrate. Bioavailability radar analysis showed that galactitol has all the drug likeness properties within optimal range (pink colour web part) (Fig. 10C). In contrast, quercetin-3-rutinoside-7-glucoside and rutin possessed slightly higher size and polarity (white portion of the egg) (Fig. 10B, D). Further, we predicted the physiochemical properties and toxicological profile of the lead phytochemicals and result are summarized in Table 3.
Fig. 10.

Boiled egg diagram and bioavailability radar map of W. somnifera compounds. A Boiled egg diagram of compound M1, M2, M3, M6, M7, M8, M9, M11, M12, M13 and M14. Bioavailability radar map of B Rutin, C Galactitol and D Quercetin-3-rutinoside-7-glucoside depicting the LIPO (lipophilicity), SIZE (molecular weight), POLAR (polarity), INSOLU (insolubility) INSATU (insaturation) and FLEX (rotatable bond flexibility) parameters. M1 Anahygrine, M2 Cuscohygrine, M3 (-)-Anaferine, M6 Sitoindoside IX, M7 Caffeoyl quinic acid, M8 Dulcitol, M9 Sitoindoside X, M11 2,3-Didehydrosomnifericin, M12 Chlorogenic acid, M13 Galactitol, M14 Quercetin
Table 3.
Physiochemical properties and toxicological parameters of lead W. somnifera phytochemicals against SARS-CoV-2 and human host target protein
| S.No. | Properties | M1 | M2 | M3 | M4 | M5 | M6 | M7 | M8 | M9 | M10 | M11 | M12 | M13 | M14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | MW | 224.34 | 224.34 | 224.34 | 610.52 | 772.66 | 632.74 | 354.31 | 182.1 | 871.15 | 516.45 | 488.61 | 354.31 | 182.1 | 302.24 |
| 2 | NHA | 16 | 16 | 16 | 43 | 54 | 45 | 25 | 12 | 62 | 37 | 35 | 25 | 12 | 22 |
| 3 | NAHA | 0 | 0 | 0 | 16 | 16 | 0 | 6 | 0 | 0 | 12 | 0 | 6 | 0 | 16 |
| 4 | FCsp3 | 0.92 | 0.92 | 0.92 | 0.44 | 0.55 | 0.82 | 0.38 | 1 | 0.86 | 0.24 | 0.79 | 0.38 | 1 | 0 |
| 5 | NH-BA | 3 | 3 | 3 | 16 | 21 | 11 | 9 | 6 | 12 | 12 | 7 | 9 | 6 | 7 |
| 6 | NH-BD | 1 | 0 | 2 | 10 | 13 | 5 | 6 | 6 | 4 | 7 | 4 | 6 | 6 | 5 |
| 7 | MR | 74.1 | 74.2 | 74.01 | 141.38 | 173.5 | 159.87 | 83.5 | 37.93 | 236.91 | 126.9 | 130.84 | 83.5 | 37.93 | 78.03 |
| 8 | TPSA | 32.34 | 23.55 | 41.13 | 269.43 | 348.58 | 175.51 | 164.75 | 121.3 | 181.58 | 211.28 | 124.29 | 164.75 | 121.3 | 131.36 |
| 9 | GIA | High | High | High | Low | Low | Low | Low | Low | Low | Low | High | Low | Low | High |
| 10 | BBB-P | Yes | Yes | Yes | No | No | No | No | No | No | No | No | No | No | No |
| 11 | Pgp-S | No | No | Yes | Yes | Yes | Yes | No | No | Yes | Yes | Yes | No | No | No |
| 12 | BS | 0.55 | 0.55 | 0.55 | 0.17 | 0.17 | 0.17 | 0.11 | 0.55 | 0.17 | 0.11 | 0.55 | 0.11 | 0.55 | 0.55 |
| 13 | ESOL-C | VS | VS | VS | S | S | MS | VS | HS | PS | S | MS | VS | HS | S |
| 14 |
ESOL-S (mg/ml) |
6.66 | 5.68 | 7.81 | 3.08 | 9.56 | 2.38 | 8.50 | 3.75 | 2.90 | 1.17 | 1.72 | 8.50 | 3.75 | 2.11 |
M1 = Anahygrine; M2 = Cuscohygrine; M3= (-)-Anaferine; M4 = Rutin; M5 = Quercetin-3-rutinoside-7-glucoside; M6 = Sitoindoside IX ; M7 = Caffeoyl quinic acid; M8 = Dulcitol; M9 = Sitoindoside X; M10 = Isochlorogenic acid B; M11 = 2,3-Didehydrosomnifericin; M12 = Chlorogenic acid; M13 = Galactitol; M14 = Quercetin; MF = Molecular formula; MW = Molecular weight; NHA = Number of heavy atoms; NAHA = Number of aromatic heavy atoms; FCsp3 = Fraction Csp3; NH-BA = Number of H-bond acceptors; NH-BD = Number of H-bond donors; MR = Molar Refractivity; TPSA = The total surface are; GIA = GI absorption; BBB-P = BBB permeant; Pgp-S = Pgp substrate; BS = Bioavailability score; ESOL-Estimated aqueous solubility; ESOL-C = Estimated aqueous solubility class; ESOL-S = ESOL solubility; VS = Very soluble; S = Soluble; MS = Moderately soluble; HS = Highly soluble; PS = Poorly soluble
Discussion
In the present study, we found that lead W. somnifera phytochemicals interacted with two of the critical amino acids (Gln493 and Ser494) by forming hydrogen bonding (Table 2). Beside these, the phytochemicals showed hydrophobic interaction with other amino acids (Tyr495, Asn422, and Pro491) involved in ACE-2 binding (Gordon et al. 2020; Shang et al. 2020) reported that residue 455, 482–486, 493, 494, and 501 are critical SARS-CoV-2 S1 domain amino acids involved in interaction with human ACE-2 protein. Lead phytochemicals also showed hydrophobic interaction (Leu492, Pro491, Asp442, Asn422, Ser494, Tyr495, and Gln493) with SARS-CoV-2 receptor binding domain (RBD) and ACE-2 protein-protein interaction interface residues. The results indicate that W. somnifera phytochemicals have potential to disrupt spike glycoprotein-ACE-2 protein-protein interaction by binding at SARS-CoV-2 RBD. Thus, the lead molecules might inhibit the viral entry into the cell.
The results demonstrate that quercetin 3-rutinoside-7-glucoside and other lead compounds (except sitoindoside X) exhibited greater hydrogen bond (˃ 5) interaction with the CD26 protein in comparison to standard inhibitor. Although, quercetin 3-rutinoside-7-glucoside and caffeoylquinic acid formed same number of hydrogen bond with the target active site of the protein, but former showed comparatively tight binding which might be due to the involvement of more hydrophobic interaction in the binding (Table 2). Different binding efficacy of various lead phytochemicals towards CD26-SARS-CoV-2 S-protein-protein interface and CD26 active site further corroborate the previous reporting that these two binding sites on CD26 are different (Lu et al. 2013; Vankadari and Wilce 2020). Several in vitro studies reported CD26 or DPP4 activity inhibition potential in W. somnifera extracts which corroborate with our present findings that phytochemical present in the plant possess CD26 inhibitors (Singh et al. 2020a, b). CD26/DPP4 possesses dipeptidyl peptidase activity and protein interface for viral protein attachment. Inhibition of peptidase activity by small molecules is known to decrease inflammation and associated disease occurrence. Moreover, disruption of viral-host protein-protein interaction by inhibitors has suggested a treatment strategy against SARS-CoV-2 (Fig. 11). Binding of W. somnifera phytochemicals to active/protein-protein interaction interface sites propose it as potential candidate inhibiting SARS-CoV-2 entry into the cell.
Fig. 11.

Lead phytochemicals present in W. somnifera and their SARS-CoV-2 and human host protein targets. The red color arrow in front of dulcitol, rutin, and QRG indicates their potential binding with the target SARS-CoV-2 and human host proteins involve in viral entry and replication. The detailed function of each protein is discussed in introduction part. Blue color text indicates the target proteins. QRG-quercetin-3-rutinoside-7-glucoside
Human TMPRSS2 is an important protease involved in the priming of SARS-CoV-2 S protein that makes it a probable drug target against viral virulence. Similar to standard inhibitor, isochlorogenic acid B and caffeoyl-quinic acid showed hydrophobic interaction with Arg277 residue of TMPRSS2 protein (Table 2). Tight binding of W. sominfera phytochemicals with critical amino acid residues, in comparison to standard inhibitor(s) indicate the SARS-CoV-2 virulence inhibitory potential of the plant. Present study corroborate with the finding of Hoffmann et al. (2020) which reported the TMPRSS2 inhibition mediated decreased SARAS-CoV-2 viral virulence by using clinically proven protease inhibitor. Gordon et al. (2020) reported that remdesivir, a nucleotide analogue possess RNA synthesis inhibition potential in SARS-CoV-2 by delaying the RNA chain termination mechanism. Several clinical trials were conducted to study the efficacy of RdRp inhibitors in SARS-CoV-2 patients. Binding of W. somnifera lead molecules at RdRp active site showed interaction with similar amino acids (Arg553, Asp623, Tyr619, Lys621, Asp618, and Asp760) which indicated their SARS-CoV-2 RdRp inhibition potential.
The present study showed no significant deviation in RMSD values after binding of the lead molecules to RdRP and TMPRSS2 proteins indicating the stable protein ligand complex formation. RMSF results indicate that lead inhibitor binding provide more flexibility to both proteins under the 10 ns simulation period investigation which means that the bound confirmations of both proteins were more stable during simulation. SASA analysis explores the surface of proteins that can be assessed by the solvent molecules. RQ and TR complex showed favorable SASA value in comparison to unbound and standard inhibitor bound protein indicating the stable binding of lead molecule to the respective protein. Radius of gyration provides valuable information about protein conformation and its stability during the simulation period. Relatively stable Rg value during and at the end of the simulation indicate favorable binding of ligand to respective proteins without affecting its stability. Hydrogen bonds are an important type of interaction and play significant role in providing the stability to the molecular complexes. Increased number of hydrogen bonds in the RdRP protein after lead molecule binding indicates that binding of inhibitor has made structure more stable in comparison to unbound RdRP protein and RdRP–favipiravir complex (Fig. 8E). Analysis of hydrogen bonding network between protein and inhibitor can provide clear indication of stability of binding between protein and inhibitor. Our results indicate that more number of hydrogen bonds were formed by the lead inhibitors in comparison to standard inhibitors, which might be responsible for their more negative docking scores. Over, these MD simulation analysis (RMSD, RMSF, SASA, Rg, HBond and Secondary structure) and free energy calculations showed that the binding of W. somnifera lead phytochemicals to their respective targeted proteins stabilized the protein structure.
Three phytochemicals viz. quercetin-3-rutinoside-7-glucoside, galactitol and rutin possess anti-SARS-CoV-2 potential by targeting viral and host proteins involved in disease infection and virulence (Fig. 7). Rutin, a plant flavonoid, contain flavonol (quercetin) and a disaccharide (rutinose). Recently, in silico SARS-CoV-2 main protease inhibitory potential of rutin has been reported (Das et al. 2021). Galactitol is a sugar alcohol found in plants. Recently, in silico antiviral activity of galactitol against Ebola virus has been reported (Nagrajan et al. 2019). Quercetin 3-rutinoside-7-glucoside is a flavonoid glycoside identified in W. somnifera aerial part (Mundkinajeddu et al. 2014). Antiviral potential of flavonoid glycoside has been reported against human coronaviruses (Schwarz et al. 2014). Furthermore, chlorogenic acid, isochlorogenic acid B, caffeoyl-quinic acid, sitoindosite X, and 4-2-3-didehydrosomnifericin phytochemicals showed better binding efficacy than respective standard inhibitors of the targeted proteins (Tables 1 and 2). Published literature showed potent antiviral efficacy of these phytochemicals against herpes simplex virus 1 and 2; human immunodeficiency virus, adenovirus, respiratory syncytial virus, and Ebola virus (Li et al. 2005; Naveed et al. 2018). Sitoindosite X is a glycowithanolides identified in W. somnifera known to possess anti-stress, and memory boosting properties in vivo. In pharmacological parameters, galactitol showed better potential (such as low molecular weight) in comparison to rutin and quercetin-3-rutinoside-7-glucoside. Toxicological profile prediction of lead molecules is an important step in in silico drug discovery. The result showed that all the three lead compounds are not able to pass blood brain barrier. Galactitol was predicted as substrate for P-Glycoprotein which indicates its possible efflux by the Pgp expressing cells. The low absorption through the gastrointestinal tract of the lead compounds reflects the general limitation in therapeutic natural compounds. Over all better pharmacological, ADMET and toxicological profile (Fig. 10; Table 3) of the lead molecules further substantiate their potential as anti SARAS-CoV-2 drug agents.
Fig. 7.

MM-GBSA analysis of the lead W. somnifera phytochemical complexed with the SARS-CoV-2 (Spike-glycoprotein and RdRp) and Human host (CD26 and TMPRSS2) test proteins. The graph represents the contribution of different types of interaction energy and the algebraic sum of the total energy of the ligand-protein complex. The total binding free energy involves different types of interactions (coulomb, covalent, lipo (lipophilic), Hbond (hydrogen bond), SolvGB (generalized born electrostatic solvation energy), vdW (Van deer Waals), and packaging interactions). A Binding free energies of CD26 and Quercetin-3-rutinoside-7-glucoside complex. B Binding free energies of TMPRSS2 and Rutin complex. C Binding free energies of RdRp and Quercetin-3-rutinoside-7-glucoside complex. D Binding free energies of Spike-protein and Galactitol complex
Conclusions
The COVID-19 pandemic has challenged the scientific community for development of effective molecules and/or herbal formulation for their use as antiviral remedy against SARS-CoV-2. Computer aided drug designing approaches such as virtual screening, molecular docking, and molecular dynamics simulation could provide information on such lead compounds in isolated form and/or present in herbal formulations. Our results uncovers that phytochemicals present in W. somnifera have potential to bind at active/protein-protein interaction interface of the SARA-CoV-2 (Spike glycoprotein, and RdRP) and human host (CD26 and TMPRSS2) target proteins involved in the viral entry and pathogenesis. The MD simulation investigation established that the protein-lead compound complex possess stable conformation and lower protein-ligand interaction energy. MM-GBSA analysis also approves the docking and simulation study of protein-lead compound complex. Over, these studies show that quercetin-3-rutinoside-7-glucoside is a potent inhibitor of human CD26 and SARS-CoV-2 RdRp while galactitol and rutin showed highest binding efficacy against viral spike glycoprotein and human TMPRSS2 protein. We conclude that W. somnifera compounds possess inhibitory potential against viral attachment, host immune hijacking, and virulence. We further recommend that the in vitro and in vivo studies should be performed to establish the anti SARS-CoV-2 potential of the W. somnifera compounds.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Author contribution
SK conceptualized and designed the study and prepared the draft manuscript; KSP, AKS, PPK and MS performed the experiments and analyzed the data; SG, SS and SPS prepared the manuscript and were involved in scientific discussion.
Funding
The authors did not receive support from any organization for the submitted work.
Availability of data and material
All data generated or analyzed during this study are included in this published article.
Declarations
Conflict of interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Ethics approval and consent to Participate
Not applicable.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Berger JP, SinhaRoy R, Pocai A, Kelly TM, Scapin G, Gao YD, Pryor KAD, Wu JK, Eiermann GJ, Xu SS, Zhang X, Tatosian DA, Weber AE, Thornberry NA, Carr RD. A comparative study of the binding properties, dipeptidyl peptidase-4 (DPP-4) inhibitory activity and glucose-lowering efficacy of the DPP-4 inhibitors alogliptin, linagliptin, saxagliptin, sitagliptin and vildagliptin in mice. Endocrinol Diabetes Metab. 2017;1:1–8. doi: 10.1002/edm2.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Zhang G, Tang B, Liu Y, Fu X, Zhang X. Promising Anti-influenza Properties of Active Constituent of Withania somnifera Ayurvedic Herb in Targeting Neuraminidase of H1N1 Influenza: Computational Study. Cell Biochem Biophys. 2015;72:727–739. doi: 10.1007/s12013-015-0524-9. [DOI] [PubMed] [Google Scholar]
- Casrouge A, Sauer AV, Barreira da Silva R, Tejera-Alhambra M, Sánchez-Ramón S, ICAReB, Cancrini C, Ingersoll MA, Aiuti A, Albert ML. Lymphocytes are a major source of circulating soluble dipeptidyl peptidase 4. Clin Exp Immunol. 2018;194:166–179. doi: 10.1111/cei.13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:1–13. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan S (2008) PyRx-python prescription v. 0.8. The Scripps Research Institute. 2008; 2010
- Das S, Sarmah S, Lyndem S, Singha Roy A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. J Biomol Struct Dyn. 2021;39:3347–3357. doi: 10.1080/07391102.2020.1763201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke JA. Database of phytochemical constituents of GRAS herbs and other economic plants. Boca Raton: CRC Press; 1992. [Google Scholar]
- El Khatabi K, Aanouz I, El-Mernissi R, Singh AK, Ajana MA, Lakhlifi T, Kumar S, Bouachrine M. Integrated 3D-QSAR, molecular docking, and molecular dynamics simulation studies on 1,2,3-triazole based derivatives for designing new acetylcholinesterase inhibitors. Turk J Chem. 2021;45:647–660. doi: 10.3906/kim-2010-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezebuo FC, Kushwaha PP, Singh AK, Kumar S, Singh P. In-silico methods of drug design: molecular simulations and free energy calculations. In: Kumar S, Egbuna C, editors. Phytochemistry: an in-silico and in-vitro update 2019. Singapore: Springer; 2019. pp. 521–533. [Google Scholar]
- Ganguly B, Umapathi V, Rastogi SK. Nitric oxide induced by Indian ginseng root extract inhibits Infectious Bursal Disease virus in chicken embryo fibroblasts in vitro. J Anim Sci Technol. 2018;60:1–5. doi: 10.1186/s40781-017-0156-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CJ, Tchesnokov EP, Feng JY, Porter DP, Götte M. The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J Biol Chem. 2020;295:4773–4779. doi: 10.1074/jbc.AC120.013056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Singh AK, Kushwaha PP, Prajapati KS, Shuaib M, Senapati S, Kumar S. Identification of potential natural inhibitors of SARS-CoV2 main protease by molecular docking and simulation studies. J Biomol Struct Dyn. 2021;39:4334–4345. doi: 10.1080/07391102.2020.1776157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Müller MA, Drosten C, Pöhlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata-Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol. 2019;93:1–15. doi: 10.1128/JVI.01815-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain J, Narayanan V, Chaturvedi S, Pai S, Sunil S. In vivo evaluation of Withania somnifera-based Indian traditional formulation (Amukkara Choornam), against Chikungunya virus-induced morbidity and arthralgia. J Evid Based Integr Med. 2018;23:11–25. doi: 10.1177/2156587218757661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempegowda PK, Zameer F, Narasimashetty CK, Kollur SP, Murari SK. Inhibitory potency of Withania somnifera extracts against DPP-4: an in vitro evaluation. Afr J Tradit Complement Altern Med. 2018;15:11–25. doi: 10.21010/ajtcam.v15i1.2. [DOI] [Google Scholar]
- Kim SC, Schneeweiss S, Glynn RJ, Doherty M, Goldfine AB, Solomon DH. Dipeptidyl peptidase-4 inhibitors in type 2 diabetes may reduce the risk of autoimmune diseases: a population-based cohort study. Ann Rheum Dis. 2015;74:1968–1975. doi: 10.1136/annrheumdis-2014-205216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar G, Patnaik R. Exploring neuroprotective potential of Withania somnifera phytochemicals by inhibition of GluN2B-containing NMDA receptors: an in silico study. Med Hypotheses. 2016;92:35–43. doi: 10.1016/j.mehy.2016.04.034. [DOI] [PubMed] [Google Scholar]
- Kumar S, Kushwaha PP, Gupta S. Emerging targets in cancer drug resistance. Cancer Drug Resist. 2019 doi: 10.20517/cdr.2018.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha PP, Vardhan PS, Kapewangolo P, Shuaib M, Prajapati SK, Singh AK, Kumar S. Bulbine frutescens phytochemical inhibits notch signaling pathway and induces apoptosis in triple negative and luminal breast cancer cells. Life Sci. 2019;234:1–15. doi: 10.1016/j.lfs.2019.116783. [DOI] [PubMed] [Google Scholar]
- Kushwaha PP, Singh AK, Bansal T, Yadav A, Prajapati KS, Shuaib M, Kumar S. Identification of natural inhibitors against SARS-CoV-2 Drugable targets using molecular docking, molecular dynamics Simulation, and MM-PBSA approach. Front Cell Infect Microbiol. 2020;11:1–17. doi: 10.3389/fcimb.2021.730288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha PP, Singh AK, Prajapati KS, Shuaib M, Fayez S, Bringmann G, Kumar S. Induction of apoptosis in breast cancer cells by naphthylisoquinoline alkaloids. Toxicol Appl Pharmacol. 2020;409:1–12. doi: 10.1016/j.taap.2020.115297. [DOI] [PubMed] [Google Scholar]
- Kushwaha PP, Singh AK, Shuaib M, Prajapati KS, Vardhan PS, Gupta S, Kumar S. 3-O-(E)-p-Coumaroyl betulinic acid possess anticancer activity and inhibit Notch signaling pathway in breast cancer cells and mammosphere. Chem Biol Interact. 2020;328:1–12. doi: 10.1016/j.cbi.2020.109200. [DOI] [PubMed] [Google Scholar]
- Kushwaha PP, Kumar R, Neog PR, Behara MR, Singh P, Kumar A, Prajapati KS, Singh AK, Shuaib M, Sharma AK, Pandey AK. Characterization of phytochemicals and validation of antioxidant and anticancer activity in some Indian polyherbal ayurvedic products. Vegetos 2021. 2021;34:286–299. doi: 10.1007/s42535-021-00205-1. [DOI] [Google Scholar]
- Kushwaha PP, Maurya SK, Singh A, Prajapati KS, Singh AK, Shuaib M, Kumar S. Bulbine frutescens phytochemicals as novel ABC-transporter inhibitor: a molecular docking and molecular dynamics simulation study. J Cancer Metastasis Treat. 2021;7:1–13. doi: 10.20517/2394-4722.2020.92. [DOI] [Google Scholar]
- Kushwaha PP, Singh AK, Prajapati KS, Shuaib M, Gupta S, Kumar S. Phytochemicals present in Indian ginseng possess potential to inhibit SARS-CoV-2 virulence: a molecular docking and MD simulation study. Microb Pathog. 2021;157:1–11. doi: 10.1016/j.micpath.2021.104954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Li Y, But PP, Ooi VE. Antiviral activity and mode of action of caffeoylquinic acids from Schefflera heptaphylla (L.) Frodin. Antiviral Res. 2005;68:1–9. doi: 10.1016/j.antiviral.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Lu G, Hu Y, Wang Q, Qi J, Gao F, Li Y, Zhang Y, Zhang W, Yuan Y, Bao J, Zhang B, Shi Y, Yan J, Gao GF. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature. 2013;500:227–231. doi: 10.1038/nature12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurya SK, Maurya AK, Mishra N, Siddique HR. Virtual screening, ADME/T, and binding free energy analysis of anti-viral, anti-protease, and anti-infectious compounds against NSP10/NSP16 methyltransferase and main protease of SARS CoV-2. J Recept Signal Transduct Res. 2020;40:605–612. doi: 10.1080/10799893.2020.1772298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundkinajeddu D, Sawant LP, Koshy R, Akunuri P, Singh VK, Mayachari A, Sharaf MH, Balasubramanian M, Agarwal A. Development and validation of high performance liquid chromatography method for simultaneous estimation of flavonoid glycosides in Withania somnifera aerial parts. Int Sch Res Notices. 2014;2014:1–6. doi: 10.1155/2014/351547. [DOI] [Google Scholar]
- Nagarajan N, Yapp EKY, Le NQK, Yeh HY. In silico screening of sugar alcohol compounds to inhibit viral matrix protein VP40 of Ebola virus. Mol Biol Rep. 2019;46:3315–3324. doi: 10.1007/s11033-019-04792-w. [DOI] [PubMed] [Google Scholar]
- Naveed M, Hejazi V, Abbas M, Kamboh AA, Khan GJ, Shumzaid M, Ahmad F, Babazadeh D, FangFang X, Modarresi-Ghazani F, WenHua L, XiaoHui Z. Chlorogenic acid (CGA): a pharmacological review and call for further research. Biomed Pharmacother. 2018;97:67–74. doi: 10.1016/j.biopha.2017.10.064. [DOI] [PubMed] [Google Scholar]
- O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:1–14. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Schüttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Schwarz S, Sauter D, Wang K, Zhang R, Sun B, Karioti A, Bilia AR, Efferth T, Schwarz W. Kaempferol derivatives as antiviral drugs against the 3a channel protein of coronavirus. Planta Med. 2014;80:177–182. doi: 10.1055/s-0033-1360277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senapati S, Kumar S, Singh AK, Banerjee P, Bhagavatula S. Assessment of risk conferred by coding and regulatory variations of TMPRSS2 and CD26 in susceptibility to SARS-CoV-2 infection in human. J Genet. 2020;99:1–5. doi: 10.1007/s12041-020-01217-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A, Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Singh B, Sharma A, Kaur K, Gupta AP, Salar RK, Hallan V, Pati PK. Leaf spot disease adversely affects human health-promoting constituents and withanolide biosynthesis in Withania somnifera (L.) Dunal. J Appl Microbiol. 2017;122:153–165. doi: 10.1111/jam.13314. [DOI] [PubMed] [Google Scholar]
- Singh AK, Kushwaha PP, Prajapati KS, Shuaib M, Gupta S, Kumar S. Identification of FDA approved drugs and nucleoside analogues as potential SARS-CoV-2 A1pp domain inhibitor: an in silico study. Comput Biol Med. 2020;130:1–10. doi: 10.1016/j.compbiomed.2020.104185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AK, Patel PK, Choudhary K, Joshi J, Yadav D, Jin JO (2020b) Quercetin and Coumarin Inhibit Dipeptidyl Peptidase-IV and Exhibits Antioxidant Properties: In Silico, In Vitro, Ex Vivo. Biomolecules 10:1–14. https://doi.org/10.3390/biom1002020b7 [DOI] [PMC free article] [PubMed]
- Singh AK, Shuaib M, Kushwaha PP, Prajapati KS, Sharma R, Kumar S. In silico updates on lead identification for obesity and cancer. In: Kumar S, Gupta S, editors. Obesity and Cancer. Singapore: Springer; 2021. pp. 257–277. [Google Scholar]
- Solerte SB, Di Sabatino A, Galli M, Fiorina P. Dipeptidyl peptidase-4 (DPP4) inhibition in COVID-19. Acta Diabetol. 2020;57:779–783. doi: 10.1007/s00592-020-01539-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Vankadari N, Wilce JA. Emerging WuHan (COVID-19) coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect. 2020;9:601–604. doi: 10.1080/22221751.2020.1739565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma AK, Maurya SK, Kumar A, Barik M, Yadav V, Umar B, Lawal M, Usman ZA, Adam MA, Awal B. Inhibition of multidrug resistance property of Candida albicans by natural compounds of parthenium hysterophorus L. An in-silico approach. J Pharmacogn Phytochem. 2020;9:55–64. doi: 10.22271/phyto.2020.v9.i3a.11480. [DOI] [Google Scholar]
- Waseem M, Ahmad MK, Srivatava VK, Rastogi N, Serajuddin M, Kumar S, Mishra DP, Sankhwar SN, Mahdi AA. Evaluation of miR-711 as novel biomarker in prostate cancer progression. Asian Pac J Cancer Prev. 2017;18:2185–2191. doi: 10.22034/APJCP.2017.18.8.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Liu Y, Yang Y, Zhang P, Zhong W, Wang Y, Wang Q, Xu Y, Li M, Li X, Zheng M, Chen L, Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10:766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article.