

Abstract
The synthesis of triarylmethanes via Pd-catalyzed Suzuki-Miyaura reactions between diarylmethyl 2,3,4,5,6-pentafluorobenzoates and aryl boronic acids is described. The system operates at mild conditions and has a broad substrate scope, including the coupling of diphenylmethanol derivatives that do not contain extended aromatic substituents. This is significant as these substrates, which result in the types of triarylmethane products that are prevalent in pharmaceuticals, have not previously been compatible with systems for diarylmethyl ester coupling. Further, the reaction can be performed stereospecifically to generate stereo-inverted products. On the basis of DFT calculations, it is proposed that the oxidative addition of the diarylmethyl 2,3,4,5,6-pentafluorobenzoate substrate occurs via an SN2 pathway, which results in the inverted products. Mechanistic studies indicate that oxidative addition of the diarylmethyl 2,3,4,5,6-pentafluorobenzoate substrates to (IPr)Pd(0) results in the selective cleavage of the O–C(benzyl) bond in part because of a stabilizing η3-interaction between the benzyl ligand and Pd. This is in contrast to previously described Pd-catalyzed Suzuki-Miyaura reactions involving phenyl esters, which involve selective cleavage of the C(acyl)–O bond, because there is no stabilizing η3-interaction. It is anticipated that this fundamental knowledge will aid the development of new catalytic systems, which use esters as electrophiles in cross-coupling reactions.
Graphical Abstract
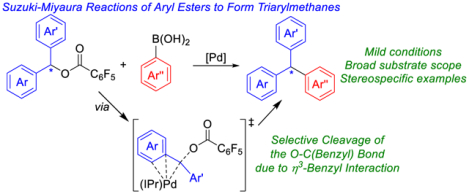
Introduction
Triarylmethanes are common motifs in natural products,1 fluorescent probes,2 organic dyes,3 metal ion sensors,4 and active pharmaceutical ingredients.5 For example, triarylmethanes have anti-viral,6 anti-tuberculosis,7 and anti-breast cancer8 properties (Figure 1). The most common method to synthesize triarylmethanes is through Lewis or Brønsted acid catalyzed Friedel-Crafts reactions of diarylmethanols or their derivatives with arenes, however, these reactions often have limited substrate scopes and poor selectivity.5b In recent years, a number of transition metal-catalyzed C(sp2)–C(sp3) cross-coupling reactions have been developed for the synthesis of triarylmethanes. These methods typically utilize non-classical electrophiles, such as diarylmethyl -ammonium salts,9 -sulfones,10 and -alcohol derivatives.11 In particular, alcohol derivatives, for example esters, are attractive as electrophiles for the synthesis of triarylmethanes because alcohols are abundant, stable, and diverse building blocks, which can easily be derivatized.12
Figure 1.

Examples of medicinally relevant triarylmethanes.
In 2005, Kuwano et al. demonstrated that benzyl acetates can be used in Pd-catalyzed Suzuki-Miyaura reactions to generate diarylmethanes (Figure 2a).13 Although this work was not extended to diarylmethyl esters, subsequent studies by Watson et al. and Jarvo et al. showed that diarylmethyl esters can be used in Ni-catalyzed Suzuki-Miyaura reactions to generate triarylmethanes stereospecifically (Figure 2b).11,14 The later reactions are currently limited to electrophiles with extended aromatic systems, such as naphthyls and biaryls, and are not compatible with diphenylmethanol derivatives, which are prevalent in natural products and pharmaceuticals.7,8b Nevertheless, they are the first examples of a synthetically valuable transformation and are fundamentally interesting because of their selectivity. Specifically, the high yields of triarylmethane products suggest that the diarylmethyl esters undergo oxidative addition exclusively across the O–C(benzyl) bond (Figure 3a).11,13,15 In contrast, depending on the catalyst and exact nature of the substrate some aryl esters can undergo oxidative addition across either the O–C(aryl) (Figure 3a)16 or C(acyl)–O bond (Figure 3b),17 to give either biaryl or ketone products, respectively, after transmetallation and reductive elimination. Further, in some cases following C(acyl)–O bond cleavage a decarbonylative step can occur, which ultimately leads to biaryl products (Figure 3c).18 At this stage, the exact reasons for the differences in selectivity are not clear, especially in regard to why the O–C(benzyl) bonds of diarylmethyl esters are cleaved and not the C(acyl)–O bonds.
Figure 2.

Summary of previous work on Suzuki-Miyaura reactions of diarylmethyl esters (a, b) and comparison to this work (c).
Figure 3.
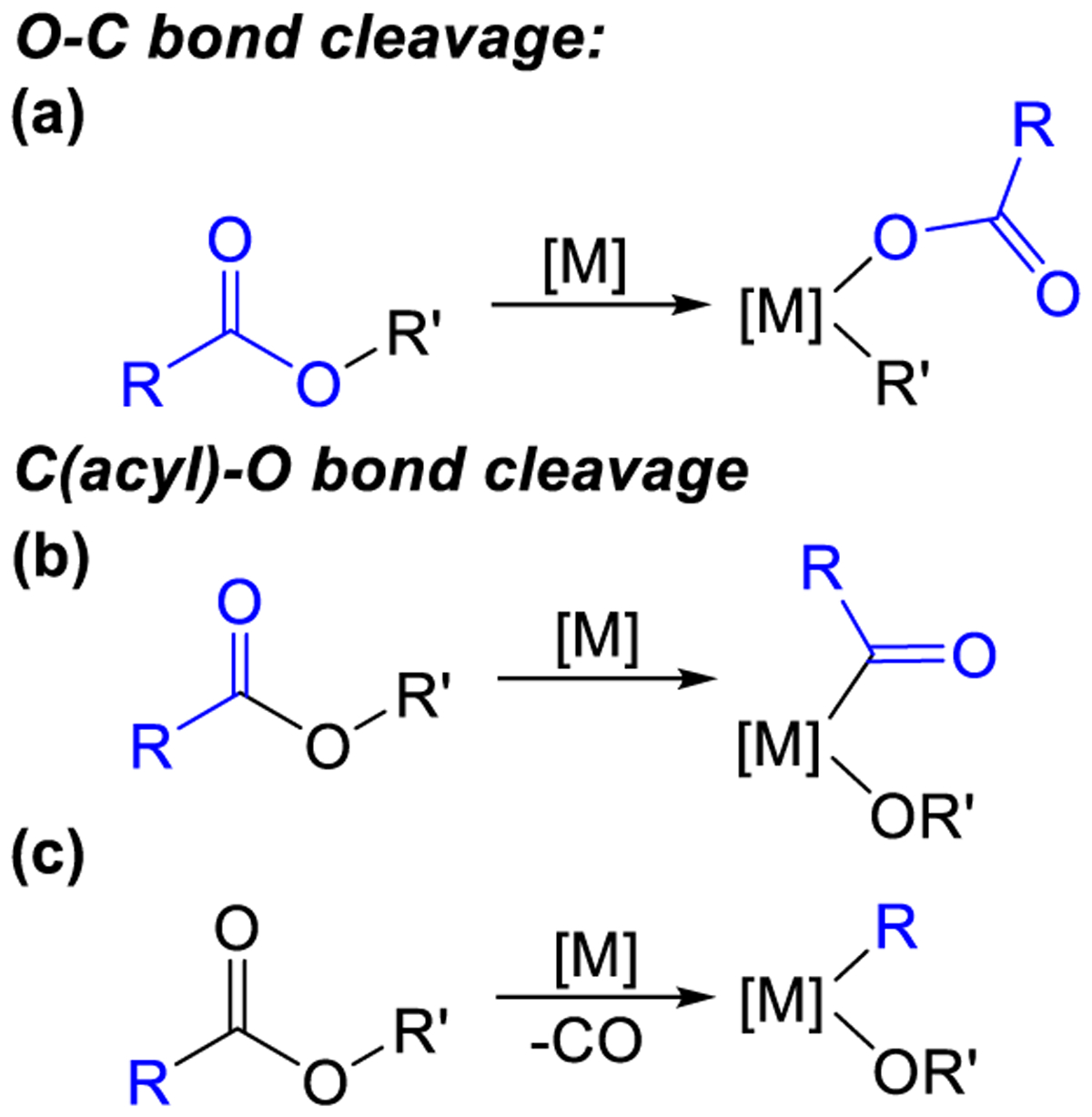
Bonds that can be cleaved in oxidative addition reactions involving esters.
Recently, our group and others have reported Pd-catalyzed Suzuki-Miyaura reactions of phenyl esters in which the C(acyl)–O bond is selectively cleaved to generate ketones in high yields.18c,19 Based in part on Kuwano et al.’s work with benzyl acetates,13 we hypothesized that if the substrate was changed to a diarylmethyl ester we could instead selectively cleave the O–C(benzyl) bond, which would ultimately lead to the formation of triarylmethanes. Herein, we report the first examples of Pd-catalyzed Suzuki-Miyaura reactions of diarylmethyl 2,3,4,5,6-pentafluorobenzoates to selectively generate triarylmethanes (Figure 2c). Importantly, the reaction is compatible with diphenylmethanol derivatives that do not contain extended aromatic substituents and enantioenriched diarylmethyl 2,3,4,5,6-pentafluorobenzoates can be coupled with high stereospecificity to give stereo-inverted products. The latter observation, supported by DFT calculations, suggests that oxidative addition of the substrate to the (IPr)Pd(0) active catalyst occurs via an SN2-type mechanism and we were able to isolate the oxidative addition product from the reaction of a diarylmethyl 2,3,4,5,6-pentafluorobenzoate to an (IPr)Pd(0) complex. Additional DFT calculations demonstrate it is kinetically and thermodynamically more favorable to cleave the O–C(benzyl) bond of benzyl benzoate during oxidative addition to (IPr)Pd(0) in part due to an η3-interaction between the benzyl group and the Pd. In contrast, it is kinetically and thermodynamically more favorable to cleave the C(acyl)–O bond of phenyl benzoate during oxidative addition to (IPr)Pd(0), which proceeds via a concerted mechanism. Overall, this work provides a notable synthetic advance due to the improved substrate scope for the formation of triarylmethanes and valuable fundamental information about selectivity in the cleavage of benzoates.
Results and Discussion
Reaction Discovery and Development
In preliminary work, we demonstrated that we could couple benzyl benzoate with phenyl boronic acid to selectively generate a diarylmethane product using (η3-1-tBu-indenyl)Pd(IPr)(Cl) (IPr = 1,3-bis(2,6-diisopropyl-phenyl)-1,3-dihydro-2H-imidazol-2-ylidene) as a precatalyst (Figure 4). This is in agreement with Kuwano et al.’s results13 and is indicative of selective cleavage of the O–C(R) (R = benzyl) bond. It is notable that when benzyl benzoate is replaced with phenyl benzoate under the same reaction conditions we selectively form benzophenone, which is consistent with selective cleavage of the C(acyl)–O bond (Figure 4). Our results with benzyl benzoate suggested that we could leverage the observed cleavage of the O–C(R) (R = benzyl) bond to develop a selective method for the synthesis of synthetically valuable triarylmethanes from diphenylmethyl esters through the careful selection of the leaving group on the ester and the optimization of the reaction conditions.
Figure 4.

Comparison of Pd-catalyzed Suzuki-Miyaura reactions of phenyl and benzyl benzoate using (η3-1-tBu-indenyl)Pd(IPr)(Cl) as the precatalyst under identical conditions.
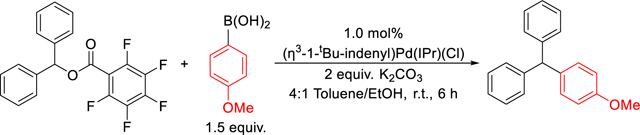
We initially synthesized diphenylmethyl-acetate, -benzoate, and −2,3,4,5,6-pentafluorobenzoate via a straightforward reaction between diarylmethanol and the appropriate carboxylic acid (see SI).17a Subsequently, using 1 mol% of (η3-1-tBu-indenyl)Pd(IPr)(Cl) as the precatalyst, a 4:1 mixture of toluene/ethanol as the solvent, and 2 equivalents of K2CO3 as the base, we assessed these esters as substrates in Suzuki-Miyaura reactions with 4-methoxyphenylboronic acid at room temperature (Figure 5a). After 6 hours, Suzuki-Miyaura reactions with diphenylmethyl-acetate and -benzoate give 28% and 35% of (diphenyl)(4-methoxyphenyl) methane, respectively, while the reaction with diphenylmethyl 2,3,4,5,6-pentafluorobenzoate yields 85% of the desired product. The observed trend in reactivity of the diphenylmethyl esters correlates with the pKas of the corresponding carboxylic acids, with acetic acid and benzoic acid having similar pKa values and 2,3,4,5,6-pentafluorobenzoic acid having a much lower pKa value.20 We suggest that the pKa values are reflective of the substrates relative ability to be activated by the Pd center via an SN2-type mechanism, as well as the strength of the interaction between Pd and the anion (vide infra).15i These reactions are some of the first examples of Suzuki-Miyaura reactions of diphenylmethanol derivatives that do not contain extended aromatic substituents.15k,21 We also compared the reactions using esters as electrophiles to those using diphenylmethyl chloride and diphenylmethyl bromide as substrates (Figure 5a). Although diphenylmethyl chloride gives a high yield (77%), the reaction results in the formation of (ethyl)(diphenylmethyl)ether as a side product (~5%) (Figure 5b), presumably as a result of base mediated nucleophilic substitution of the alcohol solvent on the halide. In agreement with this proposal, the more reactive diphenylmethyl bromide gives an even lower yield (21%), and an increased quantity of the ether side-product (~33%).
Figure 5.
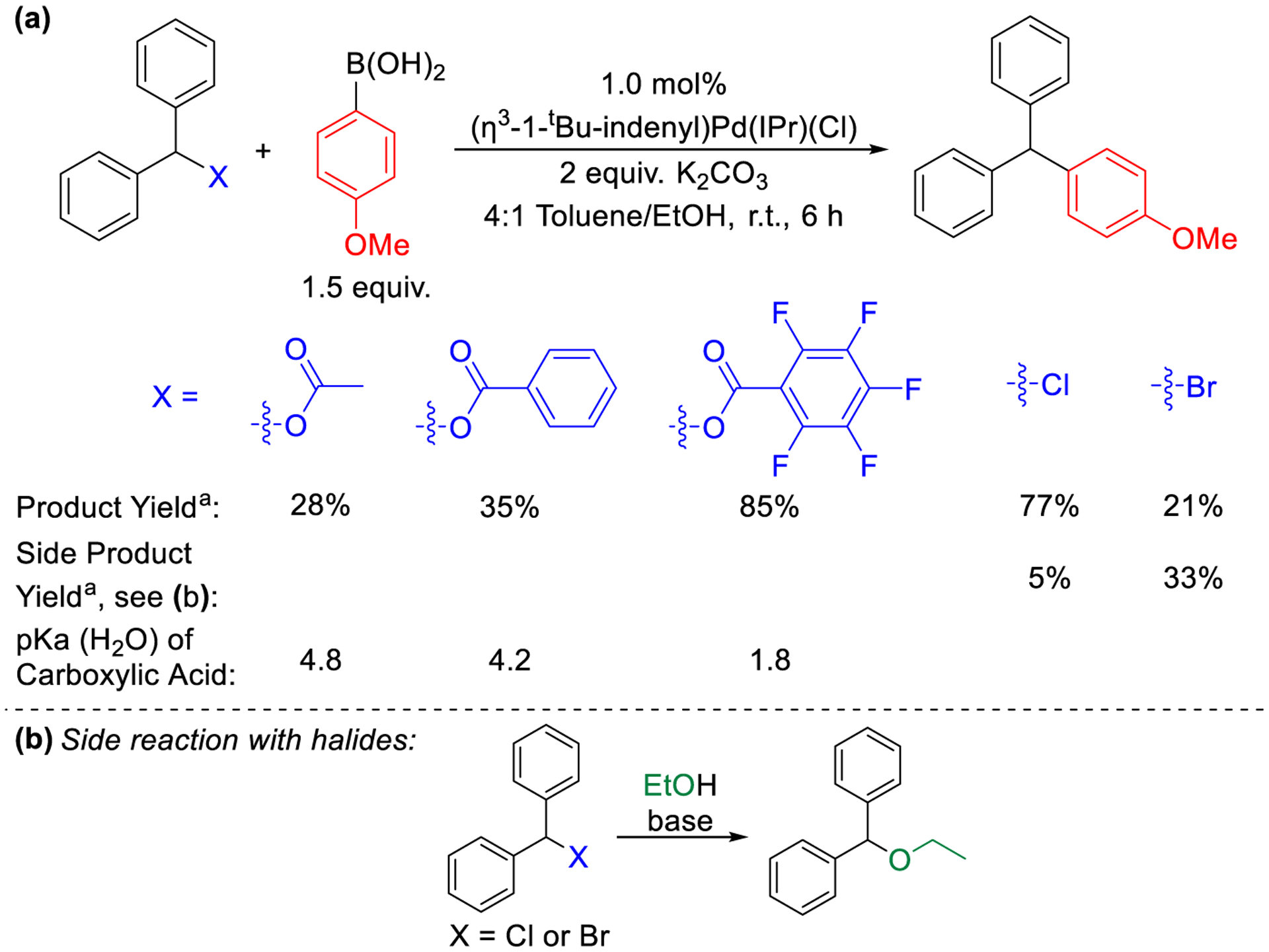
(a) Pd-catalyzed Suzuki-Miyaura reactions of diphenylmethyl esters and halides and (b) side reaction observed with aryl halides. aConditions: diphenylmethyl-X (0.05 mmol), 4-methoxyphenylboronic acid (0.075 mmol), K2CO3 (0.1 mmol), (η3-1-tBu-indenyl)Pd(IPr)(Cl) (0.0005 mmol), toluene (0.4 mL), and EtOH (0.1 mL). aYields are an average of two runs determined by GC-FID using the conversion of starting material to product.
We performed a series of Suzuki-Miyaura reactions between diphenylmethyl 2,3,4,5,6-pentafluorobenzoate and 4-methoxyphenylboronic acid to evaluate the factors that are important in obtaining high yields. In the absence of precatalyst no product is observed, and the majority of the starting material is converted to ethyl 2,3,4,5,6-pentafluorobenzoate, indicating that the diphenylmethyl 2,3,4,5,6-pentafluorobenzoate is not stable under the reaction conditions (Table 1, Entry 2). In the presence of (η3-1-tBu-indenyl)Pd(IPr)(Cl), the desired coupling outcompetes the side-reaction and quantitative conversion occurs in 24 hours (Entry 3). Increasing the temperature to 40 °C, results in a quantitative yield after 4 hours (Entry 4). Other common precatalysts, such as (η3-cinnamyl)Pd(IPr)Cl and PEPPSI-IPr, give moderate but reduced yields relative to (η3-1-tBu-indenyl)Pd(IPr)(Cl) likely due to their slower rates of activation under our reaction conditions (Entries 5 & 6).22 A similar yield was obtained when (η3-1-tBu-indenyl)Pd(IPr)Cl was replaced with [Pd(IPr)(μ-Cl)Cl]2 but this precatalyst was not pursued further, as it lacks a precursor that can be readily used for ligand screening (Entry 7).23 Changing the ancillary ligand to other frequently utilized NHC ligands, such as SIPr, IMes, and IPr*OMe (Entries 8–10), or phosphine ligands, such as PCy3, XPhos, or SPhos (Entries 11–13), gives reduced yields compared to IPr. We have observed similar trends in related Suzuki-Miyaura reactions involving phenyl benzoates.19f The choice of base is also crucial and although high yields are obtained with K2CO3, lower yields are observed with other common inorganic bases, such as K3PO4 or Na2CO3 (see SI). In the absence of a protic co-solvent no product is observed (Entry 14). Although the reaction worked comparably in the presence of methanol or ethanol (Entries 16 & 1), faster transesterification occurs in methanol to generate the methyl 2,3,4,5,6-pentafluorobenzoate by-product, which unnecessarily consumes starting material. Further, the yield suffered when water or isopropanol are used instead of ethanol (Entries 15 & 17). The role of the alcohol co-solvent may be in solubilizing the reagents, assisting in precatalyst activation,22b or facilitating transmetallation by displacing the carboxylate with an alkoxy ligand.13
Table 1.
Optimization table for the Pd-catalyzed Suzuki-Miyaura reaction of diarylmethyl 2,3,4,5,6-pentafluorobenzoate with 4-methoxyphenylboronic acid.
| ||
|---|---|---|
| Entry | Deviation from optimized conditions | GC conversiona |
| 1 | No change | 85% |
| 2 | No precatalyst | 0%b |
| 3 | 24 h instead of 6 h | >99% |
| 4 | 40 °C instead of r.t. and 4 h instead of 6 h | >99% |
| 5 | (η3-cinnamyl)Pd(IPr)Cl instead of (η3-1-tBu-indenyl)Pd(IPr)Cl | 73% |
| 6 | PEPPSI-IPr instead of (η3-1-tBu-indenyl)Pd(IPr)Cl | 64% |
| 7 | 0.5 mol% [Pd(IPr)(μ-Cl)Cl]2 instead of (η3-1-tBu-indenyl)Pd(IPr)Cl | 86% |
| 8 | SIPr instead of IPr | 62% |
| 9 | IMes instead of IPr | 16% |
| 10 | IPr*OMe instead of IPr | 40% |
| 11 | PCy3 instead of IPr | 2%c |
| 12 | XPhos instead of IPr | 3% |
| 13 | SPhos instead of IPr | 46%d |
| 14 | Toluene only | <1% |
| 15 | H2O instead of EtOH | 69% |
| 16 | MeOH instead of EtOH | 89%e |
| 17 | iPrOH instead of EtOH | 56% |
Conditions: diphenylmethyl ester (0.05 mmol), 4-methoxyphenylboronic acid (0.075 mmol), base (0.1 mmol), precatalyst (0.0005 mmol), solvent (0.5 mL).
Yields are an average of two runs determined by GC-FID using the conversion of starting material to product.
64% conversion to F5C6COOC2H5 and 36% starting material remaining.
67% conversion to F5C6COOC2H5.
32% conversion to F5C6COOC2H5.
9% conversion to F5C6COOCH3.
Substrate Scope
Given the success of Suzuki-Miyaura reactions involving diphenylmethyl 2,3,4,5,6-pentafluorobenzoate, we explored the substrate scope for the coupling of a range of diarylmethyl 2,3,4,5,6-pentafluorobenzoates with phenylboronic acid under our optimized conditions (Figure 6). Electron neutral diarylmethyl esters (6a & 6b), including fluorene-9-ester (6b), result in high yields, 96 and 94% respectively, of the desired product. The latter result is notable because fluorenes are important motifs in biologically active molecules,24 as monomers in polymer chemistry,25 and as dyes for solar cell applications, and our system provides a facile method for functionalization.26 Additionally we were able to generate 6a on a 1 mmol scale in 95% isolated yield (see SI), which demonstrates that our method is scalable. The reaction is also compatible with esters containing electron-donating (6c) and -withdrawing (6d & 6e) substituents. This includes the doubly cyano substituted ester (6e) which is coupled in 86% yield. This is significant because cyano groups often coordinate to Pd and inhibit catalysis. It is more difficult to couple substrates with ortho-substitution on an aryl group of the diarylmethyl ester, such as 6f. Even with higher precatalyst loading (4 mol%) and longer reactions times (16 h), when ethanol is used as the co-solvent, transesterification to generate the ethyl 2,3,4,5,6-pentafluorobenzoate competes with the desired cross-coupling reaction and only a 40% NMR yield of cross-coupled product is observed. However, an 87% yield is obtained by increasing the precatalyst loading to 4 mol%, the temperature to 80 °C, the time to 16 h, and switching to water as the co-solvent to prevent transesterification. Consistent with the challenges in coupling ortho-substituted substrates, (1-napthyl)(phenyl)methyl 2,3,4,5,6-pentafluorobenzoate (6g) gives minimal yield under the optimized conditions (6% NMR yield). When more forcing conditions are utilized, with the use of water in place of ethanol as a co-solvent, a yield of 77% is obtained. Under the same conditions, the even more sterically bulky bis(1-naphthyl)methyl 2,3,4,5,6-pentafluorobenzoate (6h) gives an 87% yield. In contrast, we are able to couple the less sterically bulky substrate (2-napthyl)(phenyl)methyl 2,3,4,5,6-pentafluorobenzoate (6i) without major modification to our optimized conditions (16 h instead of 4 h). Pyridyl groups are common in pharmaceuticals but are often difficult substrates for cross-coupling reactions due to coordination of the heterocycle to Pd.27 Using our more forcing conditions, including the replacement of ethanol with water as a co-solvent, we can couple a 2-pyridyl containing electrophile (6j) in 49%.
Figure 6.

Isolated and NMR yields for Pd-catalyzed Suzuki-Miyaura reactions of diarylmethyl 2,3,4,5,6-pentafluorobenzoates with (4-methoxyphenyl)boronic acid. Conditions for isolated yields: Ester (0.2 mmol), phenylboronic acid (0.3 mmol), K2CO3 (0.4 mmol), (η3-1-tBu-indenyl)Pd(IPr)Cl (0.0002 mmol), toluene (1.6 mL), ethanol (0.4 mL). Conditions for NMR yields: Ester (0.05 mmol), phenylboronic acid (0.075 mmol), K2CO3 (0.1 mmol), (η3-1-tBu-indenyl)Pd(IPr)Cl (0.0005 mmol), toluene (0.4 mL), ethanol (0.1 mL). NMR yields were determined using 1,2,4,5-tetramethyl benzene as an internal standard. a4 mol% (η3-1-tBu-indenyl)Pd(IPr)Cl instead of 1 mol%. b16 h instead of 4 h. c80 °C instead of 40 °C. dWater instead of ethanol.
Enantiomers of chiral molecules often have different biological activity due to variations in their binding affinity with chiral targets.28 Therefore, there are advantages to accessing single enantiomers of triarylmethanes.1 We hypothesized that our method could be compatible with producing single enantiomers of triarylmethanes if enantioenriched diarylmethyl 2,3,4,5,6-pentafluorobenzoates esters were utilized as substrates. The enantioenriched substrates, (S)-(2-naphthyl)(phenyl)methyl 2,3,4,5,6-pentafluorobenzoate and (S)-(4-methylphenyl)(phenyl)methyl 2,3,4,5,6-pentafluorobenzoate, were readily synthesized from the appropriate (S)-diarylmethanol by following a literature procedure involving the reaction of an in situ generated phenylzinc species and an aldehyde catalyzed by a chiral pyrrolidine ligand (see SI).29 The subsequent coupling of (S)-(2-naphthyl)(phenyl)methyl 2,3,4,5,6-pentafluorobenzoate with 3-furylboronic acid under our optimized conditions results in the formation of triarylmethane 7a in 97% yield and 99% es, indicating the reaction proceeds with high enantiospecificity (Figure 7). Comparison of the polarimetry data of isolated 7a ([α]20D −23.25 (c 0.400, CDCl3)) with literature data ([α]29D −22.0 (c 1.00, CDCl 3))11a suggests the formation of the (R)-isomer, which is consistent with a pathway involving inversion. Similarly, the reaction of (S)-(4-methylphenyl)(phenyl)methyl 2,3,4,5,6-pentafluorobenzoate with (4-methoxy)phenylboronic acid generates the enantioenriched triarylmethanes 7b in 88% yield and 90% es. Given that we propose the mechanism of the reaction involves oxidation addition, transmetallation and reductive elimination (vide infra), the stereochemical inversion of the (S)-diarylmethyl 2,3,4,5,6-pentafluorobenzoates to the (R)-triarylmethanes suggests that oxidative addition proceeds via an SN2 pathway. This is because transmetallation and reductive elimination typically occur with retention of configuration in Suzuki-Miyaura reactions,30 and therefore the stereochemistry of the product is likely based on oxidative addition. Our work is consistent with previous studies utilizing NHC-Ni catalysts for the stereoselective coupling of naphthyl and biaryl diarylmethyl esters, which also proceed with inversion and propose that oxidative addition is responsible for the observed stereochemistry.11a,15i
Figure 7.
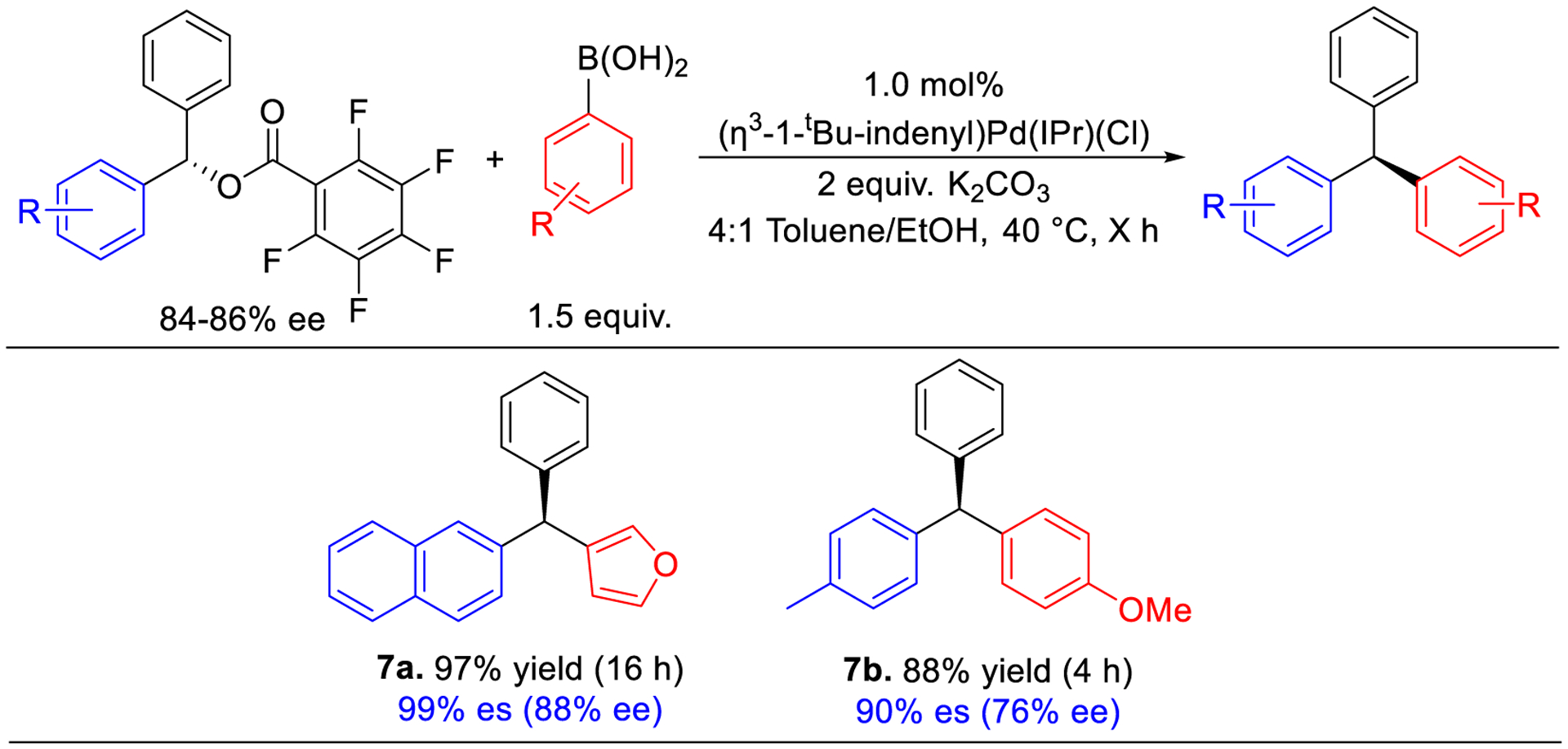
Isolated yields and enantioselectivity for Pd-catalyzed stereospecific Suzuki-Miyaura reactions of diarylmethyl 2,3,4,5,6-pentafluorobenzoates. Conditions for isolated yields: Enantioenriched ester (0.05 mmol), arylboronic acid (0.075 mmol), K2CO3 (0.1 mmol), (η3-1-tBu-indenyl)Pd(IPr)Cl (0.0005 mmol), toluene (0.4 mL), ethanol (0.1 mL).
The scope of the boronic acid coupling partner was evaluated using diphenylmethyl 2,3,4,5,6-pentafluorobenzoate as the electrophile (Figure 8). The reaction is compatible with electron-neutral, -donating, and -withdrawing aryl boronic acids (8a-h). In a noteworthy result, 4-methylester- (8f) and 4-acetyl- (8g) phenylboronic acids give yields of 87% and 70%, respectively, which are not only electron-withdrawing but can be further functionalized, for example via nucleophilic addition reactions.31 The reaction is tolerant of ortho-substituted aryl boronic acids (8i-j). However, while 2,6-dimethoxyphenyl boronic acid (8l) could be coupled in high yield (although difficulty in isolation resulted in a lower isolated yield), minimal product is observed with 2,4,6-trimethylphenyl boronic acid (8m) even with higher precatalyst loadings. 1- and 2-napthyl boronic acids (8j & 8k) are coupled in yields of 83% and 80%, respectively. Heteroaryl-substituted triarylmethanes derivatives are common in pharmaceutical compounds,7b,32 but cross-coupling reactions of diarylmethanol derivatives with heteroaryl boronic acids are limited,11a,33 possibly due to the instability of heteroaryl nucleophiles.34 Our system is able to couple 2-furyl (8n) and 3-thiophene (8o) boronic acids in yields of 98% and 97%, respectively, but 2-substituted thienyl boronic acid gives low conversion (see SI). Benzo[b]thien-2-yl- (8p) and boc-protected pyrrole- (8q) boronic acids give yields of 80% and 70%, respectively, but require longer reaction times. Unfortunately, pyridyl boronic acids could not be coupled (see SI), likely due to protodeborylation or coordination of the nitrogen atom of the pyridine ring to the catalyst.
Figure 8.
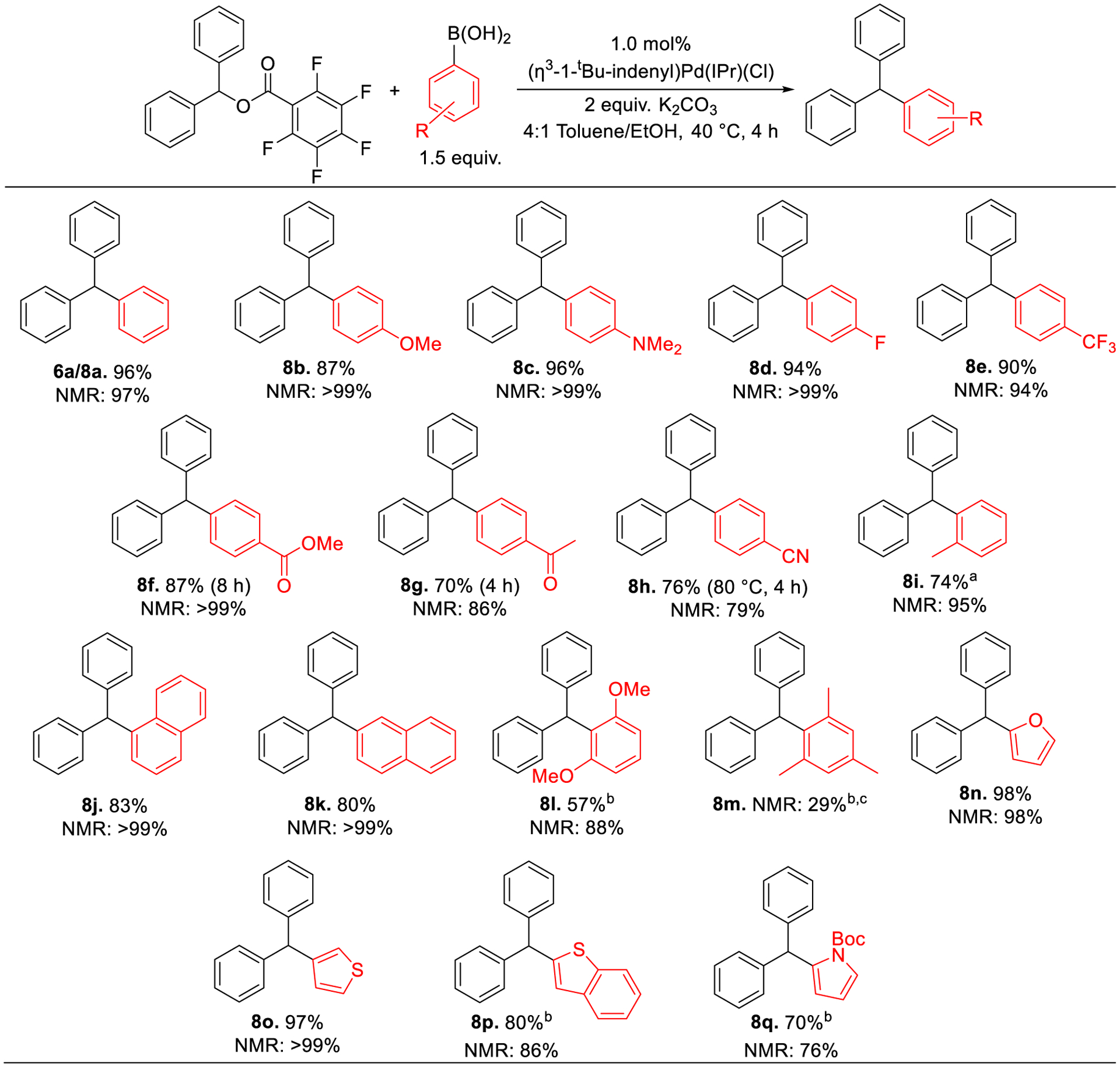
Isolated and NMR yields for Pd-catalyzed Suzuki-Miyaura reactions of diphenylmethyl 2,3,4,5,6-pentafluorobenzoate with boronic acids. Conditions for isolated yields: Ester (0.2 mmol), boronic acid (0.3 mmol), K2CO3 (0.4 mmol), (η3-1-tBu-indenyl)Pd(IPr)Cl (0.0002 mmol), toluene (1.6 mL), ethanol (0.4 mL. Conditions for NMR yields: Diphenylmethyl 2,3,4,5,6-pentafluorobenzoate ester (0.05 mmol), (4-methoxyphenyl)boronic acid (0.075 mmol), K2CO3 (0.1 mmol), (η3-1-tBu-indenyl)Pd(IPr)Cl (0.0005 mmol), toluene (0.4 mL), ethanol (0.1 mL). NMR yields were determined using 1,2,4,5-tetramethyl benzene as an internal standard. a8 h instead of 4 h. b16 h instead of 4 h. c4 mol% (η3-1-tBu-indenyl)Pd(IPr)Cl instead of 1 mol%.
Overall, our substrate scope demonstrates that we have developed a mild and general method for the synthesis of triarylmethanes, containing both extended and non-extended aromatic groups, from readily available diarylmethyl esters. In particular, the ability of our system to couple diphenylmethyl esters that do not contain extended aromatic substrates is significant, as these substrates have not previously been utilized. Further, our system is tolerant of a variety of different functional groups, is compatible with heterocyclic substrates, and is stereospecific. As a result, we expect that it will be valuable for the synthesis of medicinally relevant triarylmethanes.
Mechanistic Studies
The high yields of triarylmethanes that are observed in Suzuki-Miyaura reactions involving diarylmethyl 2,3,4,5,6-pentafluorobenzoates indicate that the reaction must be proceeding with high selectivity, consistent with the selective cleavage of the O–C(benzyl) bond. Further, there is no evidence for cleavage of the C(acyl)–O bond, which presumably occurs readily when a phenyl ester is utilized instead of a benzyl ester (Figure 4). Previous studies have hypothesized that oxidative addition to Pd(0) is responsible for the differences in selectivity between phenyl- and benzyl-esters but there are few well-defined examples of the oxidative addition of these substrates and the reaction pathways (e.g. concerted versus SN2) have not be probed.35 In seminal work, Yamamoto et al. demonstrated differences in oxidative addition for aryl- and benzyl-trifluoroacetates to phosphine ligated Pd(0) complexes.15b,35b,35c Specifically, aryl trifluoroacetates were shown to oxidatively add across the C(acyl)–O bond, while benzyl trifluoroacetates underwent oxidative addition across the O–C(benzyl) bond. However, the mechanistic origins for this difference were not elucidated and their applicability to our system, which features a different ester and ancillary ligand, was unclear. Therefore, we studied the oxidative addition of the type of esters used in this work, such as phenyl- and benzyl-benzoates, to the proposed active catalytic species (IPr)Pd(0).36
Initial evaluation of the oxidative addition of phenyl benzoate with the Pd(0) source (IPr)Pd(0)(styrene)2 resulted in slow decomposition to Pd black and (IPr)2Pd. The use of more electron withdrawing electrophiles is known to result in more facile oxidative addition and also stabilize the resulting transition metal complexes.37 In this case, treatment of phenyl 2,3,4,5,6-pentafluorobenzoate with (IPr)Pd(styrene)2 at room temperature resulted in the slow formation of new product(s) as determined by NMR spectroscopy, but this was followed by decomposition, which prevented full characterization. We hypothesized that an ortho-coordinating ligand on the benzoate group, such as diphenylphosphine, would stabilize the putative three-coordinate oxidative addition complex by binding to the open coordination site on the metal center. The reaction of phenyl 2-(diphenylphosphino)benzoate with (IPr)Pd(styrene)2 at room temperature resulted in the clean formation of a product, 1, with a single resonance in the 31P NMR spectrum at 50.4 ppm, which was isolated in 75% yield (Figure 9a). X-ray crystallography confirmed that 1 is the result of oxidative addition of the C(acyl)–O bond to (IPr)Pd(0) with coordination of the phosphine (Figure 9b). Interestingly, the crystal structure shows that the C(acyl) ligand is trans to the OPh ligand and the phosphine ligand is trans to the IPr ligand. It would be expected, however, that after concerted oxidative addition the C(acyl) ligand and OPh ligand would be cis to one another, which suggests that the ligands can rearrange on the metal center either by decoordination of the phosphine or OPh ligands. Nevertheless, the formation of 1 indicates that (IPr)Pd(0) is capable of facile cleavage of the C(acyl)–O bond of a phenyl ester. Notably, this is the first example of a well-defined oxidative addition product from the addition of a phenyl ester to Pd(0) that does not undergo decarbonylation.
Figure 9.
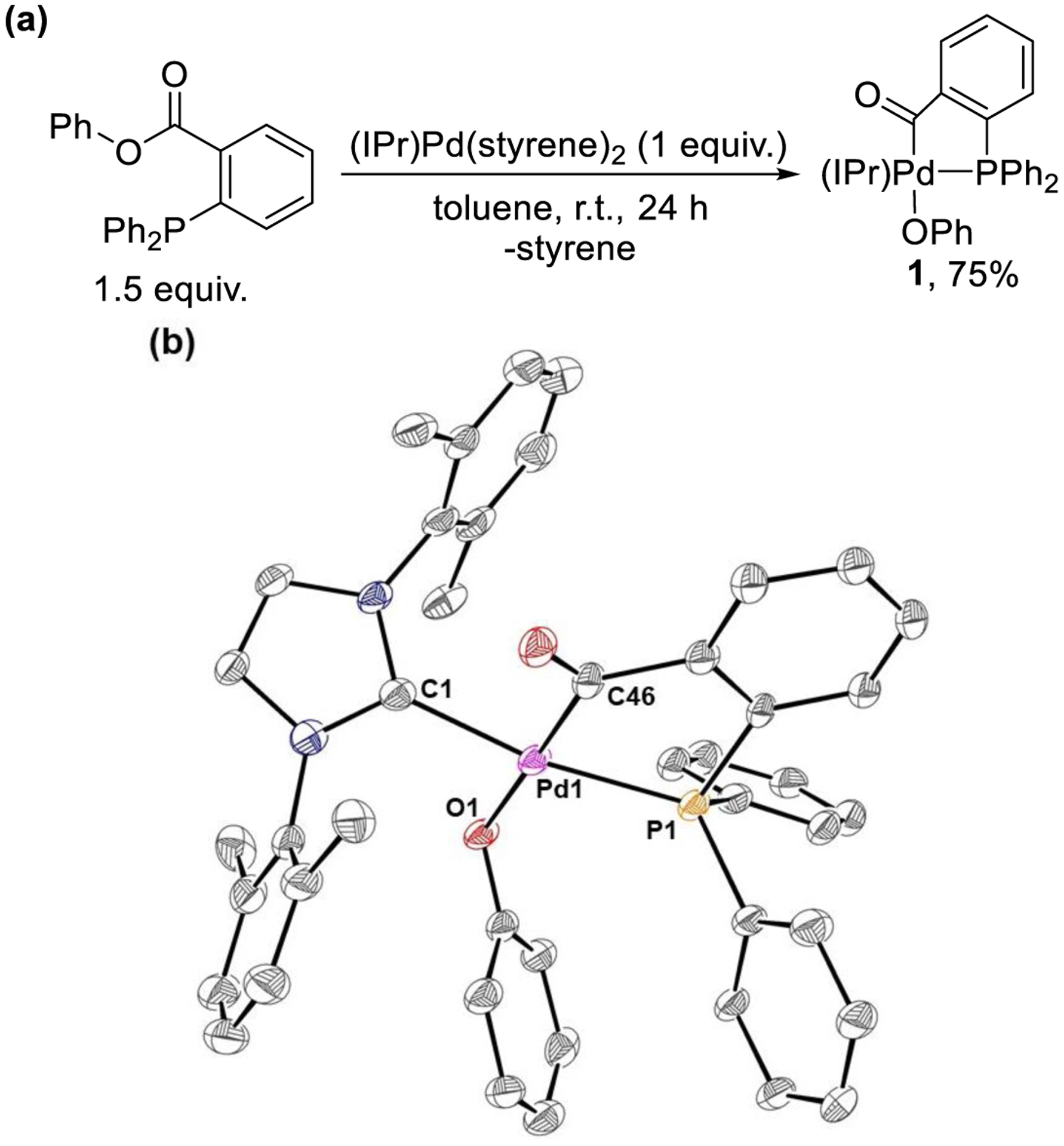
(a) Oxidative addition of phenyl 2-(diphenylphosphino)benzoate to (IPr)Pd(0) to form 1. (b) ORTEP (30% probability) of 1. Hydrogen atoms and methyl groups associated with the iso-propyl substituents of IPr omitted for clarity. Selected bond distances and angles Pd1-C1 1.971(9) Å, Pd1-C46 1.975(7) Å, Pd1-P1 2.287(2) Å, Pd1-O1 2.107(6) Å; C46-Pd1-O1 90.2(2)°, C1-Pd1-C46 89.5(3)°, P1-Pd1-O1 96.0(1)°, P1-Pd1-C1 84.0(2)°.
Next, we investigated the reaction of benzyl benzoate derivatives with (IPr)Pd(styrene)2. In an analogous fashion to the reaction with the unsubstituted phenyl benzoate, the reaction of benzyl benzoate with (IPr)Pd(styrene)2 resulted in slow decomposition to Pd black and (IPr)2Pd. However, when we performed the reaction of (IPr)Pd(styrene)2 with 2,3,4,5,6-pentafluorobenzoate, a substrate used in catalysis, one new product, 2, is formed and isolated in a 65% yield (Scheme 1).38 In the 1H NMR spectrum of 2, the resonance associated with the methine proton of the benzyl group is shifted up-field relative to that of the free ester (4.78 vs 7.19 ppm), consistent with the presence of a metal center in close proximity to the methine proton.39 The 13C NMR spectrum includes a resonance at 177.0 ppm and two new peaks are observed in the IR spectrum at 1633 cm−1 and 1343 cm−1. This is indicative of the presence of a carbonyl group. Overall, our NMR data suggests that 2 is the oxidative addition product from cleavage of the O–C(benzyl) bond of 2,3,4,5,6-pentafluorobenzoate. However, we were unable to identify the exact structure of 2 as despite repeated attempts single crystals for X-ray diffraction could not be obtained. Possible structures of 2 include those with the diarylmethyl ligand binding in either an η1 or η3-fashion, and the 2,3,4,5,6-pentafluorobenzoate ligand binding in a κ1 or κ2-manner (Scheme 1). Further, it is possible that the 2,3,4,5,6-pentafluorobenzoate anion is not coordinated to Pd and an ion pair is generated. Nevertheless, our results indicate that oxidative addition of 2,3,4,5,6-pentafluorobenzoate with cleavage of the O–C(benzyl) bond is facile and we suggest that this is the first step in catalysis. Consistent with this proposal, when 2 is used as a catalyst in the coupling of diphenylmethyl 2,3,4,5,6-pentafluorobenzoate with (4-methoxy)phenylboronic acid under our optimized conditions (Equation 1), complete conversion to (2-methoxyphenyl)(diphenyl)methane is observed, indicating that 2 is a kinetically competent catalyst.
Scheme 1.

Formation of oxidative addition complex (IPr)Pd(CHPh2)(OOCC6F5) (2) and possible structures of 2.
 |
(eq 1) |
DFT calculations were performed to help understand the observed selectivity in the oxidative addition of phenyl- and benzyl-benzoates to (IPr)Pd(0).40 Phenyl and benzyl-benzoate were used as model substrates given our results in Figure 4, showing that they undergo selective cleavage of the C(acyl)–O bond and O–C(benzyl) bond, respectively. For both substrates, the concerted oxidative addition pathway was computed for the heterolytic cleavage of the C(acyl)–O and the O–C(aryl) bonds. In the case of benzyl benzoate, an SN2 pathway was also calculated for the cleavage of the O–C(benzyl) bond.
The oxidative addition transition state for C(acyl)–O cleavage in phenyl benzoate, TSPh(CAc–O), is shown in Figure 10. In addition to the C(acyl)–O bond cleavage (1.71 Å), this transition state also involves the concerted formation of the Pd–C(acyl)Ph (2.08 Å) and Pd–OPh (2.16 Å) bonds. The three atoms involved in the bond rearrangement form a 3-membered Pd–O–C metallacycle, consistent with literature precedent.17a In contrast, the transition state for O–C(aryl) bond cleavage, in phenyl benzoate TSPh(O–CAr), has a 5-membered metallacycle geometry, in which one of the two carboxylate O atoms breaks the bond with the Ph ring (2.04 Å), whereas the other forms a new bond with Pd (2.20 Å). The full relaxation of these two transition states to the oxidative addition products yields the complexes PPh(CAc–O) and PPh(O–CAr) with the expected Ph(O)C–Pd–OPh and Ph(O)CO–Pd–Ph moieties, respectively (Figure 10).41 Importantly, the cleavage of the C(acyl)–O bond in phenyl benzoate is both kinetically (8.8 vs 19.0 kcal/mol) and thermodynamically (−4.0 vs 4.6 kcal/mol) preferred compared with cleavage of the O–C(aryl) bond, consistent with our experimental results (Figure 4).
Figure 10.
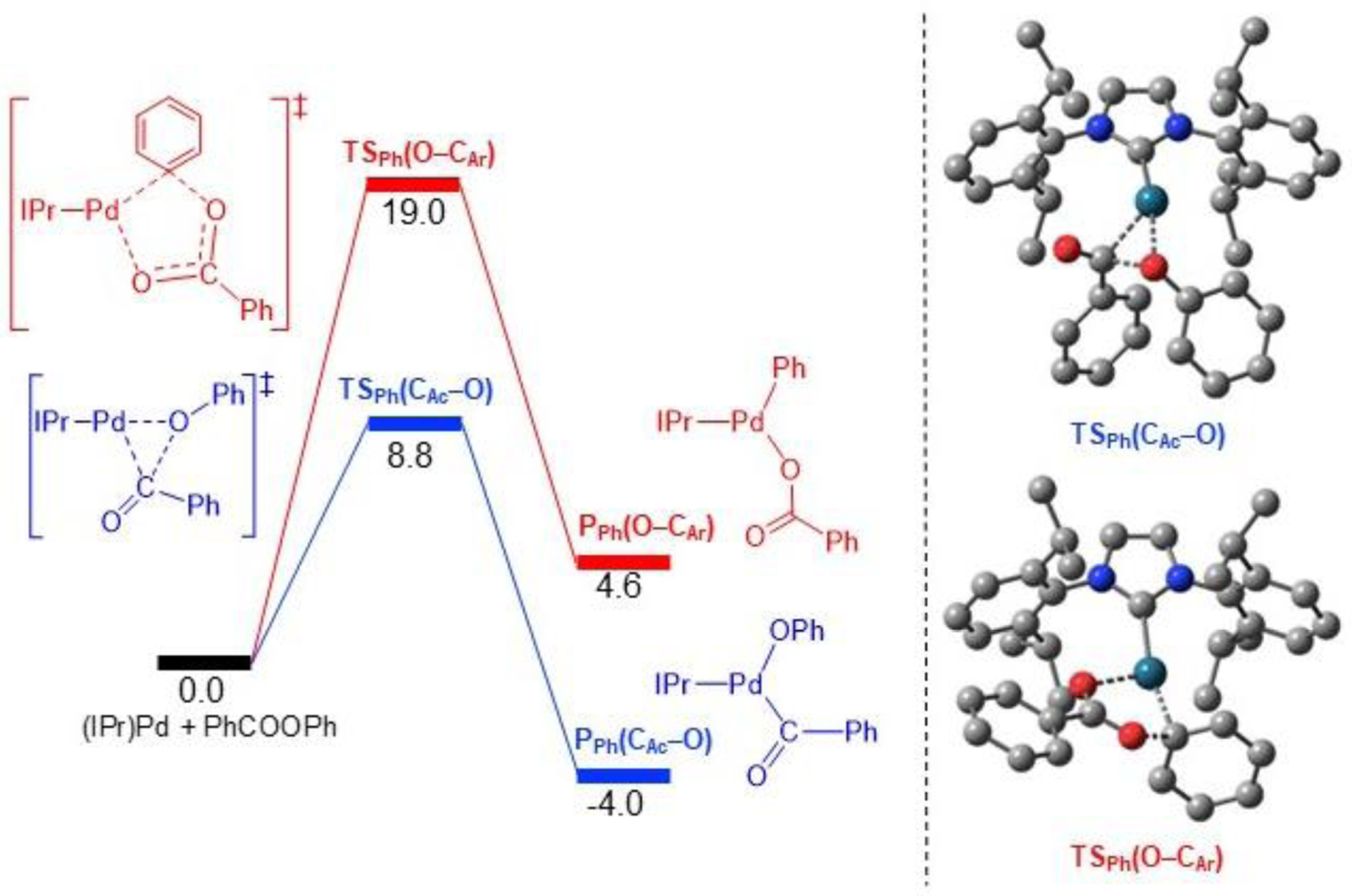
DFT calculations comparing the oxidative addition of phenyl benzoate via C(acyl)–O and O–C(aryl) bond cleavage. Relative energies in kcal/mol.
The concerted transition states associated with cleavage of the C(acyl)–O and O–C(benzyl) bond of benzyl benzoate are shown in Figure 11, along with the transition state associated with cleavage of the O–C(benzyl) bond via an SN2 mechanism. The geometry of TSBz(CAc–O) is very similar to TSPh(CAc–O), with the cleavage and formation of the C(acyl)–O (1.96 Å), Pd–C(acyl)Ph (2.02 Å) and Pd–OBz (2.11 Å) bonds in a 3-membered palladacycle. The transition state for O–C(aryl) bond cleavage, TSBz(O–CBz), is also similar to TSPh(O-CAr), involving both O atoms of the carboxylate group, one cleaving the bond to the benzyl (1.99 Å), and the other forming a new bond with Pd (2.41 Å). However, TSBz(O–CBz) has a distinct feature. Specifically, there is an η3-interaction between the metal center and the benzyl moiety, with Pd–C interatomic distances of 2.71, 2.34 and 2.17 Å for the CBz, Cipso and Cortho atoms, respectively.42 This interaction becomes a covalent η3-bond in the PBz(O–CBz) product, with distances shortened to 2.09, 2.27 and 2.39 Å, respectively. This is likely the main reason why the PBz(O–CBz) product is lower in energy than the PBz(CAc–O) product (−9.7 vs 6.0 kcal/mol), although the transition state energy associated with TSBz(CAc–O) is still lower than that observed for TSBz(O–CBz) (12.5 vs 20.7 kcal/mol).
Figure 11.
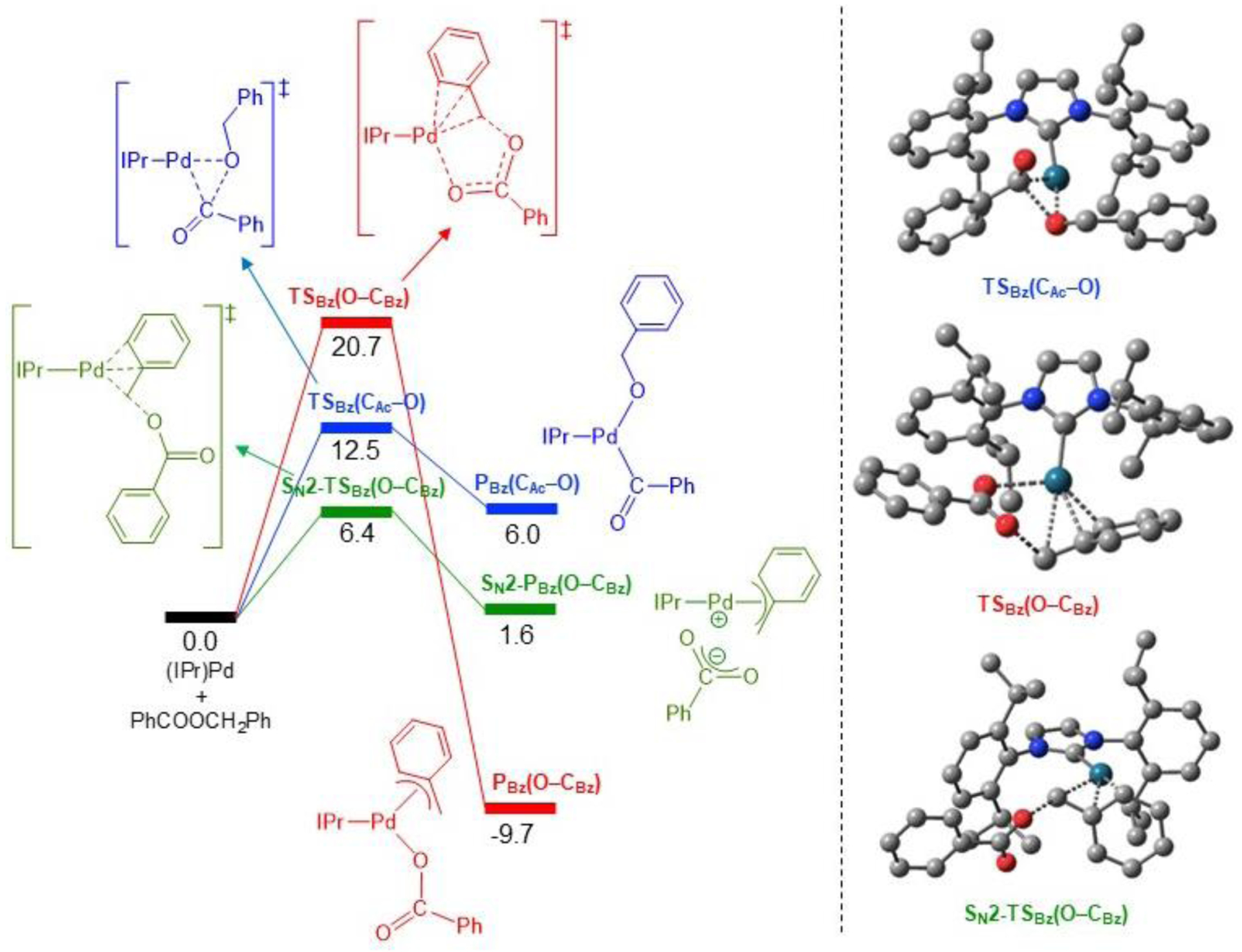
DFT calculations comparing the oxidative addition of benzyl benzoate via concerted C(acyl)–O and O–C(aryl) bond cleavage and also an SN2 pathway. Relative energies in kcal/mol.
The cleavage of the O–C(benzyl) bond can also occur via an SN2 pathway. The SN2 mechanism follows a lower energy pathway than either the concerted O–C(benzyl) or C(acyl)–O bond cleavage, as the barrier associated with the SN2-TSBz(O–CBz) transition state, 6.4 kcal/mol, is the lowest found for this substrate (Figure 11). The two main structural features of SN2-TSBz(O–CBz) are: (1) the η3-interaction between the metal center and the CBz (2.32 Å), Cipso (2.29 Å), and Cortho (2.25 Å) atoms, similar to that of the concerted TSBz(O–CBz) transition state, and (2) the linear arrangement between the forming Pd–CBz (2.32 Å) and the breaking CBz–O (2.08 Å) bonds, with a Pd–CBz–O angle of 173.6°, and CBz adopting a distorted trigonal bipyramid geometry. The preference for the SN2 pathway is in agreement with the inversion of configuration observed in the Pd-catalyzed Suzuki-Miyaura reactions of enantioenriched (diaryl)methyl 2,3,4,5,6-pentafluorobenzoate (Figure 7), as well as that seen in the literature with (NHC)Ni catalysts.11b,15i The product of the SN2 pathway, SN2-PBz(O–CBz), is an ion pair between the (IPr)Pd(η3-Bz)+ cation and the PhCO2− anion (with a natural charge q = ±0.78 value). This ion pair is more stable than the PBz(CAc–O) complex (1.6 vs 6.0 kcal/mol) and can be further stabilized by rearranging into the PBz(O–CBz) complex (1.6 vs −9.7 kcal/mol).43 Thus, O–C(benzyl) cleavage is both kinetically and thermodynamically preferred over C(acyl)–O cleavage. Overall, the computational results explain the experimental observation that for phenyl benzoate the C(acyl)–O bond is cleaved whereas for benzyl benzoate the O–C(benzyl) bond is cleaved.
In our experimental work, we demonstrated that a Suzuki-Miyaura reaction with diphenylmethyl 2,3,4,5,6-pentafluorobenzoate is more efficient than the corresponding reaction with diphenylmethyl benzoate (Figure 5). To investigate this observation, the transition state for cleavage of O–C(benzyl) bond of benzyl 2,3,4,5,6-pentafluorobenzoate via an SN2-type oxidative addition pathway was calculated using DFT. The barrier is 2.1 kcal/mol lower in energy than that of the benzyl benzoate consistent with our experimental results (Figure 12). However, in both cases the barrier for oxidative addition is very low (4.3 and 6.4 kcal/mol), suggesting that oxidative addition is facile. The SN2 product of the oxidative addition of benzyl 2,3,4,5,6-pentafluorobenzoate to (IPr)Pd(0), PFBz(O–CBz), is also an ion pair (i.e. (IPr)Pd(η3-FBz)+PhCO2−, with q = ± 0.82e), which is significantly more stable than that formed by the non-fluorinated substrate (−6.1 vs 1.6 kcal/mol), likely due to the fluorides promoting charge separation.44 We propose that the outersphere 2,3,4,5,6-pentafluorobenzoate anion in PFBz(O–CBz) may lead to faster transmetallation, the likely next elementary step in the mechanism after oxidative addition (Figure 13), in part because the formation of PFBz(O–CBz) is not as thermodynamically favorable as the formation of PBz(O–CBz) (see SI). Thus, utilizing the 2,3,4,5,6-pentafluorobenzoate as the leaving group may result in improvements to multiple elementary steps on the catalytic cycle. By analogy to other cross-coupling reactions,45 we expect that the final step reductive elimination is facile, suggesting that transmetallation is the turnover-limiting step.
Figure 12.
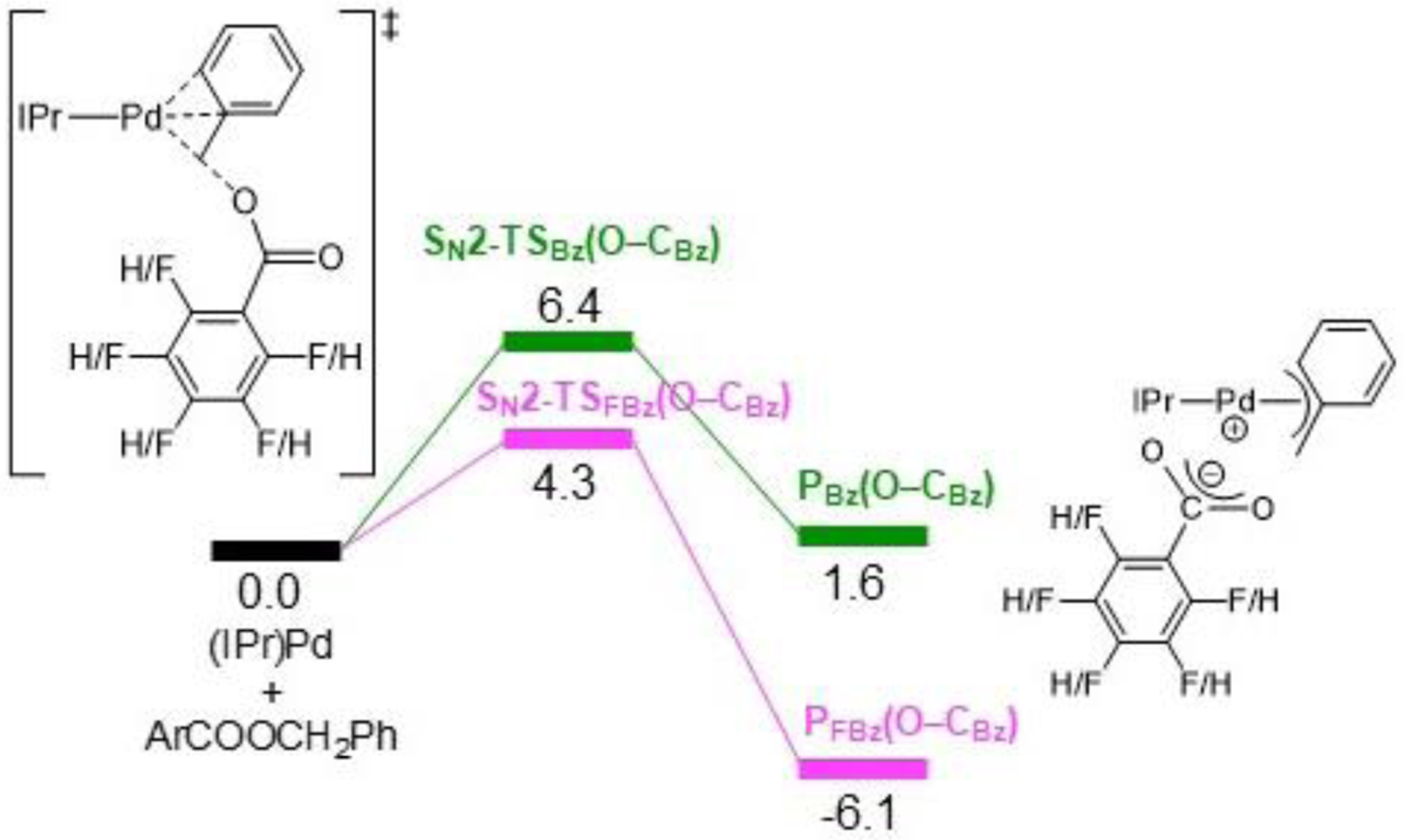
DFT calculations comparing the oxidative addition of the O–C(benzyl) bond in benzyl benzoate and benzyl 2,3,4,5,6-pentafluorobenzoate to (IPr)Pd(0). Relative energies are given in kcal/mol.
Figure 13.
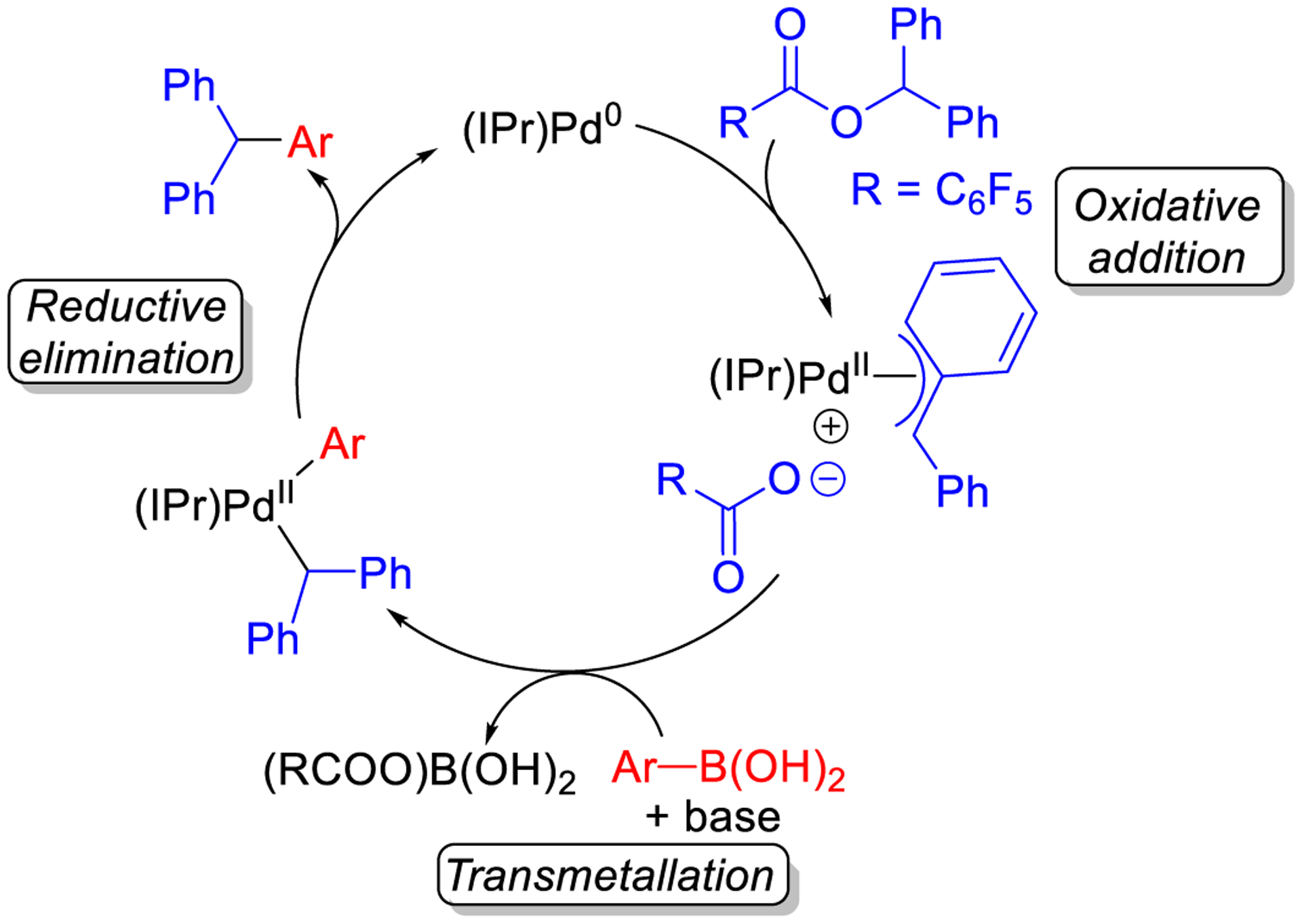
Proposed catalytic cycle for Pd-catalyzed Suzuki-Miyaura reactions of diphenylmethyl 2,3,4,5,6-pentafluorobenzoate.
To further probe the nature of the turnover-limiting step in catalysis we investigated the oxidative addition of (diphenyl)methyl-2,3,4,5,6-pentafluorobenzoate (14a) and (2-methylphenyl)(phenyl)methyl-2,3,4,5,6-pentafluorobenzoate (14b) to (IPr)Pd(styrene)2. Our substrate scope showed that the use of diarylmethylesters with ortho-substitution, such as 14b, on the aryl groups results in reduced yields under optimized conditions and necessitates the use of harsher reaction conditions to obtain satisfactory yields (Figure 6). However, the rates of oxidative addition of 14a and 14b are comparable (Figure 14), with both occurring at room temperature. This is not consistent with oxidative addition being the reason that harsher conditions are required for the coupling of 14b and suggests that a subsequent step in the catalytic cycle is turnover-limiting. It also provides further evidence that oxidative addition is facile.
Figure 14.

NMR yields of oxidative addition product over time for the reaction of diarylmethyl pentafluorobenzoate with (IPr)Pd(styrene)2.
Conclusions
We have developed a broad method for synthesizing triarylmethanes under mild conditions via Pd-catalyzed Suzuki-Miyaura coupling reactions involving 2,3,4,5,6-pentafluorobenzoate electrophiles. Importantly, our method is not limited to electrophiles containing extended aromatic systems, such as naphthyl or biaryl groups, and as a result represents a significant advance over previous methods. Further, the reaction is stereospecific and is able to generate chiral triarylmethanes with inversion of configuration. Intriguingly, while the Suzuki-Miyaura reaction of diarylmethyl esters involves the cleavage of the O–C(benzyl) bond, the reaction featuring closely related phenyl ester electrophiles involve selective cleavage of the C(acyl)–O bond. DFT calculations show that cleavage of the O–C(benzyl) bond in benzyl electrophiles is both kinetically and thermodynamically preferred. This is because oxidative addition of benzyl electrophiles to (IPr)Pd(0) via an SN2 mechanism provides a low barrier pathway for cleavage of the O–C(benzyl) bond, while the formation of products with an η3-benzyl interaction thermodynamically stabilizes the Pd(II) products. In fact, in our reactions the oxidative addition of the 2,3,4,5,6-pentafluorobenzoate electrophiles is so facile that transmellation is likely the turnover-limiting step in catalysis. Phenyl ester electrophiles cannot readily undergo oxidative addition via an SN2 pathway or form products which are stabilized via chelation and as a result cleavage of the C(acyl)–O bond is kinetically and thermodynamically preferred. Overall, apart from the development of a more general method for the synthesis of triarylmethanes, our work provides fundamental information on the selectivity of oxidative addition of ester electrophiles. Given the high level of current interest in using esters as electrophiles in cross-coupling, this is likely to be valuable for the design of new and improved synthetic methods.
Supplementary Material
Acknowledgements
NH acknowledges support from the NIHGMS under Award Number R01GM120162. ICR acknowledges the Basque Government for funding through a predoctoral fellowship (PRE_2016_1_0159). D.B. acknowledges the support from the Research Council of Norway through its Centers of Excellence Scheme (project number 262695) and the Norwegian Supercomputing Program (NOTUR; project number NN4654K). We thank Dr. Scott J. Miller for access to his group’s chiral HPLC and Orven Mallari for the synthesis of diphenylmethyl benzoate.
Footnotes
Supporting Information
Additional information about selected experiments and calculations, characterizing data including NMR spectra, and other details are available via the Internet.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Zhou B; Liu DX; Yuan XJ; Li JY; Xu YC; Li J; Li Y; Yue JM (−)- and (+)-Securidanes A and B, Natural Triarylmethane Enantiomers: Structure and Bioinspired Total Synthesis. AAAS Research 2018, 2018, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Miura T; Urano Y; Tanaka K; Nagano T; Ohkubo K; Fukuzumi S Rational Design Principle for Modulating Fluorescence Properties of Fluorescein-Based Probes by Photoinduced Electron Transfer. J. Am. Chem. Soc 2003, 125, 8666–8671; [DOI] [PubMed] [Google Scholar]; (b) Urano Y; Kamiya M; Kanda K; Ueno T; Hirose K; Nagano T Evolution of Fluorescein as a Platform for Finely Tunable Fluorescence Probes. J. Am. Chem. Soc 2005, 127, 4888–4894; [DOI] [PubMed] [Google Scholar]; (c) Beija M; Afonso CAM; Martinhp JMG Synthesis and Applications of Rhodamine Derivatives as Fluorescent Probes. Chem. Soc. Rev 2009, 38, 2410–2433. [DOI] [PubMed] [Google Scholar]
- 3.(a) Mason CD; Nord FF Studies on the Chemistry of Heterocycles. XIII. Triarylmethanes Dyes Containing a Thiophene Ring. J. Org. Chem 1951, 16, 722–727; [Google Scholar]; (b) Duxbury DF The Photochemistry and Photophysics of Triphenylmethane Dyes in Solid and Liquid Media. Chem. Rev 1993, 93, 381–433; [Google Scholar]; (c) Muthyala R; Katritzky AR; Lan X A Synthetic Study on the Preparation of Triarylmqthanes Dyes Pigm 1994, 25, 303–324. [Google Scholar]
- 4.Nolan EM; Lippard SJ Tools and Tactics for the Optical Detection of Mercuric Ion. Chem. Rev 2008, 108, 3443–3480. [DOI] [PubMed] [Google Scholar]
- 5.(a) Nambo M; Crudden CM Recent Advances in the Synthesis of Triarylmethanes by Transition Metal Catalysis. ACS Catal 2015, 5, 4734–4742; [Google Scholar]; (b) Kshatriya R; Jejurkar VP; Saha S Advances in The Catalytic Synthesis of Triarylmethanes (TRAMs). Eur. J. Org. Chem 2019, 2019, 3818–3841. [Google Scholar]
- 6.Bossche HV; Willemsens G; Roels I; Bellens D; Moereels H; Coene M-C; Jeune LL; Lauwers W; Janssen PAJR 76713 and Enantiomers: Selective, Nonsteroidal Inhibitors of the Cytochrome P450-Dependent Oestrogen Synthesis. Biochem. Pharmacol 1990, 40, 1707–1718. [DOI] [PubMed] [Google Scholar]
- 7.(a) Panda G; Shagufta; Mishra JK; Chaturvedi V; Srivastava AK; Srivastava R; Srivastava BS Diaryloxy Methano Phenanthrenes: a New Class of Antituberculosis Agents. Bioorg. Med. Chem 2004, 12, 5269–5276; [DOI] [PubMed] [Google Scholar]; (b) Parai MK; Panda G; Chaturvedi V; Manju YK; Sinha S Thiophene Containing Triarylmethanes as Antitubercular Agents. Bioorg. Med. Chem. Lett 2008, 18, 289–292. [DOI] [PubMed] [Google Scholar]
- 8.(a) Bhatnagar AS; Hausler A; Schieweck K; Lang M; Bowman R Highly Selective Inhibition of Estrogen Biosynthesis by CGS 20267, a New Non-steroidal Aromatase Inhibitor. J. Steroid Biochem. Mol. Biol 1990, 37, 1021–1027; [DOI] [PubMed] [Google Scholar]; (b) Shagufta; Srivastava AK; Sharma R; Mishra R; Balapure AK; Murthy PSR; Panda G Substituted Phenanthrenes with Basic Amino Side Chains: a New Series of Anti-breast Cancer Agents. Bioorg. Med. Chem 2006, 14, 1497–1505. [DOI] [PubMed] [Google Scholar]
- 9.(a) Shacklady-McAtee DM; Roberts KM; Basch CH; Song Y-G; Watson MP A General, Simple Catalyst for Enantiospecific Cross Couplings of Benzylic Ammonium Triflates and Boronic Acids: No Phosphine Ligand Required. Tetrahedron 2014, 70, 4257–4263; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang Z; Wang H; Qiu N; Kong Y; Zeng W; Zhang Y; Zhao J Synthesis of Triarylmethanes via Palladium-Catalyzed Suzuki Coupling of Trimethylammonium Salts and Arylboronic Acids. J. Org. Chem 2018, 83, 8710–8715. [DOI] [PubMed] [Google Scholar]
- 10.(a) Cao L-L; Ye ZS; Jiang G-F; Zhou Y-G Rhodium-Catalyzed Addition of Boronic Acids to Vinylogous Imines Generated in situ from Sulfonylindoles Adv. Synth. Catal 2011, 353, 3352–3356; [Google Scholar]; (b) Cao L-L; Li X-N; Meng F-Y; Jiang G-F Pd-Catalyzed Addition of Boronic Acids to Vinylogous Imines: a Convenient Approach to 3-sec-Alkyl Substituted Indoles. Tetrahedron Lett 2012, 53, 3873–3875; [Google Scholar]; (c) Nambo M; Crudden CM Modular Synthesis of Triarylmethanes through Palladium-Catalyzed Sequential Arylation of Methyl Phenyl Sulfone. Angew. Chem. Int. Ed 2014, 53, 742–746. [DOI] [PubMed] [Google Scholar]
- 11.(a) Harris MR; Hanna LE; Greene MA; Moore CE; Jarvo ER Retention or Inversion in Stereospecific Nickel-Catalyzed Cross- Coupling of Benzylic Carbamates with Arylboronic Esters: Control of Absolute Stereochemistry with an Achiral Catalyst. J. Am. Chem. Soc 2013, 135, 3303–3306; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhou Q; Srinivas HD; Dasgupta S; Watson MP Nickel-Catalyzed Cross-Couplings of Benzylic Pivalates with Arylboroxines: Stereospecific Formation of Diarylalkanes and Triarylmethanes. J. Am. Chem. Soc 2013, 135, 3307–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zarate C; van-Gemmeren M; Somerville RJ; Martin R Phenol Derivatives: Modern Electrophiles in Cross-Coupling Reactions. Adv. Organomet. Chem 2016, 66, 143–222. [Google Scholar]
- 13.Kuwano R; Yokogi M Cross-Coupling of Benzylic Acetates with Arylboronic Acids: One-pot Tansformation of Benzylic Alcohols to Diarylmethanes. Chem. Commun 2005, 5899–5901. [DOI] [PubMed] [Google Scholar]
- 14. In reference 11a, the primary focus is the coupling of benzylic carbamates with arylboronic esters. However, a number of examples of reactions with benzylic esters are also described.
- 15.(a) Yokogi M; Kuwano R Use of Acetate as a Leaving Group in Palladium-Catalyzed Nucleophilic Substitution of Benzylic Esters. Tetrahedron Lett 2007, 48, 6109–6112; [Google Scholar]; (b) Narahashi H; Shimizu I; Yamamoto A Synthesis of Benzylpalladium Complexes Through C–O Bond Cleavage of Benzylic Carboxylates: Development of a Novel Palladium-Catalyzed Benzylation of Olefins. J. Organomet. Chem 2008, 693, 283–296; [Google Scholar]; (c) Mendis SN; Tunge JA Palladium-Catalyzed Stereospecific Decarboxylative Benzylation of Alkynes. Org. Lett 2015, 17, 5164–5167; [DOI] [PubMed] [Google Scholar]; (d) Chen Q; Fan X-H; Zhang L-P; Yang L-M Ni(II) Source as a Pre-Catalyst for the Cross-Coupling of Benzylic Pivalates with Arylboronic Acids: Facile Access to Tri- and Diarylmethanes. RSC Adv 2015, 5, 15338–15340; [Google Scholar]; (e) Stewarta GW; Maligres PE; Baxter CA; Junker EM; Krska SW; Scott JP An Approach to Heterodiarylmethanes via sp2–sp3 Suzuki–Miyaura Cross-Coupling. Tetrahedron 2016, 72, 3701–3706; [Google Scholar]; (f) Zhou Q; Cobb KM; Tan T; Watson MP Stereospecific Cross Couplings To Set Benzylic, All-Carbon Quaternary Stereocenters in High Enantiopurity. J. Am. Chem. Soc 2016, 138, 12057–12060; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Makida Y; Matsumoto Y; Kuwano R Palladium-Catalyzed Decarboxylation of Benzyl Fluorobenzoates. Synlett 2017, 28, 2573–2576; [Google Scholar]; (h) Ohsumi M; Nishiwaki N Selective Synthesis of (Benzyl)biphenyls by Successive Suzuki–Miyaura Coupling of Phenylboronic Acids with 4-Bromobenzyl Acetate under Air Atmosphere. ACS Omega 2017, 2, 7767–7771; [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Zhang S-Q; Taylor BLH; Ji C-L; Gao Y; Harris YR; Hanna LE; Jarvo ER; Houk KN; Hong X Mechanism and Origins of Ligand-Controlled Stereoselectivity of Ni-Catalyzed Suzuki–Miyaura Coupling with Benzylic Esters: A Computational Study. J. Am. Chem. Soc 2017, 139, 12994–13005; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Masato Ohsumi; Ito A; Nishiwaki N Substrate Switchable Suzuki–Miyaura Coupling for Benzyl Ester vs. Benzyl Halide. RSC Adv 2018, 8, 35056–35061; [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Tsuji H; Hashimoto K; Kawatsura M Nickel-Catalyzed Benzylic Substitution of Benzyl Esters with Malonates as a Soft Carbon Nucleophile. Org. Lett 2019, 21, 8837–8841. [DOI] [PubMed] [Google Scholar]
- 16.(a) Quasdorf KW; Tian X; Garg NK Cross-Coupling Reactions of Aryl Pivalates with Boronic Acids. J. Am. Chem. Soc 2008, 130, 14422–14423; [DOI] [PubMed] [Google Scholar]; (b) Guan B-T; Wang Y; Bi-Jie; Yu D-G; Shi Z-J Biaryl Construction via Ni-Catalyzed C–O Activation of Phenolic Carboxylates. J. Am. Chem. Soc 2008, 130, 14468–14470; [DOI] [PubMed] [Google Scholar]; (c) Li BJ; Li YZ; Lu XY; Liu J; Guan BT; Shi ZJ Cross-Coupling of Aryl/Alkenyl Pivalates with Organozinc Reagents through Nickel-Catalyzed C-O Bond Activation under Mild Reaction Conditions. Angew. Chem. Int. Ed 2008, 47, 10124–10127; [DOI] [PubMed] [Google Scholar]; (d) Gooßen LJ; Gooßen K; Stanciu C C(aryl)-O Activation of Aryl Carboxylates in Nickel-Catalyzed Biaryl Syntheses. Angew. Chem. Int. Ed 2009, 48, 3569–3571; [DOI] [PubMed] [Google Scholar]; (e) Shimasaki T; Tobisu M; Chatani N Nickel-Catalyzed Amination of Aryl Pivalates by the Cleavage of Aryl C-O Bonds. Angew. Chem. Int. Ed 2010, 49, 2929–2932; [DOI] [PubMed] [Google Scholar]; (f) Li W; Gao JJ; Zhang Y; Tang W; Lee H; Fandrick KR; Lu B; Senanayake CH A Mild Palladium-Catalyzed Suzuki Coupling Reaction of Quinoline Carboxylates with Boronic Acids. Adv. Synth. Catal 2011, 353, 1671–1675; [Google Scholar]; (g) Ehle AR; Zhou Q; Watson MP Nickel(0)-Catalyzed Heck Cross-Coupling via Activation of Aryl C–OPiv Bonds. Org. Lett 2012, 14, 1202–1205; [DOI] [PubMed] [Google Scholar]; (h) Cornella J; Jackson EP; Martin R Nickel-Catalyzed Enantioselective C-C Bond Formation through C-O Cleavage in Aryl Esters. Angew. Chem. Int. Ed 2015, 54, 4075–4078; [DOI] [PubMed] [Google Scholar]; (i) Guo L; Hsiao C-C; Yue H; Liu X; Rueping M Nickel-Catalyzed Csp2–Csp3 Cross-Coupling via C–O Bond Activation. ACS Catal 2016, 6, 4438–4442; [Google Scholar]; (j) Liu X; Jia J; Rueping M Nickel-Catalyzed C-O Bond-Cleaving Alkylation of Esters: Direct Replacement of the Ester Moiety by Functionalized Alkyl Chains. ACS Catal 2017, 7, 4491–4496; [Google Scholar]; (k) Becica J; Gaube G; Sabbersb WA; Leitch DC Oxidative Addition of Activated Aryl-Carboxylates to Pd(0): Divergent Reactivity Dependant on Temperature and Structure. Dalton Trans 2020, 49, 16067–16071. [DOI] [PubMed] [Google Scholar]
- 17.(a) Ben-Halima T; Zhang W; Yalaoui I; Hong X; Yang Y-F; Houk Kendall N.; Newman SG Palladium-Catalyzed Suzuki–Miyaura Coupling of Aryl Esters. J. Am. Chem. Soc 2017, 139, 1311–1318; [DOI] [PubMed] [Google Scholar]; (b) Takise R; Muto K; Yamaguchi J Cross-Coupling of Aromatic Esters and Amides. Chem. Soc. Rev 2017, 46, 5864–5888; [DOI] [PubMed] [Google Scholar]; (c) Shi S; Nolan SP; Szostak M Well-Defined Palladium(II)–NHC Precatalysts for Cross-Coupling Reactions of Amides and Esters by Selective N–C/O–C Cleavage. Acc. Chem. Res 2018, 51, 2589–2599. [DOI] [PubMed] [Google Scholar]
- 18.(a) Muto K; Yamaguchi J; Musaev DG; Itami K Decarbonylative Organoboron Cross-Coupling of Esters by Nickel Catalysis. Nat. Commun 2015, 6, 7508; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) LaBerge NA; Love JA Nickel-Catalyzed Decarbonylative Coupling of Aryl Esters and Arylboronic Acids. Eur. J. Org. Chem 2015, 5546–5553; [Google Scholar]; (c) Masson-Makdissi J; Vandavasi JK; Newman SG Switchable Selectivity in the Pd-Catalyzed Alkylative Cross-Coupling of Esters. Org. Lett 2018, 20, 4094–4098. [DOI] [PubMed] [Google Scholar]
- 19.(a) Shi S; Lei P; Szostak M Pd-PEPPSI: A General Pd-NHC Precatalyst for Suzuki–Miyaura Cross-Coupling of Esters by C–O Cleavage. Organometallics 2017, 36, 3784–3789; [Google Scholar]; (b) Lei P; Meng G; Shi S; Ling Y; An J; Szostak R; Szostak M Suzuki–Miyaura Cross-Coupling of Amides and Esters at Room Temperature: Correlation with Barriers to Rotation Around C–N and C–O Bonds. Chem. Sci 2017, 8, 6525–6530; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shi S; Szostak M Pd–PEPPSI: A General Pd–NHC Precatalyst for Buchwald–Hartwig Cross-Coupling of Esters and Amides (transamidation) Under the Same Reaction Conditions. Chem. Commun 2017, 53, 10584–10587; [DOI] [PubMed] [Google Scholar]; (d) Ben-Halima T; Vandavasi JK; Shkoor M; Newman SG A Cross-Coupling Approach to Amide Bond Formation from Esters. ACS Catal 2017, 7, 2176–2180; [Google Scholar]; (e) Li G; Shi S; Lei P; Szostak M Pd-PEPPSI: Water-Assisted Suzuki Miyaura Cross-Coupling of Aryl Esters at Room Temperature using a Practical Palladium- NHC (NHC = N-Heterocyclic Carbene) Precatalyst. Adv. Synth. Catal 2018, 360, 1538–1543; [Google Scholar]; (f) Dardir AH; Melvin PR; Davis RM; Hazari N; Mohadjer-Beromi M Rapidly Activating Pd-Precatalyst for Suzuki–Miyaura and Buchwald–Hartwig Couplings of Aryl Esters. J. Org. Chem 2018, 83, 469–477; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Dardir AH; Hazari N; Miller SJ; Shugrue CR Palladium-Catalyzed Suzuki–Miyaura Reactions of Aspartic Acid Derived Phenyl Esters. Org. Lett 2019, 21, 5762–5766; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhou T; Li G; Nolan SP; Szostak M [Pd(NHC)(acac)Cl]: Well-Defined, Air-Stable, and Readily Available Precatalysts for Suzuki and Buchwald–Hartwig Cross-coupling (Transamidation) of Amides and Esters by N–C/O–C Activation. Org. Lett 2019, 21, 3304–3309; [DOI] [PubMed] [Google Scholar]; (i) Lei P; Ling Y; An J; Nolan SP; Szostak M 2-Methyltetrahydrofuran (2-MeTHF): A Green Solvent for Pd NHC-Catalyzed Amide and Ester Suzuki-Miyaura Cross- Coupling by N-C/O-C Cleavage. Adv. Synth. Catal 2019, 361, 5654–5660. [Google Scholar]
- 20.Dissociation Constants of Organic Acids and Bases. https://sites.chem.colostate.edu/diverdi/all_courses/CRC%20reference%20data/dissociation%20constants%20of%20organic%20acids%20and%20bases.pdf.
- 21.Umeda R; Jikyo T; Toda K; Osaka I; Nishiyama Y Rhenium Complex-Catalyzed Carbon-Carbon Formation of Alcohols and Organosilicon Compounds. Tetrahedron Lett 2018, 59, 1121–1124. [Google Scholar]
- 22.(a) Melvin PR; Nova A; Balcells D; Dai W; Hazari N; Hruszkewycz DP; Shah HP; Tudge MT Design of a Versatile and Improved Precatalyst Scaffold for Palladium-Catalyzed Cross-Coupling: (η3-1-tBu-indenyl)2(μ-Cl)2Pd2. ACS Catal 2015, 5, 3680–3688; [Google Scholar]; (b) Melvin PR; Balcells D; Hazari N; Nova A Understanding Precatalyst Activation in Cross-Coupling Reactions: Alcohol Facilitated Reduction from Pd(II) to Pd(0) in Precatalysts of the Type (η3-allyl)Pd(L)(Cl) and (η3-indenyl)Pd(L)(Cl). ACS Catal 2015, 5, 5596–5606. [Google Scholar]
- 23.Zhou T; Ma S; Nahra F; Obled AMC; Poater A; Cavallo L; Cazin CSJ; Nolan SP; Szostak M [Pd(NHC)(μ-Cl)Cl]2: Versatile and Highly Reactive Complexes for Cross-Coupling Reactions that Avoid Formation of Inactive Pd(I) Off-Cycle Products. iScience 2020, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hussein EM; Alsantali RI; El-Galil SMA; Obaid RJ; Alharbi A; Abourehab MAS; Ahmed SA Bioactivefluorenes. Part I. Synthesis, Pharmacological Study and Molecular Docking of Novel Dihydrofolate Reductase Inhibitors Based-2,7-dichlorofluorene. Heliyon 2019, e01982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leclerc M Polyfluorenes: Twenty Years of Progress. J. Polym. Sci. Part A: Polym. Chem 2001, 39, 2867–2873. [Google Scholar]
- 26.(a) Baheti A; Tyagi P; Thomas KRJ; Hsu Y-C; T’suen-Lin J Simple Triarylamine-Based Dyes Containing Fluorene and Biphenyl Linkers for Efficient Dye-Sensitized Solar Cells. J. Phys. Chem. C 2009, 113, 8541–8547; [Google Scholar]; (b) Shaya J; Fontaine-Vive F; Michel BY; Burger A Rational Design of Push–Pull Fluorene Dyes: Synthesis and Structure–Photophysics Relationship. Chem. Eur. J 2016, 22, 10627–10637. [DOI] [PubMed] [Google Scholar]
- 27.Kudo K; Miyazaki C; Kadoya R; Imamura T; Jitsufuchi N; Ikeda N Laxative Poisoning: Toxicological Analysis of Bisacodyl and Its Metabolite in Urine, Serum, and Stool J. Anal. Toxicol 1998, 22, 274–278. [DOI] [PubMed] [Google Scholar]
- 28.Brooks WH; Guida WC; Daniel KG The Significance of Chirality in Drug Design and Development. Curr. Top. Med. Chem 2011, 11, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braga AL; Paixão MW; Westermann B; Schneider PH; Wessjohann LA Acceleration of Arylzinc Formation and Its Enantioselective Addition to Aldehydes by Microwave Irradiation and Aziridine-2-methanol Catalysts. J. Org. Chem 2008, 73, 2879–2882. [DOI] [PubMed] [Google Scholar]
- 30.(a) Milstein D; Stille JK Mechanism of Reductive Elimination. Reaction of Alkylpalladium(II) Complexes with Tetraorganotin, Organolithium, and Grignard Reagents. Evidence for Palladium(IV) Intermediacy. J. Am. Chem. Soc 1979, 101, 4981–4991; [Google Scholar]; (b) Ridgway BH; Woerpel KA Transmetalation of Alkylboranes to Palladium in the Suzuki Coupling Reaction Proceeds with Retention of Stereochemistry. J. Org. Chem 1998, 63, 458–460; [DOI] [PubMed] [Google Scholar]; (c) Rodríguez N; de-Arellano CR; Asensio G; Medio-Simón M Palladium-Catalyzed Suzuki–Miyaura Reaction Involving a Secondary sp3 Carbon: Studies of Stereochemistry and Scope of the Reaction. Chem. Eur. J 2007, 13, 4223–4229; [DOI] [PubMed] [Google Scholar]; (d) Lennox AJJ; Lloyd-Jones GC Transmetalation in the Suzuki–Miyaura Coupling: The Fork in the Trail. Angew. Chem. Int. Ed 2013, 52, 7362–7370. [DOI] [PubMed] [Google Scholar]
- 31.Magano J; Dunetz JR Large-Scale Carbonyl Reductions in the Pharmaceutical Industry. Org. Process Res. Dev 2012, 16, 1156–1184. [Google Scholar]
- 32.Jaratjaroonphong J; Tuengpanya S; Saeeng R; Udompong S; Srisook K Green Synthesis and Anti-inflammatory Studies of a Series of 1,1-bis(heteroaryl)alkane Derivatives. Eur. J. Med. Chem 2014, 83, 561–568. [DOI] [PubMed] [Google Scholar]
- 33.Taylor BLH; Harris MR; Jarvo ER Synthesis of Enantioenriched Triarylmethanes by Stereospecific Cross- Coupling Reactions. Angew. Chem. Int. Ed 2012, 51, 7790–7793. [DOI] [PubMed] [Google Scholar]
- 34.Molander GA; Biolatto B Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling Reactions of Potassium Aryl- and Heteroaryltrifluoroborates. J. Org. Chem 2003, 68, 4302–4314. [DOI] [PubMed] [Google Scholar]
- 35.(a) Yamamoto T; shizu J; Kohara T; Komiya S; Yamamoto A Oxidative Addition of Aryl Carboxylates to Ni(0) Complexes Involving Cleavage of the Acyl-O Bond. J. Am. Chem. Soc 1980, 102, 3758–3764; [Google Scholar]; (b) Kazuhiro Nagayama IS, Akio Yamamoto Oxidative Addition of Aryl and Benzyl Trifluoroacetates to Zerovalent Palladium Complexes with Two Modes of C-O Bond Cleavage Processes. Bull. Chem. Soc. Jpn 1999, 72, 799–803; [Google Scholar]; (c) Kakino R; Shimizu I; Yamamoto A Synthesis of Trifluoromethyl Ketones by Palladium-Catalyzed Cross-Coupling Reaction of Phenyl Trifluoroacetate with Organoboron Compounds. Bull. Chem. Soc. Jpn 2001, 74, 371–376; [Google Scholar]; (d) Muto K; Yamaguchi J; Lei A; Itami K Isolation, Structure, and Reactivity of an Arylnickel(II) Pivalate Complex in Catalytic C–H/C–O Biaryl Coupling. J. Am. Chem. Soc 2013, 135, 16384–16387; [DOI] [PubMed] [Google Scholar]; (e) Xu H; Muto K; Yamaguchi J; Zhao C; Itami K; Musaev DG Key Mechanistic Features of Ni-Catalyzed C–H/C–O Biaryl Coupling of Azoles and Naphthalen-2-yl Pivalates. J. Am. Chem. Soc 2014, 136, 14834–14844; [DOI] [PubMed] [Google Scholar]; (f) Desnoyer AN; Friese FW; Chi W; Drover MW; Patrick BO; Love JA Exploring Regioselective Bond Cleavage and Cross-Coupling Reactions using a Low-Valent Nickel Complex Chem. Eur. J 2016, 22, 4070–4077. [DOI] [PubMed] [Google Scholar]
- 36. In catalysis the Pd(II) precatalyst needs to be activated to form the (IPr)Pd(0) active species. Under our reaction conditions this is likely an efficient process, which occurs via the mechanism described in reference 22b.
- 37.Kégl TR; Kollár L; Kégl T DFT Study on the Oxidative Addition of 4-Substituted Iodobenzenes on Pd(0)-Phosphine Complexes. Adv. Phys. Chem 2015, 2015, 1–6. [Google Scholar]
- 38.The oxidative addition occurs at room temperature. However, the reaction mixture is heated to 40 °C when the reaction is essentially complete as this makes purification of the product easier because it causes the decomposition of any residual (IPr)Pd(styrene)2.
- 39.(a) Andrews MA; Chang TC-T; Cheng C-WF; Emge TJ; Kelly KP; Koetzle TF Synthesis, Characterization, and Equilibria of Palladium(II) Nitrile, Alkene, and Heterometallacyclopentane Complexes Involved in Metal Nitro Catalyzed Alkene Oxidation Reactions. J. Am. Chem. Soc 1984, 106, 5913–5920; [Google Scholar]; (b) Herbert DE; Lara NC; Agapie T Arene C-H Amination at Nickel in Terphenyl–Diphosphine Complexes with Labile Metal–Arene Interactions. Chem. Eur. J 2013, 19, 16453–16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. For details about the computational methods utilized please refer to the SI. However, calculations were performed with solvent corrections due to the large influence of solvent on SN2 reactions.
- 41. For the O–C(aryl) bond cleavage, one additional transition state was found on the potential energy surface, although at a higher energy level (Figure S2). A pre-reaction complex was also found.
- 42. One additional transition state was also found in the O–C(aryl) bond cleavage pathway, although at a higher energy level (Figure S2). A pre-reaction complex was also found.
- 43. Figure S3 provides a complete list of possible isomers of this complex with their relative energies.
- 44. Figure S4 provides a complete list of possible isomers of this complex with their relative energies.
- 45.(a) Barder TE; Buchwald SL Insights into Amine Binding to Biaryl Phosphine Palladium Oxidative Addition Complexes and Reductive Elimination from Biaryl Phosphine Arylpalladium Amido Complexes via Density Functional Theory. J. Am. Chem. Soc 2007, 129, 12003–12010; [DOI] [PubMed] [Google Scholar]; (b) Zhang H; Luo X; Wongkhan K; Duan H; Li Q; Zhu L; Wang J; Batsanov AS; Howard JA; Marder TB Acceleration of Reductive Elimination of [Ar-Pd-C] by a Phosphine/Electron-Deficient Olefin Ligand: A Kinetic Investigation. Chem. Eur. J 2009, 15, 3823–3829; [DOI] [PubMed] [Google Scholar]; (c) Arrechea PL; Buchwald SL Biaryl Phosphine Based Pd(II) Amido Complexes: The Effect of Ligand Structure on Reductive Elimination. J. Am. Chem. Soc 2016, 138, 12486–12493; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Melvin PR; Nova A; Balcells D; Hazari N; Tilset M DFT Investigation of Suzuki–Miyaura Reactions with Aryl Sulfamates Using a Dialkylbiarylphosphine-Ligated Palladium Catalyst. Organometallics 2017, 36, 3664–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.