

Abstract
Cardiovascular complications of pregnancy have risen substantially over the past decades, and now account for the majority of pregnancy-induced maternal deaths, as well as having substantial long-term consequences on maternal cardiovascular health. The causes and pathophysiology of these complications remain poorly understood, and therapeutic options are limited. Pre-clinical models represent a crucial tool for understanding human disease. We review here advances made in preclinical models of cardiovascular complications of pregnancy, including preeclampsia and peripartum cardiomyopathy, with a focus on pathologic mechanisms elicited by the models, and on relevance to human disease.
Subject Terms: Animal Models of Human Disease, Basic Science Research, Cardiovascular Disease, Pregnancy, Women, Sex, Gender
Introduction
Pregnancy and delivery are a high-risk period in a woman’s life. As recently as 1920, 600 of every 100,000 women died in or around childbirth, usually from sepsis, hemorrhage, or hypertension1. The development of aseptic technique, antibiotics, and blood transfusions in the 20th century have dramatically decreased maternal mortality from these causes. Today, in the US, the main causes of maternal mortality are cardiovascular in nature, including cardiomyopathy, preeclampsia, thromboembolic disease, strokes, and others2. Strikingly, maternal mortality in the US has increased three-fold in the last 40 years, likely a reflection of advancing maternal age, preexisting morbid conditions including obesity and diabetes, improved management of congenital heart disease allowing these patients to become pregnant, and environmental factors2. The US is one of only two countries in which maternal mortality has increased since 2000. Equally strikingly, there are profound socioeconomic, geographic, and racial inequalities in outcomes. For example black women are three times as likely to die during or after childbirth than are white women3. In addition, there is an emerging recognition that pregnancy complications presage substantial long-term risk of cardiovascular disease. For example, the life-long risk of CVD in women who have had preeclampsia is as much as tripled, on par with the risk conferred by smoking or diabetes4, and the AHA now recommends that eliciting a history of preeclampsia be an integral component of routine clinic visits5. Pregnancy-associated maternal disease also frequently affects the fetus or the newborn, both short and long-term. Therapeutic options for treating or preventing cardiovascular complications of pregnancy remain limited, with no existing disease-specific therapies for most of them. There is thus an urgent need to better understand the causes and pathophysiology of cardiovascular complications of pregnancy.
General considerations
Before describing specific models of cardiovascular complications of pregnancy, it is useful to consider certain general concepts.
Placental pregnancy is restricted to mammals (rudimentary placenta-like organs do exist in some fish)6. Models of cardiovascular complications of pregnancy must therefore be in mammals. To date, most models have used rodents, predominantly mice. Numerous parallels between rodent and human development and physiology, as well as the availability of genetic manipulations in mice, make rodents ideal model systems. Comparative systems biology of human and mouse placental studies suggest that there is close to 90% concordance between proteins expressed in placental tissue from humans and mice7. Most known hormonal changes of human pregnancy are recapitulated in the mouse and rat, though not all8. Maternal changes in physiology, such as calorie retention via expansion of fat stores, and development of relative insulin resistance, also mirror those seen in human pregnancy9. Many cardiovascular changes are also recapitulated, including decreased blood pressure throughout most of pregnancy, increase in plasma volume with consequent decrease in hematocrit, increase in cardiac output, and the development of cardiac hypertrophy10, 11. However, it is also important to recognize the many limitations of the rodent models. Unlike human pregnancies, rodent pregnancies are always multiparous, typically yielding 3–12 pups. Although primates and rodents share a hemochorial type of placentation, placentation in rodents is significantly different than in humans8. For example, the invasion of the uterine wall by fetal trophoblasts is substantially shallower in mice, and trophoblast invasion in mice follows a perivascular route rather than an endovascular route noted in humans12. In addition, gestational length is rodents is significantly shorter (21 days) in contrast to ~9 months in humans, and genome-wide expression profiling of mouse and human placentae revealed clusters of genes with distinct co-expression patterns across gestation, suggesting that the developmental timeline in mouse runs parallel to the first half of human placental development13. Rodent hemodynamics are also significantly different from humans: the rodent heart, for examples, beats 600–700 times per minute, an order of magnitude faster than the human heart11.
Hormones play a major role in disease. Pregnancy profoundly changes maternal homeostasis, and does so rapidly, creating numerous vulnerabilities. Parturition and the immediate post-partum period reverses many of these changes, and introduces new ones, at an even faster rate14. These rapid changes are largely elicited and controlled by hormones such as progesterone emanating from the placenta. A simple way to conceptualize these profound effects is that the placenta, a tissue mostly of fetal origin, uses a panoply of hormones to developmentally reprogram maternal physiology to suit the needs of the fetus first, and then, anticipatorily at the end of gestation, of the newborn second. It is therefore not surprising that most uncovered mechanisms of cardiovascular complications of pregnancy involve aberrant hormonal effects, as discussed in detail below.
Pregnancy and the post-partum period represent a unique relationship between two genetically distinct organisms. During pregnancy, this interaction largely occurs within the placenta. The placenta thus must be an immune-privileged setting, where neither maternal nor fetal immune system rejects the other15. For the same reason, maternal immunity is generally suppressed during gestation, e.g., studies have suggested that pregnancy is characterized by upregulation of T-regulatory cells that suppress immunity and maintain tolerance16. This immune suppression, and the rebound immunological activation post-partum, can also contribute to disease.
Disease models of cardiovascular complications of pregnancy
Preeclampsia
Preeclampsia typically presents during the third trimester with hypertension and proteinuria and if left untreated can progress to end-organ damage in the kidney (acute kidney injury), liver (HELLP syndrome), and brain (eclampsia)17. While delivery of placenta is the definitive treatment for this disease, preeclamptic women can have both-short term and long-term cardiovascular complications18. In the short-term, severe hypertension in the post-partum period can occur and is often associated with repeat hospitalization and rarely seizures. Preeclampsia is also a significant risk factor for peripartum cardiomyopathy (see below) and for heart failure with preserved ejection fraction (HFpEF)19. In the long-term, preeclampsia is associated with 3-fold excess of cardiovascular disease (CVD)18. Many pathways related to angiogenesis, inflammation, syncytiotrophoblast stress, and autoantibodies have been proposed to play a role in the pathogenesis of preeclampsia17. Pathogenesis likely involves two distinct stages. The first, asymptomatic phase of the disease is associated with shallow invasion of the placenta into the decidua and myometrium, and decreased uteroplacental blood flow. The second stage occurs during the third trimester, when women present with clinical features of hypertension, proteinuria, and microangiopathy in various organs such as kidney, liver and brain. During this phase, there is syncytiotrophoblast stress that can be measured by high circulating levels of anti-angiogenic factors such as soluble fms-like tyrosine kinase-1 (sFLT1), soluble endoglin (sENG)20, and increased urinary levels of prolactin and its anti-angiogenic 16kDa fragment21. Animal models are critical to study the cause/effect of the various pathways that have been proposed to play a role in preeclampsia. Tools to genetically modify animals, radiotelemetry to measure blood pressures non-invasively, and ultrasound/dopplers to evaluate uteroplacental blood flow, have enabled the continuous monitoring of healthy and preeclamptic pregnancies in rodents22. Several animal models that recapitulate the first and/or second phase of the syndrome have been characterized. However, no single model fully recapitulates all the features of humans with preeclampsia, and therefore extrapolation of the experimental findings to the clinical situation should be interpreted with caution. Nevertheless, animal models have been invaluable in dissecting the pathogenesis of preeclampsia and validating potential targets for therapeutic intervention23. Here, we will summarize validated animal models currently used to study the pathophysiology of this disease (See Table 1).
Table 1:
Animal models of preeclampsia.
| Animal Models | Species | Pathways | Limitations |
|---|---|---|---|
| Surgical ischemia of placenta24–28, 31, 33, 34, 43 | Rats, Mice and Baboons | Hypoxia, Oxidative stress, Inflammation, Angiogenic Imbalance | Lack of severe disease Cannot study trophoblast invasion |
| Angiotensinogen/Renin transgenics38, 39, 41, 44 | Rats, MIce | Oxidative stress, Angiotensin receptor autoantibodies, Inflammation | Less relevant to humans as preeclampsia is characterized by suppressed renin and angiotensin II |
| sFLT1 overexpression46–51, 53 | Rats, Mice | Angiogenic imbalance, Endothelial dysfunction | Short gestation and not optimal to study early placentation events |
| BPH/5 model58, 61 | Mice | Complement activation, Angiogenic imbalance | Lack of severe disease Short gestation |
| CBA/J x DBA/2 | Mice | Complement activation, Angiogenic imbalance, immune dysfunction | Lack of severe disease Short gestation |
| COMT KO66 | Mice | Hormonal and placental hypoxia | Lack of severe disease; no phenotype in rats; short gestation |
| STOX1 transgenic73 | Mice | Inflammation, Angiogenic Imbalance | Relevance to humans unclear as loss of function mutations variable depending on populations |
| RGS2 KO81 | Mice | Histone deacetylation, cyclic AMP pathway | Relevance to humans unclear as no evidence of placental ischemia |
| AVP infusion82, 83 | Mice, Rats | Immune dysfunction, G protein signaling | Lack of severe disease; short gestation |
| L-NAME infusion85 | Mice, Rats | Endothelial Dysfunction | Partial phenotype |
| Dahl Salt-sensitive Rats86–88 | Rats | Hypoxia, Endothelial Dysfunction | Lack of severe disease; not a model for de novo preeclampsia |
Acute placental ischemia models
Since the major defect in preeclampsia is placental ischemia related to poor placentation, researchers have modeled placental ischemia in rodent and primates using surgical approaches. The most common model is induction of partial ischemia in pregnant rats using surgical clips of the uterine artery, referred to as reduced uterine perfusion pressure (RUPP) model24. Pregnant rats entering the RUPP procedure undergo clipping procedure at day 14 of gestation. Typically, a silver clip is placed around the aorta above the iliac bifurcation to reduce uterine perfusion pressure in the gravid rat by approximately 40%. Since compensation of blood flow to the placenta occurs in pregnant rats through an adaptive increase in ovarian blood flow, additional clips on both right and left uterine arcades at the ovarian end right before the first segmental artery helps maintain the placental ischemia25 (see Figure 1). RUPP rats develop reduced GFR, third trimester hypertension, and variable degree of fetal growth restriction. Importantly, pathways implicated in humans with preeclampsia such as angiogenic imbalance, autoantibodies against angiotensin receptor, and pro-inflammatory factors have all been noted in this model26–29. Many therapeutic compounds such as recombinant endothelial growth factors have been tested successfully in these models and provide valuable preclinical data needed prior to testing in pregnant humans26, 27. However, there are some caveats of this model. Preterm delivery has not been observed with this model and therefore it is difficult to assess prolongation of pregnancy with specific therapies. Also, RUPP is a model of non-severe preeclampsia, as these animals have not been reported to develop HELLP syndrome or seizures to suggest eclampsia.
Fig 1:
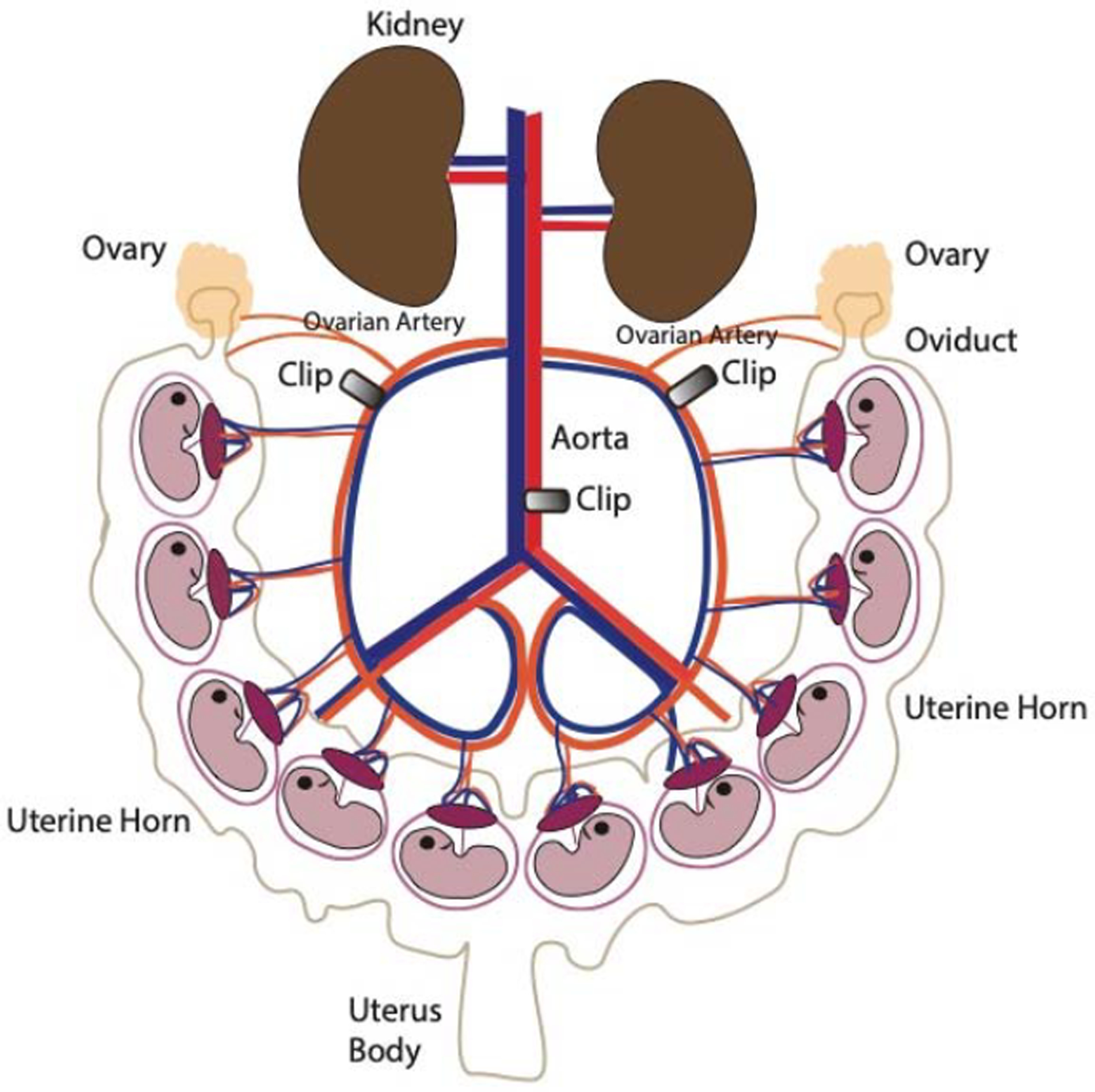
Schematic of the Reduced Uterine Perfusion Pressure (RUPP) model
The RUPP model is made by clipping the ovarian arteries and abdominal aorta with silver clips during the 3rd trimester in rats or in mice. Pregnant RUPP animals develop hypertension, decreased glomerular filtration rate, variable degrees of proteinuria, and fetal growth restriction. (Illustration credit: Ben Smith)
RUPP model has also been performed in mice by several groups, and preclinical studies to evaluate the safety and efficacy of nicotinamide and tadalafil were recently published30, 31. The advantage of using mice over rats is the availability of ready-to-use genetically modified mice from several repositories that can allow one to test the role of specific pathways in the pathogenesis of preeclampsia. To model ischemia, some groups have also exposed pregnant mice to relative hypoxia (9.5% O2) from gestational day 7 to 17, inducing preeclampsia-like features including elevated preeclampsia biomarker levels32.
Placental ischemia has also been modeled in non-human primates, typically with baboons33. Since pregnancy duration is significantly longer in baboons (180 days) than rodents, and since there is a single placenta and fetus, this surgical model closely approximates the clinical condition. Again, many therapies have been tested in this model that are now being pursued in clinical trials34, 35. While there are several advantages to using non-human primates for preclinical proof-of-concept studies, a disadvantage is the inability to study early placentation events leading to preeclampsia, because the induction of surgical ischemia may actually promote trophoblast invasion36.
Renin-Angiotensin models of preeclampsia
Preeclampsia is characterized by enhanced angiotensin II sensitivity even prior to the onset of hypertension37. Models to study enhanced angiotensin II sensitivity and signaling have largely used transgenic rodents in which the hypertension is induced by placental renin that acts on maternal angiotensinogen. When transgenic female mice expressing angiotensinogen are mated with transgenic males expressing renin, pregnant females display gestational hypertension during the 3rd trimester due to secretion of placental renin into the maternal circulation38. Generalized seizures occur in 15% of these mice and the hypertension reverses after delivery of the placenta. Subsequently other groups have reproduced this model in rats and reported that gestational hypertension was associated with the presence of agonistic antibodies against the angiotensin receptor39, 40 (see Figure 2). Follow-up studies suggested that these agonistic autoantibodies not only induce angiotensin II sensitivity41, but also may be sufficient to induce preeclampsia-like phenotypes when purified and injected into wild-type pregnant mice42, 43. While several of the features of preeclampsia such as gestational hypertension, albuminuria and angiotensin II sensitivity are similar to humans with preeclampsia, this model has very high renin and angiotensin II which is in contrast to humans with preeclampsia, where renin and angiotensin II levels are suppressed. Nevertheless, the model has been particularly useful to dissect the early events of placentation. For example, studies suggest that enhanced angiotensin II signaling may block trophoblast invasion and that neutralization of the RAS may enhance placentation44, 45.
Fig 2:
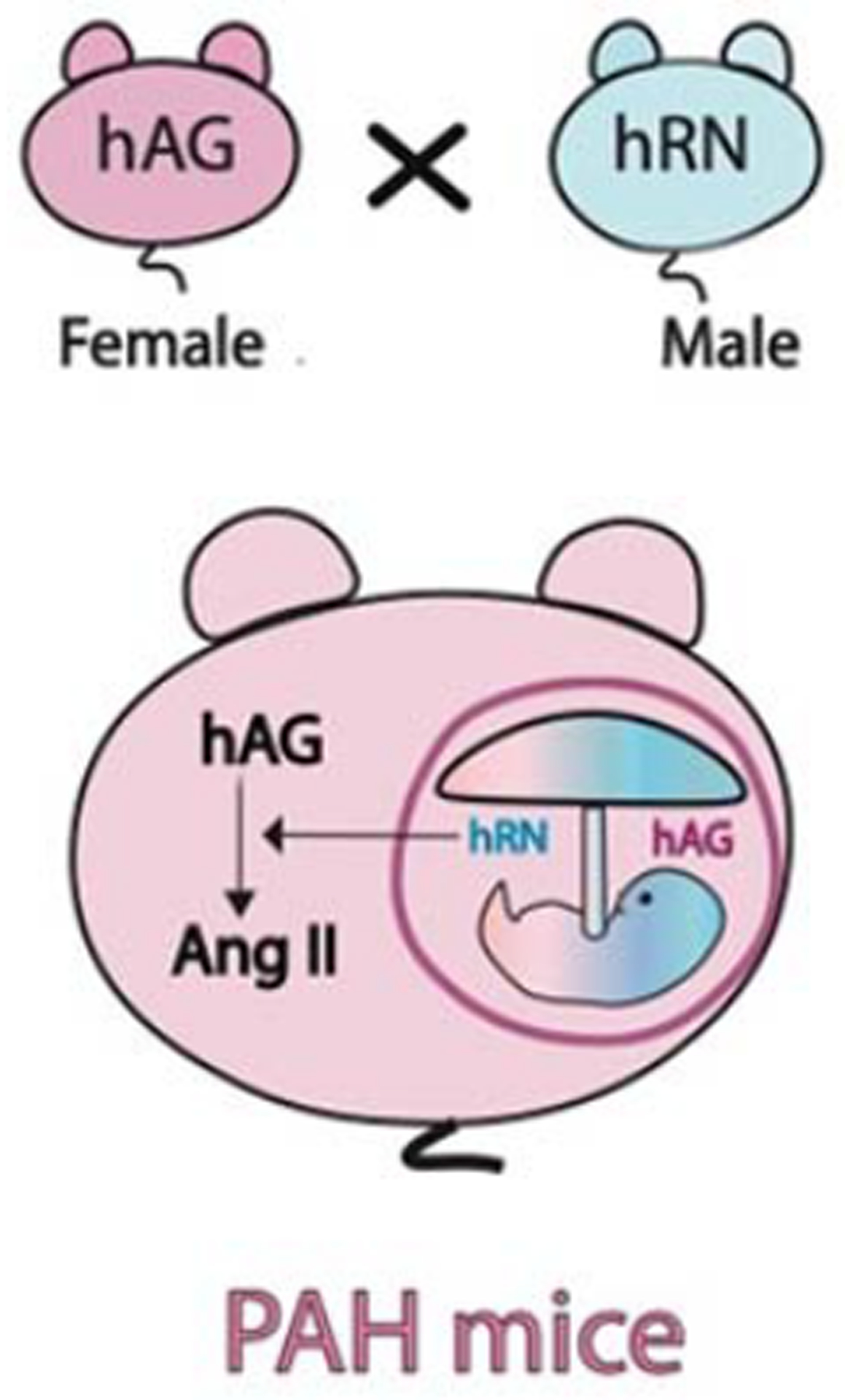
Angiotensin II model of Pregnancy-Associated Hypertension (PAH)
Female human angiotensinogen (hAogen) transgenic mice mated with male human renin (hRen) transgenic mice develop severe hypertension during pregnancy as human renin made from the fetus/placenta acts on maternal human angiotensinogen to induce excess angiotensin II that occurs only during pregnancy. Endogenous mouse renin will not cleave human angiotensinogen, making this a valuable model to study PAH. The opposite crosses (female hRen x male hAogen) can serve as controls as human angiotensinogen made in the fetus does not cross to the maternal circulation. (Illustration credit: Ben Smith)
sFLT1 overexpression model
Clinical studies suggest that high levels of sFLT1 correlate with adverse maternal and fetal outcomes related to preeclampsia. Adenoviral-mediated expression of sFLT1 in pregnant mice and rats reproduces many of the features of preeclampsia including severe hypertension, proteinuria, glomerular endotheliosis and fetal growth restriction22, 46–49 (see Figure 3). In this model, sFLT1 is expressed ectopically from the liver and circulating concentrations achieved are higher than the concentrations noted in women with preeclampsia. To evaluate a more physiologically relevant model, transgenic over-expression of sFLT1 from the placenta was recently reported50. In this model, the animals developed full-blown maternal disease as well as placental disease, including abnormal spiral artery remodeling and impaired blood flow to the feto-placental unit51. Other groups have also studied other synergistic anti-angiogenic factors, for example soluble endoglin (sENG). Transgenic expression of sENG during pregnancy leads to hypertension and fetal growth restriction52. Co-expression of sENG with sFLT1 either through adenoviral vector or as infusion of recombinant proteins has been used as a model of HELLP syndrome, a severe complication of preeclampsia53, 54. The sFLT1 over-expression model has been used to test several novel therapies, such as VEGF-121 and statins46, 55. Kumasawa et al, showed that pravastatin upregulated PlGF and rescued preeclampsia-like features in a placental lentiviral sFLT1 overexpression model56. Since clinical studies have reported that elevated circulating sFLT1 and low PlGF is present is most cases of preterm preeclampsia and that angiogenic imbalance correlates with adverse maternal and fetal outcomes related to preeclampsia57, there is strong interest in the sFLT1 model to test novel therapies, but also to understand downstream mechanisms by which sFLT1 induces vascular disease.
Fig 3:
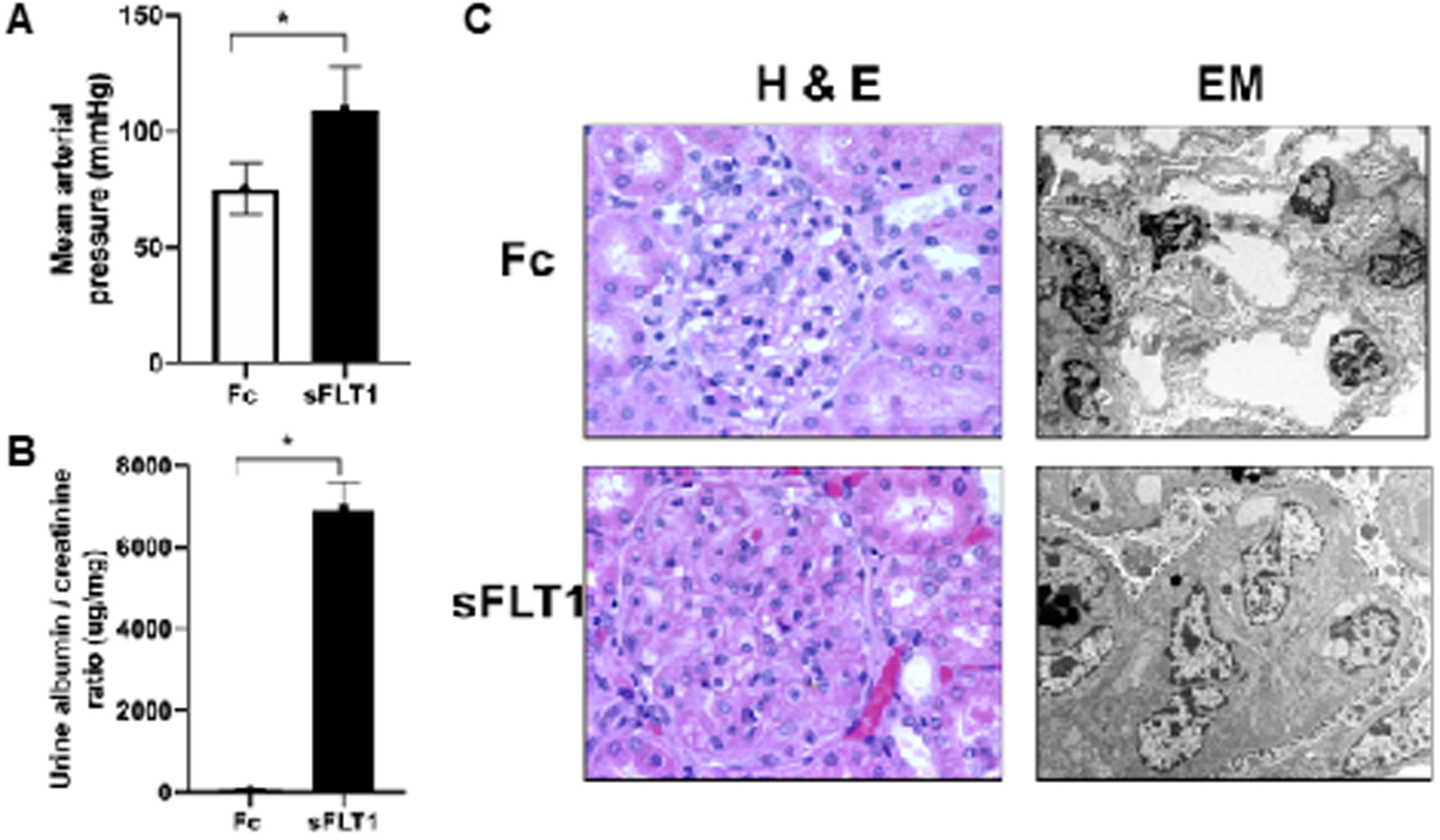
sFLT1 overexpression model of preeclampsia
Adenoviral sFLT1, versus adenoviral Fc protein as control, delivered into pregnant rats or mice induces systemic angiogenic imbalance and preeclampsia-like features including hypertension (panel A), proteinuria (panel B) and glomerular endotheliosis (panel C). Figure adapted from Maynard et al 47.
Genetic Models:
a. BPH/5 model:
BPH/5 mice are an inbred sub-line derived from 20 generations of brother-sister matings of the inbred spontaneously hypertensive strain BPH/258. During the nonpregnant states, BPH/5 has mildly elevated blood pressures when compared to the C57BL/6 strain, but does not develop proteinuria. However, during pregnancy BPH/5 strain mice develop severe hypertension during 3rd trimester that is accompanied by proteinuria and glomerular damage58. Interestingly, these maternal phenotypes are accompanied by low birth weight and smaller litter size in these mice. However, BPH/5 mice do not develop features of severe disease such as HELLP syndrome59. Many groups have attempted to study the mechanisms of why this strain develops preeclampsia-like states. Sones et al have suggested that anti-angiogenic sFLT1 protein was upregulated in the BPH/5 placentas and that aberrant sFLT1 expression was responsible for the complement activation and poor placentation in this model60. Interestingly, adenoviral delivery of VEGF121 in early pregnancy prevented the spontaneous development of preeclampsia in this model thus confirming that angiogenic imbalance was central to the pathogenesis of the maternal syndrome61.
b. CBA/J x DBA/2 model:
Immunological mechanisms have been hypothesized to play a central role in early placentation defects in preeclampsia. CBA/J x DBA/2 mating combinations has been extensively used to study miscarriages and preeclampsia62, 63. Initial studies reported embryos derived from mating CBA/J females with DBA/2 males showed an increased frequency of resorption and that surviving embryos showed consistent and significant intrauterine growth restriction. Administration of soluble CD200, an immune check point inhibitor has been shown to prevent abortions by augmenting T regulatory cells64. Ahmed et al characterized the maternal phenotypes of this mating combination and reported several features like albuminuria and glomerular fibrin deposits that are similar to humans with preeclampsia65. Although these animals at baseline did not develop hypertension, they developed exaggerated sensitivity to Angiotensin II. This animal model was explored to study several therapies such as pravastatin and pro-angiogenic factor VEGF121 with beneficial effects and without inducing toxicity.
c. COMT knockout mice:
Kanasaki et al. first reported that mice lacking Catechol-O-methyltransferase (COMT) in the placentas and in the mother developed 3rd trimester hypertension, proteinuria and kidney damage66. They further reported that the phenotype was mediated by a deficiency of 2-methoxyestradiol that is normally produced by COMT. Clinical studies suggest that genetic polymorphisms in the COMT pathway may be associated with the development of preeclampsia67. However, COMT knock-out in rats was associated with no preeclampsia-like phenotype68. Whether this discrepancy is related to differences in placentation between mice and rats has not been resolved.
d. ASB4 Knock-out mice:
Rownley-Tilson et al. reported that mice genetically deficient in ubiquitin ligase Ankyrin repeat, SOCS box-containing 4 (ASB4) develop preeclampsia-like states including 3rd trimester hypertension and proteinuria69. They further demonstrated that ASB4 promotes trophoblast differentiation through the degradation of inhibitor of DNA binding (ID2). Aberrant expression of ID2 has been observed in preeclamptic placentas, suggesting a role for this pathway in the human condition70. Li et al. reported that nicotinamide rescues the preeclampsia phenotypes and improved fetal survival in mice lacking ASB471.
e. STOX1 transgenic mice:
Storkhead box 1 (STOX1) is a transcription factor belonging to the enlarged Forkhead Box gene family that has been associated with preeclampsia, and is able to modulate trophoblast proliferation and migration. STOX1 is maternally expressed in a specific cell type of the placenta, the column extravillous trophoblasts. Overexpression of STOX1 in human choriocarcinoma cells induces transcriptional alterations that mimic those of preeclamptic placentas72. Wildtype females crossed with males expressing STOX1 under a constitutive promoter were reported to develop severe gestational hypertension, proteinuria, and increased plasma level of soluble antiangiogenic factors, as well as kidney and placenta histological alterations73. Interestingly, aspirin use in this model was shown to reverse the hypertension phenotype, resembling the human situation74. However, the role of STOX1 in human preeclampsia is still controversial. While mutations in STOX1 gene were first reported in Dutch families75, follow-up studies have not reported increased prevalence of STOX1 mutations in preeclampsia in other populations76, 77.
f. Corin knockout mice:
Corin, also known as atrial natriuretic peptide (ANP) converting enzyme, has been linked with trophoblast invasion and spiral artery remodeling78. Consistent with this hypothesis, pregnant corin mice developed preeclampsia-like features including hypertension, proteinuria and impaired spiral artery remodeling. However, mice lacking ANP, the Corin target, do not express any of the phenotypes of preeclampsia including hypertension or fetal growth restriction79. Furthermore, human studies have suggested that Corin levels may be elevated during preeclampsia80 suggesting that deficiency of Corin is unlikely to play significant role in preeclampsia.
g. RGS2 Knockout mice:
Regulator of G protein signaling (RGS) proteins have been demonstrated to contribute to numerous cardiovascular diseases. Genetically modified mice with fetal and placental deficiency of RGS2 develop preeclampsia-like features including hypertension and proteinuria during late gestation81. Interestingly, the RGS2 knockout model of preeclampsia does not have evidence of placental hypoxia or features of anti-angiogenic factor upregulation. Reduced expression of RGS2 has been reported in some cases of human preeclampsia81. RGS2 deficient mice may thus be useful to study sub-classes of preeclampsia that are not mediated by placental hypoxia or anti-angiogenic factors.
Other models of preeclampsia:
a. Pharmacological compounds:
Timed administration of compounds during pregnancy has been reported by some groups to model features of preeclampsia. Infusion of arginine vasopressin (AVP) during pregnancy was reported to exhibit robust elevations in systolic blood pressure, but without changes in diastolic blood pressure82. Changes in blood pressure were associated with increase in proteinuria and glomerular endotheliosis, both hallmarks of preeclampsia. Mechanistically, AVP was shown to induce pro-inflammatory Th1 interferon gamma and Th17 associated IL17 in the placenta83. Human studies have suggested that co-peptin, a surrogate biomarker of vasopressin, is elevated in humans with preeclampsia, but the differences in alterations were more apparent closer to the diagnosis of clinical preeclampsia84. Administration of L-NAME, an inhibitor of endothelial nitric oxide synthase (eNOS), in pregnant rats also led to the development of hypertension, fetal growth restriction and variable degrees of proteinuria85.
b. Dahl-salt sensitive rats:
Pregnant Dahl salt-sensitive rats have been used as a model of superimposed preeclampsia86. Mean arterial blood pressures were elevated in the Dahl S rats before pregnancy87. However, during pregnancy these rats have elevated blood pressures that are not seen in other hypertensive rats (for example, SHR). Dahl-S rats exhibited proteinuria, glomerulomegaly and elevated sFLT1, all features of superimposed preeclampsia in humans. Sildenafil, a phosphodiesterase 5 inhibitor, ameliorated the maternal syndrome of preeclampsia and fetal growth restriction suggesting impaired nitric oxide signaling may be central to signs noted in this model86. Consistent with this hypothesis, L-citrulline therapy in drinking water was also shown to reduce gestational hypertension and improved fetal growth in this model88. More work is needed to better characterize the early placentation events in this model.
Preeclampsia models to study CVD
Since preeclampsia is associated with long-term hypertension and CVD, some investigators have used the preeclampsia animal models to study whether preeclampsia is directly involved in the CVD risk, in contrast to the alternative hypothesis that CVD and preeclampsia share underlying risk factors. Pruthi et al, first reported that experimental preeclampsia using sFLT1 overexpression causes an enhanced vascular response to future vessel injury89. Mice with prior exposure to preeclampsia or normal pregnancies were challenged with unilateral carotid injury. Preeclampsia-exposed mice had significantly enhanced vascular remodeling with increased vascular smooth muscle cell proliferation and vessel fibrosis compared with control pregnancy89. This new paradigm may contribute to the substantially increased risk of CVD in woman exposed to preeclampsia. Bytauteiene et al reported that animals exposed to sFLT1 during pregnancy had no changes in blood pressure or vascular function six to eight months post-partum90, but cardiovascular stressors such as angiotensin II or salt were not tested. In a follow-up study, the same group reported that sFLT1 exposure during pregnancy altered over 150 plasma proteins that could contribute to long term cardiovascular disease91. STOX1 transgenic mice develop, 8 months post-pregnancy, cardiac fibrin deposits and mildly abnormal cardiac function in response to dobutamine stress92 In summary, there is some evidence from mouse models that preeclampsia is directly involved in accentuating long-term CVD, but more work is needed in solidify this conclusion. Investigators have also used preeclampsia mouse models to study cardiovascular disease in offspring. For example, offspring of BPG/5 mice have white adipose tissue accumulation and evidence of hypertension and cardiomegaly93. Here too, more work is needed to better understand how in utero exposure to preeclampsia-like states may lead cardiovascular disease in the offspring.
Summary of Preeclampsia models
In sum, while the use of animal models to study preeclampsia has increased dramatically in recent years, no one model faithfully recapitulates all the signs and symptoms of the human syndrome. Placental ischemia models in rodents have been most informative as researchers have elucidated a pathogenic role for oxidative stress, anti-angiogenic factors, and angiotensin autoantibodies in the pathogenesis of the maternal syndrome94. However, a significant limitation of ischemia models is that features of severe disease such as HELLP syndrome, eclampsia, and abruption are not noted. Transgenic or adenoviral overexpression of sFLT1 alone or in combination with sENG develop more robust preeclampsia phenotypes, including severe hypertension, nephrotic range proteinuria, cerebral edema and HELLP like phenotypes50, 51, 53, 95. Taken together with the data that, in humans, high levels of sFLT1 and sENG correlate with adverse maternal and fetal outcomes20, there has been great interest in testing targeted therapies for preeclampsia in anti-angiogenic overexpression model. A significant limitation of the rodent model, however, is lack of expression of all the isoforms of sFLT1 expressed in humans96, making it difficult to study molecular therapies such as antibodies that preferentially bind one isoform over others. It would therefore be critical to develop transgenic non-human primate models that express human sFLT1 isoforms and in whom the duration of pregnancy approximates the human situation. Furthermore, since there are no mechanical compromises to the uterine circulation in these transgenic models, the models can be used to evaluate whether therapies with maternal benefit would also benefit fetuses. Carter et al have suggested that smaller primates such as common marmoset (a New World monkey) should be used to model preeclampsia97, however it is not known whether uterine invasion by trophoblasts occur in marmosets to the same degree as in humans.
Peripartum cardiomyopathy
Peripartum cardiomyopathy (PPCM) is characterized by systolic heart failure presenting in the last month of pregnancy or the 1st few months postpartum98–103. It affects anywhere from 1:100 to 1:3000 births, with certain geographic hot spots like Haiti and Nigeria.98–103 PPCM accounts for >50% of cases of maternal cardiogenic shock in the US104 and is a leading cause of maternal peripartum death, with mortality rates in the US are as high as 25%.101, 102, 105–107 The disease affects all socioeconomic and racial sectors, but is more common and bears significantly worse prognosis in women of African descent108–110. The precise cause of PPCM remains elusive. However, work with preclinical models over the last 20 years, as outlined below (see also Table 2), has elucidated the current model that PPCM is in large part a vascular disease, triggered by aberrant hormonal changes of late pregnancy and early post-partum.111–115 In parallel, genetic studies in human cohorts has uncovered that rare genetic variants in various genes, including TTN, FLNC, DSP, and others, can predispose to PPCM.116, 117 The implications of these observations for building of pre-clinical models are also discussed below.
Table 2:
Mouse models of peripartum cardiomyopathy.
| Model | Proposed mechanisms | Rescue experiments | Evidence for human relevance | Limitations |
|---|---|---|---|---|
| GαQ transgenic118–121 | Promotion of cardiomyocyte apoptosis | Supraphysiologic overexpression. Baseline dysfunction. | ||
| STAT3 cardiac KO112, 124–126 | Cardiac secretion of cathepsin D cleaves prolactin to vasculotoxic 16KD fragment | Suppression of prolactin with bromocriptine | Some clinical support for treatment with bromocriptine. STAT3 suppressed in cardiac PPCM tissue. | No genetic evidence for role of STAT3 in human PPCM |
| PGC-1α cardiac KO113, 133 | Blunted cardiac pro-angiogenic signals create vulnerability to vasculotoxic hormones, including sFLT1 | VEGF injections, treatment with bromocriptine | sFLT1 elevations in PPCM. Epidemiological association with preeclampsia, and cardiac dysfunction. | No genetic evidence for role of PGC-1α in human PPCM |
| TTNtv knock-in139–141 | Unknown | Incontrovertible evidence that variants in TTN contribute to human PPCM | Mild, if any, pregnancy-induced cardiac dysfunction | |
| γ-secretase inhibition150 | Suppression of Notch1 activation, causing blunted angiogenesis. | Unclear mechanism, pleiotropic effects of γ-secretase inhibition | ||
| Npr1 whole-body KO151 | Indirect mechanism leads to lactation-dependent activation of aldosterone in CNS | CNS deletion of aldosterone receptor | Largely driven by elevated BP. Npr1 signaling likely elevated in PPCM, rather than blunted. No genetic evidence in humans. | |
| HSPB6/Hsp20 transgenic152–154 | Cardiomyocyte apoptosis and suppressed autophagy | Unknown role of Hsp20 in human PPCM. High expression of mutant protein. | ||
| PERK transgenic155 | Microvascular rarefaction | Unknown if PERK or the UPR is activated in PPCM hearts | ||
| Encephalo-myocarditis virus (EMCV) infection171 | Postpartum myocarditis | Unclear role of viral infection in PPCM | ||
| Erbb4 cardiac heterozygous KO173 | ERBB4 protects maternal heart from peripartum stress | Multiple microRNAs that target Erbb4 are elevated in serum of PPCM patients | Significant baseline cardiac dysfunction. Multiple microRNAs may have unpredictable targets. No genetic evidence in humans. | |
| caAkt cardiac trangenic +/− STAT3 cardiac KO174 | Enhanced oxidative stress and hypertrophy | Treatment with bromocriptine | Exhuberant hypertrophy not reflective of normal pregnancy. | |
| Zfp36l2 cardiac KO157 | Disinhibition of mTORC1 | Treatment with rapamycin | Some evidence low Zfp36l2 in PPCM hearts | Baseline cardiac dysfunction. No genetic evidence in humans. |
Genetic models of PPCM
Cardiac expression of Gαq
The first mouse model of PPCM was described in 1998. Numerous hypertrophic signals in the heart, including angiotensin and adrenergic signals, are coupled to heterotrimeric guanine nucleotide-binding proteins of the Gq family. Transgenic models expressing various levels of Gαq in the heart were generated and shown to promote hypertrophy and subsequent systolic heart failure, a process that was markedly accelerated by repeated pregnancies.118, 119 Subsequent work demonstrated that the systolic failure was caused by cardiomyocyte loss.120, 121 This model had numerous limitations, however. First, the transgenic expression of Gαq was supraphysiologic, raising the possibility that the observed mechanism of cardiomyopathy is not relevant to human disease (transgenic lines with less induction of Gαq expression revealed no phenotype). Second, the transgenic mice revealed significant systolic dysfunction at baseline, even in the absence of pregnancy, while such dysfunction is not seen in women with PPCM122, and indeed the absence of such preexisting dysfunction is included in the definition of PPCM. This model was thus important as a first demonstration that pregnancy could accelerate cardiomyopathy in mice, but the model has not been used further. How much cardiomyocyte death contributes to PPCM in more faithful models and in the human disease remains uncertain.
Cardiac deletion of STAT3
The most extensively studied model of PPCM is the Myh6-Cre;Stat3lox/lox mouse, in which the transcription factor Signal transducer of transcription 3 (STAT3) is selectively deleted in cardiomyocytes112. This model, while revealing no apparent dysfunction at baseline, develops systolic heart failure after one or two pregnancies. STAT3 is a ubiquitously expressed transcription factor that, in cardiomyocytes, regulates expression of antioxidant enzymes and pro-angiogenic factors. STAT3 is normally activated by phosphorylation in cardiomyocytes during pregnancy, and loss of cardiac STAT3 in the Myh6-Cre;Stat3lox/lox model causes unrestrained production of reactive oxygen species (ROS) and blunted angiogenesis112. The latter is in part caused by the ROS-induced secretion of the peptidase Cathepsin D, which cleaves the circulating nursing hormone prolactin to a 16kDa fragment well-established to be vasculotoxic (often called vasoinhibin) (see Figure 4)112. The 16kDa prolactin fragment interacts with circulating PAI-1 to bind to the urokinase-type plasminogen activator receptor (uPAR) on endothelial cells123, triggering a cascade of cardiotoxic events, including endothelial apoptosis and vascular insufficiency, and endothelial secretion of exosomes containing microRNA-146a that, when taken up by adjacent cardiomyocytes, suppress pro-survival signaling by Erbb4 (Figure 4). Thus, the Myh6-Cre;Stat3lox/lox model was first to illustrate a vascular/hormonal paradigm to explain PPCM, whereby aberrant signaling by a pregnancy-induced hormone (prolactin) leads to vasculotoxicity and in turn cardiomyopathy. Most tellingly, suppression of prolactin secretion with the dopamine agonist bromocriptine prevented pregnancy-induced heart failure, demonstrating the critical role of prolactin in the pathology of the Myh6-Cre;Stat3lox/lox model. The model has been used to study the role of the pregnancy hormone relaxin-2 on PPCM progression124, the effect of the metabolic modulator perhexiline125, and the role of catecholamines, especially β1-adrenergic agonism126. In the latter case, β1-adrenergic stimulation substantially worsened cardiac function, mirroring observations in a small group of women with PPCM in whom treatment with dobutamine correlated with worse outcomes126.
Fig 4:

Vasculo-hormonal hypothesis of the pathophysiology of peripartum cardiomyopathy. Pregnancy triggers the systemic secretion of hormones from the pituitary and placenta. Some of these, including prolactin (PRL) and soluble FLT1 (sFLT1) can be toxic to cardiac microvasculature, either via proteolytic conversion to vasculotoxic 16Kd PRL or via inhibition of VEGF signaling, respectively, ultimately leading to cardiac ischemia and apoptosis. See text for further details. (Illustration credit: Ben Smith)
Numerous lines of evidence support the clinical relevance of the Myh6-Cre;Stat3lox/lox model and the pathophysiological mechanism outlined above. Levels of STAT3 protein are decreased in left ventricular tissue from patients with PPCM, compared to non-failing donors112. Circulating cathepsin D activity and levels of microRNA-146a are elevated in women with PPCM, compared to pregnancy-matched healthy women.112, 127 And there is substantial, though still somewhat controversial, support for the use of bromocriptine in women with PPCM. Three randomized studies have suggested a benefit of bromocriptine use128–130, but the studies had limitations including small size, patients lost to follow-up, and the lack of a placebo arm, respectively. There thus remains some equipoise on the use of bromocriptine for PPCM, and a large (n=200) randomized placebo-controlled study has now been initiated in the US. In sum, there is substantial support for the human relevance of the Myh6-Cre;Stat3lox/lox model of PPCM.
Cardiac deletion of PGC-1α
The next model of PPCM to garner attention was the Myh6-Cre;Ppargc1alox/lox mouse, in which the transcriptional coactivator PGC-1α is deleted selectively in cardiomyocytes113. Like the STAT3 model, Myh6-Cre;Ppargc1alox/lox have minimal cardiac dysfunction at baseline, but develop dilated cardiomyopathy after one or two pregnancies. PGC-1α is a pleitropic coactivator that coordinates multiple transcriptional programs, including the promotion of oxidative metabolism and angiogenesis, the latter in part via secretion of vascular endothelial growth factor (VEGF)131, 132. Loss of cardiac PGC-1α in the Myh6-Cre;Ppargc1alox/lox model leads to pregnancy-induced defects in cardiac microvasculature, cardiac ischemia, and cardiomyopathy113. These effects were rescued by ectopic administration of VEGF and bromocriptine, but not either alone, supporting the likely role of prolactin in pregnancy-induced cardiomyopathy, but also indicating the presence of additional anti-angiogenic factors. A search for the latter identified sFLT1, the placental-derived suppressor of VEGF activity described above (see preeclampsia section, and also Figure 4). sFLT1 was sufficient to trigger cardiomyopathy in Myh6-Cre;Ppargc1alox/lox mice in the absence of pregnancy113. Activin-A, another placental derived hormone, was also recently implicated in cardiomyopathy in this model: inhibition of Activin-A receptors partially reversed pregnancy-induced cardiomyopathy in Myh6-Cre;Ppargc1alox/lox mice133. Thus, as with the Myh6-Cre;Stat3lox/lox model, the Myh6-Cre;Ppargc1alox/lox model illustrates the vascular/hormonal paradigm of PPCM, in which pregnancy-induced hormones (sFlt1, Activin-A) lead to vasculotoxicity and cardiomyopathy.
Numerous lines of evidence also support the clinical relevance of the Myh6-Cre;Ppargc1alox/lox model and the role of sFLT1, Activin-A, and likely other placental hormones, in PPCM. PGC-1α expression has been reported to be reduced in human cardiomyopathy, although not specifically in PPCM. Circulating levels of sFlt1 and Activin-A are elevated in PPCM patients, and correlate with heart failure severity and with adverse outcomes113, 133. Preeclampsia, in which sFlt1 and Activin-A levels are markedly elevated (see above), is one of the strongest risk factors for PPCM, present in 25–50% of cases101, 102, 134. In fact, even in the absence of PPCM, women with preeclampsia exhibit subclinical cardiac dysfunction that closely correlates with plasma sFlt1 levels135. It is unknown, however, if PGC-1α is altered in cardiac tissue of human PPCM, and in fact has been reported as elevated in other forms of human cardiomyopathy136. Nevertheless, loss of cardiac PGC-1α in mice is clearly a reliable model in which the murine heart is artificially made susceptible to pregnancy-induced vasculo-hormonal insults, allowing mechanistic studies of the latter.
TTN knock-in mice
Recent work has demonstrated that 10–15% of women with PPCM harbor heterozygous truncating variants (i.e, nonsense, frameshift, or splice site variants) in various genes known also to predispose to non-ischemic dilated cardiomyopathy (DCM)116, 117. Most prominent among these, present in ~10% of women with PPCM116, 117 as well as patients with DCM137, 138, is TTN, encoding for the large sarcomeric protein titin. These observations have led to the generation of knock-in mice, bearing heterozygous Ttn truncating variants analogous to those seen in human patients. These models do not exhibit spontaneous cardiomyopathy, but cardiac stressors including minipump infusions with angiotensin II, treatment with doxorubicin, or acute volume overload, can elicit a mild decline in systolic function139–141. The impact of pregnancy of these models has not been reported. Why these models are so mild, compared to the clinical disease, is unclear, but it should be noted that TTN truncating variants in humans are poorly penetrant for disease, with 95% of carriers exhibiting no cardiac dysfunction142, strongly suggesting the requirement for “second or third hits” that may not be present in the mouse models. It is also worth noting that the truncated titin protein encoded by TTN variant-bearing allele is present in human hearts143, 144, but not in rat145 or mouse (Z.A.) hearts, suggesting that protein quality control in the mouse may be better at clearing the mutated protein and thus averting disease. Thus, even though mice bearing Ttn truncating variants reflect genetic underpinnings of the human disease more faithfully than do the Myh6-Cre;Ppargc1alox/lox and Myh6-Cre;Stat3lox/lox models, the latter recapitulate better the disease phenotype. Mouse models with heterozygous deletions of other genes known to be mutated in cases of PPCM exist (Bag3146, 147, Flnc148, Dsp149), but the impact of pregnancy on their cardiac function has not been reported.
Other mouse models of PPCM
a. Pharmacological compounds:
Several groups have recently reported other mouse models that develop cardiac failure during or after pregnancy. Treatment of pregnant wild type mice during the last 3 days of gestation and first 3 weeks postpartum with an inhibitor of γ-secretase, which is required to activate Notch1 and normal angiogenesis, led to systolic cardiac failure, accompanied by blunted angiogenesis and lower circulating levels of cathepsin D and of sFlt1150. The study did not elucidate if γ-secretase inhibition elicits this cardiac phenotype primarily by acting on endothelial cells (where Notch1 plays a strong role) versus in cardiomyocytes (where cathepsin D is regulated) versus the placenta (the primary source of sFlt1 during pregnancy). Known effects of γ-secretase inhibition beyond suppression of Notch1 may also have contributed to the phenotype. Thus this model recapitulates some mechanistic aspects of the well-established Myh6-Cre;Stat3lox/lox model, and benefits from not needing genetic modifications, but the clinical relevance of Notch1 inhibition in human PPCM is not clear, and the pleiotropic effects of γ-secretase inhibition may render mechanistic investigations difficult.
b. Loss of natriuretic peptide signaling.
Another recent model of PPCM involved mice lacking Npr1, the receptor for the natriuretic peptides ANP and BNP that are secreted by the heart to modulate kidney function and blood pressure151. Npr1−/− mice have marked elevations in blood pressure, by ~20mmHg, accompanied by significant cardiac hypertrophy. After two pregnancies, the cardiac hypertrophy is accentuated, and the mice develop systolic heart failure. Mechanistic studies with various genetic crosses implicated lactation and a neuronal circuit involving signaling by aldosterone in the central nervous system151. The model appears to be driven largely by the elevated blood pressure and the ensuing cardiac hypertrophy, which is not typically observed in human PPCM. The model is also accompanied by activation of STAT3 phosphorylation in cardiac tissue, in contrast to loss of STAT3 in Myh6-Cre;Stat3lox/lox mice or in human PPCM hearts112. Moreover, PPCM and other forms of heart failure are likely accompanied by elevated, rather than blunted, ANP/BNP signaling. Nevertheless, the study suggests that it may be worth testing aldosterone inhibition with readily available FDA-approved inhibitors in this and other models of PPCM, as well as in the human disease.
c. Protein folding defects.
Two recent studies have suggested that aberrations in protein folding and the cellular response to misfolding may contribute to PPCM. HSPB6/Hsp20 is a small heat-shock protein found primarily in muscle tissues, and protective in multiple models of heart failure. Female transgenic mice with cardiac-specific expression of a mutant form of HSPB6/Hsp20, identified in a small subset of patients with DCM, develop mild systolic failure after 2–4 pregnancies, accompanied by cardiomyocyte apoptosis and evidence of impaired autophagic flux152, 153. Male transgenic mice spontaneously develop similar dysfunction starting at 6 months of age, indicating that the developed heart failure is not specific to pregnancy154. And Myh6-Cre;Perklox/lox mice, in which PERK, one of the effectors of the unfolded protein response (UPR), is deleted selectively in cardiomyocytes, develop systolic heart failure after 3–4 pregnancies, accompanied by a marked decrease in VEGF expression, and microvascular rarefaction155. Treatment with bromocriptine did not rescue pregnancy-induced systolic dysfunction in this model, but co-treatment with VEGF or other pro-angiogenic agent was not tested155. Interestingly, treatment of hypoxic isolated cardiomyocytes with 23kD prolactin promoted PERK activation, suggesting a role for PERK activation during lactation, but in vivo evidence for this conclusion remains lacking, nor is it known if PERK is activated in human PPCM hearts. Both of these studies thus suggest that aberrations in the UPR may contribute to PPCM, but further rescue studies will be needed to test this notion. The relevance of these models to the human disease is also difficult to gauge because there is little information available on the status of the UPR or of protein folding in human PPCM.
d. mTORC hyperactivity.
During mouse pregnancy, cardiac Akt and mTORC1 activity rise, concomitant with marked physiological hypertrophy and increasing cardiac mass, and this process reverses post-partum10, 156. Kouzu et al. recently demonstrated that preventing the post-partum suppression of cardiac mTORC1 activity may predispose to PPCM157. Myh6-Cre;Zfp36l2lox/lox mice lacking cardiac expression of Zfp36l2, a mRNA-destabilizing protein that suppresses mTORC1 via a Mdm2/p53/Redd1 pathway, develop profound mTORC1 hyperactivity and systolic dysfunction post-partum. The mice do spontaneously develop cardiac dysfunction in the absence of pregnancy, although pregnancy markedly accelerates the process. Importantly, systolic function is partially restored by treatment with rapamycin, a mTORC1 inhibitor. Interestingly, persistent mTORC1 activity can promote UPR response158, 159, thus potentially linking to the mouse models described above. It will be of interest to evaluate mTORC1 signaling in other models of PPCM, as well as their response to rapamycin, to test the generalizability of this model. Vascularity or response to bromocriptine treatment was not tested in this model.
Summary of models PPCM
Developing faithful models of PPCM is imperative to better study this still poorly understood disease. The variety of models associated with PPCM supports the notion that PPCM is a syndrome with multiple origins. Most clinical and pre-clinical evidence indicates that the disease is vascular and hormonal in nature, and two mouse models, the Myh6-Cre;Stat3lox/lox and Myh6-Cre;Ppargc1alox/lox models, best capture these aspects of the disease, thereby also illustrating convergence of various pathologies on common pathways. However, both models are limited by the physiological differences between mouse and human pregnancies. Various other aspects of human PPCM are also poorly reflected in these models, including the observation that most women with PPCM recover cardiac function, while the mouse models do not. Better characterization of PPCM pheno- and genotypes in humans and the subsequent development of appropriate preclinical models of PPCM would significantly advance the field.
Models of other cardiovascular complications of pregnancy
Pregnancy-associated vascular aneurysm/dissection:
Pregnancy is associated with increased risk of accelerated aneurysm progression and aortic dissection, as well as spontaneous coronary artery dissection (SCAD). These catastrophic events often are seen late in pregnancy and in the immediate post-partum period. Habashi et al recently reported that mice with severe deficiency in fibrillin-1 expression, a model of Marfan’s syndrome, showed accelerated aortic growth during pregnancy, accompanied by high incidence of dissection and markedly decreased survival, which were blocked by oxytocin antagonists or by prevention of lactation160. Similarly, pregnancy was noted to exacerbate aortic dissection in a mouse model of vascular Ehlers-Danlos syndrome that carried a dominant mutation in a major collagen (COL3A1) in blood vessels. Recently Zekavat et al. used whole exome sequencing to identify rare variants in ten fibrillar collagen genes that predisposed to SCAD, and showed that mice with heterozygous deficiency in Col5a1 or both Col5a1 and Col3a1 were prone to spontaneous aortic dissection, but they did not report if pregnancy accentuated the phenotype161. Interestingly, similar to the Marfan mouse, oxytocin antagonisms prevented pregnancy-associated aortic dissection in the COL3A1 mutant mice162. Since oxytocin is frequently used to support the delivery process, the present model may indicate more careful use of this hormonal treatment. Thus, as in preeclampsia and PPCM, pregnancy-associated hormones appear to play a dominant role in disease progression.
Pregnancy-associated thromboembolism:
Pregnancy is characterized by a hyper-coagulable state and is associated with increased thrombosis and vascular complications such as recurrent miscarriages, venous thrombosis and fetal growth restriction. Iseramman et al. first reported that mice lacking thrombomodulin developed abortions due to activation of tissue-factor-initiated activation of a blood coagulation cascade at the maternal-fetal interface163. Similarly, in order to model thrombosis related to anti-phospholipid (aPL) antibody syndrome, several groups have performed passive transfer of the aPL antibodies purified from patients into pregnant mice. These aPL antibody-injected mice develop miscarriages or severe fetal growth restrictions due to placental thrombosis164. Other groups have suggested that mice immunized with beta-2-glycoprotein I (β2GPI) also develop aPL-like syndrome165. β2GPI is a phospholipid-binding protein and it is thought that aPL antibodies are induced by binding of β2GPI to endogenous phospholipids. Many groups have tested therapeutic compounds such as complement inhibitors and statins to ameliorate vascular thrombosis and improve adverse pregnancy outcomes in these aPL models166. Recently, aPL actions via apolipoprotein E receptor 2 in placental trophoblasts was also shown to induce hypertension and fetal growth restriction in pregnant mice167. Interestingly, in this mouse model protein phosphatase 2A inhibition was found to afford complete protection from aPL-associated pregnancy complications.
Pregnancy-associated cardiac arrhythmias:
Cardiac arrhythmias due to pre-existing conditions may be exacerbated during pregnancy due to physiologic tachycardia168. El Khouri et al. confirmed that pregnancy exacerbates supraventricular tachyarrhythmias using programmed electrical stimulation protocols in anesthetized pregnant mice169, 170. Mechanistically, cell-intrinsic differences in sinoatrial node cell activity and increased L-type Ca2+ current and spontaneous Ca2+ release from sarcoplasmic reticulum could be demonstrated between nonpregnant and pregnant mice, and chronic administration of oestrogen to nonpregnant mice was sufficient to increase heart rate, in a manner dependent on the estrogen receptor α169, 170. Thus the model suggests a hormonal, rather than hemodynamic, contribution to pregnancy-associated tachycardia.
Conclusion
Cardiovascular complications of pregnancy are frequent and long-lasting, but their pathophysiology remains poorly understood. Clinical studies are challenging, in part due to especially high regulatory hurdles in this patient population, and to frequent skepticism and even disinformation. Pre-clinical studies in animal models are thus critical to advance mechanistic insight. The placenta plays a dominant role in pathophysiology, and all animal models are thus in placental mammals. To date most of these models are surgical, pharmacological, or genetic in nature. Few have incorporated environmental factors such as air pollution, or high-altitude modeling, which are also thought to contribute to cardiovascular complications of pregnancy. A potential role for infection, e.g. myocarditis, has also been only minimally modeled171, 172. Nevertheless, animal models to date have provided substantial insight into disease pathogenesis, and have led to numerous potential treatments currently under evaluation, such as bromocriptine for PPCM (NCT05180773, NCT02590601, NCT00998556), or extracorporeal removal of sFlt1 for preeclampsia (NCT02923206, NCT03231657).
Nonstandard Abbreviations and Acronyms
- HELLP
Hemolysis, Elevated Liver Enzymes, Low Platelet syndrome
- HFpEF
Heart Failure with preserved ejection fraction
- sFLT1
Soluble fms-like tyrosine kinase 1
- sENG
Soluble Endoglin
- CVD
Cardiovascular Disease
- VEGF
Vascular Endothelial Growth Factor
- RUPP
Reduced Uterine Perfusion Pressure Model
- GFR
Glomerular Filtration Rate
- COMT
Catechol-O-Methyl Transferase
- AVP
Arginine Vasopressin
- STOX1
Storkhead box 1
- ANP
Atrial natriuretic peptide
- PPCM
Peripartum Cardiomyopathy
- STAT3
Signal transducer of transcription 3
- BP
Blood pressure
- PGC-1α
PPARγ-coactivator-1α
- MYH
Myosin heavy chain
- DCM
Dilated cardiomyopathy
Footnotes
Disclosures:
None.
References
- 1.Centers for Disease C and Prevention. Healthier mothers and babies. MMWR Morb Mortal Wkly Rep. 1999;48:849–58. [PubMed] [Google Scholar]
- 2.Mehta LS, Warnes CA, Bradley E, Burton T, Economy K, Mehran R, Safdar B, Sharma G, Wood M, Valente AM, et al. Cardiovascular Considerations in Caring for Pregnant Patients: A Scientific Statement From the American Heart Association. Circulation. 2020;141:e884–e903. [DOI] [PubMed] [Google Scholar]
- 3.Petersen EE, Davis NL, Goodman D, Cox S, Syverson C, Seed K, Shapiro-Mendoza C, Callaghan WM and Barfield W. Racial/Ethnic Disparities in Pregnancy-Related Deaths - United States, 2007–2016. MMWR Morb Mortal Wkly Rep. 2019;68:762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellamy L, Casas J-P, Hingorani AD and Williams DJ. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: systematic review and meta-analysis. Bmj. 2007;335:974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bushnell C, McCullough LD, Awad IA, Chireau MV, Fedder WN, Furie KL, Howard VJ, Lichtman JH, Lisabeth LD, Pina IL, et al. Guidelines for the prevention of stroke in women: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:1545–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gundling WE Jr. and Wildman DE. A review of inter- and intraspecific variation in the eutherian placenta. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox B, Kotlyar M, Evangelou AI, Ignatchenko V, Ignatchenko A, Whiteley K, Jurisica I, Adamson SL, Rossant J and Kislinger T. Comparative systems biology of human and mouse as a tool to guide the modeling of human placental pathology. Mol Syst Biol. 2009;5:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malassine A, Frendo JL and Evain-Brion D. A comparison of placental development and endocrine functions between the human and mouse model. Hum Reprod Update. 2003;9:531–9. [DOI] [PubMed] [Google Scholar]
- 9.Liu LX and Arany Z. Maternal cardiac metabolism in pregnancy. Cardiovascular research. 2014;101:545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung E and Leinwand LA. Pregnancy as a cardiac stress model. Cardiovascular research. 2014;101:561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong AY, Kulandavelu S, Whiteley KJ, Qu D, Langille BL and Adamson SL. Maternal cardiovascular changes during pregnancy and postpartum in mice. American journal of physiology Heart and circulatory physiology. 2002;282:H918–25. [DOI] [PubMed] [Google Scholar]
- 12.Silva JF and Serakides R. Intrauterine trophoblast migration: A comparative view of humans and rodents. Cell Adh Migr. 2016;10:88–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soncin F, Khater M, To C, Pizzo D, Farah O, Wakeland A, Arul Nambi Rajan K, Nelson KK, Chang CW, Moretto-Zita M, et al. Comparative analysis of mouse and human placentae across gestation reveals species-specific regulators of placental development. Development. 2018;145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ratajczak CK, Fay JC and Muglia LJ. Preventing preterm birth: the past limitations and new potential of animal models. Dis Model Mech. 2010;3:407–14. [DOI] [PubMed] [Google Scholar]
- 15.PrabhuDas M, Bonney E, Caron K, Dey S, Erlebacher A, Fazleabas A, Fisher S, Golos T, Matzuk M, McCune JM, et al. Immune mechanisms at the maternal-fetal interface: perspectives and challenges. Nat Immunol. 2015;16:328–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kahn DA and Baltimore D. Pregnancy induces a fetal antigen-specific maternal T regulatory cell response that contributes to tolerance. Proc Natl Acad Sci U S A. 2010;107:9299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rana S, Lemoine E, Granger JP and Karumanchi SA. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ Res. 2019;124:1094–1112. [DOI] [PubMed] [Google Scholar]
- 18.Chen CW, Jaffe IZ and Karumanchi SA. Pre-eclampsia and cardiovascular disease. Cardiovasc Res. 2014;101:579–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams D, Stout MJ, Rosenbloom JI, Olsen MA, Joynt Maddox KE, Deych E, Davila-Roman VG and Lindley KJ. Preeclampsia Predicts Risk of Hospitalization for Heart Failure With Preserved Ejection Fraction. J Am Coll Cardiol. 2021;78:2281–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, Sibai BM, Epstein FH, Romero R, Thadhani R, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355:992–1005. [DOI] [PubMed] [Google Scholar]
- 21.Leanos-Miranda A, Marquez-Acosta J, Cardenas-Mondragon GM, Chinolla-Arellano ZL, Rivera-Leanos R, Bermejo-Huerta S, Romero-Arauz JF, Alvarez-Jimenez G, Ramos-Leon JC and Ulloa-Aguirre A. Urinary prolactin as a reliable marker for preeclampsia, its severity, and the occurrence of adverse pregnancy outcomes. J Clin Endocrinol Metab. 2008;93:2492–9. [DOI] [PubMed] [Google Scholar]
- 22.Burke SD, Zsengeller ZK, Khankin EV, Lo AS, Rajakumar A, DuPont JJ, McCurley A, Moss ME, Zhang D, Clark CD, et al. Soluble fms-like tyrosine kinase 1 promotes angiotensin II sensitivity in preeclampsia. J Clin Invest. 2016;126:2561–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCarthy FP, Kingdom JC, Kenny LC and Walsh SK. Animal models of preeclampsia; uses and limitations. Placenta. 2011;32:413–9. [DOI] [PubMed] [Google Scholar]
- 24.Li J, LaMarca B and Reckelhoff JF. A model of preeclampsia in rats: the reduced uterine perfusion pressure (RUPP) model. Am J Physiol Heart Circ Physiol. 2012;303:H1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Granger JP, LaMarca BB, Cockrell K, Sedeek M, Balzi C, Chandler D and Bennett W. Reduced uterine perfusion pressure (RUPP) model for studying cardiovascular-renal dysfunction in response to placental ischemia. Methods Mol Med. 2006;122:383–92. [DOI] [PubMed] [Google Scholar]
- 26.Gilbert JS, Babcock SA and Granger JP. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension. 2007;50:1142–7. [DOI] [PubMed] [Google Scholar]
- 27.Spradley FT, Tan AY, Joo WS, Daniels G, Kussie P, Karumanchi SA and Granger JP. Placental Growth Factor Administration Abolishes Placental Ischemia-Induced Hypertension. Hypertension. 2016;67:740–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gadonski G, LaMarca BB, Sullivan E, Bennett W, Chandler D and Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of interleukin 6. Hypertension. 2006;48:711–6. [DOI] [PubMed] [Google Scholar]
- 29.LaMarca BB, Bennett WA, Alexander BT, Cockrell K and Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of tumor necrosis factor-alpha. Hypertension. 2005;46:1022–5. [DOI] [PubMed] [Google Scholar]
- 30.Intapad S, Warrington JP, Spradley FT, Palei AC, Drummond HA, Ryan MJ, Granger JP and Alexander BT. Reduced uterine perfusion pressure induces hypertension in the pregnant mouse. Am J Physiol Regul Integr Comp Physiol. 2014;307:R1353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fushima T, Sekimoto A, Minato T, Ito T, Oe Y, Kisu K, Sato E, Funamoto K, Hayase T, Kimura Y, et al. Reduced Uterine Perfusion Pressure (RUPP) Model of Preeclampsia in Mice. PLoS One. 2016;11:e0155426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai Z, Kalkunte S and Sharma S. A critical role of interleukin-10 in modulating hypoxia-induced preeclampsia-like disease in mice. Hypertension. 2011;57:505–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makris A, Thornton C, Thompson J, Thomson S, Martin R, Ogle R, Waugh R, McKenzie P, Kirwan P and Hennessy A. Uteroplacental ischemia results in proteinuric hypertension and elevated sFLT-1. Kidney Int. 2007;71:977–84. [DOI] [PubMed] [Google Scholar]
- 34.Makris A, Yeung KR, Lim SM, Sunderland N, Heffernan S, Thompson JF, Iliopoulos J, Killingsworth MC, Yong J, Xu B, et al. Placental Growth Factor Reduces Blood Pressure in a Uteroplacental Ischemia Model of Preeclampsia in Nonhuman Primates. Hypertension. 2016;67:1263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turanov AA, Lo A, Hassler MR, Makris A, Ashar-Patel A, Alterman JF, Coles AH, Haraszti RA, Roux L, Godinho B, et al. RNAi modulation of placental sFLT1 for the treatment of preeclampsia. Nat Biotechnol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y, Chiu K, Brescia RJ, Combs CA, Katz MA, Kitzmiller JL, Heilbron DC and Fisher SJ. Increased depth of trophoblast invasion after chronic constriction of the lower aorta in rhesus monkeys. Am J Obstet Gynecol. 1993;169:224–9. [DOI] [PubMed] [Google Scholar]
- 37.Gant NF, Daley GL, Chand S, Whalley PJ and MacDonald PC. A study of angiotensin II pressor response throughout primigravid pregnancy. J Clin Invest. 1973;52:2682–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takimoto E, Ishida J, Sugiyama F, Horiguchi H, Murakami K and Fukamizu A. Hypertension induced in pregnant mice by placental renin and maternal angiotensinogen. Science. 1996;274:995–8. [DOI] [PubMed] [Google Scholar]
- 39.Bohlender J, Ganten D and Luft FC. Rats transgenic for human renin and human angiotensinogen as a model for gestational hypertension. J Am Soc Nephrol. 2000;11:2056–2061. [DOI] [PubMed] [Google Scholar]
- 40.Dechend R, Muller DN, Wallukat G, Homuth V, Krause M, Dudenhausen J and Luft FC. Activating auto-antibodies against the AT1 receptor in preeclampsia. Autoimmun Rev. 2005;4:61–5. [DOI] [PubMed] [Google Scholar]
- 41.Wenzel K, Rajakumar A, Haase H, Geusens N, Hubner N, Schulz H, Brewer J, Roberts L, Hubel CA, Herse F, et al. Angiotensin II type 1 receptor antibodies and increased angiotensin II sensitivity in pregnant rats. Hypertension. 2011;58:77–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, Hicks MJ, Ramin SM, Kellems RE and Xia Y. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat Med. 2008;14:855–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LaMarca B, Parrish MR and Wallace K. Agonistic autoantibodies to the angiotensin II type I receptor cause pathophysiologic characteristics of preeclampsia. Gend Med. 2012;9:139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haase N, Foster DJ, Cunningham MW, Bercher J, Nguyen T, Shulga-Morskaya S, Milstein S, Shaikh S, Rollins J, Golic M, et al. RNA interference therapeutics targeting angiotensinogen ameliorate preeclamptic phenotype in rodent models. J Clin Invest. 2020;130:2928–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geusens N, Verlohren S, Luyten C, Taube M, Hering L, Vercruysse L, Hanssens M, Dudenhausen JW, Dechend R and Pijnenborg R. Endovascular trophoblast invasion, spiral artery remodelling and uteroplacental haemodynamics in a transgenic rat model of pre-eclampsia. Placenta. 2008;29:614–23. [DOI] [PubMed] [Google Scholar]
- 46.Bergmann A, Ahmad S, Cudmore M, Gruber AD, Wittschen P, Lindenmaier W, Christofori G, Gross V, Gonzalves A, Grone HJ, et al. Reduction of circulating soluble Flt-1 alleviates preeclampsia-like symptoms in a mouse model. J Cell Mol Med. 2010;14:1857–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu F, Longo M, Tamayo E, Maner W, Al-Hendy A, Anderson GD, Hankins GD and Saade GR. The effect of over-expression of sFlt-1 on blood pressure and the occurrence of other manifestations of preeclampsia in unrestrained conscious pregnant mice. Am J Obstet Gynecol. 2007;196:396 e1–7; discussion 396 e7. [DOI] [PubMed] [Google Scholar]
- 49.Szalai G, Romero R, Chaiworapongsa T, Xu Y, Wang B, Ahn H, Xu Z, Chiang PJ, Sundell B, Wang R, et al. Full-length human placental sFlt-1-e15a isoform induces distinct maternal phenotypes of preeclampsia in mice. PLoS One. 2015;10:e0119547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vogtmann R, Kuhnel E, Dicke N, Verkaik-Schakel RN, Plosch T, Schorle H, Stojanovska V, Herse F, Koninger A, Kimmig R, et al. Human sFLT1 Leads to Severe Changes in Placental Differentiation and Vascularization in a Transgenic hsFLT1/rtTA FGR Mouse Model. Front Endocrinol (Lausanne). 2019;10:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vogtmann R, Heupel J, Herse F, Matin M, Hagmann H, Bendix I, Kraker K, Dechend R, Winterhager E, Kimmig R, et al. Circulating Maternal sFLT1 (Soluble fms-Like Tyrosine Kinase-1) Is Sufficient to Impair Spiral Arterial Remodeling in a Preeclampsia Mouse Model. Hypertension. 2021;78:1067–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perez-Roque L, Nunez-Gomez E, Rodriguez-Barbero A, Bernabeu C, Lopez-Novoa JM and Pericacho M. Pregnancy-Induced High Plasma Levels of Soluble Endoglin in Mice Lead to Preeclampsia Symptoms and Placental Abnormalities. Int J Mol Sci. 2020;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Venkatesha S, Toporsian M, Lam C, Hanai JI, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12:642–649. [DOI] [PubMed] [Google Scholar]
- 54.Wallace K, Morris R, Kyle PB, Cornelius D, Darby M, Scott J, Moseley J, Chatman K and Lamarca B. Hypertension, inflammation and T lymphocytes are increased in a rat model of HELLP syndrome. Hypertens Pregnancy. 2014;33:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Z, Zhang Y, Ying Ma J, Kapoun AM, Shao Q, Kerr I, Lam A, O’Young G, Sannajust F, Stathis P, et al. Recombinant vascular endothelial growth factor 121 attenuates hypertension and improves kidney damage in a rat model of preeclampsia. Hypertension. 2007;50:686–92. [DOI] [PubMed] [Google Scholar]
- 56.Kumasawa K, Ikawa M, Kidoya H, Hasuwa H, Saito-Fujita T, Morioka Y, Takakura N, Kimura T and Okabe M. Pravastatin induces placental growth factor (PGF) and ameliorates preeclampsia in a mouse model. Proc Natl Acad Sci U S A. 2011;108:1451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rana S, Powe CE, Salahuddin S, Verlohren S, Perschel FH, Levine RJ, Lim KH, Wenger JB, Thadhani R and Karumanchi SA. Angiogenic factors and the risk of adverse outcomes in women with suspected preeclampsia. Circulation. 2012;125:911–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davisson RL, Hoffmann DS, Butz GM, Aldape G, Schlager G, Merrill DC, Sethi S, Weiss RM and Bates JN. Discovery of a spontaneous genetic mouse model of preeclampsia. Hypertension. 2002;39:337–42. [DOI] [PubMed] [Google Scholar]
- 59.Johnston AN, Batts TL, Langohr IM, Moeller C, Liu CC and Sones JL. The BPH/5 Mouse Model of Superimposed Preeclampsia Is Not a Model of HELLP Syndrome. Biology (Basel). 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sones JL, Merriam AA, Seffens A, Brown-Grant DA, Butler SD, Zhao AM, Xu X, Shawber CJ, Grenier JK and Douglas NC. Angiogenic factor imbalance precedes complement deposition in placentae of the BPH/5 model of preeclampsia. FASEB J. 2018;32:2574–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woods AK, Hoffmann DS, Weydert CJ, Butler SD, Zhou Y, Sharma RV and Davisson RL. Adenoviral delivery of VEGF121 early in pregnancy prevents spontaneous development of preeclampsia in BPH/5 mice. Hypertension. 2011;57:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qing X, Redecha PB, Burmeister MA, Tomlinson S, D’Agati VD, Davisson RL and Salmon JE. Targeted inhibition of complement activation prevents features of preeclampsia in mice. Kidney Int. 2011;79:331–9. [DOI] [PubMed] [Google Scholar]
- 63.Girardi G, Yarilin D, Thurman JM, Holers VM and Salmon JE. Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J Exp Med. 2006;203:2165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu G, Sun Y, Foerster K, Manuel J, Molina H, Levy GA, Gorczynski RM and Clark DA. LPS-induced murine abortions require C5 but not C3, and are prevented by upregulating expression of the CD200 tolerance signaling molecule. Am J Reprod Immunol. 2008;60:135–40. [DOI] [PubMed] [Google Scholar]
- 65.Ahmed A, Singh J, Khan Y, Seshan SV and Girardi G. A new mouse model to explore therapies for preeclampsia. PLoS One. 2010;5:e13663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanasaki K, Palmsten K, Sugimoto H, Ahmad S, Hamano Y, Xie L, Parry S, Augustin HG, Gattone VH, Folkman J, et al. Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature. 2008;453:1117–21. [DOI] [PubMed] [Google Scholar]
- 67.Roten LT, Fenstad MH, Forsmo S, Johnson MP, Moses EK, Austgulen R and Skorpen F. A low COMT activity haplotype is associated with recurrent preeclampsia in a Norwegian population cohort (HUNT2). Mol Hum Reprod. 2011;17:439–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iqbal K, Dhakal P, Pierce SH and Soares MJ. Catechol-O-methyltransferase and Pregnancy Outcome: an Appraisal in Rat. Reprod Sci. 2021;28:462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Townley-Tilson WH, Wu Y, Ferguson JE, 3rd and Patterson C. The ubiquitin ligase ASB4 promotes trophoblast differentiation through the degradation of ID2. PLoS One. 2014;9:e89451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Janatpour MJ, McMaster MT, Genbacev O, Zhou Y, Dong J, Cross JC, Israel MA and Fisher SJ. Id-2 regulates critical aspects of human cytotrophoblast differentiation, invasion and migration. Development. 2000;127:549–58. [DOI] [PubMed] [Google Scholar]
- 71.Li F, Fushima T, Oyanagi G, Townley-Tilson HW, Sato E, Nakada H, Oe Y, Hagaman JR, Wilder J, Li M, et al. Nicotinamide benefits both mothers and pups in two contrasting mouse models of preeclampsia. Proc Natl Acad Sci U S A. 2016;113:13450–13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rigourd V, Chauvet C, Chelbi ST, Rebourcet R, Mondon F, Letourneur F, Mignot TM, Barbaux S and Vaiman D. STOX1 overexpression in choriocarcinoma cells mimics transcriptional alterations observed in preeclamptic placentas. PLoS One. 2008;3:e3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Doridot L, Passet B, Mehats C, Rigourd V, Barbaux S, Ducat A, Mondon F, Vilotte M, Castille J, Breuiller-Fouche M, et al. Preeclampsia-like symptoms induced in mice by fetoplacental expression of STOX1 are reversed by aspirin treatment. Hypertension. 2013;61:662–8. [DOI] [PubMed] [Google Scholar]
- 74.Rolnik DL, Wright D, Poon LC, O’Gorman N, Syngelaki A, de Paco Matallana C, Akolekar R, Cicero S, Janga D, Singh M, et al. Aspirin versus Placebo in Pregnancies at High Risk for Preterm Preeclampsia. N Engl J Med. 2017;377:613–622. [DOI] [PubMed] [Google Scholar]
- 75.van Dijk M, Mulders J, Poutsma A, Konst AA, Lachmeijer AM, Dekker GA, Blankenstein MA and Oudejans CB. Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene family. Nat Genet. 2005;37:514–9. [DOI] [PubMed] [Google Scholar]
- 76.Iglesias-Platas I, Monk D, Jebbink J, Buimer M, Boer K, van der Post J, Hills F, Apostolidou S, Ris-Stalpers C, Stanier P, et al. STOX1 is not imprinted and is not likely to be involved in preeclampsia. Nat Genet. 2007;39:279–80; author reply 280–1. [DOI] [PubMed] [Google Scholar]
- 77.Kivinen K, Peterson H, Hiltunen L, Laivuori H, Heino S, Tiala I, Knuutila S, Rasi V and Kere J. Evaluation of STOX1 as a preeclampsia candidate gene in a population-wide sample. Eur J Hum Genet. 2007;15:494–7. [DOI] [PubMed] [Google Scholar]
- 78.Cui Y, Wang W, Dong N, Lou J, Srinivasan DK, Cheng W, Huang X, Liu M, Fang C, Peng J, et al. Role of corin in trophoblast invasion and uterine spiral artery remodelling in pregnancy. Nature. 2012;484:246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Finn-Sell SL, Renshall LJ, Cowley EJ, Dilworth MR, Wareing M, Greenwood SL, Sibley CP and Cottrell EC. The atrial natriuretic peptide (ANP) knockout mouse does not exhibit the phenotypic features of pre-eclampsia or demonstrate fetal growth restriction. Placenta. 2016;42:25–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Degrelle SA, Chissey A, Stepanian A, Fournier T, Guibourdenche J, Mandelbrot L and Tsatsaris V. Placental Overexpression of Soluble CORIN in Preeclampsia. Am J Pathol. 2020;190:970–976. [DOI] [PubMed] [Google Scholar]
- 81.Perschbacher KJ, Deng G, Sandgren JA, Walsh JW, Witcher PC, Sapouckey SA, Owens CE, Zhang SY, Scroggins SM, Pearson NA, et al. Reduced mRNA Expression of RGS2 (Regulator of G Protein Signaling-2) in the Placenta Is Associated With Human Preeclampsia and Sufficient to Cause Features of the Disorder in Mice. Hypertension. 2020;75:569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sandgren JA, Deng G, Linggonegoro DW, Scroggins SM, Perschbacher KJ, Nair AR, Nishimura TE, Zhang SY, Agbor LN, Wu J, et al. Arginine vasopressin infusion is sufficient to model clinical features of preeclampsia in mice. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scroggins SM, Santillan DA, Lund JM, Sandgren JA, Krotz LK, Hamilton WS, Devor EJ, Davis HA, Pierce GL, Gibson-Corley KN, et al. Elevated vasopressin in pregnant mice induces T-helper subset alterations consistent with human preeclampsia. Clin Sci (Lond). 2018;132:419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yeung EH, Liu A, Mills JL, Zhang C, Mannisto T, Lu Z, Tsai MY and Mendola P. Increased levels of copeptin before clinical diagnosis of preeclampsia. Hypertension. 2014;64:1362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Molnar M, Suto T, Toth T and Hertelendy F. Prolonged blockade of nitric oxide synthesis in gravid rats produces sustained hypertension, proteinuria, thrombocytopenia, and intrauterine growth retardation. Am J Obstet Gynecol. 1994;170:1458–66. [DOI] [PubMed] [Google Scholar]
- 86.Gillis EE, Mooney JN, Garrett MR, Granger JP and Sasser JM. Sildenafil Treatment Ameliorates the Maternal Syndrome of Preeclampsia and Rescues Fetal Growth in the Dahl Salt-Sensitive Rat. Hypertension. 2016;67:647–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gillis EE, Williams JM, Garrett MR, Mooney JN and Sasser JM. The Dahl salt-sensitive rat is a spontaneous model of superimposed preeclampsia. Am J Physiol Regul Integr Comp Physiol. 2015;309:R62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Man AWC, Zhou Y, Lam UDP, Reifenberg G, Werner A, Habermeier A, Closs EI, Daiber A, Munzel T, Xia N and Li H. l-Citrulline ameliorates pathophysiology in a rat model of superimposed preeclampsia. Br J Pharmacol. 2021. [DOI] [PubMed] [Google Scholar]
- 89.Pruthi D, Khankin EV, Blanton RM, Aronovitz M, Burke SD, McCurley A, Karumanchi SA and Jaffe IZ. Exposure to experimental preeclampsia in mice enhances the vascular response to future injury. Hypertension. 2015;65:863–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bytautiene E, Lu F, Tamayo EH, Hankins GD, Longo M, Kublickiene K and Saade GR. Long-term maternal cardiovascular function in a mouse model of sFlt-1-induced preeclampsia. Am J Physiol Heart Circ Physiol. 2010;298:H189–93. [DOI] [PubMed] [Google Scholar]
- 91.Bytautiene E, Bulayeva N, Bhat G, Li L, Rosenblatt KP and Saade GR. Long-term alterations in maternal plasma proteome after sFlt1-induced preeclampsia in mice. Am J Obstet Gynecol. 2013;208:388 e1–388 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miralles F, Collinot H, Boumerdassi Y, Ducat A, Duche A, Renault G, Marchiol C, Lagoutte I, Bertholle C, Andrieu M, et al. Long-term cardiovascular disorders in the STOX1 mouse model of preeclampsia. Sci Rep. 2019;9:11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sutton EF, Lob HE, Song J, Xia Y, Butler S, Liu CC, Redman LM and Sones JL. Adverse metabolic phenotype of female offspring exposed to preeclampsia in utero: a characterization of the BPH/5 mouse in postnatal life. Am J Physiol Regul Integr Comp Physiol. 2017;312:R485–R491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bakrania BA, George EM and Granger JP. Animal models of preeclampsia: investigating pathophysiology and therapeutic targets. Am J Obstet Gynecol. 2022;226:S973–S987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maharaj AS, Walshe TE, Saint-Geniez M, Venkatesha S, Maldonado AE, Himes NC, Matharu KS, Karumanchi SA and D’Amore PA. VEGF and TGF-beta are required for the maintenance of the choroid plexus and ependyma. J Exp Med. 2008;205:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Palmer KR, Tong S and Kaitu’u-Lino TJ. Placental-specific sFLT-1: role in pre-eclamptic pathophysiology and its translational possibilities for clinical prediction and diagnosis. Mol Hum Reprod. 2017;23:69–78. [DOI] [PubMed] [Google Scholar]
- 97.Carter AM. Animal models of human pregnancy and placentation: alternatives to the mouse. Reproduction. 2020;160:R129–R143. [DOI] [PubMed] [Google Scholar]
- 98.Pearson GD, Veille JC, Rahimtoola S, Hsia J, Oakley CM, Hosenpud JD, Ansari A and Baughman KL. Peripartum cardiomyopathy: National Heart, Lung, and Blood Institute and Office of Rare Diseases (National Institutes of Health) workshop recommendations and review. JAMA. 2000;283:1183–8. [DOI] [PubMed] [Google Scholar]
- 99.Elkayam U Clinical characteristics of peripartum cardiomyopathy in the United States: diagnosis, prognosis, and management. J Am Coll Cardiol. 2012;58:659–70. [DOI] [PubMed] [Google Scholar]
- 100.Sliwa K, Fett J and Elkayam U. Peripartum cardiomyopathy. Lancet. 2006;368:687–93. [DOI] [PubMed] [Google Scholar]
- 101.Arany Z and Elkayam U. Peripartum Cardiomyopathy. Circulation. 2016;133:1397–409. [DOI] [PubMed] [Google Scholar]
- 102.Davis MB, Arany Z, McNamara DM, Goland S and Elkayam U. Peripartum Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2020;75:207–221. [DOI] [PubMed] [Google Scholar]
- 103.Bauersachs J, Konig T, van der Meer P, Petrie MC, Hilfiker-Kleiner D, Mbakwem A, Hamdan R, Jackson AM, Forsyth P, de Boer RA, et al. Pathophysiology, diagnosis and management of peripartum cardiomyopathy: a position statement from the Heart Failure Association of the European Society of Cardiology Study Group on peripartum cardiomyopathy. Eur J Heart Fail. 2019;21:827–843. [DOI] [PubMed] [Google Scholar]
- 104.Banayan J, Rana S, Mueller A, Tung A, Ramadan H, Arany Z, Nizamuddin J, Novack V, Scavone B, Brown SM, et al. Cardiogenic shock in pregnancy: Analysis from the National Inpatient Sample. Hypertens Pregnancy. 2016:1–7. [DOI] [PubMed] [Google Scholar]
- 105.THE CALIFORNIA PREGNANCY- ASSOCIATED MORTALITY REVIEW Report from 2002 and 2003 Maternal Death Reviews. 2012.
- 106.Creanga AA, Berg CJ, Syverson C, Seed K, Bruce FC and Callaghan WM. Pregnancy-related mortality in the United States, 2006–2010. Obstet Gynecol. 2015;125:5–12. [DOI] [PubMed] [Google Scholar]
- 107.Arany Z Understanding Peripartum Cardiomyopathy. Annu Rev Med. 2018;69:165–176. [DOI] [PubMed] [Google Scholar]
- 108.Goland S, Modi K, Hatamizadeh P and Elkayam U. Differences in clinical profile of African-American women with peripartum cardiomyopathy in the United States. J Card Fail. 2013;19:214–8. [DOI] [PubMed] [Google Scholar]
- 109.Irizarry O, Levine L, Lewey J, Boyer T, Elovitz M and Arany Z. Comparison of clinical characteristics and outcomes of Peripartum Cardiomyopathy between African American and non-African American women. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Getz KD, Lewey J, Tam V, Irizarry OC, Levine LD, Aplenc R and Arany Z. Neighborhood education status drives racial disparities in clinical outcomes in PPCM. Am Heart J. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bello NA and Arany Z. Molecular mechanisms of peripartum cardiomyopathy: A vascular/hormonal hypothesis. Trends in cardiovascular medicine. 2015:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hilfiker-Kleiner D, Kaminski K, Podewski E, Bonda T, Schaefer A, Sliwa K, Forster O, Quint A, Landmesser U, Doerries C, et al. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell. 2007;128:589–600. [DOI] [PubMed] [Google Scholar]
- 113.Patten IS, Rana S, Shahul S, Rowe GC, Jang C, Liu L, Hacker MR, Rhee JS, Mitchell J, Mahmood F, et al. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature. 2012;485:333–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ricke-Hoch M, Hoes MF, Pfeffer TJ, Schlothauer S, Nonhoff J, Haidari S, Bomer N, Scherr M, Stapel B, Stelling E, et al. In peripartum cardiomyopathy plasminogen activator inhibitor-1 is a potential new biomarker with controversial roles. Cardiovasc Res. 2020;116:1875–1886. [DOI] [PubMed] [Google Scholar]
- 115.Hilfiker-Kleiner D and Sliwa K. Pathophysiology and epidemiology of peripartum cardiomyopathy. Nature reviews Cardiology. 2014;11:364–70. [DOI] [PubMed] [Google Scholar]
- 116.Ware JS, Li J, Mazaika E, Yasso CM, DeSouza T, Cappola TP, Tsai EJ, Hilfiker-Kleiner D, Kamiya CA, Mazzarotto F, et al. Shared Genetic Predisposition in Peripartum and Dilated Cardiomyopathies. N Engl J Med. 2016;374:233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Goli R, Li J, Brandimarto J, Levine LD, Riis V, McAfee Q, DePalma S, Haghighi A, Seidman JG, Seidman CE, et al. Genetic and Phenotypic Landscape of Peripartum Cardiomyopathy. Circulation. 2021;143:1852–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH and Dorn GW 2nd. Enhanced Galphaq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci U S A. 1998;95:10140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.D’Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB and Dorn GW 2nd. Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A. 1997;94:8121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hayakawa Y, Chandra M, Miao W, Shirani J, Brown JH, Dorn GW 2nd, Armstrong RCand Kitsis RN. Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Galpha(q) transgenic mice. Circulation. 2003;108:3036–41. [DOI] [PubMed] [Google Scholar]
- 121.Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, Aronow BJ, Lorenz JN and Dorn GW 2nd. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med. 2002;8:725–30. [DOI] [PubMed] [Google Scholar]
- 122.Tamrat R, Kang Y, Scherrer-Crosbie M, Levine LD, Arany Z and Lewey J. Women with peripartum cardiomyopathy have normal ejection fraction, but abnormal systolic strain, during pregnancy. ESC Heart Fail. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bajou K, Herkenne S, Thijssen VL, D’Amico S, Nguyen NQ, Bouche A, Tabruyn S, Srahna M, Carabin JY, Nivelles O, et al. PAI-1 mediates the antiangiogenic and profibrinolytic effects of 16K prolactin. Nat Med. 2014;20:741–7. [DOI] [PubMed] [Google Scholar]
- 124.Nonhoff J, Ricke-Hoch M, Mueller M, Stapel B, Pfeffer T, Kasten M, Scherr M, von Kaisenberg C, Bauersachs J, Haghikia A, et al. Serelaxin treatment promotes adaptive hypertrophy but does not prevent heart failure in experimental peripartum cardiomyopathy. Cardiovasc Res. 2017;113:598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pfeffer TJ, List M, Muller JH, Scherr M, Bauersachs J, Hilfiker-Kleiner D and Ricke-Hoch M. Perhexiline treatment improves toxic effects of beta-adrenergic receptor stimulation in experimental peripartum cardiomyopathy. ESC Heart Fail. 2021;8:3375–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Stapel B, Kohlhaas M, Ricke-Hoch M, Haghikia A, Erschow S, Knuuti J, Silvola JM, Roivainen A, Saraste A, Nickel AG, et al. Low STAT3 expression sensitizes to toxic effects of beta-adrenergic receptor stimulation in peripartum cardiomyopathy. Eur Heart J. 2017;38:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Halkein J, Tabruyn SP, Ricke-Hoch M, Haghikia A, Nguyen NQ, Scherr M, Castermans K, Malvaux L, Lambert V, Thiry M, et al. MicroRNA-146a is a therapeutic target and biomarker for peripartum cardiomyopathy. J Clin Invest. 2013;123:2143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sliwa K, Blauwet L, Tibazarwa K, Libhaber E, Smedema JP, Becker A, McMurray J, Yamac H, Labidi S, Struman I, et al. Evaluation of bromocriptine in the treatment of acute severe peripartum cardiomyopathy: a proof-of-concept pilot study. Circulation. 2010;121:1465–73. [DOI] [PubMed] [Google Scholar]
- 129.Yameogo N, Kagambega L, Seghda A, Owona A, Kabore O, Kabore E, Millogo G, Kologo K, Toguyeni B, Samadoulougoug A, et al. Bromocriptine in Management of Peripartum Cardiomyopathy: A Randomized Study on 96 Women in Burkina Faso. J Cardiol Clin Res. 2017;5:1098. [Google Scholar]
- 130.Hilfiker-Kleiner D, Haghikia A, Berliner D, Vogel-Claussen J, Schwab J, Franke A, Schwarzkopf M, Ehlermann P, Pfister R, Michels G, et al. Bromocriptine for the treatment of peripartum cardiomyopathy: a multicentre randomized study. Eur Heart J. 2017;38:2671–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–12. [DOI] [PubMed] [Google Scholar]
- 132.Handschin C and Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27:728–35. [DOI] [PubMed] [Google Scholar]
- 133.Roh JD, Yu A, Rana S, Shahul S, Castro C, Honigberg M, Ricke-Hoch M, Yeri A, Kitchen R, Hobson R, et al. Shared Senescence Pathophysiology in Preeclampsia and Peripartum Cardiomyopathy. Circulation. 2021;144:A12940. [Google Scholar]
- 134.Bello N, Hurtado Rendon IS and Arany Z. The relationship between Preeclampsia and Peripartum Cardiomyopathy: a Systematic Review and Meta-Analysis. J Am Coll Cardiol. 2013;62:1715–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shahul S, Medvedofsky D, Wenger JB, Nizamuddin J, Brown SM, Bajracharya S, Salahuddin S, Thadhani R, Mueller A, Tung A, et al. Circulating Antiangiogenic Factors and Myocardial Dysfunction in Hypertensive Disorders of Pregnancy. Hypertension. 2016;67:1273–80. [DOI] [PubMed] [Google Scholar]
- 136.Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F and Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. 106:1541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mazzarotto F, Tayal U, Buchan RJ, Midwinter W, Wilk A, Whiffin N, Govind R, Mazaika E, de Marvao A, Dawes TJW, et al. Reevaluating the Genetic Contribution of Monogenic Dilated Cardiomyopathy. Circulation. 2020;141:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Garcia-Pavia P, Kim Y, Restrepo-Cordoba MA, Lunde IG, Wakimoto H, Smith AM, Toepfer CN, Getz K, Gorham J, Patel P, et al. Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation. 2019;140:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gramlich M, Michely B, Krohne C, Heuser A, Erdmann B, Klaassen S, Hudson B, Magarin M, Kirchner F, Todiras M, et al. Stress-induced dilated cardiomyopathy in a knock-in mouse model mimicking human titin-based disease. J Mol Cell Cardiol. 2009;47:352–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E, Rackham OJ, van Heesch S, Pua CJ, Kui M, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2017;49:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Haggerty CM, Damrauer SM, Levin MG, Birtwell D, Carey DJ, Golden AM, Hartzel DN, Hu Y, Judy R, Kelly MA, et al. Genomics-First Evaluation of Heart Disease Associated With Titin-Truncating Variants. Circulation. 2019;140:42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.McAfee Q, Chen CY, Yang Y, Caporizzo MA, Morley M, Babu A, Jeong S, Brandimarto J, Bedi KC Jr., Flam E, et al. Truncated titin proteins in dilated cardiomyopathy. Sci Transl Med. 2021;13:eabd7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fomin A, Gartner A, Cyganek L, Tiburcy M, Tuleta I, Wellers L, Folsche L, Hobbach AJ, von Frieling-Salewsky M, Unger A, et al. Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci Transl Med. 2021;13:eabd3079. [DOI] [PubMed] [Google Scholar]
- 145.van Heesch S, Witte F, Schneider-Lunitz V, Schulz JF, Adami E, Faber AB, Kirchner M, Maatz H, Blachut S, Sandmann CL, et al. The Translational Landscape of the Human Heart. Cell. 2019;178:242–260 e29. [DOI] [PubMed] [Google Scholar]
- 146.Homma S, Iwasaki M, Shelton GD, Engvall E, Reed JC and Takayama S. BAG3 deficiency results in fulminant myopathy and early lethality. Am J Pathol. 2006;169:761–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Martin TG, Myers VD, Dubey P, Dubey S, Perez E, Moravec CS, Willis MS, Feldman AM and Kirk JA. Cardiomyocyte contractile impairment in heart failure results from reduced BAG3-mediated sarcomeric protein turnover. Nat Commun. 2021;12:2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dalkilic I, Schienda J, Thompson TG and Kunkel LM. Loss of FilaminC (FLNc) results in severe defects in myogenesis and myotube structure. Mol Cell Biol. 2006;26:6522–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vasioukhin V, Bowers E, Bauer C, Degenstein L and Fuchs E. Desmoplakin is essential in epidermal sheet formation. Nat Cell Biol. 2001;3:1076–85. [DOI] [PubMed] [Google Scholar]
- 150.Zhu RR, Chen Q, Liu ZB, Ruan HG, Wu QC and Zhou XL. Inhibition of the Notch1 pathway induces peripartum cardiomyopathy. J Cell Mol Med. 2020;24:7907–7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Otani K, Tokudome T, Kamiya CA, Mao Y, Nishimura H, Hasegawa T, Arai Y, Kaneko M, Shioi G, Ishida J, et al. Deficiency of Cardiac Natriuretic Peptide Signaling Promotes Peripartum Cardiomyopathy-Like Remodeling in the Mouse Heart. Circulation. 2020;141:571–588. [DOI] [PubMed] [Google Scholar]
- 152.Onusko E, McDermott MR, Robbins N, Liu G, Kranias EG, Rubinstein J and Koch SE. Probenecid treatment improves outcomes in a novel mouse model of peripartum cardiomyopathy. PLoS One. 2020;15:e0230386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Liu GS, Gardner G, Adly G, Jiang M, Cai WF, Lam CK, Alogaili F, Robbins N, Rubinstein J and Kranias EG. A novel human S10F-Hsp20 mutation induces lethal peripartum cardiomyopathy. J Cell Mol Med. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu GS, Zhu H, Cai WF, Wang X, Jiang M, Essandoh K, Vafiadaki E, Haghighi K, Lam CK, Gardner G, et al. Regulation of BECN1-mediated autophagy by HSPB6: Insights from a human HSPB6(S10F) mutant. Autophagy. 2018;14:80–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shimizu T, Taguchi A, Higashijima Y, Kanki Y, Nakaki R, Urade Y and Wada Y. Inhibition of cardiac PERK signaling promotes peripartum cardiac dysfunction. Sci Rep. 2021;11:18687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chung E, Yeung F and Leinwand LA. Akt and MAPK signaling mediate pregnancy-induced cardiac adaptation. J Appl Physiol. 2012;112:1564–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kouzu H, Tatekoshi Y, Chang HC, Shapiro JS, McGee WA, De Jesus A, Ben-Sahra I, Arany Z, Leor J, Chen C, et al. ZFP36L2 suppresses mTORc1 through a P53-dependent pathway to prevent peri-partum cardiomyopathy in mice. J Clin Invest. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ozcan U, Ozcan L, Yilmaz E, Duvel K, Sahin M, Manning BD and Hotamisligil GS. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Molecular cell. 2008;29:541–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Appenzeller-Herzog C and Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends in cell biology. 2012;22:274–82. [DOI] [PubMed] [Google Scholar]
- 160.Habashi JP, MacFarlane EG, Bagirzadeh R, Bowen C, Huso N, Chen Y, Bedja D, Creamer TJ, Rykiel G, Manning M, et al. Oxytocin antagonism prevents pregnancy-associated aortic dissection in a mouse model of Marfan syndrome. Sci Transl Med. 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zekavat SM, Chou EL, Zekavat M, Pampana A, Paruchuri K, Lino Cardenas CL, Koyama S, Ghazzawi Y, Kii E, Uddin MM, et al. Fibrillar Collagen Variants in Spontaneous Coronary Artery Dissection. JAMA Cardiol. 2022;7:396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bowen CJ, Calderon Giadrosic JF, Burger Z, Rykiel G, Davis EC, Helmers MR, Benke K, Gallo MacFarlane E and Dietz HC. Targetable cellular signaling events mediate vascular pathology in vascular Ehlers-Danlos syndrome. J Clin Invest. 2020;130:686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Isermann B, Sood R, Pawlinski R, Zogg M, Kalloway S, Degen JL, Mackman N and Weiler H. The thrombomodulin-protein C system is essential for the maintenance of pregnancy. Nat Med. 2003;9:331–7. [DOI] [PubMed] [Google Scholar]
- 164.Holers VM, Girardi G, Mo L, Guthridge JM, Molina H, Pierangeli SS, Espinola R, Xiaowei LE, Mao D, Vialpando CG, et al. Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J Exp Med. 2002;195:211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Blank M, Faden D, Tincani A, Kopolovic J, Goldberg I, Gilburd B, Allegri F, Balestrieri G, Valesini G and Shoenfeld Y. Immunization with anticardiolipin cofactor (beta-2-glycoprotein I) induces experimental antiphospholipid syndrome in naive mice. J Autoimmun. 1994;7:441–55. [DOI] [PubMed] [Google Scholar]
- 166.Salmon JE, Girardi G and Holers VM. Activation of complement mediates antiphospholipid antibody-induced pregnancy loss. Lupus. 2003;12:535–8. [DOI] [PubMed] [Google Scholar]
- 167.Chu H, Sacharidou A, Nguyen A, Li C, Chambliss KL, Salmon JE, Shen YM, Lo J, Leone GW, Herz J, et al. Protein Phosphatase 2A Activation Via ApoER2 in Trophoblasts Drives Preeclampsia in a Mouse Model of the Antiphospholipid Syndrome. Circ Res. 2021;129:735–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.McAnulty JH. Arrhythmias in pregnancy. Cardiol Clin. 2012;30:425–34. [DOI] [PubMed] [Google Scholar]
- 169.El Khoury N, Ross JL, Long V, Thibault S, Ethier N and Fiset C. Pregnancy and oestrogen regulate sinoatrial node calcium homeostasis and accelerate pacemaking. Cardiovasc Res. 2018;114:1605–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.El Khoury N, Mathieu S, Marger L, Ross J, El Gebeily G, Ethier N and Fiset C. Upregulation of the hyperpolarization-activated current increases pacemaker activity of the sinoatrial node and heart rate during pregnancy in mice. Circulation. 2013;127:2009–20. [DOI] [PubMed] [Google Scholar]
- 171.Yokoyama T, Kanda T, Suzuki T and Murata K. Enhancement of myocardial damage and alteration of lymphocyte subsets in murine model of postpartum myocarditis. Am J Cardiovasc Pathol. 1993;4:343–51. [PubMed] [Google Scholar]
- 172.Tan JS, Liu NN, Guo TT, Hu S and Hua L. Genetic predisposition to COVID-19 may increase the risk of hypertension disorders in pregnancy: A two-sample Mendelian randomization study. Pregnancy Hypertens. 2021;26:17–23. [DOI] [PubMed] [Google Scholar]
- 173.Feyen E, Ricke-Hoch M, Van Fraeyenhove J, Vermeulen Z, Scherr M, Dugaucquier L, Viereck J, Bruyns T, Thum T, Segers VFM, et al. ERBB4 and Multiple MicroRNAs That Target ERBB4 Participate in Pregnancy-Related Cardiomyopathy. Circulation Heart failure. 2021;14:e006898. [DOI] [PubMed] [Google Scholar]
- 174.Ricke-Hoch M, Bultmann I, Stapel B, Condorelli G, Rinas U, Sliwa K, Scherr M and Hilfiker-Kleiner D. Opposing roles of Akt and STAT3 in the protection of the maternal heart from peripartum stress. Cardiovascular research. 2014;101:587–96. [DOI] [PubMed] [Google Scholar]