

Abstract
To ascertain whether position 131 of a mesophilic protease, subtilisin BPN′, is a potential critical site for cold adaptation as screened by evolutionary engineering (S. Taguchi, A. Ozaki, and H. Momose, Appl. Environ. Microbiol. 64:492–495, 1998), a full set of subtilisin BPN′ mutants with mutations at position 131 was constructed by site-saturation mutagenesis. All mutated enzymes were measured for specific activity at 10°C by the quantitative titer microplate assay system using polyclonal antibody against subtilisin BPN′ and a synthetic chromogenic substrate. All the mutants exhibited proteolytic activities almost the same as or higher than that of the wild-type enzyme, suggesting that position 131 may be important for cold adaptation. In comparison with the wild type, purified mutants G131F, G131R, G131M, and G131W were found to acquire proteolytic activities (kcat/Km) at 10°C that were 150, 94, 84, and 50% higher, respectively. In particular, for the G131F mutant, temperature dependency in enzyme activity was shown by an increase in kcat and a decrease in Km. All of these amino acid substitution mutants, G131F, G131R, G131M, and G131W, acquired increased proteolytic activities at 10°C for three different synthetic peptide substrates but no increase in caseinolytic activity. Furthermore, they all conferred thermolability on the enzyme to differing extents in terms of the half-life of enzyme inactivation at 60°C. No significant correlation was found between the amino acids preferred for cold adaptation surveyed here and those present at position 131 of subtilisin of psychrophilic cells naturally occurring in cold environments. Based on these findings, position 131 is a contributor in artificial evolution for acquiring a cold-active character and may not be related to physiological requirements for subtilisin-producing cells living in cold environments. Therefore, saturation mutagenesis would be effective in achieving rapid improvement in protein properties via evolutionary engineering.
Attractive applications of cold-active enzymes in biotechnology would include food processing, additives in detergents (cold washing), biosynthetic processes with volatile intermediates, or environmental bioremediation. Extensive attempts to isolate various cold-active enzymes from naturally occurring psychrophilic organisms have been made by many groups (3, 5, 18). In contrast to this approach, we have been attempting to create cold-adapted forms from mesophilic enzymes by artificial evolution, called “evolutionary engineering,” based on a Darwinian sequential program of mutagenesis and selection (10, 24, 25, 28).
Although cold-active or cold-adapted enzymes can be defined from various standpoints, Gerday et al. (5) stated that psychrophilic or cold enzymes can work efficiently at low temperatures, meaning that they display a specific activity at low and moderate temperatures that is higher than that of their mesophilic counterparts. Here we want to state that, compared with mesophilic or wild-type enzymes, a cold-active enzyme can act at low temperatures independent of temperature range and a cold-adapted enzyme shows higher specific activity at low temperatures in a temperature-dependent manner. To date, the cold adaptation of subtilisin BPN′, a mesophilic and industrially useful alkaline serine protease, has been studied by us as a good model for understanding the molecular mechanism of cold adaptation based on abundant data for the structure-function relationship of this enzyme (26). However, the theoretical basis for designing the cold-adapted subtilisin BPN′ is very limited. We developed a screening program that consists of random mutagenesis, for obtaining proteases with enhanced activities at a low temperature, via multistep mutations with a combination of primary mutations causing activity loss and secondary mutations causing recovery of the activity. In fact, several artificial mutants with various types of cold-adapted characters were acquired by this experimental evolution strategy (10, 24, 25, 28). Of the cold-adapted mutant enzymes so far obtained by our evolutionary engineering program, m-63 is a triple-site mutant (V72I/A92T/G131D) with twice the activity of the wild-type enzyme at 10°C. Analysis of the individual contributions of the three mutations revealed that the V72I mutation contributed negatively to the activity but that the other two mutations, A92T and G131D, overcame the negative contribution to confer a 100% increase in activity. Therefore, we postulated that the two positions, 92 and 131, causing positive contributions might be critical sites for adaptation to cold. In our previous experiment, the same mutation, G131D, resulted in a cold-active mutant with the other two mutations (28). In this study, we first chose the residue at position 131 for mutagenesis.
Here we examined the effect of amino acid substitution, by PCR saturation mutagenesis, at position 131, which is possibly important for cold adaptation of subtilisin BPN′. Previously, using a polyclonal antibody against subtilisin BPN′, we established an assay system, termed ABEA (antibody-bound enzyme assay), allowing quantitative analysis of the specific activity of subtilisin BPN′ and its mutant enzymes (17). This assay system was applied to random screening of amino acid-substituted mutants, and four selected mutant subtilisins with much higher proteolytic activities, G131F, G131R, G131M, and G131W, have been characterized and discussed in terms of kinetic properties, substrate specificity, thermal stability, and correlation with amino acid substitution patterns at the same position of wild-type members of the subtilisin protease family (21).
MATERIALS AND METHODS
Materials, bacterial strains, and expression systems.
Polyclonal antibody against subtilisin BPN′ (kindly supplied by Nagase Biochemicals Co., Ltd.) was raised in a rabbit, and its reactivity with antigen was checked at the Medical Center of Takara Shuzo Co., Ltd., Shiga, Japan. Synthetic substrates, N-succinyl-l-Ala-l-Ala-l-Pro-l-Phe-p-nitroanilide (AAPF), N-succinyl-l-Ala-l-Ala-l-Pro-l-Leu-p-nitroanilide (AAPL), and N-succinyl-l-Ala-l-Ala-l-Val-l-Ala-p-nitroanilide (AAVA), were purchased from Sigma Co., Ltd. Polystyrene 96-well microtiter plates from Nunc Co. Ltd. (Immuno Plate MaxiSorp F96; Nunc A/S, Roskilde, Denmark) were used for immunoreaction between mutants of subtilisin BPN′ and antibody. All other chemicals were of reagent grade and were used without further purification. Escherichia coli JM109 (30) was used as the host strain for the screening of subtilisin mutants on proteolytic activity assay plates (2% skim milk, 1% lactose, 1% yeast extract, and 50 μg of ampicillin per ml) established by Tange et al. (28). The recombinant subtilisin gene on the plasmid pUC18 (30) was expressed under the original promoter of subtilisin and the lac promoter in E. coli. For secretory overproduction of the recombinant subtilisin, the host strain Bacillus subtilis UOT0999 lacking multiple protease genes was cultivated in liquid Luria-Bertani medium (20) containing 20 μg of tetracycline per ml.
Construction of plasmids containing mutant subtilisins.
For preparing a set of mutant subtilisins (except for G131D), five 5′-phosphorylated mutagenic primers were designed and synthesized as given in Table 1. The target mutation was introduced using the primer pairs MUT4 (Takara Shuzo) and each mutagenic primer described above (for the first PCR) and M13 primer RV (Takara Shuzo) and M13 primer M4 (Takara Shuzo) (for the first and second PCRs) via heteroduplex formation between the first two PCR products according to the detailed procedure developed by Ito et al. (9). PCR was carried out with a Gene Amp PCR 2400 system (Perkin-Elmer) using programs of 25 cycles of 94°C for 30 s (denaturation), 55°C for 2 min (annealing), and 72°C for 3 min (elongation) (for the first PCR) and 10 cycles under the same conditions as those for the first PCR (for the second PCR). The single-stranded region of the heteroduplex was filled in by the second PCR, followed by double digestion with EcoRI and HindIII. The double-stranded DNA fragment carrying the target mutation could, in principle, be selectively digested with both enzymes and subjected to cloning into the same restriction sites of the plasmids pUC18 and pHY300PLK, respectively. The mutation points were analyzed by dideoxynucleotide chain-termination sequencing using a BcaBEST kit (Takara Shuzo). Six sequencing primers were synthesized by the solid-phase phosphoamidite method with an Applied Biosystems 381A DNA synthesizer (23).
TABLE 1.
Oligonucleotides designed for site-specific saturation mutagenesis at position 131
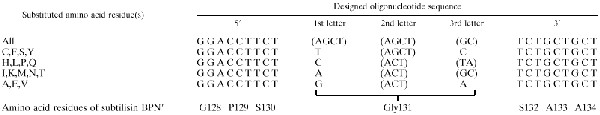 |
Plate assay of recombinant E. coli harboring subtilisin BPN′ secretion vector.
A mixture of the EcoRI-HindIII fragments including the mutagenized subtilisin gene was religated into the pUC18 plasmid to generate a mutant library. The change in proteolytic activity of mutant subtilisins was judged on the basis of the formation velocity of the cleared zone caused by proteolysis of the skim milk at the initial experimental stage, as described previously (24). For precise estimation of the catalytic properties of the mutant subtilisin, the DNA fragment including the subtilisin gene was subcloned into the EcoRI-HindIII sites of pHY300PLK (8), a shuttle expression vector between E. coli and B. subtilis, and the recombinant subtilisin was overproduced by B. subtilis UOT0999.
Assay system (ABEA) for enzyme activities of wild-type and mutant subtilisins.
The precise procedure was explained in detail in our previous paper (17).
Purification of recombinant enzymes.
A recombinant B. subtilis strain harboring the wild-type or mutated subtilisin gene (for G131F, G131R, G131M, and G131W) was cultivated in 100 ml of Luria-Bertani medium containing a final concentration of 20 μg of tetracycline per ml at 37°C for 24 h. Subtilisin excreted into the medium was recovered and purified by ammonium sulfate precipitation followed by sequential chromatographies, as described previously (24). The active protease fractions were detected by the cleared zone corresponding to caseinolytic activity on the plate containing skim milk. The purified sample was precipitated by adding a fourfold volume of acetone to the fraction containing subtilisin. The purity of the recovered samples was checked by sodium dodecyl sulfate-polyacrylamide (15%) gel electrophoresis according to the method of Laemmli (11).
Proteolytic activity assay of the purified enzymes.
Wild-type and mutant subtilisin activities were measured at various temperatures by monitoring the release of p-nitroaniline at 410 nm due to enzymatic hydrolysis of AAPF, AAPL, or AAVA (0.02 to 0.8 mM), as described previously (24). The apparent concentration of subtilisin was determined spectrophotometrically using an absorbance coefficient of E280 nm1% = 11.7 (15) and a molecular weight of 27,500 to permit calculation of kcat from the relationship kcat = Vmax/[enzyme]. The precise quantification of each purified active subtilisin was performed by active-site titration with the specific proteinaceous inhibitor Streptomyces subtilisin inhibitor (SSI) (14). The SSI concentration was determined spectrophotometrically at pH 7.0 using A276 (1 mg/ml) = 0.829 (28). The estimated value was used to correct the value of specific activity and the kinetic constant, kcat. The caseinolytic activity of each enzyme was measured by the previously described procedure (22). Briefly, 4 μg of each wild-type or mutant subtilisin BPN′ (145 pmol) was preincubated in 0.1 ml of 100 mM borate buffer (pH 9.5) at 37°C. After addition of 0.3 ml of 1.33% casein solution in the same buffer, the mixture was incubated for 10 min at 37°C, 0.4 ml of 0.44 M trichloroacetic acid was then added, and the system was allowed to stand for 20 min at 37°C. To 0.5 ml of the supernatant of the mixture, 2.5 ml of 0.44 M sodium carbonate and 0.5 ml of phenol reagent were added. After incubation for 20 min at 37°C, absorbance at 660 nm was measured.
Thermal stability of subtilisin.
A 1-ml aliquot of 1 μM purified subtilisin was incubated at 60°C, and 50 μl of each sample was taken at various times and immediately cooled on ice. The residual subtilisin activity was measured using AAPF as the substrate as described previously (28).
Molecular modeling.
The refined tertiary structure of subtilisin BPN′ (Protein Data Bank no. 2SIC) was used as a data source for computational analysis (27). In the topallh22X force field of CHARMM (1, 13), using the program X-PLOR 3.851 (2), all the hydrogen atoms were generated and added to the coordinates of subtilisin BPN′ from 2SIC, and the resultant wild-type structure was optimized by the energy minimization. Subsequently, structural models of the G131 mutants were constructed from the wild-type structure by using the mutation function provided in QUANTA97. Superposition of the structural models and calculations of the hydrogen bond positions were done by use of QUANTA97.
RESULTS
A full set of mutations at position 131 of subtilisin BPN′.
A complete set of mutations at position 131 of subtilisin BPN′ was constructed by PCR mutagenesis, as described in Materials and Methods, using a series of mutagenic primers listed in Table 1. At first, although a mutagenic primer designed for all amino acid substitutions was used, only two mutants, G131W and G131R, could be obtained. Next, the other four mutagenic primers, each of which exhaustively encodes three to five amino acids on average, were very efficient at generating all the mutants in a consistent frequency. Thus, all the mutants except for G131D (24) were isolated on the skim milk plates on the basis of detection of cleared zones appearing around E. coli transformant colonies, and their base substitutions at position 131 were analyzed by sequencing.
Comparison of specific activity among mutant subtilisins.
For enzymatic comparison, the mutant subtilisin BPN′ genes were subcloned into the B. subtilis host-vector system enabling the secretory production of subtilisins. In the protein secretion system, no significant variation in expression level (over approximately 3 μM) of recombinant subtilisins was observed between 19 mutants and the wild type under the culture condition used here, based on a densitometric scanning of the Coomassie brilliant blue-stained protein band corresponding to subtilisin BPN′ in the extracellular fraction (data not shown). Specific activity toward an authentic substrate for many subtilisin-type proteases, suc-AAPF-pNA, was measured at 10°C by applying the culture supernatant samples of wild-type and mutant subtilisins to the ABEA system. The mutant subtilisins could be ranked by their specific proteolytic activities as shown in Table 2. We succeeded in acquiring many mutant subtilisins with higher specific activities at 10°C, in particular G131F, G131R, G131M, and G131W, which exhibited activities increased over 60% from that of wild-type subtilisin. Surprisingly, all of the mutants were cold-active enzymes which had activities almost the same as or higher than that of the wild type. With respect to correlation between amino acid properties and specific activities, aromatic (F, W, and Y), basic (R, H, and K), or sulfur-containing (M) amino acids are effective for activity elevation, whereas branched aliphatic (L, V, and I) amino acids are not suitable for acquiring a cold-active character.
TABLE 2.
Specific activity in each G131 mutant subtilisin BPN′ measured by ABEAa
| Amino acid | Sp act | Character of amino acid |
|---|---|---|
| Phe | 2.6 | Aromatic |
| Arg | 2.1 | Basic |
| Met | 1.8 | S atom containing |
| Trp | 1.6 | Aromatic |
| Tyr | 1.5 | Aromatic |
| His | 1.5 | Basic |
| Ser | 1.5 | Polar |
| Asn | 1.4 | Amide containing |
| Asp | 1.4 | Acidic |
| Lys | 1.4 | Basic |
| Ala | 1.4 | Aliphatic |
| Thr | 1.3 | Polar |
| Gln | 1.3 | Amide containing |
| Glu | 1.3 | Acidic |
| Pro | 1.2 | Aliphatic |
| Cys | 1.2 | S atom containing |
| Ile | 1.1 | β-Branched aliphatic |
| Val | 1.1 | β-Branched aliphatic |
| Leu | 1.1 | Aliphatic |
| Gly (wild type) | 1.0 | Aliphatic and no side chain |
Relative activity was measured by the ABEA system described in detail in reference 17 using culture supernatant samples of subtilisins.
Purification and characterization of four G131 mutants with higher activities.
The four highly active mutant subtilisins, G131F, G131R, G131M, and G131W, were purified to electrophoretic homogeneity by two sequential steps of chromatography, i.e., ion exchange on DEAE and carboxymethyl celluloses (data not shown). Comparison of hydrolytic activity at 10°C was performed for the wild type and the four mutant enzymes based on the kcat/Km value as shown in Table 3. The precisely estimated activities of the purified enzymes were in good agreement with those obtained by the ABEA system (Table 2). Table 4 shows the temperature dependence of G131F subtilisin activity at three temperatures based on kinetic parameters using AAPF and AAPL (for 10°C alone) as synthetic substrates. The hydrolytic activity at 10°C of G131F subtilisin relative to that of the wild type exhibited a 150% increase for AAPF and a 190% increase for AAPL, when the temperature was shifted down from 50 to 10°C. In this case, temperature-dependent cold adaptation was achieved by both the increase in kcat and the marked decrease in Km value.
TABLE 3.
Kinetic parameters of purified wild-type and mutant subtilisins for hydrolysis of AAPF at 10°Ca
| Sample | kcat (s−1) | Km (μM) | kcat/Km (105 s−1 M−1) | Value relative to wild type |
|---|---|---|---|---|
| Wild type | 29.3 (±1) | 96.1 (±3) | 3.0 | 1.0 |
| G131D | 33.1 (±3) | 76.5 (±5) | 4.4 | 1.4 |
| G131W | 34.5 (±2) | 75.4 (±0) | 4.5 | 1.5 |
| G131M | 27.5 (±3) | 48.4 (±1) | 5.7 | 1.8 |
| G131R | 31.4 (±2) | 53.7 (±0) | 5.9 | 1.9 |
| G131F | 41.9 (±2) | 54.6 (±2) | 7.6 | 2.5 |
Enzyme activity was assayed using acetone-precipitated sample by the method of Wells et al. (29) with a slight modification. AAPF (Sigma) was used as the substrate. Estimation of enzyme concentration was carried out by active-site titration with SSI.
TABLE 4.
Kinetic parameters of purified wild-type and G131F mutant for hydrolysis of AAPF and AAPL at various temperaturesa
| Temp (°C) | Substrate | Sample | kcat (s−1) | Km (μM) | kcat/Km (105 s−1 M−1) | Value relative to wild type |
|---|---|---|---|---|---|---|
| 50 | AAPF | Wild type | 164.9 (±14) | 242.2 (±7) | 6.8 | 1.0 |
| G131F | 167.8 (±15) | 145.2 (±5) | 11.6 | 1.7 | ||
| 37 | AAPF | Wild type | 73.8 (±1) | 184.6 (±3) | 4.0 | 1.0 |
| G131F | 93.4 (±2) | 107.3 (±2) | 8.7 | 2.2 | ||
| 10 | AAPF | Wild type | 29.3 (±1) | 96.1 (±3) | 3.0 | 1.0 |
| G131F | 41.9 (±2) | 54.6 (±2) | 7.6 | 2.5 | ||
| AAPL | Wild type | 38.9 (±1) | 254.9 (±8) | 1.6 | 1.0 | |
| G131F | 49.8 (±1) | 109.3 (±1) | 4.6 | 2.9 |
Enzyme activity was assayed using acetone-precipitated sample by the methods of Wells et al. (29) with a slight modification. AAPF and AAPL (Sigma) were used as the substrates. Estimation of enzyme concentration was carried out by active-site titration with SSI.
In terms of substrate specificity, for G131F, G131R, G131M, and G131W mutants, Leu was the most preferred P1 residue, as shown in Fig. 1. However, enhancement in caseinolytic activity was not achieved by these mutations. Among the four mutants, the G131F mutant exhibited the highest enzyme activity toward all the substrates tested.
FIG. 1.

Comparison of relative substrate specificities of G131 mutant subtilisins and wild-type enzyme. Changes in substrate specificity (in the value of kcat/Km at 10°C) of each mutant are illustrated graphically to compare that of the wild type with those of four substrates, AAPF, AAPL, AAVA, and casein.
Moreover, we examined the thermal stability of mutant subtilisins along with the wild-type subtilisin in terms of autolysis. When the rate of thermally induced inactivation was measured at 60°C in the presence of Ca2+, the half-lives of enzyme inactivation were 25 min for G131F, 15 min for G131R, 12 min for G131M and G131W, and 65 min for the wild type, as presented in Fig. 2.
FIG. 2.

Thermal stability of the wild-type and mutant subtilisins. The residual enzyme activity after exposure to 60°C for various time intervals was assayed at 25°C by adding purified samples to 100 mM Tris-HCl buffer (pH 9.6) containing 0.1 mM AAPF and 2 mM CaCl2.
DISCUSSION
Even though there are a large number of data available for protease subtilisins, with over 450 site-directed mutants constructed for many purposes (21), it is not easy to achieve the alteration of mesophilic subtilisin to the cold-adapted form by rational-design approaches. The first use of random mutagenesis to improve the activity of a mesophilic enzyme, subtilisin BPN′, at low temperatures was recently performed by us (28). To date, seven mutants leading to the cold adaptation of subtilisin BPN′ were derived by our evolutionary engineering program based on intragenic suppression-type mutation (28). It appears that most of the activity-increase and activity-decrease mutations so far obtained (actual positions, 72, 84, 88, 92, 98, 131, and 197) fall in the N-terminal half of the subtilisin's mature portion. This suggests that the region, in particular that ranging from positions 70 to 140, of the protein might be a hot area for achieving the cold adaptation of the entire subtilisin consisting of 275 amino acid residues. Not many amino acids can be obtained by a single nucleotide substitution, i.e., Gly(GGT) can lead to Asp, Val, Ala, Cys, Arg, and Ser. Such nonconservative substitutions, obtainable solely by multiple base changes in a single codon, would be extremely rare in point-mutation variation or in natural evolution. In this context, a complete amino acid substitution at positions which would possibly be critical sites leading to the desired properties sought by evolutionary engineering would be very useful. In fact, highly cold-active mutants, G131F, G131R, G131M, and G131W, could be obtained by site-specific amino acid substitutions at position 131 of the parent subtilisin mutant, m-63.
As presented in Fig. 3, position 131 is located in the N-terminal region of the α-helix, close to, but on the reverse side of, the substrate binding area. Previously, the same mutation, Gly→Asp, at this position had been shown to be a suppressor that compensated for the defect of Ca2+ binding-mediated stabilization caused by mutation of D197N (19, 28). From the 1.8-Å refined tertiary structure, the α-carbon chain of Gly131 is oriented to the inside of the subtilisin BPN′ molecule. Therefore, it seems likely that amino acid substitution at this position, in particular by aromatic amino acids, would have a noticeable effect on a neighboring loop structure bearing aromatic amino acids Tyr167 and Tyr171. Computational modeling was conducted to predict the aromatic interactions of Phe, Trp, and Tyr at position 131 with Tyr167 and Tyr171; thus, the Tyr side chain at position 167 would be moved at the interface with the neighboring loop structure by aromatic interaction between Phe at position 131 and Tyr at position 171. Subtilisin BPN′ has two conspicuous pockets at the S1 and S4 sites. The S1 substrate-binding pocket has broad specificity and contains a large hydrophobic substrate-binding cleft that is made up of the main chain segment Ser125(S1)-Leu126(S2)-Gly127(S3) as a part of backbone segments (29). Because the loop containing Tyr167 and Tyr171 is located relatively close to the substrate-binding segment (residues 125 to 127), a subtle structural change in this loop may directly influence the structural coordination of this segment, Ser125(S1)-Leu126(S2)-Gly127(S3), further affecting the mobility of the adjacent active-site structure. Consequently, the Phe side chain conformation at position 131 may be the most finely tuned in bulkiness to confer highly enhanced catalytic efficiency at a low temperature. It may be more appropriate to examine flexibility in specific regions, such as position 131 and its related position(s), of an enzyme that govern the energetics of the conformational changes necessary for binding and catalysis.
FIG. 3.

Computational modeling of geometric coordination among the catalytic triad, the substrate-binding segment, a loop structure bearing an aromatic-aromatic interaction of 167Tyr and 171Tyr, and position 131 on the α-helix structure for amino acid substitutions. The catalytic site cleft formed by a triad of residues, Ser221, His64, and Asp32, and the substrate-binding segment, Ser125-Leu126-Gly127, are located where they would be structurally affected via movement of the neighboring loop structure triggered by amino acid substitutions at position 131 (in the case of Phe) on the α-helix.
In general, it has been shown that cold-active enzymes originating from psychrophilic microorganisms display a high thermosensitivity compared to their mesophilic counterparts (5). As presented in Fig. 2, the parameter often used to measure subtilisin stability is the rate of irreversible inactivation and autolysis at elevated temperature (16, 29). For the use of washing or bioremediation in cold areas, a cold-active and stable enzyme would be desirable. In this sense, among G131 derivatives, the G131F mutant would be the most powerful target enzyme, which should possess not only high activity but also stability in such cold environments. Giver et al. (6) have succeeded in acquiring an enzyme with significantly increased thermal stability by directed evolution without cost to its activity at lower temperatures. Considering this finding, it may be possible to create much more thermotolerant cold-active mutants through further artificial evolution using the G131F mutant as a starting molecule.
When attention is given to position 131 in the natural subtilisin protease family (members of which are generally termed subtilases), three major amino acids, Asp (30%), Ser (25%), and Gly (15%), are present at this position in this order (occupation frequency) among 125 subtilases (21). No significant correlation exists between the amino acid residue at this position and the adaptation temperature in the environment where subtilisin-producing organisms occur, i.e., psychrophilic subtilisin S41 possesses Gly (4) and its thermophilic counterpart possesses Phe (12) at position 131. From these findings, it can be concluded that position 131 is a critical site in artificial evolution for cold adaptation but may not be related to physiological requirements for subtilisin-producing cells living in cold environments.
ACKNOWLEDGMENTS
We thank Y. Miyota, H. Ohtaki, and N. Hino for useful discussions and excellent assistance. We also thank T. Nonaka, Nagaoka University, for his graphic design of Fig. 3.
This study was supported in part by Grants-in-Aid for Scientific Research (09760103 to S.T. and 10660101 to H.M.) from the Ministry of Education, Science, Sports and Culture of Japan and a grant (to S.T.) from the Nissan Science Foundation (Tokyo).
REFERENCES
- 1.Brooks B, Bruccoleri R, Olafson B, States D, Swarninathan S, Karplus A. A program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 2.Brunger A T. X-PLOR: a system for X-ray crystallography and NMR, edition 3.1. New Haven, Conn: Yale University Press; 1983. [Google Scholar]
- 3.Choo D-W, Kurihara T, Suzuki T, Soda K, Esaki N. A cold-adapted lipase of an Alaskan psychrotroph, Pseudomonas sp. strain B11-1: gene cloning and enzyme purification and characterization. Appl Environ Microbiol. 1998;64:486–491. doi: 10.1128/aem.64.2.486-491.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davail S, Feller G, Narinx E, Gerday C. Cold adaptation of proteins: purification, characterization, and sequence of the heat-labile subtilisin from the antarctic psychrophile Bacillus TA41. J Biol Chem. 1994;269:17448–17453. [PubMed] [Google Scholar]
- 5.Gerday C, Aittaleb M, Arpigny J L, Baise E, Chessa J-P, Garsoux G, Petrescu I, Feller G. Psychrophilic enzymes: a thermodynamic challenge. Biochim Biophys Acta. 1997;1342:119–131. doi: 10.1016/s0167-4838(97)00093-9. [DOI] [PubMed] [Google Scholar]
- 6.Giver L, Gershenson A, Freskgard P-O, Arnold F H. Directed evolution of a thermostable esterase. Proc Natl Acad Sci USA. 1998;95:12809–12813. doi: 10.1073/pnas.95.22.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feller G, Narinx E, Arpigny J L, Aittaleb M, Baise E, Genicot S, Gerday C. Enzymes from psychrophilic organisms. FEMS Microbiol Rev. 1996;18:189–202. [Google Scholar]
- 8.Ishiwa H, Shibahara H. New shuttle vectors for Escherichia coli and Bacillus subtilis. II. Plasmid pHY300PLK, a multi purpose cloning vector with a polylinker, derived from pHY460. Jpn J Genet. 1985;60:235–243. [Google Scholar]
- 9.Ito W, Ishiguro H, Kurosawa Y. A general method for introducing a series of mutants into cloned DNA using the polymerase chain reaction. Gene. 1991;102:67–70. doi: 10.1016/0378-1119(91)90539-n. [DOI] [PubMed] [Google Scholar]
- 10.Kano H, Taguchi S, Momose H. Cold adaptation of a mesophilic serine protease, subtilisin, by in vitro random mutagenesis. Appl Microbiol Biotechnol. 1997;47:46–51. doi: 10.1007/s002530050886. [DOI] [PubMed] [Google Scholar]
- 11.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 12.Lee J K, Kim Y O, Kim H K, Park Y S, Oh T K. Purification and characterization of a thermostable alkaline protease from Thermoactinomyces sp. E79 and the DNA sequence of the encoding gene. Biosci Biotechnol Biochem. 1996;60:840–846. doi: 10.1271/bbb.60.840. [DOI] [PubMed] [Google Scholar]
- 13.Mackerell A D, Jr, Bashford D, Bellott M, Dunbrack R L, Field M J, Fischer S, Gao J, Guo H, Ha S, Joseph D, Kuchnir L, Kuczera K, Lau F T K, Mattos C, Michnick S. Self-consistent parameterization of biomolecules for molecular modeling and condensed phase simulations. FASEB J. 1992;6:A143. [Google Scholar]
- 14.Masuda-Momma K, Shimakawa T, Inouye K, Hiromi K, Kojima S, Kumagai I, Miura K, Tonomura B. Identification of amino acid residues responsible for the changes of absorption and fluorescence spectra on the binding of subtilisin BPN′ and Streptomyces subtilisin inhibitor. J Biochem. 1993;114:906–911. doi: 10.1093/oxfordjournals.jbchem.a124275. [DOI] [PubMed] [Google Scholar]
- 15.Matsubara H, Kasper C B, Brown D M, Smith E L. Subtilisin BPN′. I. Physical properties and amino acid composition. J Biol Chem. 1965;240:1125–1130. [PubMed] [Google Scholar]
- 16.Mitchinson C, Wells J A. Protein engineering of disulfide bonds in subtilisin BPN′. Biochemistry. 1989;28:4807–4815. doi: 10.1021/bi00437a043. [DOI] [PubMed] [Google Scholar]
- 17.Miyota Y, Komada S, Momose H, Taguchi S. Quantitative assay system for specific enzyme activity using antibody: the case of protease, subtilisin BPN′. J Biotechnol. 1998;66:157–163. doi: 10.1016/s0168-1656(98)00148-5. [DOI] [PubMed] [Google Scholar]
- 18.Morita Y, Nakamura T, Hasan Q, Murakami Y, Yokoyama K, Tamiya E. Cold-active enzymes from cold-adapted bacteria. J Am Oil Chem Soc. 1997;74:441–444. [Google Scholar]
- 19.Pantoliano M W, Whitlow M, Wood J F, Rollence M L, Finzek B C, Gilliland G L, Poulos T L, Bryan P N. The engineering of binding affinity at metal ion binding sites for the stabilization of proteins: subtilisin as a test case. Biochemistry. 1988;27:8311–8317. doi: 10.1021/bi00422a004. [DOI] [PubMed] [Google Scholar]
- 20.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 21.Siezen R J, Leunissen J A M. The superfamily of subtilisin-like proteases. Protein Sci. 1997;6:501–523. doi: 10.1002/pro.5560060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taguchi S, Kumagai I, Nakayama J, Suzuki A, Miura K. Efficient extracellular expression of a foreign protein in Streptomyces using secretory protease inhibitor (SSI) gene fusions. Bio/Technology. 1989;7:1063–1066. [Google Scholar]
- 23.Taguchi S, Maeno M, Momose H. Extracellular production system of heterologous peptide driven by a secretory protease inhibitor of Streptomyces. Appl Microbiol Biotechnol. 1992;36:749–753. doi: 10.1007/BF00172187. [DOI] [PubMed] [Google Scholar]
- 24.Taguchi S, Ozaki A, Momose H. Engineering of a cold-adapted protease by sequential random mutagenesis and a screening system. Appl Environ Microbiol. 1998;64:492–495. doi: 10.1128/aem.64.2.492-495.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taguchi S, Ozaki A, Nonaka T, Mitsui Y, Momose H. A cold-adapted protease engineered by an experimental evolution system. J Biochem. 1999;126:689–693. doi: 10.1093/oxfordjournals.jbchem.a022504. [DOI] [PubMed] [Google Scholar]
- 26.Takagi H. Protein engineering on subtilisin. Int J Biochem. 1993;25:307–312. doi: 10.1016/0020-711x(93)90617-n. [DOI] [PubMed] [Google Scholar]
- 27.Takeuchi Y, Satow Y, Nakamura K T, Mitsui Y. Refined crystal structure of the complex of subtilisin BPN′ and Streptomyces subtilisin inhibitor at 1.8 angstroms resolution. J Mol Biol. 1991;221:309–325. [PubMed] [Google Scholar]
- 28.Tange T, Taguchi S, Kojima S, Miura K, Momose H. Improvement of a useful enzyme (subtilisin BPN′) by an experimental evolution system. Appl Microbiol Biotechnol. 1994;41:239–244. doi: 10.1007/BF00186966. [DOI] [PubMed] [Google Scholar]
- 29.Wells J A, Powers D B, Bott R R, Graycar T P, Estel D A. Designing substrate specificity by protein engineering of electrostatic interactions. Proc Natl Acad Sci USA. 1987;84:1219–1223. doi: 10.1073/pnas.84.5.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]