

Abstract
Studying protein complexes in vitro requires the production of a relatively pure sample that maintains the full complement, native organization, and function of that complex. This can be particularly challenging to achieve for large, multi-component, membrane embedded complexes using the traditional recombinant expression and reconstitution methodologies. However, using affinity capture from native cells, suitable whole endogenous protein complexes can be isolated. Here we present a protocol for the affinity isolation of baker’s yeast (S. cerevisiae) nuclear pore complexes, which are ~50 MDa assemblies made up of 552 distinct proteins and embedded in a double-membraned nuclear envelope. Producing this sample allowed us for the first time to perform analyses to characterize the mass, stoichiometry, morphology, and connectivity of this complex and to obtain its integrative structure with ~9 Å precision. We believe this methodology can be applied to other challenging protein complexes to produce similar results.
Keywords: Affinity capture, Native Isolation, Nuclear pore complex, Endogenous macro-molecular assembly, Structural and functional analyses, Electron microscopy, Baker’s yeast, S. cerevisiae
1. Introduction
Protein macromolecular assemblies are the workhorses of living organism; thus, deciphering their function and how they perform is crucial to understanding cellular processes. A relatively pure sample of a protein complex must be obtained to perform any in vitro biochemical, biophysical, structural, or functional studies. Two main methodologies exist to produce such a sample. One is a “bottom up” approach, namely, to overexpress each of the protein complex components in a host organism, purify them and reconstitute the assembly. The other “top down” approach is to isolate an endogenous protein or protein complex from its native organism. Both methodologies have advantages and disadvantages, depending on the specific protein complex to be produced. However, in the case of large and multi-component, or membrane embedded assemblies, overexpression in a host organism and subsequent purification and reconstitution can be extremely challenging, whereas affinity isolation from the native environment offers many advantages, both in terms of feasibility and functional quality of the sample.
The concept behind affinity isolation is quite simple – to isolate a protein complex from a cell, one of the proteins in the complex is tagged with an affinity tag (this protein is termed a “handle”), which can tightly bind to an antibody (or another binding partner). Tagging might not be necessary if there is a good (tightly binding and specific) antibody to the protein of interest itself. Cells are grown, harvested and lysed, and the clarified lysate is incubated with an insoluble matrix (e.g., agarose beads, magnetic beads) coated with the antibody. The “handle” protein binds to the antibody via the affinity tag and if the associated complex is sufficiently stable, it remains attached to the “handle” protein and thereby immobilized on the matrix. The remaining lysate is discarded, and the protein complex is eluted (natively or by denaturation). Now the protein complex can be analyzed in any desired fashion – denatured eluates are usually characterized by SDS-PAGE followed by mass spectrometry (MS), while native assemblies can be used for structural (e.g., cryo-electron microscopy (cryo-EM), cross-linking with mass spectrometric readout (CX-MS)), proteomic (e.g., quantitative MS, native MS) and functional studies (1-7).
Despite the simplicity of the overall concept behind affinity isolation, its successful application requires careful optimization of all the parameters for the protein complex in question. These parameters include performing an efficient lysis, choosing the best antibody/tag pair, choosing the correct “handle”, finding the best buffer composition and optimizing the elution. Our group has pioneered the development of methodologies for optimizing the outcomes of affinity isolations for a large variety of protein complexes in many different organisms (3,8-13). Our approach is based on several principles (Fig. 1). First, cells are lysed by flash-freezing in liquid nitrogen followed by cryo-milling to generate micron-scale cell powder. This approach, essentially keeping the lysis stage in an extremely preservative solid phase, enables excellent preservation of native protein complexes and highly efficient cell breakage leading to full access to these complexes, while minimizing proteolytic degradation (11,14,15). In addition, cell powder can be stored almost indefinitely at −80°C and portions of the milled powder can be weighed from a larger stock depending on the scale of the experiment, allowing flexibility and multiplexing of the experimental design – including making exploration of multiple subsequent buffers straightforward (below) (Fig. 1a). Next, the antibody/tag pair used for affinity isolation must be optimized for the specific complex. Important parameters include binding specificity, efficiency and speed, as well as optimal tag placement on the “handle” in terms of steric hindrance and accessibility to the antibody (13,14). The affinity isolation begins by resuspending frozen cell powder in a buffer (Fig. 1b). Once the powder is in contact with the buffer and so transitioned to the liquid phase in a (by definition) non-native environment, all protein complexes can in principle begin to destabilize and dissociate at different rates, depending on temperature, the strength of their interaction, their environmental conditions, etc. The goal is to preserve the non-covalent interactions of the “handle” with the other proteins in the complex for as long as possible by fast and efficient re-suspension into buffer conditions that have been optimized to be the most stabilizing. The physical characteristics of the resuspension process, such as buffer-to-powder ratio, temperature, and resuspension mode (vortex, shaking, brief sonication, etc.) must be empirically optimized for the specific complex. Buffer composition is crucial for maintaining complex stability throughout the affinity isolation; therefore, many conditions need to be tested to find the optimal pH and buffer type, overall ionic strength, salt type(s) and concentration, detergent type(s) and concentration, and other additives as needed. We developed a method to perform high throughput screening to find the best buffer formulations for a specific affinity purification (10). The resuspended lysate is clarified by centrifugation and/or filtration to eliminate insoluble cell debris such as cell wall fragments, followed by incubation with antibody conjugated paramagnetic beads to achieve immobilization of the complex on the beads via binding between the tag on the ”handle” and the antibody on the matrix (Fig. 1b). This process should be quick and efficient, without introducing substantial non-specific binding to the complex or to the beads, emphasizing the need to use a high affinity and specificity antibody/tag pair, beads with extremely low nonspecific binding, and appropriate buffer composition. After immobilization is complete, the lysate is discarded, and the beads are washed a few times to ensure all non-specifically bound components are removed (Fig. 1b). The washing buffer composition as well as number and mode of washing steps should also be empirically optimized to maximize removal of non-specific binders while minimizing complex destabilization. Subsequently, the complex can be eluted from the beads, either by native elution in which the complex structure and activity are preserved, or by denaturation (Fig. 1b). If native elution is required, the elution method also needs to be optimized to be quick and efficient, to preserve the complex intact, and to not introduce non-specific binding. Two options can be explored: (i) proteolytic removal via a specific protease (where a protease cleavage target sequence should be inserted between the tag and the “handle” protein) or (ii) elution by competitive binding (e.g., anti-FLAG antibody and a FLAG tag to be eluted with FLAG peptide (12,14)). The eluted complex can now be analyzed by SDS-PAGE and characterized further using any number of techniques mentioned above.
Fig. 1. Affinity isolation methodology.

An outline of a general affinity isolation protocol, which can be optimized for various protein complexes from many organisms, as described in this work. (a) Cell lysis using flash freezing and cryo-milling to obtain cell powder which can be stored at −80°C and used as needed for any scale experiment. (b) Affinity capture by resuspending cell powder in an optimized affinity capture buffer and using the clarified (centrifuged and/or filtered) lysate for incubation with antibody-coupled magnetic beads to capture the complex on the beads followed by discarding the remaining supernatant and washing steps. Complex elution is accomplished by denaturation or by native elution (proteolytic removal or competitive binding) followed by separation from the beads. Eluted complex can be analyzed by SDS-PAGE and other downstream analyses mentioned in the text.
Here, we present a protocol for affinity isolation of endogenous whole nuclear pore complexes (NPCs) from S. cerevisiae. The NPC is the sole mediator of all nucleocytoplasmic transport in eukaryotic cells and is involved in many crucial nuclear processes (16). It is a large macromolecular assembly (~50 MDa in yeast) composed of 552 distinct proteins (called nucleoporins or Nups) and embedded in the nuclear envelope which is a double-membraned barrier that surrounds the nucleus (1,17). The NPC is a cylindrical assembly made up of 8 spokes arranged around a C-8 symmetry axis perpendicular to the nuclear envelope. The spokes form concentric rings around the same C-8 axis – an inner ring and two outer rings, nuclear and cytoplasmic, in turn connected to the nuclear basket and the cytoplasmic mRNA export complex, respectively (see Fig. 4 in (1)). Recently, our group was able to determine a structure for the baker’s yeast NPC. We analyzed samples of endogenous whole NPCs prepared using the protocol presented here to determine a cryo-electron tomography (cryo-ET) map of the isolated NPC at ~28 Å average resolution, use mass spectrometric methods to determine its mass and stoichiometry, and subject the isolated NPCs to CX-MS, which provided the distance restraints within and between Nups (1). All these restraints were used to integratively calculate a structure of the yeast NPC at unprecedented detail, allowing us to draw several paradigm-shifting conclusions as to how the NPC might accomplish its functions and why it is built the way it is.
The case of the NPC is a prime example of how the ability to produce an optimized affinity-captured endogenous sample enabled significant progress in the field. Prior research of the NPC focused primarily on in-vivo or in-situ studies (18-21) or studies of isolated or endogenously overexpressed, reconstituted sub-complexes (22,23). While these are extremely valuable, a detailed picture of the complete assembly was needed to put all the data in the correct context(18,24); thus, although isolation of any macromolecular complex likely alters it to some degree, the high-resolution data obtained is nonetheless invaluable for understanding its structure and functionalities in situ. This was enabled by producing a sufficiently pure sample of whole NPCs and analyzing it using the methods described above and in (1). Using this approach on other large macro-molecular complexes may lead to similar advances.
The specific parameters of the protocol described here were optimized using the methodologies described above (3,8-10,14,15) following extensive trial and error efforts to produce the best possible sample. As a tag we used Protein A (PrA), which is a 42 kDa surface protein originally found in the cell wall of the bacteria Staphylococcus aureus. The IgG-binding domains of PrA are widely used as affinity tags since they bind to IgG via the constant (Fc) region (25,26) and therefore do not require an antigen-specific antibody for affinity capture. We used rabbit polyclonal IgG for affinity isolation of proteins and protein complexes tagged with the PrA affinity tag. Our “handle” protein is predominantly Mlp1, a nuclear basket component (1), peripherally situated at the nuclear side of the NPC which allows good steric access of the antibody to the tag for efficient binding. We explored other peripherally situated Nups as “handles” and found Nup82 (cytoplasmic mRNA export platform component (1)) and Nup84 (a component of the nuclear and cytoplasmic outer rings (1)) to be equally good. We needed intact NPCs for the downstream analyses, therefore we chose to use protease cleavage as our native elution method. PreScission Protease (PPX), a commercially available version of human rhinovirus (HRV) 3C protease was selected and its cleavage target sequence (LEVLFQ/GP) was inserted between our “handle” protein (Mlp1, Nup84, or Nup82) and the PrA tag. We routinely use Dynabeads M-270 Epoxy magnetic beads from Thermo Fisher Scientific conjugated to the antibody of choice (in our case, rabbit polyclonal IgG) for our affinity isolations (bead conjugation protocol is included here). After significant empirical exploration, we found that an optimal buffer formulation for the stabilization of the NPCs was 20 mM HEPES pH 7.4, 50 mM potassium acetate (KOAc), 20 mM NaCl, 2 mM MgCl2, 0.5% Triton X-100, 0.1% Tween-20, 1 mM DTT, 10% glycerol. The main buffer species is HEPES at pH 7.4 which was chosen after unsuccessfully trying various other options, such as Bis-Tris at pH 6.5 and sodium phosphate at pH 8. Additives KOAc, NaCl, MgCl2, and DTT were found to contribute to the stability of the intact complex (increasing the ratio of intact NPCs to dissociated subcomplexes). Many different permutations of various detergents were explored but we found that a combination of 0.5% Triton X-100 and 0.1% Tween-20 enabled efficient extraction of NPCs from the surrounding nuclear envelope (1). Even with this treatment, there are substantial remnants of the native pore membrane present in the isolated NPCs, as shown by cryo-ET analyses (1). Once the NPCs are extracted from the membrane, we continue the affinity isolation without Triton X-100, since its prolonged presence is not necessary. The addition of 10% glycerol significantly improved the yield of the affinity isolation, indicated by the ratio between the amount of the complex successfully cleaved by the protease and found in the eluate and amount of the complex successfully cleaved by the protease, but remained stuck to the beads probably via non-specific interactions with the bead surface (see Note 14). The NPC is large, flexible, and relatively fragile without the support of the nuclear envelope, therefore, once extracted it is vulnerable to harsh mechanical stresses. Consequently, our protocol employs gentle handling such as low speed centrifugation followed by filtration to eliminate insoluble cell debris from the lysate as well as a single wash step after the immobilization on the beads to remove the non-specifically bound components. We found that the optimal resuspension parameters to preserve intact NPCs are working with 1.0-1.5 g cell powder aliquots weighed out in 50 mL tubes and resuspended by vortexing in 9.0-13.5 mL of buffer (buffer-to-powder ratio 1/10 v/v). Using these conditions allows for faster and more even resuspension with less vortexing, which reduces the mechanical shear forces on the NPCs. It is very important to perform the affinity isolation as quickly as possible from the time of resuspension until the elution (in our hands this takes a few hours) and to keep everything at 4 °C for the duration of the protocol to prevent dissociation of the NPCs. This protocol was used to produce samples for the various downstream analysis described in (1) such as mass spectrometry (MS) quantitation and crosslinking and cryo-ET with sub-tomogram averaging. Therefore, it constitutes a large-scale preparation of ~175 μL of sample with a final concentration of 0.3-0.4 mg/mL, requiring 15 g of cell powder and a few adjustments in the protocol due to using such large amounts. However, downscaling is possible and encouraged for optimization tests and requires simple ratio adjustments to the protocol. We use 1-1.5 g of cell powder for testing affinity isolation performance. In general, it is not recommended to use this protocol for less than one 1-1.5 g portion of powder, due to resuspension related issues, as mentioned above.
2. Materials
2.1. Growing, harvesting, and flash-freezing yeast cells.
The yeast strain used for this protocol is MLP1-PPX-PrA (see (1), Supplementary table 5). It is a S. cerevisiae W303 strain in which the endogenous gene of interest (encoding for the “handle” protein used for the affinity isolation), in our case nucleoporin MLP1, is tagged with a PrA affinity tag (see Note 22) and a PPX cleavage target sequence inserted between MLP1 and PrA, to enable native elution. The same protocol can be used for S. cerevisiae strains in which Nup84- or Nup82-encoding genes are similarly tagged (see Nup82-PPX-PrA or Nup84-PPX-PrA in (1), Supplementary table 5). All strains can be obtained from the Rout lab upon request (http://ncdir.org/cd/).
Frozen glycerol stock of Mlp1-PPX-PrA strain (W303: MATα ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 MLP1-PPX-ProteinA::HIS5).
YPD agar plates.
YPD media: 1% yeast extract, 2% peptone, 2% glucose.
Small (100 mL) and medium (250 mL) culture flasks.
6 large culture flasks (such as 2.8 L Nalgene™ Polycarbonate Fernbach Culture Flasks from Thermo Fisher Scientific).
1 M Ampicillin.
Hemocytometer
Refrigerated centrifuge, rotor, and flasks for pelleting large cell volumes (such as Beckman Coulter Avanti J-26XP centrifuge, with JLA-8.1000 rotor and 1 L bottles).
Ice cold ddH2O
50 mL tubes.
Refrigerated centrifuge and rotor for 50 mL tubes (such as Beckman Coulter Allegra X-14R centrifuge, with SX4750A ARIES™ Swinging-Bucket Rotor and 50 mL tube adapters).
12% w/v polyvinylpyrrolidone (PVP), an extracellular cryoprotectant. Dissolve 1.2 g PVP in 10 mL ddH2O.
200 mM HEPES, pH 7.4: For 10 mL, dissolve 476.6 mg HEPES in 9 mL ddH2O. Adjust pH by adding 1 M KOH. Add ddH2O to 10 mL.
Resuspension buffer: 20 mM HEPES pH 7.5, 1.2% PVP. For 10 mL, combine 1 mL 12% w/v PVP, 1 mL 200 mM HEPES pH 7.4 and 8 mL ddH2O.
Protease Inhibitor Cocktail (Sigma Aldrich P8340).
Solution P: dissolve 2 mg Pepstatin A and 90 mg PMSF in 5 mL absolute ethanol. Store at 4 °C and discard after 3 weeks. Note that PMSF is highly poisonous, use appropriate caution (wear gloves, work in a fume hood, etc.).
1M Dithiothreitol (DTT) solution: weigh 1.54 g of DTT, dissolve completely in a final volume of 10 mL ddH2O. Filter sterilize, aliquot (1-1.5 mL) and store at −20 °C.
Vacuum aspirator.
Liquid nitrogen.
Cryoprotective gloves.
Styrofoam box.
Syringe needle.
25 mL or 50 mL syringe.
2.2. Cryo-milling of yeast cells
Liquid nitrogen.
Cryoprotective gloves.
Styrofoam box.
Retsch Planetary Ball Mill PM100.
Retsch cryomilling jar assembly: stainless steel jar (50 mL or 125 mL), lid, and 20 mm balls.
Tongs.
Spatula.
50 mL tubes.
2.3. Antibody conjugation to magnetic beads
Dynabeads M-270 Epoxy (Thermo Fisher Scientific 14302D).
Rabbit polyclonal IgG - purified from normal Rabbit Serum using Protein A affinity chromatography (Innovative Research) – this product comes as a solution with a 10-20 mg/mL concentration. Equivalent products from other sources can also be used.
15 mL tubes
Magnetic separator for 15 mL tubes (e.g., DynaMag™-15 from Thermo Fisher Scientific).
Vacuum aspirator.
Vortex mixer.
Parafilm.
2 mL round-bottom tubes.
Tube shaker or rocker.
0.1 M sodium phosphate, pH 7.4: For 1 L, dissolve 2.6 g NaH2PO4·H2O and 21.7 g Na2HPO4·7H2O in 1 L. Check the pH. Filter and store at RT.
3 M ammonium sulfate: For 100 mL, dissolve 39.6 g (NH4)2SO4 in 50 mL of 0.1 M sodium phosphate buffer, pH 7.4. Filter and store at RT.
Rotating wheel or tube shaker/rocker in an incubator (temperature set to 30°C).
0.1 M glycine–HCl pH 2.5.
10 mM Tris–HCl pH 8.8.
0.1 M trimethylamine (TEA)—make fresh. For 5mL, mix 70 μL of TEA with 4.93 mL ddH2O.
Phosphate-buffered saline (PBS).
PBS with 0.5% (w/v) Triton X-100.
PBS with 0.02% sodium azide (NaN3).
2.4. Affinity isolation of yeast NPCs
15 g cryo-milled cell powder (from section 3.1 of this protocol) (see Note 6).
750 μL conjugated IgG Dynabeads slurry (from section 3.2 of this protocol) (see Note 5)
0.22 μm filters.
50 mL tubes.
15 mL tubes.
1.5 mL tubes.
200 mL freshly made affinity capture (AC) buffer: 20 mM HEPES pH 7.4, 50 mM potassium acetate, 20 mM NaCl, 2 mM MgCl2, 0.5% Triton X-100, 0.1% Tween-20, 1 mM DTT, 10% glycerol. To prepare, combine in a 250 mL cylinder: 4 mL 1 M HEPES pH 7.4., 2 mL 5 M potassium acetate, 0.8 mL 5 M NaCl, 0.4 mL 1 M MgCl2, 10 mL 10% Triton X-100, 1 mL 20% Tween-20, 0.2 mL 1 M DTT, 20 mL 100% glycerol, 161.6 mL ddH2O. Mix, filter sterilize, and store on ice.
50 mL freshly made native elution (NE) buffer: 20 mM HEPES pH 7.4, 50 mM potassium acetate, 20 mM NaCl, 2 mM MgCl2, 0.1% Tween-20, 1 mM DTT, 10% glycerol. To prepare, combine in a 50 mL tube: 1 mL 1 M HEPES pH 7.4., 0.5 mL 5 M potassium acetate, 0.2 mL 5 M NaCl, 0.1 mL 1 M MgCl2, 0.25 mL 20% Tween-20, 0.05 mL 1 M DTT, 5 mL 100% glycerol, 42.9 mL ddH2O. Mix, filter sterilize, and store on ice.
Protease Inhibitor Cocktail (Sigma Aldrich P8340).
Solution P: dissolve 2 mg Pepstatin A and 90 mg PMSF in 5 ml absolute ethanol. Store at 4 °C and discard after 3 weeks. Note that PMSF is highly poisonous, use appropriate caution (wear gloves, work in a fume hood, etc.).
1.6 μm Whatman glass microfiber syringe filters (Cytiva Life Sciences 6882-2516).
Laboratory syringes (various sizes).
Magnetic separator for 15 and 1.5 mL tubes (e.g., DynaMag™-15, DynaMag™-2 from Thermo Fisher Scientific).
Vacuum aspirator.
Vortex mixer.
Laboratory balance.
Liquid nitrogen
Refrigerated centrifuge and rotor for 50 mL tubes (such as Beckman Coulter Allegra X-14R centrifuge, with SX4750A ARIES™ Swinging-Bucket Rotor and 50 mL tube adapters).
Refrigerated micro-centrifuge and rotor (such as Eppendorf 5417R tabletop centrifuge with an F-45-30-11 rotor).
Rotating wheel mixer (or shaker) set at 4°C.
Styrofoam box.
Spatula.
PreScission protease (Cytiva Life Sciences 27084301).
Benchtop mini centrifuge (such as mySPIN™ 6 Mini Centrifuge from ThermoFisher Scientific).
2.5. SDS-PAGE analysis
XCell SureLock Mini-Cell Electrophoresis System (ThermoFisher Scientific) or equivalent.
Electrophoresis power supply (such as PowerPac™ Basic Power Supply from Bio-Rad).
NuPAGE™ 4 to 12%, Bis-Tris, 1.0 mm, Mini Protein Gel, 10-well (ThermoFisher Scientific) (see Note 16).
MES running buffer (such as NuPAGE™ MES SDS Running Buffer (20X) from ThermoFisher Scientific).
NuPAGE™ LDS Sample Buffer (4X) (ThermoFisher Scientific).
1M DTT solution (see section 2.1, 10).
1.5 mL tubes
NuPAGE™ Antioxidant.
Protein marker (such as Color Prestained Protein Standard, Broad Range (10-250 kDa) from New England Biolabs).
10 mg/mL Bovine Serum Albumin (BSA) stock solution: add 100 mg of BSA (Sigma-Aldrich or similar) to a 15 mL tube containing 9.5 mL of ddH2O. Gently rock the capped tube until the BSA is completely dissolved. Do not vortex. Adjust the final volume to 10 mL with ddH2O. Divide into 1 mL aliquots and store at −20 °C.
0.25 mg/ml BSA stock solution: add 25 uL of the 10 mg/ml BSA stock solution to 975 uL ddH2O. Gently mix, then divide into 100 uL aliquots and store at −20 °C.
Sturdy spatula or “gel knife”.
Gel staining box or container.
Orbital shaker at room temperature.
Imperial™ Protein Stain (ThermoFisher Scientific).
Standard image scanner or gel scanner (such as ImageQuant LAS 4000 from Cytiva Life Sciences).
2.6. Negative stain electron microscopy (EM) analysis
TEM grids for negative stain EM – 200 mesh continuous carbon Cu TEM grids (such as CF200-Cu Electron Microscopy Sciences). 300 or 400 mesh continuous carbon Cu TEM grids are also suitable.
A set of fine-tipped tweezers: straight, self-closing and curved. Dumont biology grade tweezers with style 5 tips are recommended (such as 72701-01, 0202-N5-PO and 0208-7-PO, respectively, from Electron Microscopy Sciences). However, any tweezers suited for extra-fine handling are acceptable.
Transmission electron microscope - 80–120 keV or 80–200 keV side entry microscope (such as JEOL 1400 Plus or FEI TECNAI G2 Spirit BioTwin), equipped with a digital camera.
2% (w/v) Uranyl Acetate stain solution (UA stain) – combine 100 mg Uranyl Acetate powder (such as 22400 from Electron Microscopy Sciences) with 5 mL ddH2O. Place the solution on a rocker and incubate for 2 hours, then sonicate for 5 minutes. Filter the solution with a 0.22 μm syringe filter (or centrifuge and retain the supernatant). Store the UA stain wrapped in aluminum foil in a dark place (it is light sensitive) and at RT (it aggregates at lower temperatures) up to one year.
Glow discharge system or plasma cleaner (such as PELCO easiGlow™ Glow Discharge Cleaning System).
Parafilm.
1 mL syringes
0.2 μM filters for 1 mL syringes (such as 4 mm Cellulose Acetate Nalgene™ Syringe Filters, 171-0020, from Thermo Fisher Scientific).
Blot paper (e.g., Whatman grade 1 qualitative filter paper, standard grade, 90 mm circle from Cytiva Life Sciences).
EM grid storage box (e.g., 71150 from Electron Microscopy Sciences or use empty boxes from purchased grids).
3. Methods
3.1. Growing, harvesting, and flash-freezing yeast cells.
This section involves growing, harvesting, and flash-freezing of yeast cells. The affinity isolation protocol described in this manuscript requires 15 g of cryo-milled cell powder (see section 3.4). The typical yield of S. cerevisiae cell cultures harvested at 3.0 x 107 cells/mL is 3-4 g per liter, but it may vary depending on the strain and the final cell density. We usually grow 6-12 L of S. cerevisiae cell culture at a time, producing roughly 20-50 g of cryo-milled cell powder (the protocol described here is for 12 L S. cerevisiae cell culture). Cell culture volume can be adjusted according to need (see Note 1). The protocols for harvesting and flash-freezing of cells are extensively described on the National Center for Dynamic Interactome Research website (https://www.ncdir.org/protocols/), including a video tutorial (titled ‘Harvesting Cells and Making Noodles’)(27). Please check the link for updated versions before you perform these procedures. It is crucial to follow the prescribed safety procedures and take extreme care when handling liquid nitrogen.
Streak the strain from the frozen glycerol stock onto a YPD agar plate (for this strain, a regular YPD plate is used; for other strains use an appropriate selection plate, if needed). Incubate the plate at 30°C until single yeast colonies reach ~1-2 mm in diameter (this usually takes 2-3 days). Seal plates with Parafilm and store at 4°C for up to one month.
Day One
-
2.
Starter culture - prepare a small culture flask with 15 mL pre-sterilized YPD media. Add 1:1000 (v/v) 1M Ampicillin to prevent bacterial contamination. Inoculate with a single colony from the agar plate. Grow overnight at 30°C with shaking at ~200 rpm.
-
3.
Prepare 12 L of YPD media divided equally between 6 large flasks (2 L per flask). Cover flasks with aluminum foil, autoclave to sterilize and allow to cool overnight.
Day Two
-
4.
Estimate cell density of the starter culture using a hemocytometer (following manufacturer’s instructions). At this point the culture should be very dense (in stationary phase).
-
5.
Pre-culture - prepare a flask with 50 mL pre-sterilized YPD media. Add 1:1000 (v/v) 1M Ampicillin. Inoculate with 0.3 mL of the starter culture (1/50 v/v). Grow at 30°C with shaking at ~200 rpm until mid-log phase (~3 x 107 cells/mL) is reached (estimate cell density using a hemocytometer). This usually takes ~5 hours assuming a doubling time of ~1.5 hours.
-
6.
Large scale cultures – add 1:1000 (v/v) 1M Ampicillin to the 6 autoclaved flasks with YPD media. Calculate inoculation volume to yield a final 2 L culture at mid-log phase (~3 x 107 cells/mL) after an overnight incubation (for example: ~18 hours incubation requires ~0.58 mL inoculation volume from a 2.5 x 107 cells/mL pre-culture assuming a doubling time of 1.5 hours). Inoculate the large flasks with the calculated inoculation volume from the actively growing pre-culture.
-
7.
Grow the large cultures overnight (for the calculated incubation time) at 30°C with shaking at ~200 rpm.
Day Three
-
8.
Estimate cell density of the large cultures using a hemocytometer to make sure cells are at mid-log phase (~3 x 107 cells/mL).
-
9.
Harvest cells by spinning down at 5000 X g for 5 minutes at 4°C (for centrifuge and rotor options see section 2.1, 8). Discard the supernatant.
-
10.
Gently resuspend the cell pellet in 25 mL of ice-cold ddH2O. Pool the mixtures into 50 mL tubes and spin down at 2000 x g for 5 minutes at 4°C (for centrifuge and rotor options see section 2.1, 11). Repeat once more.
-
11.
Add 1/100 Protease Inhibitor Cocktail (v/v), 1/100 Solution P and 1/1000 1M DTT (v/v) to the resuspension buffer (see section 2.1, 14).
-
12.
Note the volume of the cell pellet in the tubes and resuspend in an equal volume of resuspension buffer with the Protease Inhibitor Cocktail, Solution P, and DTT. Spin down at 2500 X g for 20 minutes. Aspirate the supernatant, leave the pellet as dry as possible.
-
13.
Fill a clean Styrofoam box with liquid nitrogen. Submerge a conical tube rack for 50 mL tubes into the bath. Take a 50 mL tube and poke its cap with a syringe needle to form tiny holes. Unscrew the cap and place the tube into the tube rack inside the liquid nitrogen. Fill the tube with liquid nitrogen.
-
14.
With a spatula, transfer the cell paste into a 20 mL or 50 mL syringe depending on the cell pellet volume.
-
15.
Gently press the plunger and extrude the cell paste directly into the cryo-cooled 50-mL tube forming frozen strings of cell paste or “noodles”.
-
16.
When the tube is full, screw on the cap with holes and pour off the excess liquid nitrogen. Store the frozen noodles at −80°C until cryo-milling. Typically, for 12L of yeast cell culture, the yield is roughly two 50 mL tubes of noodles.
3.2. Cryo-milling of yeast cells.
This protocol is extensively described on the National Center for Dynamic Interactome Research website (https://www.ncdir.org/protocols/), including a video tutorial (Cryogenic Disruption Using PM100 Ball Mill). Please check the link for updated versions before you perform this procedure. It is crucial to follow the prescribed safety procedures and take extreme care when handling liquid nitrogen. Protective lab clothing and cryoprotective gloves must be used at all times during cryo-milling.
For the cryo-milling process, every component that will be in touch with the frozen cells must be kept in liquid nitrogen temperatures. Pre-cool the components for cryo-milling (stainless steel jar, lid, and balls) by immersion in a liquid nitrogen bath (e.g., Styrofoam box). Once the vigorous boiling in the bath ceases, take out the pre-cooled milling jar and add the frozen yeast noodles (<20 mL of noodles/50 mL jar or 20-50 mL of noodles/125 mL jar). Place three balls in the 50 mL jar or seven to eleven balls in the 125 mL jar.
Replace the lid of the milling jar. Weigh the complete jar assembly and adjust the corresponding counterbalance weight. Ensure all liquid nitrogen inside the jar has evaporated before milling to prevent the buildup of excess pressure.
Place the assembled milling jar in the planetary ball mill and clamp down as per manufacturers’ instructions. Ensure that the jar is securely clamped to prevent accidents or damages.
Set up the milling cycle. For the 125 mL jar, set 400 rpm rotation speed, 3 minutes grinding time, and 1 minute interval for reverse rotation with no breaks between rotations. For the 50 mL jar, set 500 rpm speed with the same timing parameters as the 125 mL jar.
Start the milling sequence. You must hear the balls rattling around inside the jar. If there is no rattling sound, wait for the cycle to finish, unclamp the jar, and add or remove balls (see Note 2).
When the milling sequence is complete, unclamp and place the milling jar in the liquid nitrogen bath. Do not remove the lid and do not completely submerge the jar to prevent the liquid nitrogen from entering the jar. Using tongs and a 50 mL Falcon tube, pour liquid nitrogen on the lid to cool the unsubmerged portion of the jar. Once the whole assembly is fully cooled (no vigorous bubbling upon pouring of nitrogen), re-clamp the jar as before and start another cycle.
Perform eight milling cycles (repeat steps 11 – 13).
After the eighth cycle, open the jar and assess the powder quality – if compacted or stuck to the side of the jar (caked), remove all the balls and scrape the inner walls of the jar with a pre-chilled spatula to dislodge the caked material and perform an additional milling cycle. Keep checking the powder quality until it is loose and uniform. Repeat milling cycles if it is not.
Remove the steel balls and transfer the yeast powder into a pre-cooled, pre-weighed 50 mL tube using a pre-chilled spatula. Work quickly to avoid thawing the cell powder. Record the weight of the cryo-milled cell powder. The typical yield is 3-4 g per liter of culture harvested at 3.0 x 107 cells/mL, but it may vary depending on the strain and the final cell density. In our case, for 12L of cell culture, the yield is roughly 50 g of powder.
Store the cell powder at −80°C until needed.
3.3. Antibody conjugation to paramagnetic beads
This protocol is available on the NCDIR website (https://www.ncdir.org/protocols/), so please check the link for updated versions before you perform this procedure. Typically, this conjugation requires ~0.167 mg antibody (rabbit polyclonal IgG) per 1 mg magnetic beads (Dynabeads) to make our final working suspension of 15% w:v conjugated Dynabeads in PBS (150 mg beads per 1 mL slurry). The affinity isolation protocol described in this manuscript requires a total of 0.75 mL of the slurry (equivalent to ~112 mg of conjugated Dynabeads). We usually conjugate one whole vial of Dynabeads (300 mg) to 50 mg of rabbit polyclonal IgG to obtain a 2 mL final Dynabeads slurry (see Note 3 for scaling options). The protocol takes two consecutive days.
Day One – Preparing the beads followed by conjugation
Resuspend entire vial of Dynabeads (300 mg) in 16 mL of 0.1 M sodium phosphate buffer, pH 7.4. Vortex for 30 seconds.
Divide bead suspension into four 15 ml tubes (4 mL suspension in each tube).
Collect any remaining beads in the glass vial with an additional 2 mL of 0.1 M sodium phosphate buffer, pH 7.4. Divide equally amongst the four 15 mL tubes.
Mix bead suspension slowly for 10 minutes on a tube shaker or rocking platform.
- While the bead suspension is mixing, prepare the antibody conjugation solution (AB). The AB solution contains the IgG at a concentration of 2.5 mg/mL, 3 M ammonium sulfate, and 0.1 M sodium phosphate pH 7.4. Total solution volume and 3 M ammonium sulfate volume must remain constant at 20 and 6.65 mL, respectively. The volumes of IgG and 0.1 M sodium phosphate may change depending on the concentration of the IgG. Typically, our IgG concentration is 14 mg/mL, so for a total of 50 mg IgG we need 3.57 mL, leaving the volume of 0.1 M sodium phosphate to be 9.78 mL (see Note 3 for scaling options).
- Take an IgG solution volume equivalent to 50 mg (here we assume this will be 3.57 mL), spin down at 14k rpm for 10 minutes, at 4°C. Save supernatant and discard pellet.
- Add 9.78 mL 0.1 M sodium phosphate pH 7.4.
- Slowly, while slightly shaking the tube, add 6.65 mL 3 M ammonium sulfate (you will see brief cloudiness appear upon addition).
- Filter using 0.22 μm syringe filter.
Continue from 5 - place tubes with the beads in the magnetic separator, wait until all the beads are attached to the tube wall. Carefully aspirate supernatant.
Wash again with 4 mL 0.1 M sodium phosphate buffer, pH 7.4 per tube. Vortex for 15 seconds. Place tubes in the magnetic separator, wait until all the beads are attached to the tube wall. Carefully aspirate supernatant.
Add 5 mL of AB solution to each tube, vortex until a homogeneous mixture is achieved.
Wrap tops of tubes with Parafilm and place on rotating wheel or tube shaker/rocker at 30°C overnight (incubation must last at least 18 hours but no more than 24 hours).
Day Two - Washing the beads after conjugation
After incubation is over, wash beads as described below for each 15 mL tube. After each wash place tubes in the magnetic separator, wait until all the beads are attached to the tube wall and carefully aspirate supernatant.
-
10.
Wash once with 3 mL of 100 mM Glycine HCL pH 2.5. Add, mix well and aspirate as quickly as possible. Prolonged incubation in highly acidic conditions may lead to IgG denaturation.
-
11.
Wash once with 3 mL of 10 mM Tris pH 8.8.
-
12.
Wash once with 3 mL of a freshly made 0.1 M triethylamine solution. Add, mix well and aspirate as quickly as possible. Prolonged exposure to highly alkaline conditions may lead to IgG denaturation.
-
13.
Wash once with PBS, then mix for 5 minutes on a tube shaker or rocking platform. Repeat four more times.
-
14.
Wash once with PBS with 0.5% TritonX-100, then mix for 5 minutes on a tube shaker or rocking platform. Then wash again with PBS with 0.5% TritonX-100, followed by 15 minutes mixing.
-
15.
Resuspend in PBS with 0.02% sodium azide to a final volume of ~2 mL (collect the beads with ~1 mL buffer, transfer slurry to a 2 mL round-bottom tube, then bring up the volume to ~2 mL).
-
16.
Store at 4 °C for up to 6 months.
3.4. Affinity isolation of yeast NPCs
This protocol has 3 stages, which all need to be completed on the same day.
Stage 1: Buffer, beads, and powder preparation
Add 0.4 mL (1:500 v/v) Protease Inhibitor Cocktail and 0.8 mL (1:250 v/v) Solution P to 200 mL freshly made and filter sterilized AC buffer (see section 2.3, 7) to make AC(+) buffer. Store on ice.
Take 10 mL of freshly made and filter sterilized NE buffer (see section 2.3, 8), add 20 μL (1:500 v/v) Protease Inhibitor Cocktail and 40 μL (1:250 v/v) Solution P to make NE(+) buffer. The remaining 40 mL of NE buffer will be NE(−) buffer (see Note 4). Store on ice.
Take out 750 μL conjugated IgG Dynabeads slurry from the stock (see Note 5). Wash the 750 μL conjugated IgG Dynabeads slurry with 1 mL of AC(+) buffer 3 times. Resuspend in 750 μL of AC(+) buffer (see Note 6). Store on ice.
Pre-chill ten 50 mL tubes and a spatula in a styrofoam box with liquid nitrogen. Weigh ~1.5 g of powder into each tube (see Note 7). Store in liquid nitrogen.
Stage 2: Affinity isolation (this part should be done as fast as possible).
-
5.
Place the tubes with the powder on ice for ~2 minutes to let the powder thaw a little.
-
6.
While on ice, add 13.5 mL of AC(+) buffer to each tube (1:10 w/v).
-
7.
Vortex each tube for 20 seconds and return to ice. Check for visible powder clumps. Repeat until a homogeneous suspension is achieved.
-
8.
Clarify lysate by centrifugation at 2500 X g for 8 minutes.
-
9.
Gently separate supernatant from each tube (try to not disturb the pellet) by slowly pouring it into a 20 mL syringe and filter using 1.6 μm Whatman glass microfiber syringe filters into a 15 mL tube (see Note 8). Discard pellet. At this point, there should be ten 15 mL tubes each with ~14 mL lysate placed on ice.
-
10.
Add to each tube 75 μL washed conjugated IgG Dynabeads slurry (from 3). Gently mix by inverting the tube several times.
-
11.
Incubate with gentle agitation (on a rotating wheel or a shaker) at 4°C for 30 minutes (see Note 9).
-
12.
After incubation, place the tubes with the lysate-Dynabeads suspension in the magnetic separator and aspirate supernatant, while combining Dynabeads from ten 15 mL tubes into two 15 mL tubes (see Note 10).
-
13.
Resuspend Dynabeads in each 15 mL tube using 0.5 mL of NE(−) buffer and transfer into a 1.5 mL tube. Use another 0.5 mL of NE(−) buffer to collect the remaining Dynabeads in each 15 mL tube and transfer to the same 1.5 mL tube (see Note 11). At this point there should be two 1.5 mL tubes each with ~1 mL Dynabeads slurry. Gently mix by inverting the tube several times. Place in the magnetic separator and aspirate supernatant. If there are more residual beads in the two 15 mL tubes, use 1 mL of NE(−) buffer to collect the remaining Dynabeads in each tube, then add to each of the 1.5 tubes while they remain in the magnetic separator. Wait for the beads to adhere to the walls of the tubes and aspirate supernatant. Take off the magnet, place on ice.
Stage 3: Native elution by protease cleavage –
PreScission protease (see section 2.4, 23) is used to cleave at the PPX target sequence between MLP1 and the affinity tag PrA, thereby releasing the NPCs from the Dynabeads. The final concentration of PreScission protease used in this protocol is ~0.1 units/μl (1:20 v/v of the commercial stock solution of 2 units/μL). Generally, it is recommended to use ~0.08 units protease per 5 μL of initial Dynabead slurry added to achieve full cleavage(3).
-
14.
Add 50 μL of NE(−) buffer to each of the two 1.5 mL tubes with Dynabeads from step 14. Gently mix by tapping the tube (do not invert, shake or vortex tubes).
-
15.
Add 2.5 μL of PreScission protease stock solution (1:20 v/v). Gently mix by tapping the tube (do not invert, shake or vortex tubes).
-
16.
Incubate with gentle agitation (on a rotating wheel or a shaker) at 4°C for 45 minutes (see Note 12).
-
17.
Spin tubes in a benchtop mini centrifuge for ~10 seconds. Place in the magnetic separator, carefully collect the supernatant from each tube and combine in a new 1.5 mL tube. Keep on ice.
-
18.
Add 20 μL of NE(+) buffer to each of the two 1.5 mL tubes with Dynabeads and gently mix by tapping the tube and inverting it a few times. This is done to collect the remaining protein complex from the Dynabeads.
-
19.
Spin tubes in a benchtop mini centrifuge for ~10 seconds. Place in the magnetic separator, carefully collect the supernatant from each tube and add to the same single 1.5 mL tube from step 18. Keep on ice.
-
20.
At this point, there should be one 1.5 mL tube containing ~150-180 μL supernatant with the natively eluted complex as well as two 1.5 mL tubes containing the Dynabeads pellet. The Dynabeads pellet should be kept for SDS-PAGE analysis (see section 3.4) and can be stored at −20°C until then.
-
21.
Centrifuge the supernatant in a micro-centrifuge at 14,000 rpm at 4°C for 5 minutes to spin down the remainder of the Dynabeads (see Note 13). Place in the magnetic separator, carefully collect the supernatant and transfer to a new 1.5 mL tube. Estimate the volume using a pipette tip.
-
22.
Add 1:1000 v/v Protease Inhibitor Cocktail to the supernatant.
-
23.
Take two ~5 μL aliquots (for SDS-PAGE and negative stain EM analyses - section 3.4 and 3.5) and transfer into two new 1.5 mL tubes. Dilute each aliquot twice with NE(+) buffer. Plunge freeze in liquid nitrogen and store at −80°C.
-
24.
At this point, you should have one 1.5 mL tube with ~150-180 μL final sample at a concentration of 0.3-0.4 mg/mL. We recommended aliquoting it into ~30-40 μL aliquots and then plunge freezing in liquid nitrogen and storing at −80°C until use.
3.5. SDS-PAGE analysis
SDS-PAGE is used both qualitatively to assess the efficiency of the affinity isolation protocol as well as quantitatively to estimate the yield and concentration of the final sample before downstream analyses such as CX-MS or cryo-EM (1). The samples analyzed are the Dynabeads pellet (see section 3.4, 21) to visualize proteins remaining on the beads after protease cleavage (see Note 14) as well as a fraction of the elution (usually ~3% v/v or the equivalent of 0.5 g of powder used for the affinity isolation). Protein concentration in samples is estimated by running a series of 3-5 BSA standards (~25-500 ng range) on the same gel, followed by densitometry analysis (see Note 15). We use the NuPAGE SDS polyacrylamide gel system (Thermo Fisher Scientific) with 4-12% Bis-Tris precast gels and MES running buffer. To visualize proteins on the gel, we use Imperial™ Protein Stain (Thermo Fisher Scientific). However, any comparable SDS-PAGE setup should yield similar results (Fig. 2).
Fig. 2. SDS-PAGE analysis.
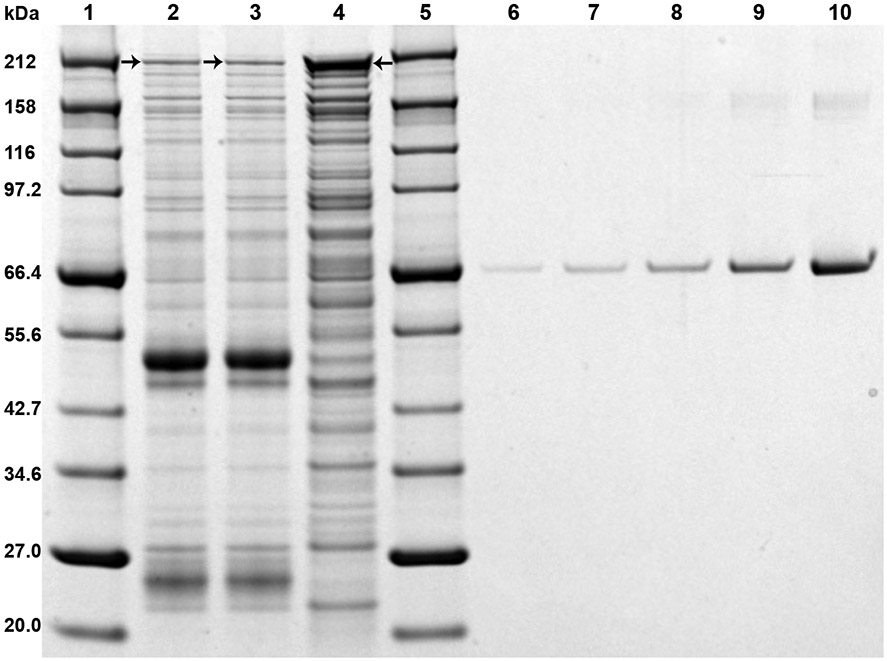
Typical results using the exact protocol described are shown (see section 3.5). Gel is loaded as described in section 3.5, 10 and scanned as described in section 3.5, 18. Lanes 1 and 5 contain a protein marker (molecular weights are indicated on the left side). Lanes 2 and 3 contain two Dynabeads pellet samples. Lane 4 contains the native elution sample. Lanes 6-10 contain BSA standards (25, 50, 100, 250, 500 ng, respectively). Black arrows indicate the “handle” protein MLP1 (see section 3.5, 19 and Note 14).
Take out the two 1.5 mL tubes containing the Dynabeads pellet (see section 3.3, 21) as well as the one tube containing the elution aliquot taken as a sample for SDS-PAGE analysis. Place on ice, allow to defrost for a few minutes.
Prepare 0.4 mL of 1x loading buffer – combine 100 μL 4x NuPAGE™ LDS Sample Buffer, 20 μL 1 M DTT and 280 μL ddH2O. Keep at room temperature.
To each of the two Dynabeads pellet tubes, add 1x loading buffer equivalent to the estimated volume of Dynabeads elution (see 3.3, 22) – it should be ~150-180 μL. Vortex to mix. Keep at room temperature.
To the elution aliquot (10 μL), add 6 μL 4x NuPAGE™ LDS Sample Buffer, 1.2 μL 1M DTT and 6.8 μL ddH2O, making the total volume 24 μL which is suitable for loading into a 10-well gel (see Note 16). Vortex to mix. Keep at room temperature.
Prepare 5 BSA standards – 25, 50, 100, 250, 500 ng (see Note 17): make a fresh 50 ng/μL BSA stock by adding 10 μL 0.25 mg/mL BSA (see 2.4, 11) to 40 μL ddH2O. Combine 0.5, 1, 2, 5, 10 μL of the 50 ng/μL BSA stock with 9.5, 9, 8, 5, 0 μL ddH2O, respectively, to obtain the corresponding standards. Gently mix, keep on ice. Discard unused 50 ng/μL BSA stock.
Add to all the BSA standards (10 μL) 6 μL 4x NuPAGE™ LDS Sample Buffer, 1.2 μL 1M DTT and 6.8 μL ddH2O, making the total volume 24 μL. Vortex to mix. Keep at RT.
Prepare protein marker sample: combine 5 μL protein marker stock (see 2.4, 9) with 20 μL 1x loading buffer. Vortex to mix. Keep at RT.
Incubate all gel samples (there should be 9 total) at 70-98°C for 10 minutes. Spin down in a tabletop centrifuge at 14,000 rpm for 1 minute at room temperature.
Assemble a 10-well 4-12% Bis-Tris gel (see 2.4, 3) in the XCell SureLock apparatus (see 2.5, 1) according to the manufacturer’s protocol. Prepare 1 L 1x MES buffer by combining 50 mL 20x MES running buffer stock with 950 mL ddH2O (see 2.4, 4) and mixing thoroughly. Use 1x MES running buffer to fill the inner chamber of the gel tank until all the loading wells of the gel are submerged (~200 mL) and then fill the outer chamber to roughly the same height (~600 mL).
Load the gel in the following order (see Note 18) from left to right: 25 μL (all) protein marker, 10 μL of each of the two Dynabeads pellet samples (place the tubes in a magnetic separator first to avoid magnetic beads in the loaded volume), 24 μL (all) elution sample, 25 μL 1x loading buffer (empty lane) or another sample of protein marker, 24 μL (all) 25, 50, 100, 250, 500 ng BSA standards in this order (Fig. 2).
Add 0.5 mL NuPAGE™ Antioxidant to the inner chamber of the gel box.
Run the gel at 180 V for ~70 minutes (see Note 19).
Prepare a clean gel staining box with enough ddH2O to cover the gel (~50 mL or more, depending on the box size).
After the gel run is finished, remove the gel cassette from the apparatus, disassemble using a sturdy spatula or a gel knife (the gel will remain on one of the open cassette plates), and gently transfer the gel into the ddH2O filled gel box (spatula or gel knife can be used for gel transfer).
Incubate on an orbital shaker for 5 minutes at room temperaturewith gentle agitation. Discard the water, repeat wash two more times (according to the protocol of the Imperial™ Protein Stain).
Discard the water, add enough Imperial™ Protein Stain to cover the gel and incubate on an orbital shaker with gentle agitation for at least 1 hour up to overnight at room temperature.
To destain the gel, discard the Imperial™ Protein Stain, wash with ddH2O and incubate in ddH2O for least 1 hour up to overnight at room temperature. After destaining, the gel should appear clear (little or no background staining) with protein bands easily visible by eye, at least in the protein marker and BSA standards lanes (Fig. 2).
Once the gel has fully destained, scan it on a gel scanner or a standard image scanner to digitize. We use ImageQuant LAS 4000 to expose gels using white trans illumination (operate according to manufacturer’s instructions) for the densitometry analysis (see Note 15).
Assessment of sample quality from SDS-PAGE analysis entails looking at the pattern of proteins on the gel and the relative intensity of the elution and Dynabeads pellet lanes (Fig. 2). A good quality sample will show the correct pattern of proteins in the elution lane (Fig. 2) and the intensity of the Dynabeads pellet lanes will be significantly lower than the elution lane (Fig. 2), signifying that most of the complex has been eluted from the beads (see Note 14). Densitometry analysis should show final sample concentration of 0.3-0.4 mg/mL for the exact affinity isolation protocol described here (see Note 15).
3.6. Negative stain EM analysis
Negative stain EM analysis is used to visualize the affinity purified NPCs and qualitatively assess their general appearance and concentration, thereby ensuring their quality is suitable for the downstream analyses such as CX-MS or cryo-EM (1). We use 2% uranyl acetate in ddH2O as our negative stain coupled with standard grid preparation and imaging procedures (28-33). Uranyl acetate is a mildly radioactive and toxic material and should be handled in accordance with all applicable radiation safety policies and precautions suitable for heavy metal salts. Dispose of unused uranyl acetate as liquid radioactive waste. Always wear appropriate PPE when handling it. The protocol outlined here is an example, however any equivalent protocol should yield similar results (see video protocols in (32,33)).
Glow discharge a desired number of carbon-coated TEM grids to render the carbon support hydrophilic – we use an in-house glow discharge apparatus with its specific parameters, however any glow discharge system or plasma cleaner (see 2.5, 5) should yield the same results, if used according to the manufacturer’s instructions or reputable literature sources (28-31). Place grids in the glow discharge system with the carbon coated side up (the carbon coated side is dull grey in color whereas the non-carbon coated copper side is shiny orange-brown). Close the chamber, wait for the appropriate vacuum to be reached. Set the desired glow discharge time (30 seconds to a few minutes). Note that the carbon coating can be compromised if the glow discharge settings are too aggressive. Grids should be used within 2 hours of the glow discharge procedure. Do not glow discharge the same grid more than once.
Prepare ddH2O and 2% UA stain, for washing and staining the grids, respectively. Keep the UA stain at room temperaturesince it precipitates at lower temperatures. Take 0.5 mL ddH2O and 0.5 mL UA stain per one grid, transfer to 1.5 mL tubes, centrifuge at 14,000 rpm in a tabletop micro-centrifuge for 10 minutes at room temperature (see Note 20). Keep tubes at room temperatureand do not agitate until use to avoid disturbing any dust or aggregate pellet.
Cut Parafilm into a rectangle and place on a clean working surface, such as the laboratory bench.
For each grid, place 3 ~0.15 mL drops of centrifuged ddH2O (extract from tube gently to avoid disturbing the pellet) onto the parafilm (for an example, see video protocols in (32,33)).
Take out a freshly glow discharged grid with self-closing tweezers and place the tweezers with the grid onto the parafilm (the tweezers should grip the grid by the very edge of it to not damage the carbon coating). Make sure the grid is facing carbon side up.
Apply to the grid 5 μL of the elution aliquot taken as a sample for negative stain EM analysis (see section 3.3, 24). Incubate on the grid for 1-5 minutes (the grid must remain wet – watch it carefully for the entire incubation time and if the drop on it starts to decrease in size and disappear, quickly move to the next step). The longer the incubation time, the more particles will be absorbed to the carbon (see Note 21).
Transfer the grid to the first ddH2O drop, carbon side facing down, such that the grid is floating on top of the drop. Incubate for 1 minute.
Use the straight or curved tweezers to transfer the grid to the second ddH2O drop. Incubate for 1 minute, then transfer to the third ddH2O drop and incubate for 1 minute.
Draw up ~0.5 mL of centrifuged UA stain using a 1 mL syringe. Place the syringe filter on it.
Use the self-closing tweezers to capture the grid and hold it at a ~45-degree angle over a receptacle (such as a large weighing boat or a plastic box). Apply the UA stain onto the grid drop by drop from the syringe with the filter. Once all 0.5 mL is applied, gently shake off the excess and blot with the Whatman filter paper by touching it to the side of the grid until all the stain has been wicked off.
Place the tweezers on the parafilm and allow the stained grid to air-dry for 5 minutes. Then store in a grid storage box in a dark and dry environment until use (we usually store our grid boxes wrapped in aluminum foil in a desiccator – the grids are viable for at least one year).
TEM imaging – make sure you receive detailed instruction on how to use the TEM available to you. When everything is ready for imaging, survey the grid and make sure it is of good quality (most carbon in the grid squares is intact, staining is not too deep and not too shallow, grid is not too contaminated etc.). Make sure you can see NPCs on the grid – they are usually very readily observed on the grid due to their very large size of ~100 nm in diameter (Fig. 3). If the grid is not good enough or there are no discernable NPCs on it, make a new grid (see (28-33) for tips and troubleshooting of negative stain grid preparation). With this sample, the grid will have quite a bit of background (proteins, etc.) in addition to NPCs since it is derived from a “crude” preparation (has not been purified further on a sucrose gradient or otherwise). However, due to their large size, the NPCs usually are the dominant species on the grid, enough for the evaluation of the quality of the sample for downstream analyses (Fig. 3). After the initial grid evaluation, survey the grid at relatively low magnification to find an area with good contrast and sufficient concentration of particles, increase magnification to the desired value, adjust focus and take an image. Repeat the process until enough images have been acquired (this is an individual estimate, since only a qualitative assessment is required; we usually take 10-20 images from different areas of the grid.
Assessment of sample quality from negative stain analysis looking at the concentration of intact NPCs on the grid, their shape, and their general appearance. A good quality sample will contain a moderate to dense field of NPCs in each image (Fig. 3), although this can be affected greatly by grid preparation (we usually rely more on the concentration estimate from the densitometry analysis). The NPCs will be mostly intact, almost perfectly round and some of the C-8 symmetric spoke structure (1) will be discernable at the proper magnification (Fig. 3). In most NPCs, the central transporter will be present filling the middle of the NPC “donut” shape (1) (Fig. 3). If most NPCs are broken or deformed, the affinity purification was not successful enough, and this sample cannot be used for any downstream analyses.
Fig. 3. Negative stain EM images.

Four representative images at different magnifications taken using grids prepared and imaged as described in section 3.6. Black boxes indicate a selection of individual NPCs. Magenta arrows point to central transporters within selected NPCs. Scale bar indicated in black in the bottom left side of every image.
NOTES:
As explained in the introduction, we recommend working with 1-1.5 g cell powder portions. Therefore, if the goal of the protocol to perform several small-scale tests, it may be that 5-15 g of cell powder is sufficient. In that case, cell culture volume can be 2-4 L, which should yield 6-16 g powder.
In some cases, the rattling sound disappears after a few milling cycles. One possible reason is that the cell powder has compacted inside the jar and stuck to the side (caked). If caking is suspected, open the jar assembly, remove all the balls (balls and lid should be kept in liquid nitrogen), and scrape the inner walls of the jar with a pre-chilled spatula to dislodge the caked material, add back the balls and close the jar lid prior to proceeding with the next milling cycle.
The amount of Dynabeads to conjugate can be scaled according to need. 50 μL of conjugated Dynabeads slurry is needed per 1 g of cell powder, so if the goal is to perform several small-scale tests in 1-1.5 g cell powder portions, it may be that 0.5 mL or less of conjugated Dynabeads slurry is sufficient. In that case, 75 mg of Dynabeads can be conjugated by weighing the needed amount of Dynabeads and using the same protocol with all other variables adjusted according to the ratio of the Dynabeads (1/4). For example, for 75 mg Dynabeads, 12.5 mg of rabbit polyclonal IgG is required. The new AB solution volume should be 5 mL (adjusted to maintain IgG concentration of 2.5 mg/mL) and the volume of 3 M ammonium sulfate added should be 1.66 mL.
The need for buffers with and without protease inhibitors (Protease Inhibitor Cocktail and Solution P) is because we are performing a proteolytic cleavage for our native elution. The cleavage will be less efficient if it takes place in a buffer containing protease inhibitors, therefore, only for the cleavage part of the protocol, the buffer is exchanged to one not containing protease inhibitors. However, in all other stages, protease inhibitors must be present to prevent degradation of proteins.
Magnetic beads settle in solution after being stationary for a while. Therefore, before use, a homogeneous suspension needs to be re-created to ensure that accurate and reproducible amounts of beads are being used. We recommend resuspending the beads by a combination of pipetting up and down and tapping, and not by inverting the tube or vortexing it. Once a homogeneous slurry has been achieved, the proper volume of beads can be taken out and used.
Washing beads: place tube with bead slurry in the magnetic separator, wait until all the beads are attached to the tube wall; carefully aspirate supernatant with a vacuum aspirator placing the tip on the side of the tube opposite the one to which the beads adhere; take tube out of the magnetic separator, add 1 mL of wash buffer, resuspend by tapping on the tube, gently shaking and inverting it a few times until the slurry looks homogeneous; place in the magnet, wait until all the beads are attached to the tube wall; invert the magnet with the tubes a couple of times to recover material stuck on the lid and let settle again; carefully aspirate supernatant with a vacuum aspirator – this constitutes one wash cycle.
Cell powder must be kept at liquid nitrogen temperatures anytime it is out of its storage at −80°C. To weigh out a portion, the tube is moved from the −80°C storage into a tube rack in liquid nitrogen in a Styrofoam box. The cell powder needs to be examined before use for possible compaction or clumping, which tends to happen if the cell powder has been in −80°C storage for too long or if, for some reason, it has been subjected to partial thawing or significant changes in storage temperature. Compacted or clumped cell powder is not optimal for use – cell powder needs to be loose and uniform to ensure proper resuspension in buffer. However, it is possible to salvage clumped or compacted powder by using a pre-chilled spatula to try and break down the clumps. If possible, one can use the powder after testing in a small-scale affinity capture, but if the compaction is severe, the powder is not usable.
At this point the lysate still contains a fair amount of insoluble debris, due to the relatively mild centrifugation. Therefore, filtering, even through a relatively large pore (1.6 μm) filter, can be difficult. Typically, we use several syringe filters (2-3) for one lysate tube.
The incubation time can be changed if needed, but we recommend keeping it within 30 – 60 minutes, which is sufficient to capture the target protein complexes with minimal accumulation of non-specific proteins[25].
Place the first two 15 mL tubes into the magnetic separator, wait until the beads are accumulated on the sides of tube, next to the magnet. Aspirate supernatant. Take two additional tubes, mix by inverting a few times and pour content into the two tubes on the magnet. Wait until the beads are accumulated on the sides of tube, aspirate supernatant. Repeat with the rest of the 15 mL tubes until you are left with only the tubes in the magnet.
Remove the two 15 mL tubes from the magnet, put on ice. Add 0.5 mL NE(−) buffer to each tube. Mix by rotating the tube such that the buffer is slowly resuspending the beads on the sides of the tube. Do not invert and try not to let the buffer get into the cap of the tube. After most of the beads have been washed off the sides of the tube, place it back on ice, take up the bead suspension with a pipette (volume will be more than 0.5 mL, closer to 1 mL) and transfer it to a 1.5 mL tube. Use the second 0.5 mL NE(−) buffer to repeat the above for the beads still left in the 15 mL tubes.
The incubation time can be changed if needed, but we found that the PreScission protease is quite efficient at 4°C, so most of the cleavage process usually takes place within the first 20-30 minutes.
It is quite important to remove all magnetic beads from the elution, since they are detrimental to performing an SDS-PAGE analysis (if even a small amount of beads is present, the gel runs unevenly) as well as the negative stain EM analysis and probably any downstream analysis in general.
One of the major issues interfering with the native elution process is the “sticky-ness” of the beads, meaning the bead coating propensity for non-specific protein binding under certain conditions. While the protease successfully cleaves and releases the complex, some of the cleaved complex does not reach the actual eluate but instead remains stuck to the beads probably through interaction with the bead coating. This can be easily corroborated by the SDS-PAGE analysis, where it is clearly seen that the “handle” protein MLP1 in the lane of the Dynabeads pellet is the same size as MLP1 in the lane of the elution, meaning it has been separated from its PrA tag (Fig. 2). The amount of such binding almost always depends on the buffer conditions used. Some buffer components can be very stabilizing for a complex but unsuitable for native elution due to non-specific adsorption of the complex to the beads (for example, high concentrations (0.5-1 M) of potassium acetate, ammonium acetate, sodium citrate, and other cosolvents tend to cause such effect). For this reason, it is very important to make sure the chosen affinity isolation buffer allows efficient, high yield native elution without too much residual complex stuck to the beads.
Densitometric analysis is a convenient way to estimate protein quantities from SDS-PAGE. It involves determining band intensities from stained gels and quantifying them using a standard curve (in our case BSA standards) (34,35). We use the Gel Analyzer tool of ImageJ (NIH), a public domain program from the National Institutes of Health that allows image processing. More details about protein densitometry analysis can be found in (34,35) as well as on this website: https://openwetware.org/wiki/Protein_Quantification_Using_ImageJ. To estimate sample concentration in the Dynabeads pellet and the elution lanes, we summarize the intensities of all the bands in the specific lane and use that as the total protein content of that sample.
Other gel configurations can be used; however, a 10-well gel allows for better densitometry analysis.
A minimum of 3 BSA standards can be used (to allow for proper signal calibration during the densitometry analysis) in the desired range. We recommend using a range of 25-500 ng, since it covers almost all interpretable band intensities in the dynamic range of the densitometry analysis.
The reasoning for this specific order of gel lanes is that placing the Dynabeads pellet samples and the elution sample in neighboring lanes of the gel allows better comparison between them, which is important for assessing the quality of the sample (see section 3.5, 19).
This running time is longer than the time necessary for the protein marker to reach the bottom of the gel. However, we found that for the configuration described here, this run time is best for the optimal separation of the higher molecular weight components of the NPC and allows for better visualization, quality, and quantity assessment.
UA stain and ddH2O used for making negative stain EM grids must be clear of any large aggregates, such as dust particles or sediment, to prevent grid contamination.
The drop placed on the EM grid is subject to both capillary forces which may aspirate part or all of it into the tweezers as well as to evaporation, therefore must be watched closely during the incubation to prevent drying out of the grid. If the number of particles adsorbed to the grid during this incubation is insufficient, another drop may be applied following the first one and so on. Alternatively, the grid can be incubated on top of a larger drop of sample (10-15 μL) inside a high humidity chamber. If the number of particles adsorbed to the grid is too high, reduce sample concentration or incubation time.
-
This is the DNA sequence of the PrA tag we used:
GGTGAAGCTCAAAAACTTAATGACTCTCAAGCTCCAAAAGCTGATGCGCAACAAAATAACTTCAACAAAGATCAACAAAGCGCCTTCTATGAAATCTTGAACATGCCTAACTTAAACGAAGCGCAACGTAACGGCTTCATTCAAAGTCTTAAAGACGACCCAAGCCAAAGCACTAACGTTTTAGGTGAAGCTAAAAAATTAAACGAATCTCAAGCACCGAAAGCTGATAACAATTTCAACAAAGAACAACAAAATGCTTTCTATGAAATCTTGAATATGCCTAACTTAAACGAAGAACAACGCAATGGTTTCATCCAAAGCtTAAAAGATGACCCAAGCCAAAGTGCTAACCTTTTGTCAGAAGCTAAAAAGTTAAATGAATCTCAAGCACCGAAAGCTGACAACAAATTCAACAAAGAACAACAAAATGCTTTCTATGAAATTTTACATTTACCTAACTTAACTGAAGAACAACGTAACGGCTTCATCCAAAGCCTTAAAGACGATCCTTAA
This is the amino acid sequence of the PrA tag we used:
GEAQKLNDSQAPKADAQQNNFNKDQQSAFYEILNMPNLNEAQRNGFIQSLKDDPSQSTNVLGEAKKLNESQAPKADNNFNKEQQNAFYEILNMPNLNEEQRNGFIQSLKDDPSQSANLLSEAKKLNESQAPKADNKFNKEQQNAFYEILHLPNLTEEQRNGFIQSLKDDP
ACKNOWLEDGEMENTS:
This work was supported by National Institutes of Health grants U54 GM103511, R01 GM112108, P41 GM109824 and U54 DK107981. We thank all members of the Laboratory of Cellular and Structural Biology (Rout lab) and the Laboratory of Mass Spectrometry and Gaseous Ion Chemistry (headed by Prof. Brian Chait), especially Wenzhu Zhang and Erica Jacobs, for their assistance with the development and testing of this protocol as well as for their feedback and helpful discussions. We thank Azraa S. Chaudhury for her help with protocol optimization. We gratefully acknowledge Kunihiro Uryu and the EMRC Resource Center (Rockefeller University) for teaching and supporting our negative stain electron microscopy analysis. We are grateful to Dan Simon for support, invaluable discussions, and critical reading of the manuscript.
REFERENCES:
- 1.Kim SJ, Fernandez-Martinez J, Nudelman I, Shi Y, Zhang W, Raveh B, Herricks T, Slaughter BD, Hogan JA, Upla P, Chemmama IE, Pellarin R, Echeverria I, Shivaraju M, Chaudhury AS, Wang J, Williams R, Unruh JR, Greenberg CH, Jacobs EY, Yu Z, de la Cruz MJ, Mironska R, Stokes DL, Aitchison JD, Jarrold MF, Gerton JL, Ludtke SJ, Akey CW, Chait BT, Sali A, Rout MP (2018) Integrative structure and functional anatomy of a nuclear pore complex. Nature 555 (7697):475–482. doi: 10.1038/nature26003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olinares PD, Dunn AD, Padovan JC, Fernandez-Martinez J, Rout MP, Chait BT (2016) A Robust Workflow for Native Mass Spectrometric Analysis of Affinity-Isolated Endogenous Protein Assemblies. Anal Chem 88 (5):2799–2807. doi: 10.1021/acs.analchem.5b04477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LaCava J, Fernandez-Martinez J, Rout MP (2016) Native Elution of Yeast Protein Complexes Obtained by Affinity Capture. Cold Spring Harb Protoc 2016 (7):pdb prot087940. doi: 10.1101/pdb.prot087940 [DOI] [PubMed] [Google Scholar]
- 4.Fernandez-Martinez J, Kim SJ, Shi Y, Upla P, Pellarin R, Gagnon M, Chemmama IE, Wang J, Nudelman I, Zhang W, Williams R, Rice WJ, Stokes DL, Zenklusen D, Chait BT, Sali A, Rout MP (2016) Structure and Function of the Nuclear Pore Complex Cytoplasmic mRNA Export Platform. Cell 167 (5):1215–1228 e1225. doi: 10.1016/j.cell.2016.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Algret R, Fernandez-Martinez J, Shi Y, Kim SJ, Pellarin R, Cimermancic P, Cochet E, Sali A, Chait BT, Rout MP, Dokudovskaya S (2014) Molecular architecture and function of the SEA complex, a modulator of the TORC1 pathway. Mol Cell Proteomics 13 (11):2855–2870. doi: 10.1074/mcp.M114.039388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandez-Martinez J, Phillips J, Sekedat MD, Diaz-Avalos R, Velazquez-Muriel J, Franke JD, Williams R, Stokes DL, Chait BT, Sali A, Rout MP (2012) Structure-function mapping of a heptameric module in the nuclear pore complex. The Journal of cell biology 196 (4):419–434. doi: 10.1083/jcb.201109008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olinares PDB, Chait BT (2020) Native Mass Spectrometry Analysis of Affinity-Captured Endogenous Yeast RNA Exosome Complexes. Methods Mol Biol 2062:357–382. doi: 10.1007/978-1-4939-9822-7_17 [DOI] [PubMed] [Google Scholar]
- 8.LaCava J, Fernandez-Martinez J, Hakhverdyan Z, Rout MP (2016) Optimized Affinity Capture of Yeast Protein Complexes. Cold Spring Harb Protoc 2016 (7):pdb prot087932. doi: 10.1101/pdb.prot087932 [DOI] [PubMed] [Google Scholar]
- 9.LaCava J, Fernandez-Martinez J, Hakhverdyan Z, Rout MP (2016) Protein Complex Purification by Affinity Capture. Cold Spring Harb Protoc 2016 (7):pdb top077545. doi: 10.1101/pdb.top077545 [DOI] [PubMed] [Google Scholar]
- 10.Hakhverdyan Z, Domanski M, Hough LE, Oroskar AA, Oroskar AR, Keegan S, Dilworth DJ, Molloy KR, Sherman V, Aitchison JD, Fenyo D, Chait BT, Jensen TH, Rout MP, LaCava J (2015) Rapid, optimized interactomic screening. Nature methods 12 (6):553–560. doi: 10.1038/nmeth.3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Obado SO, Field MC, Chait BT, Rout MP (2016) High-Efficiency Isolation of Nuclear Envelope Protein Complexes from Trypanosomes. Methods Mol Biol 1411:67–80. doi: 10.1007/978-1-4939-3530-7_3 [DOI] [PubMed] [Google Scholar]
- 12.Winczura K, Domanski M, LaCava J (2020) Affinity Proteomic Analysis of the Human Exosome and Its Cofactor Complexes. Methods Mol Biol 2062:291–325. doi: 10.1007/978-1-4939-9822-7_15 [DOI] [PubMed] [Google Scholar]
- 13.Fridy PC, Li Y, Keegan S, Thompson MK, Nudelman I, Scheid JF, Oeffinger M, Nussenzweig MC, Fenyo D, Chait BT, Rout MP (2014) A robust pipeline for rapid production of versatile nanobody repertoires. Nature methods 11 (12):1253–1260. doi: 10.1038/nmeth.3170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LaCava J, Molloy KR, Taylor MS, Domanski M, Chait BT, Rout MP (2015) Affinity proteomics to study endogenous protein complexes: pointers, pitfalls, preferences and perspectives. BioTechniques 58 (3):103–119. doi: 10.2144/000114262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oeffinger M, Wei KE, Rogers R, DeGrasse JA, Chait BT, Aitchison JD, Rout MP (2007) Comprehensive analysis of diverse ribonucleoprotein complexes. Nature methods 4 (11):951–956. doi: 10.1038/nmeth1101 [DOI] [PubMed] [Google Scholar]
- 16.Ptak C, Aitchison JD, Wozniak RW (2014) The multifunctional nuclear pore complex: a platform for controlling gene expression. Current opinion in cell biology 28:46–53. doi: 10.1016/j.ceb.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alber F, Dokudovskaya S, Veenhoff L, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait B, Sali A, Rout M (2007) The molecular architecture of the nuclear pore complex. Nature 450 (7170):695–701 [DOI] [PubMed] [Google Scholar]
- 18.Allegretti M, Zimmerli CE, Rantos V, Wilfling F, Ronchi P, Fung HKH, Lee CW, Hagen W, Turonova B, Karius K, Bormel M, Zhang X, Muller CW, Schwab Y, Mahamid J, Pfander B, Kosinski J, Beck M (2020) In-cell architecture of the nuclear pore and snapshots of its turnover. Nature 586 (7831):796–800. doi: 10.1038/s41586-020-2670-5 [DOI] [PubMed] [Google Scholar]
- 19.Mosalaganti S, Kosinski J, Albert S, Schaffer M, Strenkert D, Salome PA, Merchant SS, Plitzko JM, Baumeister W, Engel BD, Beck M (2018) In situ architecture of the algal nuclear pore complex. Nat Commun 9 (1):2361. doi: 10.1038/s41467-018-04739-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kosinski J, Mosalaganti S, von Appen A, Teimer R, DiGuilio AL, Wan W, Bui KH, Hagen WJ, Briggs JA, Glavy JS, Hurt E, Beck M (2016) Molecular architecture of the inner ring scaffold of the human nuclear pore complex. Science 352 (6283):363–365. doi: 10.1126/science.aaf0643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eibauer M, Pellanda M, Turgay Y, Dubrovsky A, Wild A, Medalia O (2015) Structure and gating of the nuclear pore complex. Nat Commun 6 (May):7532. doi: 10.1038/ncomms8532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stuwe T, Bley CJ, Thierbach K, Petrovic S, Schilbach S, Mayo DJ, Perriches T, Rundlet EJ, Jeon YE, Collins LN, Huber FM, Lin DH, Paduch M, Koide A, Lu V, Fischer J, Hurt E, Koide S, Kossiakoff AA, Hoelz A (2015) Architecture of the fungal nuclear pore inner ring complex. Science 350 (6256):56–64. doi: 10.1126/science.aac9176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nordeen SA, Turman DL, Schwartz TU (2020) Yeast Nup84-Nup133 complex structure details flexibility and reveals conservation of the membrane anchoring ALPS motif. Nat Commun 11 (1):6060. doi: 10.1038/s41467-020-19885-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beck M, Mosalaganti S, Kosinski J (2018) From the resolution revolution to evolution: structural insights into the evolutionary relationships between vesicle coats and the nuclear pore. Curr Opin Struct Biol 52:32–40. doi: 10.1016/j.sbi.2018.07.012 [DOI] [PubMed] [Google Scholar]
- 25.Lindmark R, Thoren-Tolling K, Sjoquist J (1983) Binding of immunoglobulins to protein A and immunoglobulin levels in mammalian sera. J Immunol Methods 62 (1):1–13. doi: 10.1016/0022-1759(83)90104-7 [DOI] [PubMed] [Google Scholar]
- 26.Moks T, Abrahmsen L, Nilsson B, Hellman U, Sjoquist J, Uhlen M (1986) Staphylococcal protein A consists of five IgG-binding domains. Eur J Biochem 156 (3):637–643. doi: 10.1111/j.1432-1033.1986.tb09625.x [DOI] [PubMed] [Google Scholar]
- 27.LaCava J, Jiang H, Rout MP (2016) Protein Complex Affinity Capture from Cryomilled Mammalian Cells. J Vis Exp (118). doi: 10.3791/54518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gallagher JR, Kim AJ, Gulati NM, Harris AK (2019) Negative-Stain Transmission Electron Microscopy of Molecular Complexes for Image Analysis by 2D Class Averaging. Curr Protoc Microbiol 54 (1):e90. doi: 10.1002/cpmc.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frechard A, Sharov G, Werderer M, Schultz P (2021) Optimization of Sample Preparation for the Observation of Macromolecular Complexes by Electron (cryo-)Microscopy. Methods Mol Biol 2247:243–256. doi: 10.1007/978-1-0716-1126-5_13 [DOI] [PubMed] [Google Scholar]
- 30.Brillault L, Landsberg MJ (2020) Preparation of Proteins and Macromolecular Assemblies for Cryo-electron Microscopy. Methods Mol Biol 2073:221–246. doi: 10.1007/978-1-4939-9869-2_13 [DOI] [PubMed] [Google Scholar]
- 31.Arthur CP, Ciferri C (2019) High-Throughput Protein Analysis Using Negative Stain Electron Microscopy and 2D Classification. Methods Mol Biol 2025:477–485. doi: 10.1007/978-1-4939-9624-7_22 [DOI] [PubMed] [Google Scholar]
- 32.Scarff CA, Fuller MJG, Thompson RF, Iadanza MG (2018) Variations on Negative Stain Electron Microscopy Methods: Tools for Tackling Challenging Systems. J Vis Exp (132). doi: 10.3791/57199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Booth DS, Avila-Sakar A, Cheng Y (2011) Visualizing proteins and macromolecular complexes by negative stain EM: from grid preparation to image acquisition. J Vis Exp (58). doi: 10.3791/3227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rehbein P, Schwalbe H (2015) Integrated protocol for reliable and fast quantification and documentation of electrophoresis gels. Protein Expr Purif 110:1–6. doi: 10.1016/j.pep.2014.12.006 [DOI] [PubMed] [Google Scholar]
- 35.Alonso Villela SM, Kraiem H, Bouhaouala-Zahar B, Bideaux C, Aceves Lara CA, Fillaudeau L (2020) A protocol for recombinant protein quantification by densitometry. Microbiologyopen 9 (6):1175–1182. doi: 10.1002/mbo3.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]