

PURPOSE
Biliary tract cancers (BTCs) are aggressive cancers that carry a poor prognosis. An enhanced understanding of the immune landscape of anatomically and molecularly defined subsets of BTC may improve patient selection for immunotherapy and inform immune-based combination treatment strategies.
METHODS
We analyzed deidentified clinical, genomic, and transcriptomic data from the Tempus database to determine the mutational frequency and mutational clustering across the three major BTC subtypes (intrahepatic cholangiocarcinoma [IHC], extrahepatic cholangiocarcinoma, and gallbladder cancer). We subsequently determined the relationship between specific molecular alterations and anatomical subsets and features of the BTC immune microenvironment.
RESULTS
We analyzed 454 samples of BTC, of which the most commonly detected alterations were TP53 (42.5%), CDKN2A (23.4%), ARID1A (19.6%), BAP1 (15.5%), KRAS (15%), CDKN2B (14.2%), PBRM1 (11.7%), IDH1 (11.7%), TERT (8.4%), KMT2C (10.4%) and LRP1B (8.4%), and FGFR2 fusions (8.7%). Potentially actionable molecular alterations were identified in 30.5% of BTCs including 39.1% of IHC. Integrative cluster analysis revealed four distinct molecular clusters, with cluster 4 predominately associated with FGFR2 rearrangements and BAP1 mutations in IHC. Immune-related biomarkers indicative of an inflamed tumor-immune microenvironment were elevated in gallbladder cancers and in cluster 1, which was enriched for TP53, KRAS, and ATM mutations. Multiple common driver genes, including TP53, FGFR2, IDH1, TERT, BRAF, and BAP1, were individually associated with unique BTC immune microenvironments.
CONCLUSION
BTC subtypes exhibit diverse DNA alterations, RNA inflammatory signatures, and immune biomarkers. The association between specific BTC anatomical subsets, molecular alterations, and immunophenotypes highlights new opportunities for therapeutic development.
INTRODUCTION
Biliary tract cancers (BTCs) are a group of cancers that arise from the biliary tract and are historically subcategorized by their anatomical site of origin into intrahepatic cholangiocarcinomas (IHCs), extrahepatic cholangiocarcinomas (EHCs), and gallbladder (GB) cancers.1,2 BTC is the second most common primary hepatic malignancy after hepatocellular carcinoma and comprises approximately 3% of all gastrointestinal cancers.2-4 Although it is a rare cancer, the incidence of BTCs (0.3-6 per 100,000 individuals per year, with large geographic variation) and overall mortality burden have been increasing over the past few decades worldwide, representing a growing global health challenge.2,5 Across nearly all countries, BTC is frequently diagnosed during advanced stages of the disease, which limits therapeutic options and results in a poor prognosis.6 Significant improvements in long-term survival for patients with advanced disease have not occurred over the past decade, and 5-year survival rates remain low (7%-20%). Surgical resection is the most effective treatment, but tumor recurrence rates even after resection remain high.2
CONTEXT
Key Objective
Biliary tract cancers (BTCs) are increasingly subtyped by the presence of specific genomic alterations, but little is known about the immune microenvironment of molecularly defined subsets of BTC. The objective of this study was to characterize the immune landscape of molecularly defined subsets of BTC using genomic and transcriptomic data.
Knowledge Generated
We analyzed 454 samples of BTC and found that BTC subtypes exhibit diverse DNA alterations, RNA inflammatory signatures, and immune biomarkers. Potentially actionable molecular alterations were identified in 30.5% of BTCs including 39.1% of intrahepatic cholangiocarcinoma. We identified four clusters of BTC, each containing uniquely altered genes and a distinctive tumor-immune microenvironment.
Relevance
BTC clusters have distinct clinical and biologic features, and such clusters may provide opportunities for therapeutic development. We also identify relationships between certain molecular alterations and distinctive immunophenotypes, which may provide new opportunities for therapeutic development.
BTC is increasingly subtyped by the presence of specific genomic alterations identified through next-generation sequencing analyses. Prior efforts to sequence BTC have revealed that targetable genomic alterations occur with moderately high frequency in this cancer.7 These include alterations in well-studied genes including but not limited to FGFR2, IDH1, BRAF, and ERBB2 (HER2).7,8 The molecular profiles are known to vary between the different anatomical subtypes of the disease, with a higher rate of potentially actionable mutations identified in IHC than other anatomical subsets of BTC.7,9,10 Other common BTC alterations are not therapeutically targetable but nonetheless provide important prognostic information. A previous integrative clustering analysis of nearly 500 cholangiocarcinomas defined four clusters, with the cluster enriched in BAP1, IDH1/2, and FGFR2 alterations having a significantly better survival than the cluster enriched in TP53, ARID1A, and BRCA1/2 mutations.11
Immunotherapies targeting the programmed cell death protein 1 (PD1) axis and other immune checkpoints have transformed the management of many cancers, but have thus far demonstrated limited clinical activity in BTC. For example, the phase II KEYNOTE-158 trial enrolled 104 patients with BTCs, of whom six had an objective response to therapy (objective response rate 5.8%).12 Other phase II trials targeting the PD1 axis in BTC have similarly reported response rates of 2.9%-11%.13 Enhanced comprehension of the immune landscape in molecularly defined subsets of BTCs may specifically enhance patient selection for immunotherapy and inform the development of novel combination strategies for distinct molecularly defined subsets of this cancer.
A number of biomarkers related to immune system function have been discovered and developed to better predict responses to immunotherapy-based treatments. These biomarkers include quantities that can be calculated from either genomic or RNA expression–level data. Tumors with large numbers of genomic mutations (having a high tumor mutational burden [TMB]) appear to be particularly susceptible to immune-checkpoint blockade therapy.14,15 Similarly, programmed death-ligand 1 (PD-L1) protein levels are partially predictive of the response to anti–PD-L1 therapies across numerous cancer types.16-18 In most tumor types, TMB and PD-L1 expression each provide independent information and are minimally correlated with one another.19 Various other metrics describing immune cell infiltration and/or the expression of particular subsets of genes have been developed and are predictive of immunotherapy responses in particular contexts.20-24 A better understanding of the overall landscape of immune-related biomarkers in BTCs and possible subtype-specific differences may thus help guide future research and therapeutic options.
Here, we present real-world data for the mutational patterns of BTC subtypes from the Tempus clinicogenomic database, which contains clinical, genomic, and transcript-level data. Critically, we leverage the availability of RNA-based gene expression data to calculate several immune-related biomarkers of immunotherapy response and compare these differences across subtypes and mutational landscapes to provide insight into possible subtype-specific differences in immunotherapy treatment responses. Our study thus presents an overview of the intersection between particular genomic mutations, BTC subtypes, and immune-related biomarkers.
METHODS
Cohort Selection and Data Processing
The Tempus clinicogenomic database consists of deidentified clinical data and DNA and RNA sequencing, performed for the care of oncology patients in standard clinical practice. Patients were eligible for our cohort if they had a confirmed histologic diagnosis of cholangiocarcinoma, with paired clinical demographic data and RNA sequencing. Records were included in the cohort regardless of sex, race, stage, treatment status, or tissue sample site. Of 1,500 potential BTC records in the Tempus database, we identified BTC records with matched RNA and clinical data (N = 454). Of these 454 records, most had matched DNA sequencing data as well (n = 367). These data were derived from the Tempus xT solid tumor LDT assay (DNA-seq of 595-648 genes at 500× coverage, full transcriptome RNA-seq).25,26 The primary mutations identified by this assay include germline and/or somatic single-nucleotide polymorphisms (SNPs), insertions/deletions (Indels), fusions, and copy number variations. Intrahepatic, extrahepatic, and gallbladder cancer designations were derived from curated clinical data. All specimens undergo pathologist assessment of the hematoxylin and eosin slide for overall tumor amount and percent tumor cellularity as a ratio of tumor to normal nuclei. A minimum tumor cellularity of 20% is required to proceed for xT and RNA fusion analysis and 30% for RNA expression. Approval for this study was obtained from the Advarra Institutional Review Board Protocol (Pro00042950).
Sequencing and Processing of RNA Samples
RNA-seq gene expression data were generated from formalin-fixed, paraffin-embedded tumor samples using an exome capture–based RNA-seq protocol previously published.25,26 In brief, RNA-seq data were aligned to GRCh38 using STAR (2.4.0.1)27 and expression quantification per gene was computed using featureCounts (1.4.6).28 Normalized gene expression data for cancer types were log10-transformed and used for all subsequent analyses.
Mutation-Based Cluster Generation
We applied the agglomerative clustering method (part of the scikit-learn Python package) to cluster patients on the basis of the presence or absence of driver mutations in the following genes: IDH1, PBRM1, FGFR2, BRAF, ERBB2, KRAS, NRAS, TP53, PIK3CA, BRCA1, BRCA2, ATM, POLE, MET, BAP1, ARID1A, CDKN2A, CDKN2B, KMT2C, TERT, KMT2D, and LRP1B. The number of clusters n was determined by manually evaluating a range of possible values and observing that the most stable clusters occurred with n = 4. The other important clustering parameters that we set include the affinity (Euclidean) and linkage (ward).
TMB Estimation
TMB was derived from targeted genomic sequencing. The TMB was calculated by dividing the count of all nonsilent mutations (including missense SNPs/indels) by the total size of the panel coding region. For this analysis, we include 367 records (from the larger subset of 454) for which we had DNA-level information.
Immune-Related Biomarkers
All reported immune-related biomarkers were derived from RNA expression data. PD-L1 expression levels—calculated from the CD274 gene—were extracted for each sample from RNA expression data and are displayed as the log of the normalized abundances, following mean and variance transformation. Immune cytolytic activity (CYT) is derived from transcript levels of two key cytolytic effectors, granzyme A and perforin, and was calculated as described by Rooney et al.29 In a pan-cancer cohort, higher CYT scores were associated with a modest but improved long-term survival benefit. The neoadjuvant response signature (NRS) was calculated as described by Huang et al.30 In that study, higher NRS scores were associated with improved outcomes during anti–PD-1 therapy in stage III/IV melanoma. The immuno-predictive score (IMPRES) was calculated as described by Auslander et al.21 Higher scores on this metric were shown to be associated with high immune response and improved outcomes after immune checkpoint blockade therapy in melanomas. Estimates of immune cell infiltration were derived from an RNA deconvolution model, as previously described.25,31
Statistical Analysis
For all pairwise comparisons on continuous, normally distributed variables, we used the two-sided Student's t-test for P value calculation. Similarly, we used one-way analysis of variance for comparisons involving more than two groups. We applied the nonparametric Mann-Whitney U test or Kruskal-Wallis H test for statistical comparisons involving non-normally distributed continuous variables (TMB, RNA signatures, and immune infiltration) and Fisher's exact test for assessing significant differences between categorical variables. When multiple pairwise comparisons were performed, we used Bonferroni-corrected P value thresholds to ensure statistical robustness of our findings. All statistical tests were performed using the SciPy package in Python.
RESULTS
Clinical Characteristics, Demographic Features, and Mutational Patterns
Our retrospective study leveraged deidentified real-world data records from the Tempus clinicogenomic database, selecting records with matched RNA and clinical data (N = 454) as well as a subset with matched DNA, RNA, and clinical data (n = 367). Intrahepatic, extrahepatic, and gallbladder cancer data were included regardless of stage, treatment, or tumor site. Using this rich data set, we assessed associations between subtypes and a range of features including demographic (age, sex, and smoking status) and clinical characteristics (stage; Table 1).
TABLE 1.
Comparison of Clinical Characteristics of Data Records in the Overall Cohort and Across BTC Subtypes

Integrated Clustering of BTC Reveals Four Distinct Genomic Clusters
Across all subtypes, where available, we analyzed mutational patterns and detected alterations in TP53 (42.5%), CDKN2A (23.4%), ARID1A (19.6%), BAP1 (15.5%), CDKN2B (14.2%), KRAS (15%), PBRM1 (11.7%), IDH1 (11.7%), TERT (8.4%), KMT2C (10.4%), and LRP1B (8.4%), along with FGFR2 fusions (8.7%). We assessed associations between driver gene mutations and BTC subtypes. Consistent with previous studies,9,10 FGFR2 fusions and mutations in BAP1, IDH1, and PBRM1 were enriched in intrahepatic BTC. Mutations in TP53 and ERBB2 were observed at similar frequencies across gallbladder and extrahepatic BTCs but were significantly less common in intrahepatic BTC (Appendix Table A1).
To determine the potential benefit of molecular testing in BTC, we assessed the percentage of BTC samples for which testing identified potentially actionable alterations. For this analysis, we restricted our analysis to biomarkers for which tumor-agnostic drug approvals exist in the United States (eg, TMB > 10 mutations per megabase [m/MB], microsatellite instability or mismatch repair deficiency, or NTRK gene fusions) or phase II-III trials have demonstrated clear evidence of therapeutic benefit in biliary tract cancers (FGFR2 fusions or rearrangements, and IDH1, ERBB2, or BRAF [V600E] mutations). Using these criteria, we identified potentially actionable biomarkers in 30.5% of all BTCs, including 39.1% of IHC, 29.6% of EHC, and 15% of GB cancers.
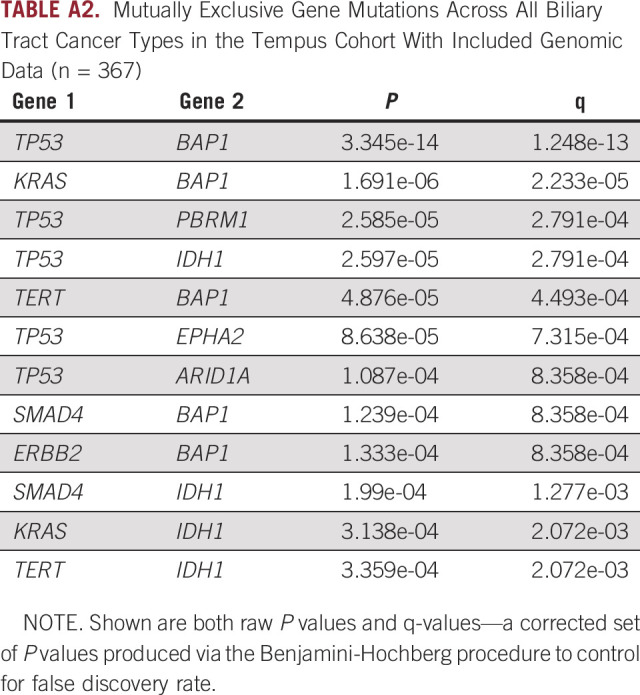
We observed a number of mutually exclusive mutational pairs (tested across all subtypes), where the presence of comutations is significantly lower than that expected on the basis of single mutation frequencies. The most notable example of mutual exclusivity that we observed was between TP53 and BAP1 (chi-squared test, P < .001). Furthermore, one of TP53, BAP1, or IDH1 was involved in all significantly identified mutually exclusive pairings that we identified (Appendix Table A2), consistent with previous studies.10
Clustering analysis on the basis of driver mutation status (22 genes in total) revealed four distinct clusters, with cluster 4 predominately associated with FGFR2 and BAP1 mutations in intrahepatic BTC (Fig 1). The rest of the clusters consisted of a mix between all three subtypes. Cluster 1 was enriched for TP53, KRAS, and ATM mutations, cluster 2 was enriched for CDKN2A/B alterations, cluster 3 was enriched for mutations in the chromatin-remodeling genes ARID1A and PBRM1, as well as IDH1 mutations.
FIG 1.
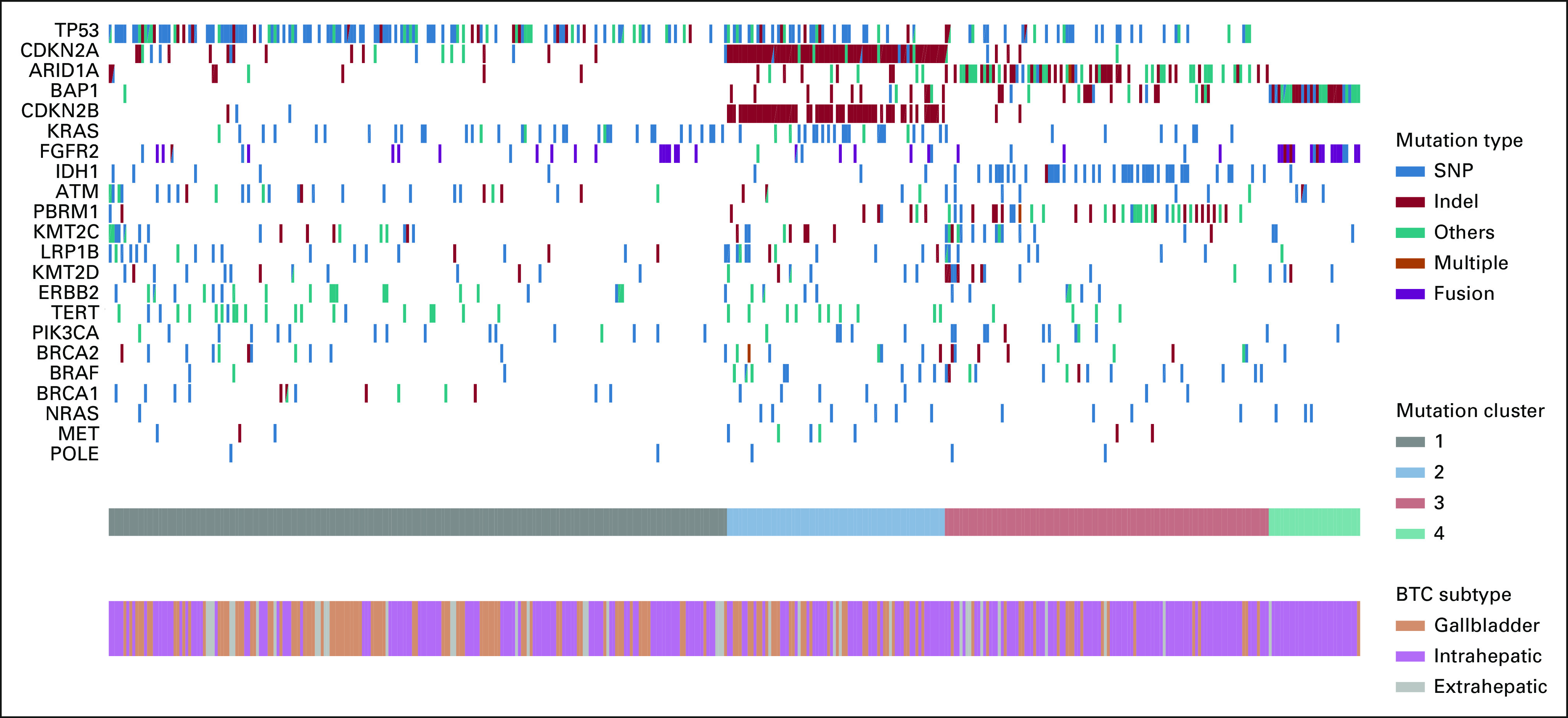
Mutational frequency and clustering across BTC subtypes. We detected alterations in TP53 (42.5%), CDKN2A (23.4%), ARID1A (19.6%), BAP1 (15.5%), CDKN2B (14.2%), KRAS (15%), PBRM1 (11.7%), IDH1 (11.7%), TERT (8.4%), KMT2C (10.4%), and LRP1B (8.4%), along with FGFR2 fusions (8.7%). Note that the others category may contain a variety of changes including splice variants, stop gain, start loss, amplification, or promoter region variants. Four distinct clusters on the basis of driver mutation status (22 genes) were detected. Clusters 1-3 crossed anatomical subsets of BTC, whereas cluster 4 was highly enriched for IHC (IHC: 96.7%, EHC: 3.3%, GB: 0%). Cluster 1 (IHC: 47.5%, EHC: 9%, and GB: 43.5%) was enriched for TP53, KRAS, ERBB2 (HER2), and ATM mutations, cluster 2 (IHC: 55%, EHC: 5%, and GB: 40%) was most enriched for CDKN2A/B alterations, and cluster 3 (IHC: 74.5%, EHC: 6.1%, and GB: 19.4%) was enriched for mutations in the chromatin-remodeling genes ARID1A and PBRM1 as well as IDH1 mutations, whereas cluster 4 consists primarily of FGFR2 fusions and BAP1 mutations. BTC, biliary tract cancer; EHC, extrahepatic cholangiocarcinoma; GB, gallbladder; IHC, intrahepatic cholangiocarcinoma; indel, insertions/deletions; SNP, single nucleotide polymorphism.
From our rich DNA-seq and RNA-seq data set, we quantified CD274 (PD-L1) gene expression levels and a variety of other immune-related biomarkers for each record. We subsequently examined the variation in these biomarkers across the four identified clusters and found a number of significant differences. Cluster 1—enriched in TP53, KRAS, and ATM variants—had the highest PD-L1 gene expression and was significantly higher than cluster 4—enriched in FGFR2 and BAP1 variants (Fig 2A). In addition, clusters 1 and 3 had significantly higher CYT scores than cluster 2 (Fig 2B). There were significant differences between clusters for a number of other immune-related biomarkers (Figs 2C and 2D); cluster 1 was generally associated with the highest scores, which is indicative of an inflamed tumor-immune microenvironment. TMB was similar across all four clusters.
FIG 2.

Cluster-biomarker associations (N = 454). (A) PD-L1 gene expression and (B) CYT scores for each of the four driver gene–defined clusters identified in Figure 1 (significant pairwise associations were assessed via the Mann-Whitney U test; *P < .01 and **P < .001 after correcting for multiple comparisons). (C) Immune-related biomarkers for which the Mann-Whitney U test showed significant differences across clusters (P < .01). For visualization, each box is colored according to the log-fold change between the single cluster when compared across aggregated values from all other clusters. Red denotes that the cluster had higher biomarker scores, whereas blue denotes lower. (D) Summary of cluster-defining features. BTC, biliary tract cancer; CYT, cytolytic activity; GEP, T-cell inflamed gene expression profile; IHC, intrahepatic cholangiocarcinoma; PD-L1, programmed death-ligand 1; NK, natural killer; NRS, neoadjuvant response signature.
Differences in TMB, PD-L1, and Immune-Related Signatures Across BTC Subtypes
Previous analyses show that BTC subtypes exhibit a range of possible mutations and generally fail to cluster according to subtype when looking only at driver gene mutations. However, we discovered that these clusters have significant variation across a number of immune-related biomarkers. We next wanted to assess whether subtypes themselves exhibit distinct immune-related features to get a better understanding of subtype diversity at the molecular level.
Within the intrahepatic, extrahepatic, and gallbladder BTC subtypes, we observed that both TMB and PD-L1 gene expression are highly variable (Figs 3A and 3B). TMB was higher in extrahepatic and gallbladder cancers than in intrahepatic cancers with median TMB values of 2.5 m/MB for gallbladder, 1.92 m/MB for intrahepatic, and 2.63 m/MB for extrahepatic subtypes. Across all possible pairwise comparisons, we observed significant differences (Mann-Whitney U test, P < .01) between gallbladder and intrahepatic subtypes and between intrahepatic and extrahepatic subtypes. Median PD-L1 gene expression values (log of the normalized abundances, following mean and variance transformation) were 0.997 for gallbladder, 0.875 for intrahepatic, and 1.01 for extrahepatic subtypes. Across all possible pairwise comparisons, the only significant difference we observed was between gallbladder and intrahepatic subtypes.
FIG 3.

Subtype-specific differences in immune-related biomarkers: (A) TMB (n = 367), (B) PD-L1 gene expression (N = 454), (C) CYT (N = 454), (D) NRS (N = 454), and (E) immuno-predictive (IMPRES, N = 454) scores for different BTC subtypes. All pairwise comparisons were performed, and significant comparisons—after correcting for multiple testing—are shown (*P < .01, **P < .001). Boxes show the 25th-75th percentiles, and red lines denote the sample medians. BTC, biliary tract cancer; CYT, cytolytic activity; EHC, extrahepatic cholangiocarcinoma; GB, gallbladder; IHC, intrahepatic cholangiocarcinoma; NRS, neoadjuvant response signature; TMB, tumor mutational burden.
We next used our RNA-seq data set to investigate subtype-specific differences in immune-related biomarkers that have been previously described, notably CYT, NRS, and immuno-predictive (IMPRES) scores21,29,30 (Figs 3C-3E). Scores were relatively similar across these major BTC subsets, but with a general observed trend of higher immune scores in gallbladder and lower immune scores in intrahepatic. Median CYT scores were 1.89 for gallbladder, 1.75 for intrahepatic, and 1.86 for extrahepatic subtypes. Median NRS scores were 2.09 for gallbladder, 1.98 for intrahepatic, and 2.02 extrahepatic subtypes. For both CYT and NRS scores, the differences between gallbladder and intrahepatic subtypes were significant (Student's t-test, P < .01 after correcting for multiple comparisons), whereas all other pairwise comparisons were insignificant. Median IMPRES scores were 9.0 for gallbladder, 9.0 for intrahepatic, and 10.0 for extrahepatic subtypes. None of the possible pairwise comparisons were significantly different for this score.
Immune-Related Features Vary Significantly Across Genotypes
Previous analyses show that BTC subtypes significantly vary according to numerous biomarkers of immune function. In addition, biomarkers of immune function were unique across the four clusters that we have identified. Interestingly, each of these clusters were enriched for a distinct set of genetic alterations. However, our real-world data set consists of hundreds of tumors with a variety of genomic mutations. We wondered if the differences in immune function biomarkers were driven by specific individual genetic alterations found within each cluster. We thus assessed gene-biomarker associations across the subset of patients for whom we have matched DNA, RNA, and clinical data (n = 367). We expanded our set of immune biomarkers to encompass a broad range of features that have been shown in various studies to play a role in either predicting immune function or responses to immunotherapies. We display our findings as a heat map where colored blocks indicate significantly correlated gene-biomarker pairs (log-fold change) in mutant versus wild-type groups (Mann-Whitney U test, P < .05 after correction for multiple testing; Fig 4). Shown are the subset of commonly characterized driver genes in BTC for which we observed a statistically significant interaction with at least one immune biomarker.
FIG 4.

Gene-biomarker associations across patients with matched DNA, RNA, and clinical data (n = 367). Colored blocks in the heat map indicate significantly correlated gene-biomarker pairs in mutant versus WT groups (colors represent log-fold change, Mann-Whitney U test, P < .05 multiple-testing correction). Red squares indicate examples where the given biomarker is higher in mutant (relative to WT) genotypes, whereas blue squares denote cases where it is lower. CYT, cytolytic activity; DC, dendritic cell; GEP, T-cell inflamed gene expression profile; NRS, neoadjuvant response signature; PD-L1, programmed death-ligand 1; WT, wild-type
Across all comparisons, we observed roughly equal numbers of instances where specific biomarker signatures were higher (and lower) in mutant (relative to wild-type) genotypes. Records with mutant TP53, for instance, had significantly higher TMB (red squares, Fig 4). By contrast, PD-L1 expression was significantly lower in records where BAP1 was mutated (blue squares, Fig 4). Most genes showed only a small number of significant associations, but IDH1, TP53, BRAF, and BAP1 each had several significant differences in immune biomarkers across mutant and wild-type genotypes, consistent with previous reports.
DISCUSSION
We analyzed a clinically annotated cohort of more than 400 BTCs using the Tempus molecular profiling platform. To our knowledge, this is the largest reported cohort of BTC with comprehensive genomic and transcriptomic profiling. Consistent with previous reports, we find that a high frequency of BTCs have potentially actionable molecular alterations, especially IHCs, supporting the use of molecular profiling for patients with BTC. Our estimate that 39.1% of IHCs have potentially actionable molecular alterations is a relatively conservative estimate because we only included biomarkers supported by published phase II-III studies in BTC or tumor-agonistic approvals. Inclusion of other biomarkers with the potential to be actionable on the basis of case reports in BTC, including but not limited to ROS1 fusions or BRCA1/2 mutations,32 would have resulted in a higher estimation of patients standing to benefit from multimodal genomic profiling.
BTCs are a heterogeneous group of tumors but are generally treated similarly with the exception of the subset of BTCs with potentially actionable molecular alterations. Here, we find that several biomarkers thought to be associated with anti-PD1 sensitivity—including TMB, PD-L1 expression, and other immune-related biomarkers indicative of an inflamed tumor-immune microenvironment—were elevated in GB cancer as compared with IHC and EHC. Clinical trials of PD1-targeted/PD-L1–targeted therapy in BTC have reported only modest clinical activity, but have generally recruited a relatively small number of patients with GB cancer. This makes it difficult to assess the efficacy of anti-PD1 therapy in this anatomical subset of BTC. Our data are hypothesis-generating but suggest that GB cancer could be more sensitive to anti-PD1 therapy than other anatomical subsets, and further evaluation of anti-PD1 therapy and other immunotherapies in GB cancer is warranted.
Several molecular alterations identified in our data set were mutually exclusive, suggesting that such alterations define distinct BTC subtypes. Our cluster analyses revealed four distinct clusters of BTC. Cluster 4 (consisting primarily of FGFR2 fusions and BAP1 mutations in IHC) has been identified in two other integrative clustering analyses of BTC.11,33 Conversely, other clusters identified in this work were not obviously matched with these other analyses. We acknowledge that the genomic heterogeneity of BTCs might very well reflect the diverse underlying risk factors and associated pathologies. We therefore hypothesize that differences in the patient populations may account for some of these differences, as our samples were exclusively from North America where the incidence of liver fluke–associated BTC is rare or absent, whereas clustering analyses conducted on behalf of the International Cancer Genome Consortium included a large number of fluke-associated BTCs from Asia.
Our clustering analyses demonstrate that the molecular profiling may provide distinct information about the biology of BTC, even for tumors for which targeted therapies are not yet available. With the exception of cluster 4, all the other subsets comprise a mix of IHC, EHC, and GB tumors, indicating that the molecular data provide information beyond what can be learned from the anatomical site. Although cluster 4 appears to be driven by abrupt genomic events (eg, FGFR2 rearrangements or fusions), the biology of cluster 3 appears to be driven by genes that regulate transcription, DNA repair, and the epigenetic landscape, which, in turn, lead to tumor progression. Specifically, IDH1 mutations are enriched in this cluster and have been shown to increase 2-hydroxyglutarate oncometabolite production, leading to widespread epigenetic dysregulation.34,35
Our retrospective analysis of a real-world data set is advantageous because of the scale and complexity of data that we are able to obtain, but it nevertheless has several possible limitations. First and most foremost is that real-world data are heterogeneous. We investigated possible factors that could confound our analyses (Table 1), but differences across a range of other demographic or clinical features—such as prior therapies—may obscure important effects or bias our results. We also focused a portion of our analyses on RNA, but RNA and protein abundances do not exhibit a one-to-one correspondence. Finally, the objective of our study was to assess differences in molecular-level and genome-level features across subtypes, and it is important to note that we did not consider clinical outcomes or end points.
In summary, we identified a high frequency of potentially actionable molecular alterations in BTC, and we believe that molecular profiling should be considered for all patients who may stand to benefit from the discovery of a potentially actionable mutation in this population. With the exception of a few biomarker-indicated therapies (eg, FGFR2 inhibitors for FGFR2 fusion–positive BTC), most BTC is treated without regard to molecular drivers and most therapeutic trials are conducted across anatomical and molecular clusters. Our results indicate that specific BTC clusters have distinct clinical and biologic features and such clusters may provide opportunities for therapeutic development. We also identify relationships between individual driver genes and certain immune-related features, including enhanced M2 polarization of macrophages in IDH1-mutated BTC and low immune infiltration in BTC with FGFR2 fusions or rearrangements, providing initial evidence for combining targeted inhibition of specific drivers to reprogram the tumor-immune microenvironment in combination with systemic immunotherapy.
ACKNOWLEDGMENT
The authors thank Alexandria Bobe and members of the Tempus Scientific Communications team for feedback on the manuscript and the Cholangiocarcinoma Foundation for support for this project.
APPENDIX
TABLE A1.
Driver Gene Associations With Particular Biliary Tract Cancer Subtypes (n = 367)

TABLE A2.
Mutually Exclusive Gene Mutations Across All Biliary Tract Cancer Types in the Tempus Cohort With Included Genomic Data (n = 367)
Kabir Mody
Stock and Other Ownership Interests: CytoDyn, ONCOtherapeutics
Consulting or Advisory Role: Celgene, Genentech/Roche, Merrimack, Eisai, AstraZeneca, Vicus Therapeutics, Ipsen, Boston Scientific, BTG, BTG, Exelixis, Incyte (Inst), QED Therapeutics
Research Funding: FibroGen (Inst), Senhwa Biosciences (Inst), ARIAD (Inst), TRACON Pharma (Inst), MedImmune (Inst), Agios (Inst), ArQule (Inst), Taiho Pharmaceutical (Inst), Gritstone Bio (Inst), Incyte (Inst), Merck (Inst), Vyriad (Inst), Turnstone Bio (Inst), AstraZeneca (Inst), Basilea (Inst)
Prerna Jain
Employment: Tempus, Concerto HealthAI
Travel, Accommodations, Expenses: Tempus
Sherif M. El-Refai
Employment: Tempus
Stock and Other Ownership Interests: Tempus
Nilofer S. Azad
Consulting or Advisory Role: QED Therapeutics, Merck, Incyte, Helsinn Therapeutics/QED Therapeutics, AstraZeneca, Mirati Therapeutics
Research Funding: Celgene (Inst), Genentech (Inst), Astex Pharmaceuticals (Inst), Agios (Inst), Merck (Inst), Bristol Myers Squibb (Inst), Syndax (Inst), Array BioPharma (Inst), Intensity Therapeutics (Inst), Bayer (Inst), EMD Serono, Debiopharm Group (Inst), Incyte (Inst), Loxo/Lilly (Inst), AtlasMedx (Inst)
Daniel J. Zabransky
Honoraria: Remedy Health Group, LLC
Research Funding: Roche/Genentech
Patents, Royalties, Other Intellectual Property: Under separate licensing agreements between Horizon Discovery, Ltd and The Johns Hopkins University, I am entitled to a share of royalties received by the university on sales of products
Rachna T. Shroff
Consulting or Advisory Role: Exelixis, Merck, QED Therapeutics, Incyte, AstraZeneca, Taiho Pharmaceutical, Boehringer Ingelheim, Servier, Genentech, Basilea
Research Funding: Pieris Pharmaceuticals, Taiho Pharmaceutical, Merck, Exelixis, QED Therapeutics, Rafael Pharmaceuticals, Bristol Myers Squibb, Bayer, Immunovaccine, Seattle Genetics, Novocure, NuCana, Loxo/Lilly, Faeth Therapeutics
R. Katie Kelley
Consulting or Advisory Role: Agios (Inst), AstraZeneca (Inst), Bristol Myers Squibb (Inst), Genentech/Roche, Merck (Inst), Gilead Sciences, Exact Sciences, Kinnate Biopharma, Exelixis/Ipsen (Inst)
Research Funding: Lilly (Inst), Exelixis (Inst), Novartis (Inst), Bristol Myers Squibb (Inst), MedImmune (Inst), Merck Sharp & Dohme (Inst), Agios (Inst), AstraZeneca (Inst), Adaptimmune (Inst), Taiho Pharmaceutical (Inst), Bayer (Inst), QED Therapeutics (Inst), EMD Serono (Inst), Partner Therapeutics (Inst), Genentech/Roche (Inst), Surface Oncology (Inst), Relay Therapeutics (Inst), Loxo/Lilly (Inst)
Travel, Accommodations, Expenses: Ipsen
Anthony B. El-Khouiery
Honoraria: Bayer, Bristol Myers Squibb, Roche/Genentech, EMD Serono, Eisai, Merck, Agenus, Pieris Pharmaceuticals, Exelixis, CytomX Therapeutics, Gilead Sciences, AstraZeneca/MedImmune, ABL Bio, QED Therapeutics, Servier, Tallac Therapeutics
Consulting or Advisory Role: CytomX Therapeutics, Bristol Myers Squibb, Bayer, Eisai, Roche, EMD Serono, Merck, Exelixis, Pieris Pharmaceuticals, Agenus, Gilead Sciences, AstraZeneca/MedImmune, ABL Bio, QED Therapeutics, Servier, Tallac Therapeutics
Research Funding: AstraZeneca, Astex Pharmaceuticals, Fulgent Genetics
Adam J. Hockenberry
Employment: Tempus
Stock and Other Ownership Interests: Tempus
Denise Lau
Employment: Tempus
Stock and Other Ownership Interests: Tempus
Patents, Royalties, Other Intellectual Property: Tempus
Gregory B. Lesinski
Consulting or Advisory Role: ProDa
Research Funding: Merck (Inst), Bristol Myers Squibb (Inst), Novartis (Inst), Boehringer Ingelheim (Inst), Vaccinex (Inst)
Mark Yarchoan
Consulting or Advisory Role: Eisai, Exelixis, AstraZeneca, Genentech/Roche, Replimune, Hepion Pharmaceuticals
Research Funding: Bristol Myers Squibb (Inst), Merck (Inst), Exelixis (Inst), Incyte (Inst)
Uncompensated Relationships: Merck
No other potential conflicts of interest were reported.
SUPPORT
Supported by The Cholangiocarcinoma Foundation (to M.Y.), the National Cancer Institute Specialized Program of Research Excellence (SPORE) in Gastrointestinal Cancers (P50 CA062924), the NIH Center Core Grant (P30 CA006973), and the American Society of Clinical Oncology Career Development Award.
K.M. and P.J. contributed equally to this work.
AUTHOR CONTRIBUTIONS
Conception and design: Kabir Mody, Prerna Jain, Nilofer S. Azad, Anthony B. El-Khouiery, Gregory B. Lesinski, Mark Yarchoan
Administrative support: Sherif M. El-Refai, Mark Yarchoan
Provision of study materials or patients: Sherif M. El-Refai, Mark Yarchoan
Collection and assembly of data: Kabir Mody, Prerna Jain, Sherif M. El-Refai, Rachna T. Shroff, R. Katie Kelley, Adam J. Hockenberry
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Kabir Mody
Stock and Other Ownership Interests: CytoDyn, ONCOtherapeutics
Consulting or Advisory Role: Celgene, Genentech/Roche, Merrimack, Eisai, AstraZeneca, Vicus Therapeutics, Ipsen, Boston Scientific, BTG, BTG, Exelixis, Incyte (Inst), QED Therapeutics
Research Funding: FibroGen (Inst), Senhwa Biosciences (Inst), ARIAD (Inst), TRACON Pharma (Inst), MedImmune (Inst), Agios (Inst), ArQule (Inst), Taiho Pharmaceutical (Inst), Gritstone Bio (Inst), Incyte (Inst), Merck (Inst), Vyriad (Inst), Turnstone Bio (Inst), AstraZeneca (Inst), Basilea (Inst)
Prerna Jain
Employment: Tempus, Concerto HealthAI
Travel, Accommodations, Expenses: Tempus
Sherif M. El-Refai
Employment: Tempus
Stock and Other Ownership Interests: Tempus
Nilofer S. Azad
Consulting or Advisory Role: QED Therapeutics, Merck, Incyte, Helsinn Therapeutics/QED Therapeutics, AstraZeneca, Mirati Therapeutics
Research Funding: Celgene (Inst), Genentech (Inst), Astex Pharmaceuticals (Inst), Agios (Inst), Merck (Inst), Bristol Myers Squibb (Inst), Syndax (Inst), Array BioPharma (Inst), Intensity Therapeutics (Inst), Bayer (Inst), EMD Serono, Debiopharm Group (Inst), Incyte (Inst), Loxo/Lilly (Inst), AtlasMedx (Inst)
Daniel J. Zabransky
Honoraria: Remedy Health Group, LLC
Research Funding: Roche/Genentech
Patents, Royalties, Other Intellectual Property: Under separate licensing agreements between Horizon Discovery, Ltd and The Johns Hopkins University, I am entitled to a share of royalties received by the university on sales of products
Rachna T. Shroff
Consulting or Advisory Role: Exelixis, Merck, QED Therapeutics, Incyte, AstraZeneca, Taiho Pharmaceutical, Boehringer Ingelheim, Servier, Genentech, Basilea
Research Funding: Pieris Pharmaceuticals, Taiho Pharmaceutical, Merck, Exelixis, QED Therapeutics, Rafael Pharmaceuticals, Bristol Myers Squibb, Bayer, Immunovaccine, Seattle Genetics, Novocure, NuCana, Loxo/Lilly, Faeth Therapeutics
R. Katie Kelley
Consulting or Advisory Role: Agios (Inst), AstraZeneca (Inst), Bristol Myers Squibb (Inst), Genentech/Roche, Merck (Inst), Gilead Sciences, Exact Sciences, Kinnate Biopharma, Exelixis/Ipsen (Inst)
Research Funding: Lilly (Inst), Exelixis (Inst), Novartis (Inst), Bristol Myers Squibb (Inst), MedImmune (Inst), Merck Sharp & Dohme (Inst), Agios (Inst), AstraZeneca (Inst), Adaptimmune (Inst), Taiho Pharmaceutical (Inst), Bayer (Inst), QED Therapeutics (Inst), EMD Serono (Inst), Partner Therapeutics (Inst), Genentech/Roche (Inst), Surface Oncology (Inst), Relay Therapeutics (Inst), Loxo/Lilly (Inst)
Travel, Accommodations, Expenses: Ipsen
Anthony B. El-Khouiery
Honoraria: Bayer, Bristol Myers Squibb, Roche/Genentech, EMD Serono, Eisai, Merck, Agenus, Pieris Pharmaceuticals, Exelixis, CytomX Therapeutics, Gilead Sciences, AstraZeneca/MedImmune, ABL Bio, QED Therapeutics, Servier, Tallac Therapeutics
Consulting or Advisory Role: CytomX Therapeutics, Bristol Myers Squibb, Bayer, Eisai, Roche, EMD Serono, Merck, Exelixis, Pieris Pharmaceuticals, Agenus, Gilead Sciences, AstraZeneca/MedImmune, ABL Bio, QED Therapeutics, Servier, Tallac Therapeutics
Research Funding: AstraZeneca, Astex Pharmaceuticals, Fulgent Genetics
Adam J. Hockenberry
Employment: Tempus
Stock and Other Ownership Interests: Tempus
Denise Lau
Employment: Tempus
Stock and Other Ownership Interests: Tempus
Patents, Royalties, Other Intellectual Property: Tempus
Gregory B. Lesinski
Consulting or Advisory Role: ProDa
Research Funding: Merck (Inst), Bristol Myers Squibb (Inst), Novartis (Inst), Boehringer Ingelheim (Inst), Vaccinex (Inst)
Mark Yarchoan
Consulting or Advisory Role: Eisai, Exelixis, AstraZeneca, Genentech/Roche, Replimune, Hepion Pharmaceuticals
Research Funding: Bristol Myers Squibb (Inst), Merck (Inst), Exelixis (Inst), Incyte (Inst)
Uncompensated Relationships: Merck
No other potential conflicts of interest were reported.
REFERENCES
- 1.Rizvi S, Khan SA, Hallemeier CL, et al. : Cholangiocarcinoma—evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 15:95-111, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banales JM, Cardinale V, Carpino G, et al. : Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol 13:261-280, 2016 [DOI] [PubMed] [Google Scholar]
- 3.DeOliveira ML, Cunningham SC, Cameron JL, et al. : Cholangiocarcinoma: Thirty-one-year experience with 564 patients at a single institution. Ann Surg 245:755-762, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakeeb A, Pitt HA, Sohn TA, et al. : Cholangiocarcinoma. A spectrum of intrahepatic, perihilar, and distal tumors. Ann Surg 224:463-473, 1996; discussion 473-475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertuccio P, Malvezzi M, Carioli G, et al. : Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J Hepatol 71:104-114, 2019 [DOI] [PubMed] [Google Scholar]
- 6.Charbel H, Al-Kawas FH: Cholangiocarcinoma: Epidemiology, risk factors, pathogenesis, and diagnosis. Curr Gastroenterol Rep 13:182-187, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Valle JW, Lamarca A, Goyal L, et al. : New horizons for precision medicine in biliary tract cancers. Cancer Discov 7:943-962, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudley JC, Zheng Z, McDonald T, et al. : Next-generation sequencing and fluorescence in situ hybridization have comparable performance characteristics in the analysis of pancreaticobiliary brushings for malignancy. J Mol Diagn 18:124-130, 2016 [DOI] [PubMed] [Google Scholar]
- 9.Weinberg BA, Xiu J, Lindberg MR, et al. : Molecular profiling of biliary cancers reveals distinct molecular alterations and potential therapeutic targets. J Gastrointest Oncol 10:652-662, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowery MA, Ptashkin R, Jordan E, et al. : Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: Potential targets for intervention. Clin Cancer Res 24:4154-4161, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jusakul A, Cutcutache I, Yong CH, et al. : Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov 7:1116-1135, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piha-Paul SA, Oh D-Y, Ueno M, et al. : Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: Results from the KEYNOTE-158 and KEYNOTE-028 studies. Int J Cancer 147:2190-2198, 2020 [DOI] [PubMed] [Google Scholar]
- 13.Kim RD, Chung V, Alese OB, et al. : A phase 2 multi-institutional study of nivolumab for patients with advanced refractory biliary tract cancer. JAMA Oncol 6:888-894, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yarchoan M, Hopkins A, Jaffee EM: Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 377:2500-2501, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan TA, Yarchoan M, Jaffee E, et al. : Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann Oncol 30:44-56, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reck M, Rodríguez-Abreu D, Robinson AG, et al. : Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med 375:1823-1833, 2016 [DOI] [PubMed] [Google Scholar]
- 17.Bellmunt J, de Wit R, Vaughn DJ, et al. : Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med 376:1015-1026, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferris RL, Blumenschein G Jr, Fayette J, et al. : Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med 375:1856-1867, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yarchoan M, Albacker LA, Hopkins AC, et al. : PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 4:e126908, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cristescu R, Mogg R, Ayers M, et al. : Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362:eaar3593, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Auslander N, Zhang G, Lee JS, et al. : Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat Med 24:1545-1549, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang P, Gu S, Pan D, et al. : Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24:1550-1558, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fridman WH, Pagès F, Sautès-Fridman C, et al. : The immune contexture in human tumours: Impact on clinical outcome. Nat Rev Cancer 12:298-306, 2012 [DOI] [PubMed] [Google Scholar]
- 24.Tumeh PC, Harview CL, Yearley JH, et al. : PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568-571, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beaubier N, Bontrager M, Huether R, et al. : Integrated genomic profiling expands clinical options for patients with cancer. Nat Biotechnol 37:1351-1360, 2019 [DOI] [PubMed] [Google Scholar]
- 26.Beaubier N, Tell R, Lau D, et al. : Clinical validation of the tempus xT next-generation targeted oncology sequencing assay. Oncotarget 10:2384-2396, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobin A, Davis CA, Schlesinger F, et al. : STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29:15-21, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao Y, Smyth GK, Shi W: featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923-930, 2014 [DOI] [PubMed] [Google Scholar]
- 29.Rooney MS, Shukla SA, Wu CJ, et al. : Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160:48-61, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang AC, Orlowski RJ, Xu X, et al. : A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med 25:454-461, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newman AM, Liu CL, Green MR, et al. : Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12:453-457, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jakubowski CD, Mohan AA, Kamel IR, et al. : Response to crizotinib in ROS1 fusion–positive intrahepatic cholangiocarcinoma. JCO Precis Oncol 4:825-828, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farshidfar F, Zheng S, Gingras M-C, et al. : Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep 18:2780-2794, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sturm D, Witt H, Hovestadt V, et al. : Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425-437, 2012 [DOI] [PubMed] [Google Scholar]
- 35.Mazor T, Chesnelong C, Pankov A, et al. : Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc Natl Acad Sci USA 114:10743-10748, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]