

Introduction
Pilocytic astrocytomas are primary CNS WHO grade I glial tumors and are the most common among childhood brain tumors.1 Although surgery remains as a primary treatment modality, novel nonsurgical options continue to be investigated as these tumors frequently harbor genomic alterations within the mitogen-activated protein kinase (MAPK) signaling pathway.2,3 Although the majority of pilocytic astrocytoma harbor an identified MAPK pathway alteration, in particular, KIAA1549-BRAF fusions, BRAFV600E mutations, and BRAFinsVLR, along with other BRAF fusions, a subset of these tumors are driven by alterations outside of these common drivers.2,4-6 Additional alterations identified by whole-genome sequencing with paired whole-transcriptome sequencing included fibroblast growth factor receptor 1 (FGFR1) mutations, NTRK2 fusions, NF1 mutations, and PTPN11 mutations. Development of targeted treatments focused on these alterations and the MAPK pathway continues to be an area of interest in the treatment of glial tumors.
Here, we present a case of FGFR1N546K-mutated juvenile pilocytic astrocytoma successfully treated with a pan-FGFR inhibitor pemigatinib illustrating the intracranial activity.
Case Report
A 32-year-old male patient presented to The University of Texas MD Anderson Cancer Center after treatment at an outside hospital to discuss additional advanced treatment options. The patient was diagnosed with juvenile pilocytic astrocytoma diagnosed at age 13 years. At that time, the patient presented with headache and double vision. On imaging, the patient was noted to have obstructive hydrocephalus because of a third ventricular lesion and underwent a bilateral ventriculoperitoneal shunt placement and biopsy of the mass. Pathology was diagnostic for pilocytic astrocytoma. The decision was made to monitor the patient with serial imaging, which showed interval progression at 4 months postbiopsy, after which the patient was referred to radiation oncology. That patient underwent hypothalamic intensity-modulated radiation therapy at a dose of 5,400 cGy in 30 fractions. The patient was followed with serial imaging for the next 11 years with no tumor progression noted, after which the patient left the country and was unable to obtain imaging. Several years later, the patient presented with worsening visual deficits described as a binasal hemianopia, imaging showed tumor progression of the third ventricular lesion, and the patient underwent a near gross total resection. Pathology at this time showed a recurrent pilocytic astrocytoma with anaplasia, noted to have Rosenthal fibers, granular bodies, and scattered mitotic figures. Immunohistochemical studies showed retained expression of ATRX, and few cells were positive for wild-type p53 and negative for BRAFV600E and histone H3K27M. Fluorescence in situ hybridization studies noted sporadic amplification of PDGFR in the form of double minutes. Additional studies showed no duplication or rearrangement of BRAF or deletion of CDKN2A. The patient's postoperative course was complicated by hypopituitarism and fluctuating sodium levels, including episodes of both hyponatremia and hypernatremia, ultimately leading to an admission to the intensive care unit. Once stabilized, additional radiation treatment was not considered because of potential cognitive side effects.
On presentation, the patient denied any neurologic or cognitive deficits but was found to be oriented only to person with a fund of knowledge that was impaired from that expected for his age and level of education. Physical examination was grossly unremarkable with cranial nerves II-XII intact, the muscle strength was 5/5 bilaterally in all extremities, all deep tendon reflexes were two bilaterally, and there were no abnormal reflexes (Babinski, Hoffman, and jaw jerk were all negative). The patient had no cerebellar symptoms including nystagmus, finger-to-nose test, heel-to-shin test, and tremors, and the patient's gait was normal.
Comprehensive next-generation sequencing was performed using the FoundationOne panel (Foundation Medicine; Cambridge, MA), which identified an FGFR1N546K mutation (summarized in Table 1). Given his worsening deficits, after discussion with patient and consensus in the multidisciplinary tumor board, the patient was enrolled in an ongoing dose-escalation phase I/II clinical trial of pemigatinib in advanced malignancies with FGFR mutations (NCT02393248). After screening and baseline tests, the patient received 13.5 mg once daily oral dosing continuously on a 21-day cycle. Serial imaging was compared with the patient's baseline magnetic resonance imaging, and response was graded by RECIST V 1.1 (Figs 1A and 1B).7 The patient achieved a partial response with a 52% reduction at first restaging, which deepened to a 91% reduction as the best response in 13 cycles (Figs 1C and 1D) and sustained this response for a total of 18 months before a slight interval increase of the tumor on magnetic resonance imaging. The patient tolerated the treatment reasonably well for the first 6 months and had to undergo dose reduction to 9 mg once daily because of elevated liver function tests (grade 2 ALT and grade 1 AST from a history of fatty liver), mucositis (grade 1), and hand-foot syndrome (grade 2). Shortly thereafter, the imaging showed progressive disease approximately 19 months after treatment initiation. At this time, the decision was made to continue this course of treatment because of the continuous clinical benefit, despite radiographic progression up to 27 cycles at which time the patient was taken off the trial.
TABLE 1.
Clinical Next-Generation Sequencing Results From FoundationOne Testing
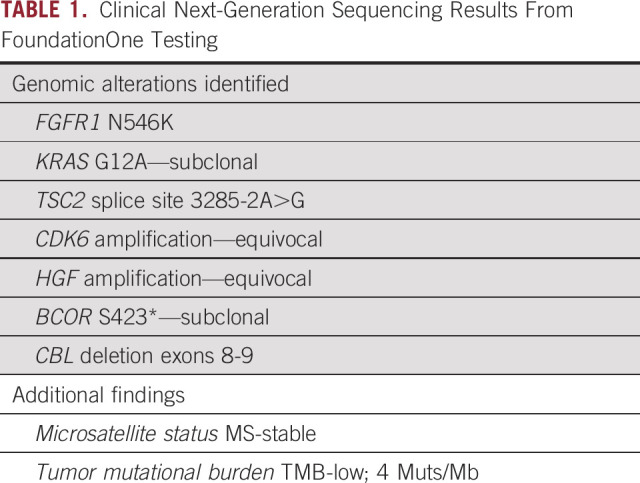
FIG 1.

Baseline MRI images after intensity-modulated radiation therapy and near gross total resection of hypothalamic pilocytic astrocytoma. (A) Axial FLAIR and (B) postcontrast images demonstrating the residual enhancing tumor. After 10 months of treatment, hypothalamic tumor still demonstrates (C) good response with a significant reduction in tumor size in axial FLAIR and (D) only minimal residual focus of enhancement present in postcontrast MR images. MRI, magnetic resonance imaging.
Consent
Informed consent to publish information and/or images from the patient was obtained for this study.
Discussion
Pemigatinib, a selective pan-fibroblast growth factor receptor (FGFR) inhibitor now US Food and Drug Administration–approved for use in locally advanced or metastatic cholangiocarcinoma with an FGFR2 fusion or rearrangement, continues to be investigated in various neoplasms.8-17 This pan-FGFR inhibitor is a small-molecule kinase inhibitor that exerts its main effects in FGFR1, FGFR2, and FGFR3 and a minor effect on FGFR4 with a half maximal inhibitory concentration (IC50) of < 2 nM.17-19 In vitro preclinical data using cancer cell lines, including those from lung, gastric, endometrial, bladder, and hematologic malignancies, showed that pemigatinib can effectively inhibit phosphorylation of FGFR1-3, which decreased downstream signaling and cell viability.18,19 These lines all harbored various FGFR alterations, including amplifications, mutations, fusions, and translocations, all of which showed a response to pemigatinib.18 Related FGFR alterations in human cancers can lead to constitutive activation of the FGFR pathway, leading to increased survival and malignant transformation. In vivo studies using a mouse xenograft model implanted with FGFR1-3–altered human tumors all showed antitumor activity of pemigatinib, including models of cholangiocarcinoma expressing the FGFR2-Transformer-2 beta homolog (TRA2b) fusion protein, FGFR2-amplified gastric cancer, FGFROP2-FGFR2 fusion–positive leukemia, and FGFR3-TACC fusion bladder carcinoma.18,19 Taken in combination, the preclinical in vitro and in vivo data show efficacy of pemigatinib across a wide variety of alterations within the FGFR pathway.
FGFR1N546K is a hot spot mutation in the tyrosine kinase domain (Fig 2), known to be activating and oncogenic, and is predominantly seen in CNS tumors.20,21 This mutation resides within the kinase binding domain of the FGFR1 gene, unlike other oncogenic mutations within FGFR1, and does not appear to alter the tertiary structure of the protein, but does alter the surface charge.22 The N546K mutation has been implicated in the in vitro transformation of cells and has shown altered autophosphorylation, leading to increased catalytic activity and downstream activation of the MAPK pathway.2,23 FGFR1N546K is present in 0.11% of AACR GENIE cases, with low-grade glioma not otherwise specified (NOS), conventional glioblastoma multiforme, glioblastoma, high-grade glioma NOS, and rosette-forming glioneuronal tumor of the fourth ventricle having the greatest prevalence (Dataset v10.0, available via AACR Project GENIE cBioPortal).24,25 Although it shows the prevalence of this mutation within CNS tumors, this data set does not represent the totality of the genetic landscape.24 Intriguingly, FGFR1N546K has been described as a resistance mutation to four ATP-competitive inhibitors: ponatinib, dovitinib, PD173074, and BGJ-398.26 Intracranial activity of pemigatinib shows that not only it is able to cross the blood-brain barrier, but also it is able to do so at a concentration that preserves its efficacy and inhibition of the FGFR pathway. Subsequent dose reductions because of toxicity could have led to loss of efficacy in addition to contribution by co-occurring alterations, in particular, KRASG12A. This mutation although subclonal is downstream of the FGFR1N546K mutation and might have contributed to the resistance to pemigatinib. Although this is not an actionable alteration at this time, it highlights the need to continue developing additional targeted therapies and to comprehensively characterize the molecular alterations in primary tumors and track alterations that arise or become clonally dominant throughout treatment and recurrence.
FIG 2.
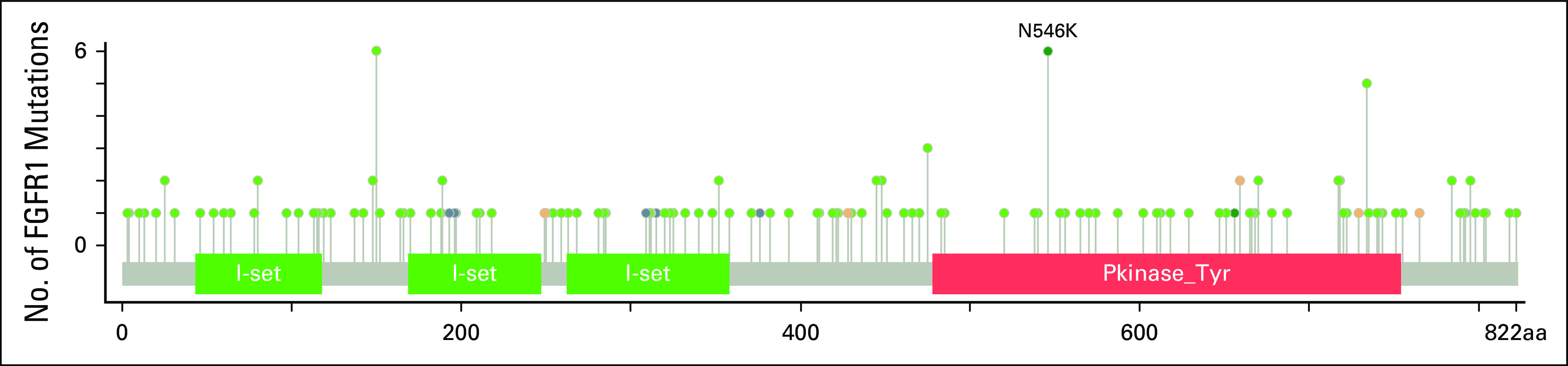
Lollipop figure of FGFR1 from cBioPortal showing FGFR1N546K mutation in the tyrosine kinase domain.
This is of particular interest in pilocytic astrocytoma, as FGFR alterations are well-established drivers in a subset of patients.2,3,27 Although not the most common drivers of disease, this subset of tumors lend themselves to systemic treatment if not able to be surgically cured. Interestingly, this cohort showed that all FGFR1-mutated tumors were extracerebellar and commonly appeared in midline locations.2 These tumors that arise in deep midline locations are challenging surgical candidates, with significant difficulties in accessing the lesion and achieving a gross total resection. The opportunity to avoid surgery in a subset of patients or to treat postoperative patients with residual tumor using adjuvant pemigatinib has the potential to decrease the associated morbidity of these tumors.28 These deep midline lesions tend to have decreased progression-free survival and increased rates of visual deficits, endocrine dysfunction, hearing abnormalities, and cranial nerve deficits.28
This observed intracranial and preserved antitumor activity in a glial tumor suggests that pemigatinib and other pan-FGFR inhibitors should be explored in higher-grade gliomas, including anaplastic astrocytoma, glioblastoma, and gliosarcomas, all of which have few treatment options at this time. Of note, glioblastoma multiforme has been shown to also harbor FGFR-driving alterations in both adult and pediatric patients,29 including a relatively high prevalence of FGFR-TACC fusions.30,31 This FGFR-TACC fusion has also shown sensitivity to FGFR inhibition,31 and as previously shown, pemigatinib has in vitro antitumor efficacy when targeting these fusion proteins.18
This signal of activity and duration warrants a prospective study assessing the use of pemigatinib and other pan-FGFR inhibitors in primary CNS tumors with an underlying FGFR alteration as monotherapy and in combination.
ACKNOWLEDGMENT
The authors would like to acknowledge the American Association for Cancer Research and its financial and material support in the development of the AACR Project GENIE registry, as well as members of the consortium for their commitment to data sharing. Interpretations are the responsibility of study authors.
V.S. acknowledges support of The Jacquelyn A. Brady Fund.
Shiao-Pei Weathers
Research Funding: Genentech/Roche (Inst), Mundipharma (Inst), Exelixis
Vivek Subbiah
Consulting or Advisory Role: MedImmune, Helsinn Therapeutics, Loxo, R-Pharm, QED Therapeutics
Research Funding: Novartis (Inst), GlaxoSmithKline (Inst), NanoCarrier (Inst), Northwest Biotherapeutics (Inst), Genentech/Roche (Inst), Berg Pharma (Inst), Bayer (Inst), Incyte (Inst), Fujifilm (Inst), PharmaMar (Inst), D3 Oncology Solutions (Inst), Pfizer (Inst), Amgen (Inst), AbbVie (Inst), MultiVir (Inst), Blueprint Medicines (Inst), LOXO (Inst), Vegenics (Inst), Takeda (Inst), Alfasigma (Inst), Agensys (Inst), Idera (Inst), Boston Biomedical (Inst), Inhibrx (Inst), Exelixis (Inst), Amgen (Inst), Turning Point Therapeutics (Inst)
Travel, Accommodations, Expenses: PharmaMar, Bayer, Novartis, Helsinn Therapeutics
Other Relationship: Medscape
No other potential conflicts of interest were reported.
SUPPORT
V.S. is an Andrew Sabin Family Foundation Fellow at The University of Texas MD Anderson Cancer Center. V.S. was supported by National Institutes of Health grant R01CA242845. The MD Anderson Cancer Center, Department of Investigational Cancer Therapeutics was supported by the Cancer Prevention and Research Institute of Texas (RP1100584); the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy, 1U01 CA180964, NCATS Grant UL1 TR000371 (Center for Clinical and Translational Sciences); and the MD Anderson Cancer Center Support Grant (P30 CA016672).
AUTHOR CONTRIBUTIONS
Conception and design: Stephen Capone, Vivek Subbiah
Financial support: Vivek Subbiah
Administrative support: Vivek Subbiah
Provision of study materials or patients: Leena Ketonen, Vivek Subbiah
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Shiao-Pei Weathers
Research Funding: Genentech/Roche (Inst), Mundipharma (Inst), Exelixis
Vivek Subbiah
Consulting or Advisory Role: MedImmune, Helsinn Therapeutics, Loxo, R-Pharm, QED Therapeutics
Research Funding: Novartis (Inst), GlaxoSmithKline (Inst), NanoCarrier (Inst), Northwest Biotherapeutics (Inst), Genentech/Roche (Inst), Berg Pharma (Inst), Bayer (Inst), Incyte (Inst), Fujifilm (Inst), PharmaMar (Inst), D3 Oncology Solutions (Inst), Pfizer (Inst), Amgen (Inst), AbbVie (Inst), MultiVir (Inst), Blueprint Medicines (Inst), LOXO (Inst), Vegenics (Inst), Takeda (Inst), Alfasigma (Inst), Agensys (Inst), Idera (Inst), Boston Biomedical (Inst), Inhibrx (Inst), Exelixis (Inst), Amgen (Inst), Turning Point Therapeutics (Inst)
Travel, Accommodations, Expenses: PharmaMar, Bayer, Novartis, Helsinn Therapeutics
Other Relationship: Medscape
No other potential conflicts of interest were reported.
REFERENCES
- 1.Ostrom QT, Patil N, Cioffi G, et al. : CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol 22:iv1-iv96, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones DTW, Hutter B, Jäger N, et al. : Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45:927-932, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones DTW, Gronych J, Lichter P, et al. : MAPK pathway activation in pilocytic astrocytoma. Cell Mol Life Sci 69:1799-1811, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kordacka J, Zakrzewski K, Gruszka R, et al. : Sensitive detection of FGFR1 N546K mosaic mutation in patient with encephalocraniocutaneous lipomatosis and pilocytic astrocytoma. Am J Med Genet A 179:1622-1627, 2019 [DOI] [PubMed] [Google Scholar]
- 5.Louis DN, Wesseling P, Aldape K, et al. : cIMPACT‐NOW update 6: new entity and diagnostic principle recommendations of the cIMPACT‐Utrecht meeting on future CNS tumor classification and grading. Brain Pathol 30:844-856, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Louis DN, Perry A, Wesseling P, et al. : The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol 23:1231-1251, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenhauer EA, Therasse P, Bogaerts J, et al. : New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 45:228-247, 2009 [DOI] [PubMed] [Google Scholar]
- 8.Merz V, Zecchetto C, Simionato F, et al. : A phase II trial of the FGFR inhibitor pemigatinib in patients with metastatic esophageal-gastric junction/gastric cancer trastuzumab resistant: The FiGhTeR trial. Ther Adv Med Oncol 12:1758835920937889, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pederzoli F, Bandini M, Marandino L, et al. : Targetable gene fusions and aberrations in genitourinary oncology. Nat Rev Urol 17:613-625, 2020 [DOI] [PubMed] [Google Scholar]
- 10.Manur R, Sung PJ, Loren AW, et al. : Leukemic lineage switch in a t(8;22)(p11.2;q11.2)/BCR-FGFR1-rearranged myeloid/lymphoid neoplasm with RUNX1 mutation—Diagnostic pitfalls and clinical management including FGFR1 inhibitor pemigatinib. Leuk Lymphoma 61:450-454, 2020 [DOI] [PubMed] [Google Scholar]
- 11.Bekaii-Saab TS, Valle JW, Van Cutsem E, et al. : FIGHT-302: First-line pemigatinib vs gemcitabine plus cisplatin for advanced cholangiocarcinoma with FGFR2 rearrangements. Futur Oncol 16:2385-2399, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Féliz L, Asatiani E, Lihou C, et al. : FIGHT-207: Phase 2 study of pemigatinib in patients with previously treated, locally advanced/metastatic or unresectable solid tumor malignancies harboring activating fibroblast growth factor receptor (FGFR) gene alterations. Mol Cancer Ther 18, 2019. (suppl 12; abstr A077) [Google Scholar]
- 13.Gutierrez M, Subbiah V, Nemunaitis JJ, et al. : Safety and efficacy of pemigatinib plus pembrolizumab combination therapy in patients (pts) with advanced malignancies: Results from FIGHT-101, an open-label phase I/II study. J Clin Oncol 38, 2020. (suppl; abstr 3606) [Google Scholar]
- 14.Kuboki Y, Furukawa M, Takahashi Y, et al. : Preliminary results from fight-102: A phase 1 study of pemigatinib in Japanese patients with advanced malignancies. Ann Oncol 30:vi125, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subbiah V, Iannotti NO, Gutierrez M, et al. : FIGHT-101, a first-in-human study of potent and selective FGFR 1-3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann Oncol 10.1016/j.annonc.2022.02.001 [epub ahead of print on February 14, 2022] [DOI] [PubMed] [Google Scholar]
- 16.Verstovsek S, Vannucchi AM, Rambaldi A, et al. : Interim results from Fight-203, a phase 2, open-label, multicenter study evaluating the efficacy and safety of pemigatinib (INCB054828) in patients with myeloid/lymphoid neoplasms with rearrangement of fibroblast growth factor receptor 1 (FGFR1). Blood 132:690, 2018 [Google Scholar]
- 17.Abou-Alfa GK, Sahai V, Hollebecque A, et al. : Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol 21:671-684, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu PCC, Koblish H, Wu L, et al. : INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS One 15:e0231877, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Incyte Corporation : Pemazyre (Pemigatinib) [package insert]. US Food and Drug Administration website. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213736s000lbl.pdf [Google Scholar]
- 20.Bale TA: FGFR- gene family alterations in low-grade neuroepithelial tumors. Acta Neuropathol Commun 8:21, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fisher MJ, Jones DTW, Li Y, et al. : Integrated molecular and clinical analysis of low-grade gliomas in children with neurofibromatosis type 1 (NF1). Acta Neuropathol 141:605-617, 2021 [DOI] [PubMed] [Google Scholar]
- 22.Rand V, Huang J, Stockwell T, et al. : Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc Natl Acad Sci USA 102:14344-14349, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lew ED, Furdui CM, Anderson KS, et al. : The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci Signal 2:ra6, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.AACR Project GENIE : Powering precision medicine through an International consortium. Cancer Discov 7:818-831, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.AACR Project GENIE cBioPortal : genie.cbioportal.org
- 26.Yoza K, Himeno R, Amano S, et al. : Biophysical characterization of drug-resistant mutants of fibroblast growth factor receptor 1. Genes Cells 21:1049-1058, 2016 [DOI] [PubMed] [Google Scholar]
- 27.Fomchenko EI, Reeves BC, Sullivan W, et al. : Dual activating FGFR1 mutations in pediatric pilomyxoid astrocytoma. Mol Genet Genomic Med 9:1-8, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Armstrong GT, Conklin HM, Huang S, et al. : Survival and long-term health and cognitive outcomes after low-grade glioma. Neuro Oncol 13:223-234, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartzentruber J, Korshunov A, Liu X-Y, et al. : Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226-231, 2012 [DOI] [PubMed] [Google Scholar]
- 30.Singh D, Chan JM, Zoppoli P, et al. : Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337:1231-1235, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lasorella A, Sanson M, Iavarone A: FGFR-TACC gene fusions in human glioma. Neuro Oncol 19:475-483, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]