

SUMMARY
Incomplete lineage sorting (ILS) makes ancestral genetic polymorphisms persist during rapid speciation events, inducing incongruences between gene trees and species trees. ILS has complicated phylogenetic inference in many lineages, including hominids. However, we lack empirical evidence that ILS leads to incongruent phenotypic variation. Here, we performed phylogenomic analyses to show that the South American monito del monte is the sister lineage of all Australian marsupials, although over 31% of its genome is closer to the Diprotodontia than to other Australian groups due to ILS during ancient radiation. Pervasive conflicting phylogenetic signals across the whole genome are consistent with some of the morphological variation among extant marsupials. We detected hundreds of genes that experienced stochastic fixation during ILS, encoding the same amino acids in non-sister species. Using functional experiments, we confirm how ILS may have directly contributed to hemiplasy in morphological traits that were established during rapid marsupial speciation ca. 60 mya.
In brief
The majority of marsupial genomes are affected by incomplete lineage sorting, which causes unique morphological traits in descendant species. Through in-depth phylogenetic analyses and in vivo validation, these data resolve the marsupial lineage at high resolution and suggest a strong impact of ILS on phylogenic relationships.
Graphical abstract
INTRODUCTION
A central goal of comparative genomics is to understand the relationship between genomic and phenotypic divergence during speciation. However, evolutionary events such as introgressive hybridization, convergent evolution, and incomplete lineage sorting (ILS) often complicate our inferences of phenotypic evolution by causing phylogenetic incongruence between morphological and molecular data (Dávalos et al., 2012; Gaubert et al., 2005; Larson, 1998; Olsson et al., 2010; Zou and Zhang, 2016). Although hybridization events and episodes of convergent evolution occur after speciation, ILS happens during speciation and is particularly likely in rapid successive speciation events, which implies that ancestral polymorphisms persist when descendant lineages have irreversibly diverged (Avise and Robinson, 2008; Bravo et al., 2019; Degnan and Rosenberg, 2009; Szöllősi et al., 2015). ILS, also called hemiplasy, has been an enigmatic source of topological discordance between gene trees and species trees (Avise and Robinson, 2008). This has been reported in insects (Pollard et al., 2006), birds (Jarvis et al., 2014), marine mammals (Lopes et al., 2021), Australian marsupials (Gallus et al., 2015; Nilsson et al., 2018), and great apes (Mailund et al., 2014), offering major challenges in reconstructing phylogenetic relationships. ILS events are most likely to occur when new lineages rapidly descend from ancestors with large effective population sizes (Ne), conditions that have likely applied in many lineages during at least some part of their evolutionary history (Pease et al., 2016; Suh et al., 2015). However, understanding the general impact of ILS has remained elusive because reliable detection and quantification require fully sequenced reference genomes from an entire phylogeny and the application of computationally demanding algorithms for comparative bioinformatics analyses.
The early diversification of the marsupial mammals is a classic example of a rapid radiation that resulted in a long-standing interpretational controversy of the phylogeny (Nilsson et al., 2010; Szalay, 1994). The phylogenetic position of the enigmatic Microbiotheria, represented by only a single extant species, the South American monito del monte (Dromiciops gliroides), has played a key role in this debate (Amrine-Madsen et al., 2003; Burk et al., 1999; Duchêne et al., 2018; Mitchell et al., 2014; Nilsson et al., 2003, 2010; Springer et al., 1998) because it shares many characteristics with Australian marsupials (see below). Although the most recent molecular phylogenetic analyses suggested that D. gliroides is the sister taxon of Australasian marsupials, which is a monophyletic group that reached Australasia by a single migration event from South America (Nilsson et al., 2010), earlier analyses based on mtDNA and morphology suggested alternate scenarios, hinging on the phylogenetic placement of D. gliroides, that marsupials colonized Australia twice via Antarctica/South America (Nilsson et al., 2004).
D. gliroides is a special lineage because it shares various anatomical characters with all, or some, Australian marsupials. For example, their ankle bone articulation is more similar to that of Australian marsupials, especially the diprotodontians (Szalay, 1982, 1994), than to that of South American marsupials. D. gliroides also has unpaired sperm and lacks mammary glands in the males, similar to Australian marsupials, but in contrast to all other South American marsupials (Frankham and Temple-Smith, 2012; Renfree et al., 1990; Temple-Smith, 1987, 1994; Tyndale-Biscoe and Renfree, 1987). Also, their chromosome morphology more closely resembles that of the Australian marsupials (Sharman, 1982), and there is mosaicism in the D. gliroides male sex chromosomes, similar to Australian petaurids and peramelids but unlike other South American marsupials (Gallardo and Patterson, 1987). Finally, recent comparisons of the brain structure of D. gliroides and other marsupials showed greater similarity to Australian marsupials, especially the diprotodontians (Gurovich and Ashwell, 2020).
Here, we present a draft genome of D. gliroides and detailed phylogenomic comparisons with five other marsupial species. Our analyses confirm that exceptionally high frequencies of ILS must have contributed to the controversy over the early geographic speciation among ancestral marsupials. After identifying a series of genes with strong signatures of ILS, we used transgenic techniques to demonstrate that ILS can induce morphological similarity across non-sister lineages. Our results underline the likely pervasiveness of ILS and the urgency of quantifying its general impact on phylogenetic reconstruction and trait evolution.
RESULTS
Phylogenomic analyses support the monito del monte as a sister lineage to the Australian marsupials
Several studies have demonstrated the power of using whole genome data for addressing deep evolutionary relationships (Jarvis et al., 2014; Rokas et al., 2003; Wolf et al., 2002). To resolve the marsupial tree of life, we obtained a draft genome assembly of monito del monte (Microbiotheria, Micr) using Illumina short read sequencing for an assembly length of 3.4 Gb with a scaffold N50 size of 17.8 Mb, a contig N50 size of 10.2 kb, and 20,639 protein-coding genes (Tables S1 and S2). We performed comparative phylogenomic analyses with two diprotodontian marsupials (Dipr): a phascolarctid, the koala (Phascolarctos cinereus, RefSeq: GCF_002099425.1) (Johnson et al., 2018), and a macropodid, the tammar wallaby (Macropus eugenii, GenBank: GCA_000004035.1) (Renfree et al., 2011); two dasyuromorphian marsupials (Dasy): Tasmanian devil (Sarcophilus harrisii, RefSeq: GCF_902635505.1), and brown antechinus (Antechinus stuartii, GenBank: GCA_016696395.1) (Brandies et al., 2020); and a didelphimorphian, a didelphid, the gray short-tailed opossum (Monodelphis domestica, RefSeq: GCF_000002295.2, Mono) (Mikkelsen et al., 2007) as the outgroup.
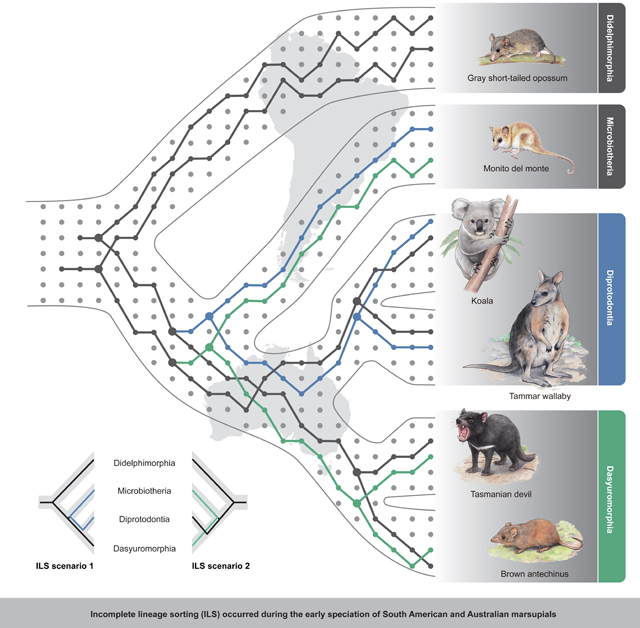
We extracted approximately 984 Mb of orthologous regions from the whole genome alignments (WGAs) of these six species and used these regions for phylogenetic analyses with both the coalescence-based method, ASTRAL-III (Zhang et al., 2018), and the concatenation-based method, randomized axelerated maximum likelihood (RAxML) (Stamatakis, 2006). These two approaches resulted in an identical tree topology that placed the monito del monte outside the Australasian group, as sister clade to the common ancestor of Diprotodontia and Dasyuromorphia (this topology is hereafter referred to as the “Dipr_Dasy tree”) (Figures 1 and S1). Our Dipr_Dasy tree conflicts with previously published phylogenies, including one inferred from mitochondrial data (“Dasy_Micr tree”) (Nilsson et al., 2004) and one based on morphological characters (“Dipr_Micr tree”) (Horovitz and Sánchez-Villagra, 2003). The Dasy_Micr tree combined Dasyuromorphia and monito del monte as closest relatives, whereas the Dipr_Micr tree recovered Diprotodontia and monito del monte as closest relatives. We also obtained the Dipr_Dasy tree when using only the coding regions, the 4-fold degenerate sites, the first and second codon positions (C12) and the third codon positions (C3) of 9,227 orthologous genes identified in all six species (Figure S1). Finally, given that transposable elements (TEs) are generally free of homoplasy (Springer et al., 2020), we also constructed a species tree from the retroelement bipartitions converted from a presence/absence matrix of 401 informative markers and once more obtained a Dipr_Dasy tree. Thus, irrespective of the tree-building method and data used, we recovered a consistent topology that supports the monophyly of our sampling of Australian marsupials (Figure 1), and we, therefore, used the Dipr_Dasy tree as the marsupial speciation tree in downstream analyses.
Figure 1. Phylogenetic tree based on WGAs using the RAxML concatenation-based method.

The best-scoring ML tree had 100% bootstrap node-support values. The branch length scale refers to the expected number of substitutions per site. The sketch map indicates the geographic distribution of monito del monte obtained from Oda et al. (2019) and the distribution data for the other five marsupials obtained from International Union for Conservation of Nature (IUCN). See also Figure S1 and Tables S1 and S2.
Pervasive incomplete lineage sorting throughout marsupial genomes
We produced individual gene trees based on ca. 569 Mb WGAs using non-overlapping 100 bp windows and 22,743 exon blocks of orthologs after removing the low-quality blocks. These phylogenetic analyses revealed substantial discordances between the species tree and the gene trees, with the latter generating two alternative topologies, the Dasy_Micr tree and the Dipr_Micr tree. Overall, we found that 59.53% of the WGAs and 62.17% of the exon blocks supported a topology placing the monito del monte together with the Australasian group (Figure 2A). Such high proportions suggest pervasive marsupial gene tree incongruence in the ancestral population of Diprotodontia and Dasyuromorphia, which might have been significantly affected by either ILS or hybridization. We also compared the numbers of TEs showing presence and absence patterns following the three topologies. We found 80 TEs supporting the Dipr_Dasy partition with the retroelement insertion occurring in the ancestor branch of Diprotodontia and Dasyuromorphia, and a similar number of TEs supported each of the other two partitions (Dipr_Micr and Dasy_Micr) as expected under ILS (Figure 2B). By applying a multi-directional Kuritzin-Kischka-Schmitz-Churakov (KKSC) insertion significance test on the number of markers shared by the different lineages (Kuritzin et al., 2016), we found that polytomy could be convincingly rejected (p value = 2.0437e–16) and that hybridization could not be accepted (p value = 0.5966) as alternative explanation, which implies that the symmetric conflicting TEs are likely to be the result of ILS.
Figure 2. Pervasive signatures of incomplete lineage sorting in marsupial genomes.

(A) Discordance between gene trees and species tree in WGAs and exon blocks. Red branches and sectors represent genomic regions for which the gene tree topology is same as the species tree (Dipr_Dasy), blue branches and sectors represent regions supporting Diprotodontia and Microbiotheria as sister taxa (Dipr_Micr), and green branches and sectors represent regions whose topology supports Dasyuromorphia and Microbiotheria as closest relatives (Dasy_Micr).
(B) Presence/absence status of retroelements in marsupials. The horizontal bars represent the number of retroelements that were exclusively found in a specific combination of species shown on the left. The gray circles indicate absence of retroelements markers, whereas colored circles indicate presence. The red bar indicates that the presence pattern of retroelements markers supported the species tree, whereas the blue bar supported the Dipr_Micr tree and the green bar supported the Dasy_Micr tree. A similar number of TEs supported Dipr_Micr or Dasy_Micr as expected when created by ILS.
(C) Four potential genealogy scenarios for each locus in CoalHMM analysis.
The standard relationship (species tree, non-ILS) was designated as type0 (without deep coalescence) and type1 (with deep coalescence), and two alternative genealogies, type2 (Dipr and Micr are closest relatives) and type3 (Dasy and Micr are closest relatives), represent two alternative ILS scenarios. Color coding is the same as in (A). Dasy, Dasyuromorphia; Dipr, Diprotodontia; Micr, Microbiotheria (monito del monte); Mono, M. domestica.
See also Figures S2 and S3.
Genome-wide signatures of ILS and hybridization can be distinguished because coalescence times for regions under ILS should be older than the speciation events, whereas hybridization occurs after speciation is completed (Figure S2A). To test the ILS and hybridization models, we partitioned the genomic sequences into three paired-topology categories (Dipr_Dasy, Dasy_Micr, and Dipr_Micr) and reconstructed phylogenetic trees using concatenated genome sequences for each category. We assumed that the Dipr_Dasy genomic sequences that generated the species tree were less affected by ILS or hybridization, so we expected that estimated divergence time (t) between Diprotodontia and Dasyuromorphia would approximately reflect the time of speciation. Likewise, the estimated divergence time between monito del monte and Diprotodontia or Dasyuromorphia from the other two categories of genomic data should correspond to a longer expected coalescence time under ILS (ti) or to a shorter expected divergence time under hybridization (th). Our divergence time estimates with MCMCTree (Yang, 1997) for these three alignments produced longer divergence times between monito del monte and either Dasyuromorphia (Dasy_Micr tree) or Diprotodontia (Dipr_Micr tree) compared with the ones obtained from the Dipr_Dasy tree (Figure S2A). This strongly suggests that ILS, and not hybridization, has been the main cause of the pervasive signatures of incongruence across the marsupial genomes.
We further applied a tree-based method, quantifying introgression via branch lengths (QuIBL) (Edelman et al., 2019), to evaluate whether ILS is the prime explanation of the mismatch between the species tree and the gene trees across the marsupial species. QuIBL distinguishes between ILS and introgression based on the distribution of internal branch lengths for a given three-taxon subtree (triplet). For the internal branch of Microbiotheria and two Australian groups, the Bayesian information criterion (BIC) test indicated that the phylogenetic discordances were caused by ILS only (Figures S2B–S2E). This conclusion was also supported by the four-taxon D-statistic test (Green et al., 2010). By running this test on each 5-kb window, we verified that up to 95% of windows had a statistically equal number of ABBA and BABA sites, a symmetry that corroborates the conclusion that the observed discordances across entire genomes are more likely to have been produced by ILS than by post-speciation gene flow.
We next applied an updated version of coalescence hidden Markov model (CoalHMM) (Dutheil et al., 2009; Hobolth et al., 2007) that directly models heterogeneous substitution rates across lineages to reduce the long-branch attraction effects when detecting ILS signals from whole-genome alignments of species with different evolutionary rates. A posterior decoding approach was used to reconstruct the most likely genealogy for each genomic position (Figure 2C): Dipr_Dasy relationship without (type0) or with deep coalescence (type1); Dipr_Micr relationship (type2); and Dasy_Micr relationship (type3). The last two genealogies represent the consequences of ILS in the speciation period of Diprotodontia, Dasyuromorphia, and Microbiotheria. The CoalHMM analysis was applied on four different quartet combinations of species and showed that over half of the orthologous genome alignments in marsupials were affected by ILS (Figure S3A). Across the four quartet combinations, we detected on average 31.32% of genomic regions showing type2 ILS and 26.57% type3 ILS. The slightly higher proportion of type2 ILS was attributed to the relatively higher evolutionary rate in the common ancestor of Dasyuromorphia (Figure 1). This long-branch attraction effect is slightly more pronounced in the quartet with the koala, which had a relatively slower evolutionary rate than the tammar wallaby (Figure 1). The orthologous coding regions offered more consistent support to the species tree when compared with the WGAs level in all four combinations (chi-square test, p value < 2.2e–16, Figure S3A). This pattern is consistent with coding regions being under stronger purifying selection than the rest of the genome, reducing Ne for these regions so that less ILS would be expected. In concordance with this, we found that the whole genome conservation scores (Haeussler et al., 2019; Pollard et al., 2010) were significantly higher in non-ILS regions when compared with ILS regions (Welch two sample t test, p value < 2.2e–16). Less ILS in regions expected to be under stronger purifying selection was also reported for comparisons between humans, chimpanzees, and gorillas (Scally et al., 2012). On average, the lengths of the genomic segments affected by ILS are very short (∼80 bp) (Figure S3B). However, we also detected hundreds of long ILS segments (>1 kb), with about 16% of these long ILS segments overlapping coding regions, which is significantly higher than expected (chi-square test, p value < 2.2e–16).
Molecular dating with awareness of ILS unveils marsupial speciation times
The single base-pair map of ILS given by CoalHMM allowed us to increase the resolution in the alignment partition. We estimated the divergence times from concatenation of the genomic loci that supported the canonical phylogenetic Dipr_Dasy state and the two ILS states, respectively (Figure 3A; Table S3). These estimations indicated that divergence of D. gliroides and the ancestor of Diprotodontia and Dasyuromorphia occurred 59.7 mya, much later than the time the sea-floor spreading began between Australia and Antarctica ca. 84 mya, suggested by most studies (Tikku and Cande, 1999, 2000; White et al., 2013; Williams et al., 2019; Figure 3B). This eliminates the possibility that the ancestral monito del monte population could have arrived to Australia from South America via Antarctica. Our analyses confirmed that the ILS regions had an older divergence time between monito del monte and the Dasyuromorphia (52.3 mya) or the Diprotodontia (54.0 mya) than the genomic regions that corresponded with the actual species differentiation (45.8 mya) (Figure 3A). Moreover, the biogeographic data show that the final separation of Australia and Antarctica along the South Tasman Rise occurred at ca. 45 mya (Van Den Ende et al., 2017; White et al., 2013), i.e., at the time that early diversification of the Australian marsupials began according to our estimation (Figure 3B). This more accurate evolutionary reconstruction is supported by the fossil record. The oldest Microbiotherid fossil from South America was dated to 59.2–64.5 mya (Goin and Abello, 2013), consistent with our estimated divergence time of D. gliroides from the Australian marsupials (59.7 mya). This scenario is also consistent with strong evidence that the four Australian marsupials in our study all originated in, and remained restricted to, Australia, as fossils of these lineages have only been found on the Australian plate and were dated to be younger than the separation between South America and Antarctica ca. 35 mya, i.e., when Australia became an island continent (Behrensmeyer and Turner, 2013; Livermore et al., 2005; Figure 3B). This combined evidence makes the alternative scenario of hybridization after Microbiotheria had separated from the Australian marsupials highly unlikely.
Figure 3. Dating genetic divergence times and speciation events in early marsupials.

(A) The reconstructed evolutionary history of the three-lineage bifurcation among the six extant marsupials using three types of concatenated alignments. The Dipr_Dasy alignment (red) containing the type0 loci, where ILS was assumed to be absent, were supported by all four species combinations. The Dipr_Micr ILS alignment (blue) and the Dasy_Micr ILS alignment (green) contained the loci identified as, respectively, type2 and type3 in all four combinations. Names were labeled with plate colors.
(B) The known movements of the South American, Antarctic, and Australian continental plates during the crucial time window of ca. 84–35 mya. The sea-floor spreading began between Australia and Antarctica at ca. 84 mya (left panel). The final separation of Australia and Antarctica along the South Tasman Rise at ca. 45 mya created a shallow marine crust region between Australia and Antarctica that gradually opened further (gray area in middle panel). This process resulted in Australia and Antarctica being fully separated ca. 35 mya at which point also the connection between South America and Antarctica was severed (right panel). This pattern of continental drift excludes the possibility of continuing hybridization between monito del monte and Australian marsupials after their divergence had been completed. South America, Antarctica, and Australia are represented in blue, yellow, and brown, respectively. The contours represent the reconstructed continental plate and the black lines outside the plates represent the submerged continental plate crust areas (Vizcaíno et al., 1998; Williams et al., 2019).
See also Table S3.
Incomplete lineage sorting affected phenotypic diversification
The stochastic persistence of ancestral genetic polymorphisms can by chance cause two phylogenetically distant species to inherit the same ancestral genotypes, and, if the alleles encode specific morphological traits, this can lead to discordance between species trees and morphological trees. Previous studies have shown that the monito del monte and the diprotodontian marsupials share a range of similar morphological characters in several anatomical systems, including the skeleton (Horovitz and Sánchez-Villagra, 2003), the reproductive organs (Frankham and Temple-Smith, 2012), and the brain (Gurovich and Ashwell, 2020), for which some Australian marsupial groups are remarkably different–in many cases mismatches with the true phylogenetic relationships.
In particular, we found many genes related to skeletal functions affected by ILS significantly, inspiring us to focus on the skeletal system where we could obtain data for all studied species. As expected, some examples of skeletal differences that might be attributable to hemiplasy, according to their variable expression across the marsupials sampled in this study, concerned the curvature of the humerus, the relative length of the spinous processes of thoracic vertebrae, and the morphology of the incisors (Figure 4A). The curvature of the humerus in the monito del monte and both diprotodontian marsupials is much shallower than that in both dasyuromorphian marsupials and the gray short-tailed opossum. To compare the curvature pattern, we placed the bone in the side view and added a line segment from the posterior margin of the head to the first intersection at which the bone made contact with the vertical plane. By comparing with the middle point of the line, we found that the position where the curvature changed most significantly in the monito del monte and both diprotodontian marsupials was closer to the top of the line, whereas the other species changed curvature in the middle. Furthermore, in the gray short-tailed opossum and the two dasyuromorphian marsupials, the spinous process of the first thoracic vertebra (T1) is shorter than the spinous process of the second thoracic vertebra (T2), but they are of equal length in the monito del monte and the diprotodontian marsupials (Figures S4A and S4B). Finally, in side view, the central and lateral maxillary incisors have different angles and a distinct gap between them in the gray short-tailed opossum and dasyuromorphian marsupials, but this feature is lacking in the monito del monte, koala, and tammar wallaby.
Figure 4. ILS genes and their potential phenotypic effects.

(A) Three representative morphological traits showing similar patterns of variation between the monito del monte and diprotodontian species (blue branches) although they are not sister lineages. For the humerus, curvature of the humerus in the monito del monte and both diprotodontian species is much shallower than in both dasyuromorphian species and the gray short-tailed opossum. Red dashed lines highlight the position of the position where the curvature changed most significantly compared with the middle point of the line segment. Here, the line segment is from the posterior margin of the head to the first intersection at which the bone made contact with the vertical plane. For the vertebrae, the spinous process of T1 thoracic vertebra is shorter than the spinous process of T2 in the brown antechinus, Tasmanian devil, and the gray short-tailed opossum but similar in the monito del monte, koala, and tammar wallaby. For incisors, the red arrows indicate the presence and absence of a gap.
(B) Distribution of ILS sites across the three types of genes (6,425 Dipr_Dasy, 1,310 Dipr_Micr, and 803 Dasy_Micr), expressed in proportion to the Dipr_Micr ILS loci in the coding regions of each gene. Compared with the other two types of genes, Dipr_Micr genes (blue) enriched more Dipr_Micr ILS loci. The two Dipr_Micr genes (WFIKKN1 and PAPSS2) used in our transgenic experiments are labeled and the number of genes in each category is given below the axis.
(C) Among 1,310 Dipr_Micr genes, the sequence identity between the diprotodontian species and monito del monte (Micr) was significantly higher than between Australian groups (Dasy) (Welch two sample t test, p value < 2.2e–16).
(D) Among 803 Dasy_Micr genes, the sequence identity between the dasyuromorphian species and monito del monte (Micr) was significantly higher than between Australian groups (Dipr) (Welch two sample t test, p value < 2.2e–16).
(E) Frequencies of ILS genes based on their expression patterns at the organ level (annotated by the Gene Expression Database), shown as a proportion of the total number of genes with matchings in database annotated for each organ. Chi-square test showed that the frequencies between ILS genes and all orthologs were significantly different for most organs, except for Dasy_Micr genes in the gland organ.
(F) Frequencies of ILS genes related to 17 organ-specific phenotypes, expressed as a proportion of the total number of genes annotated as being instrumental for these organ systems. Chi-square test showed that the gene frequencies between ILS genes and orthologs were not significantly different for most organ systems, except for Dipr_Micr genes in the brain, craniofacial system, and ear, and Dasy_Micr genes in the brain and craniofacial system. The center lines in boxplots (B–D) represent the median with the 25th and 75th percentiles marked by the box limits. The bars extend to the farthest data points and the dots outside of the bars are outliers. *p value < 0.05; **p value < 0.01; ***p value < 0.001. See also Figure S4 and Table S4.
To identify candidate ILS genes that might be responsible for these putative examples of morphological hemiplasy, we extracted the ILS signals of orthologous coding regions from the whole genome CoalHMM’s results, which produced 6,425 genes free from ILS effects (Dipr_Dasy genes) and 2,113 genes significantly affected by ILS (Figure 4B). The sequence identities of the ILS genes between monito del monte and Diprotodontia or Dasyuromorphia were significantly higher than between the Australasian group (Welch two sample t test, p value < 2.2e–16, Figures 4C and 4D). We also measured the frequencies of ILS genes based on their expression patterns at the organ level, which showed differences compared with identified orthologs, but without obvious tissue-specific expression features (e.g., lowest frequency is as high as 80%; Figure 4E). The widespread expression of these genes suggests that ILS events can potentially have a broad functional impact on phenotypic variation across many organ systems. To infer the possible functional impact of ILS events on phenotypic variation across species, we annotated the ILS genes with the Mammalian Phenotype Ontology Database (Smith and Eppig, 2009) and confirmed that they influence a wide spectrum of phenotypes in the associated organ systems (Figure 4F; Table S4). Interestingly, we found that the brain, reproductive organs, and skeletons were among the systems with the highest number of ILS genes, consistent with the phenotypic differences observed.
Experimental evidence supporting that expressed ILS genes affect diagnostic morphological traits
To date, there is no empirical evidence that specific allelic variation associated with ILS can induce morphological hemiplasy in descendant non-sister lineages. Technical constraints precluded genetic manipulation experiments in marsupial species to validate this possible effect, but the overall evolutionary conservation of mammalian protein-coding genes allowed us to use mice to examine the morphological impact of alternative alleles at two focal ILS loci. Here, we reviewed the annotation of all ILS genes and identified the candidates associated with the skeleton anatomy that showed hemiplasy in these marsupial species. WFIKKN1 and PAPSS2 were among the top candidates showing significant ILS signals between the monito del monte and two diprotodontian marsupials; so, we focused our validations on these two genes.
WFIKKN1 is known to play a role in axial skeleton patterning, particularly of the thoracic vertebrae (Lee and Lee, 2013; Monestier and Blanquet, 2016). Over 80% of its orthologous coding region in the monito del monte was influenced by Dipr_Micr ILS, including four continuous Dipr_Micr regions longer than 100 bp (Figure 5A). This implies that several amino acids are shared between the monito del monte and both diprotodontian marsupials, but not with the two dasyuromorphian species and the gray short-tailed opossum. For example, the monito del monte, koala, and tammar wallaby shared glutamine (Q), whereas the gray short-tailed opossum, Tasmanian devil, and brown antechinus had arginine (R) at amino acid position 76 (based on Mus musculus gene ENSMUSG00000071192). This site locates in the Whey acidic protein (WAP) domain of WFIKKN1, belonging to a long ILS region (Figures 5B and S4C). The vertebrae in these marsupial species show a possible hemiplasy pattern, where species carrying Q have a spinous process of similar height on the first (T1) and second thoracic vertebra (T2), whereas marsupials carrying R have a shorter T1 spinous process than T2 (Figures 4A and 5B). To test whether the stochastic fixation of AA76 contributes to this vertebral hemiplasy pattern and whether the change from Q to R leads to the decreased T1/T2 ratio, we assessed the morphological effects produced by the 2-aa types in AA76 of Wfikkn1 using a transgenic mice model subjected to CRISPR-Cas9 point substitution technology (Figures S5A and S5B).
Figure 5. Transgenic evidence for ILS alleles affecting skeletal traits used in morphological phylogenies.

(A) Distribution of Dipr_Micr ILS signals estimated by CoalHMM (blue bar) in the genomic regions of the WFIKKN1 gene. The gene model of the monito del monte is shown.
(B) The target site in our transgenic experiment used to validate the phenotypic effects of Wfikkn1 gene expression located at c.227A (AA76) in mouse. The protein domain composition in monito del monte is annotated by Pfam (Mistry et al., 2021). The alignment of nucleotide sequences and amino acid sequences in the WAP domain are plotted for all six marsupial species and the mouse, showing the variation at the target Dipr_Micr ILS site marked with a box. The dotted line describes the correspondence of the target ILS site and its surrounding regions in the protein domain and coding region.
(C) Micro-CT scan and comparison of thoracic vertebrae spinous process in Wfikkn1Q76R/Q76R mice and wild-type mice, showing the relative size differences between the spinous process of T1 and T2. The accompanying plot shows the logarithmic T1/T2 ratios of Wfikkn1Q76R/Q76R mice (n = 11) were significantly lower than wild-type mice (n = 10) (Welch two sample t test, p value = 0.0004). Scale bars, 1,000 μm.
(D) Curvature differences of the right humeral bone among Papss2Mono/Mono (Mono, n = 9), Papss2Dipr/Dipr (Dipr, n = 11), and Papss2Dasy/Dasy (Dasy, n = 11) mice lines based on 3D geometric morphometric analysis. Paired t test showed that the Euclidean distances between Mono samples and the Dasy mice line were significantly less than the Euclidean distances between Mono samples and the Dipr mice line (p value = 0.0054).
(E) Curvature differences of the right humeral bone among Papss2Dasy/Dasy (n = 11), Papss2Dipr/Dipr (n = 11), Papss2Mono/Mono (n = 9), and Papss2Mono-Micr/Mono-Micr (Mono-Micr, n = 11) mice lines based on 3D geometric morphometric analysis. Paired t test showed that the Euclidean distances between Dipr samples and the Mono-Micr line were significantly less than the Euclidean distances between Dipr samples and the Mono line (p value = 0.0029). The center lines in boxplots (C–E) represent the median with the 25th and 75th percentiles marked by the box limits. The bars extend to the farthest data points and the dots outside of the bars are outliers. CV: canonical variate. **p value < 0.01; ***p value < 0.001.
See also Figures S4 and S5 and Table S5.
The AA76 of Wfikkn1 in wild-type mice is decoded as glutamine as in the monito del monte and the diprotodontian species. Thus, we generated transgenic mice with the alternative amino acid (arginine) in the AA76 position of this gene. We scanned the bodies of Wfikkn1Q76R/Q76R mice and wild-type mice using MicroXCT 400 and measured the height of the spinous process of their first and second thoracic vertebrates (Figure S4D). The scanning result showed that the T1/T2 ratio was significantly reduced in Wfikkn1Q76R/Q76R mice compared with WT mice (Welch two sample t test for T1/T2 ratio, p value = 0.0004, Figure 5C), due to a shorter T1 spinous process and a higher T2 spinous process (Welch two sample t test, p value = 0.0004 for T1, p value = 0.0490 for T2; Figure S4E). These results confirmed our hypothesis that changing from glutamine to arginine at this site was sufficient to cause a decrease in the T1 spinous process, consistent with phenotypic differences observed in marsupials. This experiment, thus, provides proof-of-concept that an ancestral genetic polymorphism can produce different morphologies through ILS that might create a mismatch between morphological and genomic phylogenies.
We also produced transgenic mice to validate the function of another ILS gene PAPSS2, which encompassed a large ILS region. This gene is known to play a role in humeral morphology, a syndrome of traits also subject to possible hemiplasy across the six marsupials (Figure 4A). The humerus is slightly curved in marsupials, which might be associated with terrestrial and arboreal lifestyles requiring different types of habitual muscle function (Henderson et al., 2017). The humerus of the monito del monte and both diprotodontian species is curved at the upper part of the arm bone, whereas it is curved almost in the middle for the gray short-tailed opossum and both dasyuromorphian species (Figure 4A). PAPSS2 is one of the top Dipr_Micr ILS genes showing high expression in the long bones (Stelzer et al., 2007) with 55% of its coding region exhibiting Dipr_Micr ILS according to CoalHMM’s analyses. The protein identity of this gene between the monito del monte and both diprotodontian species (92%) is higher than that within Australian groups (85%). We first examined whether the sequence variations of PAPSS2 could affect morphology. To do so, we replaced the entire mice ortholog with cDNA sequences of the gray short-tailed opossum (Papss2Mono/Mono), tammar wallaby (Papss2Dipr/Dipr), and Tasmanian devil (Papss2Dasy/Dasy) by homologous recombination (Figures S5C and S5D). We then measured the curvature of humerus morphology of these three mutant lines by constructing the 3D landmarks on the humerus bone surface and evaluated the morphological similarity of the three mutant lines by calculating Euclidean distances in a canonical variate analysis (CVA) (Figure S4F). We found that the morphological features of the three mutant lines were clearly separated according to their genotypes (Figure 5D), confirming that humerus morphology is directly associated with the genotype of PAPSS2. We also observed that the humerus morphology of the gray short-tailed opossum mutant line was significantly closer to the Tasmanian devil mutant line than to the tammar wallaby mutant line in the CVA (paired t test, p value = 0.0054, Figure 5D), consistent with expectation according to the gene sequence ILS pattern.
To further validate whether the amino acids affected by ILS have morphological impact, we synthesized a modified gray short-tailed opossum PAPSS2 cDNA by replacing 4-aa sites affected by Dipr_Micr ILS with genotypes shared by monito del monte and tammar wallaby and then generated a mutant mice line with this modified cDNA (Papss2Mono-Micr/Mono-Micr) (Figures S4G and S4H). We observed that the tammar wallaby mutant line was significantly closer to the Papss2Mono-Micr/Mono-Micr mutant line than to the gray short-tailed opossum mutant line in CVA (paired t test, p value = 0.0029, Figure 5E). Considering that the only difference between the Papss2Mono-Micr/Mono-Micr mutant line and the gray short-tailed opossum mutant line was the amino acid changes corresponding to the loci affected by Dipr_Micr ILS, this result confirms that introduction of these ILS amino acids results in humerus shapes similar to the inferred hemiplastic morphological differences in our sampled marsupials.
DISCUSSION
Adaptive radiations accompanied by substantial diversification of morphology, physiology, and ecological niche requirements have shaped extant biodiversity at all levels of complexity (Losos, 2010; Moen and Morlon, 2014; Schluter, 2000). However, the reconstruction of phylogenetic bifurcation processes has often been compromised when speciation happened in such short evolutionary time windows that genome-wide signatures of reproductive isolation cannot be distinguished from signals of later hybridization or ILS, the two evolutionary processes that have been inferred to overwrite the foundational signatures of lineage divergence in phylogenomic reconstructions. In contrast to other groups, such as Darwin’s finches (Lamichhaney et al., 2015), East African cichlids (Salzburger et al., 2002), Anopheles mosquitoes (Fontaine et al., 2015), and Heliconius butterflies (Edelman et al., 2019), which are characterized by widespread hybridization and introgression, our study documents how ILS can explain widespread discordance between gene trees and species trees. Our genome-wide analyses of six key marsupial species showed that ILS affected more than 50% of the genome-wide sequences examined. This percentage exceeds earlier figures for the great apes where genomic analyses showed that 30% of the human genome showed signatures of ILS (Scally et al., 2012). Similar indications of ILS as a dominant factor during rapid adaptive radiation have been reported in birds (Jarvis et al., 2014), other primates (Mailund et al., 2014), and red algae (Lee et al., 2018).
ILS can result in identical genotypes across species that are scattered throughout phylogenetic trees independent of speciation order. It is important to appreciate that ILS or hemiplasy is fundamentally different from evolutionary convergence or homoplasy, another process that can produce morphological and other similarities between phylogenetically distant lineages (Darwin, 1859; Muschick et al., 2012; Sackton and Clark, 2019; Stern, 2013; Sun et al., 2018). Convergence produces similar phenotypes from different and often unknown genetic encoding, whereas ILS produces similar phenotypes from the same alleles due to paraphyletic descent. Convergence is widely acknowledged as a phenomenon affecting and explaining morphological similarities between phylogenetically unrelated lineages. The potential phenotypic consequences of ILS, on the other hand, are generally ignored, quite likely because of the insurmountable methodological challenges that applied until recently. However, ignoring ILS might lead to incorrect interpretations of phenotypic evolutionary history in lineages that experienced speciation events in rapid succession. Although consistent positive selection is generally assumed to explain convergent evolution, ILS requires large Ne in ancestral lineages and the absence of strong directional selection during subsequent speciation events. Once reproductive isolation and speciation have occurred, the maintenance of ILS regions across species does not request strong natural selection. It is also possible that some of the ILS regions might be under selection in descendant lineages, but, at that point, the signatures of ILS have become irreversibly established throughout genomes post speciation. Discriminating between convergence and ILS explanations of morphological similarity between non-sister taxa, thus, represents a profound challenge, which our proof-of-concept results highlight as an important future priority. Although we cannot fully exclude the possibility that convergent evolution may have affected specific morphological traits shared by non-sister marsupial branches, the ILS interpretation is far more plausible because convergence would never result in genome-wide mismatches. Although highly significant, our conclusions are obviously based on a limited number of marsupial genomes. A greater variety of morphological traits across a wider range of marsupial species will be needed to further clarify the impact of ILS across the marsupial phylogeny.
It has often been assumed that ILS signatures should be represented by short sequences because of frequent recombination in a large ancestral population (Scally et al., 2012). It is, therefore, of particular interest that our results document that extant marsupials also maintained a substantial number of longer ILS regions in their genomes, suggesting that complex adaptive ancestral traits came under divergent and recombination-averse selection after speciation events were completed. A long continuous ILS fragment such as WFIKKN1 might, thus, have been favored by selection over sufficient evolutionary time to fix phenotypic hemiplasy in multiple marsupials. Whether such positive selection would have continued until the present day is unknown but, adaptive or not, phylogenetic reconstructions need to take ILS into account as a possible mechanism for explaining mismatches between genomic variation used to construct phylogenies and phenotypic variation mapped onto such trees.
Limitations of the study
Our study only includes six marsupial species, which is sparse compared with the high species diversity in this mammalian group. A larger set of marsupial species genomes would enable more detailed reconstruction of the rapid radiation of this lineage and more precise analysis of the evolutionary consequences of genomic regions affected by ILS for descendant species. Additionally, given the fragmented nature of genome assemblies based on short-reads, our current analyses might have missed many long TEs that could further enhance ILS signal detection. This might be overcome by producing more complete genome assemblies based on long-read sequencing technology (Rhie et al., 2021; Zhou et al., 2021). Furthermore, our functional verifications of phenotypic ILS effects using an indirect transgenic mice approach have constrained us to focus on skeletal traits that are relatively conserved in mammals. Focused development of a marsupial animal model will be needed to allow more direct validations (Kiyonari et al., 2021).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact: Guojie Zhang (guojie.zhang@bio.ku.dk).
Materials availability
This study did not generate any new unique reagents.
Data and code availability
Genome sequencing data and the genome assembly generated in this study have been deposited in the NCBI SRA under accession PRJNA639670. The above data have also been deposited in the CNSA (https://db.cngb.org/cnsa/) of CNGBdb with accession number CNP0000563. WGAs generated by LASTZ+MULTIZ, the orthologous gene table, high-definition morphological photos and other relevant data can be found in Mendeley data https://doi.org/10.17632/2n7jt8mvgb.1.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
CRISPR/Cas9 knock-in mice lines
To verify our hypothesis of the phenotypic effects of ILS genes, we used the CRISPR/Cas9 system to produce transgenic mice lines. We designed two genetic manipulation experiments targeting the point substitution of the Wfikkn1 gene (Figures S5A and S5B) and a whole gene knock-in for Papss2 gene (Figures S5C and S5D) as described below.
Wfikkn1 point substitution
A c.227A>G mutation (Q76R) was induced into the mouse Wfikkn1 gene using CRISPR/Cas9. A guide RNA targeting this mutation site was designed using Geneious (Kearse et al., 2012), transcribed in vitro with the mMESSAGE mMACHINE T7 Ultra Kit (Ambion, TX, USA) according to the manufacturer’s instructions, and subsequently purified using the MEGAclear™ Kit (ThermoFisher, USA). The guide RNA (gRNA) was: 5’ GTGTGGGCTGCAGAGCTGCG 3’. Our target sequence in exon2 of Wfikkn1 was: GACTGTGCGG CATCCGAGAAGTGCTGCACCAATGTGTGTGGGCTGCaGAGCTGCGTgGCTGCCCGCTTTCCCAGTGGTGGCCCAGCTGTACCTG AGACAGCAGCCTCCTGTGAAG. A single-stranded DNA was synthesized as donor oligo, comprising 68bp upstream and 46bp downstream of the mutation site. The sequence of the oligo donor DNA was: GACTGTGCGGCATCCGAGAAGTGCTGCACCAA TGTGTGTGGGCTGCgGAGCTGCGTcGCTGCCCGCTTTCCCAGTGGTGGCCCAGCTGTACCTGAGACAGCAGCCTCCTGTGAAG. The boldfaced lowercase base “g” represents the target-point mutation, while base “c” is the synonymous mutation for the gRNA PAM blocking mutation. Cas9 mRNA (10 ng/μl), gRNA (4.0 ng/μl) and the donor oligo (50 ng/μl) were co-injected into zygotes of C57BL/6J mice to obtain F0 knock-in mice. Genotypes of F0 point substitution mice were identified by PCR. To do this, we extracted genomic DNA from mouse tails, designed a pair of primers (Primer I: 5’ GAAGGGGACAAAGAGCTCCC 3’ and Primer II: 5’ TACAACGTGCAGGTGGAGAC 3’) to bind to flanking regions of the target site, and performed PCR tests (Figure S5B). PCR products were Sanger-sequenced to confirm the precise replacement of the target mutation A>G.
PAPSS2 knock-in experiment
The goal of this experiment was to knock-in the gray short-tailed opossum PAPSS2 cDNA, the modified gray short-tailed opossum PAPSS2 cDNA, the tammar wallaby PAPSS2 Cdna (manual inspection), and the Tasmanian devil PAPSS2 cDNA (all with protein sequence only) at the start codon position in the mouse ortholog, respectively.
The specific experiment steps to obtain F0 knock-in mice are described as follows using the gray short-tailed opossum as an example. Two guide RNAs targeting the knock-in site were designed by Geneious (Kearse et al., 2012), in vitro transcribed with the mMESSAGE mMACHINE T7 Ultra Kit (Ambion, TX, USA) according to the manufacturer’s instructions, and subsequently purified using the MEGAclear™ Kit (ThermoFisher, USA). A targeting vector constructed for homologous recombination of the target fragment consisted of a 4.7kb 5’ homology arm, 2.7kb KI of gray short-tailed opossum PAPSS2 cDNA, WPRE-BGHpA, a 4.7kb 3’ homology arm, and other necessary components. Guide sequences (gRNA1: 5’ GTAAGTAAGCCCTTGAAATC 3’ and gRNA2: 5’ AGGGCTTACTTACTCTTTTA 3’), Cas9 mRNA (5.0 ng/μl), gRNA (1.0 ng/μl) and the donor plasmid (10 ng/μl) were co-injected into zygotes of C57BL/6J mice to obtain F0 knock-in mice. The same experiment protocol and verification procedure was used to generate three transgenic mice lines with the modified gray short-tailed opossum PAPSS2 cDNA, the tammar wallaby PAPSS2 cDNA, and the Tasmanian devil PAPSS2 cDNA, respectively. The modification of the gray short-tailed opossum PAPSS2 cDNA sequence refers to changing the amino acid type at the sites from the original type to the shared type, where monito del monte shares the same amino acid type only with tammar wallaby (Figures S4G and S4H). For example, the gray short-tailed opossum and Tasmanian devil had alanine (A) at amino acid position 93 (using ENSMODG00000016494 as the reference coordinate), monito del monte and tammar wallaby shared threonine (T), and this amino acid site would be threonine in the synthesized sequence.
Genotypes of F0 knock-in mice were verified by long overlapped PCR. We extracted genomic DNA from mouse tails and performed PCR tests. Two pairs of primers were designed to bind to flanking regions of the mouse sequence outside the homology arms and to the target KI sequence for PAPSS2 knock-in mice lines (Figure S5D). The primers for the candidate F0 knock-in mice were as follows: Primer I - 5’ homology arm forward: 5’ CTCTGTTCATTCCTATTACTGGCTCT 3’; Primer II - 5’ homology arm reverse: 5’ CAACCCACATCTTCCACCTTCT 3’; Primer III - 3’ homology arm forward: 5’ AGAGGTGGTAATGGCAAAGACAA 3’; Primer IV - 3’ homology arm reverse: 5’ ATAAAGAGCCCAAACATAAAGGAAG 3’. As shown in Figure S5D, we expected a PCR fragment length of 7.5 kb for the 5’ homology arm and a PCR fragment length of 5.2 kb for the 3’ homology arm of the F0 knock-in mice, compared with a 9.7kb PCR fragment at 5’ homology arm and a 9.5kb PCR fragment at 3’ homology arm for the WT mice. F0 mice for these four lines were selected for further experiments based on the match between the size of their PCR bands and these expectations.
Breeding of homozygous mice
F0 male mice were mated for one generation to select the individuals with the reproductive capacity. Then, IVF (in vitro fertilization) was performed on these candidates to obtain F1 female mice (heterozygous). 3–4 weeks of F1 female mice were used in the IVF with F0 or F1 male mice to produce the homozygous mice for the morphological analyses. The genotypes of homozygous mice were confirmed by PCR experiment.
METHOD DETAILS
Sequencing, assembly, and evaluation
We extracted DNA from a male monito del monte for genome sequencing. Paired-end and mate-pair DNA libraries with seven different insert sizes (250 bp, 500 bp, 800 bp, 2 kb, 5 kb, 10 kb and 20 kb) were constructed and sequenced on the Illumina HiSeq2000 platform. In total, 370 Gb of raw reads were produced. To facilitate the assembly work, a series of strict filtering steps were conducted to remove artificial duplications, adapter contaminations, and low-quality reads (Li et al., 2010). Before starting the assembly, we estimated the monito del monte genome size to be 3.2 Gb using K-mer analysis.
The program SOAPdenovo v2.04.4 (Luo et al., 2012) was used for de novo assembly of qualified reads in three main steps. First, the short-insert size library data were split into appropriate K-mer sizes to construct a de Bruijn graph. The graph was simplified by removing the tips, merged bubbles, connections with low coverage and all of the small repeats. Then, all qualified data were connected into contig sequences. Second, all of the usable reads were realigned onto the contig sequences to calculate the amount of shared paired-end relationships between each pair of contigs, and to weigh the rate of consistent and conflicting paired-ends before further constructing the scaffolds from the short-insert paired ends with the long-insert paired ends. Last, we used the GapCloser module in SOAPdenovo (Luo et al., 2012) to search through the read pairs and to identify those for which one end was mapped to the unique contig and the other was in the gap region based on the paired-end information. The gaps were then closed by the local assembly for these collected reads. The program SSPACE v2.0104 (Boetzer et al., 2011) was further applied to extend the pre-assembled scaffolds using reads from all of the long-insert (2 ∼ 20 kb) libraries with the following parameters: -x 0 -k 5 -n 20. The final size of the assembled genome was 3.4 Gb (scaffold N50 = 17.8 Mb and contig N50 = 10.2 kb). We performed a core eukaryotic gene mapping analysis (CEGMA) (Parra et al., 2007) to evaluate the quality of the monito del monte genome assembly. Overall, 236 of 248 (95.16%) complete core eukaryotic genes were identified.
Genome annotation
Across the monito del monte genome, we identified tandem repeats using Tandem Repeats Finder v4.04 (Table S2; Benson, 1999) and transposable elements (TEs) using both homology-based and de novo approaches (Table S2). For homology-based predictions, we used RepeatMasker v3.3.0 (Smit et al., 1996) for DNA level prediction and RepeatProteinMask v3.3.0 (Smit et al., 1996) for protein level prediction to identify candidates based on the Repbase database of known repeats. For de novo predictions, we used RepeatModeler v1.0.5 (Price et al., 2005) to construct a de novo repeat custom library, which was further used to search the whole genome with RepeatMasker v3.3.0 (Smit et al., 1996). LTR_FINDER v1.0.5 (Xu and Wang, 2007) was used to determine the characteristic structure of full-length long-terminal repeat retrotransposons (LTRs). Similar to previously sequenced marsupials, the monito del monte genome had a high percentage (∼61%) of transposable elements, most of which (38.97%) were long interspersed nuclear elements (LINEs). This is consistent with the relative larger genome sizes of marsupials compared with other amniotic species.
We used several approaches to predict the locations and structures of protein-coding genes in the monito del monte genome (Table S1). First, protein sequences available for three species (gray short-tailed opossum, tammar wallaby and human) from Ensembl release-75 were mapped to the genome using TBLASTN (BLASTall v2.2.23) (Altschul et al., 1990) with an e-value cutoff of 1e-5. The aligned sequences were then analyzed with GeneWise v2.2.0 (Birney et al., 2004) to search for accurate spliced alignments. We further clustered three homologous-based gene sets into a non-redundant homologous gene set. Second, we trained the optimal parameters for AUGUSTUS v2.5.5 (Stanke et al., 2006) using the gene models with high GeneWise scores from the homolog-based predictions. Third, de novo prediction was performed on the repeat-masked genome using the HMM model, AUGUSTUS, with the homologous hits in the first step and the optimal parameters in the second step. Finally, we conducted several optimization processing steps to: 1) remove the single-exon genes without any function in the known gene function databases; 2) replace split or incomplete genes or AUGUSTUS unique predictions without external evidence from corresponding homologous unique predictions; and 3) filter out pseudogenes and genes containing transposable elements.
Whole genome alignment
We generated whole genome alignments (WGAs) using the LASTZ + MULTIZ pipeline (Blanchette et al., 2004; Harris, 2007) (http://www.bx.psu.edu/miller_lab/) across the marsupial species with the gray short-tailed opossum as reference. We first carried out pairwise WGAs between genomes of the gray short-tailed opossum and five other marsupial species using the LASTZ v1.03.34 program with parameters: “–step=19 –hspthresh=2200 –gappedthresh=10000 –ydrop=3400 –inner=2000 –seed=12of19 –format=axt –scores= HoxD55”, Chain/Net package with parameters of “–minScore=5000” for the axtChain program and default parameters for other programs. To prevent the use of multiple hits from the WGAs, we used the reciprocal best matches and filtered out other multiple hits. Using the MULTIZ v11.2 program, we initially obtained ∼2.77 Gb WGAs. To meet the objectives of the subsequent analyses, only blocks longer than 100bp and containing the gray short-tailed opossum, monito del monte, at least one diprotodontian species, and at least one dasyuromorphian species were retained. These filtering steps removed about 1.14 Gb from the initial alignment.
To reduce error in phylogenetic inferences and ILS identification, we performed two rounds of correction for the above multiple sequence alignments using the method developed in Jarvis et al. (2014). The first round was to identify and remove the aberrant sequences, including 1) any alignment where only one species contributed to the alignment; and 2) any sequence for one species that was aligned to other sequences but did not appear to be homologous to any other species in that part of the alignment (regions of ≥ 36 bp window size that have < 55% sequence identity to all other species in the alignment with gaps allowed). The first case could be the insertions in one species, but such single-species sites are not useful for tree estimation and ILS identification. More often than not, these aberrant sequences reflect errors in assembly, or alignment, which would introduce errors in phylogenetic inference and ILS analysis. Thus, we removed these aberrant sequences from the segments as Jarvis et al. (2014) has suggested and a second MSA round was performed on the remaining MULTIZ segments with MAFFT v11.2 (L-INS-I, mafft –maxiterate 1000 –localpair) (Katoh and Standley, 2013). All realigned individual segments were concatenated to get a final whole genome alignment of 1.4 Gb. When only concatenating the realigned segments containing all six marsupial species, we could generate 984 Mb realigned WGAs.
Ortholog assignment
Orthologs were identified among all six species based on the sequence similarity and the synteny evidence. We first aligned protein sequences of the gene sets of the gray short-tailed opossum and another species to each other by BLASTP (BLASTall v2.2.23) with an e-value cut-off of 1e−5, and combined local alignments with the SOLAR v0.9.6 program (Almasy and Blangero, 1998). The aligned gene pairs with the homologous block lengths of ≥ 30% of length of the longest protein and identity ≥ 50% were kept as the candidate orthologs. Then, the reciprocal best hit (RBH) orthologs were identified from these candidates. To save candidate orthologs from the strict RBH method, we also included RBH orthologs from the second and third round by masking known RBH genes. RBH orthologs that were also supported by gene or genome synteny would be retained as the final pairwise orthologs between the gray short-tailed opossum and another species. Detection of gene synteny and genome synteny was done following the criteria in the published literature (Jarvis et al., 2014).
Gene synteny
The candidate RBH genes were mapped on the chromosomes according to the coordinates of gray short-tailed opossum and sorted in order. For one RBH gene (A1A2; 1 and 2 denote the gray short-tailed opossum and another species) and its nearest RBH gene (B1B2) were considered to have syntenic evidence if they met the following requirements: a) genes A1 and B1 are on same chromosome or scaffold; b) genes A2 and B2 are on same chromosome or scaffold; c) the number of genes between A1 and B1 < 5; d) the number of genes between A2 and B2 < 5. As the literature suggests (Jarvis et al., 2014), we also retained RBH genes if one of their scaffolds only has one gene.
Genome synteny
By placing the candidate RBH genes in the genomic syntenic blocks (pairwise WGAs between the gray short-tailed opossum and other species), we calculated the gene-in-synteny ratio for each gene (synteny-region-length/total-coding-region-length) and the syntenic ratio (syntenic length of the two genes/length of the shorter gene) in the coding regions. The RBH genes with gene-in-synteny ratio ≥ 0.3 and syntenic ratio ≥ 0.3 were considered to have syntenic evidence.
In this way, we built the pairwise orthologs between the gray short-tailed opossum and another five species when considering both the protein similarity and the synteny. We then constructed the orthologous genes of all six marsupial species through merging pairwise orthologs according to the reference gray short-tailed opossum gene set. There were 17,639 putative orthologs without any species restriction. When we restricted these orthologs to be present in the gray short-tailed opossum, monito del monte, at least one diprotodontian species, and at least one dasyuromorphian species, 13,320 orthologs were left. In the final list, 9,227 orthologous genes were identified in all six species.
Transposable elements (TEs) dataset
In this analysis, we focused on three major groups of retroelements: short interspersed nuclear elements (SINEs), long interspersed nuclear elements (LINEs), and long-terminal repeats (LTRs). Since the retroelement insertions are considered to be powerful markers for resolving phylogenetic relationships, we first constructed a presence/absence matrix of retroelement insertions across all six species. To achieve this, we ran all-vs-all LASTZ pairwise alignment for all six marsupials. According to this genomic synteny, we then generated the presence/absence matrix based in each pair of species on the following criteria:
For a TE in the reference species, if only the two flanking sequences of this TE (upstream and downstream 2kb) in the query species can be aligned, and there is no corresponding TE in the middle, this TE element is missing in that query species (character state “0”).
For a TE in the reference species, if the two flanking sequences in the query species can be aligned, and there are also orthologous TE pairs (length of the shorter/length of the longer TE > 50%) in the middle, this element exists in that query species (character state “1”).
If a TE in the reference species does not fall into either of these above categories, the corresponding status of this element in the query species is marked as “?”.
In this way, for each TE in the reference species, we could assign it a corresponding status in the query species. Here, we removed the TE with a “?” label. We then combined these pairwise presence/absence matrices of retroelement insertions into one matrix with 401 informative markers across all six species. Here, the informative markers are those elements with “0” in the outgroup (Mono) and simultaneously with “1” in at least two, but not all, of the five species.
Species tree inference
We converted each matrix of presence/absence characters into a set of incompletely resolved “gene trees” with each gene tree representing a single retroelement character and executed ASTRAL-III v5.6.2 (Zhang et al., 2018) in “exact” mode (-x) given these incompletely resolved “gene trees” as input, as a previous study suggested (Springer et al., 2020).
Multi-directional KKSC insertion significance test
To determine whether the cause of conflicting TEs is ILS or hybridization, we applied a multi-directional KKSC insertion significance test (Kuritzin et al., 2016). According to the algorithm of this test, for a triplet to be checked, there are three possible TE insertion modes: 1) insertions can be detected only in lineage A and B; 2) insertions can be detected only in lineage A and C; and 3) insertions can be detected only in lineage B and C. The mode with the most insertions was considered to support the true evolutionary path of the species, and the likely factors were then determined by testing whether the number of insertions in the remaining two modes was statistically symmetric or not. If they are symmetric, the ILS is more likely to be the cause; otherwise, the conflicting TEs are caused by the hybridization. In our study, lineages A, B and C are Dipr, Dasy and Micr. In total, we obtained 80 TEs that were only inserted in both two Dipr and two Dasy marsupials (named as Dipr_Dasy partition), 18 TEs that were only inserted in both two Dipr and Micr marsupials (named as Dipr_Micr partition), and 14 TEs that were only inserted in both two Dasy and Micr marsupials (named as Dasy_Micr partition). The number of these markers was used in the KKSC insertion significance test.
Robustness
Considering that the assembly procedure could result in a small number of Ns in some LASTZ synteny blocks, we found 61 informative markers with Ns in both upstream and downstream in the tammar wallaby and/or monito del monte genomes. By excluding these TEs in the analyses, the above conclusion was not affected. The monito del monte is still placed outside the Australasian group, while polytomy remains rejected (p-value = 1.1477e−14), and hybridization is not accepted either (p-value = 1).
Species tree inferences
We used two methods to infer the phylogeny of the six species: 1) coalescence-based method, ASTRAL-III v5.6.2 (Zhang et al., 2018), and 2) concatenation-based method, Randomized Axelerated Maximum Likelihood RAxML, (v8.2.9) (Stamatakis, 2006).
ASTRAL-III strategy
ASTRAL could estimate a species tree with the branch lengths in coalescent units given a set of individual gene trees under the multispecies coalescent model, which is useful for handling ILS events. We generated two different sets of individual gene trees from 984 Mb realigned WGAs and 9,227 orthologs of all six marsupial species using IQ-TREE v1.6.12 (Nguyen et al., 2015), respectively.
We divided WGAs into non-overlapping windows of 100bp and only those windows with the gap ratio ≤ 30% for each sequence of the six species were retained. To ensure the validity of the tree inference, the windows that consist of less than four different haplotypes were filtered out. Also, to make the variable sites adequate for the tree inference, the windows having > 70% constant sites (a site containing the same nucleotide in all sequences) were also filtered out. Then, we inferred the topology for the windows that passed the above filtering steps using IQ-TREE with the ModelFinder function (Kalyaanamoorthy et al., 2017). IQ-TREE performs a composition Chi-square test for every sequence in the alignment, the purpose of which is to test for homogeneity of character composition: a sequence is denoted as “failed” if its character composition significantly deviates from the average composition of the alignment. Thus, only the individual gene trees inferred from those windows that passed the composition Chi-square test were accepted. Further, to avoid the potential effect of the long branch attraction on the phylogeny, we ran TreeShrink (v1.3.7) (Mai and Mirarab, 2018) to detect and filter out the inferred individual gene trees with unexpectedly long branches caused by the erroneous sequences in any species of monito del monte, two diprodontian species, or two dasyuromorphian species. After the above quality control steps, 5,685,945 qualified gene trees were used as the candidate input for ASTRAL.
We aligned the complete protein-coding sequences of 9,227 orthologs with MAFFT L-ins-I, and back-translated the protein alignment into the nucleotide alignment. Then, for each alignment, we also used IQ-TREE to infer the topology for all orthologous genes with the ModelFinder function. The output gene trees were used as the input for ASTRAL.
RAxML
RAxML v8.2.9 was performed with GTRCAT model and 100 bootstrap replicates on five different datasets: 1) WGAs containing all six species after the realigned step (∼984 Mb); 2) aligned coding regions of 9,227 orthologous genes identified in all six species (∼20 Mb); 3) four-fold degenerate sites of these orthologs (∼1.3 Mb); 4) C12 of these orthologs (∼14 Mb); and 5) C3 of these orthologs (∼6.9 Mb).
Incongruence between gene trees and species trees
Results from the WGAs dataset
We used DiscoVista v1.0 to calculate phylogenetic discordance using the Dipr_Dasy tree as the species tree and 5,685,945 loci trees inferred from 984 Mb realigned WGAs of all six marsupial species (window size = 100bp) (Sayyari et al., 2018). We focused on the discordance occurring at the most recent common ancestor (MRCA) of the monito del monte, Diprotodontia and Dasyuromorphia.
Results from the orthologs dataset
Based on the coordinate information of 9,227 orthologs in all six marsupial species, we extracted the exon blocks that were shared among all marsupial species from the WGAs. In total, 7,471 of 9,227 orthologs (81%) had at least one exon block that was qualified for topology inference. We considered an exon block as qualified when it met the following criteria: a) alignment consists of more than four different haplotypes; and b) constant sites < 70% or < 70bp. IQ-TREE v1.6.12 with the ModelFinder function was used to infer the topologies from these exon blocks and we only kept the gene trees inferred from those windows that passed the composition Chi-square test. Then, we ran TreeShrink v1.3.7 (Mai and Mirarab, 2018) to detect and filter out the inferred gene trees with unexpectedly long branches caused by the erroneous sequences in any species of monito del monte, two diprotodontian species, or two dasyuromorphian species. In total, 22,743 exon blocks with an average length of 281 bp were retained and DiscoVista was then applied to measure the phylogenetic discordance at the MRCA of the monito del monte, diprotodontian and dasyuromorphian species based on these gene trees.
Divergence time estimation
Species divergence time was estimated using the MCMCTree program in the PAML package v4.5 (Yang, 1997) with the approximate likelihood calculation algorithm. Baseml in the PAML package was used to estimate alpha and the substitution rate before we used gHmatrix to produce an out.BV file containing the Hessian matrix. The MCMCTree was then used to estimate divergence times based on these parameters. We applied this pipeline to two sets of WGAs containing all six marsupials, and each with three types of alignments that supported the Dipr_Dasy tree, the Dipr_Micr tree and the Dasy_Micr tree, respectively.
Set 1: WGAs partition based on the topologies inferred from non-overlapping windows of 100bp
As mentioned above, we totally had 5,685,945 qualified windows obtained from 984 Mb realigned WGAs after a set of filters. According to the topologies inferred by IQ-TREE for each window, we concatenated the windows with the output tree that supported the Dipr_Dasy tree and repeated the process for windows supporting the other two topologies. The MCMCTree program was then used to analyze three types of alignments and the outputs are presented in Figure S2A.
Set 2: Loci partition based on the CoalHMM results
We performed CoalHMM analysis on four sets of the multiple alignment data, which were made up of different species (more details in “CoalHMM analysis” section). Based on the gray short-tailed opossum’s coordinates, we picked out the overlapped type 0 loci in four combinations and concatenated these loci into a multiple alignment that supported the Dipr_Dasy tree (∼43 Mb). Although type1 loci also supported the Dipr_Dasy tree, we exclude these loci when estimating the divergence time to eliminate interference from the loci with a deeper coalescence between diprotodontian and dasyuromorphian species. In the same way, we generated the multiple alignment supporting the Dipr_Micr tree by concatenating the overlapped type2 loci in four combinations (∼100 Mb). We also generated the multiple alignment supporting the Dasy_Micr tree by concatenating the overlapped type3 loci in four combinations (∼67 Mb). The MCMCTree program was then used to analyze three types of alignments and the outputs are presented in Figure 3A.
Three input trees corresponding to three sets of alignment used in the MCMCTree program are as follows. To improve the accuracy of estimation, we used the estimates from independent molecular dating studies as the evidence for calibration at the root node with the upper limit as 116 mya and the lower limit as 64 mya according to the literature (Hope et al., 1989; Nilsson et al., 2003), because the fossil resources for the early origin of marsupials are lacking (Luo et al., 2003, 2011).
species tree (Dipr_Dasy tree): ((((S. harrisii, A. stuartii), M. eugenii, P. cinereus)), D. gliroides), M. domestica)’>0.64<1.16’.
Dipr_Micr tree: ((((M. eugenii, P. cinereus), D. gliroides), S. harrisii, A. stuartii)), M. domestica)’>0.64<1.16’.
Dasy_Micr tree: ((((S. harrisii, A. stuartii), D. gliroides), M. eugenii, P. cinereus)), M. domestica)’>0.64<1.16’.
QuIBL analysis
We used 5,685,945 qualified loci trees inferred from 984 Mb realigned WGAs as the candidate input set for QuIBL analysis (Edelman et al., 2019). We randomly selected 5,000 individual trees from this candidate set as an input for one QuIBL estimation and repeated this random selection 100 times to generate 100 QuIBL outputs. In each QuIBL estimation, we focused on the discordance analysis in the following four triplets: 1) D. gliroides - A. stuartii - M. eugenii; 2) D. gliroides - S. harrisii - M. eugenii; 3) D. gliroides - A. stuartii - P. cinereus; and 4) D. gliroides - S. harrisii - P. cinereus. Taking the triplet, D. gliroides - A. stuartii - M. eugenii, as an example, the main steps of QuIBL analysis were as follows.
-
1
Of the 5,000 loci trees invested, each tree would be first grouped into the following three subsets based on its topology. Since the topologies of individual trees in Subset 1 corresponded to the species tree, we would not take them into account when analyzing the discordances.
Subset 1 (Dipr_Dasy tree): ((M. eugenii, A. stuartii), D. gliroides).
Subset 2 (Dipr_Micr tree): ((M. eugenii, D. gliroides), A. stuartii).
Subset 3 (Dasy_Micr tree): ((A. stuartii, D. gliroides), M. eugenii).
-
2
QuIBL then calculated the likelihood values (Bayesian Information Criterion test, BIC) that the inner branch lengths in Subset 2 and Subset 3 were best described by a simple exponential distribution as expected under ILS (scenario 1) or a mixture of ILS and introgression (scenario 2). The difference in BIC values (Delta.BIC) was calculated as the BIC value of scenario 2 minus the BIC value of scenario 1. Since the BIC value is less than 0, when Delta.BIC is greater than 10, the scenario of ILS only with the lower BIC value is preferable. However, when Delta.BIC is less than −10, the scenario of mixture of ILS and introgression with the lower BIC value is preferable. In other cases, the two scenarios are indistinguishable.
-
3
QuIBL also inferred the theoretical distributions of inner branches under ILS or introgression for Subset 2 and Subset 3. After plotting these two theoretical distributions, they could be compared visually with the observed distribution of inner branches.
All 100 QuIBL outputs were summarized in Figures S2B–S2E. In all four target triplets of these repetitions, the ILS only scenario had a lower BIC value than the mixture scenario. Further, the overall Delta.BIC values obtained from the subset supporting Dipr_Micr tree or the subset supporting Dasy_Micr tree in four target triplets were greater than 10. Thus, as the interpretation suggested by QuIBL, the discordances observed in the early evolutionary period of marsupials are caused by ILS only.
Four-taxon D-statistic test
We performed a four-taxon D-statistic test, also known as the ABBA-BABA statistic test, to detect gene flow despite the existence of ILS (Green et al., 2010). This method compares the number of parsimony-informative sites, ABBA and BABA, which support two genealogies discordant with the species tree. If the two types of sites are not statistically different, they are likely to be produced by ILS. Otherwise, gene flow is present and causes two non-sister species to be more similar to each other than expected. We used Dfoil software (Pease and Hahn, 2015) with the mode as “dstat” to conduct this D-statistic method. To ensure that each window examined had adequate parsimony-informative sites for the test, we combined 50 adjacent 100bp-windows into a single 5kb window. By doing this, all windows met the requirements of statistical testing, and each window contained an average of 61 ABBA sites and 68 BABA sites. We then did the four-taxon D-statistic test on these 5kb windows in four different combinations of species, each consisting of Mono, Micr, one species from Dipr and one species from Dasy.
For Macr_Ante_Micr_Mono, 95.2% of the windows had no significant difference in the number of ABBA and BABA sites. 4.4% of the windows were thought to have gene flow between Macr and Micr. The remaining 0.4% of the windows were thought to have gene flow between Ante and Micr.
For Macr_Sarc_Micr_Mono, 95.2% of the windows had no significant difference in the number of ABBA and BABA sites. 4.3% of the windows were thought to have gene flow between Macr and Micr. The remaining 0.5% of the windows were thought to have gene flow between Sarc and Micr.
For Phas_Ante_Micr_Mono, 90.9% of the windows had no significant difference in the number of ABBA and BABA sites. 9.0% of the windows were thought to have gene flow between Phas and Micr. The remaining 0.1% of the windows were thought to have gene flow between Ante and Micr.
For Phas_Sarc_Micr_Mono, 90.9% of the windows had no significant difference in the number of ABBA and BABA sites. 9.0% of the windows were thought to have gene flow between Phas and Micr. The remaining 0.1% of the windows were thought to have gene flow between Sarc and Micr.
These outputs showed that up to 95% windows with equal ABBA and BABA sites, which indicated that two genealogies discordant with the species tree, ABBA and BABA, were more likely produced by ILS across almost the entire whole genome. On closer examination of the supposed windows of gene flow, we found that such windows had the fewer identical sites shared between Micr and Dasy than other windows, but had the similar identical sites shared between Micr and Dipr as other windows. Rather than gene flow between species Micr and Dipr, there is another possibility for this scenario: the faster substitution rate in Dasy the longer branch length in Figure 1 had cleaned the identical sites between Micr and Dasy.
CoalHMM analysis
To clarify the extent of ILS in the marsupial genomes at the site level, we used a coalescent inference model, CoalHMM, to identify ILS regions on 1.4 Gb realigned WGAs, which contained the gray short-tailed opossum, monito del monte, at least one diprotodontian species, and at least one dasyuromorphian species (Hobolth et al., 2007, 2011). Here, we allowed the aligned blocks where some species were absent, because the input alignments in CoalHMM analysis consisted of data for four species. Thus, for example, an aligned block containing the gray short-tailed opossum, monito del monte, tammar wallaby (a diprotodontian species) and brown antechinus (a dasyuromorphian species) is valid for the CoalHMM analysis.
As we focused on the ILS occurring in the speciation of monito del monte, Diprotodontia and Dasyuromorphia, we had four combinations:
Combination 1 (Macr_Ante): M. domestica - D. gliroides - A. stuartii - M. eugenii.
Combination 2 (Macr_Sarc): M. domestica - D. gliroides - S. harrisii - M. eugenii.
Combination 3 (Phas_Ante): M. domestica - D. gliroides - A. stuartii - P. cinereus.
Combination 4 (Phas_Sarc): M. domestica - D. gliroides - S. harrisii - P. cinereus.
Thus, we first filtered the species from the specified branch to produce four sets of the alignments. Each alignment was processed as follows:
Columns where all rows were gaps were removed.
After merging the consecutive blocks (in the gray short-tailed opossum’s coordinates) of less than 50 nt apart, we further removed blocks with less than 500 nt.
We separated the alignment blocks into sets of blocks containing roughly 1 Mb.
We ran CoalHMM with the unclock model, which allows one species to have a longer terminal branch. The assignment of the longest branch was based on the inferred species tree (Figure 1), and in all cases it was the branch leading to the dasyuromorphian species (S. harrisii or A. stuartii).
To obtain the optimized starting parameters, we randomly selected three 1 Mb windows from the alignment, ran CoalHMM under unclock model with default parameters for each 1 Mb window separately. From the parameters estimated by CoalHMM, we calculated the optimized starting parameters as the mean of the three runs for tau1, tau2, theta1 and theta2.
Finally, we ran CoalHMM under the unclock model in each 1 Mb window individually, setting the starting parameters as the ones estimated in the previous step. A posterior decoding approach was used in CoalHMM to reconstruct the most likely genealogy for each locus: either the standard Dipr_Dasy relationship (non-ILS, type 0 and type 1) or the alternatives Dipr_Micr (type2) or Dasy_Micr (type3), which represent the consequences of ILS in the speciation period of Diprotodontia, Dasyuromorphia, and monito del monte. Depending on whether there was a deeper coalescence between Diprotodontia and Dasyuromorphia, the non-ILS sites could be further distinguished as type 0 (without deep coalescence) and type1 (with deep coalescence). The assigned genealogy of a locus is the type with the highest posterior probability.
For each combination, we collected the posterior probabilities per 1 Mb run based on the gray short-tailed opossum’s coordinates. To obtain ILS results in the orthologous coding regions, we extracted the posterior probabilities in the coding regions based on the coordinates of gray short-tailed opossum.
We then used the detected ILS patterns to explore the selection forces during the early period of Australian marsupial speciation. We downloaded the phyloP score of 100 vertebrate species including the gray short-tailed opossum, Tasmanian devil and tammar wallaby from the UCSC database (Haeussler et al., 2019; Pollard et al., 2010) to check whether these ILS sites overlapped with any conserved regions. Here, we only used the sites that were assigned the identical genealogy in four combinations with different species, which resulted in 43 Mb type0 loci, 95 Mb type1 loci, 100 Mb type2 loci and 67 Mb type3 loci. After converting the human - gray short-tailed opossum coordinates, 41 Mb genomic regions with phyloP scores were extracted. Chi-square test was used to detect differences in the distribution of conserved sites among non-ILS (type0 and type1), Dipr_Micr ILS (type2) and Dasy_Micr ILS (type3) regions. Moreover, the degree of conservation of non-ILS regions was significantly higher than that of ILS regions (Welch Two Sample t-test, p-value < 2.2e−16 for both ILS types). This indicates that the ILS regions were less constrained by selection. Moreover, less-constrained regions are also more likely to be more divergent, which increases their likelihood of being categorized as having been under ILS in the CoalHMM model.
ILS candidate gene identification
When defining ILS candidate genes in 13,320 orthologs, we integrated the evidence of CoalHMM’s results and the topology inference in four combinations. These are the steps taken:
For each orthologous gene, we extracted the posterior probabilities of the coding regions from the corresponding whole-genome level CoalHMM’s results based on the gray short-tailed opossum’s coordinates in all possible combinations that this orthologous gene could form. For example, if an orthologous gene can be found in M. domestica, D. gliroides, A. stuartii, S. harrisii, and M. eugenii, we would extract the posterior probabilities from Combination 1 (Macr_Ante) and Combination 2 (Macr_Sarc), respectively. Based on the extracted information, we could further calculate four values: the total number of extracted sites; the number of non-ILS sites (type0 and type1); the number of Dipr_Micr ILS sites (type2); and the number of Dasy_Micr ILS sites (type3). Of the last three values, the type corresponding to the maximum value was considered to be the ILS type of this combination. For each orthologous gene, depending on the type of combination it could form, there should be at least one and at most four sets of these values.
For each orthologous gene, we extracted the species from the alignment of the coding regions based on the possible combinations that this orthologous gene could form. For example, if an orthologous gene can be found in M. domestica, D. gliroides, A. stuartii, S. harrisii, and M. eugenii, we would produce two sets of the alignment: M. domestica - D. gliroides - A. stuartii - M. eugenii; and M. domestica - D. gliroides - S. harrisii - M. eugenii. Next, we used RAxML to calculate the likelihood values of each alignment under three candidate topologies: Dipr_Dasy tree, Dipr_Micr tree, and Dasy_Micr tree (with the command “-z”). The topology with the highest likelihood value would then be considered as the best tree for this alignment. For each orthologous gene, each combination it formed would thus have its own best topology.
Then, we integrated the evidence from the above steps. For any combination of each orthologous gene, it would be considered valid only if the following two criteria are met: a) the total number of extracted sites/the coding regions ≥30%; and b) the best topology assigned by RAxML is as same as the ILS type assigned by CoalHMM.
For an orthologous gene, if all of its valid combinations supported Dipr_Dasy, the gene was inferred to be a Dipr_Dasy gene. We used the same criterion for Dipr_Micr genes and Dasy_Micr genes.
In total, we identified 6,425 Dipr_Dasy genes, 1,310 Dipr_Micr genes, and 803 Dasy_Micr genes by this method.
Functional annotation of Dipr_Micr and Dasy_Micr candidate genes
First, the bi-directional best hit method was applied to generate the orthologous relationship between the monito del monte predicted genes and the mouse Ensembl genes. Then, we assessed the relative breadth of gene expression at the organ level based on data from the Gene Expression Database (Smith et al., 2019). By searching the mouse’s counterparts in the database, we located 11,718 of 13,320 orthologs with hits, including 1,099 Dipr_Micr candidate genes, and 670 Dasy_Micr candidate genes. Together, there was adequate evidence to suggest that 1,092 of 1,301 Dipr_Micr candidate genes, 666 of 803 Dasy_Micr candidate genes and 11,685 of 13,320 orthologs were expressed in at least one of the following organs: sensory organ, testis, brain, gland, ovary, metanephros, liver, heart, skin, lung, and pancreas. The detailed frequencies of these three gene sets in each organ are presented in Figure 4E. In addition, we searched the orthologous genes in the mouse of these ILS genes in the Mammalian Phenotype Ontology Database (Smith and Eppig, 2009) to annotate them at the phenotypic level focusing on the 17 phenotypic systems listed in Table S4. In total, 613 of 1,310 Dipr_Micr candidate genes, 335 of 803 Dasy_Micr candidate genes, and 6,710 of 13,320 orthologs with the counterparts in mouse were involved in at least one of these 17 systems. The detailed distribution of these genes in each system is shown in Table S4 and we further calculated the gene frequencies of each system in these three sets of genes, which are shown in Figure 4F. The gene frequency of a phenotype system is the proportion of the total number of genes annotated as being instrumental for these organ systems. Next, to identify candidate genes associated with the skeleton anatomy used in the transgenic experiments, we required that the candidates should contain the same amino acids shared between monito del monte and the diprotodontian marsupials, and that alignment across all investigated species showed no insertions or deletions near the shared amino acid sites. We also required that the candidate genes showed expression signals or with knockout phenotypes on the relevant tissues in mice.
Morphological analysis of knock-in mice
Entire mouse individuals at 1∼2 months of age were scanned with a MicroXCT 400 (Carl Zeiss X-ray Microscopy Inc., Pleasanton, USA) at the Institute of Zoology, Chinese Academy of Sciences, using a beam energy of 60 kV, 133 mA, absorption contrast and a spatial resolution of 34.014∼46.296 μm. From the image stacks, morphological structures, including the thoracic vertebrae and the humerus of each specimen were reconstructed and separated with Amira 5.4 (Visage Imaging, San Diego, USA). Morphological information of each specimen was measured with Geomagic Studio 2013 (3D Systems, South Carolina, USA). Subsequent volume rendering and animations were performed with VGStudio MAX 2.1 (Volume Graphics, Heidelberg, Germany) (Bai et al., 2016, 2018). The final figures were prepared with PhotoshopCS5 (Adobe, San Jose, USA).
In total, we had 11 Wfikkn1Q76R/Q76R mice and 10 wild-type mice for the measurement. For each individual from the Wfikkn1Q76R/Q76R mice line, four sets of values for the morphological information of the vertebrae were measured under the mice actual size (parity proportions) by using the measurement tool of Geomagic studio 2013 (Katz and Friess, 2014): 1) the height of the spinous process on T1; 2) the width of the centrum of T1; 3) the height of the spinous process on T2; and 4) the width of the centrum of T2 (Figure S4D). To be specific, the height of the spinous process was measured as the straight-line distance between the vertex and the midpoint on the base of a spinous process, and the width of the centrum was measured as the straight-line distance between the front and rear endpoints of the inner side of the centrum. The ratio of the spinous process (T1/T2) were compared by the Welch Two Sample t-test between the wild type mice and the Wfikkn1Q76R/Q76R mice after log-transformation (Figure 5C). To further measure the changes of the spinous process of T1 and T2 independently, we used the width of the centrum of the vertebrae to standardize the height of the spinous process, and compared the ratio of the spinous_process and centrum_width between the wild type mice and the Wfikkn1Q76R/Q76R mice after log-transformation (Figure S4E).
For mice samples from the PAPSS2 knock-in experiment, we used 3D geometric morphometric analyses to compare the differences among Papss2Mono/Mono, Papss2Mono-Micr/Mono-Micr, Papss2Dipr/Dipr, and Papss2Dasy/Dasy mice lines based on the curvature of the humerus bone. We extracted one curve from the humerus of each individual to represent its specific external form, and then resampled each curve into 41 equally spaced semi-landmarks the curve tip position is highlighted with a black-dotted line on the 3D model of the humerus just above (Figure S4F; MacLeod, 2017). These curves and semi-landmarks were then digitized using the IDAV Landmark software package (Wiley et al., 2005). Next, the datasets used for the subsequent morphological analysis was obtained by converting semi-landmarks into landmarks (MacLeod, 2017) in text file format: the curve number and point number for each sample were deleted, and then landmark numbers were replaced by point numbers (Tong et al., 2021; Zhang et al., 2019). The landmark configurations were scaled, translated, and rotated against the consensus configuration using the Procrustes superimposition method in advance (Bai et al., 2014; MacLeod, 2017). Finally, Canonical Variate Analysis (CVA) and the degree of differentiation in mathematical spaces formed by the first two CV axes were used to visualize the discreteness of the humerus between mice test lines in Mathematica (MacLeod, 2007). Figure 5D has plotted the first two canonical variables of Papss2Mono/Mono, Papss2Dipr/Dipr, and Papss2Dasy/Dasy mice lines, with CV1 representing 87.61% and CV2 representing 12.39% of the weighted sample variables, respectively. Figure 5E has plotted the first two canonical variables of Papss2Dipr/Dipr, Papss2Dasy/Dasy, Papss2Mono/Mono, and Papss2Mono-Micr/Mono-Micr mice lines with CV1 representing 66.84% and CV2 representing 24.97% of the weighted sample variables. Next, the Euclidean distance, i.e. the absolute distance between two points in multidimensional space (all CVs were considered in our study), meant to digitize the differences between these mice lines, was calculated based on the CVA. In the comparison of Papss2Mono/Mono, Papss2Dipr/Dipr, and Papss2Dasy/Dasy mice lines in Figure 5D, we calculated the Euclidean distances from each sample in the Papss2Mono/Mono mice line to the cluster center of Papss2Dipr/Dipr mice line and to the cluster center of Papss2Dasy/Dasy mice line, respectively. In the comparison of Papss2Dipr/Dipr, Papss2Dasy/Dasy, Papss2Mono/Mono, and Papss2Mono-Micr/Mono-Micr mice lines in Figure 4E, we calculated the Euclidean distances from each sample in the Papss2Dipr/Dipr mice line to the cluster center of Papss2Mono/Mono mice line and to the cluster center of Papss2Mono-Micr/Mono-Micr mice line, respectively.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification approaches and statistical analyses of the genome sequencing, quality assessment of the assembly, phylogeny, QuIBL analysis, four-taxon D-statistic test, CoalHMM analysis, as well as the morphological comparative analyses can be found in the relevant sections of the method details.
Supplementary Material
(A) ASTRAL output based on WGAs. Lengths of internal branches estimated by ASTRAL-III are labeled on the tree in coalescent units (scale bar at the bottom).
(B) ASTRAL output based on 9,227 orthologs.
(C) RAxML output based on the concatenated coding alignment of 9,227 orthologs. Branch length scale bar refers to the expected number of substitutions per site. Bootstrap value is labeled at the node in parentheses if the value is less than 100.
(D) RAxML output based on the 4-fold degenerate sites of 9,227 orthologs.
(E) RAxML output based on the alignment of 1st and 2nd codon positions of 9,227 orthologs.
(F) RAxML output based on the alignment of 3rd codon positions of 9,227 orthologs.
{kind=link}
(A) Using the estimated divergence times to distinguish between ILS and hybridization scenarios. As the upper schematic trees illustrate, the coalescence time under ILS (ti) should be earlier than the speciation event (t), whereas the expected divergence time under hybridization (th) should be later than the speciation event (t). The next trees show the divergence times across six species estimated by MCMCTree with three alternative topologies and the corresponding genomic regions. Dipr_Dasy: the topology and the regions supporting the species tree. Dipr_Micr: the topology and the regions supporting Dipr and Micr as sister species. Dasy_Micr: the topology and the regions supporting Dasy and Micr as closest relatives. The minimum and maximum calibration dates labeled at the root are 64 and 116 MYA, respectively, based on the literature.
(B) QuIBL output of the triplet, D. gliroides-A. stuartii-M. eugenii. (Upper) Distribution of Delta. BIC values of the Dipr_Micr subset (blue) and the Dasy_Micr subset (green) calculated by QuIBL. Delta. BIC value is calculated as the BIC value of scenario 2 minus the BIC value of scenario 1 (see STAR Methods). (Bottom) The distribution of the internal branch lengths (gray) is more in line with the inferred ILS distribution (red) than the inferred introgression distribution (black) for these two subsets. As shown in (B), we also present the results of D. gliroides-S. harrisii-M. eugenii triplet in (C), D. gliroides-A. stuartii-P. cinereus triplet in (D), and D. gliroides-S. harrisii-P. cinereus triplet in (E). Dasy, Dasyuromorphia; Dipr, Diprotodontia; Micr, monito del monte.
{kind=link}
(A) The proportion of ILS cases observed in WGAs (left) and orthologous coding regions (right) inferred by CoalHMM in four combinations. In each combination, the proportions of the loci belonging to the four candidate genealogies (x axis) were calculated. Four candidate genealogies are: 0 represents the species tree (non-ILS) without deep coalescence; 1 represents the species tree (non-ILS) with deep coalescence between Dipr and Dasy; 2 represents that Dipr and Micr are closest relatives; and 3 represents that Dasy and Micr are closest relatives. The last two represent the consequences of ILS in the speciation period of Diprotodontia, Dasyuromorphia, and Microbiotheria. The overall proportion of ILS was equal to the sum of 2 and 3, which was marked in the bar plot with a red dotted line and number.
(B) In the distribution of fragment lengths, the red dotted line indicates the distribution of fragments whose state is 0. The red, blue, and green solid lines represented 1, 2, and 3, respectively. The vertical dotted line represented the average of the corresponding distributions. Dasy, Dasyuromorphia; Dipr, Diprotodontia; Micr, monito del monte; Mono, gray short-tailed opossum; Macr, tammar wallaby; Ante, brown antechinus; Sarc, Tasmanian devil; Phas, koala.
(A) Measurements of the spinous process of the first (T1) and the second (T2) thoracic vertebra in koala.
(B) Measurements of the spinous process of T1 and T2 in Tasmanian devil.
(C) Status of the p.76 site of the mouse WFIKKN1 protein (ENSMUSP00000093141) in mammals. This alignment included six marsupials in this study and nine other mammals and used platypus as an outgroup. In this alignment, Q and R are two ancestral alleles at the target site, whereas allele H in humans is a species-specific mutation. All sequences were obtained from the Ensembl database, except monito del monte, tammar wallaby, and brown antechinus.
(D) Schematic of the measured values in the thoracic vertebrae. For each individual, we measured four sets of values: (1) the height of the spinous process of T1; (2) the width of the centrum of T1; (3) the height of the spinous process of T2; and (4) the width of the centrum of T2.
(E) Comparison of the relative ratio of the spinous process to the width of the centrum of the thoracic vertebrae between mice carrying the alternative amino acid R and the WT mice with Q. To measure the changes in the spinous process of T1 and T2 independently, we used the width of the centrum of the T1 and T2 vertebrae to standardize the height of the spinous process of T1 and T2, and then obtained the spinous_process/centrum_width ratio. The log-transformed ratios of T1 were significantly lower in Wfikkn1Q76R/Q76R mice than in WT mice, whereas T2 increased significantly.
(F) The 3D morphometrical black dot landmarks mapped on the rigth humeral bone surface were used in the CVA analysis to represent the overall curvature characteristics.
(G) Dipr_Micr ILS signals of PAPSS2 gene estimated by CoalHMM were shown with the gene structure of gray short-tailed opossum (ENSMODG00000016494).
(H) Four amino acid sites (red star) were replaced from the original type to the shared type between monito del monte and tammar wallaby in the gray short-tailed opossum cDNA to generate the Papss2Mono-Micr/Mono-Micr mutant line. Non-ILS (Dipr_Dasy), and two ILS patterns were shown above the alignments.
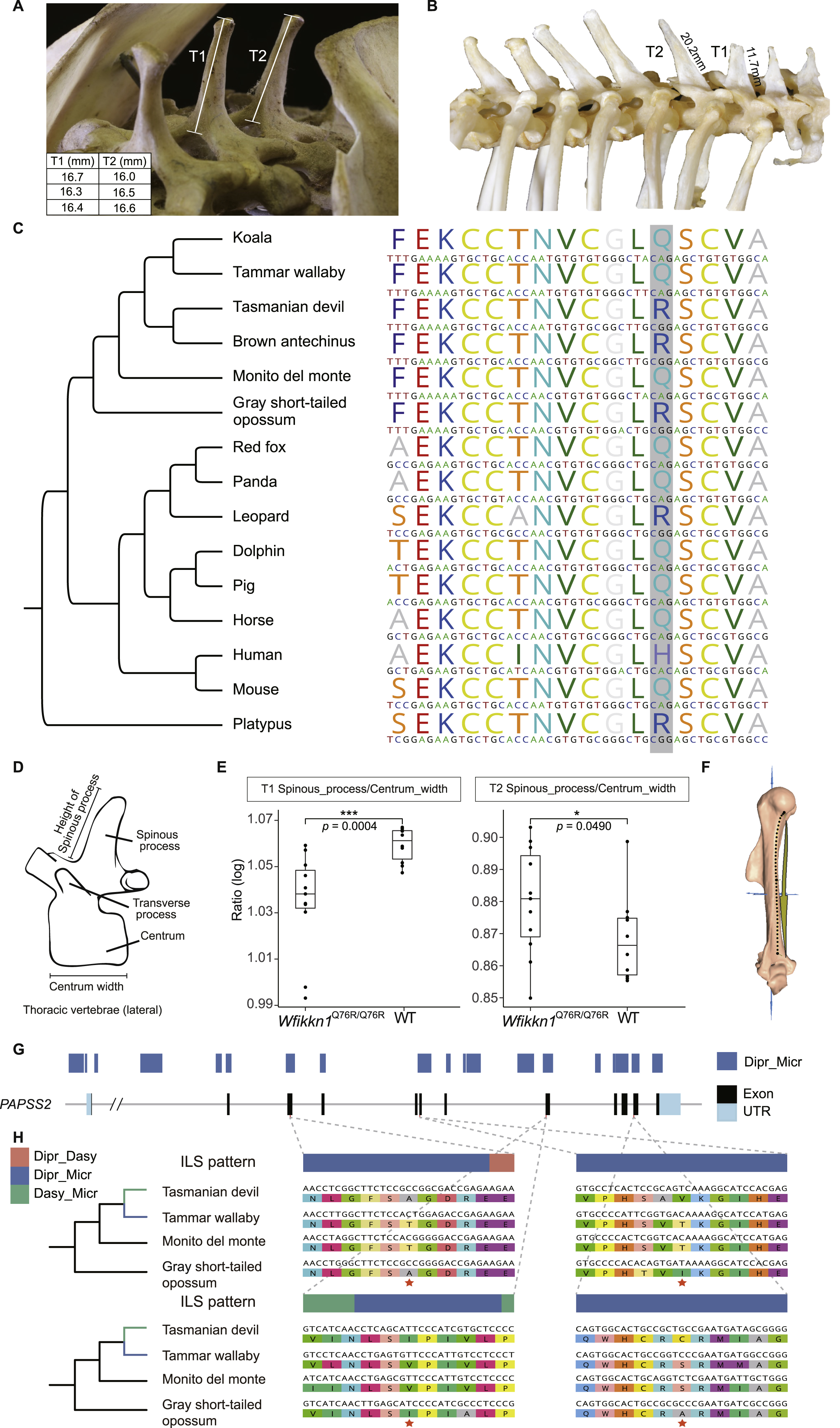{kind=link}
(A) The Wfikkn1 point-mutation mice line. The targeted mutation is c.227A > G (p.Q76R) of the mouse Wfikkn1 gene (ENSMUSG00000071192) located in exon 2. Cas9 mRNA, gRNA, and the donor oligo were co-injected into zygotes of C57BL/6J mice to obtain F0 knockin mice. Site c.237G > C is the synonymous mutation for the gRNA PAM blocking mutation. Only parts of the targeted sequence and the donor oligo sequence are shown. See STAR Methods for the complete sequences.
(B) Genotyping of F0 Wfikkn1 point-mutation mice by PCR. A pair of primers was designed to bind to flanking regions of the target site A > G.
(C) The PAPSS2 knockin mice lines. Gray short-tailed opossum PAPSS2 cDNA, modified gray short-tailed opossum PAPSS2 cDNA, tammar wallaby PAPSS2 cDNA, and Tasmanian devil PAPSS2 cDNA were introduced at the ATG start codon located in the 5′ end of mouse Papss2 gene (ENSMUSG00000024899), respectively. The targeting vector was constructed for homologous recombination of the target fragment, comprising the homologous sequences, KI fragment, and polyA, etc. Cas9 mRNA, gRNA, and the donor plasmid were co-injected into zygotes of C57BL/6J mice to obtain F0 knockin mice.
(D) Genotyping of F0 PAPSS2 knockin mice lines by PCR. Two pairs of primers were designed to bind to flanking regions of the mouse sequence outside the homology arms and to the target KI sequence for PAPSS2 knockin mice lines.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Male Dromiciops gliroides tissue sample | Valdivia, Chile | NCBI Biosample ID: SAMN15244861 |
| Deposited data | ||
| Whole-genome sequencing data of Dromiciops gliroides | This paper | NCBI project ID: PRJNA639670 CNSA project ID: CNP0000563 |
| Dromiciops gliroides reference genome | This paper | NCBI project ID: PRJNA639670 |
| Phascolarctos cinereus reference genome | Johnson et al., 2018 | RefSeq: GCF_002099425.1 |
| Macropus eugenii reference genome | Renfree et al., 2011 | GenBank: GCA_000004035.1 |
| Sarcophilus harrisii reference genome | N/A | RefSeq: GCF_902635505.1 |
| Antechinus stuartii reference genome | Brandies et al., 2020 | GenBank: GCA_016696395.1 |
| Monodelphis domestica reference genome | Mikkelsen et al., 2007 | RefSeq: GCF_000002295.2 |
| Mammalian Phenotype Ontology Database | Smith and Eppig, 2009 | https://www.ebi.ac.uk/ols/ontologies/mp |
| Gene Expression Database | Smith et al., 2019 | http://www.informatics.jax.org/expression.shtml |
| Whole genome alignment of six studied marsupials generated by LASTZ+MULTIZ | This paper; Mendeley Data | https://doi.org/10.17632/2n7jt8mvgb.1 |
| Gene annotation of Dromiciops gliroides | This paper; Mendeley Data | Table S1; https://doi.org/10.17632/2n7jt8mvgb.1 |
| Orthologous gene table of six studied marsupials | This paper; Mendeley Data | https://doi.org/10.17632/2n7jt8mvgb.1 |
| Reannotation results of gene WFIKKN1 in Macropus eugenii and Antechinus stuartii |
This paper; Mendeley Data | https://doi.org/10.17632/2n7jt8mvgb.1 |
| Reannotation results of gene PAPSS2 in Macropus eugenii |
This paper; Mendeley Data | https://doi.org/10.17632/2n7jt8mvgb.1 |
| Morphological HD images | This paper; Mendeley Data | Figure 4A; https://doi.org/10.17632/2n7jt8mvgb.1 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | Cat#000664 |
| Mouse: Wfikkn1Q76R/Q76R: C57BL/6J- Wfikkn1em1(Q76R)Smoc |
This paper | N/A |
| Mouse: Papss2Mono/Mono: C57BL/6J- Papss2em1(PAPSS2(Mono)-Wpre-pA)Smoc |
This paper | N/A |
| Mouse: Papss2Dipr/Dipr: C57BL/6J- Papss2em1(PAPSS2(Dipr)-Wpre-pA)Smoc |
This paper | N/A |
| Mouse: Papss2Dasy/Dasy:C57BL/6J- Papss2em1(PAPSS2(Dasy)-Wpre-pA)Smoc |
This paper | N/A |
| Mouse: Papss2Mono-Micr/Mono-Micr: C57BL/6J- Papss2em1(PAPSS2(Mono-Micr)-Wpre-pA)Smoc |
This paper | N/A |
| Oligonucleotides | ||
| Primers for genotypes of Wfikkn1Q76R F0 point substitution mice | This paper | Primer I: 5’ GAAGGGGACAAAGAGCTCCC 3’; Primer II: 5’ TACAACGTGCAGGTGGAGAC 3’ |
| Primers for genotypes of PAPASS2 F0 knock-in mice | This paper | Primer I - 5’ homology arm forward: 5’ CTCTGTTCATTCCTATTACTGGCTCT 3’; Primer II - 5’ homology arm reverse: 5’ CAACCCACATCTTCCACCTTCT 3’; Primer III - 3’ homology arm forward: 5’ AGAGGTGGTAATGGCAAAGACAA 3’; Primer IV - 3’ homology arm reverse: 5’ ATAAAGAGCCCAAACATAAAGGAAG 3’. |
| Software and algorithms | ||
| SOAPdenovo v2.04.4 | Luo et al., 2012 | https://github.com/aquaskyline/SOAPdenovo2 |
| SSPACE v2.0104 | Boetzer et al., 2011 | http://www.baseclear.com/bioinformatics-tools/ |
| Tandem Repeats Finder v4.04 | Benson, 1999 | https://tandem.bu.edu/trf/trf.html |
| RepeatMasker v3.3.0 | Smit et al., 1996 | http://repeatmasker.org |
| RepeatProteinMask v3.3.0 | Smit et al., 1996 | http://repeatmasker.org |
| RepeatModeler v1.0.5 | Price et al., 2005 | http://www.repeatmasker.org/RepeatModeler/ |
| LTR_FINDER v1.0.5 | Xu and Wang, 2007 | https://github.com/xzhub/LTR_Finder |
| BLASTall v2.2.23 | Altschul et al., 1990 | http://nebc.nox.ac.uk/bioinformatics/docs/blastall.html |
| GeneWise v2.2.0 | Birney et al., 2004 | https://www.ebi.ac.uk/seqdb/confluence/display/THD/GeneWise |
| AUGUSTUS v2.5.5 | Stanke et al., 2006 | http://bioinf.uni-greifswald.de/augustus/ |
| LASTZ v1.03.34 | Harris, 2007 | https://github.com/lastz/lastz |
| MULTIZ v11.2 | Blanchette et al., 2004 | https://github.com/multiz/multiz |
| SOLAR v0.9.6 | Almasy and Blangero, 1998 | https://doi.org/10.1086/301844 |
| ASTRAL-III v5.6.2 | Zhang et al., 2018 | https://github.com/smirarab/ASTRAL |
| IQ-TREE v1.6.12 | Nguyen et al., 2015 | http://www.iqtree.org/ |
| RAxML v8.2.9 | Stamatakis, 2006 | https://github.com/stamatak/standard-RAxML |
| TreeShrink v1.3.7 | Mai and Mirarab, 2018 | https://github.com/uym2/TreeShrink |
| DiscoVista v1.0 | Sayyari et al., 2018 | https://github.com/esayyari/DiscoVista |
| CoalHMM | Hobolth et al., 2007; Hobolth et al., 2011 | https://github.com/jydu/coalhmm |
| MAFFT v7.402 | Katoh and Standley, 2013 | https://mafft.cbrc.jp/alignment/software/ |
| MCMCTree program in PAML package v4.5 | Yang, 1997 | http://abacus.gene.ucl.ac.uk/software/ |
| QuIBL | Edelman et al., 2019 | https://github.com/michaelmiyagi/QuIBL |
| Dfoil | Pease and Hahn, 2015 | https://github.com/jbpease/dfoil |
| Amira 5.4 | Stalling et al., 2005 | https://www.thermofisher.com/amira-avizo |
| Geomagic Studio 2013 | Katz and Friess, 2014 | https://www.3dsystems.com/press-releases/geomagic/announces-studio-2013 |
| VGStudio MAX 2.1 | Volume Graphics | https://www.volumegraphics.com/ |
| Landmark | Institute for Data Analysis and Visualization, University of California, Davis |
http://ice.ucdavis.edu/partner/idav |
| Mathematica, Canonical Variates Analysis Program (Version 1.38) | MacLeod, 2007 | https://www.wolfram.com/mathematica/online |
| Geneious Version 2020.2.4 | Kearse et al., 2012 | https://www.geneious.com/download/ |
| R version 4.1.2 | R Core Team, 2021 | https://www.r-project.org/ |
| KKSC insertion significance test | Kuritzin et al., 2016 | http://retrogenomics.uni-muenster.de:3838/KKSC_significance_test/ |
| Other | ||
| Geo-schematic diagram of Dromiciops gliroides | Oda et al., 2019 | N/A |
| Geo-schematic diagram of Phascolarctos cinereus | Woinarski and Burbidge, 2021 | https://dx.doi.org/10.2305/IUCN.UK.2020-1.RLTS.T16892A166496779.en |
| Geo-schematic diagram of Macropus eugenii | Burbidge and Woinarski, 2016 | https://dx.doi.org/10.2305/IUCN.UK.2016-2.RLTS.T41512A21953803.en |
| Geo-schematic diagram of Sarcophilus harrisii | Hawkins et al., 2008 | https://dx.doi.org/10.2305/IUCN.UK.2008.RLTS.T40540A10331066.en |
| Geo-schematic diagram of Antechinus stuartii | Burnett and Dickman, 2016 | https://dx.doi.org/10.2305/IUCN.UK.2016-2.RLTS.T40526A21946655.en |
| Geo-schematic diagram of Monodelphis domestica | Flores and de la Sancha, 2016 | https://dx.doi.org/10.2305/IUCN.UK.2016-2.RLTS.T40514A22171137.en |
| Micro-XCT 400 (Located in Institute of Zoology, Chinese Academy of Sciences) | Carl Zeiss X-ray Microscopy, Inc., Pleasanton, USA | https://www.zeiss.com/microscopy/us/products/x-ray-microscopy.html |
Highlights.
Whole genome data support Dromiciops as a sister lineage of all Australian marsupials
More than 50% of marsupial genomes are affected by incomplete lineage sorting (ILS)
ILS is likely to have affected complex morphological traits in extant species
Functional experiments validated representative phenotypic effects suggested by ILS
ACKNOWLEDGMENTS
We thank Yun Ding (University of Pennsylvania) for helpful discussions of the transgenic experiments in mice. This work was supported by grants from the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB31020000); the International Partnership Program of the Chinese Academy of Sciences (152453KYSB20170002); the Carlsberg Foundation (CF16–0663); the Villum Foundation (25900) to G.Z.; a National Natural Science Foundation of China grant (31901214) to S.F.; grants from the National Natural Science Foundation of China (31961143002), Bureau of International Cooperation, Chinese Academy of Sciences, the First-class discipline of Prataculture Science of Ningxia University (NXYLXK2017A01), Hainan Yazhou Bay Seed Lab (B21HJ0102), and Guizhou Science and Technology Planning Project (General support-2022-173) to M.B.; GDAS Special Project of Science and Technology Development (2020GDASYL-20200301003) to H.Y.; a NIH grant (OD022988) to K.E.S.; a FONDECYT grant (1180917) to R.F.N.; and a grant from the Novo Nordisk Foundation (NNF18OC0031004) to M.H.S. We thank the Beijing Synchrotron Radiation Facility (BSRF) and Shanghai Synchrotron Radiation Facility (SSRF) for beam time, staff 4W1A and 4W1B of the BSRF, and staff BL13W1 of the SSRF for analytical assistance. Parts of this manuscript were prepared while Warren E. Johnson held a National Research Council Research Associateship Award at the Walter Reed Army Institute of Research (WRAIR). The material has been reviewed by WRAIR and there is no objection to its presentation and/or publication. The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting true views of the Department of the Army or the Department of Defense. We thank Associate Professor Stephen Johnston (University of Queensland), the Queensland Museum, and the Australian Museum for making photographs of marsupial skeletal material available.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2022.03.034.
REFERENCES
- Almasy L, and Blangero J (1998). Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 62, 1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Amrine-Madsen H, Scally M, Westerman M, Stanhope MJ, Krajewski C, and Springer MS (2003). Nuclear gene sequences provide evidence for the monophyly of australidelphian marsupials. Mol. Phylogenet. Evol. 28, 186–196. [DOI] [PubMed] [Google Scholar]
- Avise JC, and Robinson TJ (2008). Hemiplasy: a new term in the lexicon of phylogenetics. Syst. Biol. 57, 503–507. [DOI] [PubMed] [Google Scholar]
- Bai M, Beutel RG, Klass K-D, Zhang W, Yang X, and Wipfler B (2016). Alienoptera–a new insect order in the roach–mantodean twilight zone. Gondwana Res. 39, 317–326. [Google Scholar]
- Bai M, Beutel RG, Zhang W, Wang S, Hörnig M, Gröhn C, Yan E, Yang X, and Wipfler B (2018). A new Cretaceous insect with a unique cephalo-thoracic scissor device. Curr. Biol. 28, 438–443.e1. [DOI] [PubMed] [Google Scholar]
- Bai M, Yang X, Li J, and Wang W (2014). Geometric morphometrics, a super scientific computing tool in morphology comparison. Sci. Bull. 59, 887–894. [Google Scholar]
- Behrensmeyer AK, and Turner A (2013). Taxonomic occurrences of Suidae recorded in the Paleobiology Database (Fossilworks). http://fossilworks.org.
- Benson G (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Clamp M, and Durbin R (2004). GeneWise and Genomewise. Genome Res. 14, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. (2004). Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14, 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, and Pirovano W (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. [DOI] [PubMed] [Google Scholar]
- Brandies PA, Tang S, Johnson RSP, Hogg CJ, and Belov K (2020). The first Antechinus reference genome provides a resource for investigating the genetic basis of semelparity and age-related neuropathologies. Gigabyte 1, 7. 10.46471/gigabyte.46477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo GA, Antonelli A, Bacon CD, Bartoszek K, Blom MPK, Huynh S, Jones G, Knowles LL, Lamichhaney S, Marcussen T, et al. (2019). Embracing heterogeneity: coalescing the tree of life and the future of phylogenomics. PeerJ 7, e6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbidge AA, and Woinarski J (2016). Macropus eugenii. The IUCN Red List of Threatened Species 2016. 10.2305/IUCN.UK.2016-2.RLTS.T41512A21953803.en. [DOI] [Google Scholar]
- Burk A, Westerman M, Kao DJ, Kavanagh JR, and Springer MS (1999). An analysis of marsupial interordinal relationships based on 12S rRNA, tRNA valine, 16S rRNA, and cytochrome b sequences. J. Mamm. Evol. 6, 317–334. [Google Scholar]
- Burnett S, and Dickman C (2016). Antechinus stuartii. The IUCN Red List of Threatened Species 2016. 10.2305/IUCN.UK.2016-2.RLTS.T40526A21946655.en. [DOI] [Google Scholar]
- Darwin C (1859). The Origin of Species (John Murray). [Google Scholar]
- Dávalos LM, Cirranello AL, Geisler JH, and Simmons NB (2012). Understanding phylogenetic incongruence: lessons from phyllostomid bats. Biol. Rev. Camb. Philos. Soc. 87, 991–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan JH, and Rosenberg NA (2009). Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24, 332–340. [DOI] [PubMed] [Google Scholar]
- Duchêne DA, Bragg JG, Duchêne S, Neaves LE, Potter S, Moritz C, Johnson RN, Ho SYW, and Eldridge MDB (2018). Analysis of phylogenomic tree space resolves relationships among marsupial families. Syst. Biol. 67, 400–412. [DOI] [PubMed] [Google Scholar]
- Dutheil JY, Ganapathy G, Hobolth A, Mailund T, Uyenoyama MK, and Schierup MH (2009). Ancestral population genomics: the coalescent hidden Markov model approach. Genetics 183, 259–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman NB, Frandsen PB, Miyagi M, Clavijo B, Davey J, Dikow RB, García-Accinelli G, Van Belleghem SM, Patterson N, Neafsey DE, et al. (2019). Genomic architecture and introgression shape a butterfly radiation. Science 366, 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores D, and de la Sancha N (2016). Monodelphis domestica. The IUCN Red List of Threatened Species. Version 2016.2. 10.2305/IUCN.UK.2016-2. [DOI] [Google Scholar]
- Fontaine MC, Pease JB, Steele A, Waterhouse RM, Neafsey DE, Sharakhov IV, Jiang X, Hall AB, Catteruccia F, Kakani E, et al. (2015). Mosquito genomics. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 347, 1258524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankham GJ, and Temple-Smith PD (2012). Absence of mammary development in male Dromiciops gliroides: another link to the Australian marsupial fauna. J. Mammal. 93, 572–578. [Google Scholar]
- Gallardo MH, and Patterson BD (1987). An additional 14-chromosome karyotype and sex-chromosome mosaicism in South American marsupials. Fieldiana Zool. 39, 111–116. [Google Scholar]
- Gallus S, Janke A, Kumar V, and Nilsson MA (2015). Disentangling the relationship of the Australian marsupial orders using retrotransposon and evolutionary network analyses. Genome Biol. Evol. 7, 985–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaubert P, Wozencraft WC, Cordeiro-Estrela P, and Veron G (2005). Mosaics of convergences and noise in morphological phylogenies: what’s in a viverrid-like carnivoran? Syst. Biol. 54, 865–894. [DOI] [PubMed] [Google Scholar]
- Goin FJ, and Abello MA (2013). Los Metatheria sudamericanos de comienzos del Neógeno (Mioceno temprano, edad mamífero Colhuehuapense): Microbiotheria y Polydolopimorphia. Ameghiniana 50, 51–78. [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, et al. (2010). A draft sequence of the Neandertal genome. Science 328, 710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurovich Y, and Ashwell KW (2020). Brain and behavior of Dromiciops gliroides. J. Mamm. Evol. 27, 177–197. [Google Scholar]
- Haeussler M, Zweig AS, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Hinrichs AS, Gonzalez JN, et al. (2019). The UCSC Genome Browser database: 2019 update. Nucleic Acids Res. 47, D853–D858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS (2007). Improved Pairwise Alignment Of Genomic DNA (The Pennsylvania State University). [Google Scholar]
- Hawkins C, McCallum H, Mooney N, Jones M, and Holdsworth M (2008). Sarcophilus harrisii. In IUCN red list of threatened species. Version 2009.1. www.iucnredlist.org. [Google Scholar]
- Henderson K, Pantinople J, McCabe K, Richards HL, and Milne N (2017). Forelimb bone curvature in terrestrial and arboreal mammals. PeerJ 5, e3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobolth A, Christensen OF, Mailund T, and Schierup MH (2007). Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model. PLoS Genet. 3, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobolth A, Dutheil JY, Hawks J, Schierup MH, and Mailund T (2011). Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Res. 21, 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope R, Cooper S, and Wainwright B (1989). Globin macromolecular sequences in marsupials and monotremes. Aust. J. Zool. 37, 289–313. [Google Scholar]
- Horovitz I, and Sánchez-Villagra MR (2003). A morphological analysis of marsupial mammal higher-level phylogenetic relationships. Cladistics 19, 181–212. [Google Scholar]
- Jarvis ED, Mirarab S, Aberer AJ, Li B, Houde P, Li C, Ho SY, Faircloth BC, Nabholz B, Howard JT, et al. (2014). Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 346, 1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RN, O’Meally D, Chen Z, Etherington GJ, Ho SYW, Nash WJ, Grueber CE, Cheng Y, Whittington CM, Dennison S, et al. (2018). Adaptation and conservation insights from the koala genome. Nat. Genet. 50, 1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, and Jermiin LS (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, and Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz D, and Friess M (2014). Technical note: 3D from standard digital photography of human crania-a preliminary assessment. Am. J. Phys. Anthropol. 154, 152–158. [DOI] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyonari H, Kaneko M, Abe T, Shiraishi A, Yoshimi R, Inoue KI, and Furuta Y (2021). Targeted gene disruption in a marsupial, Monodelphis domestica, by CRISPR/Cas9 genome editing. Curr. Biol. 31, 3956–3963.e4. [DOI] [PubMed] [Google Scholar]
- Kuritzin A, Kischka T, Schmitz J, and Churakov G (2016). Incomplete lineage sorting and hybridization statistics for large-scale retroposon insertion data. PLoS Comput. Biol. 12, e1004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhaney S, Berglund J, Almén MS, Maqbool K, Grabherr M, Martinez-Barrio A, Promerová M, Rubin C-J, Wang C, Zamani N, et al. (2015). Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518, 371–375. [DOI] [PubMed] [Google Scholar]
- Larson A (1998). The comparison of morphological and molecular data in phylogenetic systematics. Molecular Approaches to Ecology and Evolution (Springer; ), pp. 275–296. [Google Scholar]
- Lee JM, Song HJ, Park SI, Lee YM, Jeong SY, Cho TO, Kim JH, Choi HG, Choi CG, Nelson WA, et al. (2018). Mitochondrial and plastid genomes from coralline red algae provide insights into the incongruent evolutionary histories of organelles. Genome Biol. Evol. 10, 2961–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, and Lee SJ (2013). Regulation of GDF-11 and myostatin activity by GASP-1 and GASP-2. Proc. Natl. Acad. Sci. USA 110, E3713–E3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Fan W, Tian G, Zhu H, He L, Cai J, Huang Q, Cai Q, Li B, Bai Y, et al. (2010). The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livermore R, Nankivell A, Eagles G, and Morris P (2005). Palaeogene opening of Drake Passage. Earth Planet. Sci. Lett. 236, 459–470. [Google Scholar]
- Lopes F, Oliveira LR, Kessler A, Beux Y, Crespo E, Cárdenas-Alayza S, Majluf P, Sepúlveda M, Brownell RL, Franco-Trecu V, et al. (2021). Phylogenomic discordance in the eared seals is best explained by incomplete lineage sorting following explosive radiation in the Southern hemisphere. Syst. Biol. 70, 786–802. [DOI] [PubMed] [Google Scholar]
- Losos JB (2010). Adaptive radiation, ecological opportunity, and evolutionary determinism. American Society of Naturalists E.O. Wilson award address. Am. Nat. 175, 623–639. [DOI] [PubMed] [Google Scholar]
- Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZX, Ji Q, Wible JR, and Yuan CX (2003). An Early Cretaceous tribosphenic mammal and metatherian evolution. Science 302, 1934–1940. [DOI] [PubMed] [Google Scholar]
- Luo ZX, Yuan CX, Meng QJ, and Ji Q (2011). A Jurassic eutherian mammal and divergence of marsupials and placentals. Nature 476, 442–445. [DOI] [PubMed] [Google Scholar]
- MacLeod N (2007). Automated Taxon Identification in Systematics: Theory, Approaches and APPLICATIONs (CRC Press; ). [Google Scholar]
- MacLeod N (2017). Morphometrics: history, development methods and prospects. Syst. Zool. 42, 4–33. [Google Scholar]
- Mai U, and Mirarab S (2018). TreeShrink: fast and accurate detection of outlier long branches in collections of phylogenetic trees. BMC Genom. 19, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailund T, Munch K, and Schierup MH (2014). Lineage sorting in apes. Annu. Rev. Genet. 48, 519–535. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Wakefield MJ, Aken B, Amemiya CT, Chang JL, Duke S, Garber M, Gentles AJ, Goodstadt L, Heger A, et al. (2007). Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature 447, 167–177. [DOI] [PubMed] [Google Scholar]
- Mistry J, Chuguransky S, Williams L, Qureshi M, Salazar GA, Sonnhammer ELL, Tosatto SCE, Paladin L, Raj S, Richardson LJ, et al. (2021). Pfam: the protein families database in 2021. Nucleic Acids Res. 49, D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell KJ, Pratt RC, Watson LN, Gibb GC, Llamas B, Kasper M, Edson J, Hopwood B, Male D, Armstrong KN, et al. (2014). Molecular phylogeny, biogeography, and habitat preference evolution of marsupials. Mol. Biol. Evol. 31, 2322–2330. [DOI] [PubMed] [Google Scholar]
- Moen D, and Morlon H (2014). From dinosaurs to modern bird diversity: extending the time scale of adaptive radiation. PLoS Biol. 12, e1001854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monestier O, and Blanquet V (2016). WFIKKN1 and WFIKKN2: “Companion” proteins regulating TGFB activity. Cytokine Growth Factor Rev. 32, 75–84. [DOI] [PubMed] [Google Scholar]
- Muschick M, Indermaur A, and Salzburger W (2012). Convergent evolution within an adaptive radiation of cichlid fishes. Curr. Biol. 22, 2362–2368. [DOI] [PubMed] [Google Scholar]
- Nguyen LT, Schmidt HA, von Haeseler A, and Minh BQ (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson MA, Arnason U, Spencer PB, and Janke A (2004). Marsupial relationships and a timeline for marsupial radiation in south Gondwana. Gene 340, 189–196. [DOI] [PubMed] [Google Scholar]
- Nilsson MA, Churakov G, Sommer M, Tran NV, Zemann A, Brosius J, and Schmitz J (2010). Tracking marsupial evolution using archaic genomic retroposon insertions. PLoS Biol. 8, e1000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson MA, Gullberg A, Spotorno AE, Arnason U, and Janke A (2003). Radiation of extant marsupials after the K/T boundary: evidence from complete mitochondrial genomes. J. Mol. Evol. 57 (Suppl. 1 ), S3–S12. [DOI] [PubMed] [Google Scholar]
- Nilsson MA, Zheng Y, Kumar V, Phillips MJ, and Janke A (2018). Speciation generates mosaic genomes in kangaroos. Genome Biol. Evol. 10, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda E, Rodríguez-Gómez GB, Fontúrbel F, Soto-Gamboa M, and Nespolo R (2019). Southernmost records of Dromiciops gliroides: extending its distribution beyond the Valdivian rainforest. Gayana 83, 145–149. [Google Scholar]
- Olsson U, Alström P, Svensson L, Aliabadian M, and Sundberg P (2010). The Lanius excubitor (Aves, Passeriformes) conundrum–Taxonomic dilemma when molecular and non-molecular data tell different stories. Mol. Phylogenet. Evol. 55, 347–357. [DOI] [PubMed] [Google Scholar]
- Parra G, Bradnam K, and Korf I (2007). CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Pease JB, Haak DC, Hahn MW, and Moyle LC (2016). Phylogenomics reveals three sources of adaptive variation during a rapid radiation. PLoS Biol. 14, e1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease JB, and Hahn MW (2015). Detection and polarization of introgression in a five-taxon phylogeny. Syst. Biol. 64, 651–662. [DOI] [PubMed] [Google Scholar]
- Pollard DA, Iyer VN, Moses AM, and Eisen MB (2006). Widespread discordance of gene trees with species tree in Drosophila: evidence for incomplete lineage sorting. PLoS Genet. 2, e173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, and Siepel A (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Jones NC, and Pevzner PA (2005). De novo identification of repeat families in large genomes. Bioinformatics 21 (Suppl. 1), i351–i358. [DOI] [PubMed] [Google Scholar]
- R Core Team (2021). R: A language and environment for statistical computing (R Foundation for Statistical Computing). [Google Scholar]
- Renfree MB, Papenfuss AT, Deakin JE, Lindsay J, Heider T, Belov K, Rens W, Waters PD, Pharo EA, Shaw G, et al. (2011). Genome sequence of an Australian kangaroo, Macropus eugenii, provides insight into the evolution of mammalian reproduction and development. Genome Biol. 12, R81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renfree MB, Robinson ES, Short RV, and Vandeberg JL (1990). Mammary glands in male marsupials: I. Primordia in neonatal opossums Didelphis virginiana and Monodelphis domestica. Development 110, 385–390. [DOI] [PubMed] [Google Scholar]
- Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, Koren S, Uliano-Silva M, Chow W, Fungtammasan A, Kim J, et al. (2021). Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas A, Williams BL, King N, and Carroll SB (2003). Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425, 798–804. [DOI] [PubMed] [Google Scholar]
- Sackton TB, and Clark N (2019). Convergent evolution in the genomics era: new insights and directions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374, 20190102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzburger W, Baric S, and Sturmbauer C (2002). Speciation via introgressive hybridization in East African cichlids? Mol. Ecol. 11, 619–625. [DOI] [PubMed] [Google Scholar]
- Sayyari E, Whitfield JB, and Mirarab S (2018). DiscoVista: interpretable visualizations of gene tree discordance. Mol. Phylogenet. Evol. 122, 110–115. [DOI] [PubMed] [Google Scholar]
- Scally A, Dutheil JY, Hillier LW, Jordan GE, Goodhead I, Herrero J, Hobolth A, Lappalainen T, Mailund T, Marques-Bonet T, et al. (2012). Insights into hominid evolution from the gorilla genome sequence. Nature 483, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter D (2000). The Ecology of Adaptive Radiation (OUP Oxford). [Google Scholar]
- Sharman G (1982). Karyotypic similarities between Dromiciops australis (Microbiotheriidae, marsupialia) and some Australian marsupials. In Carnivorous Marsupials, Archer M, ed. (Royal Society of New South Wales; ), pp. 711–714. [Google Scholar]
- Smit AFA, Hubley R, and Green P (1996). RepeatMasker. http://repeatmasker.org. [Google Scholar]
- Smith CL, and Eppig JT (2009). The mammalian phenotype ontology: enabling robust annotation and comparative analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 1, 390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CM, Hayamizu TF, Finger JH, Bello SM, McCright IJ, Xu J, Baldarelli RM, Beal JS, Campbell J, Corbani LE, et al. (2019). The mouse Gene Expression Database (GXD): 2019 update. Nucleic Acids Res. 47, D774–D779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer MS, Molloy EK, Sloan DB, Simmons MP, and Gatesy J (2020). ILS-aware analysis of low-homoplasy retroelement insertions: inference of species trees and introgression using quartets. J. Hered. 111, 147–168. [DOI] [PubMed] [Google Scholar]
- Springer MS, Westerman M, Kavanagh JR, Burk A, Woodburne MO, Kao DJ, and Krajewski C (1998). The origin of the Australasian marsupial fauna and the phylogenetic affinities of the enigmatic monito del monte and marsupial mole. Proc. Biol. Sci. 265, 2381–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalling D, Westerhoff M, and Hege H-C (2005). Amira: a highly interactive system for visual data analysis. Vis. Handb. 38, 749–767. [Google Scholar]
- Stamatakis A (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. [DOI] [PubMed] [Google Scholar]
- Stanke M, Keller O, Gunduz I, Hayes A, Waack S, and Morgenstern B (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer C, Brimmer A, Hermanns P, Zabel B, and Dietz UH (2007). Expression profile of Papss2 (3′-phosphoadenosine 5′-phosphosulfate synthase 2) during cartilage formation and skeletal development in the mouse embryo. Dev. Dyn. 236, 1313–1318. [DOI] [PubMed] [Google Scholar]
- Stern DL (2013). The genetic causes of convergent evolution. Nat. Rev. Genet. 14, 751–764. [DOI] [PubMed] [Google Scholar]
- Suh A, Smeds L, and Ellegren H (2015). The dynamics of incomplete lineage sorting across the ancient adaptive radiation of neoavian birds. PLoS Biol. 13, e1002224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YB, Fu TT, Jin JQ, Murphy RW, Hillis DM, Zhang YP, and Che J (2018). Species groups distributed across elevational gradients reveal convergent and continuous genetic adaptation to high elevations. Proc. Natl. Acad. Sci. USA 115, E10634–E10641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalay FS (1982). A new appraisal of marsupial phylogeny and classification. In Carnivorous Marsupials, Archer M, ed. (Royal Zoological Society; ), pp. 621–640. [Google Scholar]
- Szalay FS (1994). Evolutionary History of the Marsupials and an Analysis of Osteological Characters (Cambridge University Press; ). [Google Scholar]
- Szöllősi GJ, Tannier E, Daubin V, and Boussau B (2015). The inference of gene trees with species trees. Syst. Biol. 64, e42–e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple-Smith PD (1987). Sperm structure and marsupial phylogeny. Possums Opossums Stud. Evol. 1, 171–193. [Google Scholar]
- Temple-Smith PD (1994). Comparative structure and function of marsupial spermatozoa. Reprod. Fertil. Dev. 6, 421–435. [DOI] [PubMed] [Google Scholar]
- Tikku AA, and Cande SC (1999). The oldest magnetic anomalies in the Australian–Antarctic Basin: are they isochrons? J. Geophys. Res. Solid Earth 104, 661–677. [Google Scholar]
- Tikku AA, and Cande SC (2000). On the fit of broken ridge and Kerguelen Plateau. Earth Planet. Sci. Lett. 180, 117–132. [Google Scholar]
- Tong Y-J, Yang H-D, Jenkins Shaw J, Yang X-K, and Bai M (2021). The relationship between genus/species richness and morphological diversity among subfamilies of jewel beetles. Insects 12, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyndale-Biscoe CH, and Renfree MB (1987). Reproductive Physiology of Marsupials (Cambridge University Press; ), p. 476. [Google Scholar]
- Van Den Ende C, White LT, and van Welzen PC (2017). The existence and break-up of the Antarctic land bridge as indicated by both amphi-Pacific distributions and tectonics. Gondwana Res. 44, 219–227. [Google Scholar]
- Vizcaíno SF, Pascual R, Reguero MA, and Goin FJ (1998). Antarctica as background for mammalian evolution. In Paleógeno de América del sur y de la Península Antártica (Asociación Paleontológica Argentina, Publicación Especial), pp. 199–209. [Google Scholar]
- White LT, Gibson GM, and Lister GS (2013). A reassessment of paleogeographic reconstructions of eastern Gondwana: bringing geology back into the equation. Gondwana Res. 24, 984–998. [Google Scholar]
- Wiley DF, Amenta N, Alcantara DA, Ghosh D, Kil YJ, Delson E, Harcourt-Smith W, Rohlf FJ, St John K, and Hamann B (2005). Evolutionary Morphing (IEEE). [Google Scholar]
- Williams SE, Whittaker JM, Halpin JA, and Müller RD (2019). Australian-Antarctic breakup and seafloor spreading: balancing geological and geophysical constraints. Earth Sci. Rev. 188, 41–58. [Google Scholar]
- Woinarski J, and Burbidge A (2021). Phascolarctos cinereus (amended version of 2016 assessment). The IUCN Red List of Threatened Species 2020: e.T16892A166496779. 10.2305/IUCN.UK.2020-1.RLTS.T16892A166496779.en. [DOI] [Google Scholar]
- Wolf YI, Rogozin IB, Grishin NV, and Koonin EV (2002). Genome trees and the tree of life. Trends Genet. 18, 472–479. [DOI] [PubMed] [Google Scholar]
- Xu Z, and Wang H (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556. [DOI] [PubMed] [Google Scholar]
- Zhang C, Rabiee M, Sayyari E, and Mirarab S (2018). ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Ruan Y, Wan X, Tong Y, Yang X, and Ming Bai B (2019). Geometric morphometric analysis of the pronotum and elytron in stag beetles: insight into its diversity and evolution. ZooKeys 833, 21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Shearwin-Whyatt L, Li J, Song Z, Hayakawa T, Stevens D, Fenelon JC, Peel E, Cheng Y, Pajpach F, et al. (2021). Platypus and echidna genomes reveal mammalian biology and evolution. Nature 592, 756–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z, and Zhang J (2016). Morphological and molecular convergences in mammalian phylogenetics. Nat. Commun. 7, 12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) ASTRAL output based on WGAs. Lengths of internal branches estimated by ASTRAL-III are labeled on the tree in coalescent units (scale bar at the bottom).
(B) ASTRAL output based on 9,227 orthologs.
(C) RAxML output based on the concatenated coding alignment of 9,227 orthologs. Branch length scale bar refers to the expected number of substitutions per site. Bootstrap value is labeled at the node in parentheses if the value is less than 100.
(D) RAxML output based on the 4-fold degenerate sites of 9,227 orthologs.
(E) RAxML output based on the alignment of 1st and 2nd codon positions of 9,227 orthologs.
(F) RAxML output based on the alignment of 3rd codon positions of 9,227 orthologs.
(A) Using the estimated divergence times to distinguish between ILS and hybridization scenarios. As the upper schematic trees illustrate, the coalescence time under ILS (ti) should be earlier than the speciation event (t), whereas the expected divergence time under hybridization (th) should be later than the speciation event (t). The next trees show the divergence times across six species estimated by MCMCTree with three alternative topologies and the corresponding genomic regions. Dipr_Dasy: the topology and the regions supporting the species tree. Dipr_Micr: the topology and the regions supporting Dipr and Micr as sister species. Dasy_Micr: the topology and the regions supporting Dasy and Micr as closest relatives. The minimum and maximum calibration dates labeled at the root are 64 and 116 MYA, respectively, based on the literature.
(B) QuIBL output of the triplet, D. gliroides-A. stuartii-M. eugenii. (Upper) Distribution of Delta. BIC values of the Dipr_Micr subset (blue) and the Dasy_Micr subset (green) calculated by QuIBL. Delta. BIC value is calculated as the BIC value of scenario 2 minus the BIC value of scenario 1 (see STAR Methods). (Bottom) The distribution of the internal branch lengths (gray) is more in line with the inferred ILS distribution (red) than the inferred introgression distribution (black) for these two subsets. As shown in (B), we also present the results of D. gliroides-S. harrisii-M. eugenii triplet in (C), D. gliroides-A. stuartii-P. cinereus triplet in (D), and D. gliroides-S. harrisii-P. cinereus triplet in (E). Dasy, Dasyuromorphia; Dipr, Diprotodontia; Micr, monito del monte.
(A) The proportion of ILS cases observed in WGAs (left) and orthologous coding regions (right) inferred by CoalHMM in four combinations. In each combination, the proportions of the loci belonging to the four candidate genealogies (x axis) were calculated. Four candidate genealogies are: 0 represents the species tree (non-ILS) without deep coalescence; 1 represents the species tree (non-ILS) with deep coalescence between Dipr and Dasy; 2 represents that Dipr and Micr are closest relatives; and 3 represents that Dasy and Micr are closest relatives. The last two represent the consequences of ILS in the speciation period of Diprotodontia, Dasyuromorphia, and Microbiotheria. The overall proportion of ILS was equal to the sum of 2 and 3, which was marked in the bar plot with a red dotted line and number.
(B) In the distribution of fragment lengths, the red dotted line indicates the distribution of fragments whose state is 0. The red, blue, and green solid lines represented 1, 2, and 3, respectively. The vertical dotted line represented the average of the corresponding distributions. Dasy, Dasyuromorphia; Dipr, Diprotodontia; Micr, monito del monte; Mono, gray short-tailed opossum; Macr, tammar wallaby; Ante, brown antechinus; Sarc, Tasmanian devil; Phas, koala.
(A) Measurements of the spinous process of the first (T1) and the second (T2) thoracic vertebra in koala.
(B) Measurements of the spinous process of T1 and T2 in Tasmanian devil.
(C) Status of the p.76 site of the mouse WFIKKN1 protein (ENSMUSP00000093141) in mammals. This alignment included six marsupials in this study and nine other mammals and used platypus as an outgroup. In this alignment, Q and R are two ancestral alleles at the target site, whereas allele H in humans is a species-specific mutation. All sequences were obtained from the Ensembl database, except monito del monte, tammar wallaby, and brown antechinus.
(D) Schematic of the measured values in the thoracic vertebrae. For each individual, we measured four sets of values: (1) the height of the spinous process of T1; (2) the width of the centrum of T1; (3) the height of the spinous process of T2; and (4) the width of the centrum of T2.
(E) Comparison of the relative ratio of the spinous process to the width of the centrum of the thoracic vertebrae between mice carrying the alternative amino acid R and the WT mice with Q. To measure the changes in the spinous process of T1 and T2 independently, we used the width of the centrum of the T1 and T2 vertebrae to standardize the height of the spinous process of T1 and T2, and then obtained the spinous_process/centrum_width ratio. The log-transformed ratios of T1 were significantly lower in Wfikkn1Q76R/Q76R mice than in WT mice, whereas T2 increased significantly.
(F) The 3D morphometrical black dot landmarks mapped on the rigth humeral bone surface were used in the CVA analysis to represent the overall curvature characteristics.
(G) Dipr_Micr ILS signals of PAPSS2 gene estimated by CoalHMM were shown with the gene structure of gray short-tailed opossum (ENSMODG00000016494).
(H) Four amino acid sites (red star) were replaced from the original type to the shared type between monito del monte and tammar wallaby in the gray short-tailed opossum cDNA to generate the Papss2Mono-Micr/Mono-Micr mutant line. Non-ILS (Dipr_Dasy), and two ILS patterns were shown above the alignments.
(A) The Wfikkn1 point-mutation mice line. The targeted mutation is c.227A > G (p.Q76R) of the mouse Wfikkn1 gene (ENSMUSG00000071192) located in exon 2. Cas9 mRNA, gRNA, and the donor oligo were co-injected into zygotes of C57BL/6J mice to obtain F0 knockin mice. Site c.237G > C is the synonymous mutation for the gRNA PAM blocking mutation. Only parts of the targeted sequence and the donor oligo sequence are shown. See STAR Methods for the complete sequences.
(B) Genotyping of F0 Wfikkn1 point-mutation mice by PCR. A pair of primers was designed to bind to flanking regions of the target site A > G.
(C) The PAPSS2 knockin mice lines. Gray short-tailed opossum PAPSS2 cDNA, modified gray short-tailed opossum PAPSS2 cDNA, tammar wallaby PAPSS2 cDNA, and Tasmanian devil PAPSS2 cDNA were introduced at the ATG start codon located in the 5′ end of mouse Papss2 gene (ENSMUSG00000024899), respectively. The targeting vector was constructed for homologous recombination of the target fragment, comprising the homologous sequences, KI fragment, and polyA, etc. Cas9 mRNA, gRNA, and the donor plasmid were co-injected into zygotes of C57BL/6J mice to obtain F0 knockin mice.
(D) Genotyping of F0 PAPSS2 knockin mice lines by PCR. Two pairs of primers were designed to bind to flanking regions of the mouse sequence outside the homology arms and to the target KI sequence for PAPSS2 knockin mice lines.
Data Availability Statement
Genome sequencing data and the genome assembly generated in this study have been deposited in the NCBI SRA under accession PRJNA639670. The above data have also been deposited in the CNSA (https://db.cngb.org/cnsa/) of CNGBdb with accession number CNP0000563. WGAs generated by LASTZ+MULTIZ, the orthologous gene table, high-definition morphological photos and other relevant data can be found in Mendeley data https://doi.org/10.17632/2n7jt8mvgb.1.