

Abstract
Vasopressin regulates renal water excretion by binding to a Gαs-coupled receptor (V2R) in collecting duct cells, resulting in increased water permeability through regulation of the aquaporin-2 (AQP2) water channel. This action is widely accepted to be associated with cAMP-mediated activation of protein kinase A (PKA). Here, we use phosphoproteomics in collecting duct cells in which PKA has been deleted (CRISPR-Cas9) to identify PKA-independent responses to vasopressin. The results show that V2R-mediated vasopressin signaling is predominantly, but not entirely, PKA-dependent. Upregulated sites in PKA-null cells include Ser256 of AQP2, which is critical to regulation of AQP2 trafficking. In addition, phosphorylation changes in the protein kinases Stk39 (SPAK) and Prkci (an atypical PKC) are consistent with PKA-independent regulation of these protein kinases. Target motif analysis of the phosphopeptides increased in PKA-null cells indicates that vasopressin activates one or more members of the AMPK/SNF1-subfamily of basophilic protein kinases. In vitro phosphorylation assays using recombinant, purified SNF1-subfamily kinases confirmed postulated target specificities. Of interest, measured IBMX-depdendent cAMP levels were an order of magnitude higher in PKA-null than in PKA-intact cells, indicative of a PKA-dependent feedback mechanism. Overall, the findings support the conclusion that V2-receptor mediated signaling in collecting duct cells is in part PKA-independent.
Keywords: mpkCCD, Phosphoproteomics, cAMP, PRM-MS, AMPK, AQP2
INTRODUCTION
Body water balance is regulated chiefly by the peptide hormone vasopressin, which acts in the kidney to control the rate of water excretion (1). Vasopressin regulates water excretion chiefly by controlling the osmotic water permeability of collecting duct cells through changes in the abundance and cellular distribution of the water channel aquaporin-2 (AQP2) (2). The permeability changes, therefore, determine whether luminal water is returned to the general circulation (high vasopressin) or excreted in the urine (low vasopressin). The regulation of osmotic water transport in the renal collecting duct is accomplished largely through two mechanisms: a) control of membrane-trafficking, which carries AQP2 water channels to and from the apical plasma membrane (3, 4); and b) control of transcription of the Aqp2 gene (5–7), which codes for AQP2 (8).
Signaling pathways responsible for vasopressin action in renal collecting duct cells are incompletely understood. What is known is that vasopressin binds to a G-protein coupled receptor (GPCR), namely the vasopressin V2 receptor (V2R), which activates adenylyl cyclase through the heterotrimeric G-protein α subunit Gαs, thereby increasing intracellular cyclic-AMP (cAMP). Although cAMP can act in the cell via other effectors, it is generally believed that protein kinase A (PKA) is responsible for vasopressin’s effects on AQP2 trafficking and transcription. This view was supported by recent studies in which both genes coding for PKA catalytic subunits were deleted in cultured collecting duct cells (mpkCCD) (9). Although these PKA-null cells grew well and maintained their normal epithelial polarity, both apical trafficking of AQP2 and transcription of the Aqp2 gene were virtually completely ablated, implying that PKA is important for AQP2 regulation by vasopressin. However, whether vasopressin exerts some of its actions through PKA-independent signaling pathways is thus far unclear.
In our previous paper (9), we compared vasopressin-treated PKA-null cells with vasopressin-treated PKA-intact cells, but did not investigate phosphoproteomic responses to vasopressin in these two cell lines. To identify vasopressin-dependent signaling pathways in PKA-intact and PKA-null mpkCCD cells, we have now carried out large scale phosphoproteomic analysis using quantitative protein mass spectrometry. The data allowed us to create two publicly accessible online databases (for vasopressin responses in PKA-intact and PKA-null cells, respectively) to serve as resources for future studies of V2R signaling and signaling mediated by other Gαs-coupled receptors. The data in PKA-null cells allowed identification of several protein kinases, viz., SPAK (Stk39), PKC-iota (Prkci) and one or more members of the SNF1-subfamily of protein kinases, as regulated targets of vasopressin in the absence of PKA. In vitro nLC-MS/MS-based phosphorylation assays using recombinant, purified protein SNF1-subfamily kinases confirmed postulated target specificities.
MATERIALS AND METHODS
Reagents
Unless mentioned otherwise, all chemicals and reagents except the one used for proteomics sample preparation were purchased from Sigma (St. Louis, MO). The reagents used for proteomics sample preparation and nLC-MS/MS analysis were purchased from Thermo Fisher Scientific (Waltham, MA) unless stated otherwise.
Cell Lines
Three clones each of PKA-null and PKA-intact mouse kidney collecting duct cell lines, derived from mpkCCD clone 11–38 (mpkCCDC11–38) using CRISPR-Cas9 to introduce mutations were used for this study (9). All experiments were performed with passages from 10 to 20 relative to the production of the original CRISPR clones.
Cell Culture
Cells were maintained in complete medium containing DMEM/Ham’s F-12 medium (DMEM/F-12), 2% (v/v) FBS plus supplements (5 μg/mL insulin; 50 nM dexamethasone; 1 nM triiodotyrosine; 10 ng/mL epidermal growth factor; 60 nM sodium selenite; 5 μg/mL transferrin) at 37°C and 5% (v/v) CO2. For all experiments, cells were seeded onto permeable membrane supports (Transwell, 0.4 μm Polyester membrane; cat. no. 3450)(Corning Costar, Corning, NY) with complete media for 4–7 days to allow formation of a confluent epithelial monolayer with cell polarization. Then, the media were changed to serum-free simple media (DMEM/F12 containing 50 nM dexamethasone, 60 nM sodium selenite, and 5 μg/mL transferrin) for 3 days to induce cellular differentiation and to ensure complete polarization. Except where indicated, the basolateral medium was identical with the apical media except for the addition of 0.1 nM of the vasopressin analog, D-amino D-arginine vasopressin (dDAVP). Transepithelial resistances were measured by voltmeter (EVOM2, WPI, Sarasota, FL). The apical and basolateral media were changed daily.
Stable-Isotopic Labeling with Amino Acids in Cell Culture (SILAC) and Short-term dDAVP Stimulation
The cells were labeled by growing in complete SILAC medium (Thermo Fisher) containing either heavy (13C615N4 arginine and 13C6 lysine) and light (12C614N4 arginine and 12C6 lysine) amino acids. The cells were cultured for 17 days (five passages) to reach >99.9% labeling (10). Heavy- or light-labeled cells were then grown on separate 6-well Transwell plates (containing labeled amino acids) for 7 days in complete SILAC medium and for 3 days in simple SILAC medium in the presence of dDAVP (0.1 nM) in basolateral media. On the 10th day, following a 2 h dDAVP wash-out period, light-labeled and heavy-labeled plates were treated with 0.1 nM dDAVP or vehicle, respectively, for 30 min. The cells were washed with PBS and stored immediately at −80°C until further processing.
Sample Preparation for Total Proteomics and Phosphoproteomics
The cells were thawed on ice and treated with urea buffer (8 M urea, 50 mM Tris·HCl, 75 mM NaCl, 1× Halt protease and phosphatase inhibitors), scraped and sonicated (duration: 2 min, pulse/pause: 2 sec/2 sec, output level = 1) to solubilize proteins. Following measurement of protein concentration (BCA reagent), equal amounts of heavy- and light-labeled protein extracts were mixed (total, 2.5 ± 0.4 mg). The mixed samples were reduced with 20 mM DTT for 1 h at 37 °C, and then alkylated with 40 mM iodoacetamide for 1 h at 25 °C in the dark. The samples were diluted with 20 mM triethylammonium bicarbonate buffer (pH 8.5) to 1 M urea, and then digested with trypsin/LysC (Promega, Madison, WI) (1:20 wt/wt) overnight at 37 °C. The peptides were desalted using hydrophilic–lipophilic–balanced extraction cartridges (Oasis, 1 cc, 30 mg). The peptide sample was divided into three parts: total peptide analysis (4%), heretofore referred to as ‘Total’ and phosphopeptide enrichments (48% × 2) using Fe-NTA (High-Select™ Fe-NTA Phosphopeptide Enrichment Kit, Cat. No.: 88300) and TiO2 columns (High-Select™ TiO2 Phosphopeptide Enrichment Kit, Cat No.: A32993). The enrichment protocols were as described in the manufacturer’s instructions with minor modifications. The enriched peptides from Fe-NTA columns were desalted using graphite columns (Pierce™ Graphite Spin Columns, Cat. No.: 88302). All three samples (Total, TiO2, Fe-NTA) were fractionated in a 96-well plate with off-line high-pH reverse-phase chromatography (Agilent 1200 HPLC system, Santa Clara, CA) using a XBridge BEH C18 column (Waters, Milford, MA; 130Å, 5 μm, 2.1 mm X 30 mm). A flow rate of 0.5 ml/min was used to generate the gradient with buffer A (10 mM TEAB) and buffer B (10 mM TEAB, 90% acetonitrile (ACN)). For each SILAC experiment, eluted samples were concatenated using a discontinuous scheme to 24 (Total), 12 (TiO2) and 12 (Fe-NTA) fractions, vacuum-dried and stored at −80 °C. The dried peptides were reconstituted with 0.1% (v/v) formic acid (FA) in LC-MS–grade water (J.T. Baker, Fisher Scientific) before mass spectrometry analysis.
Nano Liquid Chromatography-Tandem Mass Spectrometry (nLC-MS/MS)
Reversed-phase capillary HPLC separations were performed using a Dionex UltiMate 3000 RSLC nano system coupled in-line with an Orbitrap Fusion Lumos Tribrid mass spectrometer. Approximately 1.2 μg of peptides (calculated from digested cell lysate) were loaded onto the trapping column (PepMap100, C18, 75 μm × 2 cm), for 8 min at a flow rate of 5 μL/min with buffer A (0.1% (v/v) FA). The trapped peptides were fractionated on an analytical column (PepMap RSLC C18, 2 μm, 100Å, 75 μmi.d. × 50 cm) using a gradient of buffer B (ACN, 0.1% (v/v) FA) as follows: 4–22% for 83 min; 22–32% for 17 min; 32–90% for 3 min; followed by 90% buffer B for 7 min. The method duration was 120 min. Phosphopeptide-enriched samples were injected one each for higher-energy collisional dissociation (HCD) and electron-transfer/higher-energy collision dissociation (EThcD) fragmentation while ‘Total’ samples were analyzed by HCD fragmentation only.
HCD Fragmentation
MS(/MS) data were acquired on an Orbitrap Fusion as follows: All MS1 spectra were acquired over m/z 375−1500 in the orbitrap with a resolution of 120,000 (FWHM) at m/z 200; automatic gain control was set to accumulate 4 × 105 ions, with a maximum injection time of 50 ms. The intensity threshold for fragmentation was set to 25,000 and included charge states +2 to +5. A dynamic exclusion of 15 s was applied with a mass tolerance of 7 ppm. Data-dependent tandem MS analysis was performed using a top-speed approach and the dynamic parallelization using “ADAPT” technology. The isolation window (m/z) was set at 1.6. HCD collision energy was set at 30%. MS2 spectra were acquired with Orbitrap resolution of 30 000 and a fixed first m/z of 110. Automatic gain control target was set to 5 × 104, with a maximum injection time of 60 ms.The cycle time was 3 sec.
EThcD Fragmentation
Most of the MS(/MS) parameters for EThcD was identical as HCD fragmentation except the following: The charge states for fragmentation range from +3 to +7. ETD calibrated charge-dependent parameters and ETD supplemental activation were applied for data-dependent tandem MS. Supplemental activation collision energy for EThcD was set at 15%. Automatic gain control was set to 5 × 104, with a maximum injection time of 120 ms.
MS Raw Data Analysis
Raw data were analyzed using Proteome Discoverer 1.4 (version 1.4.0.288) in conjunction with two search engines; Mascot and SequestHT. The protein database used for SequestHT was the mouse Swiss-Prot (downloaded on April 16, 2017). The Mascot search engine and database were maintained by NIH Mascot server (https://biospec.nih.gov). The search criteria for all HCD raw files were set as follows: 1) precursor mass tolerance = 10 ppm, 2) fragment mass tolerance = 0.02 Da, 3) enzyme specificity was set as trypsin with two missed cleavages allowed. Carbamidomethylation of cysteine (C+57.021 Da) was set as a fixed modification. Variable modifications were as follows: a) isotope labeling of lysine (K+6.020 Da) and arginine (R+10.008 Da), b) oxidation of methionine (M+15.995 Da), c) deamination on glutamine and asparagine (N, Q+0.984 Da), d) phosphorylation of serine, threonine and tyrosine (S, T, Y+79.966 Da) and e) acetylation of protein N-terminal. The FDR was calculated by the target-decoy algorithm.
The search parameters for raw data files generated through EThcD fragmentation were kept identical except following modifications : 1) precursor selection: ‘use MS(n - 1) precursor’ instead of ‘use MS1 precursor’; 2) maximum precursor mass: 8000 Da instead of 5000 Da; 3) total intensity threshold: 0 instead of 1000; 3) maximum missed cleavage sites: 3. In addition, for SequestHT search, ‘c’ and ‘z’ ions were used for spectral matching instead of ‘b’ and ‘y’ ions that were used for HCD raw files.
The probabilities of the phosphorylation site localizations were calculated based on the given MS2 data using the PhosphoRS 3.0 module within Proteome Discoverer. The following data reduction filters was used; peptide confidence: high, peptide rank = 1. The FDR was calculated by the target decoy PSM validator. The FDR was set <0.01.
Data Integration across Biological Replicates
For ‘Total’ proteomics, the SequestHT and Mascot search results of three SILAC experiments corresponding to three clones each (biological replicate = 3) of PKA-intact and PKA-null cells were integrated in Proteome Discoverer after disabling the ‘protein grouping’ option. The ratio (dDAVP/Vehicle) obtained by SequestHT was used when available. Otherwise the ratio generated by Mascot was taken. The proteins that were quantified in all three biological replicates were used for further analysis. Paired t tests were used to calculate p values for comparison of log2[dDAVP/Vehicle] ratios versus log2[1] (null hypothesis). The median of log-ratios was reported along with corresponding p value.
For phosphoproteomics of PKA-intact and PKA-null cells, ‘.msf’ files of TiO2 and Fe-NTA samples searched with Mascot and SequestHT for all three SILAC experiments were integrated in Proteome Discoverer that resulted in two combined files, one each for HCD and EThcD fragmentation. The peptides in the combined file were grouped by ‘mass and sequence’ available with Proteome Discoverer. The phosphopeptides having an area (MS1 scan) of at least 1.0E7 in each of the replicate cell clones along with a corresponding median ratio calculated from TiO2 and Fe-NTA results were considered for further analysis. The phosphopeptides with a greater than 80% of phosphorylation site-assignment probability were selected. Amino acid sequences of these phosphopeptides were centralized around the phosphorylation site using PTM Centralizer (https://hpcwebapps.cit.nih.gov/ESBL/PtmCentralizer/). To account for biological variation across three replicates, i) the phosphopeptides having three ‘dDAVP/Vehicle’ ratios out of three SILAC experiments ii) showing consistent direction of regulation were considered for further analysis. The data files obtained from HCD and EThcD fragmentation were combined and median phosphorylation of site-specific ratios were calculated from all monophosphopeptides representing the same phopsho-site. Dual criteria were used to identify phospho-sites with altered abundance, namely, |log2(dDAVP/vehicle)| >0.378 and Pfp< 0.005. A value of 0.378 was assigned to encompass 95% of log2(control/control) values (95% confidence interval for control-to-control comparisons) (11). Pfp (a measure of false positive probability) for phosphopeptide quantification was calculated as a product of the p value from the t-statistic using all three pairs of dDAVP-vehicle replicates and the area under the Gaussian distribution curve outside the range [−Z, Z], where Z is the value divided by the SD of log2(control/control) values. PTM-Logo was used to generate sequence logos (12). The inputs were 13-mer centralized sequences around the phosphorylation site.
Cyclic AMP Measurement
For short-term vasopressin stimulation, dDAVP-conditioned cells on 6-well Transwell plates in simple media in the absence of dDAVP for 2 h (wash-out phase) on the day of experiment were treated with IBMX (3-isobutyl-1-methylxanthine, 0.5 mM on both apical and basolateral media) and concurrently with either 0.1 nM dDAVP or vehicle for 30 min (in the basolateral medium only). The experiments were performed in triplicate for each of three clones of PKA-intact and PKA-null cells. After incubation, the cells were rapidly washed with ice-cold PBS and frozen at −80°C. For another set of parallel experiment, IBMX was omitted.
Cyclic-AMP content was measured using the cAMP enzyme immunoassay kit (Cayman Biochemical) according to the manufacturer’s instructions. Briefly, the cells were lysed using 0.1 M HCl and centrifuged to collect the supernatant. The protein content of the samples was measured by BCA assay. The samples were diluted 10 times with ELISA buffer (provided in kit) before estimating the cAMP concentration in duplicate using standards provided in the kit. The amount of cAMP was inversely proportional to the optical density measured. Results are expressed as pmol/mg protein. Data is represented as mean ± standard deviation.
Protein Kinase Prediction
The Group-based Prediction System (GPS 3.0, http://gps.biocuckoo.org) was used to predict probable kinases based on the substrate sequence motifs (13). The FASTA sequences of the proteins were uploaded using the ‘batch predictor’ option in GPS 3.0 – species specific, where ‘mouse’ was selected as ‘organism’. ‘Tyrosine Kinase’ was excluded from the analysis. The most stringent threshold (i.e. ‘High’) for the cut off for false positive rate was chosen. This corresponds to 2% false positive rates for kinases while making predictions on large scale data. The site-specific scores for predicted kinases were calculated as the ‘score difference’ between the actual score and cut-off value. For prediction of multiple probable kinases for a phospho-site, only the top hit having the maximum ‘score difference’ was used for subsequent analysis.
Immunoblot Analysis
Laemmli buffer (2% SDS,63 mM Tris, pH 6.8, protease and phosphatase inhibitors) was used to solubilize the cellular proteins. Protein concentration was measured using the BCA assay. The denatured samples were subjected to SDS/PAGE. The proteins were transferred to nitrocellulose membranes and probed with primary antibodies from Cell Signaling Technology (Danvers, MA) at indicated dilutions: anti-AMPKα (1:1000, isotype: Rabbit, Cat. No. 5832), anti-phospho AMPKα (Thr172) (1: 1000, isotype: Rabbit IgG, Cat. No. 50081). REVERT total protein stain, blocking buffer and infrared fluorescence-conjugated secondary antibodies were obtained from LI-COR. Fluorescence images were visualized by a LI-COR Odyssey System (ODY-0428). Band intensities were analyzed by LI-COR Image Studio software.
In Vitro Phosphorylation Assay
Synthetic and nonphosphorylated 13-mer peptides centralized around the increased phosphorylation sites in Crtc1 (S151, SWRRTNS*DSALHQ), Lipe (S559, SMRRSVS*EAALAQ) and Arhgef2 (S151, SLAKSVS*TTNIAG) were synthesized (FDA, White Oak, MD). AMARA (ab204852, Abcam, Cambridge, MA) and SAMS-tide (ab120182) were used as standard peptides for SIK and AMPK phosphorylation. All SNF1-subfamily enzymes (AMPK, MARK3, QIK (SIK2), NUAK2, and MELK) were purchased from Carna Biosciences (Chuo-ku, Kobe, Japan). Recombinant AMPK (AMPKα1β1γ1, AMPKα2β2γ1, AMPKα2β1γ1) was custom-made without phosphorylation at T183 (in AMPKα1) or T172 (in AMPKα2). All peptides (custom peptides, 20 nmol; standard peptides, 10 nmole) were incubated with various purified enzymes individually (i.e. AMPK, SIK2) in kinase reaction buffer (25 mM MOPS, 25 mM MgCl2, 2 mM EDTA, 12.5 mM β-glycerol-phosphate) and supplemented with 100 μM ATP and 0.25 mM DTT for 1 h at 30 °C (all components were from Signal Chem, Richmond, BC, Canada). AMP (0 – 100 μM) (Signal Chem) and cAMP (0 – 1000 μM) were added in different concentrations as mentioned in Figure legands. The enzyme concentration was 2–4 ng/μl. The optimum concentration was determined based on the enzyme activity data provided by the manufacturer. The enzymatic reaction was terminated by centrifuging (14,000g, 10 min) the reaction mixture in a 30K molecular weight cut-off filter (Amicon Ultra, Milipore) to retain the enzymes, while the phosphorylated peptides are collected as filtrate. The filtrate was diluted in 0.1% FA, 2% ACN for MS analysis in a Parallel Reaction Monitoring (PRM) mode.
Parallel Reaction Monitoring Mass Spectrometry
The Orbitrap Fusion Lumos Tribrid MS instrument was used in a PRM mode. The total method duration was 52 min. The data type was set as ‘profile’ with ‘positive’ polarity and source fragmentation was disabled for both MS1 and MS2 scans. All MS1 master scans were acquired over a scan range (m/z) 375−850 in the orbitrap with a resolution of 120,000 (FWHM) at m/z 200. Automatic gain control was set 1.0e6, with a maximum injection time of 100 ms.
For targeted MS2 (tMS2) scan, the pre-defined precursor ions were selected in quadrupole mode with an isolation window of 0.7 m/z. The ions were fragmented by CID with a CID collision energy 35% and CID activation time 10 ms. The fragment ions were detected using orbitrap mass analyzer with following settings: orbitrap resolution: 50,000, mass range: normal, scan range (m/z): 200–1300, RF lens (%): 30, automatic gain control: 2.0e5. The cycle time was set as 3 sec. The precursor m/z and charge (z) for all peptides with and without phosphorylation can be found in the Supplemental Table 2.
PRM Data Analysis with Skyline
Raw data were processed on an open-source software, Skyline (version 4.2.0. 19072) (https://skyline.ms/project/home/software/Skyline/begin.view) (14). Briefly, the peptide sequences were added manually to skyline. The peptide and transition settings were updated as mentioned. In transition setting, peptides with a precursor charge of 2–4, ion charge of 1–3 and ion types of b, y, and p (precursor) were used for filtering all ions. The ion match tolerance was set as 0.02 m/z for peak extraction. In full scan tab, orbitrap was selected as precursor and product mass analyzer. The acquisition method was set as targeted under MS/MS filtering. All matching scans were included for retention time filtering. Subsequently, MS data files were imported into the software. Based on the incorporated parameters, the software extracts the chromatogram peaks and autocalculates the charge state, retention time, background-substracted chromatogram area, and background for all precursor and corresponding product ions. The retention time window of used chromatograms was manually verified and adjusted among different samples to avoid sample-specific bias, if any. The corrected chromatogram area of precursors and diagnostic product ions was used for final analysis. Phosphorylation was measured from the chromatogram area of the mono-phosphorylated precursor ion of specific peptides. The site of phosphorylation was determined by measuring the intensity of the site-specific fragment ion (i.e. diagnostic product ion) and/or corresponding neutral loss (i.e. diagnostic neutral loss). Phosphorylation increases the precursor mass by 79.96, while neutral loss decreases the product ion mass by 98 (15).The product ions from the y-series were used for the area measurement.
Data Availability
Mass spectrometer generated raw (.raw) and analysis (.msf) files for SILAC experiment have been uploaded to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD015719 (Username: reviewer89424@ebi.ac.uk; Password: HKPizc3T). These data are accessible at www.ebi.ac.uk/pride/archive/. The curated files have been made available through publicly available webpages: (1) PKA-null dataset: https://hpcwebapps.cit.nih.gov/ESBL/Database/PKA-null/index.html; (2) PKA-intact dataset: https://hpcwebapps.cit.nih.gov/ESBL/Database/PKA-Intact/index.html.
Statistical Analysis
Data were analyzed with GraphPad Prism and ‘data analysis’ add-in of Microsoft excel software. The statistical methods used, indexed to figures in the main text, are as follows. Data for cAMP and immunoblot measurement was analyzed using a two-tailed independent samples t-test (*, p<0.05). Over-representation of a specific amino acid (amino acid frequency) was subjected to chi-square analysis (chi-square alpha < 0.01).
RESULTS
Quantitative Phosphoproteomics of PKA-Null and PKA-Intact Collecting Duct Cells
The mpkCCD cell line has been previously shown to exhibit vasopressin-responses (AQP2 trafficking and AQP2 transcriptional regulation) typical of native collecting duct cells (16). CRISPR-Cas9 genome editing was previously used to create multiple lines of PKA-null cells and control PKA-intact cells allowing us to generate true biological replicate experiments using the different lines (9).To identify phosphorylation changes in the PKA-null and PKA-intact mpkCCD cells in response to the V2-selective vasopressin analog dDAVP, we carried out quantitative phosphoproteomics using SILAC for quantification (see Methods). In general, total protein abundances did not change in response to dDAVP (30 min) in either PKA-null or -intact cells, based on analysis of aliquots prior to phosphopeptide enrichment (Supplemental Figure 1). To maximize the depth of phosphopeptide identification, enriched samples were subjected to two different fragmentation methods (HCD and EThcD). In the PKA-null cells, we quantified 13,275 unique phosphopeptides (2,479 phosphoproteins) using HCD and 5,789 unique phosphopeptides (1,502 phosphoproteins) with EThcD fragmentation, based on filtering criteria described in Methods (Figure 1). For PKA-intact cells, using an identical criterion, 17,336 unique phosphopeptides (2,930 phosphoproteins) were quantified using HCD and 7,173 unique phosphopeptides (1,722 phosphoproteins) were quantified with EThcD fragmentation (Figure 1). Overall, addition of EThcD increased the coverage of unique phosphopeptide identification by about 10% beyond what would have been obtained with HCD alone. Single-, double-,triple-, and higher-phosphorylated peptides represented 79%, 18%, 3% and <1% of the total phosphopeptides respectively. We observed mostly serine phosphorylation (PKA-null, 91.5%; PKA-intact, 90.7%), followed by threonine (PKA-null, 8.3%; PKA-intact, 9%) and tyrosine (PKA-null, 0.2%; PKA-intact, 0.3%) phosphorylation. The curated data set for quantified phosphopeptides in the PKA-null cells can be viewed at https://hpcwebapps.cit.nih.gov/ESBL/Database/PKA-null/index.html. Curated data set for quantified phosphopeptides in PKA-intact cells can be viewed at https://hpcwebapps.cit.nih.gov/ESBL/Database/PKA-Intact/index.html.
Figure 1.
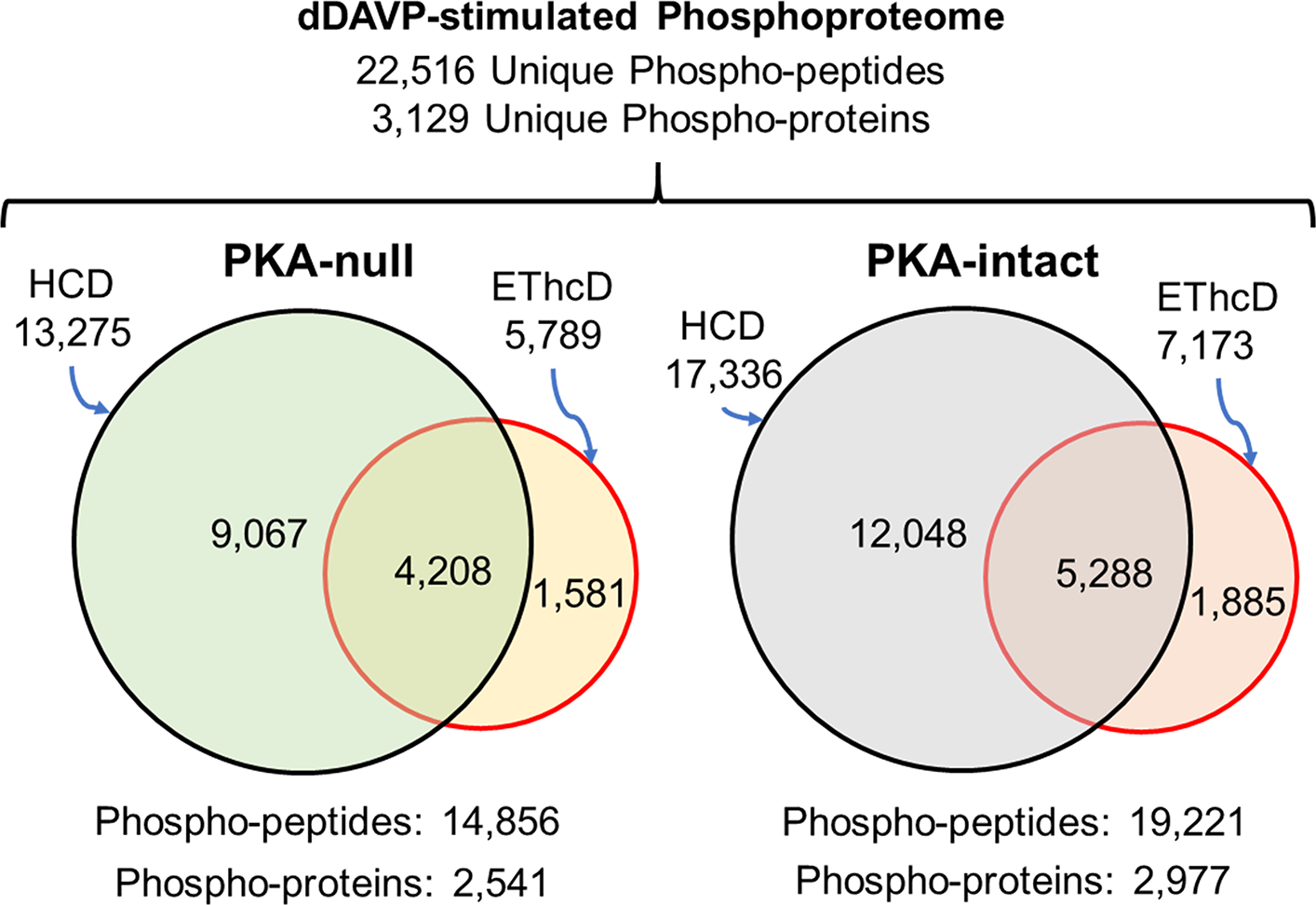
Venn diagrams comparing unique phosphopeptides and phosphoproteins identified by SILAC-based quantitative phosphoproteomics analysis in dDAVP-stimulated PKA-null and -intact cells using HCD and EThcD fragmentation. These phosphopeptides were identified 1) in all three biological replicates for receptive cell-types 2) with a minimum spectral area (MS1 scan) of at least 1.0E7. Peptide FDR was <0.01.
AQP2 Phosphorylation in Response to Vasopressin in PKA-Null cells
Prior studies indicate that vasopressin stimulation increases phosphorylation of AQP2 at S256 (17). To provide confirmation that the cells are responding appropriately to vasopressin in the current experiments, we show the AQP2-S256 MS1 spectra from one SILAC experimental PKA-intact pair before (heavy) and after (light) dDAVP exposure (Figure 2A). Overall, dDAVP caused a consistent increase in phosphorylation at S256 [log2(dDAVP/Vehicle) = 1.44, p <0.05, n=3]. Thus, the PKA-intact cells appear to be responding to dDAVP stimulation as expected. Interestingly, AQP2 was seen to be phosphorylated at S256 in the PKA-null cells as well (Figure 2B). Furthermore, the phosphorylation increased in response to dDAVP [log2(dDAVP/Vehicle) = 0.63, p <0.05] for all mono-S256 phosphopeptides [n=6 pairs](Supplemental Table 1). Taken together, we conclude that 1) both PKA-intact and -null cells respond to short-term vasopressin stimulation; and 2) S256 of AQP2 can be phosphorylated and regulated in response to vasopressin even in the absence of PKA.
Figure 2.
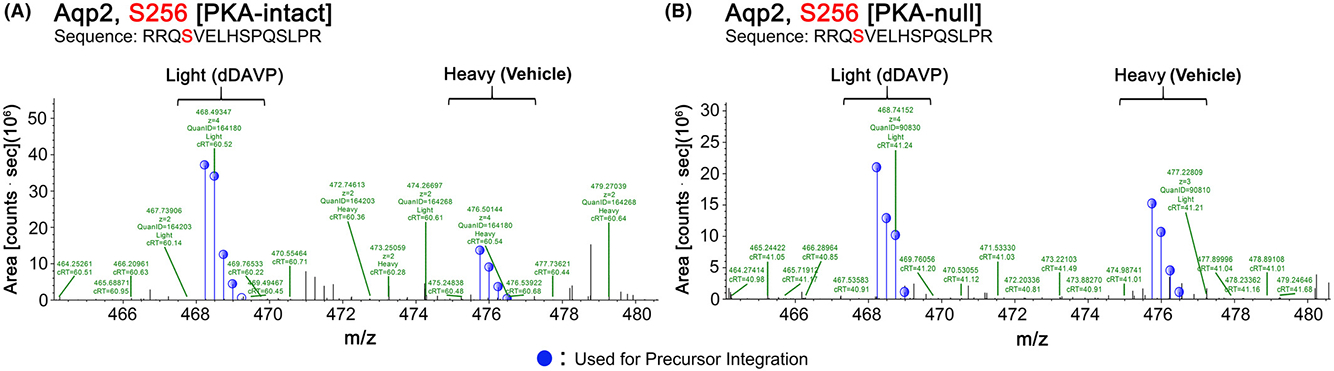
Representative MS1 spectra of Ser256 phosphopeptide of AQP2 showing an increase in dDAVP-treated (labeled with light amino acids) samples compared to vehicle treated or control (labeled with heavy amino acids) samples of (A) PKA-intact and (B) PKA-null cells. The phosphorylation site was marked in red in the peptide sequence. The vertical axis shows peptide ion intensity, and the horizontal axis shows the mass-to-charge ratio (m/z). Each peptide has several m/z peaks due to the presence of natural isotopes.
Major Elements of V2 Vasopressin Receptor Signaling are PKA-Dependent
Figure 3A–B summarizes abundance changes for phosphorylation sites ascertained from monophosphopeptides in response to the V2-selective agonist dDAVP in PKA-intact cells and PKA-null cells. The threshold for identifying a phosphopeptide as ‘changed’ is defined as Pfp <0.01 (calculated as described in Methods). In the PKA-intact cells, dDAVP addition resulted in significant perturbations at 452 sites (in 366 distinct proteins) out of 3748 quantified (12%). Of these, 205 were increased and 247 were decreased. In the PKA-null cells, there were also changes in the phosphoproteome although there were fewer altered phosphorylation sites than in PKA-intact cells. Overall, in the PKA-null cells, there were changes in 2.2% (61/2842) of quantified phospho-sites (Figure 3). Of these, 32 were increased and 29 were decreased. Analysis of multiply phosphorylated peptides also showed many more regulated phosphopeptides in the PKA-intact cells than in PKA-null cells (Supplemental Figure 2).
Figure 3.
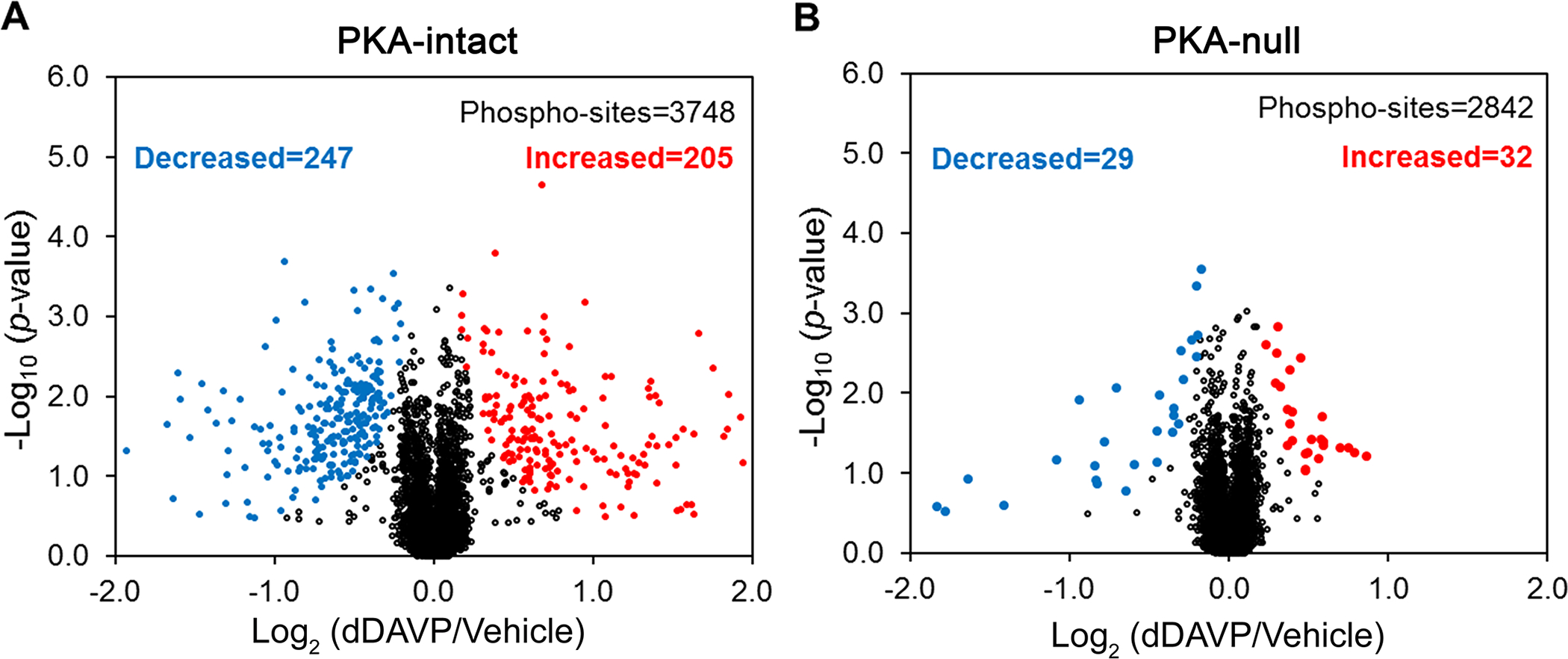
Volcano plot for single and unique phosphorylation sites quantified with a consistent trend across all three pairs (dDAVP vs. vehicle) of (A) PKA-intact and (B) PKA-null clones. Phosphosites having a site-assignment probability >80% as determined by PhosphoRS 3.0 were used for this analysis. The red and blue dots indicate increased and decreased phosphorylation sites with Pfp ≤0.01. The plots were presented with an x-axis value of −2.0 to +2.0 for better visualization.
Although the preceding data establish that most of the phosphorylation changes in mpkCCD cells in response to vasopressin are PKA-dependent, PKA-independent changes were also observed. To assess the quality of these data, we manually examined the MS1 and MS2 spectra of all phosphopeptides that were preliminarily assessed as being changed in PKA-null cells. This resulted in a list of high confidence responses to vasopressin in the PKA-null cells (Table 1, https://hpcwebapps.cit.nih.gov/ESBL/Database/PKA-null/sites.html). Some example MS1 spectra corresponding to phospho-sites reported in Table 1 are shown in Supplemental Figure 3.
Table 1.
Vasopressin responsive phosphorylation sites in PKA-null cells
| Accession | Gene Symbol | Site(s) | Protein Name | Centralized Sequence | Median Log2(dDAVP/ Vehicle) | −log10 (Pfp) |
|---|---|---|---|---|---|---|
|
| ||||||
| P59729 | Rin3 | S411 | Ras and Rab interactor 3 | GISRTAS*LNLPPQ | 0.86 | 3.52 |
| Q8R4H2 | Arhgef12 | S41 | Rho guanine nucleotide exchange factor 12 | KVERSSS*HDFDPT | 0.79 | 3.28 |
| Q8C1B1 | Camsap2 | S439 | Calmodulin-regulated spectrin-associated protein 2 | GIIRSVS*NEGLTL | 0.75 | 3.16 |
| Q9Z1E4 | Gys1 | S711¶ | Glycogen [starch] synthase, muscle | GSKRSNS*VDTGPS | 0.70 | 2.98 |
| P45481 | Crebbp | S120 | CREB-binding protein | LGAMGKS*PLNQGD | 0.59 | 2.63 |
| Q9D279 | Misp | S364 | Mitotic interactor and substrate of PLK1 | GLQRSLS*SDCILS | 0.59 | 2.67 |
| Q9ERG2 | Strn3 | S202 | Striatin-3 | RSQRVRS*LLGLSN | 0.58 | 2.95 |
| Q3UHI0 | Ccser2 | S222 | Serine-rich coiled-coil domain-containing protein 2 | KMVRSQS*FSHSIQ | 0.58 | 2.66 |
| P19001 | Krt19 | S59 | Keratin, type I cytoskeletal 19 | VTSSSGS*YGGVRG | 0.52 | 2.47 |
| Q68ED7 | Crtc1 | S151 | CREB-regulated transcription coactivator 1 | SWRRTNS*DSALHQ | 0.49 | 2.24 |
| Q3TWF6 | Wdr70 | S641 | WD repeat-containing protein 70 | MFAQVES*DDEESK | 0.49 | 2.75 |
| A6H8H2 | Dennd4c | S1321 | DENN domain-containing protein 4C | YKDRSTS*LSALVR | 0.48 | 1.98 |
| Q9CY58 | Serbp1 | S203 | Plasminogen activator inhibitor 1 RNA-binding protein | SGSDRSS*FSHYSG | 0.48 | 2.18 |
| Q8BG32 | Psmd11 | S14 | 26S proteasome non-ATPase regulatory subunit 11 | EFQRAQS*LLSTDR | 0.48 | 1.97 |
| Q3B7Z2 | Osbp | S377 | Oxysterol-binding protein 1 | GHKRTGS*NISGAS | 0.39 | 2.48 |
| Q60875 | Arhgef2 | S163 | Rho guanine nucleotide exchange factor 2 | GHFNDES*PLGLRQ | 0.39 | 2.11 |
| Q60875 | Arhgef2 | S151 | Rho guanine nucleotide exchange factor 2 | SLAKSVS*TTNIAG | 0.39 | 2.31 |
| Q9ESE1 | Lrba | T1011 | Lipopolysaccharide-responsive and beige-like anchor protein | SNTELQT*HDMSSD | 0.37 | 2.44 |
| Q6GYP7 | Ralgapa1 | S772 | RalGTPase-activating protein subunit alpha-1 | TRVRHFS*QSEDTG | 0.33 | 2.62 |
| Q9ESE1 | Lrba | S1000 | Lipopolysaccharide-responsive and beige-like anchor protein | SIGRASS*IDSASN | 0.30 | 2.75 |
| Q9Z1W9 | Stk39 | S383 | STE20/SPS1-related proline-alanine-rich protein kinase | RRVPGSS*GHLHKT | 0.29 | 2.60 |
| P54310 | Lipe | S559 | Hormone-sensitive lipase | SMRRSVS*EAALAQ | 0.25 | 3.34 |
| P99027 | Rplp2 | S79 | 60S acidic ribosomal protein P2 | VSAAPGS*AAPAAG | −0.18 | 1.33 |
| Q8BTM8 | Flna | S968 | Filamin-A | SVGVSPS*LDLSKI | −0.20 | 2.98 |
| Q9JKB3 | Ybx3 | S52 | Y-box-binding protein 3 | AALLAGS*PGGDAA | −0.20 | 2.75 |
| Q62074 | Prkci | T411 | Protein kinase C iota type | PGDTTST*FCGTPN | −0.34 | 2.39 |
| Q62074 | Prkci | T415 | Protein kinase C iota type | TSTFCGT*PNYIAP | −0.35 | 2.32 |
| Q9D1C9 | Rrp7a | S99 | Ribosomal RNA-processing protein 7 homolog A | KPDLAES*PTEPKS | −0.35 | 2.12 |
| Q8C0T5 | Sipa1l1 | S1626 | Signal-induced proliferation-associated 1-like protein 1 | HGEFSAS*DSSLTD | −0.45 | 1.99 |
| Q9Z2H5 | Epb41l1 | S378 | Band 4.1-like protein 1 | TFFRLVS*PEPPPK | −0.45 | 2.39 |
| P99027 | Rplp2 | S86 | 60S acidic ribosomal protein P2 | AAPAAGS*APAAAE | −0.59 | 3.65 |
| P39447 | Tjp1 | S280 | Tight junction protein ZO-1 | LSDSIHS*ANASER | −0.65 | 2.25 |
| Q3UHX2 | Pdap1 | S60¶ | 28 kDa heat- and acid-stable phosphoprotein | EKKSLDS*DESEDE | −0.83 | 1.25 |
| P11276 | Fn1 | S285¶ | Fibronectin | ASAGSGS*FTDVRT | −0.84 | 3.67 |
| Q99J36 | Thumpd1 | S88 | THUMP domain-containing protein 1 | DQQPSGS*EGEDDD | −0.94 | 4.60 |
| P09405 | Ncl | S145 | Nucleolin | NAKKEDS*DEDEDE | −1.64 | 16.00 |
| Q3THK3 | Gtf2f1 | S311 | General transcription factor IIF subunit 1 | SESSEES*EEEKPP | −2.46 | 15.60 |
These sites have redundant quantification evidence showing similar changes (e.g. PKA-null and PKA-intact cells, site containing peptides with or without missed cleavage, and HCD-EThcD pairs). The MS1 spectra of monophosphopeptides containing these sites can be found in Supplemental Figure 3. Eleven of the increased phosphorylation sites are putative SNF1-subfamily targets based on the presence of an (R/K)-(T/S)-X-p(S/T) motif with nonpolar amino acid in position −5 and/or position +4 relative to the phosphorylated amino acid (underlined amino acids).
Overall, these results suggest that 1) the major elements of vasopressin signaling are dependent on PKA, and 2) a smaller component of vasopressin signaling is independent of PKA.
Baseline and dDAVP Stimulated cAMP Production in PKA-Null Cells
Vasopressin signaling is generally accepted to be cAMP-mediated. The marked decrease in the number of phosphorylation events in dDAVP-stimulated PKA-null cells may be related to the failure of cells to produce cAMP due to a dysfunctional V2R or by some other mechanism involving the Gsα-adenylyl cyclase complex. To rule out such possibility, we compared vasopressin-dependent cAMP-levels in PKA-intact versus PKA-null cells measured in the presence of the broad spectrum phosphodiesterase inhibitor IBMX (Figure 4). The PKA-null cells responded to dDAVP with a large increase in cAMP in the presence of IBMX. The surprising aspect of these data was a large increase in the baseline (vasopressin-independent) cAMP levels amounting to more than a 10-fold increase compared to PKA-intact cells (Figure 4A). It also shows an equivalent 10-fold increase of cAMP level in dDAVP-treated PKA-null cells when compared to dDAVP-treated PKA-intact cells. When the cells were not treated with IBMX, cAMP levels were still readily measurable, but values were not different between PKA-intact and PKA-null mpkCCD cells (Figure 4B). This indicates that under the conditions of the phosphoproteomic studies, cAMP production is likely to be markedly increased although high levels in the cells are not sustained due to rapid degradation by cyclic nucleotide phosphodiesterases. The measurements however do not rule out localized increases in cAMP levels. Overall, these measurements 1) confirmed the responsiveness of the PKA-null cells to vasopressin at the level of cAMP production, 2) suggest the presence of a PKA-controlled feedback inhibition of cAMP generation in PKA-intact cells.
Figure 4.
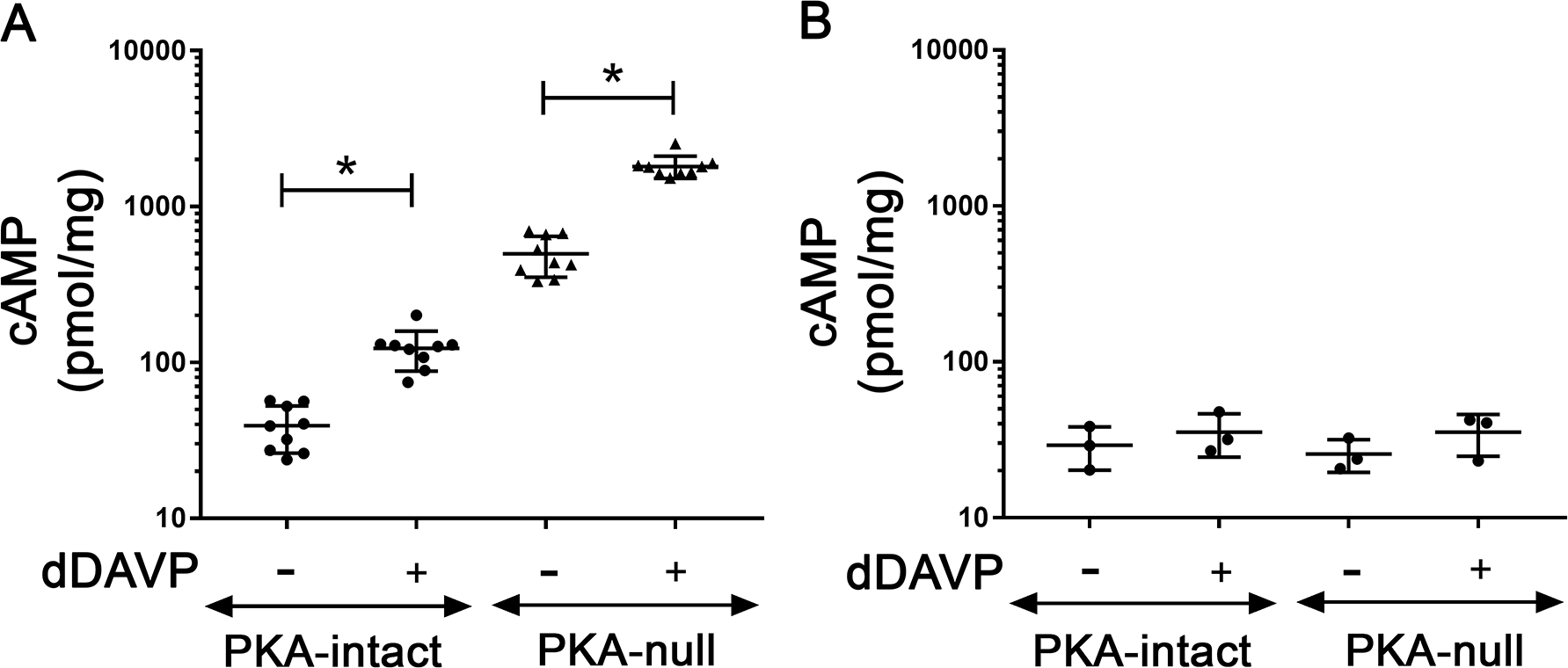
Effect of dDAVP on cAMP generation in PKA-intact and PKA-null cells. The experiment was performed (A) in presence or (B) in absence of IBMX. The measured cAMP values in pmol/ml were normalized with the protein concentration of the respective samples. Data were presented as mean ± SD. A two-tailed independent samples t-test was used. n ≥ 3; *, p<0.05; SD = standard deviation
Vasopressin Activates non-PKA Basophilic Kinases in PKA-Null Cells
Among the sites that were increased by dDAVP in PKA-null cells, there was a preponderance of sequences with a basic amino acid (e.g. R or K) in position −3 relative to the phosphorylated amino acid (Chi-square = 20.9, p = 5.00E-06, versus all quantified monophosphopeptides). This implies that one or more basophilic protein kinases (AGC or CAMK group) were activated in response to dDAVP in the PKA-null cells. To narrow down the possibilities, we used prior transcriptomic data to identify basophilic kinases expressed in PKA-null mpkCCD cells (9), augmenting this list with protein kinases found by nLC-MS/MS analysis in the present study. In Figure 4A, the circle shows the 47 basophilic kinases (out of 126 basophilic kinases in the mouse kinome) that are expressed at both transcript and protein levels in PKA-null cells. This list contains 25 kinases from the AGC group and 22 kinases from the CAMK group. The CAMK group of kinases was predicted to account for a majority of (14/22, 64%) elevated phospho-sites in PKA-null cells (Figure 5B) based on the Group-based Prediction System (GPS 3.0) (13). This program uses machine-learning approaches to optimize the match between individual phosphorylation sites and kinase preference motifs across the kinome to identify the kinase group, kinase family, kinase subfamily and individual kinase level matches. Additional analysis revealed preferences for serine or threonine in position −2 (Chi-square = 14.4, p = 1.45E-04), hydrophobic amino acids (isoleucine, methionine and tryptophan) in postion −5 (Chi-square = 23.4, p = 1.00E-06) and in position +4 (glycine, leucine, isoleucine and valine, Chi-square = 7.4, p = 6.41E-03). Figure 4C shows the preferred motif that was created using PTM-Logo (12) from the list of increased phospho-sites. Overall, among the increased phospho-sites there were eleven with either of the two motifs: ([I/M/W]-X-[R/K]-[S/T]-X-p[S/T], or [R/K]-[S/T]-X-p[S/T]-X-X-X-[G/L/I/V]), including some with hydrophobic amino acids in both −5 and +4 position (Table 1). These motifs are not a general feature of the CAMK group, but correspond to the CAMK branch known as the AMPK-related kinase subfamily (or SNF1-subfamily), which includes the AMP-activated protein kinases Prkaa1 and Prkaa2, the MAP/microtubule affinity-regulating kinases Mark1 through Mark4, the salt-inducible kinases Sik1 through Sik3, the SNF1-like kinases Nuak1 and Nuak2, the BR serine/threonine kinases Brsk1 and Brsk2, and the maternal embryonic leucine zipper kinase Melk (18). Further, at least three of the increased phosphorylation sites in Table 1 have been experimentally characterized in other cell types and linked to kinases from the SNF1-subfamily. These sites are: S151 of Arhgef2 (Rho guanine nucleotide exchange factor 2, upstream kinase: Mark3 (19)); S151 of Crtc1 (CREB-regulated transcription coactivator 1, upstream kinase: Sik2 or Prkaa1 (20)); and S559 of Lipe (Hormone-sensitive lipase, upstream kinase: Prkaa1 (21)). Together, these results indicate probable activation of one or more members of SNF1 (AMPK-related) kinase subfamily in PKA-null cells following dDAVP stimulation. It is noteworthy that two other protein kinases with no obvious relationship to AMPK/SNF1-subfamily kinases undergo changes at activity-altering phosphorylation sites in response to vasopressin in the PKA-null cells. These kinases are Stk39 (SPAK, at S383) which is increased, and Prkci (PKC-iota, at T411 and T415) which shows decreased phosphorylation.
Figure 5.
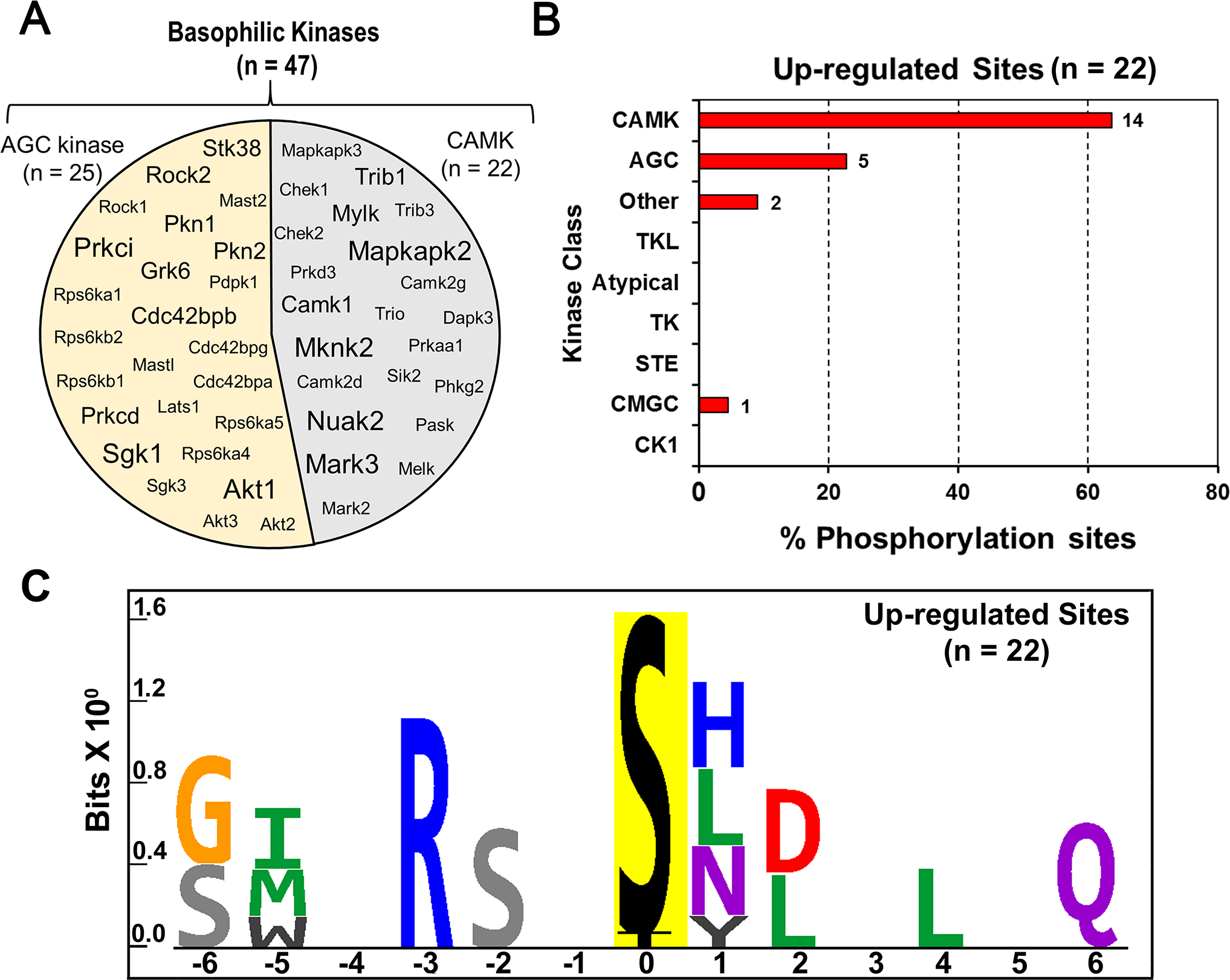
(A) The circle contains gene symbols of 47 basophilic kinases expressed in PKA-null cells. The font-size corresponds to their expression at the RNA level as determined earlier by Isobe et al. (9). (B) Histogram to predict relative frequency of various kinase classes upstream of altered phospho-sites in dDAVP treated PKA-null cells. A total of 22 elevated phospho-sites were analyzed using GPS 3.0 to predict the class of individual kinases. A frequency distribution analysis was performed to determine the % of phospho-sites (x-axis) that belong to certain class of kinase (y-axis). CAMK (14/22, 63%) were the most frequently predicted kinase class among upregulated sites in PKA-null cells. (C) Sequence logo for the up-regulated phosphorylation sites in dDAVP treated PKA-null cells. PTM-Logo was used for motif analysis. The background contains all unique and mono-phosphorylated peptides.
In Vitro Phosphorylation by Recombinant SNF1-Subfamily Protein Kinases
Several SNF1-subfamily protein kinases are expressed in mpkCCD cells, namely Prkaa1 (AMPKα1), Sik2, Mark2, Mark3, Nuak2 and Melk (Figure 5A). We asked which among these are responsible for increased phosphorylation in PKA-null cells following dDAVP treatment. First we focused on AMPK as it is the major and most well-studied member of SNF1-subfamily of basophilic kinase. Phosphorylation of conserved T172 present within the activation loop of the α-subunit kinase domain of AMPKα2 (Prkaa2, analogous to T183 of Prkaa1/AMPKα1) is known to activate this kinase (18). However, immunoblot results do not show an increase in the phosphorylation on T172 of AMPKα following dDAVP treatment in PKA-null or intact cells when compared to vehicle-treated cells (Figure 6). AMPK can also be activated allosterically by AMP that may not necessarily need a pre-phosphorylation of AMPKα in its activation loop (18). To test this possibility, we designed an in vitro phosphorylation assay with custom-made human recombinant AMPKα that is not phosphorylated at its activation loop. AMPK heterotrimers consist of catalytic α subunit (α1 or α2) along with a scaffolding β (β1 or β2) subunit and aregulatory γ (either γ1, or γ2, or γ3) subunit. Here,we used three different versions of heterotrimeric AMPK; namely AMPKα1β1γ1, AMPKα1β2γ1, and AMPKα2β2γ1. Centralized 13-mer synthetic peptides around the elevated phosphorylation sites of Arhgef2 (S151), Crtc1 (S151) and Lipe (S559) protein with a matching SNF1 motif were used as substrates for this in vitro assay. Because it seemed possible that cAMP could substitute for AMP as an allosteric activator of AMPK, we tested varying concentrations of cAMP (10 −1000 μM) in addition to AMP (10–100 μM) in the reaction mixture. PRM-MS was used to measure phosphopeptides and to quantify site-specific phosphorylation using a diagnostic transition ion or neutral loss ion as described in Methods (Supplemental Table 2). Standard AMPK/Sik2 substrates AMARA and SAMS-tide were also included as positive controls. As shown in Figure 7A, increasing concentrations of cAMP (10 −1000 μM) were unable to increase the phosphorylation of any of the above-mentioned three sites or the target sites in standard peptides. Instead, cAMP appears to inhibit phosphorylation at higher concentrations. In contrast, AMP increased phosphorylation of all three sites at the tested concentrations that were elevated in mpkCCD cells in response to dDAVP (Figure 7B). Standard peptides also showed similar trends. Thus, AMPK is a candidate for a role in vasopressin-induced phosphorylation changes in PKA-null mpkCCD cells, but it cannot be allosterically activated by cAMP.
Figure 6.
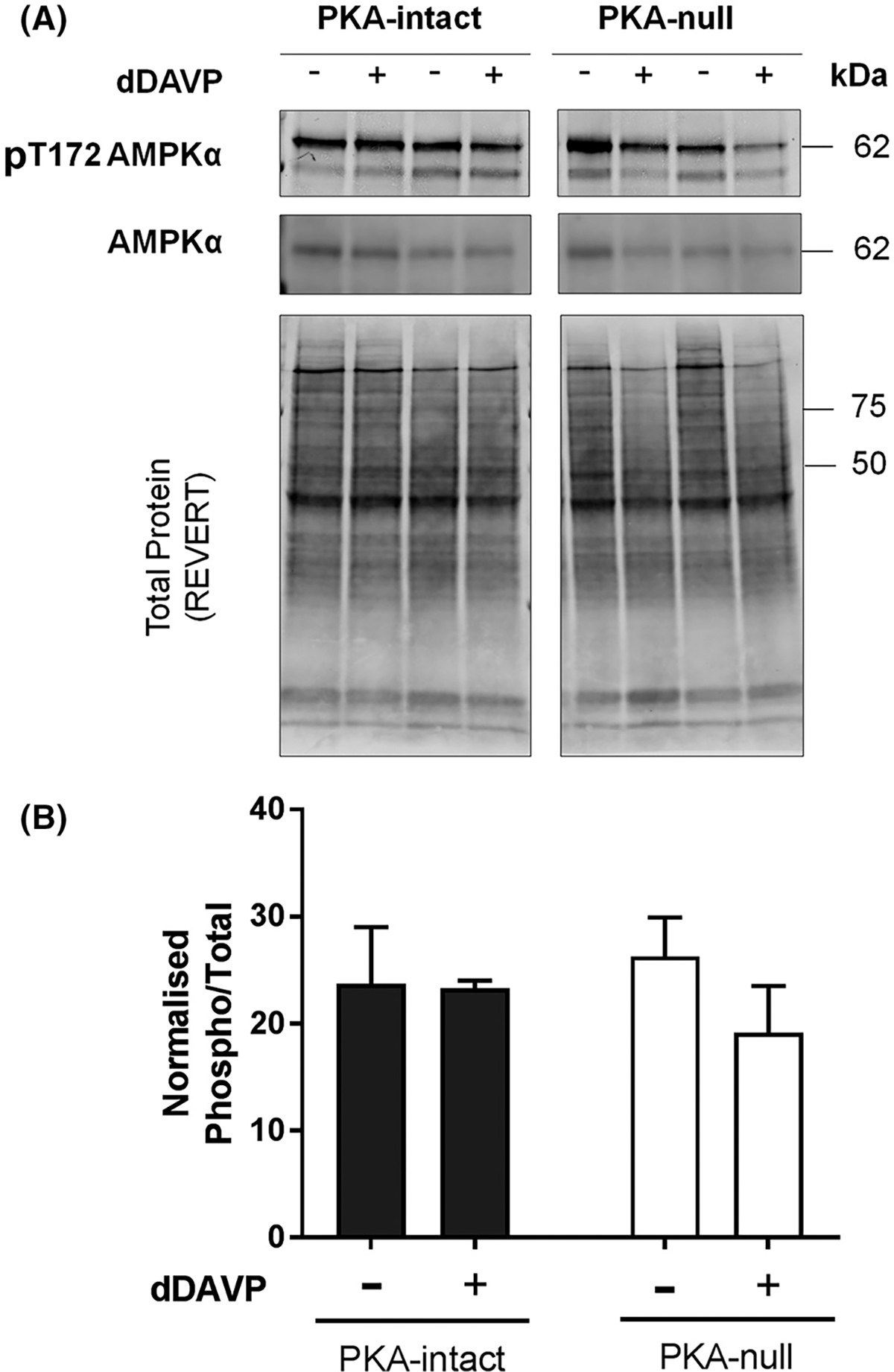
Immunoblot analysis to detect differential phosphorylation at the active site of AMPKα. (A) AMPKα and pT172-AMPKα immunoblot from dDAVP- and vehicle-treated PKA-intact and PKA-null cells. The gels were stained with REVERT (Li Cor) stain to check equal loading among different samples. (B) Bar chart of normalized immuno-reactivities (mean ± SEM in arbitrary units) of pT172 AMPKα. Unpaired t-test was used to determine significant difference between dDAVP and vehicle treated samples. dDAVP did not cause a significant difference in phosphorylation of T172 of AMPKα in PKA-intact or PKA-null cells. *, p< 0.05
Figure 7.
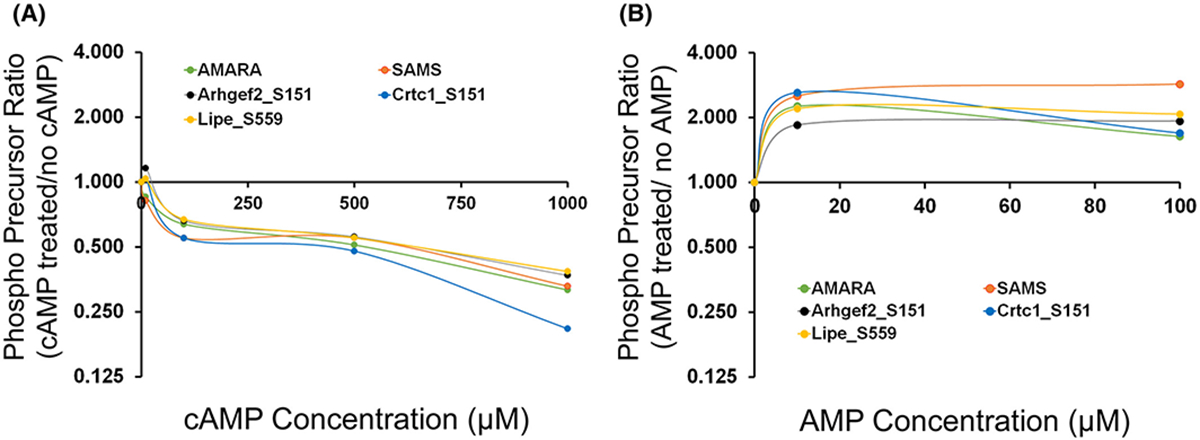
PRM-MS measurement of mono-phosphorylated form of 13-mer synthetic peptides centralized around S151 of Arhgef2, S151 of Crtc1 and S559 of Lipe. Human recombinant AMPK (e.g. AMPKα2β2γ1) that was not phosphorylated at its active site, was used for the in vitro phosphorylation reaction. AMARA and SAMS were used as standard peptide substrates for AMPK phosphorylation. (A) Effect of increasing concentrations of cAMP (0, 10, 100, 500, 1000 μM) on AMPK-induced phosphorylation. (B) Effect of increasing concentrations of AMP (0, 10, 100 μM) on AMPK-induced phosphorylation. X-axis indicates the ratio of chromatographic area of phosphorylated peptides (based on precursor ion chromatogram area) following incubation with various concentrations of (A) cAMP (B) AMP when compared to (A) ‘no cAMP’ or (B) ‘no AMP’ (0 μM). An x-axis value of 1.0 indicates no change.
Non-AMPK Members of SNF1-Subfamily can Phosphorylate Selected Sites of Arhgef2, Crtc1, and Lipe
To search for potential alternate mechanisms behind the elevation of phospho-sites with the SNF1 recognition motif, we extended the in vitro phosphorylation assay on the same set of target substrates to include additional members of the SNF1-subfamily of kinases, namely Mark3, Nuak2, Melk, and Sik2. The choice of these enzymes was driven by the objective to include multiple kinases from different branches of the SNF1-subfamily that are expressed in PKA-null cells. Our results show that under in vitro conditions, Mark3, Sik2, Nuak2, and Melk can all phosphorylate the sites in Arhgef2, Crtc1, and Lipe (Figure 8) and that the activity of these enzymes were not increased by AMP addition (Figure 9).
Figure 8.
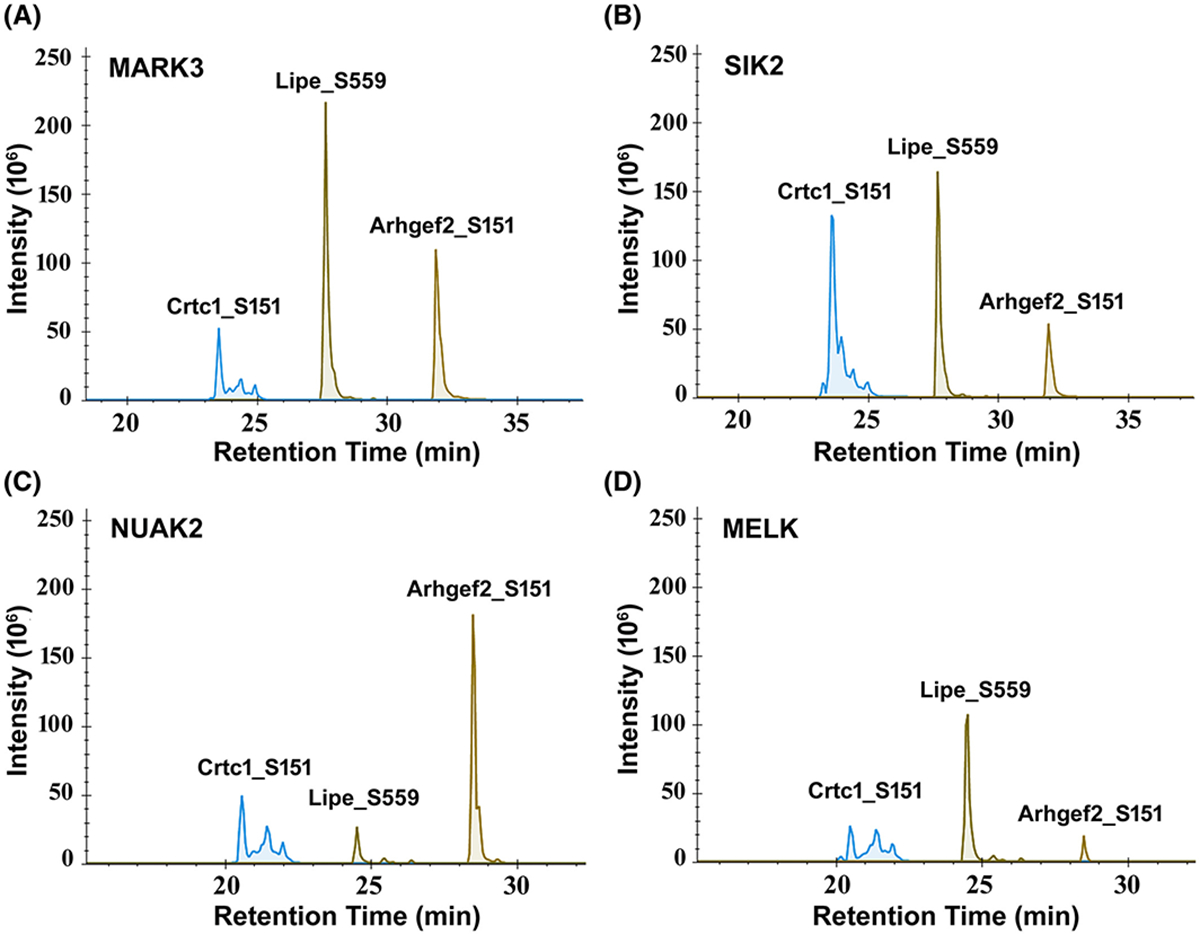
Precursor ion chromatogram of phosphorylated 13-mer synthetic peptides centralized around S151 of Arhgef2, S151 of Crtc1 and S559 of Lipe. The synthetic peptides were phosphorylated following an in vitro phosphorylation reaction in presence of various human recombinant SNF1-subfamily of kinases; (A) MARK3, (B) SIK2, (C) NUAK2, (D) MELK. The phosphorylation occurs predominantly at the target site of the respective peptides as determined by the chromatogram area of site-specific product ions. All SNF1-subfamily enzymes can phosphorylate the three selected phosphorylation sites on Arhgef2 (S151), Crtc1 (S151) and Lipe (S559) to different extent under in vitro condition.
Figure 9.
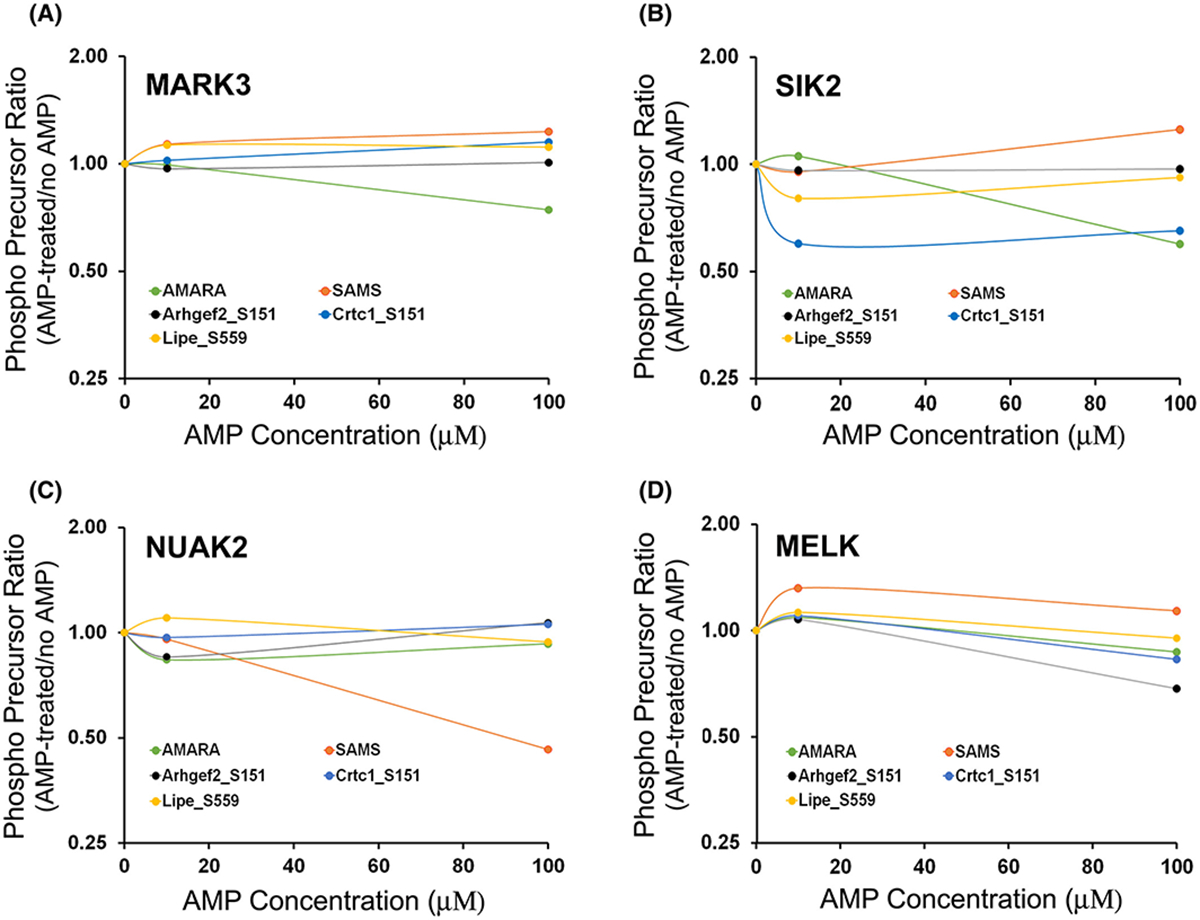
Effect of increasing concentration of AMP on the phosphorylation of 13-mer synthetic peptides centralized around S151 of Arhgef2, S151 of Crtc1 and S559 of Lipe in presence of various SNF1-subfamily protein kinases. Human recombinant (A) MARK3, (B) SIK2, (C) NUAK2, (D) MELK were used for the in vitro phosphorylation reaction. PRM-MS assay was used to measure the chromatogram area of phosphorylated peptide precursors. X-axis indicates the ratio of chromatogram area of phosphorylated peptides (based on precursor mass) following incubation with various concentrations of AMP (10, 100 μM) when compared to ‘no AMP’ (0 μM). An x-axis value of 1.0 indicates no change. AMARA and SAMS were included as control peptides.
DISCUSSION
We used quantitative phosphoproteomics to show that V2R-mediated vasopressin signaling in cultured collecting duct cells is predominantly PKA-dependent. In addition, there is a smaller component that is PKA-independent, likely involving activation of one or more members of the AMPK/SNF1-subfamily of the CAMK-like family of kinases. Among the PKA-independent phosphorylation targets that were detected were three known targets of SNF1-subfamily kinases, namely Arhgef2 (S151), Crtc1 (S151) and Lipe (S559)(19–21). Further, we show that the vasopressin-induced increase in phosphorylation of AQP2 at Ser256 can occur in the absence of PKA, although the specific kinase involved was not identified.
Prior evidence for PKA-independent vasopressin responses in collecting duct.
A number of prior studies in collecting duct cells have provided evidence for cAMP- or PKA- independent effects of vasopressin. Olesen et al. (22) have questioned the role of cAMP in vasopressin-mediated trafficking to the plasma membrane, based on differences in the time courses of intracellular cAMP changes and immunochemical detection of AQP2 redistribution in collecting duct cells. Nickols et al. (23) found that V2-receptor mediated mobilization of intracellular Ca2+ was blocked by mutation of a calmodulin-binding site in the COOH-terminal tail of the V2 receptor, without affecting cAMP production. The presence of cAMP-independent mobilization of intracellular Ca2+ in collecting duct cells was also supported by Naruse (24), Star et al. (25), and Champigneulle et al. (26). On the other hand, the finding that vasopressin-mediated Ca2+ mobilization was replicated in collecting ducts in response to exposure to cAMP analogs suggests that Ca2+ mobilization is at least in part cAMP dependent (27). Beyond this, Yip (28) has provided evidence that cAMP-mediated Ca2+ mobilization, and concomitant AQP2 trafficking to the plasma membrane, could be the result of cAMP activation of the Rap1 guanine-nucleotide exchange factor, Epac1 (Rapgef3) and not PKA. Another study provided evidence for a role for Epac1, but not PKA, in the effect of long-term exposure to vasopressin to increase Aqp2 gene transcription (29). Activation of Epac1 is predicted to activate Rap1 through its guanine-nucleotide exchange factor (GEF) activity. Rap1, among other functions, competes with Ras to reduce MAP kinase signaling (30). However, there was no evidence in the present study of altered phosphorylation of any MAP kinase in response to vasopressin in the PKA-null cells. Beyond this, in our prior study, deletion of both PKA catalytic genes abrogated the effect of vasopressin to stimulate Aqp2 gene transcription and AQP2 trafficking, and these effects were rescued by transfection with PKA (9), strongly supporting a central role for PKA in both actions of vasopressin.
Possible mechanisms of accelerated cAMP production in PKA-null cells.
A potentially important new finding in this paper is the observation that IBMX-dependent cAMP levels are around 10-fold higher in PKA-null cells than in PKA-intact cells. Since the difference in cAMP levels between PKA-null cells and PKA-intact cells was not seen in the absence of the phosphodiesterase inhibitor IBMX, we conclude that PKA-null cells likely have accelerated cAMP production. The most direct interpretation of this finding is that there may be a PKA-dependent feedback mechanism at the level of the V2R-adenylyl cyclase pathway that is interrupted with PKA deletion. There are several possibilities that could account for this feedback inhibition based on the various components of the coupling mechanism connecting receptor occupation and cAMP production, namely the V2R receptor (Avpr2), the heterotrimeric G-protein stimulatory alpha subunit Gsα (Gnas) or its inhibitory counterparts, the RGS protein Rgs3 (a GTPase activating protein), and adenylyl cyclase 6 (Adcy6). Among these, only Rgs3 showed a change in phosphorylation in response to dDAVP in the PKA-intact cells, viz. an increase in phosphorylation at a basophilic site (S686), although the effect of this modification on its activity is currently unknown based on the data on PhosphoSitePlus. In general, protein function can be regulated by processes other than phosphorylation and it will require a systematic examination of multiple competing hypotheses to identify the feedback mechanism responsible for the high cAMP levels in the PKA-null cells.
Possible mechanisms of vasopressin signaling in the absence of PKA.
The mechanism by which vasopressin activates downstream signaling in the absence of PKA has not yet been identified. As vasopressin signals through the Gαs-coupled GPCR V2R, it is likely that the activation involves cAMP. We propose four possible mechanisms of PKA-independent cAMP effects: (a) cAMP could be working through effectors other than PKA, e.g. Rapgef3 (Epac1); (b) cAMP could act allosterically to increase AMPK activity, mimicking AMP; (c) high AMP concentration could be achieved locally in the cell due to increased production and degradation of cAMP that could activate AMPK locally; and (d) high cAMP could directly activate other kinases such as Protein Kinase G. The first possibility seemed to be a viable choice because of observations in the literature that RapGEF3/Epac1 can activate the protein kinase Lkb1 (Stk11), which activates AMPK through active site phosphorylation (31). However, immunoblotting for active site phosphorylation of AMPKα failed to show any tendency toward an increase in phosphorylation, ruling out this specific mechanism (Figure 6). The second possibility was seemingly ruled out by in vitro phosphorylation assays showing that cAMP, even at very high concentrations, inhibits rather than activates AMPK (Figure 7A). The third possibility was supported in part by the results of in vitro phosphorylation assays that confirm that AMP can indeed enhance the ability of AMPK to phosphorylate three specific targets increased by vasopressin in PKA-null cells (Figure 7B). In addition, we found that several other SNF1-subfamily protein kinases can also target the regulated phosphorylation sites in vitro (Figure 8). Consistent with the fourth possibility, cGMP-activated kinases (Protein Kinase G) have been shown to be activated by high levels of cAMP, similar to those measured in PKA-null cells in the present study (32, 33). Prior studies have found that AMPK can be activated by nitric oxide working through cGMP (34, 35) pointing to a role for Protein Kinase G in the regulation of AMPK. It is not clear whether activation of Protein Kinase G can cause activation of other SNF1-subfamily kinases, however.
Downstream effects of SPAK, atypical PKC, and SNF1-subfamily protein kinases in vasopressin action.
The phosphoproteomics data in this study indicate that, in the absence of PKA, vasopressin activates the protein kinase SPAK (Stk39) and one or more members of SNF1 (AMPK-related) kinase subfamily. It also decreases activation of an atypical protein kinase C (Prkci) through a decrease in active site phosphorylation (T411). SPAK has previously been implicated in the regulation of blood pressure and NaCl balance through effects in the distal nephron (36). SPAK phosphorylation was increased by vasopressin in PKA-null cells at a site in its regulatory domain (S383), a known target of the “With-No-K’ protein kinase WNK4 (37). Vasopressin activates the thiazide-sensitive Na-Cl cotransporter in association with increased SPAK phosphorylation (38) and the regulation is ablated by SPAK deletion (39), indicating that the vasopressin effect on NaCl transport is dependent on SPAK activation. Although SPAK has been best characterized in non-AQP2 expressing cells, it has been found to be strongly expressed in collecting duct principal cells (40), although a specific role in AQP2 regulation has not yet been explicitly assigned.
Activation of one or more SNF1-subfamily kinases by vasopressin in PKA-null mpkCCD cells may have implications with regard to regulation of AQP2. Prior studies implicated AMPK in the regulation of AQP2 trafficking (41, 42) and osmotic water transport (41, 43) in the renal collecting duct. Furthermore, data from a cultured collecting duct cell line, MDCK, provided strong evidence for a central role of another SNF1-subfamily kinase, viz. Mark2 (Par-1), in the regulation of collecting duct cell polarity and vesicular trafficking (44–54). PKC-iota is an ‘atypical’ protein kinase C, i.e. it is calcium- and diacylglycerol-independent, that plays an important role in creating and maintaining epithelial polarity (55).
A key finding was that, in both PKA-intact and PKA-null cells, vasopressin increased phosphorylation of AQP2 at a key residue, S256, which is believed to be critical to AQP2 trafficking to the plasma membrane (56). Thus, some non-PKA protein kinase can phosphorylate S256 of AQP2. A Bayesian analysis of AQP2 phosphorylation identified the protein kinases (among the 523 protein kinases in the mouse genome) that are most likely to phosphorylate S256, based on integration of multiple data types (57). The top ranked kinases included several SNF1-subfamily kinases including Mark2, Mark3, and Nuak2. It also included non-SNF1-subfamily kinases, Prkci and Protein Kinase G (Prkg2), but not SPAK. Evidence for a role for Protein Kinase G has previously been presented by Bouley and colleagues (58). Other studies have also proposed S256 phosphorylation by other non-PKA protein kinases including AKT (59), casein kinase (60), and protein kinase C-δ (PKCδ) (59), none of which derive support from the present study.
An additional important question is whether the activation of one or more SNF1-subfamily kinases seen in PKA-null cells also occur in PKA-intact cells. In the present study, there are two sites (S711 of Gys1 and S377 of Osbp) that responded to V2R stimulation with an increase in phosphorylation in both PKA-null and PKA-intact cells (Supplemental Table 3). Prats et al. had showed that PKA phosphorylates human muscle Gys1 on S710 (analogous to S711 in mouse) following exhausting exercise that alters its intracellular distribution (61). Mohammadi et al. reported S381 of rabbit Osbp (analogous to S377 in mouse) as a cholesterol-sensitive phosphorylation site (62). Notably, in PKA-intact cells, no changes in phosphorylation in Arhgef2 at S151, Crtc1 at S151 or Lipe at S559 was observed. In our previous study in PKA-intact cells, vasopressin exposure was shown to trigger Crtc1 translocation into the nucleus (9), although a change in S151 phosphorylation was not tested. Overall, majority of the putative SNF1-subfamily kinase targets that increased in PKA-null cells due to V2R stimulation, remained unchanged in PKA-intact cells. We, therefore, speculate that the activation of SNF1-subfamily kinases may be dependent on the very high cAMP production rate in the PKA-null cells and may not be a general feature of PKA signaling.
Data sharing.
Although this paper focuses on signaling processes related to the action of vasopressin to regulate water transport in the kidney, the experimental system that we used in this study can be viewed as a model of Gsα-coupled GPCR signaling in general. To facilitate the use of the data in the future, we have provided the curated datasets in the form of two publically accessible databases (PKA-null and PKA-intact). In general, our own analysis only ‘scratched the surface’ regarding the bioinformatic analysis of these data and hope that the provision of these publicly accessible data resources will facilitate its utilization.
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by the Division of Intramural Research, National Heart, Lung and Blood Institute (Projects ZIA-HL-001285 and ZIA-HL-006129, to M. A. Knepper). We thank Angel Aponte, Guanghui Wang and Marjan Gucek of the NHLBI Proteomics Core Facility for mass spectrometry assistance. We thank Kiyoshi Isobe, Hyun Jun Jung, Lihe Chen, and Anika Kao for helpful discussions.
ABBREVIATIONS
- PKA
Protein Kinase A
- GPCR
G-protein Coupled Receptor
- V2R
Vasopressin V2 Receptor
- AQP2
Aquaporin-2
- dDAVP
D-amino D-arginine Vasopressin
- SILAC
Stable-Isotopic Labeling with Amino Acids in Cell Culture
- PRM
Parallel Reaction Monitoring
- HCD
Higher-energy Collisional Dissociation
- EThcD
Electron-Transfer/Higher-energy Collision Dissociation
- FA
Formic Acid
- nLC-MS/MS
Nano Liquid Chromatography-Tandem Mass Spectrometry
- IBMX
3-isobutyl-1-methylxanthine
Footnotes
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
REFERENCES
- 1.Knepper MA, Kwon T-H, and Nielsen S (2015) Molecular physiology of water balance. N Engl J Med 372, 1349–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nielsen S, Frokiaer J, Marples D, Kwon TH, Agre P, and Knepper MA (2002) Aquaporins in the kidney: from molecules to medicine. Physiological reviews 82, 205–244 [DOI] [PubMed] [Google Scholar]
- 3.Nielsen S, Chou CL, Marples D, Christensen EI, Kishore BK, and Knepper MA (1995) Vasopressin increases water permeability of kidney collecting duct by inducing translocation of aquaporin-CD water channels to plasma membrane. Proceedings of the National Academy of Sciences of the United States of America 92, 1013–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown D (2003) The ins and outs of aquaporin-2 trafficking. American journal of physiology. Renal physiology 284, F893–901 [DOI] [PubMed] [Google Scholar]
- 5.Matsumura Y, Uchida S, Rai T, Sasaki S, and Marumo F (1997) Transcriptional regulation of aquaporin-2 water channel gene by cAMP. Journal of the American Society of Nephrology : JASN 8, 861–867 [DOI] [PubMed] [Google Scholar]
- 6.Hasler U, Leroy V, Martin PY, and Feraille E (2009) Aquaporin-2 abundance in the renal collecting duct: new insights from cultured cell models. American journal of physiology. Renal physiology 297, F10–18 [DOI] [PubMed] [Google Scholar]
- 7.Sandoval PC, Claxton JS, Lee JW, Saeed F, Hoffert JD, and Knepper MA (2016) Systems-level analysis reveals selective regulation of Aqp2 gene expression by vasopressin. Scientific reports 6, 34863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uchida S, Sasaki S, Fushimi K, and Marumo F (1994) Isolation of human aquaporin-CD gene. The Journal of biological chemistry 269, 23451–23455 [PubMed] [Google Scholar]
- 9.Isobe K, Jung HJ, Yang CR, Claxton J, Sandoval P, Burg MB, Raghuram V, and Knepper MA (2017) Systems-level identification of PKA-dependent signaling in epithelial cells. Proc. Natl. Acad. Sci. U. S. A 114, E8875–E8884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loo CS, Chen CW, Wang PJ, Chen PY, Lin SY, Khoo KH, Fenton RA, Knepper MA, and Yu MJ (2013) Quantitative apical membrane proteomics reveals vasopressin-induced actin dynamics in collecting duct cells. Proceedings of the National Academy of Sciences of the United States of America 110, 17119–17124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schenk LK, Bolger SJ, Luginbuhl K, Gonzales PA, Rinschen MM, Yu MJ, Hoffert JD, Pisitkun T, and Knepper MA (2012) Quantitative proteomics identifies vasopressin-responsive nuclear proteins in collecting duct cells. Journal of the American Society of Nephrology : JASN 23, 1008–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saethang T, Hodge K, Yang CR, Zhao Y, Kimkong I, Knepper MA, and Pisitkun T (2019) PTM-Logo: a program for generation of sequence logos based on position-specific background amino-acid probabilities. Bioinformatics (Oxford, England) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xue Y, Ren J, Gao X, Jin C, Wen L, and Yao X (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Molecular & cellular proteomics : MCP 7, 1598–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics (Oxford, England) 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dephoure N, Gould KL, Gygi SP, and Kellogg DR (2013) Mapping and analysis of phosphorylation sites: a quick guide for cell biologists. Molecular biology of the cell 24, 535–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu MJ, Miller RL, Uawithya P, Rinschen MM, Khositseth S, Braucht DW, Chou CL, Pisitkun T, Nelson RD, and Knepper MA (2009) Systems-level analysis of cell-specific AQP2 gene expression in renal collecting duct. Proceedings of the National Academy of Sciences of the United States of America 106, 2441–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishimoto G, Zelenina M, Li D, Yasui M, Aperia A, Nielsen S, and Nairn AC (1999) Arginine vasopressin stimulates phosphorylation of aquaporin-2 in rat renal tissue. The American journal of physiology 276, F254–259 [DOI] [PubMed] [Google Scholar]
- 18.Hardie DG, Schaffer BE, and Brunet A (2016) AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends in cell biology 26, 190–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sandi MJ, Marshall CB, Balan M, Coyaud E, Zhou M, Monson DM, Ishiyama N, Chandrakumar AA, La Rose J, Couzens AL, Gingras AC, Raught B, Xu W, Ikura M, Morrison DK, and Rottapel R (2017) MARK3-mediated phosphorylation of ARHGEF2 couples microtubules to the actin cytoskeleton to establish cell polarity. Science signaling 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altarejos JY, and Montminy M (2011) CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nature reviews. Molecular cell biology 12, 141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donsmark M, Langfort J, Holm C, Ploug T, and Galbo H (2004) Contractions induce phosphorylation of the AMPK site Ser565 in hormone-sensitive lipase in muscle. Biochemical and biophysical research communications 316, 867–871 [DOI] [PubMed] [Google Scholar]
- 22.Olesen ET, Moeller HB, Assentoft M, MacAulay N, and Fenton RA (2016) The vasopressin type 2 receptor and prostaglandin receptors EP2 and EP4 can increase aquaporin-2 plasma membrane targeting through a cAMP-independent pathway. American journal of physiology. Renal physiology 311, F935–f944 [DOI] [PubMed] [Google Scholar]
- 23.Nickols HH, Shah VN, Chazin WJ, and Limbird LE (2004) Calmodulin interacts with the V2 vasopressin receptor: elimination of binding to the C terminus also eliminates arginine vasopressin-stimulated elevation of intracellular calcium. The Journal of biological chemistry 279, 46969–46980 [DOI] [PubMed] [Google Scholar]
- 24.Naruse M (1992) Arginine vasopressin increases intracellular calcium ion concentration in isolated mouse collecting tubule cells: distinct mechanism of action through V2 receptor, but independent of adenylate cyclase activation. Nihon Jinzo Gakkai shi 34, 337–347 [PubMed] [Google Scholar]
- 25.Star RA, Nonoguchi H, Balaban R, and Knepper MA (1988) Calcium and cyclic adenosine monophosphate as second messengers for vasopressin in the rat inner medullary collecting duct. The Journal of clinical investigation 81, 1879–1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Champigneulle A, Siga E, Vassent G, and Imbert-Teboul M (1993) V2-like vasopressin receptor mobilizes intracellular Ca2+ in rat medullary collecting tubules. The American journal of physiology 265, F35–45 [DOI] [PubMed] [Google Scholar]
- 27.Chou CL, Yip KP, Michea L, Kador K, Ferraris JD, Wade JB, and Knepper MA (2000) Regulation of aquaporin-2 trafficking by vasopressin in the renal collecting duct. Roles of ryanodine-sensitive Ca2+ stores and calmodulin. The Journal of biological chemistry 275, 36839–36846 [DOI] [PubMed] [Google Scholar]
- 28.Yip KP (2006) Epac-mediated Ca(2+) mobilization and exocytosis in inner medullary collecting duct. American journal of physiology. Renal physiology 291, F882–890 [DOI] [PubMed] [Google Scholar]
- 29.Kortenoeven ML, Trimpert C, van den Brand M, Li Y, Wetzels JF, and Deen PM (2012) In mpkCCD cells, long-term regulation of aquaporin-2 by vasopressin occurs independent of protein kinase A and CREB but may involve Epac. American journal of physiology. Renal physiology 302, F1395–1401 [DOI] [PubMed] [Google Scholar]
- 30.Zwartkruis FJ, and Bos JL (1999) Ras and Rap1: two highly related small GTPases with distinct function. Experimental cell research 253, 157–165 [DOI] [PubMed] [Google Scholar]
- 31.Fu D, Wakabayashi Y, Lippincott-Schwartz J, and Arias IM (2011) Bile acid stimulates hepatocyte polarization through a cAMP-Epac-MEK-LKB1-AMPK pathway. Proceedings of the National Academy of Sciences of the United States of America 108, 1403–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lorenz R, Bertinetti D, and Herberg FW (2017) cAMP-Dependent Protein Kinase and cGMP-Dependent Protein Kinase as Cyclic Nucleotide Effectors. Handbook of experimental pharmacology 238, 105–122 [DOI] [PubMed] [Google Scholar]
- 33.Kim JJ, Casteel DE, Huang G, Kwon TH, Ren RK, Zwart P, Headd JJ, Brown NG, Chow DC, Palzkill T, and Kim C (2011) Co-crystal structures of PKG Ibeta (92–227) with cGMP and cAMP reveal the molecular details of cyclic-nucleotide binding. PloS one 6, e18413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deshmukh AS, Long YC, de Castro Barbosa T, Karlsson HK, Glund S, Zavadoski WJ, Gibbs EM, Koistinen HA, Wallberg-Henriksson H, and Zierath JR (2010) Nitric oxide increases cyclic GMP levels, AMP-activated protein kinase (AMPK)alpha1-specific activity and glucose transport in human skeletal muscle. Diabetologia 53, 1142–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy BA, Fakira KA, Song Z, Beuve A, and Routh VH (2009) AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. American journal of physiology. Cell physiology 297, C750–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richardson C, and Alessi DR (2008) The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. Journal of cell science 121, 3293–3304 [DOI] [PubMed] [Google Scholar]
- 37.Gagnon KB, and Delpire E (2010) On the substrate recognition and negative regulation of SPAK, a kinase modulating Na+-K+−2Cl− cotransport activity. American journal of physiology. Cell physiology 299, C614–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pedersen NB, Hofmeister MV, Rosenbaek LL, Nielsen J, and Fenton RA (2010) Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney international 78, 160–169 [DOI] [PubMed] [Google Scholar]
- 39.Saritas T, Borschewski A, McCormick JA, Paliege A, Dathe C, Uchida S, Terker A, Himmerkus N, Bleich M, Demaretz S, Laghmani K, Delpire E, Ellison DH, Bachmann S, and Mutig K (2013) SPAK differentially mediates vasopressin effects on sodium cotransporters. Journal of the American Society of Nephrology : JASN 24, 407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L, Lee JW, Chou CL, Nair AV, Battistone MA, Paunescu TG, Merkulova M, Breton S, Verlander JW, Wall SM, Brown D, Burg MB, and Knepper MA (2017) Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proceedings of the National Academy of Sciences of the United States of America 114, E9989–e9998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klein JD, Wang Y, Blount MA, Molina PA, LaRocque LM, Ruiz JA, and Sands JM (2016) Metformin, an AMPK activator, stimulates the phosphorylation of aquaporin 2 and urea transporter A1 in inner medullary collecting ducts. American journal of physiology. Renal physiology 310, F1008–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Bataineh MM, Li H, Ohmi K, Gong F, Marciszyn AL, Naveed S, Zhu X, Neumann D, Wu Q, Cheng L, Fenton RA, Pastor-Soler NM, and Hallows KR (2016) Activation of the metabolic sensor AMP-activated protein kinase inhibits aquaporin-2 function in kidney principal cells. American journal of physiology. Renal physiology 311, F890–f900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liwang JK, Ruiz JA, LaRocque LM, Rianto F, Ma F, and Wang Y (2019) Role of PKC and AMPK in hypertonicity-stimulated water reabsorption in rat inner medullary collecting ducts. American journal of physiology. Renal physiology 316, F253–f262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cohen D, and Musch A (2003) Apical surface formation in MDCK cells: regulation by the serine/threonine kinase EMK1. Methods (San Diego, Calif.) 30, 269–276 [DOI] [PubMed] [Google Scholar]
- 45.Cohen D, Brennwald PJ, Rodriguez-Boulan E, and Musch A (2004) Mammalian PAR-1 determines epithelial lumen polarity by organizing the microtubule cytoskeleton. The Journal of cell biology 164, 717–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki A, Hirata M, Kamimura K, Maniwa R, Yamanaka T, Mizuno K, Kishikawa M, Hirose H, Amano Y, Izumi N, Miwa Y, and Ohno S (2004) aPKC acts upstream of PAR-1b in both the establishment and maintenance of mammalian epithelial polarity. Current biology : CB 14, 1425–1435 [DOI] [PubMed] [Google Scholar]
- 47.Elbert M, Cohen D, and Musch A (2006) PAR1b promotes cell-cell adhesion and inhibits dishevelled-mediated transformation of Madin-Darby canine kidney cells. Molecular biology of the cell 17, 3345–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ducharme NA, Hales CM, Lapierre LA, Ham AJ, Oztan A, Apodaca G, and Goldenring JR (2006) MARK2/EMK1/Par-1Balpha phosphorylation of Rab11-family interacting protein 2 is necessary for the timely establishment of polarity in Madin-Darby canine kidney cells. Molecular biology of the cell 17, 3625–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cohen D, Tian Y, and Musch A (2007) Par1b promotes hepatic-type lumen polarity in Madin Darby canine kidney cells via myosin II- and E-cadherin-dependent signaling. Molecular biology of the cell 18, 2203–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zeaiter Z, Cohen D, Musch A, Bagnoli F, Covacci A, and Stein M (2008) Analysis of detergent-resistant membranes of Helicobacter pylori infected gastric adenocarcinoma cells reveals a role for MARK2/Par1b in CagA-mediated disruption of cellular polarity. Cellular microbiology 10, 781–794 [DOI] [PubMed] [Google Scholar]
- 51.Lapierre LA, Avant KM, Caldwell CM, Oztan A, Apodaca G, Knowles BC, Roland JT, Ducharme NA, and Goldenring JR (2012) Phosphorylation of Rab11-FIP2 regulates polarity in MDCK cells. Molecular biology of the cell 23, 2302–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lazaro-Dieguez F, Cohen D, Fernandez D, Hodgson L, van Ijzendoorn SC, and Musch A (2013) Par1b links lumen polarity with LGN-NuMA positioning for distinct epithelial cell division phenotypes. The Journal of cell biology 203, 251–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lapierre LA, Manning EH, Mitchell KM, Caldwell CM, and Goldenring JR (2017) Interaction of phosphorylated Rab11-FIP2 with Eps15 regulates apical junction composition. Molecular biology of the cell 28, 1088–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McRae R, Lapierre LA, Manning EH, and Goldenring JR (2017) Rab11-FIP1 phosphorylation by MARK2 regulates polarity in MDCK cells. Cellular logistics 7, e1271498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ohno S (2001) Intercellular junctions and cellular polarity: the PAR-aPKC complex, a conserved core cassette playing fundamental roles in cell polarity. Current opinion in cell biology 13, 641–648 [DOI] [PubMed] [Google Scholar]
- 56.Katsura T, Gustafson CE, Ausiello DA, and Brown D (1997) Protein kinase A phosphorylation is involved in regulated exocytosis of aquaporin-2 in transfected LLC-PK1 cells. The American journal of physiology 272, F817–822 [PubMed] [Google Scholar]
- 57.Bradford D, Raghuram V, Wilson JL, Chou CL, Hoffert JD, Knepper MA, and Pisitkun T (2014) Use of LC-MS/MS and Bayes’ theorem to identify protein kinases that phosphorylate aquaporin-2 at Ser256. American journal of physiology. Cell physiology 307, C123–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bouley R, Breton S, Sun T, McLaughlin M, Nsumu NN, Lin HY, Ausiello DA, and Brown D (2000) Nitric oxide and atrial natriuretic factor stimulate cGMP-dependent membrane insertion of aquaporin 2 in renal epithelial cells. The Journal of clinical investigation 106, 1115–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Douglass J, Gunaratne R, Bradford D, Saeed F, Hoffert JD, Steinbach PJ, Knepper MA, and Pisitkun T (2012) Identifying protein kinase target preferences using mass spectrometry. American journal of physiology. Cell physiology 303, C715–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Procino G, Carmosino M, Marin O, Brunati AM, Contri A, Pinna LA, Mannucci R, Nielsen S, Kwon TH, Svelto M, and Valenti G (2003) Ser-256 phosphorylation dynamics of Aquaporin 2 during maturation from the ER to the vesicular compartment in renal cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 17, 1886–1888 [DOI] [PubMed] [Google Scholar]
- 61.Prats C, Helge JW, Nordby P, Qvortrup K, Ploug T, Dela F, and Wojtaszewski JF (2009) Dual regulation of muscle glycogen synthase during exercise by activation and compartmentalization. The Journal of biological chemistry 284, 15692–15700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mohammadi A, Perry RJ, Storey MK, Cook HW, Byers DM, and Ridgway ND (2001) Golgi localization and phosphorylation of oxysterol binding protein in Niemann-Pick C and U18666A-treated cells. Journal of lipid research 42, 1062–1071 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometer generated raw (.raw) and analysis (.msf) files for SILAC experiment have been uploaded to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD015719 (Username: reviewer89424@ebi.ac.uk; Password: HKPizc3T). These data are accessible at www.ebi.ac.uk/pride/archive/. The curated files have been made available through publicly available webpages: (1) PKA-null dataset: https://hpcwebapps.cit.nih.gov/ESBL/Database/PKA-null/index.html; (2) PKA-intact dataset: https://hpcwebapps.cit.nih.gov/ESBL/Database/PKA-Intact/index.html.