

Abstract
Background:
Depression is associated with higher risk for Alzheimer’s disease (AD) in several prospective studies; however, mechanisms underlying this association remain unclear.
Methods:
We examined genetic correlation between depression and AD using LDSC regression. We then tested for evidence of causality between depression and AD using Mendelian randomization and genome-wide association study (GWAS) results. Subsequently, cis and trans quantitative trait locus (QTL) analyses for the depression-GWAS signals were performed to resolve the genetic signals to specific DNA-methylation sites, brain transcripts, and proteins. These transcripts and proteins were then examined for associations with AD and its endophenotypes. Lastly, associations between depression polygenic risk score (PRS) and AD endophenotypes were examined.
Results:
We detected a significant genetic correlation between depression and AD suggesting that they have a shared genetic basis. Furthermore, we found that depression has a causal role in AD through Mendelian randomization but did not find evidence for a causal role of AD on depression. Moreover, we identified 75 brain transcripts and 28 brain proteins regulated by the depression GWAS signals through QTL analyses. Among these, 46 transcripts and 7 proteins were associated with rates of cognitive decline over time, AD pathologies, and AD diagnosis in two separate cohorts, implicating them in AD. Additionally, we found that higher depression PRS was associated with faster decline of episodic memory over time.
Conclusions:
Depression appears to have a causal role in AD, and this causal relationship is likely driven, in part, by the 53 brain transcripts and proteins identified in this study.
Keywords: Depression, Alzheimer’s disease, genetic correlation, Mendelian randomization, brain protein, quantitative trait locus
Depression and Alzheimer’s dementia (AD) are commonly comorbid in individuals over age 65 (1–3). Depression at any age (early-, mid-, or late-life) has been found to be associated with an increased risk for AD in large prospective epidemiological studies (4–7) and is arguably a modifiable target for the prevention of cognitive decline and dementia (8).
The genetic link between depression and AD has been explored using candidate genes (9–11) or genome-wide association study (GWAS) results (12–14) with mixed to negative results. Those early studies were likely limited by power since the latest published AD GWAS found a nominal genetic correlation between AD and depressive symptoms (15). The two most salient differences between the earlier and recent studies are the availability of full GWAS summary statistics and larger sample sizes (14). Thus, with these new data, we undertook the present study to identify shared molecular changes between these two illnesses using functional genomic approaches that capitalize on newly available brain transcriptomic and proteomic data. Here, we performed LDSC regression, Mendelian randomization, cis and trans QTL analyses using brain DNA methylation, transcriptomic, and proteomic data to identify brain transcripts and proteins that link depression to AD in order to elucidate the molecular basis behind the association between depression and elevated AD risk.
Methods
GWAS summary statistics: We used results from the GWAS of depression among 807,553 individuals (16) and GWAS of AD among 455,258 individuals (15), all of European ancestry, in this study (Table S1A). SNP-based estimates of heritability were similar to those reported in the primary papers (depression: hSNP = 0.080 ± 0.003; AD hSNP = 0.012 ± 0.002). For sensitivity analyses, we used results of two additional AD GWAS (17, 18) (Table S1A).
Religious Orders Study (ROS) and Rush Memory and Aging Project (MAP) are two prospective clinical-pathologic cohorts of aging and dementia (19, 20). All ROS/MAP participants are cognitively normal at enrollment, undergo annual clinical evaluations, and agree to brain donation. Both studies were approved by an Institutional Review Board of Rush University Medical Center. We used the DNA methylation, transcriptomes and proteomes profiled from the dorsolateral prefrontal cortex (dPFC) of post-mortem brain samples donated by these participants (Table S1B). Details of the clinical and pathologic assessments performed in ROS/MAP are provided in supplementary methods.
Banner Sun Health Research Institute (Banner) is a longitudinal study of healthy aging, Alzheimer’s and Parkinson’s disease (21). Participants were enrolled as cognitively healthy volunteers and underwent standardized medical, neurological, and neuropsychological assessments during life (21) (Table S1B). We used brain proteomes profiled from the dPFC donated by Banner participants (Table S1B).
Proteomic quantification and quality control: Proteomic profiles of 400 ROS/MAP and 200 Banner participants were generated using tandem mass tag isobaric labelling and mass spectrometry as described previously (22). Using previously described procedures (22), we performed quality control as followed: (i) excluding proteins with missing values in more than 50% of the subjects, (ii) dividing each protein abundance by a sample-specific total protein abundance to remove effects of protein loading differences, (iii) performing log2 transformation of protein abundance, and iv) detecting and removing outlier samples. We then removed effects of batch, post-mortem interval, age, and study from the proteomic profiles using regression. After quality control, 8,356 proteins in 391 ROS/MAP subjects and 7,854 proteins in 196 Banner subjects remained for analysis (Table S1B).
Transcriptomic quantification and quality control: Transcriptomes were profiled from the dPFC of 638 ROS/MAP participants as described previously (23). Reads were aligned using STAR (24). We removed genes with <1 count per million in at least 50% of the samples and with missing gene length and percent GC content. Outlier samples were removed. Effects of batch, RIN, post-mortem interval, and age were regressed from the transcriptomic profiles. After quality control, 15,822 transcripts in 630 individuals remained (Table S1B).
DNA methylation was profiled from the dPFC of 737 ROS/MAP participants using the Illumina HumanMethylation450 Beadchip array as described before (23). Color channel normalization and background removal were performed using the Illumina GenomeStudio. Quality control and normalization were performed using previously described procedures (25). Briefly, we removed probes (i) with detection p-value > 0.01 in any sample, (ii) annotated to the X or Y chromosome, (iii) cross-hybridized with other probes, (iv) non-CpG site probes, and (v) overlapped with common SNPs. The remaining CpG sites were then normalized using BMIQ in Watermelon R package (26) and ComBat in the SVA package (27). After quality control, there were 340,516 CpGs in 664 individuals for analysis (Table S1B).
Genotyping of ROS/MAP participants were profiled using either the Illumina OmniQuad Express, Affymetrix GeneChip 6.0, or whole genome sequencing (WGS) (23, 28). WGS was prioritized when multiple data sources were available. Banner participants were genotyped using the Affymetrix Precision Medicine Array (29). Quality control of WGS and each array-based genotyping source was performed separately using Plink (30). We removed individuals with genotyping missing rate >5%, variants with Hardy Weinberg equilibrium p-value < 10−5, variants with missing genotype rate >5%, variants with minor allele frequency <1%, and variants that are not single nucleotide polymorphisms (SNPs). KING was used to remove individuals estimated to be closer than second-degree kinship (31). Genotyping was imputed to the 1000 Genome Project Phase 3 (32) using the Michigan Imputation Server (33) and SNPs with imputation R2 > 0.3 were retained for analysis. After quality control, 580 ROS/MAP subjects had complete genetic and transcriptomic data, 372 ROS/MAP and 97 Banner subjects had complete genetic and proteomic data, and 664 ROS/ MAP subjects had complete genetic and methylation data to be included in the analysis (Table S1B).
Statistical analysis
Linkage disequilibrium score (LDSC) regression (34) was performed to estimate the genetic correlation (rg) between depression and AD using their GWAS summary statistics. Pre-estimated LD scores were obtained from the 1000 Genomes European reference population and the genetic correlation was calculated using HapMap3 SNPs only (LD reference panel SNPs) to minimize potential bias by differences in LD structure (34). Additionally, following the LDSC framework, we removed SNPs with extremely large effect sizes (X2 > 80) since these can unduly influence LDSC regression (34).
Mendelian randomization (MR): Bidirectional MR was conducted to investigate the causal relationship between depression and AD using Generalized Summary data-based Mendelian Randomization (GSMR) approach (35). To meet the assumptions of MR, we used clumping with parameter of r2 <0.05, removed SNPs with large allele-frequency differences from the 1000 Genomes reference samples, and only considered SNPs with the strongest effect on the exposures (P-value < 5 × 10−8) as the instrument variables in the forward and reverse models, respectively. We used the HEIDI-outlier approach (35) to remove SNPs that have pleiotropic effects on both the exposure and the outcome. To address potential horizontal pleiotropy, we used the MR Pleiotropy Residual Sum and Outlier (MR-PRESSO) (36) to detect and correct for potential horizonal pleiotropy. Both MR methods detected the same outlier SNPs, and the retained SNPs were then tested for the association with the outcome for causal effect. Here, we conducted the forward GSMR analysis on the 115 depression SNPs as the instrument variables and the reverse GSMR analysis on 61 AD SNPs as the instrument variables.
Surrogate variable analysis (SVA) (37) was performed to identify hidden confounders in the expression data using the SVA package (27). For both transcriptomic and proteomic analyses, we included the first ten significant surrogate variables as covariates where relevant.
Quantitative trait locus (QTL) analyses were performed to identify associations between genetic variants and DNA methylation (mQTL), transcript (eQTL), and protein level (pQTL), respectively. To that end, the transcript and protein levels were regressed against the genotype for each SNP, assuming additive genetic effects and including depressive symptoms, AD status, ten genetic principal components, and ten surrogate variables as covariates (38). We used the Benjamini-Hochberg approach to control false discovery rate (FDR) and set a threshold of 5% to declare a QTL statistically significant. QTLs were categorized as proximal if the SNP is located within 1Mb of a gene’s transcription start site (TSS). Those outside that window were defined as distal. All statistical analyses were performed in R v.4.0.2.
Depression-associated transcripts/proteins versus AD features: We examined associations between transcripts and proteins regulated by the depression-SNPs and AD diagnosis and its endophenotypes. These AD characteristics include (i) rates of change of cognitive performance over time; (ii) beta-amyloid; (iii) neurofibrillary tangles; and (iv) clinical diagnosis of AD. To this end, we used linear regression in which a specific AD feature was the outcome, transcript/protein was the predictor, and age at death, sex, and depressive symptoms were the covariates.
Meta-analysis was performed with METAL (39) to combine results from the discovery (ROS/MAP) and replication (Banner) cohorts using test statistics and standard errors.
Polygenic risk score (PRS) for depression was estimated for ROS/MAP participants using results from the depression GWAS (16) following the PRSice-2 approach (40, 41). Subsequently, associations between the depression PRS and AD features were examined, adjusting for sex, 10 genetic principal components, and genotype platform using regression modeling.
Results
Shared genetic risk between depression and AD
To investigate whether depression and AD have shared genetic risk, we performed LDSC regression and found a significant positive genetic correlation (rg = 0.17, p = 5.54×10−5), suggesting that they have a shared genetic basis. Since the Jansen et al AD GWAS we used (15) included both AD-by-diagnosis and AD-by-proxy, we conducted a sensitivity analysis to examine whether this shared genetic basis was driven by AD-by-proxy cases. To that end, we performed LDSC regression using the same depression GWAS and an AD GWAS that only included AD-by-diagnosis cases (17) and an AD GWAS that only included AD-by-proxy cases (18) (Table S1A). Notably, the number of AD-by-proxy cases was relatively similar between the Jansen GWAS (n=47,793) and the AD-by-proxy-cases-only GWAS (18) (n=42,034; Table S1A). We found no significant genetic correlation for the AD-by-diagnosis GWAS (rg = − 0.02, p = 0.73), likely due to a smaller sample size and thus lower power. We found a nominal and weaker correlation for the AD-by-proxy GWAS (rg = 0.12, p = 0.02). Together, these findings suggest that the genetic correlation between depression and AD was not substantially driven by the AD-by-proxy cases.
Evidence for a causal genetic effect of depression on AD
Genetic correlation may arise from pleiotropy (i.e., genes independently affecting both depression and AD) or from the causal effect of depression on AD or vice versa. To identify a potential causal effect of depression on AD, we performed Mendelian randomization using GSMR (42). We used the 115 SNPs found to be associated with depression at genome-wide significant level from the depression GWAS as the instruments, depression as the exposure, and AD as the outcome. We found a significant causal effect of depression on AD (effect size β = 0.029, PGSMR = 0.001, Figure 1A, B; Table S2). Next, to test for the probability of a causal effect of AD on depression, we used the 61 AD GWAS-significant SNPs as the instruments, AD as the exposure, and depression as the outcome. We did not find a causal effect of AD on depression (β = −0.001, PGSMR = 0.954; Figure 1C; Table S2).
Figure 1. Generalized summary data-based Mendelian randomization (GSMR) analysis to test for effect of depression on AD and vice versa.
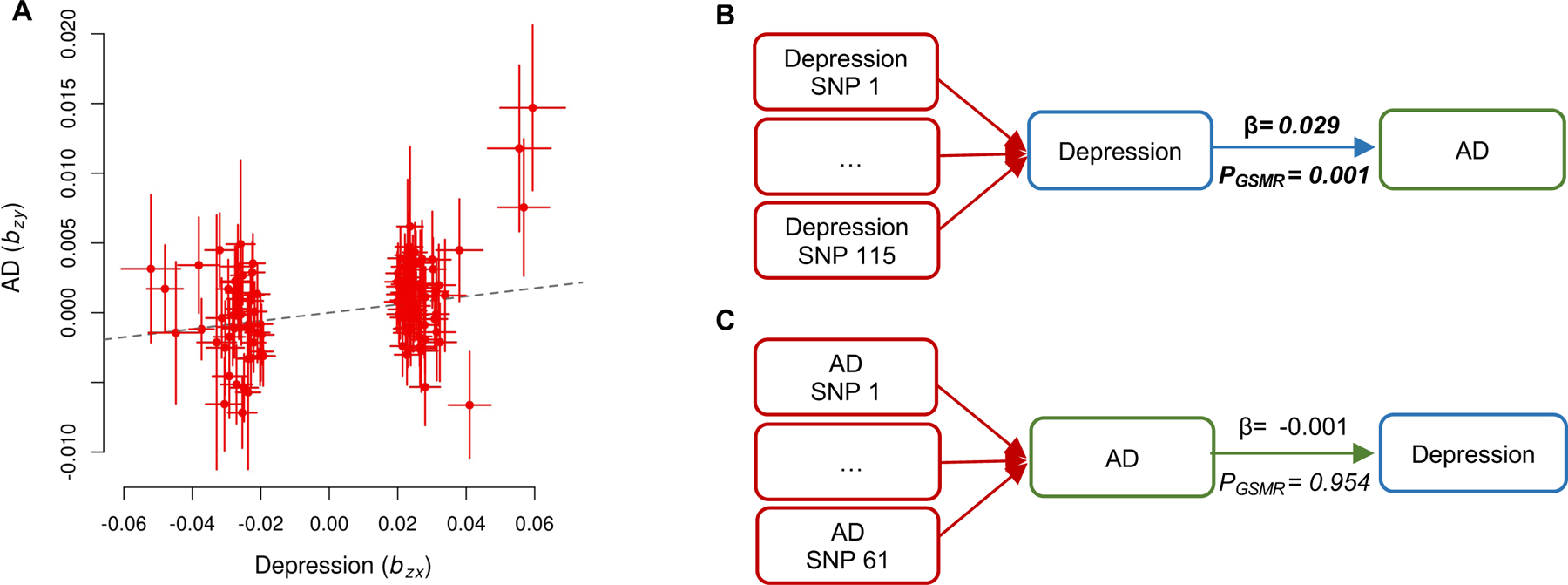
(A) GSMR plot of effect sizes of all the genetic instruments from GWAS for depression against those for AD with HEIDI-outlier filtering. (B) GSMR results of depression liability on AD. The forward GSMR analysis included 115 SNPs associated with depression at a genome-wide significance level (i.e., p < 5e-8). (C) GSMR results of AD liability on depression. The reverse GSMR analysis on 61 AD SNPs (at p < 5e-8) as the instrumental variables. Bonferroni-corrected significance threshold for 2 tests: p < 0.05/2. Bold represents a significant p-value. Abbreviations: AD: Alzheimer’s disease; SNP: number of single nucleotide polymorphisms included in each GSMR analysis; β: effect size; pGSMR: p-value for the causal estimates.
To address potential horizontal pleiotropy, we conducted a sensitivity analysis using MR-PRESSO (36). We found no evidence of pleiotropy based on the MR-PRESSO Global Test (P = 0.38). The MR-PRESSO also detected a significant causal effect of depression on AD (β = 0.029, P MR-PRESSO = 0.002; Table S2).
Depression-associated variants are also brain eQTLs and pQTLs
To investigate how the 115 depression-associated SNPs underlie the potential causal effect of depression on AD, we performed brain eQTL and pQTL analyses for these SNPs. We identified 31 depression-SNPs associated with 80 brain transcripts at FDR <0.05 (Figure 2A; Table S3). Among the 80 SNP-transcript pairs, 67% of the SNPs (21 of 31 SNPs) had a proximal effect on 33 corresponding transcripts, and 35% (11 of 31 SNPs) had a distal effect on 42 transcripts (Figure 2A; Table S3). Overall, we found that 31 of the 115 depression-associated SNPs regulate expression of 75 unique brain transcripts (Figure 2A; Table S3).
Figure 2. Identified brain proximal- and distal-QTLs from the 115 depression-associated SNPs.
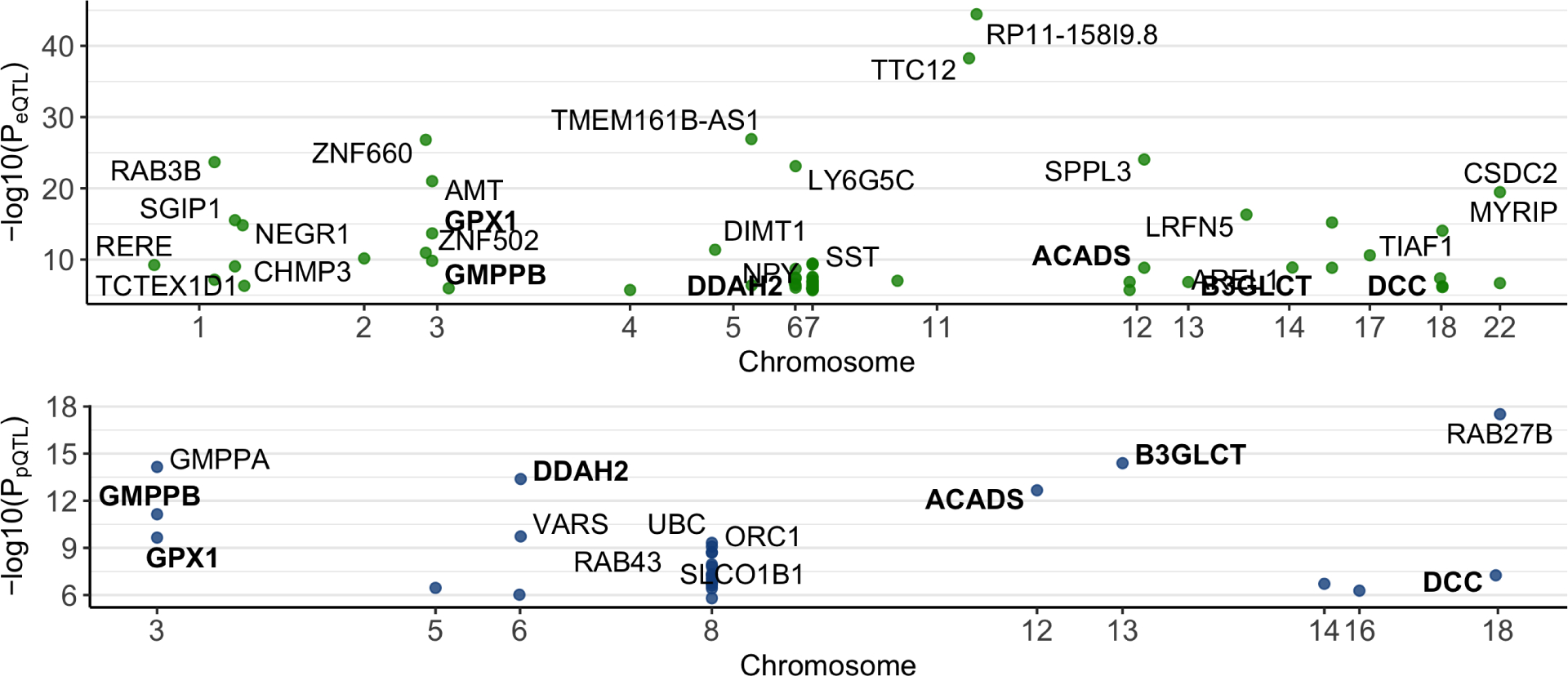
(A) 80 eQTLs were identified (green dots). Each green point represents an eQTL, which is labeled with a gene symbol of the transcript associated with it. (B) 32 pQTLs were found (blue dots). Each blue point represents a pQTL, which is labeled with a gene symbol of the protein associated with it. Bold represents sites regulating the expression of both transcript and protein of the same gene. More details are in Supplementary Tables 3–4
At the protein level, we found that 32 depression-SNPs were pQTLs for 28 brain proteins at FDR <0.05 (Figure 2B; Table S4). Among the 32 SNP-protein pairs, 69% of the SNPs (9 of 13 unique SNPs) had a proximal effect on 9 corresponding proteins, and 46% (6 of 13 SNPs) had a distal effect on 19 proteins (Figure 2B; Table S4). Furthermore, we found that 8 depression-SNPs were both eQTLs and pQTLs, and 6 of them were for the same genes (Figure 2B, bolded genes). In sum, we found that 13 of the 115 depression-SNPs regulate expression of 28 brain proteins (Figure 2B; Table S4).
Brain transcripts regulated by depression-SNPs are associated with AD features
Since we observed a causal effect of depression on AD at the genetic level, we examined whether the 75 brain transcripts regulated by the depression-SNPs are associated with AD diagnosis and endophenotypes, including trajectory of cognitive performance over time, beta-amyloid, and neurofibrillary tangles. In ROS/MAP cohorts we found that 34 brain transcripts were associated with beta-amyloid and 29 transcripts associated with tangles after adjusting for sex, age at death, and depressive symptoms (FDR p <0.05, N=587, Figure 3A, Table S5). Additionally, we found 25 transcripts associated with AD diagnosis and 30 transcripts associated with cognitive trajectory after adjusting for sex, age at death, and depressive symptoms (FDR p <0.05, N=587, Figure 3A, Table S5). Among these transcripts, higher levels of ZNF740, RERE, SAP30L, B3GLCT, and FANCE were associated with higher levels of beta-amyloid and tangles, greater probability of having AD diagnosis, and faster cognitive decline (more negative slope of cognitive trajectory, Figure 3A, Table S5). Lower levels of SST, CAP2, TRIM36, SGIP1, and AMT transcripts were associated with higher levels of beta-amyloid and tangles, greater probability of having AD diagnosis, and faster cognitive decline (Figure 3A, Table S5). In sum, we found that 46 brain transcripts (of 75 tested, or 61%) were significantly associated with at least one AD feature. These findings support the notion that the depression risk variants contribute to AD via regulating expression of their corresponding transcripts in the brain.
Figure 3. Heatmaps for associations between AD features and the brain transcripts and proteins regulated by the depression-associated SNPs.
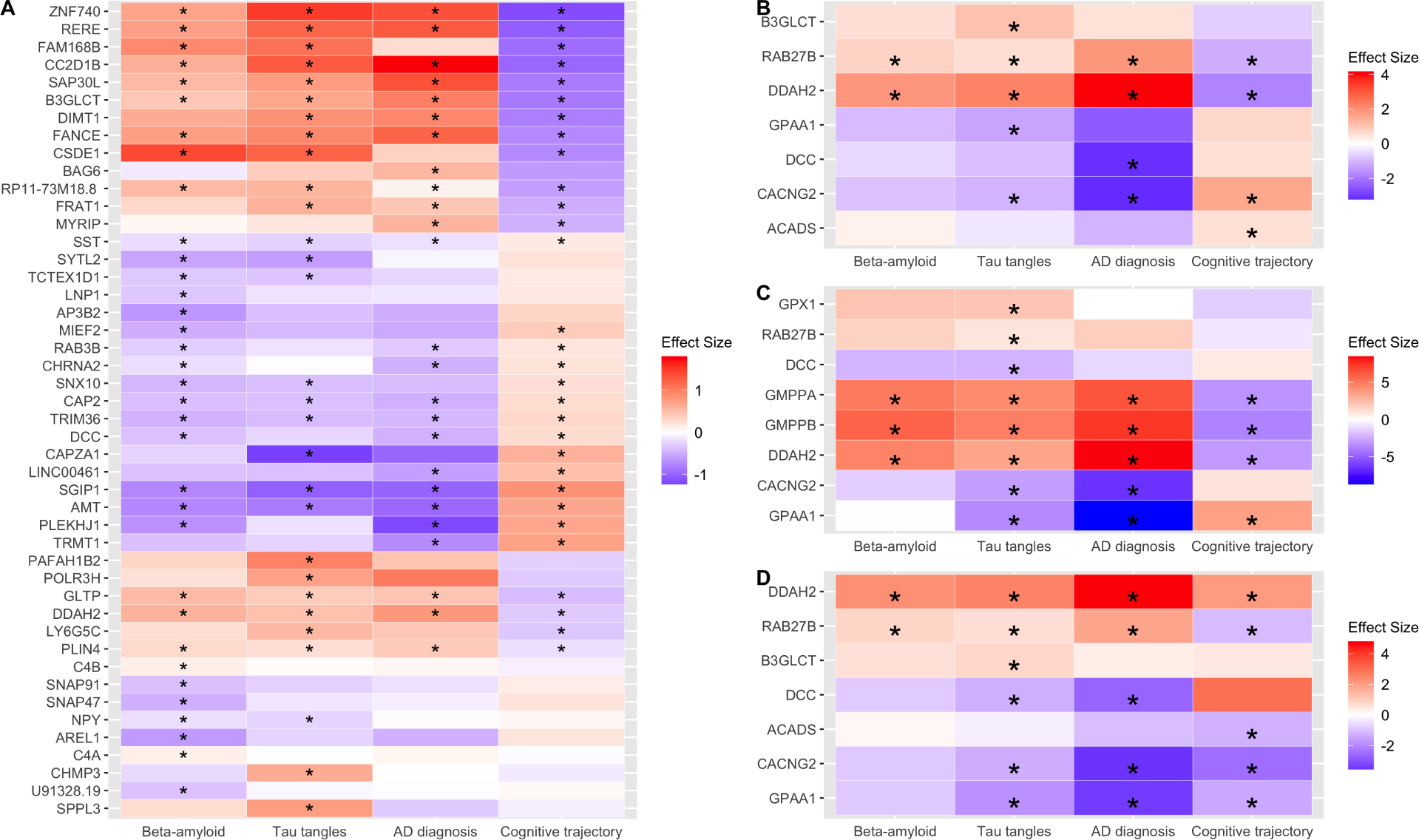
(A) For the 46 transcripts (y-axis) associated with AD-related traits (x-axis) in ROS/MAP samples. (B) For seven proteins (y-axis) in ROS/MAP samples associated with AD-related traits (x-axis). (C) For eight proteins (y-axis) in Banner samples associated with AD-related traits (x-axis). The asterisk depicts the FDR p < 0.05. (D) For seven proteins (y-axis) that replicated in both the discovery and replication analyses after a meta-analysis. The asterisk represents. The color reflects the direction of the association of expression levels with each AD-related trait.
Brain proteins regulated by depression-SNPs are associated with AD characteristics
Likewise, we examined associations between the 28 brain proteins regulated by the depression-SNPs and AD characteristics. In the ROS/MAP discovery cohorts, we identified two proteins (RAB27B and DDAH2) associated with beta-amyloid and five proteins associated with tangles after adjusting for sex, age at death, and depressive symptoms (FDR p <0.05, N=391, Figure 3B, Table S6A). Additionally, we identified four proteins associated with AD diagnosis and four proteins associated with cognitive trajectory after adjusting for the aforementioned covariates (FDR p <0.05, N=391, Figure 3B, Table S6A). Among these proteins, we found that RAB27B and DDAH2 were associated with all four AD features in consistent directions of association - higher abundance of RAB27B was associated higher levels of beta-amyloid and tangles, greater probability of having AD, and faster cognitive decline (Figure 3B; Table S6A). We also found that lower abundance of DDAH2 was associated higher levels of beta-amyloid and tangles, higher probability of having AD, and faster cognitive decline (Figure 3B; Table S6A).
In the Banner replication cohort, three proteins were associated with beta-amyloid and eight proteins were associated with tangles after adjusting for sex, age, and depressive symptoms (FDR p <0.05, N=125, Figure 3C, Table S6B). Additionally, we identified five proteins associated with AD diagnosis and four proteins associated with cognitive trajectory after adjusting for the aforementioned covariates (FDR p <0.05, N=125, Figure 3C, Table S6B). Among these proteins, we found that DDAH2, GMPPA, and GMPPB were associated with all four AD features in consistent directions of association (Figure 3C; Table S6A). Several factors may be behind the different proteins identified in the discovery and replication cohorts, including the different sample sizes and different methods of assessing AD diagnosis (considering AD pathologies or not) and pathologies (immunohistochemistry versus silver staining). Given these differences, we performed a meta-analysis of these results.
Thirteen proteins were profiled in both ROS/MAP and Banner cohorts and were included in the meta-analysis. Replication was defined as having a meta-analysis p-value smaller than those from both the discovery and replication results and having the same directions of association in both. Of the 13 proteins, two replicated in beta-amyloid, six replicated in tangles, five replicated in AD diagnosis, and five replicated in cognitive trajectory (Figure 3D, Table S6C). Two proteins (DDAH2 and RAB27B) were notable for replicating in all four AD features in consistent directions of association in both the discovery and replication cohorts (Figure 3D, Table S6C). Together, these results further support a molecular link between depression and higher risk for AD through these brain proteins.
Depression-SNPs are mQTLs for CpG sites proximal to genes underlying the association between depression and AD
Among the 115 depression-SNPs, we performed mQTL analysis and found 424 cis mQTLs after adjusting for age, depressive symptoms, cognitive diagnosis, and 10 genetic principal components (FDR p<0.05, N=664), and among these, 69 were unique mQTLs. We sought to determine how many of these 69 mQTLs are located proximal to the 46 transcripts and 7 proteins that likely underlie the contribution of depression to AD identified above. We found 17 unique mQTLs proximal to these 18 transcripts (Table S7) and 8 unique mQTLs proximal to these 4 proteins (Table S8). Together, these findings point to brain DNA methylation, transcripts, and proteins as underlying the contribution of depression to AD.
Higher depression PRS is associated with faster decline of episodic memory over time
We estimated depression PRS for ROS/MAP participants using summary statistics from the depression GWAS (16) and examined its associations with cognitive trajectory, cognitive diagnosis, amyloid, and tangles. We found a trend for the association between higher depression PRS and higher beta-amyloid after adjusting for sex and 10 genetic principal components (p = 0.07, beta = 38.5, N=1288, p-threshold = 5.0 × 10−8). We also found a trend for the association between higher depression PRS and faster decline of semantic memory over time after adjusting for similar covariates (p = 0.05, beta =−236.4, N=1817; p-threshold = 1.0 × 10−4). Notably, we found a significant association between higher depression PRS and faster decline of episodic memory over time after adjusting for sex and 10 genetic principal components (p = 0.02, beta = −271.5, N=1830; p-threshold = 1.0 × 10−4). Together, these findings further support that depression genetic variants contribute to AD.
Discussion
Prospective epidemiological studies in approximately 50,000 participants found an association between depression and higher risk for dementia (4–7). Here, we sought to elucidate the genetic and molecular basis underlying this association. We found a small but significant genetic correlation between depression and AD suggesting that they have shared genetic basis. Next, we showed that the preponderance of genetic evidence is consistent with a causal role of depression on AD but not vice versa using two-sample Mendelian randomization. Furthermore, we identified 75 brain transcripts and 28 brain proteins regulated by the depression-predisposing genetic variants. Notably, a sizable subset of these (46 transcripts and 7 proteins) were associated with either AD diagnosis, AD pathologies, or cognitive trajectory. Finally, we found that higher depression PRS was associated with faster decline of episodic memory over time. Taken together, these findings suggest that these 46 brain transcripts and 7 brain proteins likely contribute to the association between depression and higher AD risk.
There is an ever-growing body of evidence that transcript expression is not a perfect proxy for protein expression, which is the final executor of cellular processes and biological functions (43, 44). Here, we found seven proteins underlying association between depression and higher AD risk. Among these, RAB27B and DDAH2 are notable for their consistent association with all four AD features in both the ROS/MAP and Banner cohorts. RAB27B belongs to Rab GTPases protein family that regulates membrane trafficking and has been implicated in neurodegenerative diseases (45, 46). Our finding of higher RAB27B abundance being associated with AD characteristics is consistent with upregulation of RAB27B transcript and protein levels in postmortem cholinergic basal forebrain neurons of individuals with AD (47). Additionally, we note that the protein level of RAB27 was regulated by a depression-SNP while the transcript level was not, consistent with a recent study showing that certain genetic variants regulate brain proteins but not transcripts (48). DDAH2 belongs to the family of enzymes that maintain homeostatic control of nitric oxide across different tissues (49). Our finding that higher DDAH2 protein level was associated with AD characteristics is consistent with findings from another study in which DDAH2 was elevated in neurons undergoing oxidative stress in AD patients and undetectable in the neurons of age-matched healthy controls (50).
Among the 54 genes identified as contributing to the association between depression and higher AD risk, three genes (RAB27B, B3GLCT, and ACYP1) were also identified as neuroticism genes (51). Furthermore, some of these 54 genes are targets of therapeutics for neurological and non-neurological conditions (SNAP91, AP3B2, DDAH2, DPY30, PLIN4, RAB27B, and SGIP1) (52) and others have been nominated as promising targets for AD drug development (53).
Our findings, which rely on the largest and most recent GWAS of AD and depression, are consistent with prior work using similarly powered GWAS summary results (15, 16). The reasons for prior null results are likely due limited power (12, 14) and should not be viewed as inconsistent with our findings. Furthermore, our findings are also consistent with recent work showing that individuals with higher PRS for major depression had higher conversion rate from amnestic mild cognitive impairment to AD (54). Lastly, our finding are also consistent a recent study demonstrating evidence for pleiotropy between depression and AD (11).
Our findings should be interpreted in light of the study’s limitations. First, while LDSC regression is robust to sample overlap (34), there is a small possibility that the genetic correlation between depression and AD was overestimated due to overlap of samples from the UKBB in the two GWAS. We found that this is a small probability since approximately 19,200 UKBB participants reported both depression and dementia/family history of dementia, which represents only 2.4% and 4.2% of the participants of the depression GWAS and AD GWAS, respectively. Second, we analyzed 8606 proteins, which are not complete proteomic profiles, thus, deeper coverage of proteomic sequencing can advance investigation of mechanisms underlying the associations between depression and AD. Third, further mechanistic studies using model systems are needed to validate our findings. Fourth, the depression GWAS used in this study did not distinguish between early-, mid-, or late-life depression, therefore, we cannot distinguish the time window in which depression contributes to AD. Fifth, we used results from the depression GWAS that included both clinically significant depression and major depressive disorder. Likewise, we used results from the AD GWAS that included both clinically diagnosed AD and AD-by-proxy. Thus, there is a small probability that the shared heritability observed in LDSC regression or GSMR might be driven by the proxy traits of MDD and AD. Sixth, we focused on results from GWAS among participants of European ancestry due to lack of GWAS among participants of other ancestries with sufficient power; therefore, future studies should focus on participants of other ancestries. Lastly, since this is a cross-sectional study, the causal directionality between transcript and protein levels and AD-related characteristics are not conclusive.
Our study has several notable strengths. First, this is the first study to use deep human brain proteomic data to elucidate the molecular links between depression and AD to the best of our knowledge. Second, we used data from the largest and latest available GWAS. Third, we performed both proximal and distal eQTL and pQTL analyses. Fourth, we used a discovery and replication design when examining the associations between brain proteins and AD features to increase the level of confidence in our findings.
In conclusion, we demonstrated that there is a genetic basis for the association between depression and higher AD risk and identified brain transcripts and proteins that likely underlie this contribution for further mechanistic and therapeutic studies.
Supplementary Material
KEY RESOURCES TABLE.
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Add additional rows as needed for each resource type | Include species and sex when applicable. | Include name of manufacturer, company, repository, individual, or research lab. Include PMID or DOI for references; use “this paper” if new. | Include catalog numbers, stock numbers, database IDs or accession numbers, and/or RRIDs. RRIDs are highly encouraged; search for RRIDs at https://scicrunch.org/resources. | Include any additional information or notes if necessary. |
| Antibody | ||||
| Bacterial or Viral Strain | ||||
| Biological Sample | ||||
| Cell Line | ||||
| Chemical Compound or Drug | ||||
| Commercial Assay Or Kit | ||||
| Deposited Data; Public Database | https://doi.org/10.7303/syn24872746 | |||
| Genetic Reagent | ||||
| Organism/Strain | ||||
| Peptide, Recombinant Protein | ||||
| Recombinant DNA | ||||
| Sequence-Based Reagent | ||||
| Software; Algorithm | ||||
| Transfected Construct | ||||
| Other |
Acknowledgements
This work was made possible by the participants, partners, staff of the ROS, MAP, and Brain and Body Donation Program studies. We also thank the 23andMe Research Team for providing the summary statistics from the Howard et al. study. Support was provided by R01 AG056533, R01 AG053960, the Accelerating Medicine Partnership for AD (U01 046152; U01 AG046161; U01 AG061356; U01 AG061357), the Emory Alzheimer’s Disease Research Center (P50 AG025688), and the NINDS Emory Neuroscience Core (P30 NS055077), R01 AG017917, R01 AG015819, RC2 AG036547, P30 AG10161, and in part by the intramural program of the National Institute on Aging (NIA). The Brain and Body Donation Program has been supported by U24 NS072026, P30 AG19610, the Arizona Department of Health Services, the Arizona Biomedical Research Commission, and the Michael J. Fox Foundation for Parkinson’s Research. APW is also supported by NIH U01 MH115484 and VA I01 BX003853. TSW is also supported by NIH grants RF1 AG057470, U01 AG061357, P50 AG025688, R56 AG062256, and R56 AG060757. NTS is also supported by an Alzheimer’s Association, Alzheimer’s Research UK, The Michael J. Fox Foundation for Parkinson’s Research, the Weston Brain Institute Biomarkers Across Neurodegenerative Diseases Grant (11060), R01 AG061800, and R01 AG057911. The views expressed in this work do not necessarily represent the views of the Veterans Administration or the United States Government.
Footnotes
Disclosures
In the past 36 months, we have the following disclosures to make. A.I.L. received royalties that were not relevant to this manuscript.
These individuals received consulting fees: T.G.B. (Vivid Genomics from Roche Diagnosics); E.M.R. (Alzheon, Aural Analytics, Denali, Green Valley, Retromer Therapeutics, and Vaxxinity); J.A.S. (Eli Lilly, Alnylam, and National hockey League); P.A.B. (NSHAP study); D.A.B. (Takeda Inc and adjudication committee Origent Inc SBIR); J.J.L. (Roche Diagnostics); A.I.L. (Cognito Therapeutics, Biogen, GENUV, and Karuna).
D.A.B., A.I.L., E.M.R., and T.S.W. received honoraria for lectures to academic or US government organizations. J.A.S. received payment for expert testimony.
These individuals received support for attending meetings and/or travel: E.M.R. (companies while serving as a consultant), J.A.S. (NIH, Alzheimer’s Association, and foundation conferences); P.A.B. (FINRA educational and academic conferences); D.A.B. (academic and government institutions for conferences or lectures); T.S.W. (academic, Alzheimer’s Association, and PRE Alz Assoc Research Round Table conferences).
These individuals have patents planned, issued, or pending: E.M.R. (pending patent related to the role of APOE Christchurch variant in the treatment and prevention of Alzheimer’s disease); A.I.L. (pending patents for proteomics and digital biomarkers).
These individuals participated on a Data Safety Monitoring Board or Advisory Board: P.A.B. (FHS advisory board; no payment involved); D.A.B. (AbbVie inc); J.J.L. (The National Center for Complementary and Integrative Health (1R61AT009628) and Oregon Health and Science University No payments); J.A.S. (OSMB, Discovery OSMB, Framingham Study).
These individuals held a leadership or fiduciary role in any other board, society, committee, or advocacy group (paid or unpaid): T.G.B. (Vivid Genomics Scientific Advisory Board); E.M.R. (Board of Directors of the Flinn Foundation); P.A.B. (Trustee for McKnight Brain Research Foundation); A.I.L. (Clinical task force for the NIA ADRC program and the NAPA Advisory Council for the Department of HHS).
T.G.B. received personal stock options without payments from Vivid Genomics.
D.A.B. received gifts to Rush Philanthropy.
E.M.R. also reports he is a co-founder and shareholder of ALZPath, a start-up company related to the advancement of blood-based biomarkers in research, treatment development and clinical care.
All other authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Wilson RS, Barnes LL, Mendes de Leon CF, Aggarwal NT, Schneider JS, Bach J, et al. (2002): Depressive symptoms, cognitive decline, and risk of AD in older persons. Neurology. 59:364. [DOI] [PubMed] [Google Scholar]
- 2.Steffens DC, Fisher GG, Langa KM, Potter GG, Plassman BL (2009): Prevalence of depression among older Americans: the Aging, Demographics and Memory Study. Int Psychogeriatr. 21:879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saczynski JS, Beiser A, Seshadri S, Auerbach S, Wolf PA, Au R (2010): Depressive symptoms and risk of dementia: the Framingham Heart Study. Neurology. 75:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellou V, Belbasis L, Tzoulaki I, Middleton LT, Ioannidis JPA, Evangelou E (2017): Systematic evaluation of the associations between environmental risk factors and dementia: An umbrella review of systematic reviews and meta-analyses. Alzheimers Dement. 13:406–418. [DOI] [PubMed] [Google Scholar]
- 5.Diniz BS, Butters MA, Albert SM, Dew MA, Reynolds CF, 3rd (2013): Late-life depression and risk of vascular dementia and Alzheimer’s disease: systematic review and meta-analysis of community-based cohort studies. The British journal of psychiatry : the journal of mental science. 202:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mourao RJ, Mansur G, Malloy-Diniz LF, Castro Costa E, Diniz BS (2016): Depressive symptoms increase the risk of progression to dementia in subjects with mild cognitive impairment: systematic review and meta-analysis. Int J Geriatr Psychiatry. 31:905–911. [DOI] [PubMed] [Google Scholar]
- 7.Barnes DE, Alexopoulos GS, Lopez OL, Williamson JD, Yaffe K (2006): Depressive symptoms, vascular disease, and mild cognitive impairment: findings from the Cardiovascular Health Study. Arch Gen Psychiatry. 63:273–279. [DOI] [PubMed] [Google Scholar]
- 8.Dafsari FS, Jessen F (2020): Depression—an underrecognized target for prevention of dementia in Alzheimer’s disease. Translational Psychiatry. 10:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ni H, Xu M, Zhan GL, Fan Y, Zhou H, Jiang HY, et al. (2018): The GWAS Risk Genes for Depression May Be Actively Involved in Alzheimer’s Disease. J Alzheimers Dis. 64:1149–1161. [DOI] [PubMed] [Google Scholar]
- 10.Arlt S, Demiralay C, Tharun B, Geisel O, Storm N, Eichenlaub M, et al. (2013): Genetic risk factors for depression in Alzheimer`s disease patients. Curr Alzheimer Res. 10:72–81. [PubMed] [Google Scholar]
- 11.Lutz MW, Sprague D, Barrera J, Chiba-Falek O (2020): Shared genetic etiology underlying Alzheimer’s disease and major depressive disorder. Translational Psychiatry. 10:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibson J, Russ TC, Adams MJ, Clarke TK, Howard DM, Hall LS, et al. (2017): Assessing the presence of shared genetic architecture between Alzheimer’s disease and major depressive disorder using genome-wide association data. Transl Psychiatry. 7:e1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, Duncan L, et al. (2018): Analysis of shared heritability in common disorders of the brain. Science. 360:eaap8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang J, Zuber V, Matthews PM, Elliott P, Tzoulaki J, Dehghan A (2020): Sleep, major depressive disorder, and Alzheimer disease. Neurology. 95:e1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. (2019): Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nature Genetics. 51:404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howard DM, Adams MJ, Clarke TK, Hafferty JD, Gibson J, Shirali M, et al. (2019): Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nature neuroscience. 22:343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. (2019): Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nature Genetics. 51:414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marioni RE, Harris SE, Zhang Q, McRae AF, Hagenaars SP, Hill WD, et al. (2018): GWAS on family history of Alzheimer’s disease. Translational Psychiatry. 8:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS (2012): Overview and findings from the rush Memory and Aging Project. Curr Alzheimer Res. 9:646–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA (2018): Religious Orders Study and Rush Memory and Aging Project. Journal of Alzheimer’s disease : JAD. 64:S161–s189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beach TG, Adler CH, Sue LI, Serrano G, Shill HA, Walker DG, et al. (2015): Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology. 35:354–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wingo AP, Fan W, Duong DM, Gerasimov ES, Dammer EB, Liu Y, et al. (2020): Shared proteomic effects of cerebral atherosclerosis and Alzheimer’s disease on the human brain. Nature Neuroscience. 23:696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Jager PL, Ma Y, McCabe C, Xu J, Vardarajan BN, Felsky D, et al. (2018): A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Scientific data. 5:180142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Logsdon BA, Perumal TM, Swarup V, Wang M, Funk C, Gaiteri C, et al. (2019): Meta-analysis of the human brain transcriptome identifies heterogeneity across human AD coexpression modules robust to sample collection and methodological approach. bioRxiv.510420. [Google Scholar]
- 25.Hüls A, Robins C, Conneely KN, De Jager PL, Bennett DA, Epstein MP, et al. (2020): Association between DNA methylation levels in brain tissue and late-life depression in community-based participants. Translational Psychiatry. 10:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, et al. (2013): A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 29:189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD (2012): The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics (Oxford, England). 28:882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Jager PL, Shulman JM, Chibnik LB, Keenan BT, Raj T, Wilson RS, et al. (2012): A genome-wide scan for common variants affecting the rate of age-related cognitive decline. Neurobiology of aging. 33:1017.e1011–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wingo AP, Liu Y, Gerasimov ES, Gockley J, Logsdon BA, Duong DM, et al. Integrating human brain proteomes with genome-wide association data implicates new proteins in Alzheimer’s disease pathogenesis. Nature genetics. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira M, Bender D, et al. (2007): PLINK: a toolset for whole-genome association and population-based linkage analysis. American journal of human genetics. 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM (2010): Robust relationship inference in genome-wide association studies. Bioinformatics (Oxford, England). 26:2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. (2012): An integrated map of genetic variation from 1,092 human genomes. Nature. 491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, et al. (2016): Next-generation genotype imputation service and methods. Nature genetics. 48:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Patterson N, et al. (2015): LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nature genetics. 47:291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Z, Zheng Z, Zhang F, Wu Y, Trzaskowski M, Maier R, et al. (2018): Causal associations between risk factors and common diseases inferred from GWAS summary data. Nature Communications. 9:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verbanck M, Chen CY, Neale B, Do R (2018): Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nature genetics. 50:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leek JT, Storey JD (2007): Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 3:1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006): Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics. 38:904–909. [DOI] [PubMed] [Google Scholar]
- 39.Willer CJ, Li Y, Abecasis GR (2010): METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics (Oxford, England). 26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi SW, Mak TS, O’Reilly PF (2020): Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc. 15:2759–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi SW, O’Reilly PF (2019): PRSice-2: Polygenic Risk Score software for biobank-scale data. GigaScience. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang J, Lee SH, Goddard ME, Visscher PM (2011): GCTA: A Tool for Genome-wide Complex Trait Analysis. The American Journal of Human Genetics. 88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, et al. (2014): Proteogenomic characterization of human colon and rectal cancer. Nature. 513:382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Sousa Abreu R, Penalva LO, Marcotte EM, Vogel C (2009): Global signatures of protein and mRNA expression levels. Mol Biosyst. 5:1512–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan CC, Scoggin S, Wang D, Cherry S, Dembo T, Greenberg B, et al. (2011): Systematic discovery of Rab GTPases with synaptic functions in Drosophila. Curr Biol. 21:1704–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu E, Kanno E, Choi S, Sugimori M, Moreira JE, Llinás RR, et al. (2008): Role of Rab27 in synaptic transmission at the squid giant synapse. Proceedings of the National Academy of Sciences. 105:16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ginsberg SD, Mufson EJ, Alldred MJ, Counts SE, Wuu J, Nixon RA, et al. (2011): Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J Chem Neuroanat. 42:102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robins C, Liu Y, Fan W, Duong DM, Meigs J, Harerimana NV, et al. (2021): Genetic control of the human brain proteome. Am J Hum Genet. 108:400–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leiper JM, Santa Maria J, Chubb A, MacAllister RJ, Charles IG, Whitley GS, et al. (1999): Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem J. 343 Pt 1:209–214. [PMC free article] [PubMed] [Google Scholar]
- 50.Smith MA, Vašák M, Knipp M, Castellani RJ, Perry G (1998): Dimethylargininase, a nitric oxide regulatory protein, in Alzheimer disease. Free Radical Biology and Medicine. 25:898–902. [DOI] [PubMed] [Google Scholar]
- 51.Wingo TS, Liu Y, Gerasimov ES, Gockley J, Logsdon BA, Duong DM, et al. (2021): Brain proteome-wide association study implicates novel proteins in depression pathogenesis. Nature Neuroscience. 24:810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, et al. (2017): The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med. 23:405–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greenwood AK, Gockley J, Daily K, Aluthgamage D, Leanza Z, Sieberts SK, et al. (2020): Agora: An open platform for exploration of Alzheimer’s disease evidence. Alzheimer’s & Dementia. 16:e046129. [Google Scholar]
- 54.Xu J, Li Q, Qin W, Jun Li M, Zhuo C, Liu H, et al. (2018): Neurobiological substrates underlying the effect of genomic risk for depression on the conversion of amnestic mild cognitive impairment. Brain : a journal of neurology. 141:3457–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.