

Abstract
Background:
Colorectal cancer (CRC) is a leading cause of cancer mortality worldwide. Mutations in the adenomatous polyposis coli (APC) gene are pivotal in colorectal tumorigenesis. Recently, we demonstrated that aldehyde dehydrogenase 1B1 (ALDH1B1) knockdown dramatically reduced colon tumor growth in a mouse xenograft model. The purpose of the present preliminary study is to examine the effect of loss of ALDH1B1 in CRC development in an inducible colon-specific Apc mouse model.
Methods:
ApcW/FCdx2ERT2-Cre mice develop uni-allelic inactivation of Apc specifically in colon epithelial cells following tamoxifen treatment. Aldh1b1−/− KO mice were crossed with ApcW/FCdx2ERT2-Cre mice. Six-month-old male ApcW/FCdx2ERT2-Cre/Aldh1b1−/−, and ApcW/FCdx2ERT2-Cre/Aldh1b1+/+ mice were treated with tamoxifen (50 mg/kg, i.p.) for three consecutive days. ApcW/F/Aldh1b1−/− and ApcW/F/Aldh1b1+/+ mice were treated with corn oil (i.e., tamoxifen vehicle control) for three consecutive days. Eighteen days later, mice were sacrificed and their colons examined microscopically, macroscopically and histologically for the presence of adenoma.
Results:
All ApcW/FCdx2ERT2-Cre/Aldh1b1+/+ and ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice treated with tamoxifen developed colorectal adenoma. The ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice showed a significant decrease in the total volume of all ileal and colonic adenomas, and decreased incidence of large colonic adenoma compared to ApcW/FCdx2ERT2-Cre/Aldh1b1+/+ mice. Immunohistochemical analysis of p53 and β-catenin showed a trend toward decreased expression score in colonic adenomas of ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice.
Conclusion:
The present preliminary study suggests that deletion of ALDH1B1 may protect against the full development of colorectal cancer. Further mechanistic studies are required to elucidate how ALDH1B1 contributes for colorectal cancer.
Keywords: Colorectal cancer, Adenomatous Polyposis Coli, Aldehyde dehydrogenase 1B1, Mouse models, Beta-catenin
Introduction
Colorectal cancer (CRC) is the third most common cancer and the second leading cause of cancer-related death in the USA [1]. Approximately 150,000 U.S. residents are diagnosed with CRC annually, and one-third of CRC patients die from the disease [1]. The pathogenic and etiological factors underlying the development of CRC are complex and still not clearly understood [2]. The development of CRC has been shown to be influenced by diet, lifestyle, environmental factors, and acquired somatic mutations [3, 4]. CRC arises by accumulation of multiple genetic and epigenetic alterations associated with genes regulating cell growth and differentiation [5]. Such constant alterations push the colonic epithelium to progress from normal to dysplastic, then adenomatous, and finally to adenocarcinoma [6], with metastasis to other sites [7]. Inflammatory mediators, such as cytokines and chemokines, may serve as tumor growth and survival factors, as well as promoting angiogenesis and suppressing immune-mediated tumor elimination [8–10]. Colonic tumors exhibit activation of multiple transcription factors, including nuclear factor-κB (NFκB), a master regulator of inflammatory pathways [11, 12], and signal transducer and activator of transcription 3 (STAT3) [9].
Mutations in the adenomatous polyposis coli (APC) gene are responsible for the familial adenomatous polyposis coli syndrome (FAP), a condition characterized by development of colorectal adenomas [13]. Germline-inactivating APC mutations have been identified in FAP patients, and inactivation of both APC alleles has been observed in greater than 80% of sporadic colorectal adenomas [14]. These results suggest that APC is a tumor suppressor, and acts as a central gatekeeper protein in colorectal tumorigenesis. One proposed gatekeeper mechanism revolves around APC regulation of β-catenin (a dual function cell-cell adhesion and gene transcription protein) and activation of Wnt signaling [15, 16]. Multiple mouse models carrying germline Apc mutations have been developed [15]. The multiple intestinal neoplasia (MIN) mouse model (ApcMin/+) recapitulates some of the features observed in FAP patients; however, these mice develop polyps and adenomas in the small intestine [17, 18]. Recently, colon-specific deletion of Apc has been achieved through the use of the colon-specific Cre construct, CDX2P-CreERT2 [19]. These mice carry a CDX2-NLS Cre recombinase transgene and a floxed Apc allele that, upon tamoxifen treatment, produces a colon-specific APC knockout animal. This model develops tumors that are predominantly located in the distal colon and rectum, forms pedunculated-type tumors similar to human colorectal cancers, and exhibits biallelic Apc inactivation, β-catenin dysregulation, and global DNA hypomethylation [19].
Aldehyde dehydrogenase (ALDH) catalytic activity has been identified as a biomarker for many cancers and cancer stem cells [20]. The expression of ALDH1B1 in normal colon tissue is limited to the bottom of the intestinal crypts where the stem cell resides [21]. In cancerous colon tissue, there is a drastic increase of ALDH1B1 expression [22]. Importantly, we have demonstrated that ALDH1B1 knockdown dramatically reduces tumor growth in a xenograft model [23]. In order to better understand the role of ALDH1B1 in colorectal carcinogenesis, we crossed the Aldh1b1−/− (KO) mouse line with the ApcW/FCdx2ERT2-Cre mouse line. Our hypothesis is that ALDH1B1 plays a role in CRC development. As such, it is anticipated that deletion of ALDH1B1 will reduce CRC development in the colon-specific APC knockout animal.
Materials and Methods
Animals
Global Aldh1b1−/− mice were developed and originally characterized by our laboratory [23].ApcW/FCdx2ERT2-Cre mice carrying a CDX2P-NLS Cre recombinase transgene and a loxP-targeted Apc allele were provided by Dr. Yatrik M. Shah (University of Michigan, Ann Arbor, USA). For tumor studies, Aldh1b1−/− mice were bred with ApcW/FCdx2ERT2-Cre mice to generate four genotype groups: ApcW/FCdx2ERT2-Cre/Aldh1b1−/−, ApcW/FCdx2ERT2-Cre/Adh1b1+/+, ApcW/F/Aldh1b1−/−, and ApcW/F/Aldh1b1+/+ mutant mice. All animal procedures were reviewed and approved by the Yale University’s Institutional Animal Care & Use Committee (IACUC).
Adenoma induction and macroscopic evaluation
A total of thirteen 6 mo-old, male mice were examined in this study. For adenoma induction, mice were treated with tamoxifen or an equivalent volume of tamoxifen vehicle (corn oil) as shown in Fig. 1. Specifically, tamoxifen (TAM, 50 mg/kg body weight; i.p.) was administered i.p. to ApcW/FCdx2ERT2-Cre/Aldh1b1−/− (n=3) and ApcW/FCdx2ERT2-Cre/Aldh1b1+/+ (n=3) mice whereas 5 ml/kg corn oil (CON) was administered i.p. to ApcW/F/Aldh1b1−/− (n=3), and ApcW/F/Aldh1b1+/+ (n=4) mice. Tamoxifen or CON were administered on the first three consecutive days (days 1, 2 and 3) of the experiment. Mice were individually housed at room temperature under 12 h light/12 h dark cycle with ad libitum access to water and food (regular chow diet). The health of the mice was monitored closely for any deterioration, including rectal bleeding, rectal prolapse, anemia, and weight loss (>15%). On day 18, the mice were anesthetized using 5% isoflurane, exsanguinated by collecting blood from posterior vena cava and performing whole body perfusion with room temperature normal saline. The small and large intestines were then carefully removed from each mouse and flushed with normal saline. The jejunum, ileum, and colon of each mouse were surgically separated and opened longitudinally, and examined under a stereomicroscope (Nikon, C-DS, Melville, NY) for adenoma by an observer blinded to the animal genotype. The number and size (length, width) of adenomas were measured in each intestinal segment. Visually observable lesions were termed macroscopic adenomas. Later, lesions visualized in the longitudinally opened colon and ileum tissue by stereomicroscope were termed microscopic adenomas. The volume of each adenoma was quantified from its length and width as described previously [24].
Figure 1.

Schematic representation of the animal study design. Tamoxifen (TAM, 50 mg/kg, i.p.) or corn oil (CON, 5 ml/kg, i.p.) was administered to mice on three consecutive days, i.e., days 1, 2, 3. Mice were euthanized on day 18 and their intestines were removed for morphological, histological and immunohistochemical evaluation.
Histology and immunohistochemical staining
Following macroscopic examination, each intestinal tissue segment was carefully rolled-up and placed in 10% neutral buffered formalin (NBF) for fixing. After 24–48 hr, the tissues were transferred to 70% ethanol and processed for hematoxylin & eosin (H&E) and immunohistochemical (IHC) staining by the Yale Histology Core Laboratory. All tissues were fixed in phosphate-buffered 10% formalin (Fisher Scientific, Pittsburgh, PA) and embedded in paraffin. Multiple sections were cut and mounted on glass slides (Superfrost™/Plus, Fisher Scientific, Hampton, NH) and stored at 4°C. Tissue sections were subjected to H&E staining using routine techniques. Immunohistochemical staining was performed according to a standard procedure [25]. Briefly, paraffin-embedded tissue sections (5 μm) were deparaffinized and rehydrated. The section underwent heat-induced antigen retrieval using 0.01 M citrate buffer (pH 6.6) for 30 min at 95°C, was then cooled to room temperature, and subsequently rinsed and placed in Tris Buffered Saline (50 mM Tris-Cl, 150 mM NaCl; pH 7.6) containing Tween 20 (0.1%) (TBS-T). After incubation with 3% hydrogen peroxide, sections were incubated with β-catenin mouse monoclonal antibody (Biocare Medical, Pacheco, CA) or p53 mouse monoclonal antibody (Thermo Scientific, Waltham, MA) at a dilution of 1:100 dilution in Protein Blocker (Open Biosystems, Huntsville, AL) for 60 min at room temperature. Horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibody (Biocare Medical, Pacheco, CA) was used at a 1:50 dilution for 10–20 min at room temperature. Slides were incubated with diaminobenzidine (Open Biosystems, Huntsville, AL) for 10 min, followed by counterstaining with hematoxylin, dehydration, clearing and mounting using mounting media. Microscopic identification and enumeration, and quantification of the staining of the adenomas were performed by a trained histologist blinded to the mouse genotypes. To quantify the IHC signal, the relative staining intensity (I) was assigned a value from 0–3 as follows: no signal (0), weak signal (1), moderate signal (2), or intense signal (3). The staining extensiveness (E) was calculated as the percent of positively-stained cells (1–100%). The overall IHC expression score (S) was calculated as the product of I and E, yielding a number between 0 and 300 for each adenoma.
Statistical analyses
Differences between the body weight and changes in the ApcW/FCdx2ERT2-Cre/Adh1b1+/+ and ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice were determined by two way ANOVA with post-hoc Tukey’s or Sidak’s multiple comparison test and two-tailed Mann-Whitney test (respectively) using the GraphPad Prism 7 software for Windows (GraphPad Software Inc., La Jolla CA, USA). The differences were considered significant when the P value was less than 0.05.
Results
Effect of genotype on small and large intestinal adenomas
During the course of this study, no significant difference in the body weight change was observed in any of the four experimental groups (Fig. 2). None of the vehicle-treated ApcW/F/Aldh1b1−/− (Fig. 3A) or ApcW/F/Aldh1b1+/+ (Fig. 3B) mice developed macroscopic- or microscopic size intestinal adenomas. In the two TAM-treated groups (ApcW/FCdx2ERT2-Cre/Aldh1b1−/−, ApcW/FCdx2ERT2-Cre/Adh1b1+/+), adenomas were not evident at the macroscopic level in either the jejunum or ileum. Similarly, no adenomas were evident in the jejunum at the microscopic level as well. By contrast, ileal adenomas were observed microscopically in all (three out of three) ApcW/FCdx2ERT2−Cre/Aldh1b1−/− mice and two out of three ApcW/FCdx2ERT2-Cre/Adh1b1+/+ mice (Fig. 4A). All of the ileal adenomas in these two groups of mice were in the range 0.9–1.0 mm in diameter (Fig. 4B). The average and total volume of ileal adenomas in ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice (0.8 mm3 and 5.5 mm3 (respectively)) were significantly lower (P<0.001) than that from the ApcW/FCdx2ERT2-Cre/Adh1b1+/+ mice (2.9 mm3 and 17.8 mm3 (respectively)) (Fig. 4C). Adenomas were visualized at the microscopically (1.0–1.6 mm)- (n=3) and macroscopically- (n=10) ranging in size from 2.3–4.4 mm in the colons of two of the three ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice and all three of the ApcW/FCdx2ERT2-Cre/Adh1b1+/+ mice had both microscopically (0.5–1.0)- (n=3) and macroscopically- (n=13) observable adenomas ranging from 2.5–12.5 mm. The colon adenoma multiplicity (i.e., total number of adenomas per mouse in the colon) was similar between ApcW/FCdx2ERT2-Cre/Aldh1b1−/− and ApcW/FCdx2ERT2-Cre/Adh1b1+/+ mice (Fig. 4D). Interestingly, it was noted that ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice developed significantly lower (P<0.001) number of macro adenomas (i.e., ≥ 2mm) compared to ApcW/FCdx2ERT2-Cre/Aldh1b1+/+mice (Fig. 4E). However, there was no significant difference in the number of micro adenomas (i.e., ≤2mm) in these two groups of mice. The average and total volume of all macro adenomas in ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice (40.3 and 403.7 mm3 (respectively)) were significantly lower (P<0.001) than that in the ApcW/FCdx2ERT2-Cre/Adh1b1+/+ mice where average and total volume was 314.7 mm3 and 4,091 mm3 respectively (Fig. 4F). In contrast, there was no significant difference in the average and total volume of micro adenomas in these two groups of mice.
Figure 2.

Mouse genotype and body weights. The weight of each mouse was monitored daily for the duration of the experimental protocol. Data are presented as the mean ± standard deviation from n=3–4 mice.
Figure 3.

Macroscopic evaluation of gastrointestinal tissues. Representative images of tissues from ApcW/F/Aldh1b1−/− (A), ApcW/F/Aldh1b1+/+ (B), ApcW/FCdx2ERT2−Cre/Aldh1b1−/− (C) and ApcW/FCdx2ERT2-Cre/Aldh1b1+/+ (D) mice on day 18 of the experimental protocol are shown. Right panels show a low power magnification of the small and large intestine, with jejunum, ileum, caecum and colorectal regions highlighted. Left panels show a higher magnification (A 100X, B 100X, C 400X, D 400X) of the luminal surface of the colon. Macroscopically-observable growths within the colon are indicated by arrows.
Figure 4.

Effect of ALDH1B1 expression on characteristics of ileal and colonic adenomas in ApcW/FCdx2ERT2-Cre mice. The total number of ileal adenomas (multiplicity) (A), number of ileal adenomas with diameter mm (B), total ileal adenomas volume (C), total number of colon adenomas (multiplicity) (D), number of colon adenomas with diameter mm (E) and total colon adenoma volume (F) in individual ApcW/FCdx2ERT2-Cre/Aldh1b1+/+ mice (red symbols) and ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice (blue symbols) are shown. * P < 0.05, *** P < 0.001, two-tailed Mann-Whitney test or two-way ANOVA with Sidak’s multiple comparison test, compared to ApcW/FCdx2ERT2-Cre/Aldh1b1+/+ mice. The horizontal line represents the mean.
Occurrence of histologically-identifiable adenomas in the small and large intestines
In order to examine the effect of genotypic variations of Apc and Aldh1b1 genes, abnormal histologic lesions were induced in all experimental mice. Representative images of H&E-stained adenomas in the ileum and colon are shown in Figs. 5A and 5D, respectively. Colonic lesions in ApcW/FCdx2ERT2-Cre/Adh1b1−/− showed highly pleomorphic nuclei but did not appear to invade through the basement membrane and did not include any mucous secreting cells which represents adenoma characteristics. Mice from the ApcW/FCdx2ERT2-Cre/Adh1b1+/+ possess similar tumors. Eighteen days into the experimental protocol, adenomas were present in the colon (but not in the ileum) of ApcW/FCdx2ERT2-Cre/Adh1b1+/+ mice (Table 1A). In contrast, ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice developed adenomas in both the colon and ileum. As shown in the table 1A, the numbers of adenomas in the colon were equally distributed between two groups. The adenomas identified in these experiments were remarkably similar histologically to those described previously [19, 26].
Figure 5.
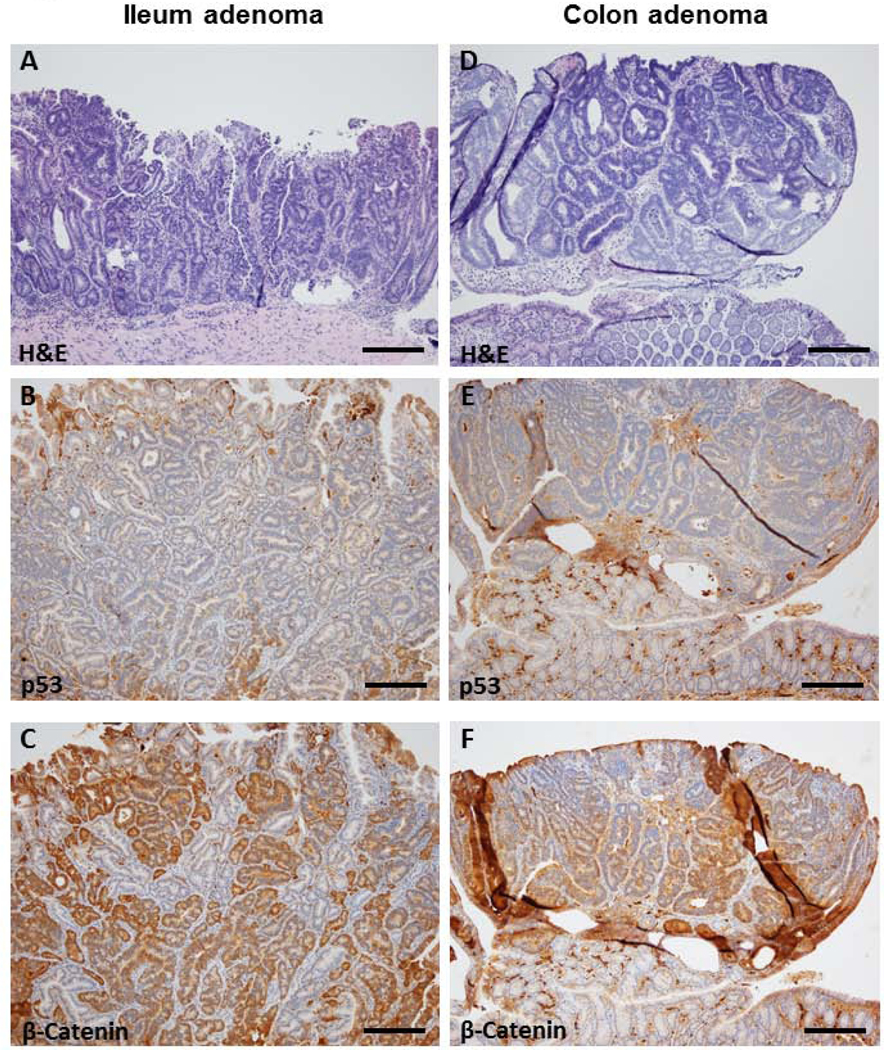
Histological analysis of intestinal adenomas in ApcW/FCdx2ERT2-Cre/Aldh1b1−/− and ApcW/FCdx2ERT2-Cre/Aldh1b1+/+ mice. Formalin-fixed sections of ileum (A, B, C) or colon (D, E, F) were subjected to H&E staining (A, D) or immunohistochemical staining for p53 (B, E) or β-catenin (C, F). Black bar = 200 microns.
Table 1.
Microscopic Analysis of the Adenomas in Tamoxifen-Treated Mice
| Genotype | Mouse ID | Ileum | Colon | ||
|---|---|---|---|---|---|
| # lesions a | Total score b | # lesions a | Total score b | ||
| 1A. Histologic adenoma quantification | |||||
| ApcW/FCdx2ERT2-Cre/Adh1b1+/+ | AA01 | 0 | 3 | ||
| AA04 | 0 | 1 | |||
| AA09 | 0 | 1 | |||
| Apc W/F Cdx2 ERT2-Cre /Aldh1b1 −/− | AA02 | 4 | 1 | ||
| AA05 | 2 | 3 | |||
| AA07 | 1 | 0 | |||
| 1B. p53 IHC quantification | |||||
| Apc W/F Cdx2 ERT2-Cre /Adh1b1 +/+ | AA01 | 0 | 0 | 3 | 105 |
| AA04 | 0 | 0 | 1 | 0 | |
| AA09 | 0 | 0 | 1 | 5 | |
| Apc W/F Cdx2 ERT2-Cre /Aldh1b1 −/− | AA02 | 4 | 16 | 1 | 0 |
| AA05 | 2 | 2 | 3 | 2 | |
| AA07 | 1 | 270 | 0 | 0 | |
| 1C. β-catenin IHC Quantification | |||||
| Apc W/F Cdx2 ERT2-Cre /Adh1b1 +/+ | AA01 | 0 | 0 | 3 | 780 |
| AA04 | 0 | 0 | 1 | 200 | |
| AA09 | 0 | 0 | 1 | 240 | |
| Apc W/F Cdx2 ERT2-Cre /Aldh1b1 −/− | AA02 | 4 | 730 | 1 | 5 |
| AA05 | 2 | 260 | 3 | 670 | |
| AA07 | 1 | 270 | 0 | 0 | |
The number of histologically-identifiable adenomas in ileum or colon
Total score = (percentage of cells that label positive the immunofluorescent antibody) × (relative intensity of IHC fluorescence signal)
Expression of p53 and β-catenin in ileum and colon adenomas
To further characterize adenomas, the content of p53 and β-catenin expression, was examined using immunohistochemistry (IHC) approach. Representative images of p53 IHC staining are provided in Figs. 5B and 5E. As noted, ileal adenomas (5B) were present only in the ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice. The p53 expression in these adenomas was variable, with the total score (272) ranging from 2 – 270 (Table 1B). In the colon adenomas (5E) of ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice, p53 expression tended to be lower (total score (2) range 0–2) than the levels observed in the ileal adenomas (total score (272) range 2–270) (Table 1B). The p53 expression (total score (110)) in colon adenomas of ApcW/FCdx2ERT2-Cre/Aldh1b1+/+ mice ranging from 5–105. Representative images of the β-catenin IHC staining are shown in Figs. 5C and 5F. The presence of ileal adenomas (5C) was found only in ApcW/FCdx2ERT2-Cre/Aldh1b1−/−, with the total expression score of 1260, ranging from 260–730. Colonic expression of β-catenin (5F) was found in both group of mice. The total expression score in ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice was 675 and ranged from 5–670. However, the expression in the ApcW/FCdx2ERT2-Cre/Aldh1b1+/+ mice group showed a trend towards increased expression score of 1220, that ranged from 200–780 (Table 1C). Although it did not reach statistical significance, the total β-catenin expression score in the ApcW/FCdx2ERT2-Cre/Aldh1b1−/− mice group was lower than those from ApcW/FCdx2ERT2-Cre/Aldh1b1+/+ group. Notably, the average score of β-catenin expression was higher than p53 in adenomas of both groups.
Discussion
Colonic ALDH1B1 activity is normally confined to the stem cell compartment found at the colonic crypt base; increased expression of ALDH1B1 was found in human colon cancer specimens [22]. Furthermore, ALDH1B1 knockdown caused dramatic reduction of colon tumor growth in xenograft models [23]. Based on this strong association towards colon tumor pathology, we hypothesized and investigated the decisive function of ALDH1B1 using APC model of colorectal carcinogenesis.
Since, normal expression of the APC gene was found to be absent in 80–90% of colon carcinomas, several mouse models were created to understand the functional role of Apc gene towards colon carcinogenesis [27]. Human colorectal tumorigenesis was accurately recapitulated in the mouse by targeted colon-specific deletion of Apc using the CDX2P-CreERT2 gene [19]. Specifically, Cre targeting of the Apc+/loxP allele with tamoxifen treatment in these mice led to loss of APC expression that was restricted to the distal small intestine, cecum, colon and rectum [19].
We examined the effects of functional loss of Aldh1b1 gene on colorectal carcinogenesis by cross breeding ApcW/FCdx2ERT2-Cre mice strain with the ALDH1B1 knockout (i.e., Aldh1b1−/−) mice. All of the resultant ApcW/FCdx2ERT2-cre/Aldh1b1+/+ and ApcW/FCdx2ERT2-cre/Aldh1b1−/− mice developed micro adenoma in the ileum and both micro and macro adenomas in the colon, and had equivalent numbers of adenomas. The distribution of the adenomas in the small and large intestine were restricted to distal region. Our results are similar to the observation of Hinoi and colleagues [19], who showed the distribution of intestinal tumors of the CDX2P-NLS Cre; Apc+/loxP (CPC;Apc) mice to the distal region of the intestines. These data suggest that differential expression of Cdx2 promoter or regulated transgenes may play a primary role in regional distribution.
The newly created ApcW/FCdx2ERT2-cre/Aldh1b1−/− mice showed ~100-fold lower colonic volume (macro adenomas) than ApcW/FCdx2ERT2-Cre/Adh1b1+/+ mice. Whereas, the volume of ileal microadenoma was ~3 fold lower than in ApcW/FCdx2ERT2-cre/Aldh1b1+/+ mice. The overall observation supports the notion that ALDH1B1 expression strongly influences colon carcinogenesis in the ApcW/FCdx2ERT2-Cre mouse. Our studies did not demonstrate an impact of ALDH1B1 expression on adenoma multiplicity between ApcW/FCdx2ERT2-cre/Aldh1b1+/+ and ApcW/FCdx2ERT2-cre/Aldh1b1−/− mice. This may have been a result of the small number of animals in each group.
p53, a potent tumor suppressor, is genetically altered in over 80% of colorectal tumors [28]. Mutations in p53 are found in 50 – 60% of human colorectal carcinomas; however, these mutations are only observed in less than 10% of colorectal adenomas [29]. We found p53 expression tend to be higher in colonic adenomas of ApcW/FCdx2ERT2-cre/Aldh1b1+/+ than ApcW/FCdx2ERT2-cre/Aldh1b1−/− mice. One study reported moderately elevated p53 expression in neoplastic cells of colonic tumors of CPC;Apc mice and absence of Trp53 missense mutations in colonic adenomas [19]. Similarly, p53 mutations have previously been observed to occur at a low frequency (15–30%) in mouse colonic adenomatous polyps [30]. The present study suggests that p53 expression does not play a role in the formation of the adenomas in ApcW/FCdx2ERT2-Cre mice nor in changes in colon tumorigenesis occurring in mice devoid of ALDH1B1.
Beta-Catenin has been observed to be upregulated in all adenomas arising on a mutant APC background [31]. A close association between ALDH1B1 and activation of Wnt/β-catenin signaling was established previously in colon cancer cells [23]. In the present study, we observed a trend towards increased expression of β-catenin in colonic adenomas in ApcW/FCdx2ERT2-cre/Aldh1b1+/+ mice. In human colorectal cancer, expression of β-catenin was closely correlated with the degree of tumor differentiation [32], suggesting that activation of β-catenin plays a significant role in cancer progression. Furthermore, in human patients with stage II or III colorectal cancer, Wnt1 expression was strongly correlated with high expression of nuclear β-catenin [33]. Curiously, the expression of Wnt/β-catenin, Notch and PI3K/Akt are all down-regulated in cultured human colon cancer cells following shRNA knockdown of ALDH1B1 expression [23].
APC mutations contributes approximately 85% of sporadic and inherited forms of human colorectal cancer cases which was reviewed extensively [34]. In addition to the contributing role of APC gene, activating missense mutations in the β-catenin gene (CTNNB1) [35] and/or human homologue of mouse conductin (Axin1 and −2) truncating mutations also associated with the development of colorectal tumors in humans [36, 37]. Binding elements for the Wnt/β-catenin signaling transcription factor T-cell factor/lymphoid enhancing factor have been identified in the human ALDH1B1 gene promoter [38]. Singh and colleagues hypothesized [23] that increased expression of ALDH1B1 in colon cancer may lead to increased levels of retinoic acid and activation of PPARβ/δ, thereby increasing expression of pro-survival genes through the PI3K/Akt pathway [39]. Loss-of-function of Apc mutation induces β-catenin signaling that was attributed for the colorectal cancer progression. The activation of Wnt/β-catenin signaling elevates the expression of ALDH1B1 as demonstrated in colon cancer cell lines [23]. Since Wnt/β-catenin signaling helps in progression of tumorigenesis, the collective effect of functional loss of Apc and expression of Aldh1b1 sophisticates to generate more aggressive colon cancer outcomes. The colon-specific inducible ApcW/FCdx2ERT2-cre/Aldh1b1+/+ and ApcW/FCdx2ERT2-cre/Aldh1b1−/− mutant lines that predominantly develop adenomas similar to human CRCs help to gain a better insight for the collective response. ApcW/FCdx2ERT2-cre/Aldh1b1−/− mice showed dramatic decrease in the β-catenin expression even with loss of Apc gene function.
In addition, these ApcW/FCdx2ERT2-cre/Aldh1b1−/−mice showed a significant decrease in total adenoma volume in the small intestine when compared to ApcW/FCdx2ERT2-cre/Aldh1b1+/+. The decrease in ileal adenoma volume might correspond to the activity of the Cdx2 promoter, which was involved in deletion of Apc. Normally, the human caudal type homeo box 2 transcription factor, CDX2, is important in the anterior to posterior patterning of the intestinal epithelium and in defining patterns of proliferation and differentiation along the crypt-villus axis [37]. Its expression is stronger in the colon than in the ileum. Further investigations are needed to identify the effect of CDX2 on the expression of ALDH1B1.
For the first time we demonstrate the effect of loss-of-function of Apc and absence of Aldh1b1, which reduces colon adenoma and in turn delays the tumorigenesis and progression of cancer. This study indicates that ALDH1B1 functions as a key player in the development of Apc associated colon cancer, making it a potential target for anticancer therapy. In summary, our findings are consistent with a modulatory role of ALDH1B1 in colorectal cancer development. Our newly created ApcW/FCdx2ERT2-cre/Aldh1b1−/−mouse may be valuable for studying the role of tumor suppressor genes or proto-oncogenes in colon tumorigenesis in vivo.
Acknowledgements
We wish to acknowledge the insight and dedication to this multi-year project by our brilliant colleague Dr. Surendra Singh, he has not been forgotten. This work was supported by the National Institutes of Health (NIH) grants AA017754, AA022057, AA021724, AA025093.
Footnotes
Jaya Prakash Golla, Ying Chen, Yatrik M. Shah, Vasilis Vasiliou: Conceptualization,
Jaya Prakash Golla, Aikaterini Kandyliari, Tan Wan Ying, Ying Chen, David J. Orlicky, Vasilis Vasiliou: Methodology
Jaya Prakash Golla, Aikaterini Kandyliari, Tan Wan Ying: Investigation
Jaya Prakash Golla: Data curation, Validation, Visualization, Writing-Original draft preparation
Jaya Prakash Golla, David J. Orlicky: Validation, Formal analysis.
Jaya Prakash Golla, David J. Orlicky, David C. Thompson, Vasilis Vasiliou: Writing-Reviewing and Editing
Vasilis Vasiliou: Supervision
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA, Jemal A, Colorectal cancer statistics, 2020, CA Cancer J Clin, (2020). [DOI] [PubMed]
- [2].Potter JD, Colorectal cancer: molecules and populations, J Natl Cancer Inst, 91 (1999) 916–932. [DOI] [PubMed] [Google Scholar]
- [3].Huxley RR, Ansary-Moghaddam A, Clifton P, Czernichow S, Parr CL, Woodward M, The impact of dietary and lifestyle risk factors on risk of colorectal cancer: a quantitative overview of the epidemiological evidence, Int J Cancer, 125 (2009) 171–180. [DOI] [PubMed] [Google Scholar]
- [4].Slattery ML, Diet, lifestyle, and colon cancer, Semin Gastrointest Dis, 11 (2000) 142–146. [PubMed] [Google Scholar]
- [5].Mundade R, Imperiale TF, Prabhu L, Loehrer PJ, Lu T, Genetic pathways, prevention, and treatment of sporadic colorectal cancer, Oncoscience, 1 (2014) 400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kinzler KW, Vogelstein B, Lessons from hereditary colorectal cancer, Cell, 87 (1996) 159–170. [DOI] [PubMed] [Google Scholar]
- [7].Yamagishi H, Kuroda H, Imai Y, Hiraishi H, Molecular pathogenesis of sporadic colorectal cancers, Chin J Cancer, 35 (2016) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Karin M, Nuclear factor-kappaB in cancer development and progression, Nature, 441 (2006) 431–436. [DOI] [PubMed] [Google Scholar]
- [9].Yu H, Pardoll D, Jove R, STATs in cancer inflammation and immunity: a leading role for STAT3, Nat Rev Cancer, 9 (2009) 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Grivennikov SI, Greten FR, Karin M, Immunity, inflammation, and cancer, Cell, 140 (2010) 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Manna SK, Golla S, Golla JP, Tanaka N, Cai Y, Takahashi S, Krausz KW, Matsubara T, Korboukh I, Gonzalez FJ, St. John’s Wort Attenuates Colorectal Carcinogenesis in Mice through Suppression of Inflammatory Signaling, Cancer Prev Res (Phila), 8 (2015) 786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sakamoto K, Maeda S, Hikiba Y, Nakagawa H, Hayakawa Y, Shibata W, Yanai A, Ogura K, Omata M, Constitutive NF-kappaB activation in colorectal carcinoma plays a key role in angiogenesis, promoting tumor growth, Clin Cancer Res, 15 (2009) 2248–2258. [DOI] [PubMed] [Google Scholar]
- [13].Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P, Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients, Science, 253 (1991) 665–669. [DOI] [PubMed] [Google Scholar]
- [14].Fearnhead NS, Britton MP, Bodmer WF, The ABC of APC, Hum Mol Genet, 10 (2001) 721–733. [DOI] [PubMed] [Google Scholar]
- [15].Su LK, Vogelstein B, Kinzler KW, Association of the APC tumor suppressor protein with catenins, Science, 262 (1993) 1734–1737. [DOI] [PubMed] [Google Scholar]
- [16].Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P, Association of the APC gene product with beta-catenin, Science, 262 (1993) 1731–1734. [DOI] [PubMed] [Google Scholar]
- [17].Moser AR, Mattes EM, Dove WF, Lindstrom MJ, Haag JD, Gould MN, ApcMin, a mutation in the murine Apc gene, predisposes to mammary carcinomas and focal alveolar hyperplasias, Proc Natl Acad Sci U S A, 90 (1993) 8977–8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Moser AR, Pitot HC, Dove WF, A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse, Science, 247 (1990) 322–324. [DOI] [PubMed] [Google Scholar]
- [19].Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, Williams BO, Cho KR, Fearon ER, Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation, Cancer Res, 67 (2007) 9721–9730. [DOI] [PubMed] [Google Scholar]
- [20].Singh S, Brocker C, Koppaka V, Chen Y, Jackson BC, Matsumoto A, Thompson DC, Vasiliou V, Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress, Free Radic Biol Med, 56 (2013) 89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Koivisto T, Salaspuro M, Aldehyde dehydrogenases of the rat colon: comparison with other tissues of the alimentary tract and the liver, Alcohol Clin Exp Res, 20 (1996) 551–555. [DOI] [PubMed] [Google Scholar]
- [22].Chen Y, Orlicky DJ, Matsumoto A, Singh S, Thompson DC, Vasiliou V, Aldehyde dehydrogenase 1B1 (ALDH1B1) is a potential biomarker for human colon cancer, Biochem Biophys Res Commun, 405 (2011) 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Singh S, Arcaroli J, Chen Y, Thompson DC, Messersmith W, Jimeno A, Vasiliou V, ALDH1B1 Is Crucial for Colon Tumorigenesis by Modulating Wnt/beta-Catenin, Notch and PI3K/Akt Signaling Pathways, PLoS One, 10 (2015) e0121648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Feldman JP, Goldwasser R, Schwartz J, Orion I, A mathematical model for tumor volume evaluation using two dimensions, J Appl Quantitative Methods, 4 (2008) 455–465. [Google Scholar]
- [25].Coskran TM, Morton D, Menniti FS, Adamowicz WO, Kleiman RJ, Ryan AM, Strick CA, Schmidt CJ, Stephenson DT, Immunohistochemical localization of phosphodiesterase 10A in multiple mammalian species, J Histochem Cytochem, 54 (2006) 1205–1213. [DOI] [PubMed] [Google Scholar]
- [26].Oshima H, Oshima M, Kobayashi M, Tsutsumi M, Taketo MM, Morphological and molecular processes of polyp formation in Apc(delta716) knockout mice, Cancer Res, 57 (1997) 1644–1649. [PubMed] [Google Scholar]
- [27].Jackstadt R, Sansom OJ, Mouse models of intestinal cancer, J Pathol, 238 (2016) 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JK, Hamilton S, Vogelstein B, p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis, Cancer Res, 50 (1990) 7717–7722. [PubMed] [Google Scholar]
- [29].Arnold CN, Goel A, Blum HE, Boland CR, Molecular pathogenesis of colorectal cancer: implications for molecular diagnosis, Cancer, 104 (2005) 2035–2047. [DOI] [PubMed] [Google Scholar]
- [30].Hao XP, Frayling IM, Sgouros JG, Du MQ, Willcocks TC, Talbot IC, Tomlinson IP, The spectrum of p53 mutations in colorectal adenomas differs from that in colorectal carcinomas, Gut, 50 (2002) 834–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kongkanuntn R, Bubb VJ, Sansom OJ, Wyllie AH, Harrison DJ, Clarke AR, Dysregulated expression of beta-catenin marks early neoplastic change in Apc mutant mice, but not all lesions arising in Msh2 deficient mice, Oncogene, 18 (1999) 7219–7225. [DOI] [PubMed] [Google Scholar]
- [32].Cao YC, Yang F, Liu XH, Xin X, Wang CC, Geng M, [Expression of Wnt5a, APC, beta-catenin and their clinical significance in human colorectal adenocarcinoma], Zhonghua Zhong Liu Za Zhi, 34 (2012) 674–678. [DOI] [PubMed] [Google Scholar]
- [33].Yoshida N, Kinugasa T, Ohshima K, Yuge K, Ohchi T, Fujino S, Shiraiwa S, Katagiri M, Akagi Y, Analysis of Wnt and beta-catenin Expression in Advanced Colorectal Cancer, Anticancer Res, 35 (2015) 4403–4410. [PubMed] [Google Scholar]
- [34].Fearon ER, Molecular genetics of colorectal cancer, Annu Rev Pathol, 6 (2011) 479–507. [DOI] [PubMed] [Google Scholar]
- [35].Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW, Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC, Science, 275 (1997) 1787–1790. [DOI] [PubMed] [Google Scholar]
- [36].Najdi R, Holcombe RF, Waterman ML, Wnt signaling and colon carcinogenesis: beyond APC, J Carcinog, 10 (2011) 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Silberg DG, Swain GP, Suh ER, Traber PG, Cdx1 and cdx2 expression during intestinal development, Gastroenterology, 119 (2000) 961–971. [DOI] [PubMed] [Google Scholar]
- [38].Daniels DL, Weis WI, Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation, Nat Struct Mol Biol, 12 (2005) 364–371. [DOI] [PubMed] [Google Scholar]
- [39].Schug TT, Berry DC, Shaw NS, Travis SN, Noy N, Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors, Cell, 129 (2007) 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]