

Abstract
Increasing evidence suggests that durable drug-free control of HIV-1 replication is enabled by effective cellular immune responses that may induce an attenuated viral reservoir configuration with a weaker ability to drive viral rebound. Here, we comprehensively tracked effects of antiviral immune responses on intact and defective proviral sequences from elite controllers (ECs), analyzing both classical escape mutations and HIV-1 chromosomal integration sites as biomarkers of antiviral immune selection pressure. We observed that, within ECs, defective proviruses were commonly located in permissive genic euchromatin positions, which represented an apparent contrast to autologous intact proviruses that were frequently located in heterochromatin regions; this suggests differential immune selection pressure on intact versus defective proviruses in ECs. In comparison to individuals receiving antiretroviral therapy (ART), intact and defective proviruses from ECs showed reduced frequencies of escape mutations in cytotoxic T cell epitopes and antibody contact regions, possibly due to the small and poorly-inducible reservoir that may be insufficient to drive effective viral escape in ECs. Approximately 15% of ECs harbored nef deletions in intact proviruses, consistent with increased viral vulnerability to host immunity in the setting of nef dysfunction. Together, these results suggest a distinct signature of immune footprints in proviral sequences from ECs.
One Sentence Summary:
Immune selection footprints in proviral sequences distinguish individuals with natural, drug-free control of HIV-1.
INTRODUCTION
Individuals who naturally control HIV-1 viremia in the absence of antiretroviral therapy (here termed “elite controllers”, ECs) can be regarded as living evidence that the human immune system is able to effectively restrict HIV-1 replication, and fuel hope that a drug-free remission of HIV-1 infection may be inducible in larger numbers of people living with HIV-1 (1–3). Immunological studies in these individuals have identified several distinguishing host features, most notably with regard to cellular immune responses mediated by HIV-1-specific cytotoxic T cells. In HIV-1 controllers, such cells frequently display a durable memory cell profile, characterized by strong proliferative activities and interleukin (IL)-2 secretion (4, 5), and preferentially target epitopes containing topologically important viral amino acid residues (6, 7). Nevertheless, a pool of virally-infected cells remains detectable in almost all of these individuals (8–10). Remarkably, recent studies have begun to unravel pronounced differences between persisting viral reservoirs in ECs and the majority of antiretroviral therapy (ART)-treated individuals. Using a near full-length proviral sequencing approach, previous work demonstrated a profound reduction of genome-intact proviruses in ECs (11), an observation in line with reduced numbers of replication-competent proviruses determined by quantitative viral outgrowth assays (12–14). Moreover, such studies also demonstrated that chromosomal integration sites of intact proviruses in ECs were biased towards heterochromatin regions and frequently located to centromeric satellite DNA and to a defined region of genes in chromosome 19 that encode for Zinc Finger Nucleases (ZNF) and are densely packed with repressive chromatin marks (11); this represented a marked contrast to proviral integration sites determined in the majority of ART-treated patients (15–18). When integrated in such heterochromatin locations, proviral gene expression seems ineffective due to poor access to host transcriptional machinery and epigenetic chromatin modifications that negatively regulate viral gene transcription. Although positioning in such repressive chromatin locations may not completely prevent proviral gene expression, it is reasonable to assume that intact proviral sequences confined to these heterochromatin positions are more conducive to control by antiviral immune responses, and unable to effectively drive viral replication and rebound viremia.
The mechanisms that lead to the distinct intact proviral reservoir profile in ECs are unclear and deserve further clarification. Initial studies concluded, based on simultaneous analysis of proviral integration sites of in vitro- and in vivo-infected cells, that heterochromatin regions in the genome of ECs do not display enhanced susceptibility to HIV-1 integration (11). Instead, the specific chromosomal integration site landscape of intact proviruses in ECs may represent the result of immune-mediated selection mechanisms. This hypothesis assumes that viral integration into more permissive chromatin regions is associated with a higher risk for exposure to immune recognition, a higher susceptibility to immune-mediated killing and a higher probability of proviral elimination by antiviral host immune responses, thus implying a selection disadvantage for intact proviral species located in permissive chromatin segments. Together, these considerations suggest intricate and reciprocal interactions between the intact proviral reservoir configuration and host immune activity, and insinuate that proviral chromosomal positioning may be regarded as an indirect biomarker of host immune selection forces.
Immune selection effects on HIV-1 have traditionally been investigated through an analysis of sequence variations that are consistent with viral mutational escape (19). The advent of technologies for analysis of intact and defective proviral sequences in conjunction with the corresponding proviral integration sites (11, 18, 20, 21) in the human genome may now allow profiling of footprints of host immune selection effects in greater detail. Here, we used a large set of viral sequencing data to conduct a detailed analysis of immune selection footprints in intact and defective proviral species from ECs and a comparison cohort of ART-treated individuals, paired with an analysis of corresponding proviral chromosomal integration site features. Our results reveal multiple layers of evidence that viral reservoir cells in ECs are subject to host immune selection pressure that can modulate viral reservoir evolution in favor of the host.
RESULTS
Integration site landscapes of intact and defective proviruses in ECs
Our recent work identified a distinct chromosomal integration site profile of intact proviruses in ECs, with a disproportionate overrepresentation in heterochromatin regions (11); however, the location of defective proviruses in these individuals has never been systematically analyzed. Defective proviruses frequently account for up to 90% of all detectable HIV-1 proviral DNA in infected individuals, and predominantly result from errors during viral reverse transcription (22). Here, we extended our prior dataset (comprising proviral sequencing data from n=50 ECs in whom at least one intact provirus was detectable) by conducting near full-length individual provirus sequencing (FLIP-Seq)(23) in 8 additional individuals, leading to a total EC study cohort of n=58. In 10 of them, we performed proviral integration site analysis of defective proviruses, resulting in successful integration site mapping for 45 individual proviral species, of which 32 represented independent (non-clonal) proviral sequences. Integration sites of intact proviruses from 14 ECs, partially described in our previous work (11), were analyzed for comparison (n=103 individual proviral species, n=37 independent proviral sequences). Integration sites of defective and intact proviruses were analyzed using matched integration site and proviral sequencing (MIP-Seq) (18), an experimental approach involving phi29-catalyzed whole genome amplification of single proviruses followed by near full-length proviral sequencing and corresponding chromosomal integration site analysis; this same technique was also applied to the analysis of chromosomal integration sites of intact proviruses from ECs in our previous work (11). For comparative purposes, we analyzed near full-length proviral sequencing data in peripheral blood mononuclear cells (PBMCs) of 42 ART-treated individuals and evaluated proviral integration sites of a total of 113 intact (n=80 independent) and 84 defective (n=76 independent) proviruses from 7 ART-treated individuals, generated by MIP-Seq and described in part previously (18).
Overall, we analyzed a total of n=433 and n=523 near full-length defective proviruses from ECs and ART-treated individuals, respectively, of which n=301 (in EC) and n=395 (in ART-treated individuals) corresponded to independent proviral species. Sequence-identical defective proviral species in ECs that were detected at least two times and derived from clonally-expanded HIV-1-infected cells accounted for 37.64% of all analyzed defective proviruses (Fig. 1A to F). This proportion was smaller relative to 65.19% of intact proviruses from ECs that were part of sequence-identical clonal clusters, but not different from the proportion of intact or defective proviral sequences detected within clonal clusters in ART-treated individuals (Fig. 1F and G). Generally, we observed that clonal genome-intact proviruses from both ECs and ART-treated individuals were frequently integrated in members of the ZNF gene family; a notable proportion of clonal defective proviruses from ECs were also integrated in ZNF genes (Fig. 1C to E, H). In contrast, intact and defective proviruses detected as singlets from either cohort showed no such bias (Fig. 1H, fig. S1A). Among intact and defective proviruses from ECs and ART-treated individuals integrated in genic regions, we did not detect differences in terms of their orientation relative to host genes or locations in exons/introns (fig. S1B to E). Notably, the fraction of defective proviruses from ECs located in non-genic chromosomal regions reached 3.13%, which was significantly lower (P = 0.0002) compared to intact proviruses from ECs, but not substantially different from the corresponding proportion of defective proviruses in ART-treated individuals (Fig. 2A and B, fig. S2A, data file S1). Moreover, we did not observe a single defective proviral sequence from ECs that was integrated in centromeric DNA or in satellite DNA (Fig. 2B), which generally is infrequently targeted for HIV-1 integration and associated with transcriptional repression of proviral gene expression (24, 25); absence of intact or defective proviruses in such regions was also observed in ART-treated individuals, but represented a notable contrast to the strong enrichment of clonal and non-clonal intact proviruses from ECs integrated in centromeric or satellite DNA (Fig. 1H, Fig. 2B, fig. S2A, data file S1). Finally, we failed to note a specific enrichment of defective proviruses from ECs in members of the ZNF gene family on Chromosome 19, which share a similar chromatin state as satellite DNA and are enriched for repressive chromatin marks (26, 27); the lower frequency of defective proviruses from ECs that were integrated in these ZNF genes on chromosome 19 represented a sharp contrast to intact proviruses from ECs, but not to intact and defective proviruses from ART-treated individuals (Fig. 1H, Fig. 2A and C, fig. S2B, data file S1). When combined, genome-defective proviral sequences located either in non-genic DNA or in ZNF genes represented 15.63% of 32 independent genome-defective proviral sequences and 33.33% of all 45 genome-defective proviral sequences in elite controllers; these proportions were lower relative to genome-intact proviral sequences in ECs, but comparable to the corresponding proportions of proviral sequences in ART-treated individuals (Fig. 2D, fig. S2C).
Fig. 1. Chromosomal positioning of defective proviruses from ECs.
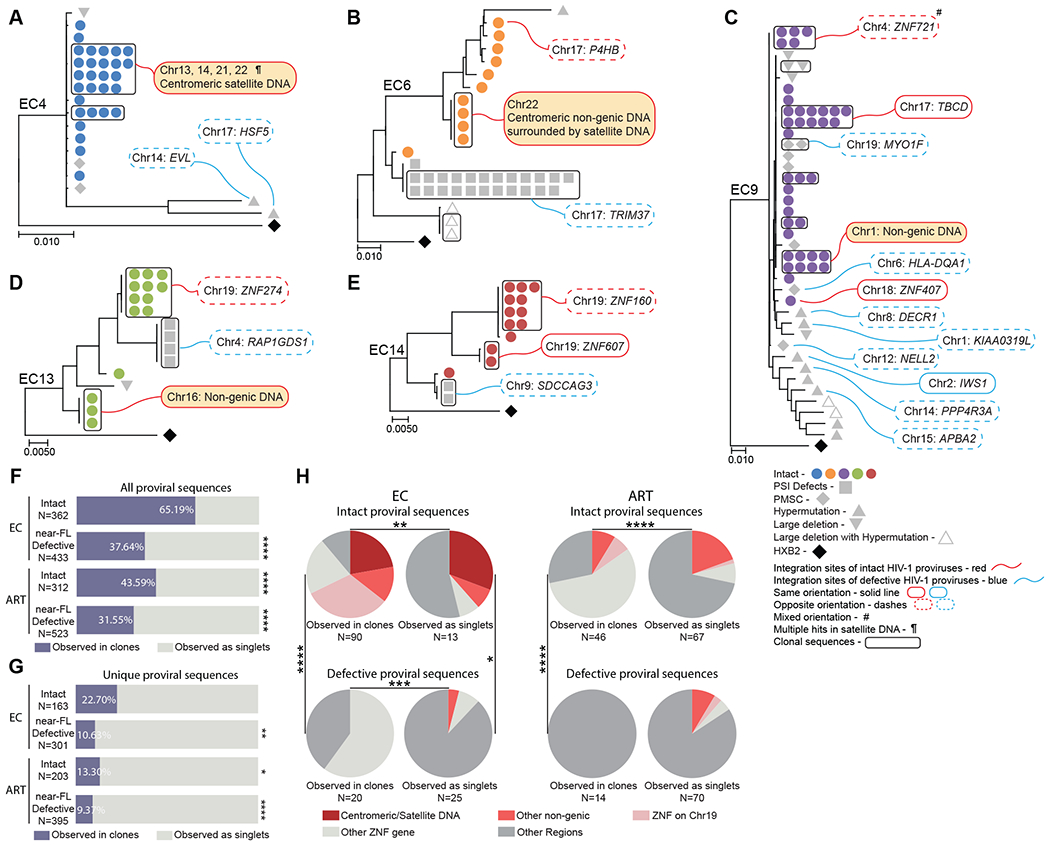
(A to E) Maximum-likelihood phylogenetic trees of near full-length proviral sequences from 5 representative elite controllers are shown. Symbol shapes indicate proviral sequence classification. Genes harboring proviral integrations were identified using Ensembl (v86); gene names are shown according to the HUGO classification (https://www.genenames.org). PSI defects: packaging-signal defects; PMSC: premature stop codons; # Integration sites with mixed orientation among multiple genes; ¶ multi-hit integration sites that cannot be definitively mapped to one genomic location due to positioning in repetitive centromeric satellite DNA present in multiple regions of the human genome. Clonally-expanded proviruses, defined by identical proviral sequences and identical corresponding integration sites, are highlighted in curved black boxes. (F and G) Proportions of intact and defective proviruses observed in clones (in purple) or as singlets (in gray) from ECs and ART-treated individuals are shown. Clonal sequences are counted individually (F) or only once (G). Statistical comparisons were made relative to intact proviruses from ECs. Near-FL: near full-length. (H) Pie charts are shown reflecting proportions of integration sites of intact and defective proviruses in defined genomic regions. Data are shown for clonal sequences (counted individually) and for sequences detected as singlets from ECs and ART-treated individuals. FDR-adjusted two-sided Fisher’s exact tests were used in (F) and (G). A chi-square test was used in (H). * P < 0.05, ** P < 0.01, *** P < 0.001, ****P < 0.0001.
Fig. 2. Genomic and epigenetic features of defective proviruses in ECs.
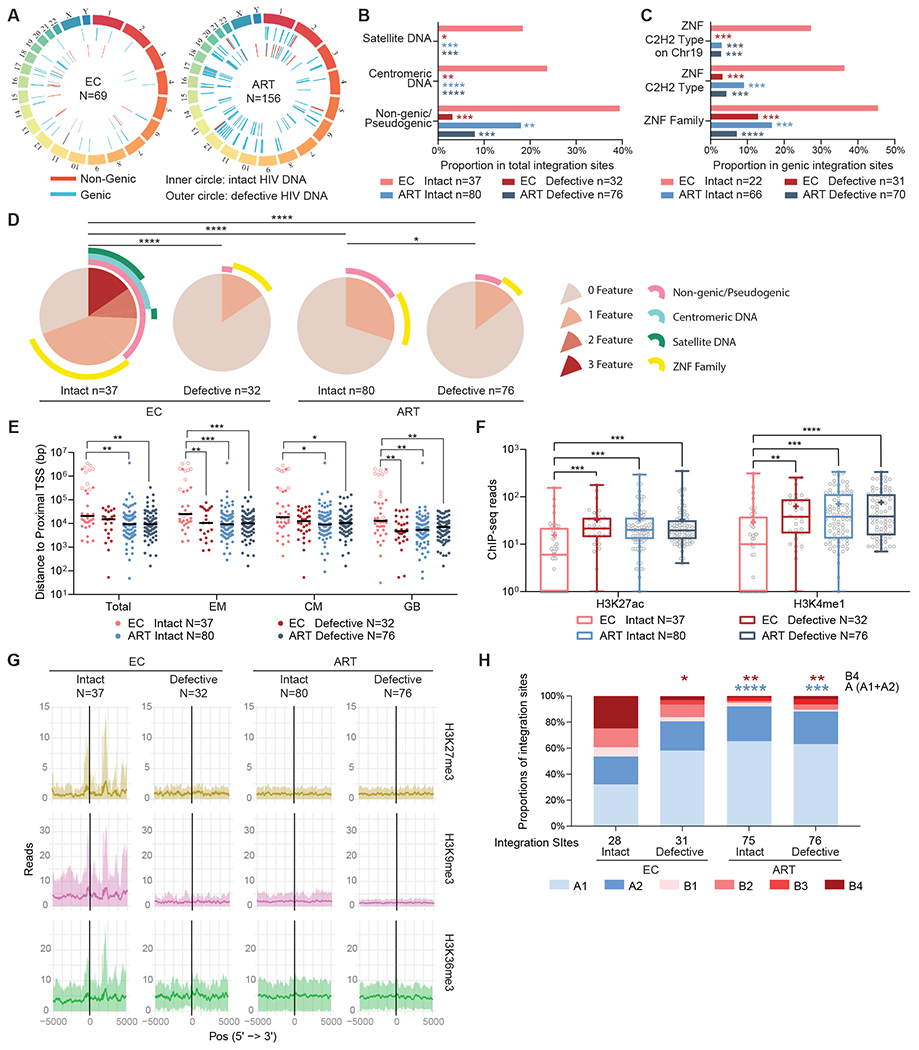
(A) Circos plots reflecting the chromosomal locations of intact and defective proviruses in genic versus non-genic DNA from ECs and ART-treated individuals are shown. (B and C) Proportions of integration sites of intact and defective proviruses are shown for ECs and ART-treated individuals located in indicated genomic regions. Statistical comparisons were made relative to intact proviruses from ECs. (D) SPICE diagrams demonstrating proportions of intact and defective proviruses with indicated integration site features are shown for ECs and ART-treated individuals. (E) Chromosomal distance between integration sites and the most proximal TSS are shown for primary total, effector memory (EM) or central memory (CM) CD4+ T-cells or from Genome Browser database (GB). Horizontal lines reflect the geometric mean. Integration sites in centromeric/satellite DNA are shown in open circles. (F and G) Numbers of DNA sequencing reads associated with activating (H3K27ac and H3K4me1) (F) or repressive (H3K27me3, H3K9me3, and H3K36me3) (G) histone protein modifications in proximity (±5k base pairs) to integration sites from intact and defective proviruses are shown for ECs and ART-treated individuals. Median and confidence intervals (one standard deviation) of ChIP-Seq data from primary memory CD4+ T cells included in the Roadmap repository (26) are shown in (G). Box-and-whisker plots reflect mean, median, minimum, maximum, and interquartile ranges. (H) Proportions of integration sites located in structural compartments A and B (and associated sub-compartments) are shown as determined by Hi-C Seq data described by Rao et al. (29). Integration sites in regions not covered in (29) were excluded. Clonal sequences are counted once in all panels. FDR-adjusted two-sided Fisher’s exact tests were used in (B to D) and (H). FDR-adjusted two-sided Kruskal-Wallis nonparametric test were used in panels (E and F). * P < 0.05, ** P < 0.01, *** P < 0.001, ****P < 0.0001.
In a subsequent analysis, we investigated the relative positioning of defective proviruses to host transcriptional start sites (TSS) as determined by RNA-Seq in CD4 T cells (Fig. 2E, fig. S2D). These studies demonstrated that the enhanced distance of intact proviruses from ECs to transcriptionally active genomic regions (described in our previous work (11)) was not recapitulated for defective proviruses from ECs. In addition, we evaluated the positioning of proviral integration sites relative to activating and inhibitory histone modifications determined by chromatin immunoprecipitation sequencing (ChIP-Seq) in primary CD4 T cells from the Roadmap Consortium (26); these evaluations indicated that, unlike intact proviruses from ECs, defective proviruses from these individuals failed to display an enrichment with the inhibitory histone marks H3K27me3, H3K9me3 and H3K36me3, and a de-enrichment with the stimulatory histone modifications H3K27ac and H3K4me1 in proximity to their chromosomal integration sites (Fig. 2F and G, fig. S2E). Chromatin marks for H3K36me3, a marker for constitutive or facultative heterochromatin (28), were also elevated at integration sites of intact proviruses from ECs, relative to defective proviruses from ECs as well as intact or defective proviruses from ART-treated individuals (Fig. 2G). Finally, aligning proviral integration sites to three-dimensional (3D) chromosomal contact data generated by Hi-C Seq (29) demonstrated that defective proviruses from ECs were predominantly located in compartments A1 and A2 (Fig. 2H, fig. S2F); these compartments mainly include actively expressed chromatin and represent the premier location of intact and defective proviruses from the majority of ART-treated individuals. In contrast, chromosomal locations of intact proviruses from ECs were disproportionately enriched in the heterochromatin compartments B2 and B4. Specifically, integration sites in ZNF genes on chromosome 19 were strongly enriched in compartment B4, consistent with prior results; this chromosomal compartment accounts for a small proportion (0.3%) of the human genome and displays a distinct signature of repressive chromatin marks (27, 30). Since Hi-C next-generation sequencing reads cannot be reliably mapped to centromeric satellite DNA regions, we excluded proviruses located in these regions from this analysis; therefore, the differential representation of intact and defective proviruses from ECs in Hi-C compartments was not driven by proviruses integrated in centromeric regions. Together, these results suggest that, contrary to intact proviruses, defective proviruses from ECs do not show strong integration site biases towards heterochromatin positions; instead, they seem to largely imitate the proviral integration site landscape typically observed in HIV-1-infected individuals undergoing suppressive antiretroviral therapy.
Footprints of T cell mediated immune pressure in intact and defective proviruses from ECs
We next interrogated near full-length proviral sequences from ECs and ART-treated individuals for signs of cytotoxic T lymphocyte (CTL)-driven immune pressure. Aligning HIV-1 amino acid sequences to previously-defined, optimal CTL epitopes restricted by autologous human leukocyte antigen (HLA) class I alleles (data file S2), we observed that the proportions of clade B wild-type CTL epitopes in intact proviruses were significantly higher (P < 0.0001) in ECs compared to ART-treated individuals (Fig. 3A and B, fig. S3A and B). A similar observation was made for proviruses with 5’ packaging-signal (PSI) defects, with premature stop codons (PMSC) or with hypermutations (Fig. 3A, fig. S3A). Correspondingly, frequencies of proviruses with sequence variations matching recognized escape mutations in CTL epitopes restricted by autologous HLA class I alleles were significantly lower (P = 0.0021) in ECs relative to ART-treated individuals (Fig. 3C and D, fig. S3C and D). Of note, in both ECs and ART-treated individuals, the lowest frequencies of wild-type CTL epitopes were detected in near full-length hypermutated sequences, likely as a result of apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) 3G/3F-induced sequence variations; however, the proportions of wild-type CTL epitopes in hypermutated sequences from ECs was still significantly higher (P = 0.0010) compared to ART-treated individuals (Fig. 3A, fig. S3A). In a subsequent, more detailed analysis, we noted that differences in the frequencies of wild-type and escaped CTL epitopes between intact proviruses from ECs and ART-treated individuals were most obvious within the viral gag and pol sequences, and less visible within optimal epitopes described in the viral env, nef and other proteins (Fig. 3B, D, and E, fig. S3B and D). Moreover, we identified a list of immunodominant optimal CTL epitopes within intact proviruses for which the proportions of wild-type sequences in ECs exceeded the corresponding proportions in ART-treated individuals; this list included frequently-described CTL epitopes located in gag, pol, and nef, such as A2-SL9 (SLYNTVATL), B8-FL8 (FLKEKGGL), B27-KK10 (KRWIILGLNK) and B57-TW10 (TSTLQEQIGW) (Fig. 3F, fig. S3E).
Fig. 3. Footprints of CTL-mediated immune pressure in intact and defective proviruses from ECs and ART-treated individuals.
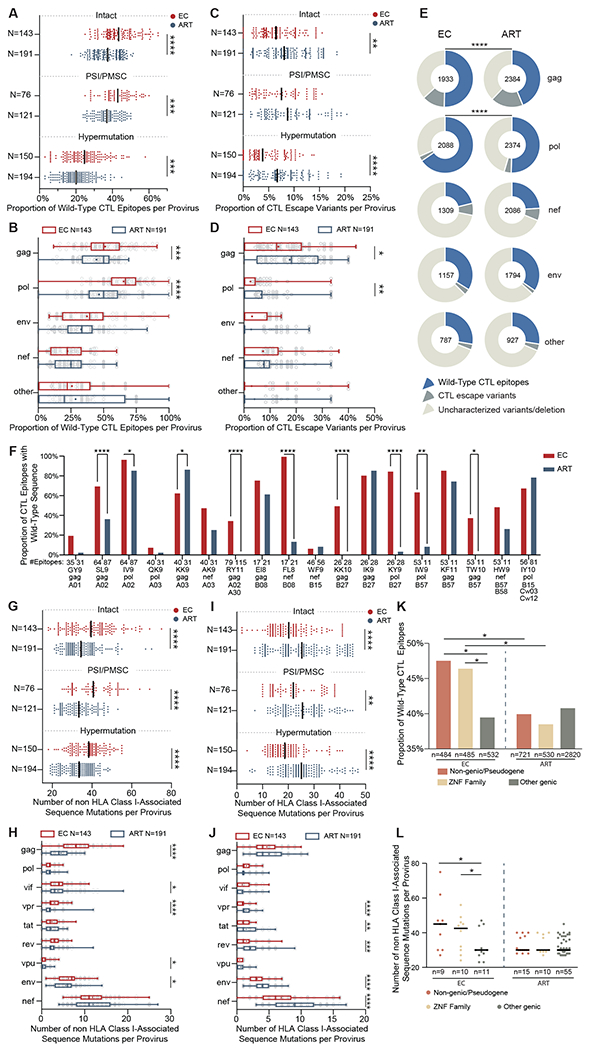
(A to D) Proportions of optimal CTL epitopes (restricted by autologous HLA class I alleles) with wild-type clade B consensus sequences (A and B) or with previously-described CTL escape mutations (C and D) are shown. Each dot represents one provirus. Intact and near full-length proviral sequences with PSI defects/PMSC or hypermutation from ECs and ART treated individuals are shown in (A) and (C). CTL epitope sequences (restricted by autologous HLA class I alleles) within gag, pol, env, nef and other HIV-1 proteins from clade B intact proviruses in ECs and ART-treated individuals are shown in (B) and (D). (E) Pie charts are shown reflecting proportions of CTL epitopes displaying the clade B consensus sequences, previously-described CTL escape variants or uncharacterized sequence variations. Data are shown separately for indicated HIV-1 products; the number within each pie chart indicates the total number of analyzed CTL epitopes restricted by autologous HLA class I alleles in each viral protein for each study cohort. (F) Proportions of epitopes with wild-type clade B consensus sequences are shown among CTL epitopes restricted by common autologous HLA class I alleles within ECs and ART-treated individuals. (G to J) Numbers of sequence variations with (I and J) or without (G and H) statistically significant associations with autologous HLA class I alleles are shown, determined as described by (31). Each dot represents one provirus. Intact proviruses and near full-length proviral sequences with PSI defects/PMSC, or hypermutation from ECs and ART treated individuals are shown in (G) and (I); sequences of nine individual HIV-1 genes from intact proviruses in ECs and ART-treated individuals are shown in (H) and (J). (K and L) Proportions of optimal CTL epitopes with wild-type clade B consensus sequences (K) and numbers of mutations non-adapted to autologous HLA class I alleles (L) are shown from HIV proviruses integrated in indicated genomic regions in ECs and ART-treated individuals. Clonal sequences were counted once. Dot plots with median are shown. Box-and-whisker plots reflect mean, median, minimum, maximum, and interquartile ranges. FDR-adjusted two-tailed Mann Whitney U tests were used in (A to D) and (G to J). Two-sided Fisher’s exact tests were used in (E and F). FDR-adjusted two-sided Kruskal-Wallis nonparametric test was used in panels (K and L). * P < 0.05, ** P < 0.01, *** P < 0.001, ****P < 0.0001.
As an additional analysis step, we evaluated proviral sequence immune adaptation to autologous HLA class I alleles by determining the frequencies of polymorphisms in clade B sequences that were statistically associated with a study participant’s expressed HLA class I isotypes, as determined by a previously-described algorithm (31). The frequencies of sequence variations unrelated to HLA class I isotypes were significantly (P < 0.0001) higher in intact proviral sequences from ECs compared to ART-treated individuals; these differences were most visible when selectively analyzing gag, vpr and env sequences within intact proviruses (Fig. 3G and H, fig. S3F and G), and were also observed in near full-length proviral sequences from ECs that harbored PSI defects, PMSC, or hypermutations (Fig. 3G, fig. S3F). On the other hand, the frequencies of sequence mutations associated with autologous HLA class I alleles were higher in intact and defective near full-length sequences from ART-treated individuals relative to ECs (Fig. 3I and J, fig. S3H and I).
Higher frequencies of wild-type clade B CTL epitope sequences, paired with more limited signs of immune adaption to HLA immune pressure, were most obvious in intact proviruses from ECs integrated in non-genic DNA or in ZNF genes (Fig. 3K and L, fig. S3J and K); although the number of such intact sequences was limited, this observation suggests that proviral species at these chromosomal locations were seeded early in the disease process (prior to substantial immune adaptation to autologous HLA class I isotypes) and subsequently persisted long-term. In contrast, intact proviruses from ECs in other genic positions showed a higher degree of immune adaption (Fig. 3K and L, fig. S3J and K), and most likely represent less ancestral sequences derived from cells infected at more advanced stages of infection. No such differences were observed within intact proviruses from ART-treated individuals. Together, our results paradoxically show that intact proviral sequences from ECs display more limited signs of CTL-driven mutational escape and sequence adjustment to HLA class I-associated immune pressure relative to ART-treated individuals. This observation is arguably best explained by the smaller number of intact proviruses in ECs and their preferential locations in heterochromatin regions. This specific reservoir configuration may be unable to drive higher plasma viral loads required for effective selection of CTL escape mutations. In the context of ECs, lower numbers of CTL escape mutations may therefore represent an indirect sign of effective viral control.
Sequence variations in contact regions of broadly-neutralizing antibodies
We subsequently extended our analysis to investigate signs of humoral immune selection in viral envelope sequences from near full-length proviruses of ECs and ART-treated individuals. We noted that intra-individual env sequence diversity, as determined by pair-wise comparisons within each study participant in whom more than one intact provirus was identified, was more limited in intact proviruses from ECs relative to ART-treated individuals (Fig. 4A); this comparison reached statistical significance for all five variable (V) domains of HIV-1 env (V1: P = 0.0009, V2: P = 0.0009, V3: P = 0.0001, V4: P = 0.0024, V5: P = 0.0003). Moreover, the lengths of the V1V2 domain and the numbers of N-glycosylation sites within env sequences from intact proviruses were smaller in ECs (Fig. 4B and C, fig. S4A and B), consistent with more restricted env sequence evolution and a lower overall resistance to immune recognition by humoral immune responses (32, 33). Notably, the proportion of intact proviruses with R5-tropic envelope sequences in ECs exceeded the corresponding proportion in ART-treated individuals when viral tropism was evaluated using previously-developed computational prediction tools (34) (Fig. 4D and E, fig. S4C).
Fig. 4. HIV-1 env sequence diversity in intact and defective proviruses from ECs and ART-treated individuals.
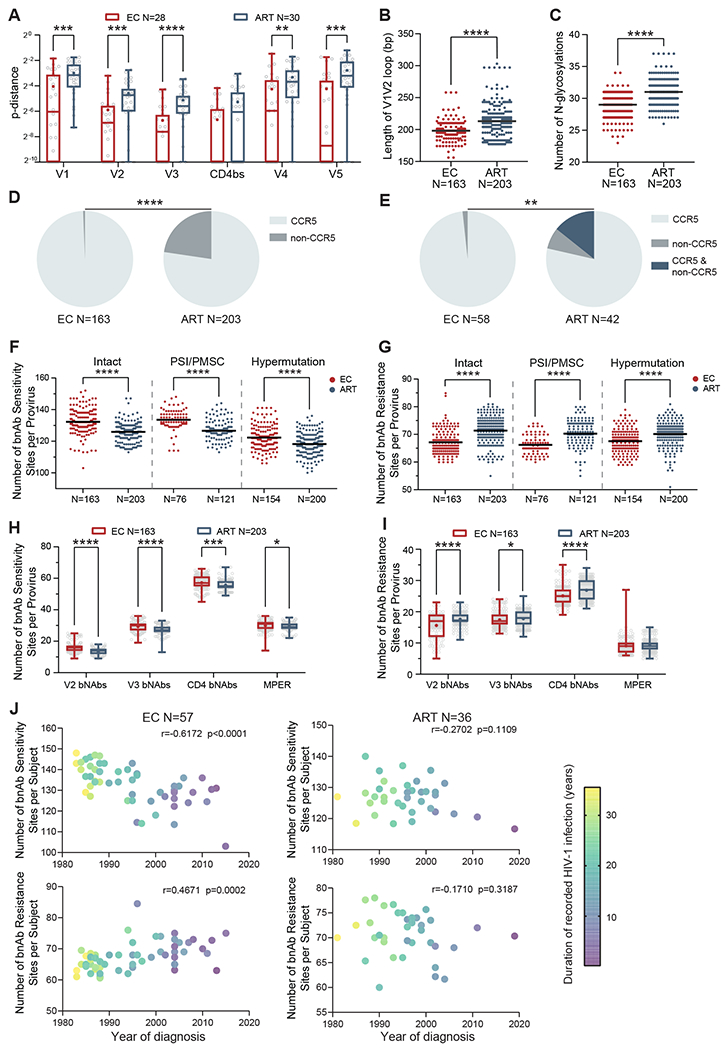
(A) Mean p-distance between indicated env sequence domains from intact proviruses is shown, calculated by pair-wise comparisons within study participants with at least two detectable intact proviruses. Each symbol represents one individual. (B) Lengths of the V1V2 loop in env sequences are shown from intact proviruses in ECs and ART-treated individuals. (C) Numbers of estimated N-glycosylation sites in env sequences are shown from intact proviruses from ECs and ART-treated individuals. (D) Pie charts indicating the proportions of intact proviruses with CCR5- or non-CCR5-tropic V3 envelope sequences are shown. (E) Pie charts reflecting the proportions of individuals harboring intact proviruses with CCR5-tropic, non-CCR5-tropic, or a combination of both CCR5-tropic and non-CCR5-tropic env sequences are shown. HIV-1 tropism was computationally inferred using Geno2pheno (https://coreceptor.geno2pheno.org/) in (D) and (E). HIV-1 tropism was classified as “CCR5” if the false positive rate (FPR) predicted by Geno2pheno was >2%, and “non-CCR5” if FPR was ≤2%. (F to I): Numbers of broadly-neutralizing antibody (bnAb) sensitivity (F and H) and resistance (G and I) signature sites in indicated proviral sequences are shown. Numbers of signature sites associated with resistance or sensitivity to four classes of bnAbs (specific for V2 domain, V3 domain, CD4 binding site, and membrane proximal external region (MPER)), identified as described before (35), were analyzed. (F) and (G) summarize data from intact proviruses and near full-length proviral sequences with PSI defects/ PMSC, or hypermutation from ECs and ART-treated individuals; (H) and (I) indicate data from intact proviruses from ECs and ART-treated individuals, stratified by bnAb class. (J) Correlation analysis is shown between the average numbers of bnAb sensitivity/resistance signature sites within intact proviruses from indicated study individuals and the corresponding HIV-1 infection diagnosis years. Color coding indicates the duration between HIV-1 diagnosis date and sample collection date. Clonal sequences are counted once; the Spearman correlation coefficient is shown for each plot. Dot plots with median are shown. Box-and-whisker plots reflect mean, median, minimum, maximum, and interquartile ranges. FDR-adjusted two-tailed Mann Whitney U tests were used in (A) and (F to I). Two-tailed Mann Whitney U tests were used in (B and C). Two-sided Fisher’s exact test was used in (D and E). * P < 0.05, ** P < 0.01, *** P < 0.001, ****P < 0.0001.
Taking advantage of an existing compendium of proviral sequence signature mutations that influence susceptibility to broadly-neutralizing antibodies (35), we evaluated the frequencies of amino acid variations associated with sensitivity or resistance to broadly-neutralizing antibodies recognizing the CD4 binding site, the V2/V3 envelope regions, or the membrane proximal external region (MPER). Overall, intact proviruses from ECs had significantly (P < 0.0001) higher numbers of amino acid positions associated with susceptibility to broadly-neutralizing antibodies recognizing these env regions, relative to ART-treated individuals; this was also true for near full-length proviruses with PSI defects, PMSC, or hypermutations (Fig. 4F, fig. S4D). Correspondingly, within intact proviruses, the numbers of amino acid changes linked to resistance to broadly-neutralizing antibodies were significantly (P < 0.0001) lower in ECs (Fig. 4G, fig. S4E). Differences in sequence variations consistent with susceptibility to broadly-neutralizing antibodies between the two cohorts were most pronounced for antibodies targeting the CD4 binding site, and for antibodies recognizing the V2 and the V3 envelope regions (Fig. 4H and I, fig. S4F and G).
Hypothesizing that the presence or absence of resistance mutations to broadly-neutralizing antibodies may possibly be associated with the timing of infection, we stratified all ECs according to the presumed year of HIV-1 acquisition and the duration of recorded HIV-1 infection. We noted that an earlier timepoint of infection and a longer duration of HIV-1 infection was associated with a higher number of amino acid positions associated with susceptibility to the four classes of broadly-neutralizing antibodies within intact proviruses from ECs (Fig. 4J). In contrast, a more recent infection timepoint and a shorter duration of infection in ECs was associated with a higher number of resistance mutations to broadly-neutralizing antibodies. This suggests that resistance to broadly-neutralizing antibodies in ECs may predominantly reflect population-level HIV-1 sequence evolution that is transmitted by founder viruses to ECs at the time of infection. Notably, there was no association between timing of HIV-1 infection and evolution of resistance to broadly-neutralizing antibodies in ART-treated individuals, likely because antibody resistance mutations in this group of individuals develop as a result of intra-individual antibody-driven immune selection during periods of high plasma viral loads prior to the institution of antiretroviral therapy. Consistent with these results, we noted that intact proviruses from ECs were phylogenetically more closely related to the most recent common ancestor (MRCA) of all intact sequences from ECs and ART-treated individuals combined (Fig. 5A and B), an observation that was mostly driven by lower frequencies of MRCA sequence divergence within env and the HIV-1 accessory genes in ECs (Fig. 5C).
Fig. 5. Phylogenetic associations between intact proviruses from ECs and ART-treated individuals.
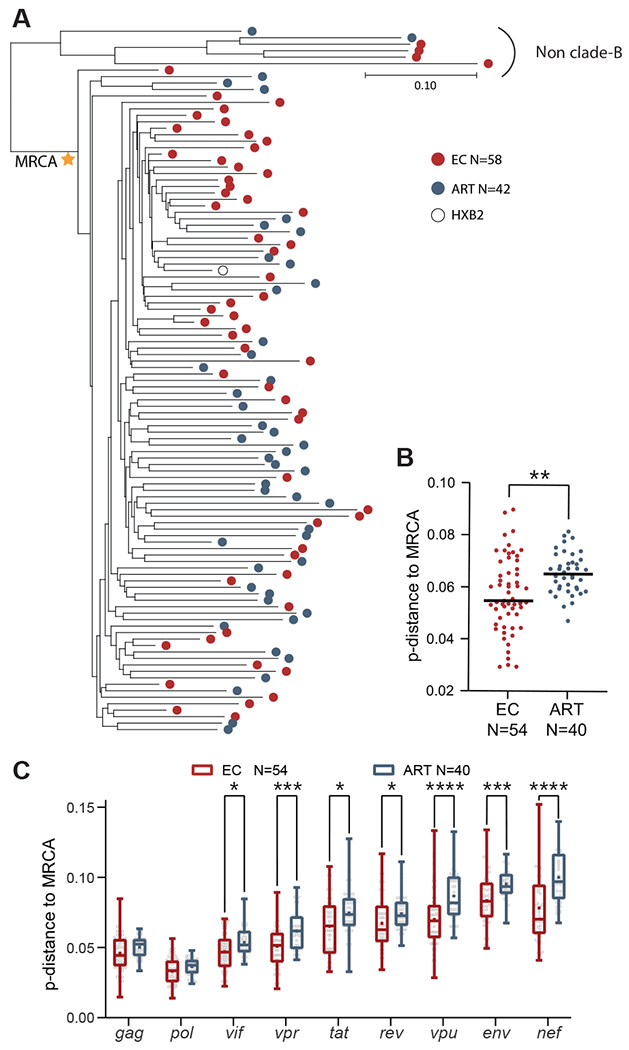
(A) Maximum likelihood phylogenetic trees of intact proviruses from ECs and ART-treated individuals are shown. The most recent common ancestor (MRCA) among clade B intact sequences is marked by a star. HXB2, reference HIV-1 clade-B sequence. (B and C) Genetic distance of all intact clade B sequences to the MRCA is shown. The MRCA and maximum likelihood-corrected distances to the MRCA were estimated in DIVEIN (71). (B) shows data for all intact proviral sequences; (C) shows data for individual HIV-1 genes within intact proviruses. An intra-individual consensus sequence reflecting the dominant base pair residue from all available intact sequences at each base pair position for each study participants was entered into the analysis in (A to C); each dot reflects one study participant. Dot plots with median are shown. Box-and-whisker plots reflect mean, median, minimum, maximum, and interquartile ranges. A two-tailed Mann Whitney U test was used in (B). An FDR-adjusted two-tailed Mann Whitney U test was used in (C). * P < 0.05, ** P < 0.01, *** P < 0.001, ****P < 0.0001.
Viral nef gene deletions in intact proviral DNA
Deletions in viral nef sequences have previously been reported in individuals with long-term nonprogressive HIV-1 infection (36–39), but the frequency of nef deletions has never been systematically studied in a larger cohort of ECs from whom near full-length proviral sequences are available. We observed that nef deletions were significantly (P < 0.0001) more common among intact proviruses from ECs relative to ART-treated individuals (Fig. 6A, fig. S5A); moreover, the proportion of ECs with shorter nef sequences (<600 bp, equal to 97% of the length of nef in HIV-1 clade-B reference sequence HXB2) within intact proviruses reached about 15%, whereas such nef deletions were virtually absent in ART-treated individuals (Fig. 6B and C). Larger deletions in accessory genes other than nef were very infrequent in all analyzed sequences, independently of the study cohort (fig. S5B). Notably, ECs (n=9) with nef deletions showed a reservoir profile that differed in several ways from the remaining EC cohort (n=49) and instead appeared to resemble ART-treated individuals in several ways: Specifically, the ratio of intact versus defective proviruses in ECs with nef deletions was lower (Fig. 6D); moreover, the proportions of epitopes with wild-type sequences or with non-HLA class I associated sequence mutations in intact proviral sequences were also reduced in this subgroup of ECs (Fig. 6E and F, fig. S5C and D); these findings suggest a replicative history with higher viral turnover in ECs with nef deletions. In addition, lower frequencies of polyfunctional HIV-1 gag-specific CD8 T cells secreting both interferon (IFN)-γ and tumor necrosis factor (TNF)-α were observed within the EC subgroup with nef deletions (fig. S5E). Notably, in two ECs (EC20 and EC21), nef deletions were detected in all intact proviruses, but not in a subgroup of defective proviruses (fig. S5F), arguing against acquisition of a nef-deleted virus at the time of viral transmission but instead suggesting de novo evolution of nef-deleted viral variants within these individuals. Together, our results imply that ECs with nef deletions represent a distinct entity of ECs in whom viral control is not primarily achieved by antiviral host mechanisms, but instead largely related through infection with or intra-host evolution of an attenuated virus.
Fig. 6: Intact provirus with large inner nef deletions in ECs.
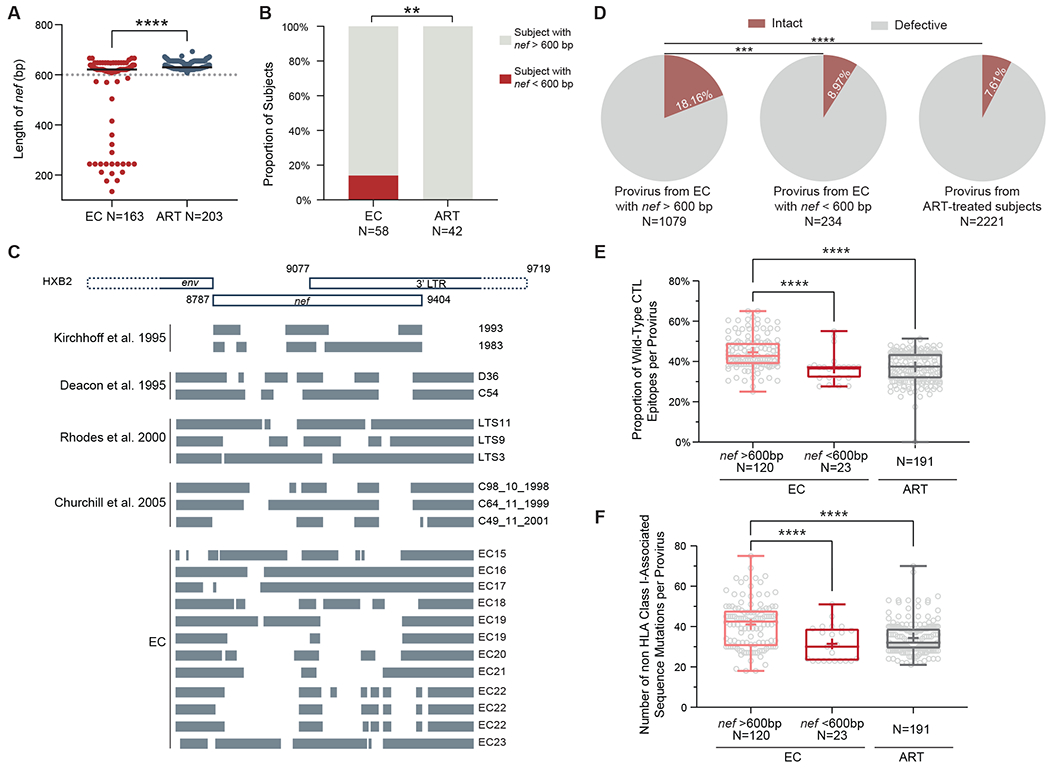
(A) Length of nef genes in intact proviruses from ECs and ART-treated individuals are shown. (B) Proportions of individuals with nef sequence length less than 600 bp within all detected intact proviruses are shown. (C) A schematic representation shows nef/long terminal repeat (LTR) sequence deletions within HIV-1 from previously reported studies (36–39) and within intact proviruses from ECs. HXB2 was used as the reference; indicated genome coordinates are in HXB2 nomenclature. Indicated nef deletions were present in all intact proviruses from each EC, except for EC19 and EC22, in whom different types of nef deletions were observed in intact proviruses. (D) Proportions of intact proviruses are shown out of total proviruses identified from ECs with or without large inner nef deletions and from ART-treated individuals. (E) Proportions of optimal CTL epitopes with wild-type clade B consensus sequences are shown for intact proviruses with or without large inner nef deletions in ECs and ART-treated individuals. (F) Numbers of non HLA class I-associated sequence mutations are shown within intact proviruses with or without large inner nef deletions in ECs and ART-treated individuals. Clonal sequences are counted once in all panels. Dot plots with median are shown. Box-and-whisker plots reflect mean, median, minimum, maximum, and interquartile ranges. A two-tailed Mann Whitney U test were used in (A). A two-sided Fisher’s exact test was used in (B). A FDR-adjusted two-sided Fisher’s exact test was used in (D). FDR-adjusted two-sided Kruskal-Wallis nonparametric tests were used in (E) and (F). ** P < 0.01, *** P < 0.001, ****P < 0.0001.
APOBEC3G/3F-induced hypermutations
Hypermutated near full-length proviral sequences result from destructive cytosine to uracil changes catalyzed by APOBEC3G/3F during reverse transcription of HIV-1 RNA into DNA (40, 41), and reveal innate immune restriction of HIV-1 replication. We observed that the absolute frequencies of near full-length proviral sequences incorporating hypermutations was smaller in ECs, likely reflecting a generally smaller number of proviral sequences in ECs compared to ART-treated individuals (fig. S6A). The relative proportions of hypermutated sequences among all near full-length proviral sequences tended to be larger in ECs (fig. S6B). Using a recently-developed algorithm (42), we observed that footprints of APOBEC3G and 3F-induced hypermutations were seen at relatively similar proportions in ECs and ART-treated individuals (fig. S6C to F); proviral hotspots that were preferentially hypermutated within specific regions of near full-length proviral sequences were also not notably different between the two cohorts (fig. S6G). Moreover, no consistent difference was found in the proportions of total guanine residues that were hypermutated to adenine in the HIV-1 DNA plus strand when autologous intact HIV-1 proviruses were used as reference (fig. S6H); however, the proportions of GG→GA hypermutations, catalyzed by APOBEC3G, were slightly higher (P = 0.0062) in ECs, although there was no difference with regard to GA→AA mutations (catalyzed by APOBEC3F) in this analysis (fig. S6I). Notably, the numbers of stop codons introduced by APOBEC3G/3F hypermutations tended to be slightly higher in ECs within the viral gag, rev and vpu regions (fig. S6J). Overall, these results fail to support a major role of APOBEC3G/3F for contributing to drug-free HIV-1 control in ECs.
DISCUSSION
A broad consensus exists that elite, drug-free control of HIV-1 replication is, in the majority of cases, mediated by host immune factors. Yet, elite control may not simply be due to the presence of potent antiviral immune responses to suppress ongoing viral replication; in addition, immune responses in ECs may select for a specific viral reservoir structure that is unable to effectively drive rebound viremia and more conducive to viral immune control. Immune selection has traditionally been analyzed by investigating viral sequence variations consistent with mutational escape; however, proviral chromosomal integration site profiling might represent a complementary, equally informative approach for unraveling immune selection effects. Here, we performed simultaneous assessments of individual proviral sequences and their corresponding chromosomal location to generate a comprehensive analysis of the proviral reservoir landscape of intact and defective proviruses from ECs. Most importantly, these investigations demonstrated an atypical reservoir profile of intact proviruses in ECs, characterized by intact proviral species that displayed very limited evidence of CTL- or antibody-driven escape mutations, but were preferentially located in heterochromatin regions that are generally disfavored for proviral integration (43–45). This specific reservoir profile represented a sharp contrast to autologous defective proviruses, and to intact and defective proviruses from ART-treated individuals, which were predominantly integrated in permissive chromatin locations and, in the case of intact proviral sequences from ART-treated individuals, frequently exhibited more evidence of mutational escape from adaptive host immune responses. We propose that this specific profile of intact proviruses in ECs is the result of host immune pressure that effectively eliminates many proviral species before viral escape mutations can be effectively selected for. The net outcome of such immune pressure in ECs appears to be a skeleton reservoir consisting of a limited collection of intact proviral species that remain fully susceptible to host immune recognition, but resist immune-mediated elimination through chromosomal integration into heterochromatin locations conferring deep latency and protecting against immune targeting.
The low frequency of HLA class I-associated sequence variations in CTL epitopes from ECs represents an unexpected and perplexing result, specifically since CTL-driven viral mutational escape has been described in circulating HIV-1 RNA sequences from ECs on numerous occasions (46–48). However, the low frequency of escape mutations in intact proviruses from ECs reported here should not be interpreted as evidence of low immune selection pressure, but could instead be viewed as a paradoxical footprint of the extraordinary antiviral immune activity in ECs. Indeed, counterintuitively, more limited signs of immune adaptation in viral sequences can be equally associated with very weak or very strong immune selection activity (49); in contrast, immune escape typically observed in HIV-1-infected individuals is best explained by an intermediate degree of immune pressure that can select for sequence variations but is unable to control the disease (49). Notably, our data are also in line with previous reports suggesting that the degree of viral adaptation to HLA class I-mediated immune pressure is critical for driving HIV-1 disease progression, possibly exceeding the impact of viral fitness (50). In ECs, the low numbers of sequences harboring escape mutations may be partially related to the lower viral reservoir population pool size; in addition, we propose that integration of intact proviruses into less permissive integration sites can restrict the viral replication rate through negative regulation of proviral transcriptional activity. In the setting of such a small, presumably weakly-inducible reservoir, viral production and turnover may be insufficient to drive effective viral escape, and, when it occurs, mutated viral species can likely be readily eliminated by HIV-specific CD8 T cells before re-seeding of a new viral reservoir; recognition of mutated viral epitopes by HIV-1-specific CD8 T cells from ECs has indeed been documented before (6, 51, 52). Higher cell-intrinsic susceptibility of CD4 T cells to CTL killing, previously hypothesized for ECs (53), may also be contributing to the fast elimination of newly-infected cells, as may other, non-CTL-dependent immune mechanisms (54). Together, these data suggest that in ECs, re-fueling of viral reservoir cells through ongoing viral replication is ineffective when viral production is low and antiviral immunity is high; instead, the proviral reservoir in ECs seems mostly or almost entirely fueled by clonal proliferation of latently-infected cells harboring early-seeded intact proviruses that display only limited signs of immune adaptation. In the future, it may be possible to evaluate specific proviral reservoir features in the slightly larger group of “viremic controllers”, who maintain low HIV-1 replication rates (typically less than 2000 copies/ml) in the absence of therapy. In such individuals, phylogenetic signs of ongoing, low viral replication rates might be more obvious (55).
An important finding here is that defective proviruses from ECs were frequently integrated in rather typical chromosomal locations in introns of highly expressed genes, in contrast to atypical location of intact proviruses. Indeed, within the 32 defective proviral species we analyzed, not a single integration site in centromeric satellite DNA was noted; moreover, no proviral integrations of defective proviruses into Krüppel-associated box (KRAB) ZNF genes on Chr 19 were noted, which are densely covered with heterochromatin proteins (27). These observations support the hypothesis that the atypical integration site landscape of intact proviruses from ECs is not the result of alternative integration site preferences in ECs. We propose that the differential integration site landscape between intact and defective proviruses from ECs is most likely related to the ability of immune cells to distinguish cells infected with intact versus defective proviruses, and to mount different degrees of immune selection pressure against cells harboring intact proviruses that are able to release fully-functional infectious virions, versus cells encoding for defective proviruses that are only able to produce viral proteins or gene transcripts (56, 57). The exact immune mechanisms that enable such a differential immune recognition will need to be clarified in the future, but it is noteworthy that evidence suggesting different degrees of immune selection pressure for cells encoding for intact versus defective proviruses has been observed in a number of recent studies: for instance, Peluso et al. (58), Falcinelli et al. (59) and Gandhi et al. (60) observed a faster longitudinal decline of intact versus defective proviruses in ART-treated adults, a finding that was also made in a longitudinal evaluation of HIV-1-infected infants started on ART immediately after birth (61). Moreover, Einkauf et al. (18) noted that within HIV-1-infected adults undergoing 8 to 10 years of antiretroviral therapy, the frequency of intact proviruses in non-genic regions and in opposite orientation to host genes tended to be higher relative to defective proviruses. An important question that deserves future consideration is the possible role of defective proviruses in ECs for maintaining antiviral immune responses. It is theoretically conceivable that intact proviruses in these individuals, due to their position in heterochromatin, are only weakly able to secrete sufficient amount of viral antigen to maintain a functional T cell response, the immunological hallmark of antiviral immunity in ECs (4, 5, 62). Therefore, persistence of a functional T cell response in ECs may largely be driven by viral antigen produced by defective proviruses, which based on their chromosomal locations seem better positioned to secrete a tailored dose of viral proteins that is sufficient to drive the maintenance of HIV-1-specific T cells, without exposing the reservoir cell to immune-mediated elimination. Thus, defective proviruses may possibly act as equivalents of a replication-defective vaccine vector that is stably integrated into host DNA and serves as a durable immunogen. However, this view should not downplay the possibility that a residual transcriptional activity may be ongoing in intact proviruses from ECs, despite their integration in heterochromatin positions, and contribute to maintaining antiviral T cell responses.
Our study is limited to an examination of immune selection markers in proviral sequences from one timepoint in each study participant; longitudinal investigations will be needed to extend this analysis in the future. Moreover, our work involved large and diverse patient cohorts (58 ECs and 42 ART-treated participants), and we cannot exclude that these differences in the duration of infection may have influenced our study results. Finally, our study included only near full-length (greater than 8,000 bp) defective HIV-1 proviruses, but it is possible that signs of immune selection may be more pronounced in smaller defective proviral sequences with larger deletions; sequencing of a larger library of defective proviruses will allow for this to be addressed in the future.
Although our results indicate distinct immune selection footprints in ECs, they also show some evidence of immune selection of viral reservoir cells in ART-treated individuals. In particular, we found that integration sites of clonally-expanded intact proviruses from ART-treated individuals were frequently located in ZNF family genes, which typically are enriched for the inhibitory histone modification H3K9me3 (63, 64); this chromatin mark facilitates epigenetic transcriptional silencing through the human silencing hub (HUSH) complex (65). Notably, within ZNF genes, repressive chromatin marks tend to cluster across the gene bodies and the 3’ end of genes (64); HIV-1 integration in close proximity to such inhibitory chromatin features may provide a preferred genomic niche for proviruses in proliferating T cells, arguably because cell proliferation does not lead to proviral reactivation when HIV-1 is integrated into the more repressive chromatin environment at ZNF gene loci. Therefore, integration into ZNF genes may offer protection against immune recognition and confer a longitudinal selection advantage. Although we observed a larger proportion of intact proviruses from ECs displaying repressive integration site features compared to ART-treated individuals, our results suggest that immune selection processes in ECs are only gradually but not fundamentally different from ART-treated individuals. Therefore, it remains possible that intensification of antiviral host immune activity through strategies such as therapeutic vaccines may be able to increase immune selection of reservoir cells in ART-treated individuals, possibly to an extent that they may approximate an EC-like “blocked and locked” reservoir profile facilitating natural control of HIV-1 replication.
MATERIALS AND METHODS
Study design
The objective of this study was to investigate classical escape mutations and HIV-1 chromosomal integration sites of intact and defective HIV-1 proviral sequences from elite controllers. HIV-1-infected study participants, including 58 elite controllers and 42 ART-treated participants were recruited at the Massachusetts General Hospital (MGH), the Brigham and Women’s Hospital (BWH, both in Boston, MA, USA), the San Francisco General Hospital (San Francisco, CA, USA), and the University Hospital of Sevilla (Spain). No formal sample size calculations, randomizations or investigator blinding operations were performed. Clinical and demographical characteristics of study participants are summarized in table S1. PBMC samples were obtained according to protocols approved by the respective Institutional Review Boards. HLA class I typing was performed using a targeted next generation sequencing method, as described previously (72).
HIV-1 DNA quantification by droplet digital polymerase chain reaction (ddPCR)
PBMCs were subjected to gDNA extraction using DNeasy Blood & Tissue Kit (Qiagen DNeasy #69504). Total HIV-1 DNA were detected using ddPCR (Bio-Rad), using primers and probes described previously(11). The droplets were subsequently read by the QX200 droplet reader (Bio-Rad) and data were analyzed using QuantaSoft software (Bio-Rad).
Whole genome amplification (WGA)
Isolated genomic DNA was diluted to single viral DNA copies according to ddPCR results, so that one HIV-1 provirus was present in approximately 20 to 30% of wells. Subsequently, DNA in each well was subjected to multiple displacement amplification (MDA) with phi29 polymerase (Qiagen REPLI-g Single Cell Kit #150345), per the manufacturer’s protocol. Following this unbiased whole genome amplification, DNA from each well was split and separately subjected to viral sequencing and integration site analysis, as described below.
HIV near full-length proviral sequencing
Proviral sequences were amplified from genomic DNA or WGA products using a near full-length PCR approach (23) or a non-multiplexed 5-amplicon approach (18) and visualized by agarose gel electrophoresis (Quantify One and ChemiDoc MP Image Lab, Bio-Rad). Near full-length (greater than 8000bp) and 5-amplicon positive products were subjected to Illumina MiSeq sequencing at the MGH DNA Core facility. Resulting short reads were de novo assembled using Ultracycler v1.0 and aligned to HXB2 using MUSCLE to identify large deletion, premature/lethal stop codons, out-of-frame indels, internal inversions, or 5’ packaging signal (PSI) defects, using an automated in-house pipeline written in Python scripting language (https://github.com/BWH-Lichterfeld-Lab/Intactness-Pipeline). Presence or absence of APOBEC3G/3F-associated hypermutations was determined using the Los Alamos National laboratory (LANL) HIV Sequence Database Hypermut 2.0 program. Viral sequences that lacked all the defects mentioned above were termed “intact”.
Integration site analysis
Integration sites associated with individual viral sequences were obtained by integration site loop amplification (ISLA), using a protocol previously described (16), or by ligation-mediated PCR (17) (Lenti-X Integration Site Analysis Kit (Takara Bio #631263)); DNA produced by whole-genome amplification was used as template. Resulting PCR products were subjected to next-generation sequencing using Illumina MiSeq. MiSeq paired-end FASTQ files were demultiplexed; small reads (142 bp) were then aligned simultaneously to human reference genome GRCh38 and HIV-1 reference genome HXB2 using bwa-mem (66). Biocomputational identification of integration sites was performed according to previously-described procedures (16, 67).
Sequence analysis
The near full-length HIV-1 proviral sequences were aligned to the clade-B reference sequence HXB2 using multiple alignment using fast fourier transform (MAFFT) (68) or Clustal W (http://www.ebi.ac.uk/Tools/msa/clustalw2). Nucleotides and translated amino acid sequences for each of the nine HIV-1 gene products were obtained using Gene Cutter (https://www.hiv.lanl.gov/content/sequence/GENE_CUTTER/cutter.html). Clades of intact HIV-1 proviral sequences were determined using the LANL HIV Sequence Database Recombinant Identification Program (https://www.hiv.lanl.gov/content/sequence/RIP/RIP.html). For each clade B proviral sequence, the optimal CTL epitope sequences restricted by autologous HLA class I alleles within nine HIV-1 genes were identified using the A-List (data file S2) (69), which includes best-defined HIV-1 CTL/CD8+ T cell epitopes from the LANL HIV Immunology Database. The “CTL/CD8+ Epitope Variants and Escape Mutations” table from the same database was used to classify epitope sequences from each provirus as wild-type, escaped, or uncharacterized according to the respective HIV-1 subtype and HLA allele (data file S2). The number of sequence mutations associated with HLA class I-mediated pressure were calculated in clade-B proviruses as previously described (31). The sensitivity of proviral species to broadly-neutralizing antibodies (bnAb) were estimated by calculating the number of amino acid signature sites associated with sensitivity to four bnAb classes within the env amino acid sequence from each provirus, as previously described (35). P-distance and length of HIV-1 genes were calculated by molecular evolutionary genetics analysis X (MEGA-X) (www.megasoftware.net). Number of N-glycosylation sites in env was predicted by N-GlycoSite (https://www.hiv.lanl.gov/content/sequence/GLYCOSITE/glycosite.html) (70). HIV-1 tropism was analyzed by Geno2pheno (https://coreceptor.geno2pheno.org/) within the V3 loop sequence from each intact provirus. HIV-1 tropism was classified as “CCR5” if the false positive rate (FPR) predicted by Geno2pheno was >2%, and “non-CCR5” if FPR was <2%. Phylogenetic trees were inferred by the Maximum-Likelihood method using PhyML and visualized in FigTree (http://tree.bio.ed.ac.uk/software/figtree). The most recent common ancestor (MRCA) and distance to the MRCA were estimated in divergence, diversity, informative sites and phylogenetic analyses (DIVEIN) (71). APOBEC3G or 3F-associated hypermutations were calculated as described in (42) and in the LANL HIV Sequence Database Hypermut 2.0 program.
Intracellular cytokine staining
PBMCs were stimulated with anti-CD28/CD49d antibody (1 μg/ml, BS Fastimmune) and 2 μg/ml of overlapping peptide pools spanning the HIV-1 clade B sequence of gag. After 1 hour of incubation, Brefeldin A (1μg/ml, BioLegend) and Golgistop (1μg/ml, BD Biosciences) were added and cultured overnight. Cells stimulated with phytohemagglutinin (PHA) (2 μg/ml) served as positive control. After stimulation, cells were stained in FACS buffer with surface antibodies against CD3 (2 µl per million cells, allophycocyanin (APC)-Cy7, HIT3a, BioLegend), CD8 (2 µl per million cells, Peridinin-Chlorophyll-protein (PerCP)-Cy5.5, SK1, BD Biosciences), and LIVE/DEAD Blue Viability Dye (1 µl per million cells, Invitrogen) at 4°C for 15 minutes. Then, cells were treated with fixation and permeabilization solutions and stained for 30 minutes at room temperature with antibodies against IFN-γ (3 µl per million cells, Phycoerythrin (PE)-Dazzle, B27, BioLegend) and TNF-α (3 µl per million cells, Brilliant Violet 711, Mab11, BioLegend). Cells were then acquired on a BD LSRFortessa cytometer (BD Bioscience) at the Ragon Institute Imaging Core Facility at MGH and analyzed using FlowJo software (BD Biosciences).
RNA-Seq, ChIP-Seq, Hi-C Seq data
RNA-Seq data generated from primary CD4 T cells and described in a previous publication (11) were used for analysis. These data are deposited in NCBI GEO (accession number GSE144334). ChIP-Seq data derived from primary human memory CD4 T cells and are included in the Roadmap Consortium portal (http://www.roadmapepigenomics.org/). Hi-C Seq data used in this study were described by Rao et al. (29).
Statistical analyses
Raw, individual-level data for experiments where n<20 are shown in data file S3. Data are shown as pie charts, simplified presentation of incredibly complex evaluations (SPICE) diagrams, Scatter plot (indicating median), Box-and-whisker plots (indicating the median, minimum, maximum, interquartile ranges and mean as “+”). Differences were tested for statistical significance using Mann Whitney U tests, Fishers’ exact tests, Chi-square tests, and two-sided Kruskal-Wallis nonparametric tests, as appropriate. P < 0.05 was considered significant; false discovery rate (FDR) correction was performed using the Benjamini-Hochberg method (73). Analyses were performed using GraphPad Prism, SPICE and R software.
Supplementary Material
Acknowledgement:
The authors gratefully acknowledge all study participants and the MGH DNA core facility.
Funding:
This work is supported by NIH grants HL134539 (to X.G.Y.), AI155171 (to X.G.Y.), AI116228 (to X.G.Y.), AI078799 (to X.G.Y.), DA047034 (to X.G.Y.), AI150396 (to X.G.Y.), the Bill and Melinda Gates Foundation (INV-002703) (to X.G.Y.), AI114235 (to M.L.), AI117841 (to M.L.), AI120008 (to M.L.), AI130005 (to M.L.), DK120387 (to M.L.), AI152979 (to M.L.), AI135940 (to M.L.), AI155233 (to M.L.) and the American Foundation for AIDS Research (amfAR#110181) (to M.L.). XGY and ML are Members of the DARE Collaboratory (UM1AI126611) and the BEAT-HIV Martin Delaney Collaboratory (UM1 AI126620). E.R.M. was supported by Consejo Superior de Investigaciones Científicas (CSIC) and by grants: PI19/01127 Instituto de Salud Carlos III, Fondos FEDER and Consejeria de Transformacion Economica, Industria, Conocimiento y Universidades Junta de Andalucia (P20_01276). Support was also provided by the Harvard University (HU) and University of California at San Francisco (UCSF)/Gladstone Institute for HIV Cure Research Centers for AIDS Research (P30 AI060354 and P30 AI027763, respectively) which are supported by the following institutes and centers co-funded by and participating with the U.S. National Institutes of Health: NIAID, NCI, NICHD, NHLBI, NIDA, NIMH, NIA, FIC, and OAR. Additional support for the SCOPE cohort was provided by the Delaney AIDS Research Enterprise (DARE; AI096109, A127966) and the amfAR Institute for HIV Cure Research (amfAR 109301). The International HIV Controller Cohort is supported by the Bill and Melinda Gates Foundation (OPP1066973), the Ragon Institute of MGH, MIT and Harvard, the NIH (R37 AI067073 to BDW) and the Mark and Lisa Schwartz Family Foundation. This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported in part by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research.
Footnotes
Competing interests: All authors declare no competing interests.
Data and materials availability:
RNA-Seq data are deposited in NCBI GEO (accession number GSE144334). Owing to study participant confidentiality concerns, full-length viral sequencing data cannot be publicly released, but will be made available to investigators upon reasonable request and after signing a data sharing agreement. Correspondence and requests for data should be addressed to Dr. Xu Yu (xyu@mgh.harvard.edu).
REFERENCES AND NOTES
- 1.Blankson JN, The study of elite controllers: a pure academic exercise or a potential pathway to an HIV-1 vaccine? Curr Opin HIV AIDS 6, 147–150 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Saez-Cirion A, Pancino G, HIV controllers: a genetically determined or inducible phenotype? Immunol Rev 254, 281–294 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Migueles SA, Connors M, Long-term nonprogressive disease among untreated HIV-infected individuals: clinical implications of understanding immune control of HIV. JAMA 304, 194–201 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA, HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107, 4781–4789 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, Van Baarle D, Kostense S, Miedema F, McLaughlin M, Ehler L, Metcalf J, Liu S, Connors M, HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol 3, 1061–1068 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Gaiha GD, Rossin EJ, Urbach J, Landeros C, Collins DR, Nwonu C, Muzhingi I, Anahtar MN, Waring OM, Piechocka-Trocha A, Waring M, Worrall DP, Ghebremichael MS, Newman RM, Power KA, Allen TM, Chodosh J, Walker BD, Structural topology defines protective CD8(+) T cell epitopes in the HIV proteome. Science 364, 480–484 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferguson AL, Mann JK, Omarjee S, Ndung’u T, Walker BD, Chakraborty AK, Translating HIV sequences into quantitative fitness landscapes predicts viral vulnerabilities for rational immunogen design. Immunity 38, 606–617 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blankson JN, Bailey JR, Thayil S, Yang HC, Lassen K, Lai J, Gandhi SK, Siliciano JD, Williams TM, Siliciano RF, Isolation and characterization of replication-competent human immunodeficiency virus type 1 from a subset of elite suppressors. J Virol 81, 2508–2518 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li JZ, Segal FP, Bosch RJ, Lalama CM, Roberts-Toler C, Delagreverie H, Getz R, Garcia-Broncano P, Kinslow J, Tressler R, Van Dam CN, Keefer M, Carrington M, Lichterfeld M, Kuritzkes D, Yu XG, Landay A, Sax PE, A. C. T. G. S. A. Team, ART reduces T cell activation and immune exhaustion markers in HIV controllers. Clin Infect Dis, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamine A, Caumont-Sarcos A, Chaix ML, Saez-Cirion A, Rouzioux C, Delfraissy JF, Pancino G, Lambotte O, Replication-competent HIV strains infect HIV controllers despite undetectable viremia (ANRS EP36 study). AIDS 21, 1043–1045 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Jiang C, Lian X, Gao C, Sun X, Einkauf KB, Chevalier JM, Chen SMY, Hua S, Rhee B, Chang K, Blackmer JE, Osborn M, Peluso MJ, Hoh R, Somsouk M, Milush J, Bertagnolli LN, Sweet SE, Varriale JA, Burbelo PD, Chun TW, Laird GM, Serrao E, Engelman AN, Carrington M, Siliciano RF, Siliciano JM, Deeks SG, Walker BD, Lichterfeld M, Yu XG, Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nature, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casado C, Galvez C, Pernas M, Tarancon-Diez L, Rodriguez C, Sanchez-Merino V, Vera M, Olivares I, De Pablo-Bernal R, Merino-Mansilla A, Del Romero J, Lorenzo-Redondo R, Ruiz-Mateos E, Salgado M, Martinez-Picado J, Lopez-Galindez C, Permanent control of HIV-1 pathogenesis in exceptional elite controllers: a model of spontaneous cure. Sci Rep 10, 1902 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noel N, Pena R, David A, Avettand-Fenoel V, Erkizia I, Jimenez E, Lecuroux C, Rouzioux C, Boufassa F, Pancino G, Venet A, Van Lint C, Martinez-Picado J, Lambotte O, Saez-Cirion A, Prado JG, Long-Term Spontaneous Control of HIV-1 Is Related to Low Frequency of Infected Cells and Inefficient Viral Reactivation. J Virol 90, 6148–6158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chun TW, Shawn Justement J, Murray D, Kim CJ, Blazkova J, Hallahan CW, Benko E, Costiniuk CT, Kandel G, Ostrowski M, Kaul R, Moir S, Casazza JP, Koup RA, Kovacs C, Fauci AS, Effect of antiretroviral therapy on HIV reservoirs in elite controllers. J Infect Dis 208, 1443–1447 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, Spindler J, Ferris AL, Mellors JW, Kearney MF, Coffin JM, Hughes SH, HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345, 179–183 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagner TA, McLaughlin S, Garg K, Cheung CY, Larsen BB, Styrchak S, Huang HC, Edlefsen PT, Mullins JI, Frenkel LM, HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 345, 570–573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohn LB, Silva IT, Oliveira TY, Rosales RA, Parrish EH, Learn GH, Hahn BH, Czartoski JL, McElrath MJ, Lehmann C, Klein F, Caskey M, Walker BD, Siliciano JD, Siliciano RF, Jankovic M, Nussenzweig MC, HIV-1 Integration Landscape during Latent and Active Infection. Cell 160, 420–432 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Einkauf KB, Lee GQ, Gao C, Sharaf R, Sun X, Hua S, Chen SM, Jiang C, Lian X, Chowdhury FZ, Rosenberg ES, Chun TW, Li JZ, Yu XG, Lichterfeld M, Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J Clin Invest 129, 988–998 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goulder PJ, Watkins DI, HIV and SIV CTL escape: implications for vaccine design. Nat Rev Immunol 4, 630–640 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Patro SC, Brandt LD, Bale MJ, Halvas EK, Joseph KW, Shao W, Wu X, Guo S, Murrell B, Wiegand A, Spindler J, Raley C, Hautman C, Sobolewski M, Fennessey CM, Hu WS, Luke B, Hasson JM, Niyongabo A, Capoferri AA, Keele BF, Milush J, Hoh R, Deeks SG, Maldarelli F, Hughes SH, Coffin JM, Rausch JW, Mellors JW, Kearney MF, Combined HIV-1 sequence and integration site analysis informs viral dynamics and allows reconstruction of replicating viral ancestors. Proc Natl Acad Sci U S A 116, 25891–25899 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halvas EK, Joseph KW, Brandt LD, Guo S, Sobolewski MD, Jacobs JL, Tumiotto C, Bui JK, Cyktor JC, Keele BF, Morse GD, Bale MJ, Shao W, Kearney MF, Coffin JM, Rausch JW, Wu X, Hughes SH, Mellors JW, HIV-1 viremia not suppressible by antiretroviral therapy can originate from large T cell clones producing infectious virus. J Clin Invest 130, 5847–5857 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, Siliciano RF, Replication-Competent Noninduced Proviruses in the Latent Reservoir Increase Barrier to HIV-1 Cure. Cell 155, 540–551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee GQ, Orlova-Fink N, Einkauf K, Chowdhury FZ, Sun X, Harrington S, Kuo HH, Hua S, Chen HR, Ouyang Z, Reddy K, Dong K, Ndung’u T, Walker BD, Rosenberg ES, Yu XG, Lichterfeld M, Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J Clin Invest 127, 2689–2696 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jordan A, Defechereux P, Verdin E, The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J 20, 1726–1738 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewinski MK, Bisgrove D, Shinn P, Chen H, Hoffmann C, Hannenhalli S, Verdin E, Berry CC, Ecker JR, Bushman FD, Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J Virol 79, 6610–6619 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M, Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogel MJ, Guelen L, de Wit E, Peric-Hupkes D, Loden M, Talhout W, Feenstra M, Abbas B, Classen AK, van Steensel B, Human heterochromatin proteins form large domains containing KRAB-ZNF genes. Genome Res 16, 1493–1504 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chantalat S, Depaux A, Hery P, Barral S, Thuret JY, Dimitrov S, Gerard M, Histone H3 trimethylation at lysine 36 is associated with constitutive and facultative heterochromatin. Genome Res 21, 1426–1437 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL, A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hahn MA, Wu X, Li AX, Hahn T, Pfeifer GP, Relationship between gene body DNA methylation and intragenic H3K9me3 and H3K36me3 chromatin marks. PLoS One 6, e18844 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlson JM, Brumme CJ, Martin E, Listgarten J, Brockman MA, Le AQ, Chui CK, Cotton LA, Knapp DJ, Riddler SA, Haubrich R, Nelson G, Pfeifer N, Deziel CE, Heckerman D, Apps R, Carrington M, Mallal S, Harrigan PR, John M, Brumme ZL, International HIVAC, Correlates of protective cellular immunity revealed by analysis of population-level immune escape pathways in HIV-1. J Virol 86, 13202–13216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Gils MJ, Bunnik EM, Boeser-Nunnink BD, Burger JA, Terlouw-Klein M, Verwer N, Schuitemaker H, Longer V1V2 region with increased number of potential N-linked glycosylation sites in the HIV-1 envelope glycoprotein protects against HIV-specific neutralizing antibodies. J Virol 85, 6986–6995 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaillon A, Braibant M, Moreau T, Thenin S, Moreau A, Autran B, Barin F, The V1V2 domain and an N-linked glycosylation site in the V3 loop of the HIV-1 envelope glycoprotein modulate neutralization sensitivity to the human broadly neutralizing antibody 2G12. J Virol 85, 3642–3648 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lengauer T, Sander O, Sierra S, Thielen A, Kaiser R, Bioinformatics prediction of HIV coreceptor usage. Nat Biotechnol 25, 1407–1410 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Bricault CA, Yusim K, Seaman MS, Yoon H, Theiler J, Giorgi EE, Wagh K, Theiler M, Hraber P, Macke JP, Kreider EF, Learn GH, Hahn BH, Scheid JF, Kovacs JM, Shields JL, Lavine CL, Ghantous F, Rist M, Bayne MG, Neubauer GH, McMahan K, Peng H, Cheneau C, Jones JJ, Zeng J, Ochsenbauer C, Nkolola JP, Stephenson KE, Chen B, Gnanakaran S, Bonsignori M, Williams LD, Haynes BF, Doria-Rose N, Mascola JR, Montefiori DC, Barouch DH, Korber B, HIV-1 Neutralizing Antibody Signatures and Application to Epitope-Targeted Vaccine Design. Cell Host Microbe 26, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC, Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N Engl J Med 332, 228–232 (1995). [DOI] [PubMed] [Google Scholar]
- 37.Deacon NJ, Tsykin A, Solomon A, Smith K, Ludford-Menting M, Hooker DJ, McPhee DA, Greenway AL, Ellett A, Chatfield C, Lawson VA, Crowe S, Maerz A, Sonza S, Learmont J, Sullivan JS, Cunningham A, Dwyer D, Dowton D, Mills J, Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270, 988–991 (1995). [DOI] [PubMed] [Google Scholar]
- 38.Rhodes DI, Ashton L, Solomon A, Carr A, Cooper D, Kaldor J, Deacon N, Characterization of three nef-defective human immunodeficiency virus type 1 strains associated with long-term nonprogression. Australian Long-Term Nonprogressor Study Group. J Virol 74, 10581–10588 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Churchill MJ, Rhodes DI, Learmont JC, Sullivan JS, Wesselingh SL, Cooke IR, Deacon NJ, Gorry PR, Longitudinal analysis of human immunodeficiency virus type 1 nef/long terminal repeat sequences in a cohort of long-term survivors infected from a single source. J Virol 80, 1047–1052 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ, Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 481, 371–375 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris RS, Liddament MT, Retroviral restriction by APOBEC proteins. Nat Rev Immunol 4, 868–877 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Ebrahimi D, Anwar F, Davenport MP, APOBEC3G and APOBEC3F rarely co-mutate the same HIV genome. Retrovirology 9, 113 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F, HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110, 521–529 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Battivelli E, Dahabieh MS, Abdel-Mohsen M, Svensson JP, Tojal Da Silva I, Cohn LB, Gramatica A, Deeks S, Greene WC, Pillai SK, Verdin E, Distinct chromatin functional states correlate with HIV latency reactivation in infected primary CD4(+) T cells. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sherrill-Mix S, Lewinski MK, Famiglietti M, Bosque A, Malani N, Ocwieja KE, Berry CC, Looney D, Shan L, Agosto LM, Pace MJ, Siliciano RF, O’Doherty U, Guatelli J, Planelles V, Bushman FD, HIV latency and integration site placement in five cell-based models. Retrovirology 10, 90 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bailey JR, Williams TM, Siliciano RF, Blankson JN, Maintenance of viral suppression in HIV-1-infected HLA-B*57+ elite suppressors despite CTL escape mutations. J Exp Med 203, 1357–1369 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mens H, Kearney M, Wiegand A, Shao W, Schonning K, Gerstoft J, Obel N, Maldarelli F, Mellors JW, Benfield T, Coffin JM, HIV-1 continues to replicate and evolve in patients with natural control of HIV infection. J Virol 84, 12971–12981 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Connell KA, Brennan TP, Bailey JR, Ray SC, Siliciano RF, Blankson JN, Control of HIV-1 in elite suppressors despite ongoing replication and evolution in plasma virus. J Virol 84, 7018–7028 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grenfell BT, Pybus OG, Gog JR, Wood JL, Daly JM, Mumford JA, Holmes EC, Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303, 327–332 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Carlson JM, Du VY, Pfeifer N, Bansal A, Tan VY, Power K, Brumme CJ, Kreimer A, DeZiel CE, Fusi N, Schaefer M, Brockman MA, Gilmour J, Price MA, Kilembe W, Haubrich R, John M, Mallal S, Shapiro R, Frater J, Harrigan PR, Ndung’u T, Allen S, Heckerman D, Sidney J, Allen TM, Goulder PJ, Brumme ZL, Hunter E, Goepfert PA, Impact of pre-adapted HIV transmission. Nat Med 22, 606–613 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pohlmeyer CW, Buckheit RW 3rd, Siliciano RF, Blankson JN, CD8+ T cells from HLA-B*57 elite suppressors effectively suppress replication of HIV-1 escape mutants. Retrovirology 10, 152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu XG, Lichterfeld M, Chetty S, Williams KL, Mui SK, Miura T, Frahm N, Feeney ME, Tang Y, Pereyra F, Labute MX, Pfafferott K, Leslie A, Crawford H, Allgaier R, Hildebrand W, Kaslow R, Brander C, Allen TM, Rosenberg ES, Kiepiela P, Vajpayee M, Goepfert PA, Altfeld M, Goulder PJ, Walker BD, Mutually exclusive T-cell receptor induction and differential susceptibility to human immunodeficiency virus type 1 mutational escape associated with a two-amino-acid difference between HLA class I subtypes. J Virol 81, 1619–1631 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buzon MJ, Yang Y, Ouyang Z, Sun H, Seiss K, Rogich J, Le Gall S, Pereyra F, Rosenberg ES, Yu XG, Lichterfeld M, Susceptibility to CD8 T-Cell-Mediated Killing Influences the Reservoir of Latently HIV-1-Infected CD4 T Cells. J Acquir Immune Defic Syndr 65, 1–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koofhethile CK, Ndhlovu ZM, Thobakgale-Tshabalala C, Prado JG, Ismail N, Mncube Z, Mkhize L, van der Stok M, Yende N, Walker BD, Goulder PJR, Ndung’u T, CD8+ T Cell Breadth and Ex Vivo Virus Inhibition Capacity Distinguish between Viremic Controllers with and without Protective HLA Class I Alleles. J Virol 90, 6818–6831 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boritz EA, Darko S, Swaszek L, Wolf G, Wells D, Wu X, Henry AR, Laboune F, Hu J, Ambrozak D, Hughes MS, Hoh R, Casazza JP, Vostal A, Bunis D, Nganou-Makamdop K, Lee JS, Migueles SA, Koup RA, Connors M, Moir S, Schacker T, Maldarelli F, Hughes SH, Deeks SG, Douek DC, Multiple Origins of Virus Persistence during Natural Control of HIV Infection. Cell 166, 1004–1015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pollack RA, Jones RB, Pertea M, Bruner KM, Martin AR, Thomas AS, Capoferri AA, Beg SA, Huang SH, Karandish S, Hao H, Halper-Stromberg E, Yong PC, Kovacs C, Benko E, Siliciano RF, Ho YC, Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host Microbe 21, 494–506 e494 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Imamichi H, Smith M, Adelsberger JW, Izumi T, Scrimieri F, Sherman BT, Rehm CA, Imamichi T, Pau A, Catalfamo M, Fauci AS, Lane HC, Defective HIV-1 proviruses produce viral proteins. Proc Natl Acad Sci U S A 117, 3704–3710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peluso MJ, Bacchetti P, Ritter KD, Beg S, Lai J, Martin JN, Hunt PW, Henrich TJ, Siliciano JD, Siliciano RF, Laird GM, Deeks SG, Differential decay of intact and defective proviral DNA in HIV-1-infected individuals on suppressive antiretroviral therapy. JCI Insight 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Falcinelli SD, Kilpatrick KW, Read J, Murtagh R, Allard B, Ghofrani S, Kirchherr J, James KS, Stuelke E, Baker C, Kuruc JD, Eron JJ, Hudgens MG, Gay CL, Margolis DM, Archin NM, Longitudinal dynamics of intact HIV proviral DNA and outgrowth virus frequencies in a cohort of ART-treated individuals. J Infect Dis, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gandhi RT, Cyktor JC, Bosch RJ, Mar H, Laird GM, Martin A, Collier AC, Riddler SA, Macatangay BJ, Rinaldo CR, Eron JJ, Siliciano JD, McMahon DK, Mellors JW, Team ACTGA, Selective Decay of Intact HIV-1 Proviral DNA on Antiretroviral Therapy. J Infect Dis 223, 225–233 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garcia-Broncano P, Maddali S, Einkauf KB, Jiang C, Gao C, Chevalier J, Chowdhury FZ, Maswabi K, Ajibola G, Moyo S, Mohammed T, Ncube T, Makhema J, Jean-Philippe P, Yu XG, Powis KM, Lockman S, Kuritzkes DR, Shapiro R, Lichterfeld M, Early antiretroviral therapy in neonates with HIV-1 infection restricts viral reservoir size and induces a distinct innate immune profile. Sci Transl Med 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, Rood JE, Berkley AM, Sacha JB, Cogliano-Shutta NA, Lloyd M, Roby G, Kwan R, McLaughlin M, Stallings S, Rehm C, O’Shea MA, Mican J, Packard BZ, Komoriya A, Palmer S, Wiegand AP, Maldarelli F, Coffin JM, Mellors JW, Hallahan CW, Follman DA, Connors M, Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity 29, 1009–1021 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Becker JS, McCarthy RL, Sidoli S, Donahue G, Kaeding KE, He Z, Lin S, Garcia BA, Zaret KS, Genomic and Proteomic Resolution of Heterochromatin and Its Restriction of Alternate Fate Genes. Mol Cell 68, 1023–1037 e1015 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tchasovnikarova IA, Kingston RE, Beyond the Histone Code: A Physical Map of Chromatin States. Mol Cell 69, 5–7 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Gottgens B, Dougan G, Dawson MA, Lehner PJ, GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 348, 1481–1485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Serrao E, Cherepanov P, Engelman AN, Amplification, Next-generation Sequencing, and Genomic DNA Mapping of Retroviral Integration Sites. J Vis Exp, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakamura T, Yamada KD, Tomii K, Katoh K, Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34, 2490–2492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Llano A, Cedeño S, Arrieta SS, Brander C. (2019). [Google Scholar]
- 70.Zhang M, Gaschen B, Blay W, Foley B, Haigwood N, Kuiken C, Korber B, Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology 14, 1229–1246 (2004). [DOI] [PubMed] [Google Scholar]
- 71.Deng W, Maust BS, Nickle DC, Learn GH, Liu Y, Heath L, Kosakovsky Pond SL, Mullins JI, DIVEIN: a web server to analyze phylogenies, sequence divergence, diversity, and informative sites. Biotechniques 48, 405–408 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Digitale JC, Callaway PC, Martin M, Nelson G, Viard M, Rek J, Arinaitwe E, Dorsey G, Kamya M, Carrington M, Rodriguez-Barraquer I, Feeney ME, HLA Alleles B(*)53:01 and C(*)06:02 Are Associated With Higher Risk of P. falciparum Parasitemia in a Cohort in Uganda. Front Immunol 12, 650028 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Benjamini YH, Yosef, Controlling the false discovery rate: A practical and powerful approach to multiple testing. . Journal of the Royal Statistical Society, Series B: Methodological 57, 289–300 (1995). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.