

Abstract

Pinocembrin, a bioflavonoid, is extensively used in complementary/alternative medicine. It turns out as a promising candidate against neurodegenerative diseases because of its multifaceted pharmacological action toward neuroprotection. However, literature evidence is still lacking for its inhibitory action on CYP1A2, which is responsible for xenobiotic metabolism leading to the generation of toxic metabolites and bioactivation of procarcinogens. In the present study, our aim was to evaluate the CYP1A2 inhibitory potential of pinocembrin via in silico, in vitro, and in vivo investigations. From the results of in vitro studies, pinocembrin is found to be a potent and competitive inhibitor of CYP1A2. In vitro–in vivo extrapolation results indicate the potential of pinocembrin to interact with CYP1A2 substrate drugs clinically. Molecular docking-based in silico studies demonstrate the strong interaction of pinocembrin with human CYP1A2. In in vivo investigations using a rat model, pinocembrin displayed a marked alteration in the plasma exposure of CYP1A2 substrate drugs, namely, caffeine and tacrine. In conclusion, pinocembrin has a potent CYP1A2 inhibitory action to cause drug interactions, and further confirmatory study is warranted at the clinical level.
1. Introduction
Pinocembrin, a bioflavonoid abundantly present in propolis and Piper genus species,1,2 has emerged as a promising candidate against neurodegenerative diseases, including Alzheimer’s disease (AD), due to its multifaceted pharmacological actions.3−6 AD is accompanied by the accumulation of amyloid-beta (Aβ) aggregates and the formation of neurofibrillary tangles in the brain that lead to neuroinflammation, cell death, and neurodegeneration.7,8 Aβ can bind to the receptor for advanced glycation end products (RAGE), whose expression gets upregulated in AD, and the Aβ–RAGE interaction induces an inflammatory response.9 Pinocembrin inhibits the RAGE expression and the Aβ–RAGE interaction to exert its protective role.10 Another protein, mitogen-activated protein kinase (MAPK), primarily performs neuronal apoptosis by initiating proinflammatory and proapoptotic signals in AD.11 Pinocembrin is reported to suppress the activation of MAPKs.12 Extracellular stress-related kinase phosphorylation (p-ERK) leads to tau phosphorylation, which orchestrates the formation of neurofibrillary tangles and senile plaques. Regulation of p-ERK by pinocembrin reverses neurotoxicity.13 The cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) and the brain-derived neurotrophic factor (BDNF) play a role in neuronal survival, plasticity, dendritic branching, formation of new synapses, and modulation in the profile of excitatory and inhibitory neurotransmitters.14,15 It is reported that pinocembrin has the capability to recover the cholinergic system in the transgenic mouse model by conserving the ERK-CREB-BDNF pathway.4 Pinocembrin maintains blood-brain barrier integrity, prevents apoptotic signals, and alleviates impaired autophagy.16−18 Its anti-inflammatory activity is mediated by modulation of interleukin-1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), prostaglandin E2 (PGE2), and nitric oxide by suppressing the phosphoinositide-3-kinase (PI3K)/protein kinase B (Akt)/nuclear factor kappa light chain enhancer of the activated B cell (NF-κB) signaling pathway.19 Pinocembrin is reported to protect against Parkinson’s disease via hindering 1-methyl-4-phenylpyridinium (MPP+)-induced oxidative damage and activating ERK1/2 signaling pathways.20
Cytochrome P450 (CYP) is a group of proteins responsible for the phase I metabolism of most drugs and various xenobiotics. Any change in the activity of these CYP enzymes can cause an alteration in the pharmacokinetic behavior of a drug that can lead to unwanted drug interactions.21,22 CYP1A2 is one of the crucial CYP isoforms majorly expressed in the hepatic region. CYP1A2 is mainly responsible for the metabolism of more than 100 clinically used drugs that particularly belong to the class of analgesic, antipsychotic, anti-Parkinson, anti-Alzheimer, antidepressant, CNS stimulator, anticancer, and so on.23−25 CYP1A2 also causes biotransformation of endogenous compounds like melatonin, bilirubin, uroporphyrinogen, etc. as well as bioactivates procarcinogens.26 Tobacco contains a procarcinogen, namely, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), which is reported to cause lung cancer due to the generation of its CYP1A2-mediated metabolite.27 Further, induction of CYP1A2 activity also occurs due to various factors, including smoking, which leads to the loss of therapeutic effects of substrate drugs as well as contributes markedly to the enhancement of the risk for lung cancer.28 Similar evidence is the formation of CYP1A2-mediated carcinogenic hydroxylamines that are generated due to the consumption of cooked muscle meat containing heterocyclic aromatic amines.29 Aflatoxin B1, a procarcinogen that is metabolized by CYP1A2 and produces B1-8,9-exo-epoxide, causes toxicity leading to the development of hepatocellular carcinoma.30 Moreover, drugs that undergo metabolism by CYP1A2 generate metabolites, which can precipitate hepatotoxicity, like in the case of tacrine.31 Therefore, research is being conducted to identify a potent CYP1A2 inhibitor that could be utilized as a chemoprotective agent or in the reduction of CYP1A2-mediated hepatotoxicity.32,33 It is also evident from the literature that fluvoxamine, a potent CYP1A2 inhibitor upon coadministration with clozapine, can prevent the generation of the toxic metabolite norclozapine.34 Therefore, information on CYP1A2 inhibition by any candidate is not only advantageous to avoid pharmacokinetic interactions but also explicitly essential to reduce the harmful effects of xenobiotic metabolism-mediated toxicities. In this context, pinocembrin is reported to have a weak inhibitory action on various CYP enzymes (CYP3A4, CYP2C9, CYP2E1, and CYP2C8).35−37 However, little information is known about the CYP1A2 inhibitory activity of pinocembrin in human liver microsomes (HLM).38
Hence, the objectives of the current research work were as follows: (a) in vitro study for the CYP1A2 inhibition activity of pinocembrin using HLM and rat liver microsomes (RLM), (b) prediction of the drug interaction potential of pinocembrin by in vitro–in vivo extrapolation, (c) in silico evaluation of the type of interaction between pinocembrin and the human CYP1A2 enzyme by molecular docking analysis, and (d) in vivo investigations for the effect of pinocembrin on pharmacokinetic alteration of CYP1A2 substrates using a rat model.
2. Results and Discussion
2.1. Pinocembrin Showed Potent CYP1A2 Inhibition in HLM
Before conducting the inhibition experiments, the stability of pinocembrin was assessed in HLM. The amount of pinocembrin remaining at the different incubation times was compared with 0 min data to evaluate any degradation of pinocembrin in HLM. Results showed a negligible disappearance of pinocembrin in HLM up to the experimental time frame (Figure 1A). The obtained results suggest that pinocembrin is stable in HLM under experimental conditions. This study helped to ensure whether pinocembrin remained available in the reaction mixture containing liver microsomes to inhibit the activity of the CYP1A2 enzyme. A similar stability study in HLM was also performed before conducting CYP2C8 inhibition experiments to assess the stability of steviol acyl glucuronide.39
Figure 1.

(A) Stability of pinocembrin in HLM, (B) Michaelis–Menten plot for the formation of paracetamol in HLM, and IC50 curves of fluvoxamine (C) and pinocembrin (D) for CYP1A2-catalyzed phenacetin O-deethylation in HLM. Data are presented as the mean ± SEM (n = 3).
Then, we examined the inhibitory potential of pinocembrin on the enzymatic activity of CYP1A2 in HLM using the probe substrate phenacetin. Initially, kinetic parameters for CYP1A2-catalyzed phenacetin O-deethylation were calculated. Results of kinetic analysis are presented in the Michaelis–Menten plot for the generation of paracetamol in HLM (Figure 1B). Kinetic parameters for the index reaction, i.e., the maximum velocity of the uninhibited reaction (Vmax), and the Michaelis constant (Km) calculated were 1584 ± 12 pmol/min/mg of protein and 160 ± 7 μΜ, respectively. The results are in line with the reported values.40,41
The next part of the study was the investigation of the CYP1A2 inhibitory potential of pinocembrin, where the concentration of the substrate was set at 145 μM, which was less than and close to its experimental Km value.42 The IC50 value for fluvoxamine (positive control) was calculated in parallel with pinocembrin and found to be 0.4 ± 0.01 μM (Figure 1C). Reported IC50 value of fluvoxamine for CYP1A2-catalyzed phenacetin O-deethylation corroborates with our present investigation results.43 Results revealed that pinocembrin strongly inhibits the activity of the CYP1A2 enzyme in HLM, depicting an IC50 value of 0.52 ± 0.07 μM (Figure 1D). From in vitro inhibition results in HLM, pinocembrin is found to be a potent inhibitor of CYP1A2 (IC50 < 1 μM). Rutaecarpine, a flavonoid, is also reported to have potent CYP1A2 inhibitory activity (IC50 of 20 nM) using 7-ethoxyresorufin O-deethylation in a bacterial membrane expressing human CYP enzymes.44 Rofecoxib, an anti-inflammatory drug, also displayed strong inhibitory action on CYP1A2-mediated phenacetin O-deethylation in HLM with an IC50 value of 4.2 μM.45 The potential of rofecoxib to inhibit the activity of CYP1A2 was also observed at a clinical level. Rofecoxib increased the plasma concentration of the CYP1A2 substrate tizanidine by 13.6-fold in humans.46
2.2. Pinocembrin Displayed Competitive CYP1A2 Inhibition in HLM
Further mechanistic studies were performed for evaluating the mode of CYP1A2 inhibition by pinocembrin. For this investigation, various concentrations of phenacetin (72.5–580 μM) and pinocembrin (0.4–3.2 μM) were used in the reaction. On the basis of paracetamol generation data, the mode of CYP1A2 inhibition was found to be of the competitive type. The calculated inhibition constant (Ki) value was 0.27 ± 0.11 μM with an R2 value of 0.9916. The mode of inhibition was evaluated on the basis of the y-intercepts, x-intercepts, and slopes for each data set obtained in the experiment during the visual investigation of the Lineweaver–Burk plot (Figure 2A) and the Dixon plot (Figure 2B), which was confirmed to be a competitive type of inhibition. Competitive inhibition occurs when the inhibitor binds at the same site on the enzyme where the substrate binds, thereby inhibiting substrate metabolism. The competitive behavior of CYP1A2 inhibition by pinocembrin can be correlated with the relationship between Ki and the IC50 value of pinocembrin. The competitive, noncompetitive, and mixed inhibitors have Ki = IC50/2, Ki = IC50, and Ki = IC50/2 to IC50, respectively.47 From the in vitro results, the Ki value obtained for pinocembrin is close to half of its IC50 value, i.e., 0.27 μM [0.52 μM (IC50)/2 = 0.26 μM]. Luteolin, a flavonoid, also competitively inhibited CYP1A2 activity in HLM with IC50 and Ki values of 6.09 and 3.18 μM, respectively. The Ki value obtained for it was also almost half of its IC50 value, i.e., 6.09/2 = 3.045.48
Figure 2.

(A) Lineweaver–Burk plot for the effect of pinocembrin on the kinetics of CYP1A2-catalyzed phenacetin O-deethylation in HLM. The paracetamol formation was evaluated at four phenacetin concentrations (72.5, 145, 290, and 580 μM) in the absence (plot represented by blue) and presence of pinocembrin (plot represented by red, green, yellow, and pink for 0.4, 0.8, 1.6, and 3.2 μM, respectively). Data are presented as the mean ± SEM (n = 3); (B) Dixon plot for the effect of pinocembrin on the kinetics of CYP1A2-catalyzed phenacetin O-deethylation in HLM. The paracetamol formation was evaluated at four pinocembrin concentrations (0, 0.4, 0.8, 1.6, and 3.2 μM) at four concentrations of phenacetin (plot represented by blue, yellow, green, and red for 72.5, 145, 290, and 580 μM, respectively). Data are presented as the mean ± SEM (n = 3).
2.3. Pinocembrin Predicted to Cause the CYP1A2 Inhibition-Mediated Drug Interaction in Humans
From the in vitro investigation in HLM, pinocembrin is found to be a potent CYP1A2 inhibitor. With the help of in vitro results, the potential of pinocembrin to cause drug interactions in in vivo (human) at the hepatic level was predicted. The highest plasma concentration (Cmax) of pinocembrin after intravenous administration was 280 ng/mL (i.e., ∼1093 nM).49 As the protein concentration of microsomes used in in vitro studies was low, the nonspecific binding with microsomes was regarded insignificant. The determined R value was 5.0 (i.e., >1.1), which represents the ratio of the area under the curve (AUC) in the absence and presence of an inhibitor, which is directly proportional to [I]/Ki. Results depict the potential of pinocembrin to severely interact with substrate drugs of CYP1A2 at the hepatic level.
From the above investigation, pinocembrin is found to be a potent CYP1A2 inhibitor in HLM and is likely to cause interactions with the CYP1A2 substrate drugs at the clinical level. The ability of a compound to inhibit the CYP enzyme can be predicted with the help of computational tools. These tools help to understand better the relation between predicted results and the experimental outcomes. Therefore, computational analysis for CYP1A2 inhibition by pinocembrin using the SwissADME server was used to predict the ability of pinocembrin to inhibit the CYP1A2 enzyme. Results demonstrated it as an inhibitor of CYP1A2 that matches our in vitro outcomes. Similarly, computational analysis regarding pinocembrin inhibition was also done for other CYP isoforms. Pinocembrin was described as a noninhibitor of CYP2D6, CYP3A4, and CYP2C9 but an inhibitor of CYP2C19 (Table S1, Supporting Information). The reported IC50 values for CYP2C9, CYP2C19, and CYP3A4 inhibition by pinocembrin are 13, 22, and 28 μM, respectively, i.e., in the category of weak or no inhibition.35,49
2.4. Pinocembrin Interacted Strongly with Human CYP1A2
Before proceeding further to evaluate the inhibitory potential of pinocembrin in the in vivo model, a molecular docking study was performed between the human CYP1A2 enzyme (target) and pinocembrin (ligand). It aids in better understanding the pinocembrin-mediated CYP1A2 interaction based on interacting amino acid residues, favorable binding orientation, significant interactions, interaction energy, binding affinity, and involved forces. According to the ligand’s comprehensive docking structure and superimposition, the conformer that comprises the best pose was selected that have a minimum −CDOCKER interaction energy of −37.285 kcal/mol and a CDOCKER energy of −28.524 kcal/mol. More negative CDOCKER interaction energy and CDOCKER energy indicate significant binding between the test candidate and the target enzyme.50,51 The analysis of the molecular interactions (Figure 3) showed that pinocembrin interacts with active amino acid residues of enzyme like ARG A: 456, ARG A: 108, and LEU A: 450 through conventional hydrogen bonding at distances of approximately 1.83, 6.29, and 2.44 Å, respectively; amino acids CYS A: 458, THR A: 385, and PHE A: 451 via carbon–hydrogen bond, at distances of approximately 2.42, 3.51, and 2.61 Å, respectively; amino acid PHE A: 451 through the amide−π stacking bond, at a distance of approximately 4.55 Å and amino acids ILE A: 386, ILE A: 459, ALA A: 317, CYS A: 458, and CYS A: 458 through the π–alkyl bond, at distances of approximately 4.95, 5.13, 3.42, 4.07, and 5.08 Å, respectively. In the analysis, we have observed that pinocembrin interacted with a maximum number of active amino acid residues present in the active site of CYP1A2 as defined by Sansen et al.(52) through H-bonding, hydrophobic, and π–π interactions. The observations obtained from the in silico analysis corroborate with the in vitro findings. In a similar study, miltirone inhibited the activity of CYP1A2 in HLM with an IC50 of 1.73 μM. Kinetic analysis revealed miltirone to be a mixed-type inhibitor of CYP1A2. Results of molecular docking analysis illustrated that miltirone interacted strongly with the active site of CYP1A2.53 Another study reported that diosmetin competitively inhibited the activity of the CYP2C9 enzyme with IC50 and Ki values of 3.55 and 1.71 μM, respectively. The molecular docking study also showed that diosmetin strongly interacted with the active cavity of the CYP2C9 enzyme.54
Figure 3.

(A) 3D stereo image and (B) 2D stereo image of the molecular docking study for the interaction of pinocembrin with the active site of the human CYP1A2 enzyme.
2.5. Pinocembrin Hindered CYP1A2 Activity in RLM
From the in vitro and in silico studies, pinocembrin is found to be a strong CYP1A2 inhibitor in HLM and interacted strongly with the human CYP1A2 enzyme, respectively. Although CYP1A2 is a conserved enzyme and is present in rats, the rate of metabolism may be different compared to humans.55 Also, the subsequent part of this study involved an in vivo investigation of pinocembrin inhibition on CYP1A2 activity in the rat model. Therefore, pinocembrin's stability and its CYP1A2 inhibition potential were also investigated in rat liver microsomes (RLM). Results showed a negligible disappearance of pinocembrin in RLM up to the experimental time frame (Figure 4A). It suggests that pinocembrin is stable in RLM under experimental conditions. The calculated IC50 value for CYP1A2 inhibition in RLM was 3.11 ± 0.09 μM (Figure 4B). On comparing the results of pinocembrin on CYP1A2 inhibition in HLM and RLM, it was found that pinocembrin is a more potent inhibitor of CYP1A2 in HLM as compared to RLM. From these results, it can be stated that there is a possibility of a difference in the extent of interactions of pinocembrin between humans and rats with CYP1A2 substrates like observed in other CYP enzymes. Similar results are reported for the CYP1A2 inhibitor furafylline. The reported IC50 values of furafylline in HLM and RLM were 0.48 and 20.80 μM, respectively.56
Figure 4.

(A) Stability of pinocembrin in RLM; (B) IC50 curve of pinocembrin for CYP1A2-catalyzed phenacetin O-deethylation in RLM; (C) inhibitory effect of pinocembrin on metabolism of tacrine in RLM. Data are presented as the mean ± SEM (n = 3).
Then, the CYP1A2 inhibitory effect of pinocembrin in RLM was assessed with tacrine (a CYP1A2 substrate) in the presence and absence of pinocembrin (0–50 μM) up to 60 min. Results displayed an increase in the remaining content of tacrine by a maximum of 45%, depending on the pinocembrin concentration (Figure 4C). These results suggest that pinocembrin has the potential to inhibit CYP1A2 enzymatic activity in RLM as well.
2.6. Pinocembrin Significantly Altered the Pharmacokinetics of CYP1A2 Substrates in a Rat Model
After determining the inhibitory potential of pinocembrin on in vitro CYP1A2 activity in RLM, an in vivo pharmacokinetic study of CYP1A2 substrate drugs was performed in the presence and absence of pinocembrin using a rat model. Pinocembrin was given through the intravenous route to observe the impact on hepatic CYP1A2 enzyme. Caffeine was administered orally, and we investigated pinocembrin’s effect on the pharmacokinetics of caffeine because it is the recommended CYP1A2 substrate by USFDA for evaluating CYP1A2-mediated drug interactions.57 The average plasma concentration versus time profiles and pharmacokinetic parameters of caffeine are presented in Figure 5 and Table 1, respectively. Overall plasma exposure (AUC0–t and AUC0–∞) of caffeine was significantly elevated by 1.6- to 1.7-fold upon concomitant treatment with pinocembrin compared to caffeine alone. Clearance of caffeine was substantially delayed (38%) in the presence of pinocembrin in comparison to caffeine alone. There was no noteworthy effect on any other pharmacokinetic parameters of caffeine due to simultaneous administration of pinocembrin. It is reported that a major portion of caffeine (>95%) is metabolized by CYP1A2, leading to the formation of three metabolites paraxanthine, theophylline, and theobromine. Paraxanthine is solely formed due to CYP1A2 activity, whereas CYP2C8, CYP2C9, CYP3A4, and CYP2E1 are CYP isoforms that have a minor role in caffeine metabolism.24 Literature evidence suggest that pinocembrin has weak or no inhibitory activity against CYP3A4, CYP2C9, CYP2C8, and CYP2E1.35−37 Therefore, these four CYPs play negligible role in the metabolism of caffeine. In the present experiment, we observed that pinocembrin reached plasma concentrations of 2034 and 621 ng/mL after 5 and 15 min of pinocembrin dosing intravenously to rats, respectively. The plasma level of pinocembrin at 5 min (7.9 μM) is higher than the IC50 of pinocembrin in RLM. The concentrations achieved in the liver at 5 and 15 min of pinocembrin administration were 3681 and 2378 ng/g, respectively. Moreover, the liver to plasma ratio is found to be 1.8–3.8 after 5–15 min upon intravenous pinocembrin dosing in rats. Therefore, the observed effect on caffeine pharmacokinetics can be correlated to the slowed-down effect of its metabolism by pinocembrin treatment. A similar enhancement in the plasma level of caffeine by 1.6-fold was observed in humans due to the inhibition of CYP1A2 by thiabendazole.58
Figure 5.

Mean plasma concentration versus time profile of caffeine after oral administration of caffeine alone (group I) and intravenous administration of pinocembrin followed by oral administration of caffeine (group II) in rats. Data are presented as the mean ± SEM (n = 5).
Table 1. Pharmacokinetic Parameters of Caffeine after Oral Administration as Alone (Group I) and Intravenous Administration of Pinocembrin Followed by Oral Administration of Caffeine (Group II) in Ratsa.
| caffeine |
||
|---|---|---|
| pharmacokinetic parameters | group I (alone) | group II (with pinocembrin) |
| Cmax (ng/mL) | 1063 ± 102 | 1319 ± 140 |
| Tmax (h) | 0.7 ± 0.1 | 1.1 ± 0.2 |
| AUC0–t (ng·h/mL) | 2838 ± 141 | 4629 ± 409** |
| AUC0–∞ (ng·h/mL) | 2851 ± 139 | 4864 ± 407** |
| T1/2 (h) | 1.1 ± 0.1 | 0.8 ± 0.1 |
| Vd/F (L/kg) | 8.4 ± 1.2 | 4.1 ± 0.9* |
| Cl/F (L/h/kg) | 5.3 ± 0.3 | 3.3 ± 0.3*** |
Data are presented as the mean ± SEM (n = 5). *p < 0.05, **p < 0.01, and ***p < 0.001 denote statistical significance when compared between group I versus group II. Cmax, the highest plasma concentration; Tmax, the time to reach Cmax; AUC0–t, the area under the curve for plasma concentrations from zero to the last measurable plasma sample time; AUC0–∞, the area under the curve for plasma concentrations from zero to infinity; T1/2, elimination half-life; Vd/F, the volume of distribution after oral administration; Cl/F, clearance after oral administration.
Further, we evaluated the effect of pinocembrin on the pharmacokinetics of tacrine (CYP1A2 substrate), which was used orally in humans, and CYP1A2 is the only CYP isoform involved in its metabolism with a contribution of >90%.25 The average plasma concentration versus time profiles and pharmacokinetic parameters of tacrine and hydroxytacrine are presented in Figure 6 and Table 2, respectively. Overall plasma exposure of tacrine, i.e., based on AUC0–t and AUC0–∞ was significantly enhanced by 2.0- to 2.1-fold, respectively, upon concomitant treatment with pinocembrin compared to tacrine alone. Clearance of tacrine was considerably delayed (42%) in the presence of pinocembrin compared to tacrine alone. There was no substantial effect on any other pharmacokinetic parameters of tacrine, except Cmax that was enhanced by 2.7-fold due to simultaneous administration of pinocembrin. The drug/metabolite ratio (tacrine/hydroxytacrine) was noticeably altered upon concomitant administration of tacrine with pinocembrin. Therefore, the observed effect on the pharmacokinetics of tacrine can be linked to slowing down of the effect of its metabolism by pinocembrin treatment similar to the pinocembrin effect on caffeine. A similar observation for a known CYP1A2 inhibitor, ciprofloxacin, is reported where it leads to a 31% decrease in the clearance of the CYP1A2 substrate ropivacaine in humans.59
Figure 6.
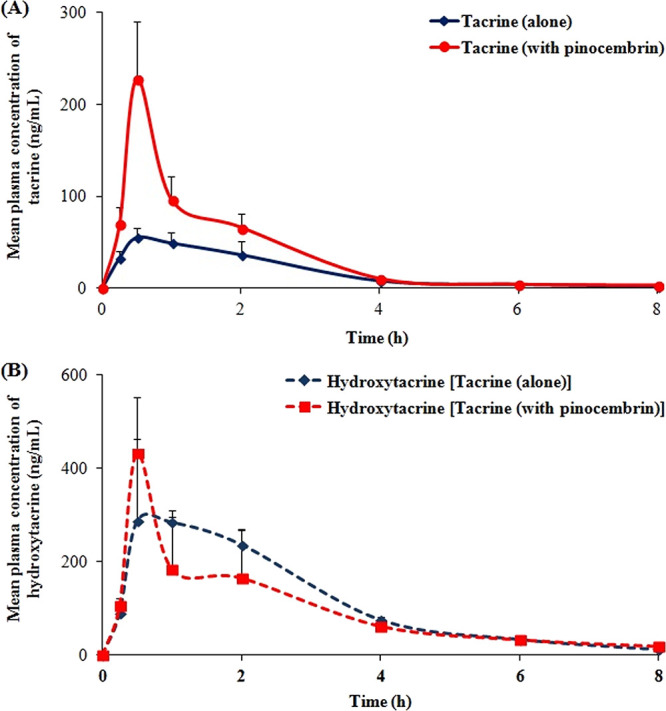
Mean plasma concentration versus time profile of (A) tacrine and (B) hydroxytacrine after oral administration of tacrine alone (group III) and intravenous administration of pinocembrin followed by oral administration of tacrine (group IV) in rats.
Table 2. Pharmacokinetic Parameters of Tacrine and Hydroxytacrine after Oral Administration of Tacrine Alone (Group III) and Intravenous Administration of Pinocembrin Followed by Oral Administration of Tacrine (Group IV) in Ratsa.
| tacrine |
hydroxytacrine |
|||
|---|---|---|---|---|
| pharmacokinetic parameters | group III (alone) | group IV (with pinocembrin) | group III (alone) | group IV (with pinocembrin) |
| Cmax (ng/mL) | 84 ± 16 | 227 ± 63 | 499 ± 126 | 450 ± 170 |
| Tmax (h) | 1.2 ± 0.4 | 0.5 ± 0.0 | 1.3 ± 0.3 | 0.8 ± 0.3 |
| AUC0–t (ng·h/mL) | 142 ± 13 | 297 ± 52* | 907 ± 129 | 763 ± 88 |
| AUC0–∞ (ng·h/mL) | 152 ± 13 | 308 ± 54* | 936 ± 135 | 808 ± 86 |
| T1/2 (h) | 1.7 ± 0.2 | 1.8 ± 0.5 | 1.5 ± 0.3 | 1.6 ± 0.6 |
| Vd/F (L/kg) | 66.2 ± 7.5 | 38.1 ± 10.5 | ||
| Cl/F (L/h/kg) | 27.1 ± 2.1 | 15.8 ± 4.1* | ||
Data are presented as the mean ± SEM (n = 5). *p < 0.05 denotes statistical significance when compared between group III versus group IV.
3. Conclusions
Based on the in vitro findings, pinocembrin is found to be a potent and competitive inhibitor of CYP1A2. Results of in vitro–in vivo extrapolation suggest that pinocembrin is likely to cause drug interactions with the CYP1A2 substrates at the clinical level. Molecular docking analysis illustrates that pinocembrin can strongly interact with the active site of the human CYP1A2 enzyme. Results of in vivo studies using a rat model demonstrate that pinocembrin has the ability to cause a marked alteration upon plasma exposure of two CYP1A2 substrate drugs, caffeine and tacrine, via a delay in their metabolism. Overall results of present investigations using alternative approaches indicate that pinocembrin is established as a potent inhibitor of CYP1A2. It should be avoided as alternative/complementary medicine during concomitant administration with prescribed drugs (CYP1A2 substrates) as it is likely to cause drug interactions. Nevertheless, the CYP1A2 inhibition-mediated beneficial action of pinocembrin has to be established before concurrent therapy to circumvent the toxic effect of metabolites or any CYP1A2 induction-mediated therapeutic failure. These preclinical observations warrant further exploration at the clinical level.
4. Materials and Methods
4.1. Chemicals and Reagents
Pinocembrin (≥95%), tacrine hydrochloride (≥98%), hydroxytacrine maleate, i.e., 1-hydroxytacrine (≥98%), and paracetamol (≥98%) were obtained from Cayman Chemical. Fluvoxamine maleate (≥97%), phenacetin (≥98%), diazepam (∼98%), chlorzoxazone (≥98%), nicotinamide adenine dinucleotide phosphate (NADPH) (≥98%), and Tween 20 were procured from Sigma-Aldrich. Caffeine was obtained from the Indian Pharmacopoeia Commission. RLM (lot no. RT059-D) and HLM (pool of 50 donors; lot no. PL050D-A) were procured from Gibco. MS-grade acetonitrile and formic acid as well as HPLC-grade methanol and acetonitrile were obtained from Thermo Fisher Scientific. Magnesium chloride and monobasic potassium phosphate were purchased from Rankem. PEG-400 and dimethylsulfoxide (DMSO) were procured from Loba Chemie. Ultrapure water was obtained from a water purification system (Direct-Q 3, Merck-Millipore). Characterization data of the test candidate (pinocembrin) are presented in Figures S1–S4.
4.2. Effect of Pinocembrin on CYP1A2 Inhibition in HLM (In Vitro)
4.2.1. Estimation of Pinocembrin Stability
Pinocembrin stability in HLM was evaluated at first. The stock solution of pinocembrin was made in DMSO and diluted further with a solvent mixture (methanol:water = 1:1, v/v) whenever used. Incubations were performed in triplicate. The incubation mixture (100 μL) was composed of phosphate buffer (0.1 M, pH 7.4) (hereafter mentioned as “phosphate buffer”), magnesium chloride (3.3 mM), and HLM (0.3 mg/mL). The reaction mixture was preincubated in a preheated shaking water bath (5 min, 37 °C). After preincubation, pinocembrin (5 μM) was added to the samples, and further incubations were done for 0, 10, 20, and 30 min in a preheated shaking water bath at 37 °C. After the specified incubation time, samples were placed in a precooled thermal block with the addition of chilled acetonitrile having chlorzoxazone (125 ng/mL) as an internal standard (IS). Samples were vortex mixed (2 min), and centrifugation was done at 3000 rpm (15 min). Samples were then transferred to vials for quantitation of pinocembrin by LC–MS/MS (Table S2). The amount of pinocembrin remaining at different incubation times was compared with 0 min data (as 100%) to evaluate any degradation of pinocembrin in HLM.
4.2.2. Measurement of Kinetic Parameters
Kinetic parameters for CYP1A2-catalyzed phenacetin O-deethylation reaction were evaluated in HLM. The stock solutions of phenacetin and paracetamol were prepared in methanol. Then, dilutions for phenacetin were done in phosphate buffer. The individual standard solutions of paracetamol were prepared in methanol. The reaction mixture consisted of phosphate buffer, magnesium chloride (3.3 mM), HLM (0.3 mg/mL), NADPH (1.2 mM) and the probe substrate, i.e., phenacetin (1–1500 μM). The final reaction mixture volume was 200 μL, where the content of the organic solvent was kept less than 0.5% (v/v). By adding NADPH, the reaction was initiated and then incubated in a preheated shaking water bath (30 min, 37 °C). The reaction was carried out in triplicate. After incubation, samples were placed in a precooled thermal block with the addition of chilled acetonitrile (200 μL) to quench the enzymatic reaction. Then, samples were vortex mixed (2 min), and centrifuged at 3000 rpm (15 min). The samples were then transferred into vials for the quantitation of paracetamol by LC–MS/MS (Table S3). The quantitation data were used to calculate Km and Vmax. The upper limit for the metabolite generation rate was limited to 20%.
4.2.3. Determination of IC50
The potential of pinocembrin to inhibit the enzymatic activity of CYP1A2 was evaluated using the optimized reaction protocol as mentioned above, except that the probe substrate concentration was chosen close to its experimental Km value. The stock solution of fluvoxamine (positive control) was made in DMSO, and further dilutions were done in phosphate buffer. CYP1A2 inhibition by fluvoxamine and pinocembrin was evaluated at concentration ranges of 0.05–5 and 0.01–25 μM, respectively. The reaction was carried out in triplicate. Samples without the presence of fluvoxamine or pinocembrin were regarded as the control. Samples were then analyzed for paracetamol by LC–MS/MS (Table S3). The data obtained were used to calculate IC50 values of fluvoxamine and pinocembrin.
4.2.4. Evaluation for the Mode of Inhibition
The next study involved the determination of Ki and the mechanism of pinocembrin inhibition toward CYP1A2. The probe substrate concentration levels of 72.5, 145, 290, and 580 μM and pinocembrin concentration levels of 0, 0.4, 0.8, 1.6, and 3.2 μM were chosen for the experimentation. The remaining reaction conditions and procedures for sample analysis of paracetamol were similar to those described above. Incubations were carried out in triplicate. The results obtained were utilized for plotting the Lineweaver–Burk plot as well as the Dixon plot.
4.2.5. Data Analysis
The software GraphPad Prism was utilized for analyzing the Vmax and Km for the index reaction; IC50 values of fluvoxamine and pinocembrin; Ki and the mode of inhibition by pinocembrin. Data regarding the metabolite formation rate versus the probe substrate concentrations were fitted to nonlinear regression analysis for estimation of Vmax and Km. The IC50 values of fluvoxamine or pinocembrin were calculated by utilizing the results obtained on percent control of the activity of the enzyme upon inhibition at the log of different concentrations of the inhibitor. The mode of CYP1A2 inhibition and apparent Ki of pinocembrin were evaluated by fitting data regarding the rate of generation of metabolites at the different concentrations of phenacetin and pinocembrin to the various models of enzyme inhibition.
4.3. In Vitro–In Vivo Extrapolation to Predict the Drug Interaction Potential of Pinocembrin
In vitro study data were utilized for predicting the potential of pinocembrin to cause drug interactions at the hepatic level. Along with this, the Cmax value of pinocembrin from a single intravenous dose (20 mg) to human subjects was used.37 CYP1A2 inhibition for pinocembrin was predicted in humans using the following equation, where AUCI/AUCUI signifies the ratio of the AUC of the substrate with an inhibitor (AUCI) or without an inhibitor (AUCUI), [I] is the Cmax of pinocembrin, and Ki represents the inhibition constant obtained in the in vitro study for prediction at the hepatic level:57,60,61
4.4. Effect of Pinocembrin on the Interaction with CYP1A2 (In Silico)
We used a molecular docking study to understand the type of interaction of pinocembrin with CYP1A2. The molecular docking method entails predicting the structure and orientation of a ligand within a certain binding site. In general, docking investigations have two goals: accurate structural modeling and precise activity prediction. However, identifying chemical characteristics that are responsible for particular biological recognition and predicting compound alterations that boost potency are challenging problems to grasp and even more difficult to model on a computer.62,63 Because of these difficulties, docking is usually designed as a multistage procedure, with each step adding one or more levels of complexity.63 The procedure starts with the use of docking algorithms to position small molecules in the active site. This is difficult in and of itself because even very basic organic molecules can have a large number of conformational degrees of freedom.62,63 Sampling these degrees of freedom will be performed with sufficient accuracy to identify the conformation that best matches the receptor structure and is fast enough to permit the evaluation of the number of compounds in a selected docking run.62 In this investigation, we performed the molecular docking study to predict the best binding and most possible trajectories within the active cavity of the human CYP1A2 enzyme. The crystal structure of the enzyme was extracted from the Protein Data Bank (PDB ID 2HI4)52 (available from https://www.rcsb.org/structure/2HI4). A rigid-based docking approach was applied using the CDOCKER (a molecular dynamics (MD) simulated-annealing-based algorithm) with a grid-based protocol for the aim of the receptor–ligand interaction,50,51 as prompted in BIOVIA Discovery Studio Client 4.1 [DS 4.1]. In rigid docking, both the ligand and protein are considered rigid entities, and just the three translational and three rotational degrees of freedom are considered during sampling. In this method, the binding site and the ligand are approximated by “hot” points, and the superposition of the matching point is evaluated.64 Before molecular docking, energy minimization of both the target and ligands was performed using the CHARMM force field with the Momany–Rone partial charge method.65 Polar hydrogen atoms were added to the structure, and all ionizable residues were set to their default protonation state at neutral pH using the option “ligand and protein preparation” protocol instigated in BIOVIA Discovery Studio Client 4.1 [DS 4.1]. The active site of the target was defined by Sansen et al.52 to contain the active amino acid residues such as ARG A: 108*, THR A: 118, THR A: 124, TRP A: 133, ARG A: 137, LEU A: 144, PHE A: 226, PHE A: 260, ASP A: 313, ILE A: 314, GLY A: 316, ALA A: 317*, GLY A: 318, ASP A: 320, THR A: 321, PHE A: 376, PHE A: 381, LEU A: 382, THR A: 385*, ILE A: 386*, HIS A: 388, GLN A: 411, LEU A: 450*, PHE A: 451*, ARG A: 456*, CYS A: 458*, ILE A: 459*, ALA A: 464, and LEU A: 497 in the active pocket of the enzyme. The grid was prepared by applying the protocol “from current selection” using the option “define and edit the binding site from receptor cavities” in the BIOVIA Discovery Studio Client 4.1 platform [DS 4.1]. At the time of molecular docking, the translation center of the ligand was stimulated to a definite position inside the active site of the receptor, forming a series of random spins.65 Therefore, random conformers of the molecule were formed, and each conformer was then relaxed by simulated-annealing molecular dynamics; this concerned “heating” the model molecule at a high temperature (700 K in 2000 steps), subsequently cooling to a target temperature of 300 K in 5000 steps.65 Molecular docking was performed inside a sphere with a radius of 18.799 Å with XYZ coordinates of 4.701, 23.623, and 22.155, respectively, in the active site pocket of the enzyme. After molecular docking, the docked inclusion complexes with the best ranked CDOCKER interaction energy, CDOCKER energy, bond formation between compounds, and active amino acid residues were chosen for the detailed interpretation and correlation.
4.5. Effect of Pinocembrin on CYP1A2 Inhibition in RLM (In Vitro)
4.5.1. Estimation of Pinocembrin Stability
Before evaluating the inhibitory potential of pinocembrin on CYP1A2 activity in RLM, the stability of pinocembrin was assessed first. The study was performed using a similar protocol to that mentioned above to assess pinocembrin stability in HLM. However, the study was conducted with the presence of RLM in place of HLM as further experimentations to investigate pinocembrin's effect on CYP1A2 inhibition were to be done in RLM.39
4.5.2. Determination of IC50
The potency of pinocembrin to inhibit the enzymatic reaction of phenacetin O-deethylation in RLM was evaluated. The protocol was the same as mentioned above to determine the IC50 value of pinocembrin in HLM, except RLM was used in place of HLM.
4.5.3. Metabolic Depletion of Tacrine
The inhibitory effect of pinocembrin on tacrine metabolism was assessed in RLM. First, pinocembrin at concentration levels of 0, 0.5, 5, 10, and 50 μM was preincubated with phosphate buffer, magnesium chloride (3.3 mM), and RLM (1 mg/mL) for 30 min. After preincubation, tacrine (1 μM) was added to the mixture, and NADPH (1.2 mM) was added to initiate the reaction, making the total volume of the reaction mixture 100 μL. Samples were incubated in a preheated shaking water bath at 37 °C for 0, 30, and 60 min. After the specified incubation time, the samples were placed in a precooled thermal block to quench the reaction with simultaneous addition of chilled acetonitrile (100 μL) followed by vortex-mixing (2 min), and centrifugation at 3000 rpm (15 min). The samples were then transferred into vials for analysis of tacrine in LC–MS/MS (Table S4). Metabolic depletion of tacrine with or without pinocembrin was evaluated.
4.6. Effect of Pinocembrin on Pharmacokinetics of CYP1A2 Substrates (In Vivo)
4.6.1. Animal Model and Ethical Approval
Healthy adult male Wistar rats were used to carry out in vivo experimentation. Animals were kept in well-ventilated cages at ambient temperature, i.e., 25 ± 2 °C, and a relative humidity of 40–60% while being in a regularly light-controlled room, i.e., 12 h exposure to light and 12 h devoid of a light source. The animals were given a standard rodent diet with free availability of water. We sought approval from the Institutional Animal Ethics Committee (IAEC approval no. 228/78/2/21) for animal experimentations. The study was performed in accordance with the approved guidelines and regulations of the “Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA)”, New Delhi, India.
4.6.2. Test Article and Dose Formulation
The current study involved the treatment of pinocembrin (10 mg/kg) with caffeine (15 mg/kg) or tacrine (4 mg/kg) where doses were selected based on literature evidence in the rat model.66,67 The dose of pinocembrin was prepared as a solution form using 1% DMSO, 5% Tween 20, 19% PEG-400, and q.s. water (v/v). Caffeine and tacrine dose formulations were made in the form of an aqueous suspension containing sodium carboxymethylcellulose (0.1%, w/v). All doses were freshly prepared on the day of experimentation, where the volume of the dose was kept as 10 mL/kg.
4.6.3. Study Arm and Blood Sampling
Before investigating the effect of pinocembrin on the pharmacokinetics of CYP1A2 substrate drugs, diet was restricted to the animals 10 h prior to the start of the experiment, but water was freely available. On the day of the experiment, animals were randomly divided into four groups with five animals each. The first (group I) and the third group (group III) of animals were treated orally with caffeine alone and tacrine alone, respectively. The second (group II) and the fourth group (group IV) of animals received intravenous administration of pinocembrin before oral treatment of caffeine and tacrine, respectively. Pinocembrin was injected 0.5 h before caffeine or tacrine treatment to achieve the highest effect.68 Blood was withdrawn in Eppendorf tubes having an anticoagulant (5% aqueous EDTA solution, w/v) at 0.25, 0.5, 1.0, 2.0, 4.0, 6.0, and 8.0 h after caffeine and tacrine dosing. Then, blood was centrifuged (8000 rpm, 10 min) to collect the plasma samples (50 μL) and kept at a deep freezer for bioanalysis.
4.6.4. Sample Processing and Bioanalysis
The stock solution at a concentration level of 1 mg/mL for caffeine, IS (phenacetin), tacrine, hydroxytacrine, and IS (diazepam) were prepared individually in DMSO. Further dilutions were done in methanol. Calibration standards were made by adding 5 μL of serially diluted standard solutions of tacrine, hydroxytacrine, and caffeine into blank plasma (45 μL) in concentration ranges from 0.9 to 500, 3.9 to 1000, and 7.8 to 4000 ng/mL, respectively. The plasma samples from the pharmacokinetic investigation (50 μL) were processed by the addition of acetonitrile (200 μL) containing 50 ng/mL phenacetin for caffeine samples and 20 ng/mL diazepam for both tacrine and hydroxytacrine samples. The samples were then vortex mixed (2 min) and centrifuged at 14,000 rpm (10 min). The samples were then transferred to vials for analysis by LC–MS/MS for caffeine (Table S5), tacrine and hydroxytacrine (Table S6).
4.6.5. Pharmacokinetic Data Evaluation and Statistical Analysis
The data obtained from the pharmacokinetic investigation were assessed to estimate pharmacokinetic parameters with the help of the PK solution software by employing a noncompartmental method. Statistical analysis was done using an unpaired Student’s t-test using the software GraphPad Prism by comparing data on alone versus combination treatment for CYP1A2 substrates. The data were regarded to be significant at p < 0.05, p < 0.01, and p < 0.001 levels.
4.7. Liver Distribution of Pinocembrin (In Vivo)
The liver to plasma ratio of pinocembrin was evaluated in the Wistar rats following the above-mentioned protocol for animal experimentation (IEAC approval no. 255/79/8/2021) and the dose formulation of pinocembrin. Briefly, 10 animals in two groups (n = 5) were treated with pinocembrin through an intravenous route at 10 mg/kg. Blood was withdrawn at 0.083 and 0.25 h after pinocembrin dosing in a microcentrifuge tube containing an anticoagulant and centrifuged at 8000 rpm (10 min). Plasma was separated and stored at −80 °C until analysis. Just after blood collection at both time points, animals were sacrificed by carbon dioxide euthanasia followed by cervical dislocation, and the liver was removed, washed using chilled normal saline, blotted dry, and stored at −80 °C until analysis. The liver sample homogenate was prepared at 500 mg/mL in phosphate buffer (10 mM, pH 7.4).69 Matrix match calibration standards were prepared by adding 5 μL of serially diluted standard solutions of pinocembrin into 45 μL of the blank liver homogenate/blank plasma in the concentration range from 0.9 to 2000 ng/mL. The samples of the current investigation (50 μL of the plasma/liver homogenate) were processed by the addition of acetonitrile (200 μL) containing chlorzoxazone (125 ng/mL) as the IS. The samples were then vortex mixed (2 min), centrifuged (14,000 rpm, 10 min), and decanted into vials for analysis by LC–MS/MS (Table S2).
Acknowledgments
S.B., D.M., and A.G. are thankful to CSIR/DST (New Delhi, India) for providing their research fellowships. IIIM publication number: CSIR-IIIM/IPR/00413.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c02315.
Characterization of pinocembrin by 1H NMR, 13C NMR, and HRMS; In silico data of pinocembrin; LC–MS/MS conditions for analysis of experimental samples (PDF)
This research was supported by the Council of Scientific and Industrial Research, New Delhi, India (MLP6006 and HCP38).
The authors declare no competing financial interest.
Supplementary Material
References
- Escriche I.; Juan-Borrás M. Standardizing the analysis of phenolic profile in propolis. Food Res. Int. 2018, 106, 834–841. 10.1016/j.foodres.2018.01.055. [DOI] [PubMed] [Google Scholar]
- López A.; Ming D. S.; Towers G. H. N. Antifungal Activity of Benzoic Acid Derivatives from Piper l anceaefolium. J. Nat. Prod. 2002, 65, 62–64. 10.1021/np010410g. [DOI] [PubMed] [Google Scholar]
- Shen X.; Liu Y.; Luo X.; Yang Z. Advances in biosynthesis, pharmacology, and pharmacokinetics of pinocembrin, a promising natural small-molecule drug. Molecules 2019, 24, 2323. 10.3390/molecules24122323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; Li J. Z.; Song J. K.; Zhou D.; Huang C.; Bai X. Y.; Du G. H. Pinocembrin improves cognition and protects the neurovascular unit in Alzheimer related deficits. Neurobiol. Aging 2014, 35, 1275–1285. 10.1016/j.neurobiolaging.2013.12.031. [DOI] [PubMed] [Google Scholar]
- Lan X.; Wang W.; Li Q.; Wang J. The natural flavonoid pinocembrin: molecular targets and potential therapeutic applications. Mol. Neurobiol. 2016, 53, 1794–1801. 10.1007/s12035-015-9125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes da Silveira C. C.; Luz D. A.; da Silva C. C.; Prediger R. D.; Martins M. D.; Martins M. A.; Fontes-Júnior E. A.; Maia C. S. Propolis: A useful agent on psychiatric and neurological disorders? A focus on CAPE and pinocembrin components. Med. Res. Rev. 2021, 41, 1195–1215. 10.1002/med.21757. [DOI] [PubMed] [Google Scholar]
- Wani A.; AI Rihani S.; Sharma A.; Weadick B.; Govindarajan R.; Khan S. U.; Sharma P. R.; Dogra A.; Nandi U.; Reddy C. N.; Bharate S. S.; Singh G.; Bharate S. B.; Vishwakarma R. A.; Kaddoumi A.; Kumar A. Crocetin promotes clearance of amyloid-β by inducing autophagy via the STK11/LKB1-mediated AMPK pathway. Autophagy 2021, 17, 3813–3832. 10.1080/15548627.2021.1872187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M. P.; LeVine H. Alzheimer’s disease and the β-amyloid peptide. J Alzheimers Dis. 2010, 19, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S. D.; Bierhaus A.; Nawroth P. P.; Stern D. M. RAGE and Alzheimer’s disease: a progression factor for amyloid-β-induced cellular perturbation?. J. Alzheimer’s Dis. 2009, 16, 833–843. 10.3233/JAD-2009-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; Wu C. X.; Zhou D.; Yang F.; Tian S.; Zhang L.; Zhang T. T.; Du G. H. Pinocembrin protects against β-amyloid-induced toxicity in neurons through inhibiting receptor for advanced glycation end products (RAGE)-independent signaling pathways and regulating mitochondrion-mediated apoptosis. BMC Med. 2012, 10, 1–21. 10.1186/1741-7015-10-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E. K.; Choi E. J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta, Mol. Basis Dis. 2010, 1802, 396–405. 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Liu R.; Li J. Z.; Song J. K.; Sun J. L.; Li Y. J.; Zhou S. B.; Zhang T. T.; Du G. H. Pinocembrin protects human brain microvascular endothelial cells against fibrillar amyloid-β1– 40 injury by suppressing the MAPK/NF-κ B inflammatory pathways. BioMed Res. Int. 2014, 2014, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; Nan G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target. Int. J. Mol. Med. 2017, 39, 1338–1346. 10.3892/ijmm.2017.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura C. A.; Valero J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Rev. Neurosci. 2011, 22, 153–169. 10.1515/RNS.2011.018. [DOI] [PubMed] [Google Scholar]
- Lima Giacobbo B.; Doorduin J.; Klein H. C.; Dierckx R. A.; Bromberg E.; de Vries E. F. Brain-derived neurotrophic factor in brain disorders: focus on neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. 10.1007/s12035-018-1283-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F.; Liu R.; Gao M.; Wang Y.; Yu X.; Xuan Z.; Sun J.; Yang F.; Wu C.; Du G. Pinocembrin attenuates blood–brain barrier injury induced by global cerebral ischemia–reperfusion in rats. Brain Res. 2011, 1391, 93–101. 10.1016/j.brainres.2011.03.010. [DOI] [PubMed] [Google Scholar]
- Liu R.; Gao M.; Yang Z. H.; Du G. H. Pinocembrin protects rat brain against oxidation and apoptosis induced by ischemia–reperfusion both in vivo and in vitro. Brain Res. 2008, 1216, 104–115. 10.1016/j.brainres.2008.03.049. [DOI] [PubMed] [Google Scholar]
- Tao J.; Shen C.; Sun Y.; Chen W.; Yan G. Neuroprotective effects of pinocembrin on ischemia/reperfusion-induced brain injury by inhibiting autophagy. Biomed. Pharmacother. 2018, 106, 1003–1010. 10.1016/j.biopha.2018.07.026. [DOI] [PubMed] [Google Scholar]
- Zhou L. T.; Wang K. J.; Li L.; Li H.; Geng M. Pinocembrin inhibits lipopolysaccharide-induced inflammatory mediators production in BV2 microglial cells through suppression of PI3K/Akt/NF-κB pathway. Eur. J. Pharmacol. 2015, 761, 211–216. 10.1016/j.ejphar.2015.06.003. [DOI] [PubMed] [Google Scholar]
- Wang H.; Wang Y.; Zhao L.; Cui Q.; Wang Y.; Du G. Pinocembrin attenuates MPP+-induced neurotoxicity by the induction of heme oxygenase-1 through ERK1/2 pathway. Neurosci. Lett. 2016, 612, 104–109. 10.1016/j.neulet.2015.11.048. [DOI] [PubMed] [Google Scholar]
- Doan J.; Zakrzewski-Jakubiak H.; Roy J.; Turgeon J.; Tannenbaum C. Prevalence and risk of potential cytochrome p450–mediated drug-drug interactions in older hospitalized patients with polypharmacy. Ann. Pharmacother. 2013, 47, 324–332. 10.1345/aph.1R621. [DOI] [PubMed] [Google Scholar]
- Rekić D.; Reynolds K. S.; Zhao P.; Zhang L.; Yoshida K.; Sachar M.; Miller M. P.; Huang S. M.; Zineh I. Clinical drug–drug interaction evaluations to inform drug use and enable drug access. J. Pharm. Sci. 2017, 106, 2214–2218. 10.1016/j.xphs.2017.04.016. [DOI] [PubMed] [Google Scholar]
- Gunes A.; Dahl M. L. Variation in CYP1A2 activity and its clinical implications: influence of environmental factors and genetic polymorphisms. Pharmacogenomics 2008, 9, 625–637. 10.2217/14622416.9.5.625. [DOI] [PubMed] [Google Scholar]
- Zhou S. F.; Wang B.; Yang L. P.; Liu J. P. Structure, function, regulation and polymorphism and the clinical significance of human cytochrome P450 1A2. Drug Metab. Rev. 2010, 42, 268–354. 10.3109/03602530903286476. [DOI] [PubMed] [Google Scholar]
- Faber M. S.; Jetter A.; Fuhr U. Assessment of CYP1A2 activity in clinical practice: why, how, and when?. Basic Clin. Pharmacol. Toxicol. 2005, 97, 125–134. 10.1111/j.1742-7843.2005.pto_973160.x. [DOI] [PubMed] [Google Scholar]
- Crettol S.; Petrovic N.; Murray M. Pharmacogenetics of phase I and phase II drug metabolism. Curr. Pharm. Des. 2010, 16, 204–219. 10.2174/138161210790112674. [DOI] [PubMed] [Google Scholar]
- Corral P. A.; Botello J. F.; Xing C. Design, synthesis, and enzymatic characterization of quinazoline-based CYP1A2 inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 126719 10.1016/j.bmcl.2019.126719. [DOI] [PubMed] [Google Scholar]
- Ma Q.; Lu A. Y. H. CYP1A induction and human risk assessment: an evolving tale of in vitro and in vivo studies. Drug Metab. Dispos. 2007, 35, 1009–1016. 10.1124/dmd.107.015826. [DOI] [PubMed] [Google Scholar]
- Koda M.; Iwasaki M.; Yamano Y.; Lu X.; Katoh T. Association between NAT2, CYP1A1, and CYP1A2 genotypes, heterocyclic aromatic amines, and prostate cancer risk: a case control study in Japan. Environ. Health Prev. Med. 2017, 22, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bbosa G. S.; Kitya D.; Ogwal-Okeng J. Aflatoxins metabolism, effects on epigenetic mechanisms and their role in carcinogenesis. Health 2013, 05, 14–34. 10.4236/health.2013.510A1003. [DOI] [Google Scholar]
- Patocka J.; Jun D.; Kuca K. Possible role of hydroxylated metabolites of tacrine in drug toxicity and therapy of Alzheimer’s disease. Curr. Drug Metab. 2008, 9, 332–335. 10.2174/138920008784220619. [DOI] [PubMed] [Google Scholar]
- Juvonen R. O.; Jokinen E. M.; Javaid A.; Lehtonen M.; Raunio H.; Pentikäinen O. T. Inhibition of human CYP1 enzymes by a classical inhibitor α-naphthoflavone and a novel inhibitor N-(3, 5-dichlorophenyl) cyclopropanecarboxamide: An in vitro and in silico study. Chem. Biol. Drug Des. 2020, 95, 520–533. 10.1111/cbdd.13669. [DOI] [PubMed] [Google Scholar]
- Kato H. Computational prediction of cytochrome P450 inhibition and induction. Drug Metab. Pharmacokinet. 2020, 35, 30–44. 10.1016/j.dmpk.2019.11.006. [DOI] [PubMed] [Google Scholar]
- Lu M. L.; Lane H. Y.; Lin S. K.; Chen K. P.; Chang W. H. Adjunctive fluvoxamine inhibits clozapine-related weight gain and metabolic disturbances. J. Clin. Psychiatry 2004, 65, 469. [DOI] [PubMed] [Google Scholar]
- Tsujimoto M.; Horie M.; Honda H.; Takara K.; Nishiguchi K. The structure–activity correlation on the inhibitory effects of flavonoids on cytochrome P450 3A activity. Biol. Pharm. Bull. 2009, 32, 671–676. 10.1248/bpb.32.671. [DOI] [PubMed] [Google Scholar]
- Li Y.; Ning J.; Wang Y.; Wang C.; Sun C.; Huo X.; Yu Z.; Feng L.; Zhang B.; Tian X.; Ma X. Drug interaction study of flavonoids toward CYP3A4 and their quantitative structure activity relationship (QSAR) analysis for predicting potential effects. Toxicol. Lett. 2018, 294, 27–36. 10.1016/j.toxlet.2018.05.008. [DOI] [PubMed] [Google Scholar]
- Bojić M.; Kondža M.; Rimac H.; Benković G.; Maleš Ž. The effect of flavonoid aglycones on the CYP1A2, CYP2A6, CYP2C8 and CYP2D6 enzymes activity. Molecules 2019, 24, 3174. 10.3390/molecules24173174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siess M. H.; Leclerc J.; Canivenclavier M. C.; Rat P.; Suschetet M. Heterogenous effects of natural flavonoids on monooxygenase activities in human and rat liver microsomes. Toxicol. Appl. Pharmacol. 1995, 130, 73–78. 10.1006/taap.1995.1010. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Zhou D.; Wang Y.; Li J.; Wang M.; Lu J.; Zhang H. CYP2C8-mediated interaction between repaglinide and steviol acyl glucuronide: In vitro investigations using rat and human matrices and in vivo pharmacokinetic evaluation in rats. Food Chem. Toxicol. 2016, 94, 138–147. 10.1016/j.fct.2016.05.024. [DOI] [PubMed] [Google Scholar]
- Walsky R. L.; Obach R. S. Validated assays for human cytochrome P450 activities. Drug Metab. Dispos. 2004, 32, 647–660. 10.1124/dmd.32.6.647. [DOI] [PubMed] [Google Scholar]
- Lin T.; Pan K.; Mordenti J.; Pan L. In vitro assessment of cytochrome P450 inhibition: Strategies for increasing LC/MS-based assay throughput using a one-point IC50 method and multiplexing high-performance liquid chromatography. J. Pharm. Sci. 2007, 96, 2485–2493. 10.1002/jps.20884. [DOI] [PubMed] [Google Scholar]
- USFDA . Drug Development and Drug Interactions. Table of Substrates, Inhibitors and Inducers; U.S. Food and Drug Administration: Silver Spring, MD, 2017. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table- substrates-inhibitors-and-inducers.
- Lee K. S.; Kim S. K. Direct and metabolism-dependent cytochrome P450 inhibition assays for evaluating drug–drug interactions. J. Appl. Toxicol. 2013, 33, 100–108. 10.1002/jat.1720. [DOI] [PubMed] [Google Scholar]
- Don M. J.; Lewis D. F. V.; Wang S. Y.; Tsai M. W.; Ueng Y. F. Effect of structural modification on the inhibitory selectivity of rutaecarpine derivatives on human CYP1A1, CYP1A2, and CYP1B1. Bioorg. Med. Chem. Lett. 2003, 13, 2535–2538. 10.1016/S0960-894X(03)00469-4. [DOI] [PubMed] [Google Scholar]
- Karjalainen M. J.; Neuvonen P. J.; Backman J. T. Rofecoxib is a potent, metabolism-dependent inhibitor of CYP1A2: implications for in vitro prediction of drug interactions. Drug Metab. Dispos. 2006, 34, 2091–2096. 10.1124/dmd.106.011965. [DOI] [PubMed] [Google Scholar]
- Backman J. T.; Karjalainen M. J.; Neuvonen M.; Laitila J.; Neuvonen P. J. Rofecoxib is a potent inhibitor of cytochrome P450 1A2: studies with tizanidine and caffeine in healthy subjects. Br. J. Clin. Pharmacol. 2006, 62, 345–357. 10.1111/j.1365-2125.2006.02653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt L. J.; Kazmi F.; Ogilvie B. W.; Buckley D. B.; Smith B. D.; Leatherman S.; Paris B.; Parkinson O.; Parkinson A. The reliability of estimating Ki values for direct, reversible inhibition of cytochrome P450 enzymes from corresponding IC50 values: a retrospective analysis of 343 experiments. Drug Metab. Dispos. 2015, 43, 1744–1750. 10.1124/dmd.115.066597. [DOI] [PubMed] [Google Scholar]
- Pouyfung P.; Sarapusit S.; Rongnoparut P. Effects of Vernonia cinerea Compounds on Drug-metabolizing Cytochrome P450s in Human Liver Microsomes. Phytother. Res. 2017, 31, 1916–1925. 10.1002/ptr.5939. [DOI] [PubMed] [Google Scholar]
- Cao G.; Ying P.; Yan B.; Xue W.; Li K.; Shi A.; Sun T.; Yan J.; Hu X. Pharmacokinetics, safety, and tolerability of single and multiple-doses of pinocembrin injection administered intravenously in healthy subjects. J. Ethnopharmacol. 2015, 168, 31–36. 10.1016/j.jep.2015.03.041. [DOI] [PubMed] [Google Scholar]
- Kumar V.; De P.; Ojha P. K.; Saha A.; Roy K. A Multi-layered Variable Selection Strategy for QSAR Modeling of Butyrylcholinesterase Inhibitors. Curr. Top. Med. Chem. 2020, 20, 1601–1627. 10.2174/1568026620666200616142753. [DOI] [PubMed] [Google Scholar]
- Kumar V.; Saha A.; Roy K. In silico modeling for dual inhibition of acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) enzymes in Alzheimer’s disease. Comput Biol Chem. 2020, 88, 107355 10.1016/j.compbiolchem.2020.107355. [DOI] [PubMed] [Google Scholar]
- Sansen S.; Yano J. K.; Reynald R. L.; Schoch G. A.; Griffin K. J.; Stout C. D.; Johnson E. F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. 10.1074/jbc.M611692200. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Wang Y.; Hu T.; Or P. M.; Wong J.; Kwan Y. W.; Wan D. C.; Hoi P. M.; Lai P. B.; Yeung J. H. Enzyme kinetic and molecular docking studies for the inhibitions of miltirone on major human cytochrome P450 isozymes. Phytomedicine 2013, 20, 367–374. 10.1016/j.phymed.2012.09.021. [DOI] [PubMed] [Google Scholar]
- Quintieri L.; Bortolozzo S.; Stragliotto S.; Moro S.; Pavanetto M.; Nassi A.; Palatini P.; Floreani M. Flavonoids diosmetin and hesperetin are potent inhibitors of cytochrome P450 2C9-mediated drug metabolism in vitro. Drug Metab. Pharmacokinet. 2010, 25, 466–476. 10.2133/dmpk.DMPK-10-RG-044. [DOI] [PubMed] [Google Scholar]
- Martignoni M.; Groothuis G. M. M.; de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug. Metab. Toxicol. 2006, 2, 875–894. 10.1517/17425255.2.6.875. [DOI] [PubMed] [Google Scholar]
- Eagling V. A.; Tjia J. F.; Back D. J. Differential selectivity of cytochrome P450 inhibitors against probe substrates in human and rat liver microsomes. Br. J. Clin. Pharmacol. 1998, 45, 107–114. 10.1046/j.1365-2125.1998.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- USFDA . In Vitro Metabolism- and Transporter-Mediated Drug–Drug Interaction Studies; U.S. Food and Drug Administration: Silver 664 Spring, MD, 2019. [Google Scholar]
- Bapiro T. E.; Sayi J.; Hasler J. A.; Jande M.; Rimoy G.; Masselle A.; Masimirembwa C. M. Artemisinin and thiabendazole are potent inhibitors of cytochrome P450 1A2 (CYP1A2) activity in humans. Eur. J. Clin. Pharmacol. 2005, 61, 755–761. 10.1007/s00228-005-0037-3. [DOI] [PubMed] [Google Scholar]
- Jokinen M. J.; Olkkola K. T.; Ahonen J.; Neuvonen P. J. Effect of ciprofloxacin on the pharmacokinetics of ropivacaine. Eur. J. Clin. Pharmacol. 2003, 58, 653–657. 10.1007/s00228-002-0540-8. [DOI] [PubMed] [Google Scholar]
- Bhatt S.; Kumar V.; Dogra A.; Ojha P. K.; Wazir P.; Sangwan P. L.; Singh G.; Nandi U. Amalgamation of in-silico, in-vitro and in-vivo approach to establish glabridin as a potential CYP2E1 inhibitor. Xenobiotica 2021, 51, 625–635. 10.1080/00498254.2021.1883769. [DOI] [PubMed] [Google Scholar]
- Bhatt S.; Manhas D.; Kumar V.; Gour A.; Sharma K.; Dogra A.; Ojha P. K.; Nandi U. Effect of Myricetin on CYP2C8 Inhibition to Assess the Likelihood of Drug Interaction Using In Silico, In Vitro, and In Vivo Approaches. ACS Omega 2022, 13260. 10.1021/acsomega.2c00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Liu G.; Zhen Z.; Shen Z.; Li S.; Li H. Molecular Docking for Ligand-Receptor Binding Process Based on Heterogeneous Computing. Sci. Program. 2022, 2022, 1. 10.1155/2022/9197606. [DOI] [Google Scholar]
- Meng X. Y.; Zhang H. X.; Mezei M.; Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmaso V.; Moro S. Bridging molecular docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 9, 923. 10.3389/fphar.2018.00923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momany F. A.; Rone R. Validation of the general purpose QUANTA 3.2/CHARMm force field. J. Comput. Chem. 1992, 13, 888–900. [Google Scholar]
- Sung J. H.; Hong S. S.; Ahn S. H.; Li H.; Seo S. Y.; Park C. H.; Park B. S.; Chung S. J. Mechanism for increased bioavailability of tacrine in fasted rats. J. Pharm. Pharmacol. 2006, 58, 643–649. [DOI] [PubMed] [Google Scholar]
- Wang Y. H.; Mondal G.; Butawan M.; Bloomer R. J.; Yates C. R. Development of a liquid chromatography-tandem mass spectrometry (LC–MS/MS) method for characterizing caffeine, methylliberine, and theacrine pharmacokinetics in humans. J. Chromatogr., B 2020, 1155, 122278 10.1016/j.jchromb.2020.122278. [DOI] [PubMed] [Google Scholar]
- Gour A.; Dogra A.; Wazir P.; Singh G.; Nandi U. A highly sensitive UPLC-MS/MS method for hydroxyurea to assess pharmacokinetic intervention by phytotherapeutics in rats. J. Chromatogr., B 2020, 1154, 122283 10.1016/j.jchromb.2020.122283. [DOI] [PubMed] [Google Scholar]
- Sharma A.; Magotra A.; Dogra A.; Rath S. K.; Rayees S.; Wazir P.; Sharma S.; Sangwan P. L.; Singh S.; Singh G.; Nandi U. Pharmacokinetics, pharmacodynamics and safety profiling of IS01957, a preclinical candidate possessing dual activity against inflammation and nociception. Regul. Toxicol. Pharmacol. 2017, 91, 216–225. 10.1016/j.yrtph.2017.10.033. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.