

Abstract

Strategies toward the total synthesis of the marine pyrroloacridine alkaloid alpkinidine have been explored, focusing on linking quinonoid CE ring-system synthons with the A ring, followed by condensation to form the B and D rings. The key Michael addition of the ester enolate derived from ethyl o-nitrophenylacetate to 2-methylisoquinoline-1,5,8(2H)-trione proceeded with the wrong regiochemistry. This issue was addressed by incorporating the D-ring nitrogen at an earlier stage, affording advanced intermediates possessing the complete carbon skeleton of alpkinidine. However, attempts to close the D and B rings were unsuccessful. The novel isoquinolinetriones reported here, and the general strategy of connecting CE- and A-ring synthons through Michael additions, may be useful in the synthesis of other pyrrolo- and pyridoacridines, in particular the anticancer lead neoamphimedine and analogues.
Introduction
In the preceding paper in this issue, we reported concise syntheses of 6-halo and 6,7-dichloroisoquinolinetriones matching the CE ring system of the marine secondary metabolite alpkinidine (10) and unsuccessful efforts to elaborate these to the natural product (Scheme 1). We previously described an efficient route to the model pentacyclic pyrroloacridine 5 by Michael substitution of 2,3-dichloronaphthoquinone (1) with the anion derived from ethyl o-nitrophenylacetate (2) to give 3, D-ring formation through nucleophilic lactamization with methylamine, and reductive cyclization of 4 to close the B ring.1 Despite this precedent, attempts to effect an analogous Michael substitution of dichloroisoquinolinetrione 6 were unsuccessful.2 Formation of the key carbon–carbon bond ultimately comprising the BD-ring fusion was achieved in the analogous reaction of monobromide 8; however, we were not able to progress adduct 9 toward alpkinidine (10).
Scheme 1. Summary of Our Previous Work toward Alpkinidine.

Please see the preceding paper for more background information on alpkinidine3 and related synthetic work by others.4−6
Herein, we report a continuation of our efforts toward the total synthesis of alpkinidine, specifically the use Michael addition (as opposed to substitution) to form the key carbon–carbon bond highlighted in red in Scheme 1.
Results and Discussion
At the outset, attempts to use Michael addition reactions to couple 1,4-naphthoquinone (11) and o-nitrophenylacetonitrile (12) had been unsuccessful, producing what appeared to be oligomeric products instead of the expected hydroquinone 13 or quinone 14 adducts (Scheme 2),1 hence our prior focus on Michael substitution of haloquinones 6 and 8 (Scheme 1).2
Scheme 2. Unsuccessful Attempted Model Michael Addition1.

In an effort to resurrect the general strategy of connecting the CE- and A-ring systems of potential precursors to alpkinidine, through the reaction of carbanions with quinone electrophiles,2 the reaction of ethyl o-nitrophenylacetate (2) with isoquinolinetrione 152 was investigated and indeed provided a hydroquinone adduct in modest yield (Scheme 3). Unfortunately, heteronuclear multiple bond correlation (HMBC) spectroscopy revealed the product to be the undesired regioisomer 16. In an attempt to elaborate 16 through to the isomer of alpkinidine with an inverted E-ring 19, the hydroquinone was oxidized, and quinone 17 was treated with methylamine in air, but the expected lactam 18 was not detected in the complex mixture of products formed.
Scheme 3. Michael Addition of 2 to Quinone 15 and Attempted Elaboration toward Alpkinidine Isomer 19.

The red double-headed arrows indicate HMBC correlations, which established the regiochemistry of the Michael addition.
The successful Michael addition to give 16 was encouraging and inspired efforts to achieve the opposite regioselectivity required for the synthesis of alpkinidine (10). Isoquinoline-1,5,8-(2H)-trione (30) had not been reported, but its hydroxypyridine tautomer 31 was assumed to be more stable by virtue of a strong intramolecular hydrogen bond (Scheme 4). It was hypothesized that this hydrogen bond might promote the desired regiochemistry by withdrawing electron density from the peri-carbonyl, thereby increasing electrophilicity at C6, and potentially provide anchimeric assistance for the Michael addition reaction, that is, through general acid catalysis under equilibrium deprotonation conditions. N-Methylation of the E-ring nitrogen could then follow at a later stage en route to alpkinidine. Hence, we set out to prepare 31.
Scheme 4. Attempted Synthesis of 1-Hydroxyisoquinoline-5,8-dione (31).

Amidation of 2,5-diacetoxybenzoic acid (20)7 with commercial aminoacetal 21 gave 22, along with the partially deacetylated product 23 (Scheme 4). The mixture was treated with concentrated H2SO4 but, while the analogous N-methylamide cyclized under these conditions,2 in this case the isoquinolone 26 could not be detected in the complex mixture of products formed. In an attempt to avoid phenol protecting groups, methyl 2,5-dihydroxybenzoate (24) was heated with neat amino acetal 21, providing the amide 25 in acceptable yield. Annulation in neat sulfuric acid was successful in this instance but provided the hydroquinone 26 in poor yield. The Pomeranz–Fritsch-like cyclization was more efficient with the dimethyl ether 28, derived from 2,5-dimethoxybenzoic acid (27), and smooth Prey demethylation8 of 29 was achieved. However, attempts to oxidize the hydroquinone 26 to the quinone 30/31 gave complex mixtures of products, and thus, this line of investigation was abandoned.
The regiochemical issues described above (and in Scheme 3) could potentially be solved by incorporation of the 7-aminomethyl group at an earlier stage of the synthesis (Scheme 5). Michael addition of ethyl o-nitrophenylacetate (2) to a suitably protected aminoquinone 32 would give hydroquinone 33, which upon deprotection of the amino group, would be expected to cyclize to give lactam 34. Reduction of the nitro group followed by aerial oxidation would then allow cyclodehydration to give alpkinidine (10).
Scheme 5. Proposed Regioselective Synthesis of Alpkinidine.

Serendipity played a role in allowing us to explore the general strategy set out in Scheme 5. During an attempt to oxidatively demethylate 35,2 Ag2O was mistakenly used instead of AgO (Scheme 6). Unsurprisingly, no quinone 15 was isolated from this reaction, but the electron-rich ring system was sufficiently reactive to undergo nitration with the nitric acid. Two regioisomers were isolated, with X-ray crystallography confirming the identities as the 7-nitroisoquinolone 36 and 4-nitroquinoline 37 isoquinolones. A similar yield of the major regioisomer 36 was obtained when the reaction was repeated in the absence of the silver(I) oxide.
Scheme 6. Serendipitous Regioselective Nitration of 35.

Astersisks (*): Yields in brackets are from the same reaction conducted several years later.
Surprisingly, when this reaction was repeated several years later, the ratio of 36:37 was inverted (see yields in brackets), as confirmed by comparison of the 1H NMR spectra of the crude product from each reaction. The only possible explanation we could posit for the change in product ratio is differing water content of the nitric acid/acetone. The variable outcomes for this reaction led us to devise alternative routes to 7-nitrogenated isoquinolones; the first is set out in Scheme 7. Formylation of p-methoxyphenol (38),9 or peri demethylation of 2,5-dimethoxybenzaldehyde10 (see the Experimental Section), gave salicylaldehyde 39, which was regioselectively nitrated providing 40.11 O-Methylation was followed by oxidation of 41 to the benzoic acid 42, which was converted to the acid chloride and amidated with secondary amine 43.2 Unsurprisingly, cyclization of benzamide 44 in sulfuric acid was not as facile as for the des-nitro analogue,2 and a temperature of 100 °C was required to achieve an acceptable yield of the isoquinolone 36.
Scheme 7. Alternative Synthesis of 7-Nitroisoquinolone 36.

Smooth reduction of 36 to the aniline 45 was effected with Fe or SnCl2, and followed by derivatization, affording the acetanilide 46 or carbamates 47 and 48 (Scheme 8). The carbamates were also N-methylated providing 49 and 50. It became apparent that these compounds could be accessed more efficiently from 352 (Scheme 8). Thus, selective demethylation peri to the carbonyl provided phenol 51, which underwent regioselective nitration affording 52, as confirmed by 2D NMR experiments. Reduction to the aniline 53 was followed by conversion to methyl carbamate 54; the use of pyridine as solvent/base avoided O-carboxymethylation, which was a competing side reaction with the stronger base triethylamine. Finally, N,O-dimethylation afforded 49.
Scheme 8. Preparation of Protected 7-Aminoisoquinolones.
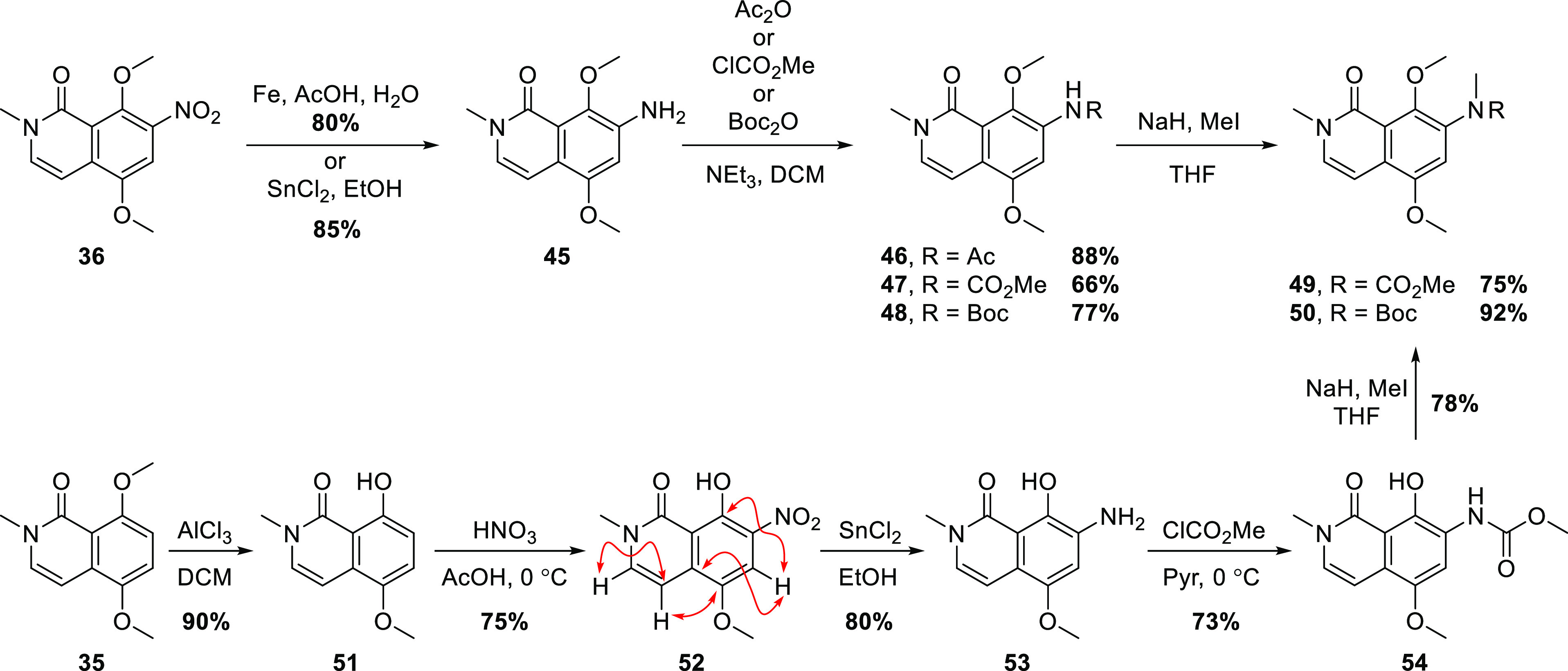
The red double-headed arrows indicate HMBC correlations, which confirmed the regiochemistry of the nitration reaction.
An attempted oxidative demethylation of 45 (Scheme 9) gave an intractable mixture, in line with previous observations from reactions of dimethoxyanilines with CAN.12 In any case, it was expected that the amino group in 55 would be so electron-donating as to preclude a subsequent Michael addition at C6, hence the protection with electron-withdrawing groups. The oxidative demethylation of the secondary acetamide 46 and carbamates 47 and 48 proceeded as expected to the give corresponding quinones 56–58, respectively. In surprising contrast, the analogous reactions of the tertiary carbamates 49 and 50 failed to give the expected quinones 59 and 60. In the case of the methyl carbamate 49, dinitration was instead observed, affording 61 in moderate yield. Related nitrations of electron-rich aromatic compounds with CAN have been reported.13−16 No reaction of 49 was observed with phenyliodine(III)-bis(trifluoroacetate) (PIFA).17 When the t-butyl carbamate 50 was treated with CAN, cleavage of the Boc group to give aniline 62 was the only reaction observed. CAN-mediated cleavage of Boc groups is well known;18 however, a sensible explanation for the difference in reactivity between the secondary (47/48) and tertiary (49/50) carbamates escapes us.
Scheme 9. Oxidations of 7-Aminoisoquinolone 45 and Derivatives.

With appropriately nitrogenated quinones in hand, the key Michael addition step was investigated (Scheme 10). The reaction of 2 with acetamide 56 gave a complex mixture of products, but encouragingly, Michael additions to the methyl (57) and t-butyl (58) carbamates were successful; the initially formed hydroquinone adducts presumably oxidized during workup to the isolated quinones 64 and 65, respectively. A single attempt to deprotect 64 failed to give the expected lactam 67, producing a complex mixture instead. More effort was devoted to the deprotection/cyclization of 65. Surprisingly, the t-butyl carbamate 65 was resistant to deprotection with TFA at rt and gave complex mixtures with BF3·OEt219 or H2SO4 in dioxane.20 Treatment with 5 M hydrochloric acid under reflux, or ethanolic HCl, gave a major product that could not be conclusively identified. The 1H NMR spectrum of this product is consistent with the ortho-quinone methide 66 or its geometric isomer, although definitive structural elucidation was precluded by material availability, and hence, this assignment is tentative. Unfortunately, attempts to cyclize this product to give lactam 67, or a tautomer, were unsuccessful.
Scheme 10. Michael Addition to 7-Nitrogenated Isoquinolinetriones and Subsequent Transformations.

Conclusions
Michael additions of ethyl o-nitrophenylacetate (2) to quinonoid electrophiles have been investigated as a means to connect fragments comprising the CE ring system and A ring of the marine pyrroloacridine alkaloid alpkinidine (10). While the reaction of the anion derived from 2 with isoquinolinetrione 15 proceeded with the wrong regiochemistry, the successful coupling of the two fragments and oxidation to give quinone 17 (Scheme 11A) encouraged further exploration of this strategy.
Scheme 11. (A–C) Summary of Major Outcomes of the Current Work.

To direct the regiochemistry of the key Michael addition, 7-nitrogentated isoquionlinetriones were targeted, and several efficient syntheses were developed (Scheme 11B). These isoquinolones and the general concept of Michael additions to such synthons may be of value in the synthesis of analogues of the closely related marine alkaloid neoamphimedine, which has a near-identical ABCE ring system to alpkinidine and promising anticancer potential.3,21−23
Proof of concept was achieved in that 2 underwent Michael addition to isquinolinetriones 57/58 and aerial oxidation to afford adducts 64/65, containing the complete carbon skeleton of alpkinidine. However, attempts to close rings B and D were unsuccessful. The key to bringing the approach outlined herein to fruition probably lies in finding the right order of redox and deprotection reactions on an advanced intermediate like 64/65, an objective that will be pursued in the future.
Experimental Section
General
General experimental details are as reported previously.1,24
Synthesis
Ethyl 2-(5,8-Dihydroxy-2-methyl-1-oxo-1,2-dihydroisoquinolin-7-yl)-2-(2-nitrophenyl)acetate (16)
Isoquinolinetrione 152 (96 mg, 0.51 mmol) was added to a stirred suspension of ethyl 2-nitrophenylacetate (2) (0.25 g, 1.2 mmol) and K2CO3 (0.17 g, 1.2 mmol) in DMF (20 mL) at 40 °C. After 1.5 h, the reaction was cooled, diluted with 1 M HCl (30 mL), and extracted with EtOAc (3 × 20 mL). The organic componenets were dried and evaporated, and the crude product was subjected to flash chromotography. Elution with 2:3 EtOAc/hexanes gave 16 (70 mg, 35%) as a pale-yellow oil. Rf (3:2 EtOAc/hexanes) 0.2. IR (ATR) νmax cm–1: 3400–2900 (OH), 1730 (C=O), 1652 (C=O). 1H NMR (600 MHz) δ 12.7 (s, 1H, OH), 8.04 (dd, J1 = 7.8, J2 = 1.2 Hz, 1H, H3″ or H6″), 7.43 (ddd, J1 = J2 = 7.8, J3 = 1.2 Hz, 1H, H4″ or H5″), 7.40 (pseudo ddd [app. t], J = 7.8 Hz, 1H, H4″ or H5″), 7.18 (pseudo dd [app. d], J = 7.8 Hz, 1H, H3″ or H6″), 6.99 (d, J = 7.2 Hz, 1H, H3′ or H4′), 6.91 (s, 1H, H6′), 6.79 (d, J = 7.2 Hz, 1H, H3′ or H4′), 6.02 (s, 1H, H2), 5.39 (s, 1H, OH), 4.27 (m, 1H, OCH2CH3), 4.21 (m, 1H, OCH2CH3), 3.56 (s, 3H, OMe), 1.26 (t, J = 7.2 Hz, 3H, OCH2CH3). 13C NMR (150 MHz) δ 172.3 (C1), 165.5 (C1′), 152.6 (C5′ or C8′), 149.1 (C5′ or C8′), 141.9 (C2″), 133.2 (C1″ or C7′), 133.1 (ArH), 131.4 (ArH), 131.1 (ArH), 128.2 (ArH), 126.6 (C4a′ or C8a′), 125.1 (ArH), 120.1 (C1″ or C7′), 119.4 (ArH), 112.3 (C4a′ or C8a′), 102.4 (ArH), 61.8 (OCH2CH3), 47.2 (C2), 36.5 (NMe), 14.1 (OCH2CH3). HRMS (APCI): calcd for C20H19N2O7+ [M + H]+ 399.1194; found 399.1187.
Ethyl 2-(2-Methyl-1,5,8-trioxo-1,2,5,8-tetrahydroisoquinolin-7-yl)-2-(2-nitrophenyl)acetate (17)
Ag2O (0.31 g, 1.34 mmol) was added to a stirred suspension of hydroquinone 16 (70 mg, 0.17 mmol) and MgSO4 (0.56 g, 4.65 mmol) in Et2O (15 mL) and DME (5 mL). After 16 h, the reaction was filtered through a plug of Celite and washed with DCM (3 × 10 mL). The volatiles were then removed to give 17 as a red-orange oil (61 mg, 88%). Rf (EtOAc) 0.1. IR (ATR) νmax cm–1: 1732 (C=O), 1686 (C=O), 1671 (C=O). 1H NMR (600 MHz) δ 8.08 (dd, J1 = 8.4, J2 = 1.2 Hz, 1H, H3″ or H6″), 7.80 (d, J = 6.6 Hz, 1H, H3 or H4), 7.65 (dt, J1 = 7.2, J2 = 1.2 Hz, 1H, H4″ or H5″), 7.53 (dt, J1 = 7.8, J2 1.8 Hz, 1H, H4″ or H5″), 7.46 (dd, J1 = 7.8, J2 = 1.2 Hz, 1H, H3″ or H6″), 6.74 (d, J = 6.6 Hz, 1H, H3′ or H4′), 6.40 (d, J = 1.2 Hz, 1H, H6′), 5.80 (s, 1H, H2), 4.25 (m, 2H, OCH2CH3), 4.20 (m, 2H, OCH2CH3), 3.66 (s, 3H, NMe), 1.25 (t, J 7.2 Hz, 3H, OCH2CH3). 13C NMR (150 MHz) δ 184.8 (C5′ or C8′), 181.2 (C5′ or C8′), 169.7 (C1), 158.4 (C1′), 149.5 (C2″), 149.1 (C1″ or C7′), 145.5 (ArH), 143.3 (C1″ or C7′), 133.8 (ArH). 133.0 (ArH), 131.1 (ArH), 130.0 (ArH), 129.4 (C4a′ or C8a′), 125.9 (ArH), 119.3 (C4a′ or C8a′), 100.3 (ArH), 62.4 (OCH2CH3), 47.9 (C2), 39.2 (NMe), 14.1 (OCH2CH3).
N-(2,2-Diethoxyethyl)-2,5-diacetoxybenzamide (22) and N-(2,2-Diethoxyethyl)-2-hydroxy-5-acetoxybenzamide (23)
A solution of 2,5-diacetoxybenzoic acid (20)7 (5.02 g, 21.1 mmol) and SOCl2 (10 mL, 0.14 mol) in PhMe (25 mL) was heated under reflux for 2 h before the solvent and excess SOCl2 were removed by distillation. The residue was cooled to 0 °C, and a solution of NEt3 (10 mL, 72 mmol) in PhMe (10 mL) was added dropwise, followed by a solution of 2,2-diethoxyethanamine (21) (3.4 g, 23 mmol) in PhMe (10 mL) dropwise. The mixture was stirred at rt for another 3 h before being diluted with H2O (50 mL) and extracted with EtOAc (3 × 20 mL). The extract was washed with sat. aq. NaHCO3 (20 mL), dried, and evaporated, and the residue was subjected to flash chromatography. Elution with 2:3 EtOAc/hexanes gave 22 (1.64 g, 22%) as a pale-yellow oil. Rf (3:2 EtOAc/hexanes) 0.35. IR (ATR) νmax cm–1: 2977 (NH), 1764 (Ac C=O), 1661 (C=O). 1H NMR (600 MHz) δ 7.59 (d, J = 2.4 Hz, 1H, H6), 7.18 (dd, J1 = 9.0, J2 = 2.4 Hz, 1H, H4), 7.11 (d, J = 9.0 Hz, 1H, H3), 6.76 (s, 1H, NH), 4.58 (t, J = 5.4 Hz, 1H, H2′), 3.74–3.66 (m, 2H, OCH2CH3), 3.60–3.52 (m, 4H, OCH2CH3 and H1′), 2.34 (s, 3H, OMe), 2.28 (s, 3H, OMe), 1.21 (t, J = 7.2 Hz, 6H, OCH2CH3). 13C NMR (150 MHz) δ 169.2 (C=O), 168.8 (C=O), 164.4 (C=O), 148.3 (ArO), 145.4 (ArO), 128.7 (C1), 125.2 (ArH), 124.4 (ArH), 123.5 (ArH), 100.5 (C2′), 62.9 (OCH2CH3), 42.5 (C1′), 21.1 (2 × COCH3), 15.4 (OCH2CH3). HRMS (APCI): calcd for C17H24NO7+ [M + H]+ 354.1547; found 354.1570.
Further elution with 3:2 EtOAc/hexanes gave 23 (2.52 g, 38%) as a pale-yellow oil. IR (ATR) νmax cm–1: 3600–2800 (OH), 2977 (NH), 1760 (Ac C=O), 1646 (C=O). 1H NMR (600 MHz) δ 12.1 (s, 1H, OH), 7.14–7.08 (m, 2H, H4 and H6), 6.96 (d, J = 8.4 Hz, 1H, H3), 6.51 (s, 1H, NH), 4.60 (t, J = 5.0 Hz, 1H, H2′), 3.78–3.68 (m, 2H, OCH2CH3), 3.61–3.53 (m, 4H, OCH2CH3 and C1′), 2.28 (s, 3H, COCH3), 1.23 (t, J = 7.0 Hz, 6H, OCH2CH3). 13C NMR (150 MHz) δ 169.6 (C=O), 169.2 (C=O), 159.2 (C2), 142.0 (C4), 127.6 (ArH), 119.3 (ArH), 118.0 (ArH), 114.0 (C1), 100.4 (C2′), 63.1 (OCH2CH3), 42.0 (C1′), 20.9 (COCH3), 15.3 (OCH2CH3). HRMS (APCI): calcd for C15H22NO6+ [M + H]+ 312.1451; found 312.1442.
N-(2,2-Diethoxyethyl)-2,5-dihydroxybenzamide (25)
Methyl 2,5-dihydroxybenzoate (24) (1.97 g, 11.8 mmol) was heated with aminoacetal 21 (5.0 mL, 35 mmol) at 100 °C under CaCl2 guard for 24 h before being diluted with H2O (20 mL) and extracted with EtOAc (3 × 10 mL). The organic extract was dried and evaporated to give 25 (2.07 g, 65%) as a pale-yellow oil. Rf (2:3 EtOAc/hexanes) 0.1. IR (ATR) νmax cm–1: 3600–2800 (OH), 2978 (NH), 1640 (C=O). 1H NMR (600 MHz) δ 11.5 (s, 1H, OH), 7.42 (br s, 1H, OH), 7.31 (s, 1H, NH), 7.07 (d, J = 2.4 Hz, 1H, H6), 6.88 (dd, J1 = 8.4, J2 = 2.4 Hz, 1H, H4), 6.78 (d, J = 8.4 Hz, 1H, H3), 4.66 (t, J = 4.8 Hz, 1H, H2′), 3.73 (m, 2H, OCH2CH3), 3.55 (m, 4H, OCH2CH3 and C1′), 1.20 (t, J = 7.2 Hz, 6H, OCH2CH3). 13C NMR (150 MHz) δ 169.8 (C=O), 154.1 (C2), 148.4 (C5), 122.3 (ArH), 118.9 (ArH), 114.6 (ArH), 112.2 (C1), 100.8 (C2′), 63.5 (OCH2CH3), 42.3 (C1′), 15.2 (OCH2CH3). HRMS (ESI–): calcd for C13H18NO5– [M–H]− 268.1167; found 268.1190.
5,8-Dihydroxyisoquinolin-1(2H)-one (26)
Method 1: Concentrated H2SO4 (5 mL) was added dropwise to neat 25 with stirring at 0 °C under CaCl2 guard. After the addition was complete, the solution was allowed to warm to rt, then stirred at 50 °C for 24 h. The reaction was diluted with H2O (20 mL) and carefully neutralized with ice cold sat. aq. NaHCO3 (∼30 mL) until effervescing ceased, then extracted with EtOAc (3 × 20 mL). The extract was dried and evaporated, and the crude product was subjected to flash chromatography. Elution with 2:3 EtOAc/hexanes gave isoquinolone 26 as an off-white solid (77 mg, 6%), mp 260–263 °C. Rf (2:3 EtOAc/hexanes) 0.1. IR (ATR) νmax cm–1: 3500–2700 (OH), 2886 (NH), 1639 (C=O). 1H NMR (600 MHz, DMSO-d6) δ 12.3 (s, 1H, OH), 11.6 (s, 1H, NH), 9.44 (s, 1H, OH), 7.09 (d, J = 7.2 Hz, 1H, H3), 7.02 (d, J = 9.0 Hz, 1H, H6 or H7), 6.72 (d, J = 7.2 Hz, 1H, H4), 6.62 (d, J = 9.0 Hz, 1H, H6 or H7). 13C NMR (150 MHz, DMSO-d6) δ 165.9 (C1), 153.0 (C5 or C8), 143.6 (C5 or C8), 126.9 (ArH), 126.8 (C4a or C8a), 119.1 (ArH), 111.6 (C4a or C8a), 111.4 (ArH), 101.7 (ArH). HRMS (ESI–): calcd for C9H6NO3– [M–H]− 176.0340; found 176.0353.
Method 2: 5,8-Dimethoxyisoquinolin-1(2H)-one (29) (0.16 g, 0.76 mmol) was added to pyridine hydrochloride,2 and the mixture was heated under reflux for 20 min before being cooled and diluted with H2O (20 mL) and extracted with EtOAc (3 × 10 mL). The extract was dried and evaporated to give hydroquinone 26 as an off-white solid (0.13 g, 95%), identical to the material described above.
N-(2,2-Diethoxyethyl)-2,5-dimethoxybenzamide (28)
A solution of 2,5-dimethoxybenzoic acid (27) (0.68 g, 3.7 mmol) and SOCl2 (5.0 mL, 69 mmol) in PhMe (15 mL) was heated under reflux for 2 h before the solvent and excess SOCl2 were removed by distillation. The residue was cooled to 0 °C, and a solution of NEt3 (5.0 mL, 36 mmol) in PhMe (5 mL) was added dropwise, followed by a solution of aminoacetal 21 (3.4 g, 23 mmol) in PhMe (5 mL) dropwise. The mixture was stirred at rt for another 3 h before being diluted with H2O (30 mL) and extracted with EtOAc (3 × 20 mL). The extract was washed with sat. aq. NaHCO3 (20 mL), dried, and evaporated, and the crude product was subjected to flash chromatography. Elution with 1:4 EtOAc/hexanes gave 28 (0.98 g, 88%) as an amber oil. Rf (2:3 EtOAc/hexanes) 0.2. IR (ATR) νmax cm–1: 2975 (NH), 1652 (C=O). 1H NMR (500 MHz) δ 8.23 (s, 1H, NH), 7.72 (d, J = 3.5 Hz, 1H, H6), 6.94 (dd, J1 = 9.0, J2 = 3.5 Hz, 1H, H4), 6.86 (d, J = 9.0 Hz, 1H, H3), 4.59 (t, J = 5.5 Hz, 1H, H2′), 3.87 (s, 3H, OMe), 3.76 (s, 3H, OMe), 3.71 (dq, J1 = J2 = 7.0 Hz, 2H, OCH2CH3), 3.60–3.50 (m, 4H, OCH2CH3 and H1′), 1.20 (t, J = 7.0 Hz, 6H, OCH2CH3). 13C NMR (125 MHz) δ 165.1 (C=O), 153.9 (ArO), 151.9 (ArO), 121.9 (C1), 119.3 (ArH), 115.6 (ArH), 113.1 (ArH), 101.0 (C2′), 62.8 (OCH2CH3), 56.9 (OMe), 55.8 (OMe), 42.4 (C1′), 15.4 (OCH2CH3). HRMS (ESI): calcd for C17H26N2NaO5+ [M + Na + MeCN]+ 361.1741; found 361.1734.
5,8-Dimethoxyisoquinolin-1(2H)-one (29)
Concentrated H2SO4 (10 mL) was added dropwise to neat 28 (0.901 g, 3.03 mmol) with stirring at 0 °C. After the addition was complete, the solution was allowed to warm to rt, then stirred at 50 °C under CaCl2 guard. After 24 h, the solution was cooled and carefully neutralized with ice-cold sat. aq. NaHCO3 (∼50 mL) until effervescing ceased, then extracted with EtOAc (3 × 30 mL). The extract was dried and evaporated to give an off-white solid (0.371 g, note: the yield was affected by a spill), which crystallized from DCM/hexanes affording isoquinolone 29 as colorless plates (0.155 g, 25%), identical to the material reported previously.25
5,8-Dimethoxy-2-methyl-7-nitroisoquinolin-1(2H)-one (36) and 5,8-Dimethoxy-2-methyl-4-nitroisoquinolin-1(2H)-one (37)a
Method A: Concentrated HNO3 (1.0 mL, 17 mmol) was added dropwise to a suspension of 352 (91 mg, 0.42 mmol) and Ag2O (0.53 g, 2.29 mmol) in acetone (15 mL) at 0 °C. After 1 h, the reaction was diluted with H2O (20 mL) and sat. aq. NaHCO3 (20 mL), then extracted with EtOAc (3 × 10 mL). The extract was dried and evaporated, and the residue was subjected to flash chromatography. Elution with 3:2 EtOAc/hexanes gave 36 as yellow rods (83 mg, 75%), mp 196–200 °C. Rf (3:2 EtOAc/hexanes) 0.25. IR (ATR) νmax cm–1: 1651 (C=O). 1H NMR (600 MHz, DMSO-d6) δ 7.70 (d, J = 7.2 Hz, 1H, H3), 7.61 (s, 1H, H6), 6.71 (d, J = 7.2 Hz, 1H, H4), 3.94 (s, 3H, OMe), 3.87 (s, 3H, OMe), 3.49 (s, 3H, NMe). 13C NMR (150 MHz, DMSO-d6) δ 158.6 (C1), 149.6 (C5 or C8), 146.4 (C5 or C8), 141.9 (C7), 137.3 (ArH), 133.7 (C4a or C8a), 119.4 (C4a or C8a), 106.7 (ArH), 97.6 (ArH), 63.6 (OMe), 56.7 (OMe), 37.0 (NMe). HRMS (APCI): calcd for C12H13N2O5+ [M + H]+ 265.0819; found 265.0824.
Further elution gave 37 as yellow plates (15 mg, 14%), mp 177–180 °C. Rf (3:2 EtOAc/hexanes) 0.1. IR (ATR) νmax cm–1: 1652 (C=O). 1H NMR (600 MHz, DMSO-d6) δ 8.41 (s, 1H, H3), 7.43 (d, J = 9.0 Hz, 1H, H6 or H7), 7.17 (d, J = 9.0 Hz, 1H, H6 or H7), 3.82 (s, 3H, OMe), 3.77 (s, 3H, OMe), 3.41 (s, 3H, NMe). 13C NMR (150 MHz, DMSO-d6) δ 158.4 (C=O), 154.4 (C5 or C8), 146.1 (C5 or C8), 135.0 (C3), 129.1 (C4), 120.7 (C4a or C8a), 117.3 (C6 or C7), 114.2 (C4a or C8a), 112.2 (C6 or C7), 56.7 (OMe), 36.7 (NMe). HRMS (APCI): calcd for C12H13N2O5+ [M + H]+ 265.0832; found 265.0819.
Method B: A solution of concentrated HNO3 (1.0 mL, 17 mmol) in acetone (10 mL) was added to a stirred solution of 352 (1.13 g, 5.17 mmol) in AcOH (10 mL) and acetone (10 mL). After 1 h, the mixture was diluted with H2O (30 mL) and sat. aq. NaHCO3 (20 mL), then extracted with EtOAc (3 × 20 mL). The extract was dried and evaporated. Precipitation from a mixture of MeOH/DCM/hexanes gave 36 as a yellow solid (0.96 g, 70%), identical to the material described above.
Method C (36 exclusively): Ice-cold conc. H2SO4 (10 mL) was added dropwise to neat 44 (5.37 g, 15.1 mmol) with stirring at 0 °C. After the addition was complete, the solution was warmed to rt, then heated to 100 °C. After 1 h, the solution was cooled and carefully neutralized with ice-cold sat. aq. NaHCO3 until effervescing ceased. The aqueous phase was extracted with EtOAc (3 × 30 mL), and the extract was dried and evaporated. The residue was purified by flash column chromatography. Elution with 3:2 EtOAc/hexanes yielded isoquinolone 36 as a yellow solid (2.59 g, 65%), identical to the material described above.
2-Hydroxy-5-methoxybenzoic acid (39)
Method A:9 Anhydrous THF (250 mL) was added to anhydrous MgCl2 (15.35 g, 161.2 mmol) and paraformaldehyde (7.26 g, 242 mmol) under a positive pressure of N2. NEt3 (22.47 mL, 161.2 mmol) was added dropwise to the stirred suspension, and after 10 min, 4-methoxyphenol (38) (10.00 g, 80.55 mmol) was added, resulting in an opaque, light-green mixture. The reaction mixture was heated under gentle reflux whereupon it rapidly turned orange/yellow color. After 6 h, the reaction mixture was cooled to rt and rinsed with ether (150 mL) into a separatory funnel. The organic phase was washed with 1 M HCl (3 × 150 mL), water (3 × 150 mL), and brine (150 mL), dried, and evaporated to leave a pale-yellow oil, which was subjected to flash column chromatography. Elution with 2:23 EtOAc/hexanes gave the benzaldehyde 39 as a pale-yellow oil (12.15 g, 99%). 1H NMR (400 MHz, CDCl3) δ 10.65 (s, 1H), 9.86 (d, J = 0.6 Hz, 1H, CHO), 7.14 (dd, J = 3.2, 9.0 Hz, 1H, H4), 7.00 (d, J = 3.2 Hz, 1H, H6), 6.93 (d, J = 9.0 Hz, 1H, H3), 3.82 (s, 3H, OMe). 13C NMR (100 MHz, CDCl3) δ 196.3 (CO), 156.2 (ArO), 152.9 (ArO), 124.4 (ArH), 119.2, 117.9 (ArH), 114.3 (ArH), 56.1 (OMe). The NMR data are consistent with the literature.26
Method B:10 Anhydrous AlCl3 (12.04 g, 90.32 mmol) was added to a stirred solution of 2,5-dimethoxybenzaldehyde (10.00 g, 60.12 mmol) in anhydrous DCM (200 mL) at 0 °C under N2. The reaction mixture was allowed to slowly warm to rt, and stirring was continued for 5 h. When TLC indicated that the reaction was complete, the mixture was diluted with ice/water (300 mL) and the DCM layer was separated. The aqueous phase was extracted with EtOAc (2 × 100 mL). The aqueous phase was then acidified with concentrated HCl and reextracted with EtOAc (2 × 100 mL). The combined organic phase was washed with brine (200 mL), dried, and evaporated to give a brown oil that was subjected to flash column chromatography. Elution with 8:92 EtOAc/hexanes gave the phenol 39 as a colorless oil (7.84 g, 86%), spectroscopically identical with the material described above.
2-Hydroxy-5-methoxy-3-nitrobenzaldehyde (40)
A solution of 70% HNO3 (5.25 mL, 33.9 mmol) in AcOH (19 mL) was added dropwise to a cooled, stirred solution of the benzaldehyde 39 (12.15 g, 79.86 mmol) in AcOH (120 mL) at such a rate as to maintain the temperature between 10 and 15 °C (50 min). After stirring for a further 45 min, water (60 mL) was added and the precipitated solid was collected by vacuum filtration, washed with water, and air-dried to afford to give 40 as a yellow solid (11.80 g, 75%), mp 132–133 °C [lit.11 132–133 °C]. Rf (1:9 EtOAc/hexanes) 0.4. IR (ATR) νmax cm–1: 3200–2800 (OH), 1691 (C=O). 1H NMR (400 MHz, CDCl3) δ 10.88 (s, 1H, OH), 10.43 (s, 1H, CHO) 7.84 (d, J = 3.3 Hz, 1H, H4 or H6), 7.70 (d, J = 3.3 Hz, 1H, H4 or H6), 3.87 (s, 3H, OMe). 13C NMR (100 MHz, CDCl3) δ 188.3 (C=O), 152.2 (ArO), 151.3 (ArO), 126.3 (C3), 123.1 (C4 or C6), 117.1 (C1), 115.3 (C4 or C6), 56.5 (OMe). The NMR data are consistent with the literature.11
2,5-Dimethoxy-3-nitrobenzaldehyde (41)
Anhydrous K2CO3 (13.68 g, 99 mmol) and MeI (6.19 mL, 99.3 mmol) were added to a solution of 40 (9.75 g, 49.5 mmol) in dry DMF (110 mL) in a stoppered flask. The mixture was stirred at 60 °C for 12 h, then poured onto ice/water (1 L), and extracted with EtOAc (3 × 200 mL). The extract was washed with water (10 × 100 mL) and brine (150 mL), dried, and evaporated to give the dimethyl ether 41 as a yellow solid (9.50 g, 90%), mp 111–113 °C [lit.27 113 °C]. Rf (1:9 EtOAc/hexanes) 0.3. 1H NMR (400 MHz, CDCl3) δ 10.36 (s, 1H, CHO), 7.62 (d, J = 3.3 Hz, 1H, H4 or H6), 7.57 (d, J = 3.3 Hz, 1H, H4 or H6), 4.03 (s, 3H, OMe), 3.88 (s, 3H, OMe). 13C NMR (100 MHz, CDCl3) δ 187.6 (C=O), 155.6 (C2), 150.2 (C5), 131.9 (C3), 117.3 (C4 or C6), 117.0 (C4 or C6), 65.9 (2-OMe), 56.5 (5-OMe). The C1 signal was not observed/coincident. The NMR data are consistent with the literature.28
2,5-Dimethoxy-3-nitrobenzoic acid (42)
A solution of KMnO4 (10.7 g, 67.7 mmol) in water (81 mL) was added to a mixture of 2,5-dimethoxy-3-nitrobenzaldehyde (41) (9.5 g, 45 mmol) and KHCO3 (9.0 g, 90 mmol) in boiling water (130 mL). When TLC indicated that the reaction was complete, the hot solution was filtered through a pad of Celite, washed through with water (100 mL), and allowed to cool. The reddish yellow filtrate was acidified with conc. HCl, and the resulting precipitate was collected by vacuum filtration, washed with water, and air-dried to give benzoic acid 42 as a pale-yellow solid (8.19 g, 80%), mp 155–160 °C [lit.29 181–183 °C]. 1H NMR (400 MHz, DMSO), δ = 7.66 (d, J = 3.3 Hz, 1H, H4 or H6), 7.51 (d, J = 3.3 Hz, H4 or H6, 1H), 3.83 (s, 3H, OMe), 3.82 (s, 3H, OMe). 13C NMR (100 MHz, DMSO) δ 165.4 (C=O), 154.5 (C2), 145.6 (C5 or C3), 144.6 (C5 or C3), 129.0 (C1), 120.1 (ArH), 112.5 (ArH), 63.9 (2-OMe), 56.4 (5-OMe). The NMR data are consistent with the literature.30
N-(2,2-Diethoxyethyl)-2,5-dimethoxy-N-methyl-3-nitrobenzamide (44)
A solution of 2,5-dimethoxy-3-nitrobenzoic acid (42) (7.59 g, 33.4 mmol) and SOCl2 (10.67 mL, 147.1 mmol) in DCM (10 mL) was heated under reflux and moisture guard for 2 h before the solvent and excess SOCl2 were evaporated under a stream of N2. The residue was cooled to 0 °C, and a solution of pyridine (10.66 mL, 132.3 mmol) in DCM (7 mL) was added dropwise, followed by dropwise addition of a solution of amine 432 (7.38 g, 50.2 mmol) in DCM (7 mL). The mixture was stirred at rt for 3 h, then diluted with H2O (50 mL), and extracted with DCM (3 × 20 mL). The extract was washed with 10% aq. CuSO4 (4 × 20 mL), sat. aq. NaHCO3 (20 mL), and brine and dried and evaporated to give the amide 44 as a reddish oil (9.50 g, 80%) sufficiently pure for the next step. Rf (1:19 MeOH/DCM) 0.35. IR (ATR) νmax cm–1: 1691 (C=O). 1H NMR (500 MHz, CDCl3; an 18*:10# mixture of rotamers) δ 7.36* (d, J = 3.2 Hz, 1H, H4 or H6), 7.34# (d, J = 3.2 Hz, 1H, H4 or H6), 7.05# (d, J = 3.1 Hz, 1H, H4 or H6), 7.00* (d, J = 3.1 Hz, 1H, H4 or H6), 4.79* (t, J = 5.5 Hz, 1H, H2′), 4.48# (t, J = 5.3 Hz, 1H, H2′), 3.89* (s, 3H, OMe), 3.87# (s, 3H, OMe), 3.83* (s, 3H, OMe), 3.82# (s, 3H, OMe), 3.81–3.69# (m, 4H, OCH2), 3.66–3.46* (m, 4H, OCH2), 3.45–3.32* (m, 2H, H1′), 3.30–3.22# (m, 2H, H1′), 3.18# (s, 3H, NMe), 2.92* (s, 3H, NMe), 1.24* (t, J = 7.0 Hz, 6H, 2 × CH3), 1.17# (t, J = 7.0 Hz, 3H, CH3), 1.12# (t, J = 6.8 Hz, 3H, CH3). 13C NMR (125 MHz, CDCl3) δ 167.6# (C=O), 167.4* (C=O), 155.6* (C2), 155.5# (C2), 144.3*, 144.2#, 143.3*, 143.0# 135.2*, 135.0#, 119.4# (C4 or C6), 118.6* (C4 or C6), 110.6*# (C4 or C6), 101.4# (CHO2), 101.0* (CHO2), 64.0* (OMe or OCH2), 63.9* OMe or OCH2), 63.8# (OMe or OCH2), 63.5# (OMe or OCH2), 63.4*# (OMe or OCH2), 56.31* (C5-OMe), 56.27# (C5-OMe), 53.8# (NCH2), 50.9* (NCH2), 38.5* (NMe), 34.7# (NMe), 15.5* (2 × CH3), 15.4# (CH3), 15.3# (CH3). HRMS (APCI): calcd for C16H25N2O7+ [M + H]+ 357.1656; found 357.1750.
7-Amino-5,8-dimethoxy-2-methylisoquinolin-1(2H)-one (45)
Method A: Iron powder (2.01 g, 35.4 mmol) was added to a vigorously stirred solution of 36 (2.32 g, 8.78 mmol) in AcOH (30 mL), H2O (30 mL), and MeOH (15 mL). After 1.5 h, the reaction mixture was diluted with H2O (50 mL) and extracted with EtOAc (3 × 25 mL). The extract was washed with sat. aq. NaHCO3 (3 × 20 mL), dried, and evaporated, and the crude product was subjected to flash chromatography. Elution with 1:19 MeOH/DCM gave aniline 45 as an amber oil (1.65 g, 80%). Rf (1:19 MeOH/DCM) 0.2. IR (ATR) νmax cm–1: 3337 (NH2), 1646 (C=O). 1H NMR (600 MHz, DMSO-d6) δ 7.04 (dd, J = 7.2, 0.6 Hz, 1H, H3), 6.73 (s, 1H, H6), 6.50 (dd, J = 7.2, 0.6 Hz, 1H, H4), 5.21 (s, 2H, NH2), 3.79 (s, 3H, OMe), 3.60 (s, 3H, OMe), 3.39 (s, 3H, NMe). 13C NMR (150 MHz, DMSO-d6) δ 159.2 (C1), 150.2 (C5), 140.9 (C8), 136.9 (C7), 128.5 (C3), 119.9 (C4a or C8a), 118.7 (C4a or C8a), 102.0 (C4 or C6), 98.8 (C4 or C6), 60.1 (C8-OMe), 55.6 (C5-OMe), 36.4 (NMe). HRMS (APCI): calcd for C12H15N2O3+ [M + H]+ 235.1081; found 235.1077.
Method B: SnCl2·2H2O (5.64 g, 25.0 mmol) was added to a solution of 36 (1.32 g, 5.00 mmol) in EtOH (30 mL), and the mixture was heated under reflux for 1.5 h. The reaction mixture was cooled to rt, then poured into ice (150 g), and the resulting suspension was made alkaline by an addition of sat. aq. NaHCO3. The aqueous phase was extracted with DCM (3 × 25 mL), and the extract was washed with brine (25 mL) and evaporated. The residue was then acidified with 1 M HCl and extracted with DCM (2 × 25 mL). The aqueous layer was made alkaline by addition of solid NaHCO3. The excess solid was filtered, and the filtrate was extracted with DCM (3 × 20 mL). The combined organic phase was washed with brine (20 mL) and evaporated to afford the aniline 45 as a yellow oil (1.00 g, 85%), spectroscopically identical to the material described above.
7-Acetylamino-5,8-dimethoxy-2-methylisoquinolin-1(2H)-one (46)
Ac2O (0.50 mL, 2.7 mmol) was added to a stirred solution of 45 (0.21 g, 0.89 mmol) and NEt3 (0.50 mL, 3.5 mmol) in DCM (15 mL). After 24 h, the reaction mixture was diluted with H2O (30 mL) and extracted with DCM (3 × 20 mL). The extract was dried and evaporated, and the residue was subjected to flash chromatography. Elution with 1:99 MeOH/DCM gave acetamide 46 as a pale-yellow solid (0.22 g, 88%), mp 177–180 °C. Rf (1:19 MeOH/DCM) 0.3. IR (ATR) νmax cm–1: 3321 (NH), 1674 (O=CNH), 1649 (O=C1). 1H NMR (600 MHz, DMSO-d6) δ 9.47 (s, 1H, NH), 8.05 (s, 1H, H6), 7.35 (d, J = 7.2 Hz, 1H, H3), 6.61 (d, J = 7.2 Hz, 1H, H4), 3.83 (OMe), 3.71 (OMe), 3.44 (NMe), 2.16 (CH3). 13C NMR (150 MHz, DMSO-d6) δ 169.2 (HNC=O), 159.0 (C1), 149.2 (C5 or C8), 142.4 (C5 or C8), 132.5 (C3 or C6), 130.6 (C3 or C6), 125.3 (C7), 119.0 (C4a or C8a), 107.3 (C4a or C8a), 98.1 (C4), 61.6 (C8-OMe), 55.9 (C5-OMe), 36.6 (NMe), 24.0 (CH3). HRMS (APCI): calcd for C14H17N2O4+ [M + H]+ 277.1183; found 277.1176.
5,8-Dimethoxy-7-methoxycarbonylamino-2-methylisoquinolin-1(2H)-one (47)
Methyl chloroformate (0.10 mL, 1.3 mmol) was added to a stirred solution of aniline 45 (65 mg, 0.28 mmol) and NEt3 (0.12 mL, 0.86 mmol) in DCM (10 mL) at 0 °C. The solution was allowed to warm to rt, and stirring was continued for 3 h; then, the reaction was diluted with H2O (20 mL) and extracted with DCM (3 × 20 mL). The extract was dried and evaporated, and the residue was subjected to flash chromatography. Elution with EtOAc gave carbamate 47 as a pale-yellow solid (54 mg, 66%), mp 160–163 °C. Rf (EtOAc) 0.3. IR (ATR) νmax cm–1: 3423 (NH), 1736 (O=COMe), 1650 (O=C1). 1H NMR (600 MHz, CDCl3) δ 8.11 (s, 1H, NH), 7.60 (s, 1H, H6), 6.97 (d, J = 7.2 Hz, 1H, H3), 6.77 (d, J = 7.2 Hz, 1H, H4), 3.94 (s, 3H, OMe), 3.89 (s, 3H, OMe), 3.82 (s, 3H, OMe), 3.55 (s, 3H, NMe). 13C NMR (150 MHz, CDCl3) δ 160.3 (C1), 154.3 (CO2Me), 150.8 (C5), 140.4 (C8), 130.8 (C3 + C6 coincident), 124.9 (C7), 119.7 (C4a or C8a), 103.8 (C4a or C8a), 100.0 (C4), 62.4 (C8-OMe), 56.1 (C5-OMe), 52.5 (CO2Me), 37.5 (NMe). HRMS (APCI): calcd for C14H17N2O5+ [M + H]+ 293.1132; found 293.1148.
7-t-Butoxycarbonylamino-5,8-dimethoxy-2-methylisoquinolin-1(2H)-one (48)
A solution of Boc2O (2.19 g, 10.0 mmol), aniline 45 (1.00 g, 4.27 mmol), and NEt3 (1.7 mL, 12 mmol) in THF (30 mL) was heated under reflux for 3 h. The solution was cooled, diluted with H2O (30 mL), and extracted with EtOAc (3 × 20 mL). The extract was dried and evaporated, and the residue was subjected to flash chromatography. Elution with 7:3 EtOAc/hexanes gave 48 as a pale-yellow oil (1.11 g, 77%). Rf (7:3 EtOAc/hexanes) 0.3. IR (ATR) νmax cm–1: 3420 (NH) 1722 (O=CO), 1650 (O=C1). 1H NMR (400 MHz, CDCl3) δ = 8.10 (s, 1H, NH), 7.43 (s, 1H, H6), 6.95 (d, J = 7.5 Hz, 1H, H3), 6.76 (d, J = 7.4 Hz, 1H, H4), 3.94 (s, 3H, OMe), 3.90 (s, 3H, OMe), 3.54 (s, 3H, NMe), 1.55 (s, 9H, t-Bu). 13C NMR (100 MHz, CDCl3) δ 160.2 (C1), 152.9 (CO2), 150.6 (C5), 140.2 (C8), 131.2 (C3 or C6), 130.4 (C3 or C6), 124.4 (C7), 119.5 (C4a or C8a), 103.8 (C4a or C8a), 99.9 (C4), 80.8 (t-Bu–O), 62.2 (C8-OMe), 56.0 (C5-OMe), 37.3 (NMe), 28.4 (3 × CH3). HRMS (APCI): calcd for C17H23N2O5+ [M + H]+ 335.1601; found 335.1604.
5,8-Dimethoxy-7-methoxycarbonyl(methyl)amino-2-methylisoquinolin-1(2H)-one (49)
Method A: NaH 60% dispersion in oil (9 mg, 0.2 mmol) and MeI (13 μL, 0.21 mmol) were added to a stirred solution of methyl carbamate 47 (50 mg, 0.17 mmol) in anhydrous THF (5 mL) at 0 °C under N2. The ice bath was removed, and stirring was continued for 3 h; then, the reaction mixture was poured into water (50 mL) and extracted with DCM (3 × 20 mL). The extract was dried and evaporated, and the residue was subjected to flash chromatography. Elution with 1:19 MeOH/DCM gave tertiary carbamate 49 as a white solid (39 mg, 75%), mp 150–155 °C. Rf (EtOAc) 0.3. IR (ATR) νmax cm–1: 1702 (O=COMe), 1671 (O=C1). 1H NMR (500 MHz, CDCl3) δ 7.04 (d, J = 6.6 Hz, 1H, H3), 6.80 (s, 1H, H6), 6.73 (d, J = 6.9 Hz, 1H, H4), 3.85 (s, 3H, OMe), 3.79 (s, 3H, OMe), 3.61 (s, 3H, OMe), 3.51 (s, 3H, N2Me), 3.23 (s, 3H, C7-NMe). 13C NMR (125 MHz, CDCl3) δ 160.3 (C1), 156.6 (CO2), 150.3 (C5 or C8), 149.7 (C5 or C8), 132.7 (C4 or C6), 130.0 (C4 or C6), 120.8 (C7), 112.5 (C4a or C8a), 107.7 (C4a or C8a), 99.6 (C4), 62.1 (C8-OMe), 56.0 (C5-OMe), 53.0 (CO2Me), 37.7 (NMe), 37.4 (NMe). HRMS (APCI): calcd for C15H19N2O5+ [M + H]+ 307.1304, found 307.1289.
Method B: MeI (0.11 mL, 1.77 mmol) and NaH 60% dispersion in oil (72 mg, 1.8 mmol) were added to a stirred solution of 54 (123 mg, 0.44 mmol) in anhydrous THF (1 mL) under N2. Stirring was continued overnight, then the reaction mixture was poured into ice/water (30 mL) and extracted with DCM (3 × 10 mL). The extract was washed with brine, dried, and evaporated, and the residue was subjected to flash chromatography. Elution with 1:19 MeOH/DCM gave tertiary carbamate 49 as a white solid (106 mg, 78%), identical with the material described above.
7-t-Butoxycarbonyl(methyl)amino-5,8-dimethoxy-2-methylisoquinolin-1(2H)-one (50)
NaH 60% dispersion in oil (140 mg, 3.5 mmol) and MeI (0.22 mL, 3.5 mmol) were added to a stirred solution of 48 (0.78 g, 2.3 mmol) in anhydrous THF (10 mL) at 0 °C under N2. The ice bath was removed, and stirring was continued for 3 h; then, the reaction mixture was poured into water (100 mL) and extracted with DCM (3 × 30 mL). The extract was washed with brine, dried, and evaporated, and the residue was subjected to flash column chromatography. Elution with 1:19 MeOH/DCM gave tertiary carbamate 50 as a pale-yellow oil (0.75 g, 92%). Rf 0.35 (1:19 MeOH/DCM). IR (ATR) νmax cm–1: 1720 (O=COMe), 1656 (O=C1). 1H NMR (400 MHz, DMSO-d6) δ 7.46 (d, J = 7.5 Hz, 1H, H3), 7.10 (s, 1H, H6), 6.64 (d, J = 7.4 Hz, 1H, H4), 3.86 (s, 3H, OMe), 3.67 (s, 3H, OMe), 3.45 (s, 3H, NMe), 3.10 (s, 3H, NMe), 1.31 (br s, 9H, t-Bu). 13C NMR (125 MHz, DMSO-d6), 159.0 (C1), 154.3 (br, CO2), 149.5 (C8), 148.8 (br, C5), 134.8 (br, C6), 134.0 (C3), 129.0 (br, C7), 119.6 (C4a or C8a), 113.3 (br, C4a or C8a), 97.9 (C4), 79.2 (br, t-Bu–O), 61.4 (C8-OMe), 56.2 (C5-OMe), 36.9 (br, 7-NMe) 36.7 (2-NMe), 28.0 (3 × CH3). One NMe signal was not observed/coincident. HRMS (APCI): calcd for C18H25N2O5+ [M + H]+ 349.1758; found 349.1764.
8-Hydroxy-5-methoxy-2-methylisoquinolin-1(2H)-one (51)
Anhydrous AlCl3 (4.56 g, 34.2 mmol) was added to a stirred solution of 35 (5.00 g, 28.8 mmol) in anhydrous DCM (100 mL) at 0 °C under N2. The reaction mixture was allowed to warm slowly to rt, and stirring was continued for 3 h. The mixture was diluted with ice/water (300 mL), and the DCM layer was separated. The aqueous phase was extracted with EtOAc (3 × 100 mL). The aqueous layer was then acidified with conc. HCl and reextracted with EtOAc (3 × 100 mL). The combined organic phase was washed with brine (300 mL), dried, and evaporated. The residue was subjected to flash column chromatography. Elution with DCM afforded phenol 51 as a white solid (4.00 g 85%), mp 180–133 °C. Rf (DCM) 0.3. IR (ATR) νmax cm–1: 3200–3000 (OH), 1676 (C=O). 1H NMR (400 MHz, CDCl3) δ 12.28 (s, 1H, OH), 6.99 (d, J = 8.8 Hz, 1H, H3), 6.94 (d, J = 7.5 Hz, 1H, H6), 6.81 (d, J = 7.3 Hz, 1H, H7), 6.79 (d, J = 8.6 Hz, 1H, H4) 3.84 (s, 3H, OMe), 3.53 (s 3H, NMe). 13C NMR (100 MHz, CDCl3) δ 164.9 (C1), 153.5 (C8), 145.6 (C5), 133.1 (C3), 127.6 (C4a or C8a), 115.4 (C8a), 111.4 (C6 or C7), 111.3 (C6 or C7), 101.2 (C4), 56.2 (OMe), 36.0 (NMe). HRMS (APCI) m/z: calcd for C11H12NO3+ [M + H] + 206.0812; found 206.0814.
8-Hydroxy-5-methoxy-2-methyl-7-nitroisoquinolin-1(2H)-one (52)
A solution of 69% HNO3 (0.19 mL, 2.9 mmol) in AcOH (3 mL) was added dropwise to a stirred solution of the phenol 51 (0.60 g, 2.9 mmol) in AcOH (3 mL) at such a rate as to maintain the temperature between 10 and 15 °C (50 min). Stirring was continued for 45 min, then water (10 mL) was added, and the precipitate was collected by vacuum filtration, washed with water, and air-dried to give 52 as a brown solid (0.67 g, 91%), mp 200–204 °C. Rf 0.3 (1:19 MeOH/DCM). IR (ATR) νmax cm–1: 1656 (C=O). 1H NMR (500 MHz, DMSO-d6) δ 14.55 (s, 1H, OH), 7.82 (d, J = 7.5 Hz, 1H, H3), 7.68 (s, 1H, H6), 6.89 (d, J = 7.4 Hz, 1H, H4), 3.92 (s, 3H, OMe), 3.60 (s, 3H, NMe). 13C NMR (125 MHz, DMSO-d6), 165.2 (C1), 151.2 (C8), 144.4 (C5), 137.9 (C3), 133.9, 130.6, 112.2, 108.5 (C6), 101.1 (C4), 56.4 (OMe), 36.6 (NMe). HRMS (APCI): calcd for C11H11N2O5+ [M + H]+ 251.0662; found 251.0661.
7-Amino-8-hydroxy-5-methoxy-2-methylisoquinolin-1(2H)-one (53)
SnCl2·2H2O (3.00 g, 13.32 mmol) was added to a stirred solution of 52 (0.66 g, 2.7 mmol) in EtOH (20 mL), and the mixture was heated under reflux for 1.5 h. The reaction mixture was cooled at rt, then poured into ice (150 g), and the resulting suspension was made alkaline by an addition of sat. aq. NaHCO3. The aqueous phase was extracted with DCM (3 × 25 mL), and the extract was dried and evaporated. The residue was diluted with 1 M HCl and washed with DCM. The aqueous layer was made alkaline by addition of NaHCO3 and reextracted with DCM (3 × 25 mL). The extract was washed with brine (25 mL), dried, and evaporated to give aniline 53 as a yellow solid (0.48 g, 81%), mp 140–143 °C. Rf (1:19 EtOAc/hexanes) 0.2. IR (ATR) νmax cm–1: 3400–2800 (OH/NH), 1652 (C=O); 1H NMR (500 MHz, DMSO-d6) δ 12.40 (s, 1H, OH), 7.04 (d, J = 7.5 Hz, 1H, H3), 6.79 (s, 1H, H6), 6.64 (d, J = 7.5 Hz, 1H, H4), 5.00 (br s, 2H, NH2), 3.77 (s, 3H, OMe), 3.46 (s, 3H, NMe). 13C NMR (500 MHz, DMSO-d6) δ 164.8 (C=O), 146.1 (C5), 138.4 (C8), 134.3, 127.5 (C3), 115.8, 110.9, 103.5 (C4 or C6), 102.0 (C4 or C6), 55.9 (OMe), 35.7 (NMe). HRMS (APCI): calcd for C11H13N2O3+ [M + H]+ 221.0921; found 251.0924.
8-Hydroxy-5-methoxy-7-methoxycarbonylamino-2-methylisoquinolin-1(2H)-one (54)
Methyl chloroformate (0.7 mL, 9 mmol) was added dropwise to a stirred solution of aniline 53 (1.98 g, 8.99 mmol) in anhydrous pyridine (30 mL) at 0 °C. After 0.5 h, the ice bath was removed and stirring was continued for 13 h. The reaction mixture was poured onto ice (∼200 mL), carefully acidified with conc. HCl, and extracted with EtOAc (4 × 50 mL). The extract was washed with water (2 × 50 mL) and brine (50 mL), dried, and evaporated to give the methyl carbamate 54 as a white solid (2.00 g, 80%), mp 132–133 °C. Rf (7:3 EtOAc/hexanes) 0.4. IR (ATR) νmax cm–1: 3200–2800 (NH/OH), 1709 (O=COMe), 1655 (O=C1). 1H NMR (500 MHz, DMSO-d6) δ 12.84 (s, 1H, OH), 8.69 (s, 1H, NH), 7.64 (br s, 1H, H6), 7.38 (d, J = 7.5 Hz, 1H, H3), 6.76 (d, J = 7.5 Hz, 1H, H4), 3.82 (s, 3H, OMe), 3.67 (s, 3H, OMe), 3.52 (s, 3H, NMe). 13C NMR (125 MHz, DMSO-d6) δ 164.8 (C1), 154.5 (CO2), 145.0 (C5 or C8), 144.4 (C5 or C8), 131.9 (C3), 126.2, 123.3, 122.1, 111.1 (C6), 101.3 (C4), 56.1 (OMe), 51.9 (CO2Me), 36.0 (NMe). HRMS (APCI): calcd for C13H15N2O5+ [M + H]+ 279.0975; found 279.0971.
7-Acetylamino-2-methylisoquinoline-1,5,8-trione (56)
A solution of CAN (0.74 g, 1.4 mmol) in H2O (5 mL) was added to a stirred solution of 46 (44 mg, 0.16 mmol) in MeCN (15 mL) at 0 °C. After 10 min, the reaction mixture was diluted with H2O (20 mL) and extracted with EtOAc (3 × 10 mL). The extract was dried and evaporated, and the residue was subjected to flash chromatography. Elution with 1:19 MeOH/DCM gave quinone 56 as a red-orange solid (30 mg, 77%), mp 230–233 °C. Rf (1:19 MeOH/DCM) 0.35. IR (ATR) νmax cm–1: 3257 (NH), 1682 (O=CNH), 1644 (O=C1). 1H NMR (600 MHz, DMSO-d6) δ 9.82 (s, 1H, NH), 8.37 (d, J = 6.6 Hz, 1H, H3), 7.54 (s, 1H, H6), 6.66 (d, J = 6.6 Hz, 1H, H4), 3.54 (s, 3H, NMe), 2.23 (s, 3H, COCH3). 13C NMR (150 MHz, DMSO-d6) δ 184.8 (C8), 177.4 (C5), 171.4 (OCNH), 157.3 (C1), 148.7 (C3), 143.2 (C4a or C7), 142.1 (C4a or C7), 115.4 (C8a), 112.8 (C6), 99.6 (C4), 38.2 (NMe), 24.7 (CH3). HRMS (ESI): calcd for C12H11N2O4+ [M + H]+ 247.0714; found 247.0738.
7-Methoxycarbonylamino-2-methylisoquinoline-1,5,8-trione (57)
A solution of CAN (0.16 g, 0.29 mmol) in H2O (1 mL) was added to a stirred solution of 47 (28 mg, 95 μmol) in MeCN (6 mL) at 0 °C. After 10 min, the reaction mixture was diluted with H2O (20 mL) and extracted with EtOAc (3 × 10 mL). The extract was dried and evaporated to give quinone 57 as a red-orange solid (24 mg, 95%), mp 189–193 °C. Rf (1:19 MeOH/DCM) 0.1. IR (ATR) νmax cm–1: 3356 (NH), 1742 (O=C), 1690 (O=C1). 1H NMR (600 MHz, DMSO-d6) δ 9.18 (s, 1H, NH), 8.38 (d, J = 6.6 Hz, 1H, H3), 7.2 (s, 1H, H6), 6.68 (d, J = 6.6 Hz, 1H, H4), 3.74 (s, 3H, OMe), 3.54 (s, 3H, NMe). 13C NMR (150 MHz, DMSO-d6) δ 184.0 (C8), 176.6 (C5), 157.3 (C1), 153.2 (CO2), 148.8 (C3), 143.4 (C4a or C7), 142.9 (C4a or C7), 115.4 (C8a), 111.5 (C6), 99.7 (C4), 52.9 (OMe), 38.2 (NMe). HRMS (APCI): calcd for C12H11N2O5+ [M + H]+ 263.0663; found 263.0675.
7-t-Butoxycarbonylamino-2-methylisoquinoline-1,5,8-trione (58)
A solution of CAN (4.8 g, 8.8 mmol) in H2O (10 mL) was added to a stirred solution of 48 (0.93 g, 2.77 mmol) in MeCN (30 mL) at 0 °C. After 10 min, the reaction mixture was diluted with H2O (50 mL) and extracted with EtOAc (3 × 20 mL). The extract was dried and evaporated to give 58 as a red-orange solid (0.59 g, 70%), mp 268–271 °C. Rf (EtOAc) 0.25. IR (ATR) νmax cm–1: 3380 (NH), 1746 (O=CO), 1621 (O=C1). 1H NMR (600 MHz, DMSO-d6) δ 8.57 (s, 1H, NH), 8.37 (d, J = 6.6 Hz, 1H, H3), 7.13 (s, 1H, H6), 6.67 (d, J = 6.6 Hz, 1H, H4), 3.54 (s, 3H, NMe), 1.48 (s, 9H, tBu). 13C NMR (150 MHz, DMSO-d6) δ 183.8 (C5), 176.6 (C8), 157.3 (C1), 151.4 (CO2), 148.9 (C3), 143.5 (C4a or C7), 142.7 (C4a or C7), 115.3 (C8a), 111.0 (C6), 99.8 (C4), 82.0 (t-Bu–O), 38.2 (NMe), 27.7 (3 × CH3). HRMS (APCI): calcd for C15H17N2O5+ [M + H]+ 305.1132; found 305.1127.
5,8-Dimethoxy-7-methoxycarbonyl(methyl)amino-2-methyl-4,6-dinitroisoquinolin-1(2H)-one (61)
A solution of CAN (0.42 g, 0.77 mmol) in H2O (1 mL) was added to a stirred solution of 49 (79 mg, 0.26 mmol) in MeCN (3 mL) at 0 °C. After 45 min, the reaction mixture was diluted with H2O (10 mL) and extracted with EtOAc (3 × 10 mL). The extract was dried and evaporated to give a residue that was purified by flash column chromatography. Elution with 9:1 EtOAc/hexanes afforded 61 as a yellow solid (67 mg, 63%), mp 160–163 °C. Rf (9:1 EtOAc/hexanes) 0.3. IR (ATR) νmax cm–1: 1687 (C=O). 1H NMR (600 MHz, CDCl3) δ 7.22 (s, 1H, H3), 3.98 (s, 3H, OMe), 3.83 (s, 3H, OMe), 3.75 (s, 3H, OMe), 3.42 (s, 3H, NMe), 3.29 (s, 3H, NMe). 13C NMR (150 MHz, CDCl3) δ 172.8, 160.6 (C1), 157.8, 157.6, 155.8, 151.4. 147.1, 124.3, 118.9, 118.4, 62.3 (OMe), 57.1 (OMe), 53.6 CO2Me), 37.5 (NMe), 27.6 (NMe).
5,8-Dimethoxy-2-methyl-7-methylaminoisoquinolin-1(2H)-one (62)
A solution of CAN (0.28 g, 0.52 mmol) in H2O (1 mL) was added to a stirred solution of carbamate 50 (60 mg, 0.17 mmol) in acetonitrile (2 mL) at 0 °C. After 10 min, the reaction mixture was diluted with H2O (3 mL) and extracted with EtOAc (3 × 10 mL). The organic extract was dried and evaporated to give quinone 62 as a pale-yellow solid (35 mg, 78%). Rf (2:23 MeOH/DCM) 0.3. IR (ATR) νmax cm–1: 3400 (NHMe), 1670 (C=O). 1H NMR (500 MHz, DMSO-d6) δ 6.77 (d, J = 7.4, Hz, 1H, H3), 6.69 (dd, J = 7.4, 0.5 Hz, 1H, H4), 6.52 (br s, 1H, H6), 4.79 (v. br s, 1H, NH), 3.91 (s, 3H, OMe), 3.86 (s, 3H, OMe), 3.51 (s, 3H, 2-NMe), 2.93 (s, 3H, 7-NMe). HRMS (APCI): calcd for C13H17N2O3+ [M + H]+ 249.1234; found 235.1236.
Ethyl (7-Methoxycarbonylamino-2-methylisoquinoline-1,5,8-trion-6-yl)-2-(2-nitrophenyl)acetate (64)
A mixture of K2CO3 (26 mg, 0.19 mmol), 57 (16 mg, 61 μmol), and ethyl o-nitrophenylacetate (2) (37 mg, 0.18 mmol) in dry DMF (15 mL) was stirred under Ar at 40 °C for 24 h, then cooled, diluted with 1 M HCl (20 mL), and extracted with EtOAc (3 × 10 mL). The extract was dried and evaporated, and the residue was subjected to flash chromatography. Elution with 1:19 MeOH/DCM gave 64 as an orange oil (20 mg, 71%). Rf (1:19 MeOH/DCM) 0.1. IR (ATR) νmax cm–1: 1736 (O=C), 1691 (O=C1). 1H NMR (600 MHz, CDCl3) δ 7.98 (d, J = 8.4 Hz, 1H, H3″), 7.87 (d, J = 7.2 Hz, 1H, H3′), 7.81 (s, 1H, NH), 7.51 (dd [app. t], J1 = J2 = 8.4 Hz, 1H, H4″ or H5″), 7.42 (dd [app. t], J1 = J2 = 8.4 Hz, 1H, H4″ or H5″), 7.28 (d, J = 8.4 Hz, 1H, H6″), 6.84 (d, J = 7.2 Hz, 1H, H4′), 6.00 (s, 1H, H2), 4.21 (m, 1H, OCH2-a), 4.14 (m, 1H, OCH2-b), 3.68 (OMe or NMe), 3.66 (OMe or NMe), 1.16 (t, J = 7.2 Hz, 3H, OCH2CH3). 13C NMR (150 MHz, CDCl3) δ 183.8 (C5′), 178.0 (C8′), 169.7 (C1), 157.8 (C1′), 153.0 (NCO2), 149.9 (C2″), 146.5 (C3′), 144.2 (C4a′), 141.7 (C6″), 132.6 (C4″ or C5″), 131.7 (C1″), 130.9 (C4″ or C5″), 128.2 (C3″ or C6″), 127.0 (C7′), 124.9 (C3″ or C6″), 116.8 (C8a′), 101.5 (C4′), 62.0 (OCH2), 53.9 (OMe), 47.0 (C2), 39.2 (NMe), 14.0 (OCH2CH3). HRMS (APCI): calcd for C22H20N3O9+ [M + H]+ 470.1195; found 470.1219.
Ethyl (7-t-Butoxycarbonylamino-2-methylisoquinoline-1,5,8-trion-6-yl)-2-(2-nitrophenyl)acetate (65)
A solution of 58 (0.56 g, 1.8 mmol) in DMF (30 mL) was added slowly to a stirred suspension of K2CO3 (1.4 g, 10 mmol), ethyl o-nitrophenylacetate (2) (1.3 g, 6.2 mmol), and 18-crown-6 (52 mg, 0.19 mmol) in dry DMF (50 mL) at 45 °C under Ar. After 24 h, the reaction mixture was cooled, diluted with 1 M HCl (50 mL), and extracted with EtOAc (3 × 25 mL). The extract was dried and evaporated, and the residue was subjected to flash chromatography. Elution with 1:19 MeOH/DCM gave 65 as an orange oil (0.43 g, 46%). Rf (1:19 MeOH/DCM) 0.2. IR (ATR) νmax cm–1: 1732 (O=CO), 1689 (O=C1). 1H NMR (600 MHz, CDCl3) δ 7.97 (d, J = 7.8 Hz, 1H, H3″), 7.89 (d, J = 6.6 Hz, 1H, H3′), 7.66 (s, 1H, NH), 7.46 (dd [app. t], J1 = J2 = 7.8 Hz, 1H, H5″), 7.39 (dd [app. t], J1 = J2 = 7.8 Hz, 1H, H4″), 7.22 (d, J = 7.8 Hz, 1H, H6″), 6.80 (d, J = 6.6 Hz, 1H, H4′), 6.01 (s, 1H, H2), 4.18 (m, 1H, OCH2-a), 4.10 (m, 1H, OCH2-b), 3.65 (s, 3H, NMe), 1.37 (s, 9H, tBu), 1.14 (t, J = 7.2 Hz, 3H, OCH2CH3). 13C NMR (150 MHz, CDCl3) δ 183.7 (C5′), 178.3 (C8′), 169.9 (C1), 157.8 (C1′), 151.2 (HNC=O), 149.7 (C2″), 146.7 (C3′), 144.2 (C4a′), 142.3 (C6′), 132.7 (C4″ or C5″), 131.8 (C1″), 130.7 (C4″ or C5″), 128.0 (C3″ or C6″), 126.4 (C7′), 124.8 (C3″ or C6″), 116.7 (C8a′), 101.5 (C4′), 83.1 (t-Bu–O), 61.9 (OCH2), 47.1 (C2), 39.1 (NMe), 27.9 (tBu), 13.9 (OCH2CH3). HRMS (ESI–): calcd for C25H24N3O9– [M–H]− 510.1518; found 510.1518.
Ethyl 2-(7-Amino-8-hydroxy-2-methyl-1,5-dioxo-1,2-dihydroisoquinolin-6(5H)-ylidene)-2-(2-nitrophenyl)acetate (66)
Ethanolic HCl was prepared by adding AcCl (5 mL, 0.3 mol) to EtOH (10 mL) at 0 °C. The resulting solution was then added dropwise to a solution of 65 (0.15 g, 0.29 mmol) in EtOH (20 mL). After 20 h, the solution was heated under reflux for 30 min before being cooled, diluted with H2O (30 mL), and extracted with EtOAc (3 × 10 mL). The extract was dried and evaporated then triturated with hot 1:9 EtOAc/hexanes, leaving what has been tentatively assigned as 66 as a purple solid (62 mg, 51%), mp 270–273 °C. Rf (1:19 MeOH/DCM) 0.15. IR (ATR) νmax cm–1: 3300–2800 (OH), 1658 (O=C1). 1H NMR (600 MHz, DMSO-d6) δ 8.25 (d, J = 6.6 Hz, 1H, H3′), 8.03 (d, J = 8.4 Hz, 1H, H3″ or H6″), 7.71 (dd [app. t], J1 = J2 = 8.4 Hz, 1H, H4″ or H5), 7.62–7.55 (m, 2H, ArH), 6.63 (d, J = 6.6 Hz, 1H, H4″), 4.20 (q, J = 7.2 Hz, 2H, OCH2), 3.52 (s, 3H, NMe), 1.16 (t, J = 7.2 Hz, 3H, OCH2CH3). 13C NMR (150 MHz, DMSO-d6) δ 179.2 (C5′), 173.0 (C1), 157.7 (C1′), 148.5 (C3′), 146.7, 145.2, 133.4 (ArH), 132.6 (ArH), 128.2, 126.0, 124.2 (ArH), 119.9, 116.3, 101.1 (ArH), 99.9 (ArH), 68.7 (OCH2), 37.9 (NMe), 14.4 (OCH2CH3).
Acknowledgments
We thank the UWA Centre for Characterisation, Microscopy and Analysis, in particular Drs. Lindsay Byrne and Gareth Nealon for assistance with NMR spectroscopy and Drs. Anthony Reeder and Michael Clarke for help with mass spectrometry. The Australian Government is gratefully acknowledged for an Australian Postgraduate Award (M.B.) and Research Training Program (RTP) Scholarships (F.D. and L.T.). Waiver of tuition fees (F.D.) and a UWA–UQ Bilateral Research Collaboration Award from the University of Western Australia facilitated this work. C.M.W. thanks the University of Queensland for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c02117.
Author Contributions
+ M.B. and F.D. contributed equally.
The authors declare no competing financial interest.
Footnotes
As noted in the Results and Discussion section, when this reaction was repeated several years later, the yields of 36:37 were inverted to 13:76%.
Supplementary Material
References
- Buccini M.; Jeow S. Y.; Byrne L.; Skelton B. W.; Nguyen T. M.; Chai C. L. L.; Piggott M. J. Bisannulation of 2,3-dichloro-1,4-naphthoquinone with o-nitrophenylacetic acid derivatives: a succinct synthesis of the ABCD ring system of alpkinidine. Eur. J. Org. Chem. 2013, 3232–3240. 10.1002/ejoc.201300227. [DOI] [Google Scholar]
- Buccini M.; Tham L.; Dhoro F.; Skelton B. W.; Williams C. M.; Piggott M. J. Towards the Total Synthesis of Alpkinidine: Synthesis of Haloquinone CE Ring-System Synthons and Attempted Nucleophilic Bisannulation. ACS Omega 2022, 10.1021/acsomega.2c02116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thale Z.; Johnson T.; Tenney K.; Wenzel P. J.; Lobkovsky E.; Clardy J.; Media J.; Pietraszkiewicz H.; Valeriote F. A.; Crews P. Structures and cytotoxic properties of sponge-derived bisannulated acridines. J. Org. Chem. 2002, 67, 9384–9391. 10.1021/jo026459o. [DOI] [PubMed] [Google Scholar]
- Kitahara Y.; Mizuno T.; Kubo A. Synthetic studies of benzo[b]pyrrolo[4,3,2-de][1,10]phenanthroline. Tetrahedron 2004, 60, 4283–4288. 10.1016/j.tet.2004.03.057. [DOI] [Google Scholar]
- Komori T.; Yokoshima S.; Fukuyama T. Synthetic studies on plakinidines. Synlett 2015, 26, 1537–1540. 10.1055/s-0034-1380689. [DOI] [Google Scholar]
- Volvoikar P. S.; Tilve S. G.; Zubkov F. I. A concise approach for the synthesis of the ABCD ring system of alpkinidine. ChemistrySelect 2019, 4, 7187–7189. 10.1002/slct.201900357. [DOI] [Google Scholar]
- Rho H. S.; Baek H. S.; Ahn S. M.; Yoo J. W.; Kim D. H.; Kim H. G. Studies on depigmenting activities of dihydroxyl benzamide derivatives containing adamantane moiety. Bioorg. Med. Chem. Lett. 2009, 19, 1532–1533. 10.1016/j.bmcl.2008.12.106. [DOI] [PubMed] [Google Scholar]
- Prey V. Cleavage of phenol ethers with pyridine hydrochloride. Ber. Dtsch. Chem. Ges. B 1941, 74, 1219–1225. 10.1002/cber.19410740715. [DOI] [Google Scholar]
- Hofslokken N. U.; Skattebol L. Convenient method for the ortho-formylation of phenols. Acta Chem. Scand. 1999, 53, 258–262. 10.3891/acta.chem.scand.53-0258. [DOI] [Google Scholar]
- Evans R. M.; Downes M.; Baiga T. J.; Noel J. P.; Embler E. K.; Fan W.; Keana J. F. W.; Bock M. G.; Kluge A. F.; Patane M. A.. Preparation of phenoxy hexanoic acid compounds as PPARγ agonists for therapy. United States patent, US20160023991 A1 2016-01-28.
- Baker R.; Castro J. L. Total synthesis of (+)-macbecin I. J. Chem. Soc., Perkin Trans. 1 1990, 47–65. 10.1039/p19900000047. [DOI] [Google Scholar]
- Godard A.; Fourquez J. M.; Tamion R.; Marsais F.; Quéguiner G. New synthesis of the streptonigrin quinoline-5,8-quinone moiety and aromatic cross-coupling. Synlett 1994, 235–236. 10.1055/s-1994-22807. [DOI] [Google Scholar]
- Chawla H. M.; Mittal R. S. Cerium (IV) induced one pot synthesis of 2,4-dihydroxy-3-nitroacetophenone - a model for biomimetic aromatic hydroxylations. Curr. Sci. 1983, 52, 1189–1190. [Google Scholar]
- Chakrabarty M.; Batabyal A. A new method of nitration of carbazoles using ceric ammonium nitrate (CAN). Synth. Commun. 1994, 24, 1–10. 10.1080/00397919408012618. [DOI] [Google Scholar]
- Ganguly N.; Sukai A. K.; De S. Cerium(IV) ammonium nitrate mediated nitration of coumarins. Synth. Commun. 2001, 31, 301–309. 10.1081/SCC-100000214. [DOI] [Google Scholar]
- Yang X.; Xi C.; Jiang Y. Regioselective nitration of N,N-dialkylanilines using cerium(IV) ammonium nitrate in acetonitrile. Tetrahedron Lett. 2005, 46, 8781–8783. 10.1016/j.tetlet.2005.10.028. [DOI] [Google Scholar]
- Tohma H.; Morioka H.; Harayama Y.; Hashizume M.; Kita Y. Novel and efficient synthesis of p-quinones in water via oxidative demethylation of phenol ethers using hypervalent iodine(III) reagents. Tetrahedron Lett. 2001, 42, 6899–6902. 10.1016/S0040-4039(01)01407-1. [DOI] [Google Scholar]
- Hwu J. R.; Jain M. L.; Tsay S.-C.; Hakimelahi G. H. Ceric ammonium nitrate in the deprotection of tert-butoxycarbonyl group. Tetrahedron Lett. 1996, 37, 2035–2038. 10.1016/0040-4039(96)00211-0. [DOI] [Google Scholar]
- Evans E. F.; Lewis N. J.; Kapfer I.; Macdonald G.; Taylor R. J. K. N-tert-Butoxycarbonyl (BOC) deprotection using boron trifluoride etherate. Synth. Commun. 1997, 27, 1819–1825. 10.1080/00397919708006783. [DOI] [Google Scholar]
- Houghten R. A.; Beckman A.; Ostresh J. M. Use of 10% sulfuric acid/dioxane for removal of N-α-tertiary-butyloxycarbonyl group during solid phase peptide synthesis. Int. J. Pept. Protein Res. 1986, 27, 653–658. [Google Scholar]
- De Guzman F. S.; Carte B.; Troupe N.; Faulkner D. J.; Harper M. K.; Concepcion G. P.; Mangalindan G. C.; Matsumoto S. S.; Barrows L. R.; Ireland C. M. Neoamphimedine: a new pyridoacridine topoisomerase II inhibitor which catenates DNA. J. Org. Chem. 1999, 64, 1400–1402. 10.1021/jo982047x. [DOI] [Google Scholar]
- Tasdemir D.; Marshall K. M.; Mangalindan G. C.; Concepcion G. P.; Barrows L. R.; Harper M. K.; Ireland C. M. Deoxyamphimedine, a new pyridoacridine alkaloid from two tropical Xestospongia sponges. J. Org. Chem. 2001, 66, 3246–3248. 10.1021/jo010153k. [DOI] [PubMed] [Google Scholar]
- Marshall K. M.; Matsumoto S. S.; Holden J. A.; Concepcion G. P.; Tasdemir D.; Ireland C. M.; Barrows L. R. The anti-neoplastic and novel topoisomerase II-mediated cytotoxicity of neoamphimedine, a marine pyridoacridine. Biochem. Pharmacol. 2003, 66, 447–458. 10.1016/S0006-2952(03)00209-0. [DOI] [PubMed] [Google Scholar]
- Dhoro F.; Parkin-Gibbs J.; McIldowie M.; Skelton B. W.; Piggott M. J. Confirmation of the revised structure of samoquasine A and a proposed structural revision of cherimoline. J. Nat. Prod. 2018, 81, 1658–1665. 10.1021/acs.jnatprod.8b00319. [DOI] [PubMed] [Google Scholar]
- Chatterjea J. N.; Jha H. C.; Banerjee B. K. Isocarbostyrils. I. Conversion of 8-alkoxyisocarbostyril-3-carboxylic acids into the related hydroxy esters by pseudo rearrangement. J. Indian Chem. Soc. 1966, 43, 633–647. [Google Scholar]
- McGarrigle E. M.; Murphy D. M.; Gilheany D. G. Ligand tuning in the chromium-salen-mediated asymmetric epoxidation of alkenes. Tetrahedron: Asymmetry 2004, 15, 1343–1354. 10.1016/j.tetasy.2004.03.010. [DOI] [Google Scholar]
- Rubenstein L. Substitution in derivatives of quinol ethers. J. Chem. Soc., Trans. 1925, 127, 1998–2004. 10.1039/CT9252701998. [DOI] [Google Scholar]
- Kashin D.; Meyer A.; Wittenberg R.; Schoening K.-U.; Kamlage S.; Kirschning A. A practical synthesis of the ansa chain of benzenic ansamycin antibiotics: total synthesis of an ansatrienol derivative. Synthesis 2007, 304–319. 10.1055/s-2006-958967. [DOI] [Google Scholar]
- Klemenc A. Nitrogentisic acid. Monatsh. Chem. 1913, 33, 1243–1254. [Google Scholar]
- Yan Y.; Qin B.; Ren C.; Chen X.; Yip Y. K.; Ye R.; Zhang D.; Su H.; Zeng H. Synthesis, structural investigations, hydrogen-deuterium exchange studies, and molecular modeling of conformationally stabilized aromatic oligoamides. J. Am. Chem. Soc. 2010, 132, 5869–5879. 10.1021/ja100579z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.