

Abstract
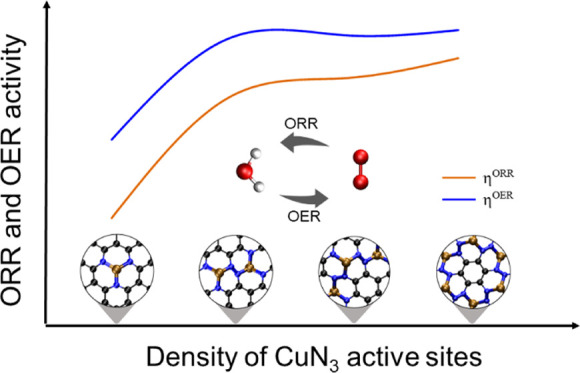
Atomically dispersed M–N–C has been considered an effective catalyst for various electrochemical reactions such as oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), which faces the challenge of increasing metal load while simultaneously maintaining catalytic performance. Herein, we put forward a strategy for boosting catalytic performances of a single Cu atom coordinated with three N atoms (CuN3) for both ORR and OER by increasing the density of connected CuN3 moieties. Our calculations first show that a single CuN3 moiety exhibiting no catalytic performance for ORR and OER can be activated by increasing the density of metal centers, which weakens the binding affinity to *OH due to the lowered d-band center of the metal atoms. These findings stimulate the further theoretical design of a two-dimensional compound of C3N3Cu with a high concentration of homogeneously distributed CuN3 moieties serving as bifunctional active sites, which demonstrates efficient catalytic performance for both ORR and OER as reflected by the overpotentials of 0.71 and 0.43 V, respectively. This work opens a new avenue for designing effective single-atom catalysts with potential applications as energy storage and conversion devices possessing high density of metal centers independent of the doping strategy and defect engineering, which deserves experimental investigation in the future.
1. Introduction
Severe energy and environmental problems have prompted research efforts into developing electrochemical energy storage and conversion devices.1−4 Rechargeable aqueous metal–air batteries,5 represented by rechargeable Zn–air batteries,6,7 possess the advantages of high theoretical energy density, low cost originating from the high-abundance anode material, and inherent safety using aqueous electrolytes.8−10 Generally, a conventional rechargeable aqueous metal–air battery is made up of a metal foil anode and a cathode using oxygen as the cathode active material. In the air cathode, the vital catalytic processes of the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) are involved during the discharge and charge processes, respectively.11−13 However, the kinetics of the cathodic oxygen redox reaction is sluggish, which restricts the conversion efficiency between O2 and H2O.14 So far, precious metals, their alloys, and their oxides are the main commercial catalysts, but the large-scale application of noble-metal-based catalysts is seriously restricted by their scarcity, stability and cost.15 In particular, their catalytic performance is closely related to the types of noble metals, and the corresponding oxygen catalytic activities of different metals are quite disparate.16 For example, RuO2/IrO2 shows excellent OER activity; unfortunately, its catalytic ORR activity is not ideal. Pt-based catalysts are considered to be the best ORR catalysts, but their OER performance is low.17 In this case, the reasonable construction of a low-cost and highly efficient bifunctional catalyst is the research hotspot of secondary metal–air batteries.18
Single-atom catalysts (SACs) containing atomically dispersed metal centers immobilized on heteroatom-doped substrates have recently sparked tremendous interest due to their high material utilization efficiency and enhanced catalytic performances.19−21 Among them, the most reported SACs are limited to transition-metal atoms (Fe, Co, Ni, Cu, etc.) supported by heteroatom N-doped carbonaceous materials, which can be applied to the catalysis of a wide range of electrochemical reactions of ORR,22−28 nitrogen reduction reaction (NRR),29−34 carbon dioxide reduction reaction (CO2RR),35−41 hydrogen reduction reaction (HER),42−44 OER,8,45,46 etc. The single metal atom and its coordination environment are usually modeled by a MN4 moiety having a metal center coordinated with four N atoms.20,47 Nevertheless, their catalytic performances toward ORR lag still behind the apex of state-of-the-art Pt. Recent studies have demonstrated that the catalytic performance of MN4 containing non-noble metals can be further enhanced by rationally tailoring the local structures of central metal atoms by introducing functional groups,45 the second metal center,48 and other heteroatoms beyond N.45,49 However, the low density of single-atomic sites is still one of the big challenges for SACs because high load and the aggregation of metal atoms have to be balanced. Recently, a metal load of up to 12.1 wt % is achieved based on a general cascade anchoring strategy for fabricating MNx (M = Fe, Mn, Co, Ni, Cu, Mo, Pt, etc.) toward ORR.26 However, an ultrahigh load of metal atoms cannot be obtained without weakening the ORR performance. For example, when the distance between two neighboring Fe atoms is lower than 0.70 nm, the strengthened interaction between two adjacent FeN4 moieties will worsen the catalytic performance for ORR.27 This indicates as well that designing high-performance FeN4 for ORR with a high concentration of active sites will be highly restricted based on the conventional heteroatom doping strategy.
The appearance of the MN3 moiety cannot be excluded based on both the complex preparation concept50 and dynamic evolution of the active sites during the reactions,51 which have recently attracted significant research efforts. Using operando XANES, Yang et al. identified that it is CuN3 instead of CuN4 that is responsible for ORR under working conditions, although ex situ characterizations show that MN4 remains before and after the reaction.51 FeN3 possesses mediocre catalytic performance toward ORR compared to FeN4,52 which could be enhanced when FeN3 was deposited over Pd particles embedded in N-doped carbons.50 Although enhanced ORR performance could be realized by increasing the concentration of the FeN3 moiety, it requires preadsorption of functional groups onto the metal atoms adjacent to the active sites.52 Such a complex strategy of introducing functional groups and/or interfaces for achieving MN3 active sites toward ORR50−52 highlights the urgency of identifying new rationality for designing high-performance SACs, which can balance the high density of active sites and facile strategy of fabricating catalysts. In this regard, two-dimensional (2D) materials containing high concentrations of homogeneously distributed metal centers will be potential candidate catalysts.53,54 Therefore, a novel designing rationality is highly required, which is in turn dependent on the knowledge of appropriate arrangements of MNx leading to competitive catalytic performance.
Herein, we propose a novel strategy of designing bifunctional catalysts for ORR and OER, which contains a high density of MN3 serving as active sites without involving additional strategies to tailor the local environment of metal atoms. Using density functional theory (DFT) calculations, we reported the first demonstration of the density effect of CuN3 on the catalytic performances for ORR and OER. Our calculations demonstrated that the systems containing three connected CuN3 moieties could give rise to significantly decreased overpotentials of 0.91 and 0.48 V for ORR and OER, respectively, compared to those larger than 1.23 V with single CuN3. These findings triggered the theoretical design of the 2D crystalline phase of C3N3Cu containing an even higher concentration of CuN3, which was then predicted to be a bifunctional catalyst for ORR and OER with lowered overpotentials of 0.71 and 0.43 V, respectively. The catalytic performances of C3N3Ni and C3N3Zn for ORR and OER were also explored. Of the two, C3N3Ni possesses good OER activity, while it shows no catalytic performance for ORR. In contrast, the performances of C3N3Zn for both ORR and OER are low.
2. Computational Details
Geometry optimizations and total energy calculations were performed based on DFT55 implemented in the Vienna ab initio simulation package (VASP).56,57 To describe the nucleus–electron interactions, the projector augmented wave (PAW)58,59 potentials were adopted. The analysis of the exchange–correlation energy was carried out by the Perdew–Burke–Ernzerhof (PBE)60,61 functional within the generalized gradient approximation (GGA).62,63 The van der Waals interactions were described by the DFT-D364 approach. The plane wave basis set57 with a cutoff energy of 520 eV was utilized throughout this work to expand the wave functions of valence electrons. To avoid interaction of periodic images along the z-direction, a vacuum layer region was set, which was larger than 15 Å. A 6 × 6 × 1 supercell of graphene was built to model these SACs with various concentrations of the CuN3 moiety. For the structural optimization, self-consistent field (SCF) calculations, and non-SCF calculations (NSCF), the corresponding Γ-centered k-mesh grids65 for sampling the Brillouin zone were set to be 2 × 2 × 1, 3 × 3 × 1 , and 6 × 6 × 1, respectively. For C3N3M (M = Cu, Zn, and Ni), a 6 × 6 × 1 k-mesh grid was used to optimize the original geometry, and a 9 × 9 × 1 k-mesh grid was set for electronic structure computations. We set up a series of 2 × 2 × 1 supercells of pristine C3N3M (containing 56 atoms) for exploring the adsorption properties of reaction intermediates of ORR and OER. A k-mesh grid of 2 × 2 × 1 was used for geometry optimization, while meshes of 3 × 3 × 1 and 6 × 6 × 1 were used for SCF calculation and NSCF calculation, respectively. All of the structure configurations were optimized until the energy and the force on each atom were less than 1 × 10–5 eV and 0.02 eV/Å, respectively. Additionally, the bonding behaviors were analyzed based on the electron localization function (ELF).66 The phonon spectra were calculated using the Phonopy code.67 The thermodynamic stabilities of a C3N3Cu monolayer and a C3N3Cu nanoflake were evaluated by ab initio molecular dynamics (AIMD) simulations in an NVT ensemble at 300 K. Note that the AIMD simulations for a C3N3Cu monolayer were carried out using a 3 × 3 × 1 supercell, which contains 126 atoms. While regarding the C3N3Cu nanoflake with 96 atoms, a simulation box with a = b = 32.00 Å, c = 21.29 Å, and α = β = γ = 90° was set up, giving rise to a vacuum layer region in x, y, and z directions of about 15, 15, and 20 Å, respectively. For the C3N3Cu monolayer and C3N3Cu nanoflake, the durations of AIMD simulations are greater than 3 ps, the time step is 3 fs, and the SMASS is set to be 2. The k-mesh grids used for both systems were set to be 1 × 1 × 1. More computational details on cohesive energies, adsorption energies, charge density difference, and Gibbs free energies68 for electrochemical reactions and surface models are given in the Supporting Information.
3. Results and Discussion
3.1. Effect of Concentrations of CuN3 Moiety on Performances of ORR and OER
We initially set up three models of SACs by introducing different numbers of CuN3 moiety in the simulation box to describe the systems containing various concentrations of active sites. As shown in Figure 1a–c, 1-CuN3, 2-CuN3, and 3-CuN3 refer to the model of SACs containing one, two, and three CuN3 moieties, respectively. Note that the reason why we utilized such arrangements of CuN3 moieties in 2-CuN3 and 3-CuN3 is claimed at the end of this section. The average bond lengths of Cu–N are 1.71, 1.89, and 1.89 Å in these three systems. Bader charge analysis shows that Cu losses 0.69, 0.69, and 0.71 |e| to its neighboring N atoms in 1-CuN3, 2-CuN3, and 3-CuN3, respectively. Calculated projected density of states (PDOSs) demonstrate that the increased density of CuN3 leads to enhanced electron conductivity as verified by the decreased band gaps following the order 1-CuN3 > 3-CuN3 > 2-CuN3. Moreover, deep analysis of PDOS demonstrates that 3dxy and 3dx2–y2 contribute mainly to those occupied states near the Fermi level in 1-CuN3, while the highest occupied orbitals become 3dyz and 3dx2–y2 when it turns to the cases of 2-CuN3 and 3-CuN3 (see Figure S1). Although these properties of bond length, charge transfer, and band gap exhibit no positive correlation with the concentration of CuN3 moieties, their significant distinctions between 1-CuN3 and 3-CuN3 still might be a hint toward tuned catalytic performances.
Figure 1.

Structural configurations, PDOS, and catalytic performances of CuN3 moieties with various concentrations. (a–c) SACs containing one, two, and three CuN3 moieties in the simulation box are labeled as 1-CuN3, 2-CuN3, and 3-CuN3, respectively. The bond lengths (Å) of C–N are listed as well. (d–f) PDOSs of 1-CuN3, 2-CuN3, and 3-CuN3. (g–i) Free energy profiles of ORR proceeding on 1-CuN3, 2-CuN3, and 3-CuN3, respectively. (j–l) Free energy profiles of OER proceeding on 1-CuN3, 2-CuN3, and 3-CuN3, respectively. In (g–l), the green arrow refers to the rate-determining step (RDS) and the numbers in green are the values of overpotentials (V).
We next systematically investigated the effect of CuN3 density on the activity toward ORR and OER based on the calculated free energies of reaction intermediates of *OOH, *OH, and *O involved in both reactions based on the equations listed in the Supporting Information. As is well known, neither too strong nor too weak interaction strength between the substrates and adsorbates is required for achieving an efficient catalytic performance of catalysts. Ideally, the free energy change for each reaction step should be 1.23 eV (when U = 0). In reality, however, the free energy steps are not distanced equally so that the reactions will be determined by the rate-determining step (RDS). As shown in Figure 1g–l, on the basis of these free energies, one can clearly see the distinct RDSs of (*OH + e– → OH–) and (*OH + OH– → *O + H2O (l) + e–) for ORR and OER, respectively, which were considered for obtaining the overpotentials of both reactions. Fully relaxed reaction intermediates adsorbed on the substrates are illustrated in Figures S2–S4. Using eq S9, we achieved the overpotentials (ηORR) of 2.30, 1.11, and 0.91 V for ORR catalyzed by 1-CuN3, 2-CuN3, and 3-CuN3, respectively. It demonstrates clearly that a single CuN3 moiety is not active for ORR, while upon increasing the number of CuN3 moieties from 1 to 3, the overpotential of ORR is gradually decreased. With order of 3-CuN3 < 2-CuN3 < 1-CuN3, the free energy change of RDS for three systems can provide us with insights into understanding the enhanced ORR performance. As shown in Figure 1g–i, when increasing the number of CuN3 there is a shift of the free energy change of RDS to a smaller value, leading to the lowest overpotential for 3-CuN3. Therefore, the enhanced ORR performance in 3-CuN3 could be attributed to the weakened interaction between reaction intermediates and substrates, especially between *OH and CuN3 moieties. The underlying mechanism will be discussed later.
DFT calculations also demonstrate the critical role of the high density of the CuN3 moiety in boosting OER activity. As described in the Supporting Information, the OER proceeds by also involving the adsorption of three reaction intermediates of *OH, *O, and *OOH. On the basis of free energy profiles of OER shown in Figure 1j–l, we obtained the corresponding overpotentials of OER (ηOER), which were 1.52, 0.52, and 0.48 V for 1-CuN3, 2-CuN3, and 3-CuN3, respectively. This implies that with an increase in the density of the CuN3 moiety, the OER performance will decrease the free energy change of the second step of OER (*OH + OH− → *O + H2O(l) + e−)), which is determined mainly by the binding strength of *OH and substrates. Therefore, similar to the case of ORR, we believed that the enhanced OER performance can also be attributed to the weakened interaction of *OH and substrates. Overall, 3-CuN3 can serve as the best bifunctional catalyst for ORR and OER, outperforming the other two systems as reflected by the overpotentials of ηORR = 0.91 V and ηOER = 0.48 V, respectively. Note that the OER performance of 3-CuN3 is even comparable to that of RuO2.8 Additional calculations for exploring the trapping capabilities for O2 and H2O were also carried out, which show that 1-CuN3, 2-CuN3, and 3-CuN3 possess strong binding strength to these two molecules as reflected by the adsorption energies and configurations (see Figure S5).
We tried four possible arrangements of a CuN3 dimer, which are shown in Figure S6 in the Supporting Information as well as their total energies and overpotentials of ORR over them, indicating their mediocre catalytic performance. Although the configuration of 2-CuN3 illustrated in Figure S6c does not outperform the other three candidates, as shown in Figure S6a,b,d, based on the overpotentials and total energies, it was still selected (see Figure 1b) due to its shortest metal distances benefiting the rational designing of 2D catalysts. Also, the corresponding adsorption configurations of reaction intermediates are shown in Figure S7. Since the selected configuration of 2-CuN3 has two connected CuN3 moieties, the model of 3-CuN3 was constructed via three connected CuN3 moieties shown in Figure 1c. Generally, increasing the concentration of CuN3 will enhance the ORR performance, which further inspired us to explore the possibilities of predicting SACs with higher loadings of metal atoms.
3.2. Structures, Stabilities and Catalytic Performances of C3N3M (M = Cu, Ni, and Zn) Monolayer
Although 3-CuN3 is proved to be active for both ORR and OER, it is rather complex to precisely control such specific distribution of CuN3 moieties during the synthesis process given the most widely accepted strategy of synthesizing SACs via one-pot pyrolysis for transition-metal precursors with N,C-containing organic precursors.69 To address such an issue, an alternative method is to rationally design two-dimensional (2D) crystalline phase compounds containing homogeneously distributed active sites, which is not dependent on the doping strategy anymore.
We theoretically designed a 2D C3N3Cu, as shown in Figure 2a, which contains not only the structural features of CuN3 connecting to each other but also possesses homogeneously distributed metal atom sites. It was found to be dynamically stable as verified by the phonon dispersion curves along high symmetry directions in the Brillouin zone, as shown in Figure 2b. Fully relaxed C3N3Cu crystallizes in space groups of P3̅ with lattice vectors of a = b = 6.51 Å. Also, more structural details are given in Table S1. Close examination of the fully relaxed geometries of C3N3Cu structures shows that the 2D compound is composed of small hexagon patches of sp2 carbons surrounded by six connected CuN3 moieties. The average Cu–N bond length is around 1.87 Å, which is slightly shorter than 1.90 Å in 3-CuN3, as shown in Figure 1c. ELF can be utilized to map the localization of electrons in the neighborhood space. As shown in Figure S8, the ELF maps imply that in C3N3Cu the interaction of N–C and C–C is characterized as covalent bonds (ELF > 0.5), while that of Cu–N is characterized as ionic bonds (ELF < 0.5).
Figure 2.

Predicated C3N3Cu monolayer and corresponding properties. (a) Structural configuration, (b) phonon dispersion curves, (c) AIMD simulations at 300 K with energy and temperature variations with respect to the simulation time as well as the snapshot of the last step of AIMD simulation, and (d) band structure and PDOS.
As shown in Figure 2c, C3N3Cu should be thermodynamically stable at room temperature as demonstrated by regular oscillations of instant temperature and kinetic energy near the equilibrium values and negligible structure distortion. Further, the stability of the C3N3Cu nanoflake was also evaluated by AIMD simulations, which indicate that nanoscale C3N3Cu should be stable at room temperature (see Figure S9). Note that in the C3N3Cu monolayer, there exists a N–N bond, as shown in Figure 2a, which has been confirmed to be stable in the graphene lattice by both experimental70 and theoretical71,72 means. Aiming to further verify the stability of the N–N bond, we calculated the free energy diagram of N2 formation by decomposition of the graphene in two different ways, which are illustrated in Figures S10 and S11. The free energy change values for the migration of the N dimer from the carbon lattice into the vacuum are around 4.62 and 1.00 eV, respectively, indicating that the N–N bond in the graphene lattice is significantly stable.
In addition, we also calculated the cohesive energies of C3N3Cu of −5.58 eV/atom, which is lower than those for Cu2Si (−3.46 eV/atom),73 FeB6 (from −5.56 to −5.79 eV/atom),74 and Be2C (−4.86 eV/atom),75 which demonstrates its thermodynamic stability as well. The band structures and density of states were also calculated, which possess a zero band gap, indicating their high conductivities, as shown in Figure 2d. More interestingly, C3N3Cu has a Dirac point located exactly at the Fermi level, which is similar to that of graphene.
ORR and OER processes in acidic solutions initialize with adsorption of O2 and H2O, and therefore, we next explored the trapping capabilities of C3N3Cu, which can be evaluated by the adsorption energies and other adsorption-induced changes of structural and electronic properties as well as charge transfer (see Figure 3). The fully relaxed geometry of O2 on the substrate is shown in Figure 3, which has an adsorption energy of −0.93 eV. As shown in Figure 3a, upon the deposition of O2, 0.43 |e| charge is transferred from C3N3Cu to an adsorbed O2 molecule, which activates the O–O bond due to the occupation of the antibonding state of the O2 molecule. This enhanced activation could be also reflected by the adsorption-induced O–O bond length change (dO–O) of 0.06 Å as well as the magnetism of the O2 molecule from 2 μB of the gas phase to 1.39 μB of the adsorbed phase. In addition, as shown in Figure 3c, upon deposition of the O2 molecule, C3N3Cu becomes spin-polarized with spin density localized mainly in the vicinity of both adsorbed O2 and CuN3 moieties. Regarding H2O adsorption, one can see also the chemisorption of H2O with a bond length of 2.13 Å between Cu and O and an adsorption energy of −0.57 eV. In contrast to the case of O2 adsorption, inverse charge transfer from the adsorbed H2O to the substrates was observed but with a smaller amount of charge of 0.06 |e|. Overall, the chemisorption of O2 and H2O is strong enough for C3N3Cu to catalyze both ORR and OER. In addition, O2 adsorption is not too strong, which guarantees the desorption of O2 involved in the OER process.
Figure 3.
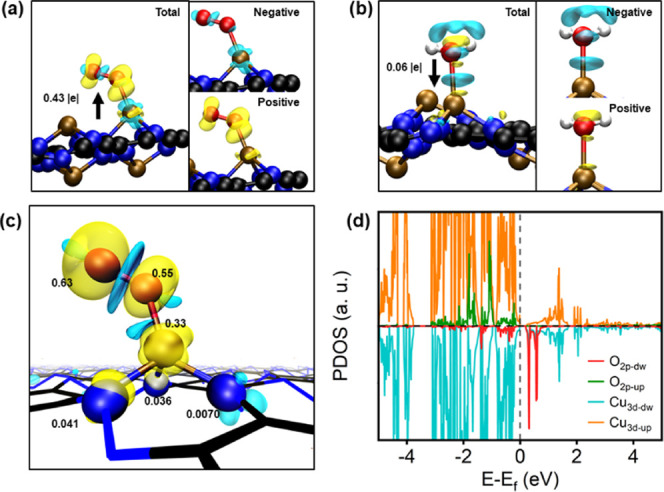
Charge redistribution induced by the adsorption of (a) H2O and (b) O2 on C3N3Cu, where the arrow refers to the direction of charge transfer and cyan and yellow denote charge depletion and accumulation, respectively. (c) Spin density distribution and local magnetism moments (μB) of O, Cu, and N atoms and (d) PDOS of O2-adsorbed C3N3Cu. In the maps of spin density distribution, the yellow and cyan surfaces describe the densities of spin-up and spin-down states, respectively.
We next explored the catalytic performance of C3N3Cu for ORR and OER by calculating Gibbs free energies of all of the related oxygen-containing reaction intermediates adsorbed on the active sites (see Figure 4a−c). As expected, the active sites are still Cu atoms bonded with three N atoms, which exhibit an enhanced catalytic performance for ORR and OER as reflected by overpotentials of 0.71 and 0.43 V, respectively. These findings indicate again that increasing the density of CuN3 will enhance simultaneously the catalytic performance for both ORR and OER. In particular, ηOER = 0.43 V is almost equal to that of 0.42 V for calculated RuO2 catalysts.76 Although the ORR performance of C3N3Cu (ηORR = 0.71 V) is not as good as Pt/C with ηORR = 0.45 V,77 it is still comparable to some SACs reported previously, such as AgN4/C (ηORR = 0.75 V)8 and FeN4/C (ηORR = 0.74 V).50 Compared to previously reported CuN3 active sites for ORR, our proposal for designing catalysts with CuN3 as active sites is more advantageous due to the fact that it requires no additional functionalization during the reaction process.51 Overall, we have shown that it is possible to achieve bifunctional catalysts for both ORR and OER with high concentrations of the CuN3 moiety.
Figure 4.
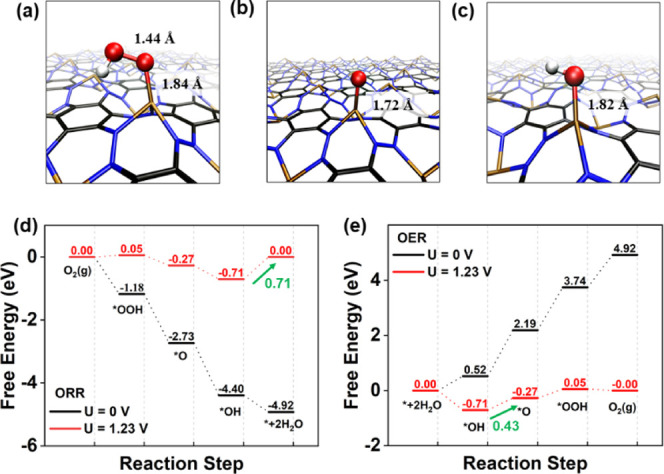
ORR and OER performance of C3N3Cu. (a–c) Adsorption configurations of reaction intermediates of *OOH, *O, and *OH where the bond length of O–O and Cu–O are also listed. (d–e) Free energy profiles of ORR and OER over C3N3Cu. The RDSs of ORR and OER are marked with green arrows as well as the values of overpotential.
As far as we know, we report for the first time that the Cu atomic center coordinated with three N atoms are active toward both ORR and OER. In particular, it is realized in a 2D crystalline phase compound containing active sites featuring high concentration and homogeneous distribution compared to SACs achieved by the doping strategy. Note that the distance between the two nearest active centers is around 0.34 nm. This is in contrast to previous work that the ORR performance of FeN4 could be enhanced when increasing the concentration of metal centers until the distance of dimmer FeN4 is less than 0.7 nm.27 It indicates that one can obtain higher concentrations of active sites by introducing MN3 instead of MN4 moieties.
Generally, our DFT calculations demonstrate the ordering of catalyst activities of 1-CuN3 < 2-CuN3 < 3-CuN3 < C3N3Cu as reflected by the overpotentials of ηORR = 2.30 V/ηOER = 1.52 V, ηORR = 1.11 V/ηOER = 0.52 V, ηORR = 0.91 V/ηOER = 0.48 V, and ηORR = 0.71 V/ηOER = 0.43 V, respectively. This is consistent with the trend of the density of the CuN3 moiety in these systems. The unambiguous structure–property correlation inspired us to dig deeper into the mechanism of the enhanced ORR/OER performance. As mentioned in Section 3.1, the improved catalytic performance for both ORR and OER can be attributed to the weakened interaction of *OH and substrates involved in the RDS. Accordingly, it is easy to speculate that increasing the CuN3 concentration weakens the interaction strength of *OH and active sites.
The d-band center theory has been widely accepted as an effective tool for analyzing the bond strength of metal centers and adsorbates. A lowered d-band center location of the metal sites downshifts the antibonding states and accordingly increases the occupancy, leading to weakened bonding strength of catalysts and reaction intermediates and vice versa.78 Accordingly, we calculated the projected density of states (PDOSs) onto Cu elements in 1-CuN3, 2-CuN3, 3-CuN3, and C3N3Cu, as shown in Figures 1d–f, 3d, and S1, which confirm the main contribution of Cu 3d orbitals to the states near the Fermi level. Based on eq S13, d-band centers of 1-CuN3, 2-CuN3 3-CuN3, and C3N3Cu were calculated to be −0.05, −0.15, −0.22, and −0.39 eV, respectively. Interestingly, there is a positive correlation between the d-band centers and overpotentials of ORR in these four systems, implying that the weakened interaction of *OH and substrates is attributed to the lowered d-band center of metal centers caused by the increased density of the CuN3 moiety (see Figure 5a).
Figure 5.

Mechanism of the weakened binding strength between *OH and substrates. (a) Linear relationship between the d-band center and ΔG*OH over 1-CuN3, 2-CuN3, 3-CuN3, and C3N3Cu, respectively. (b–e) PDOS and the negative values of crystal orbital Hamilton populations (COHPs) of *OH on substrates with different densities of CuN3. (f) Linear relationship between ICOHP and ΔG*OH over 1-CuN3, 2-CuN3, 3-CuN3, and C3N3Cu, respectively.
To unravel the underlying mechanism of the weakened binding affinity of substrates to *OH, it is straightforward to measure the intensity of Cu–O upon the adsorption of OH. To this end, we next performed crystal orbital Hamilton population (COHP) analysis79 and then integrated values of COHP (ICOHP) below the Fermi level, which enabled us to quantitatively investigate the bonding strength of Cu–*OH. A smaller value of ICOHP implies stronger binding strength and vice versa. We depicted in Figure 5b–e the COHP as well as the PDOS of the systems with *OH on 1-CuN3, 2-CuN3, 3-CuN3, and C3N3Cu with the values of ICOHP of −2.58, −2.53, −2.44, and −2.38, respectively. This demonstrates that with an increase in the concentration of CuN3, the ICOHP of Cu and *OH is increased, implying weakened interaction of the Cu site and *OH and accordingly a decrease in the free energy change of RDS, which resulted in lowered overpotentials, indicating enhanced catalytic performance. The nice correlation between ΔG*OH and ICOHP is shown in Figure 5f.
Inspired by the results of C3N3Cu, we next predicted 2D materials of C3N3M (M = Ni and Zn) and explored their catalytic performances as well. Interestingly, these two compounds are also thermodynamically and dynamically stable, which could be confirmed by the AIMD simulations and phonon dispersion curves, as shown in Figure S12. Good electron conductivity is also found in these compounds as demonstrated by their gapless band structures, which are illustrated in Figure S13. The trapping capabilities of C3N3M (M = Ni and Zn) for O2 and H2O molecules were investigated as well. O2 could be strongly adsorbed on C3N3Ni and C3N3Zn with adsorption energies of −1.57 to −1.48 eV, respectively. The chemical bonds of O2 and substrates are also reflected by the charge transfers of 0.54 and 0.63 |e| from the substrates to adsorbed O2 molecules. This leads to the ΔdO–O values of 0.07 and 0.11 Å for M = Ni and Zn, respectively. Similarly, H2O can also be chemically adsorbed on C3N3Ni and C3N3Zn with adsorption energies of −0.71 and −0.81 eV, respectively, which involves the charge transfer from H2O to the substrates by 0.07 and 0.06 |e| for C3N3Ni and C3N3Zn, respectively. Regarding the catalytic performance, the calculated overpotentials are ηORR = 1.29 V/ηOER = 0.57 V for C3N3Ni and ηORR = 2.05 V/ηOER = 1.52 V for C3N3Zn, respectively. It shows that C3N3Ni can work as an efficient catalyst toward OER, which is illustrated in Figure S14.
The relationship between the free energies of reaction intermediates (when U = 0) was examined as well, aiming to explore the possible descriptor of activity. As shown in Figure 6a,b, one can see the fitted lines of ΔG*OH vs ΔG*O and ΔG*OH vs ΔG*OOH with the linear relationships of ΔG*O = 0.77 × ΔG*OH + 1.74 and ΔG*OOH = 0.95 × ΔG*OH + 3.28, respectively. These linear relationships reflect the similar feature of chemical bonds formed between reaction intermediates (*OOH, *OH, and *O) and substrates, which characterizes as a single Cu–*O bond. The RDS for ORR is the desorption of *OH to OH–, while the RDS for OER is the formation of *O from *OH. Accordingly, we prepared the activity volcano plots by considering the relationship of overpotentials vs ΔG*OH for both ORR and OER, as shown in Figure 6c,d. The volcano plots indicate that the catalytic performances for ORR and OER of these systems containing MN3 moieties share a unique descriptor of ΔG*OH. Combined with Figure 6c,d, when ΔG*OH is less than 0.80 eV, both overpotentials for the ORR and OER are in the left leg of the volcano plot, demonstrating that the performance of both reactions could be enhanced with weaker *OH adsorption intensity. The verification of this assumption requires additional calculations about the catalytic performances of C3N3M with M representing other metals.
Figure 6.

(a, b) Correlation of free energies of reaction intermediates of ΔG*OH, ΔG*O, and ΔGOOH*, which are involved in ORR and OER. Volcano plots reflecting the relationship of overpotentials and ΔG*OH for (c) ORR and (d) OER.
4. Conclusions
The critical effect of the density of active sites on the electrocatalytic activity of CuN3 moiety toward both ORR and OER has been determined based on extensive DFT calculations. Our calculations demonstrate that the catalytic performance of CuN3 moiety can be gradually enhanced when increasing the concentration of it. We found that three connected CuN3 moieties become active for both ORR and OER as verified by the corresponding overpotentials of 0.91 and 0.48 V, respectively, much better than the performance of the single CuN3 moiety giving rise to overpotentials larger than 1.23 V. Given the fact that the conventional way of designing SACs is usually highly dependent on the doping strategy facing the challenge of precise control of high concentration and homogeneous distribution of active sites, the findings of enhanced ORR performance induced by high-density CuN3 inspired us to predict a 2D crystalline phase of C3N3Cu serving as a bifunctional catalyst for both ORR and OER exhibiting overpotentials of 0.71 and 0.43 V, respectively, which possesses superior electron conductivity and dynamical and thermodynamically stability. Furthermore, the enhanced catalytic performance is found to be attributed to the weakened interaction of OH* and substrates confirmed by the ICOHP calculations, characterizing the bonding strength. In addition, C3N3M with M = Ni and Zn has also been explored for their potential applications as catalysts for ORR and/or OER, where only C3N3Ni exhibits OER performance. The identification of bifunctional catalysts of C3N3Cu for ORR and OER containing high concentration and homogeneous distribution of CuN3 moiety without involving doping strategy and functional groups will guide the rational design of high-efficiency and low-cost electrocatalysts for other electrochemical reactions.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. U1801255, 51972350). The calculations were carried out using supercomputer “Tianhe-2” at NSCC Guangzhou.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c01562.
Computational details, structural configurations of reaction intermediates of ORR and OER, AIMD simulations, and electronic properties of C3N3Ni and C3N3Zn (PDF)
Author Contributions
§ J.T. and Z.Z. contribute equally to this work who performed the calculations and wrote the initial manuscript. Z.Y. and Y.L. proposed the idea and provided the supervision. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Material
References
- Anasori B.; Lukatskaya M. R.; Gogotsi Y. 2D metal carbides and nitrides (MXenes) for energy storage. Nat. Rev. Mater. 2017, 2, 16098 10.1038/natrevmats.2016.98. [DOI] [Google Scholar]
- Banhart F.; Kotakoski J.; Krasheninnikov A. V. Structural Defects in Graphene. ACS Nano 2011, 5, 26–41. 10.1021/nn102598m. [DOI] [PubMed] [Google Scholar]
- Chu S.; Cui Y.; Liu N. The path towards sustainable energy. Nat. Mater. 2017, 16, 16–22. 10.1038/nmat4834. [DOI] [PubMed] [Google Scholar]
- Dresselhaus M. S.; Thomas I. L. Alternative energy technologies. Nature 2001, 414, 332–337. 10.1038/35104599. [DOI] [PubMed] [Google Scholar]
- Li Y.; Lu J. Metal–Air Batteries: Will They Be the Future Electrochemical Energy Storage Device of Choice?. ACS Energy Lett. 2017, 2, 1370–1377. 10.1021/acsenergylett.7b00119. [DOI] [Google Scholar]
- Cai Z. C.; Yamada I.; Yagi S. ZIF-Derived Co9-xNixS8 Nanoparticles Immobilized on N-Doped Carbons as Efficient Catalysts for High-Performance Zinc-Air Batteries. ACS Appl. Mater. Interfaces 2020, 12, 5847–5856. 10.1021/acsami.9b19268. [DOI] [PubMed] [Google Scholar]
- Calle-Vallejo F.; Martinez J. I.; Rossmeisl J. Density functional studies of functionalized graphitic materials with late transition metals for oxygen reduction reactions. Phys. Chem. Chem. Phys. 2011, 13, 15639–15643. 10.1039/c1cp21228a. [DOI] [PubMed] [Google Scholar]
- Cao Y. H.; Zheng X. R.; Zhang H. X.; Zhang J. F.; Han X. P.; Zhong C.; Hu W. B.; Deng Y. D. Interface engineering of NiS2/CoS2 nanohybrids as bifunctional electrocatalysts for rechargeable solid state Zn-air battery. J. Power Sources 2019, 437, 226893 10.1016/j.jpowsour.2019.226893. [DOI] [Google Scholar]
- Chai G. L.; Qiu K. P.; Qiao M.; Titirici M. M.; Shang C. X.; Guo Z. X. Active sites engineering leads to exceptional ORR and OER bifunctionality in P,N Co-doped graphene frameworks. Energy Environ. Sci. 2017, 10, 1186–1195. 10.1039/C6EE03446B. [DOI] [Google Scholar]
- Cao X. C.; Zheng X. J.; Tian J. H.; Jin C.; Ke K.; Yang R. Z. Cobalt Sulfide Embedded in Porous Nitrogen-doped Carbon as a Bifunctional Electrocatalyst for Oxygen Reduction and Evolution Reactions. Electrochim. Acta 2016, 191, 776–783. 10.1016/j.electacta.2016.01.137. [DOI] [Google Scholar]
- Zhao C. X.; Liu J. N.; Wang J.; Ren D.; Li B. Q.; Zhang Q. Recent advances of noble-metal-free bifunctional oxygen reduction and evolution electrocatalysts. Chem. Soc. Rev. 2021, 50, 7745–7778. 10.1039/D1CS00135C. [DOI] [PubMed] [Google Scholar]
- Meng F.-L.; Liu K.-H.; Zhang Y.; Shi M.-M.; Zhang X.-B.; Yan J.-M.; Jiang Q. Recent Advances toward the Rational Design of Efficient Bifunctional Air Electrodes for Rechargeable Zn-Air Batteries. Small 2018, 14, 1703843 10.1002/smll.201703843. [DOI] [PubMed] [Google Scholar]
- Ai W.; Luo Z. M.; Jiang J.; Zhu J. H.; Du Z. Z.; Fan Z. X.; Xie L. H.; Zhang H.; Huang W.; Yu T. Nitrogen and Sulfur Codoped Graphene: Multifunctional Electrode Materials for High-Performance Li-Ion Batteries and Oxygen Reduction Reaction. Adv. Mater. 2014, 26, 6186–6192. 10.1002/adma.201401427. [DOI] [PubMed] [Google Scholar]
- Bai L. C.; Hsu C. S.; Alexander D. T. L.; Chen H. M.; Hu X. L. A Cobalt-Iron Double-Atom Catalyst for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2019, 141, 14190–14199. 10.1021/jacs.9b05268. [DOI] [PubMed] [Google Scholar]
- Xu H. X.; Cheng D. J.; Cao D. P.; Zeng X. C. A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 2018, 1, 339–348. 10.1038/s41929-018-0063-z. [DOI] [Google Scholar]
- Cai X. Y.; Lai L. F.; Lin J. Y.; Shen Z. X. Recent advances in air electrodes for Zn-air batteries: electrocatalysis and structural design. Mater. Horiz. 2017, 4, 945–976. 10.1039/C7MH00358G. [DOI] [Google Scholar]
- Guo Y. B.; Yao S.; Gao L. X.; Chen A.; Jiao M. G.; Cui H. J.; Zhou Z. Boosting bifunctional electrocatalytic activity in S and N co-doped carbon nanosheets for high-efficiency Zn-air batteries. J. Mater. Chem. A 2020, 8, 4386–4395. 10.1039/C9TA12762C. [DOI] [Google Scholar]
- Zhou Y. N.; Gao G. P.; Kang J.; Chu W.; Wang L. W. Transition metal-embedded two-dimensional C3N as a highly active electrocatalyst for oxygen evolution and reduction reactions. J. Mater. Chem. A 2019, 7, 12050–12059. 10.1039/C9TA01389J. [DOI] [Google Scholar]
- Wang A.; Li J.; Zhang T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2018, 2, 65–81. 10.1038/s41570-018-0010-1. [DOI] [Google Scholar]
- Zhao C.-X.; Li B.-Q.; Liu J.-N.; Zhang Q. Intrinsic Electrocatalytic Activity Regulation of M–N–C Single-Atom Catalysts for the Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2021, 60, 4448–4463. 10.1002/anie.202003917. [DOI] [PubMed] [Google Scholar]
- Xia C.; Qiu Y.; Xia Y.; Zhu P.; King G.; Zhang X.; Wu Z.; Kim J. Y.; Cullen D. A.; Zheng D.; Li P.; Shakouri M.; Heredia E.; Cui P.; Alshareef H. N.; Hu Y.; Wang H. General synthesis of single-atom catalysts with high metal loading using graphene quantum dots. Nat. Chem. 2021, 13, 887–894. 10.1038/s41557-021-00734-x. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Wang X.; Zhu M.; Leng X.; Chen W.; Wang W.; Xu Q.; Yang L.-M.; Wu Y. Structural revolution of atomically dispersed Mn sites dictates oxygen reduction performance. Nano Res. 2021, 14, 4512–4519. 10.1007/s12274-021-3823-z. [DOI] [Google Scholar]
- Zhu M.; Zhao C.; Liu X.; Wang X.; Zhou F.; Wang J.; Hu Y.; Zhao Y.; Yao T.; Yang L.-M.; Wu Y. Single Atomic Cerium Sites with a High Coordination Number for Efficient Oxygen Reduction in Proton-Exchange Membrane Fuel Cells. ACS Catal. 2021, 11, 3923–3929. 10.1021/acscatal.0c05503. [DOI] [Google Scholar]
- Luo E.; Zhang H.; Wang X.; Gao L.; Gong L.; Zhao T.; Jin Z.; Ge J.; Jiang Z.; Liu C.; Xing W. Single-Atom Cr–N4 Sites Designed for Durable Oxygen Reduction Catalysis in Acid Media. Angew. Chem., Int. Ed. 2019, 58, 12469–12475. 10.1002/anie.201906289. [DOI] [PubMed] [Google Scholar]
- Xiao M.; Zhu J.; Li G.; Li N.; Li S.; Cano Z. P.; Ma L.; Cui P.; Xu P.; Jiang G.; Jin H.; Wang S.; Wu T.; Lu J.; Yu A.; Su D.; Chen Z. A Single-Atom Iridium Heterogeneous Catalyst in Oxygen Reduction Reaction. Angew. Chem., Int. Ed. 2019, 58, 9640–9645. 10.1002/anie.201905241. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Zhang Y.; Huang L.-B.; Liu X.-Z.; Zhang Q.-H.; He C.; Wu Z.-Y.; Zhang L.-J.; Wu J.; Yang W.; Gu L.; Hu J.-S.; Wan L.-J. Cascade anchoring strategy for general mass production of high-loading single-atomic metal-nitrogen catalysts. Nat. Commun. 2019, 10, 1278 10.1038/s41467-019-09290-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Li P.; Meng Y.; Fang Z.; Xiao D.; Yu G. Understanding the inter-site distance effect in single-atom catalysts for oxygen electroreduction. Nat. Catal. 2021, 4, 615–622. 10.1038/s41929-021-00650-w. [DOI] [Google Scholar]
- Yang J.; Wang Z.; Huang C.-X.; Zhang Y.; Zhang Q.; Chen C.; Du J.; Zhou X.; Zhang Y.; Zhou H.; Wang L.; Zheng X.; Gu L.; Yang L.-M.; Wu Y. Compressive Strain Modulation of Single Iron Sites on Helical Carbon Support Boosts Electrocatalytic Oxygen Reduction. Angew. Chem., Int. Ed. 2021, 60, 22722–22728. 10.1002/anie.202109058. [DOI] [PubMed] [Google Scholar]
- Cai L.; Zhang N.; Qiu B.; Chai Y. Computational Design of Transition Metal Single-Atom Electrocatalysts on PtS2 for Efficient Nitrogen Reduction. ACS Appl. Mater. Interfaces 2020, 12, 20448–20455. 10.1021/acsami.0c02458. [DOI] [PubMed] [Google Scholar]
- Choi C.; Back S.; Kim N.-Y.; Lim J.; Kim Y.-H.; Jung Y. Suppression of Hydrogen Evolution Reaction in Electrochemical N2 Reduction Using Single-Atom Catalysts: A Computational Guideline. ACS Catal. 2018, 8, 7517–7525. 10.1021/acscatal.8b00905. [DOI] [Google Scholar]
- Huang C.-X.; Li G.; Yang L.-M.; Ganz E. Ammonia Synthesis Using Single-Atom Catalysts Based on Two-Dimensional Organometallic Metal Phthalocyanine Monolayers under Ambient Conditions. ACS Appl. Mater. Interfaces 2021, 13, 608–621. 10.1021/acsami.0c18472. [DOI] [PubMed] [Google Scholar]
- Wang X.; Yang L.-M. Efficient modulation of the catalytic performance of electrocatalytic nitrogen reduction with transition metals anchored on N/O-codoped graphene by coordination engineering. J. Mater. Chem. A 2022, 10, 1481–1496. 10.1039/D1TA08877G. [DOI] [Google Scholar]
- Zhou Y.; Gao G.; Kang J.; Chu W.; Wang L.-W. Transition metal-embedded two-dimensional C3N as a highly active electrocatalyst for oxygen evolution and reduction reactions. J. Mater. Chem. A 2019, 7, 12050–12059. 10.1039/C9TA01389J. [DOI] [Google Scholar]
- Zhao M. R.; Song B.; Yang L. M. Two-Dimensional Single-Atom Catalyst TM(3)(HAB)(2) Monolayers for Electrocatalytic Dinitrogen Reduction Using Hierarchical High-Throughput Screening. ACS Appl. Mater. Interfaces 2021, 13, 26109–26122. 10.1021/acsami.1c06414. [DOI] [PubMed] [Google Scholar]
- Wang T.; Zhao Q.; Fu Y.; Lei C.; Yang B.; Li Z.; Lei L.; Wu G.; Hou Y. Carbon-Rich Nonprecious Metal Single Atom Electrocatalysts for CO2 Reduction and Hydrogen Evolution. Small Methods 2019, 3, 1900210 10.1002/smtd.201900210. [DOI] [Google Scholar]
- Xu C.; Vasileff A.; Zheng Y.; Qiao S.-Z. Recent Progress of 3d Transition Metal Single-Atom Catalysts for Electrochemical CO2 Reduction. Adv. Mater. Interfaces 2021, 8, 2001904 10.1002/admi.202001904. [DOI] [Google Scholar]
- Zhang T.; Han X.; Yang H.; Han A.; Hu E.; Li Y.; Yang X.-q.; Wang L.; Liu J.; Liu B. Atomically Dispersed Nickel(I) on an Alloy-Encapsulated Nitrogen-Doped Carbon Nanotube Array for High-Performance Electrochemical CO2 Reduction Reaction. Angew. Chem., Int. Ed. 2020, 59, 12055–12061. 10.1002/anie.202002984. [DOI] [PubMed] [Google Scholar]
- Yang J.; Wang X.; Qu Y.; Wang X.; Huo H.; Fan Q.; Wang J.; Yang L.-M.; Wu Y. Bi-Based Metal-Organic Framework Derived Leafy Bismuth Nanosheets for Carbon Dioxide Electroreduction. Adv. Energy Mater. 2020, 10, 2001709 10.1002/aenm.202001709. [DOI] [Google Scholar]
- Liu J.-H.; Yang L.-M.; Ganz E. Efficient and Selective Electroreduction of CO2 by Single-Atom Catalyst Two-Dimensional TM–Pc Monolayers. ACS Sustainable Chem. Eng. 2018, 6, 15494–15502. 10.1021/acssuschemeng.8b03945. [DOI] [Google Scholar]
- Liu J.-H.; Yang L.-M.; Ganz E. Efficient electrocatalytic reduction of carbon dioxide by metal-doped β12-borophene monolayers. RSC Adv. 2019, 9, 27710–27719. 10.1039/C9RA04135D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.-H.; Yang L.-M.; Ganz E. Electrochemical reduction of CO2 by single atom catalyst TM–TCNQ monolayers. J. Mater. Chem. A 2019, 7, 3805–3814. 10.1039/C8TA08677J. [DOI] [Google Scholar]
- Fung V.; Hu G.; Wu Z.; Jiang D.-e. Descriptors for Hydrogen Evolution on Single Atom Catalysts in Nitrogen-Doped Graphene. J. Phys. Chem. C 2020, 124, 19571–19578. 10.1021/acs.jpcc.0c04432. [DOI] [Google Scholar]
- Hossain M. D.; Liu Z.; Zhuang M.; Yan X.; Xu G.-L.; Gadre C. A.; Tyagi A.; Abidi I. H.; Sun C.-J.; Wong H.; Guda A.; Hao Y.; Pan X.; Amine K.; Luo Z. Rational Design of Graphene-Supported Single Atom Catalysts for Hydrogen Evolution Reaction. Adv. Energy Mater. 2019, 9, 1803689 10.1002/aenm.201803689. [DOI] [Google Scholar]
- Lim J.; Back S.; Choi C.; Jung Y. Ultralow Overpotential of Hydrogen Evolution Reaction using Fe-Doped Defective Graphene: A Density Functional Study. ChemCatChem 2018, 10, 4450–4455. 10.1002/cctc.201800635. [DOI] [Google Scholar]
- Li Y.; Wu Z.-S.; Lu P.; Wang X.; Liu W.; Liu Z.; Ma J.; Ren W.; Jiang Z.; Bao X. High-Valence Nickel Single-Atom Catalysts Coordinated to Oxygen Sites for Extraordinarily Activating Oxygen Evolution Reaction. Adv. Sci. 2020, 7, 1903089 10.1002/advs.201903089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Gao G.; Li Y.; Chu W.; Wang L.-W. Transition-metal single atoms in nitrogen-doped graphenes as efficient active centers for water splitting: a theoretical study. Phys. Chem. Chem. Phys. 2019, 21, 3024–3032. 10.1039/C8CP06755D. [DOI] [PubMed] [Google Scholar]
- Xu H.; Cheng D.; Cao D.; Zeng X. C. A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 2018, 1, 339–348. 10.1038/s41929-018-0063-z. [DOI] [Google Scholar]
- Wang Z.; Jin X.; Zhu C.; Liu Y.; Tan H.; Ku R.; Zhang Y.; Zhou L.; Liu Z.; Hwang S.-J.; Fan H. J. Atomically Dispersed Co2–N6 and Fe–N4 Costructures Boost Oxygen Reduction Reaction in Both Alkaline and Acidic Media. Adv. Mater. 2021, 33, 2104718 10.1002/adma.202104718. [DOI] [PubMed] [Google Scholar]
- Shao C.; Wu L.; Zhang H.; Jiang Q.; Xu X.; Wang Y.; Zhuang S.; Chu H.; Sun L.; Ye J.; Li B.; Wang X. A Versatile Approach to Boost Oxygen Reduction of Fe-N4 Sites by Controllably Incorporating Sulfur Functionality. Adv. Funct. Mater. 2021, 31, 2100833 10.1002/adfm.202100833. [DOI] [Google Scholar]
- Lin Z.; Yang A.; Zhang B.; Liu B.; Zhu J.; Tang Y.; Qiu X. Coupling the Atomically Dispersed Fe-N3 Sites with Sub-5 nm Pd Nanocrystals Confined in N-Doped Carbon Nanobelts to Boost the Oxygen Reduction for Microbial Fuel Cells. Adv. Funct. Mater. 2022, 32, 2107683 10.1002/adfm.202107683. [DOI] [Google Scholar]
- Yang J.; Liu W.; Xu M.; Liu X.; Qi H.; Zhang L.; Yang X.; Niu S.; Zhou D.; Liu Y.; Su Y.; Li J.-F.; Tian Z.-Q.; Zhou W.; Wang A.; Zhang T. Dynamic Behavior of Single-Atom Catalysts in Electrocatalysis: Identification of Cu-N3 as an Active Site for the Oxygen Reduction Reaction. J. Am. Chem. Soc. 2021, 143, 14530–14539. 10.1021/jacs.1c03788. [DOI] [PubMed] [Google Scholar]
- Han Y.; Li Q.-K.; Ye K.; Luo Y.; Jiang J.; Zhang G. Impact of Active Site Density on Oxygen Reduction Reactions Using Monodispersed Fe–N–C Single-Atom Catalysts. ACS Appl. Mater. Interfaces 2020, 12, 15271–15278. 10.1021/acsami.0c01206. [DOI] [PubMed] [Google Scholar]
- Chen L. X.; Chen Z. W.; Jiang M.; Lu Z.; Gao C.; Cai G.; Singh C. V. Insights on the dual role of two-dimensional materials as catalysts and supports for energy and environmental catalysis. J. Mater. Chem. A 2021, 9, 2018–2042. 10.1039/D0TA08649E. [DOI] [Google Scholar]
- Chen Z. W.; Lang X. Y.; Jiang Q. Discovery of cobweb-like MoC6 and its application for nitrogen fixation. J. Mater. Chem. A 2018, 6, 9623–9628. 10.1039/C8TA03481H. [DOI] [Google Scholar]
- Kohn W.; Sham L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. 10.1103/PhysRev.140.A1133. [DOI] [Google Scholar]
- Hafner J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. 10.1002/jcc.21057. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Blöchl P. E.; Jepsen O.; Andersen O. K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223–16233. 10.1103/PhysRevB.49.16223. [DOI] [PubMed] [Google Scholar]
- Blöchl P. E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. 10.1103/PhysRevB.45.13244. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Chevary J. A.; Vosko S. H.; Jackson K. A.; Pederson M. R.; Singh D. J.; Fiolhais C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. 10.1103/PhysRevB.46.6671. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Smith D. G. A.; Burns L. A.; Patkowski K.; Sherrill C. D. Revised Damping Parameters for the D3 Dispersion Correction to Density Functional Theory. J. Phys. Chem. Lett. 2016, 7, 2197–2203. 10.1021/acs.jpclett.6b00780. [DOI] [PubMed] [Google Scholar]
- Monkhorst H. J.; Pack J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. 10.1103/PhysRevB.13.5188. [DOI] [Google Scholar]
- Becke A. D.; Edgecombe K. E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. 10.1063/1.458517. [DOI] [Google Scholar]
- Togo A.; Tanaka I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. 10.1016/j.scriptamat.2015.07.021. [DOI] [Google Scholar]
- Rossmeisl J.; Logadottir A.; Nørskov J. K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. 10.1016/j.chemphys.2005.05.038. [DOI] [Google Scholar]
- Li J.; Jiang Y.-f.; Wang Q.; Xu C.-Q.; Wu D.; Banis M. N.; Adair K. R.; Doyle-Davis K.; Meira D. M.; Finfrock Y. Z.; Li W.; Zhang L.; Sham T.-K.; Li R.; Chen N.; Gu M.; Li J.; Sun X. A general strategy for preparing pyrrolic-N4 type single-atom catalysts via pre-located isolated atoms. Nat. Commun. 2021, 12, 6806 10.1038/s41467-021-27143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao L.; Zhu J.; Zhang Y.; Yang W.; Zhou S.; Li A.; Xie C.; Zheng X.; Zhou W.; Yu S.-H.; Jiang H.-L. Non-Bonding Interaction of Neighboring Fe and Ni Single-Atom Pairs on MOF-Derived N-Doped Carbon for Enhanced CO2 Electroreduction. J. Am. Chem. Soc. 2021, 143, 19417–19424. 10.1021/jacs.1c08050. [DOI] [PubMed] [Google Scholar]
- Li T.; He C.; Zhang W. A novel porous C4N4 monolayer as a potential anchoring material for lithium–sulfur battery design. J. Mater. Chem. A 2019, 7, 4134–4144. 10.1039/C8TA10933H. [DOI] [Google Scholar]
- Jia C.; Wang Q.; Yang J.; Ye K.; Li X.; Zhong W.; Shen H.; Sharman E.; Luo Y.; Jiang J. Toward Rational Design of Dual-Metal-Site Catalysts: Catalytic Descriptor Exploration. ACS Catal. 2022, 12, 3420–3429. 10.1021/acscatal.1c06015. [DOI] [Google Scholar]
- Yang L.-M.; Bačić V.; Popov I. A.; Boldyrev A. I.; Heine T.; Frauenheim T.; Ganz E. Two-Dimensional Cu2Si Monolayer with Planar Hexacoordinate Copper and Silicon Bonding. J. Am. Chem. Soc. 2015, 137, 2757–2762. 10.1021/ja513209c. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Li Y.; Hou J.; Tu K.; Chen Z. FeB6 Monolayers: The Graphene-like Material with Hypercoordinate Transition Metal. J. Am. Chem. Soc. 2016, 138, 5644–5651. 10.1021/jacs.6b01769. [DOI] [PubMed] [Google Scholar]
- Li Y.; Liao Y.; Chen Z. Be2C Monolayer with Quasi-Planar Hexacoordinate Carbons: A Global Minimum Structure. Angew. Chem., Int. Ed. 2014, 53, 7248–7252. 10.1002/anie.201403833. [DOI] [PubMed] [Google Scholar]
- Man I. C.; Su H.-Y.; Calle-Vallejo F.; Hansen H. A.; Martínez J. I.; Inoglu N. G.; Kitchin J.; Jaramillo T. F.; Nørskov J. K.; Rossmeisl J. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem 2011, 3, 1159–1165. 10.1002/cctc.201000397. [DOI] [Google Scholar]
- Nørskov J. K.; Rossmeisl J.; Logadottir A.; Lindqvist L.; Kitchin J. R.; Bligaard T.; Jónsson H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. 10.1021/jp047349j. [DOI] [Google Scholar]
- Bligaard T.; Nørskov J. K.. Heterogeneous Catalysis. In Chemical Bonding at Surfaces and Interfaces, Nilsson A.; Pettersson L. G. M.; Nørskov J. K., Eds.; Elsevier: Amsterdam, 2008; pp 255–321. [Google Scholar]
- Tachibana M.; Yoshizawa K.; Ogawa A.; Fujimoto H.; Hoffmann R. Sulfur–Gold Orbital Interactions which Determine the Structure of Alkanethiolate/Au(111) Self-Assembled Monolayer Systems. J. Phys. Chem. B 2002, 106, 12727–12736. 10.1021/jp020993i. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.