

Abstract
Middle East respiratory syndrome coronavirus (MERS-CoV) is a highly pathogenic human coronavirus causing severe disease and mortality. MERS-CoV infection failed to elicit robust IFN response, suggesting that the virus might have evolved strategies to evade host innate immune surveillance. In this study, we identified and characterized type I IFN antagonism of MERS-CoV open reading frame (ORF) 8b accessory protein. ORF8b was abundantly expressed in MERS-CoV–infected Huh-7 cells. When ectopically expressed, ORF8b inhibited IRF3-mediated IFN-β expression induced by Sendai virus and poly(I:C). ORF8b was found to act at a step upstream of IRF3 to impede the interaction between IRF3 kinase IKKε and chaperone protein HSP70, which is required for the activation of IKKε and IRF3. An infection study using recombinant wild-type and ORF8b-deficient MERS-CoV further confirmed the suppressive role of ORF8b in type I IFN induction and its disruption of the colocalization of HSP70 with IKKε. Ectopic expression of HSP70 relieved suppression of IFN-β expression by ORF8b in an IKKε-dependent manner. Enhancement of IFN-β induction in cells infected with ORF8b-deficient virus was erased when HSP70 was depleted. Taken together, HSP70 chaperone is important for IKKε activation, and MERS-CoV ORF8b suppresses type I IFN expression by competing with IKKε for interaction with HSP70.
There are seven human coronaviruses, including the most recently identified severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes the ongoing pandemic of coronavirus disease 2019 (COVID-19), a major threat to global health and safety (1–3). The COVID-19 pandemic reminds us of the potential risks of emerging human coronaviruses such as SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV). MERS-CoV was first isolated from a 60-y-old male patient as a previously unknown human coronavirus in Saudi Arabia in 2012. The patient infected by MERS-CoV showed signs of acute respiratory distress syndrome and multiorgan dysfunction, which were clinically similar to cases of SARS-CoV infection (4). The majority of MERS cases were from sporadic outbreaks in Middle East countries (5), whereas individuals with recent travel history to those countries constituted around 10% of the reported cases and could rarely spawn an outbreak outside of the Middle East (6). According to the World Health Organization, 2357 laboratory-confirmed cases with 820 associated deaths have been reported in 27 countries with a mortality rate of around 35%. Sequence analysis revealed a close relationship of MERS-CoV to bat coronaviruses HKU4 and HKU5, all of which are classified into lineage C of betacoronaviruses (4, 7–9). MERS-CoV harbors a polycistronic positive-stranded RNA genome of around 30 kb in size, which encodes 16 nonstructural proteins at the 59-end of the genome as well as several structural and lineage-specific proteins at the 3′-end (7, 9–11). These viral proteins not only support viral replication and packaging (10) but also modulate host innate immune response such as type I IFN expression for optimal viral propagation (12–17).
Type I IFNs are of paramount importance in antiviral response, and they activate the expression of various IFN-stimulated genes (ISGs), which function to limit viral replication and spreading, activate immune cells, and induce apoptosis of infected cells (18, 19). The high mortality rate and severe outcome of MERS-CoV infection could possibly be explained by the inability of the virus to induce a robust IFN response (20–25). Upon entry into the host cell as a result of the binding of the viral spike protein to its receptor dipeptidyl peptidase 4 on the cell surface (26, 27), viral genomic RNA is released to the cytoplasm after the fusion of viral and host membranes (10). Detection of the released viral RNA by cytoplasmic sensors of the RIG-I–like receptor (RLR) family such as RIG-I and MDA5 leads to type I IFN expression and activation of the antiviral response (28). However, several proteins encoded by MERS-CoV can suppress type I IFN production induced by RNA stimuli at multiple levels (12–15). Identification and characterization of additional MERS-CoV–encoded IFN antagonists will not only shed light on viral countermeasures to evade innate immunity but also reveal new strategies to develop vaccines and antivirals.
MERS-CoV encodes five accessory proteins, namely open reading frame (ORF) 3, ORF4a, ORF4b, ORF5, and ORF8b (9–11). Accessory proteins are not directly involved in viral assembly but might be essential to immune evasion (13–15, 29, 30). Analysis of MERS-CoV accessory proteins revealed that ORF4a, ORF4b, and ORF5 but not ORF3 exhibit IFN-suppressive properties (13, 14, 29, 30). However, whether ORF8b antagonizes IFN production or signaling remains elusive. The presence of a small gene embedded within the N gene is a distinctive feature of betacoronaviruses, including SARS, MERS, human coronavirus HKU1, and mouse hepatitis virus (MHV) (9, 31). Both ORF8b and N proteins of MERS-CoV are transcribed from the same mRNA as no discrete transcription-regulatory sequence is identified immediately upstream of the start codon of ORF8b (32). ORF8b is likely translated through a leaky ribosomal scanning mechanism from the +1 frame with respect to the N gene (9). The internal (I) gene embedded in the N gene is the counterpart of MERS-CoV ORF8b in MHV. An MHV mutant virus depleted of the I gene indicates its nonessentiality for viral replication (33). In addition, the equivalent gene of MERS-CoV ORF8b in SARS-CoV named ORF9b was capable of degrading MAVS, TRAF3, and TRAF6, resulting in the dysfunction of MAVS signalosome and hence perturbation of innate immune signaling (34). However, MERS-CoV ORF8b bears no sequence homology to MHV I gene or SARS-CoV ORF9b protein. It remains to be seen whether ORF8b might possess any immunosuppressive property.
In this study, we defined the type I IFN antagonism of MERS-CoV ORF8b protein and provided the first evidence, to our knowledge, for its suppression of RIG-I–induced type I IFN expression. By mass spectrometric analysis, MERS-CoV ORF8b was found to interact with heat shock protein (HSP) 70–1A. The roles of HSPs in the regulation of innate immune response have previously been reported. Particularly, HSP90 is known to stabilize TBK1 in Sendai virus (SeV)–infected cells, leading to the activation of IRF3 phosphorylation and type I IFN expression (35). HSP70 has also been found to be the ligand of TLR2 and TLR4 to induce type I IFN expression in a measles virus–infected brain in mice (36). All these implicate a regulatory role for HSPs in innate antiviral response in the context of viral infection. In light of this, we performed further mechanistic analysis in cultured cells to show that HSP70–1A interacts with and enhances IKKε activation, which is important for IRF3 phosphorylation and hence type I IFN expression (37). An infection study using an ORF8b knockout (Δ8b) virus generated by reverse genetics further validated the IFN-antagonizing activity of ORF8b. Moreover, ectopic expression of ORF8b diminished the interaction between HSP70–1A and IKKε, lending further support to our model that the interaction between IKKε and HSP70–1A is impeded by ORF8b, resulting in the inhibition of IKKε activation and IFN-β expression.
Materials and Methods
Plasmids
RIG-I expression plasmid and luciferase construct IFN-β-luc reporter were generously provided by Prof. Takashi Fujita (Kyoto University, Kyoto, Japan) (28). TBK1, TRAF3, and IRF3 expression constructs were kind gifts from Dr. Genhong Cheng (University of California, Los Angeles, CA) (38, 39), whereas RIG-IN, MAVS, IKK, and IκB-α expression plasmids as well as IRF3-luc and κB-luc luciferase reporter constructs have been described elsewhere (40–43). RIG-IN is a constitutively active version of RIG-I containing the N-terminal CARD domain alone (28). MERS-CoV (EMC-2012) molecular clone in pBeloBAC11 backbone was kindly provided by Prof. Luis Enjuanes (Spanish National Center for Biotechnology, Madrid, Spain) (44). shHSP70–1 and shHSP70–2 were purchased from Sigma (TRCN0000342862 and TRCN0000342861).
MERS-CoV ORF8b cDNA was prepared by extracting viral RNAs from MERS-CoV–infected Vero-E6 cells using RNAiso Plus reagent purchased from Takara Bio (Otsu, Japan) followed by reverse transcription using reagents from Roche Diagnostics (Basel, Switzerland). MERS-CoV ORF8b gene was amplified from cDNA obtained from reverse transcription by PCR using reagents from Roche Diagnostics and Thermo Fisher Scientific (Grand Island, NY). PCR was run for 25 cycles with an annealing temperature of 60°C. The PCR product was cloned to expression plasmid pCAGEN with a C-terminal V5-tag (45), a gift from Dr. Connie Cepko (Harvard Medical School, Boston, MA). Human HSP cDNAs were prepared by reverse transcription of mRNAs from HEK293 cells and PCR amplified using the following primers: 5′-ATG CTT CGG TTA CCC ACA GTC-3′ (forward) and 5′-GAA CAT GCC ACC TCC CAT ACC-3′ (reverse) for HSP60, 5′-ATG GCC AAA GCC GCG GCG ATC G-3′ (forward) and 5′-ATC TAC CTC CTC AAT GGT GGG GC-3′ (reverse) for HSP70, 5′-ATG CCC CCG TGT TCG-3′ (forward) and 5′-GTC TAC TTC TTC CAT GCG TGA T-3′ (reverse) for HSP90A1 (1), and 5′-ATG CCT GAG GAA ACC CAG AC-3′ (forward) and 5′-GTC TAC TTC TTC CAT GCG TGA TG-3′ (reverse) for HSP90A1 (2). Human HSP60, HSP70, HSP90A1 (1), and HSP90A1 (2) were cloned into pCAGEN with the addition of a C-terminal myc tag.
Abs
Mouse monoclonal anti-V5 primary Ab was purchased from Thermo Fisher Scientific. Mouse monoclonal anti-FLAG and anti–β-actin primary Abs were purchased from MilliporeSigma (St. Louis, MO). Mouse anti-myc primary Ab clone 9E10 was purchased from Santa Cruz Biotechnology (Dallas, TX). Mouse monoclonal anti-HSP70 (SAB2701085) was purchased from MilliporeSigma. Rabbit polyclonal anti-ORF8b primary antiserum was raised by GenScript Biotech (Nanjing, China). Rabbit monoclonal anti–phospho-IRF3 (Ser396), anti-IKKε, and anti–phosphor-IKKε (Ser172) Abs were purchased from Cell Signaling Technology (Danvers, MA). Rabbit monoclonal anti-IRF3 and anti–phospho-IRF3 (Ser386) Abs were purchased from Immuno-Biological Laboratories (Gunma, Japan). Rabbit polyclonal Ab against HSP70–1A was purchased from Sigma (SAB2701085).
Cell culture and SeV
HEK293, Huh-7, BHK21 and Vero-E6 cells were purchased from American Type Culture Collection (Manassas, VA). The cells were cultured in DMEM supplemented with 10% FBS from Thermo Fisher Scientific at 37°C with 5% CO2. Cells were propagated and split at a ratio of 1:10 in DMEM with FBS when 80% confluence was reached. SeV Cantell strain VR907 was purchased from American Type Culture Collection. The virus was kept at −80°C and was thawed on ice slowly before use. poly(I:C) was from MilliporeSigma. Dynabeads sheep anti-mouse IgG magnetic beads were from Thermo Fisher Scientific.
Reporter and protein assays
Coimmunoprecipitation, Western blotting, and dual-luciferase assay were performed as previously described (40). Dual-luciferase assay was performed by measuring relative luciferase activity in arbitrary units. Promoter-specific firefly luciferase activity was normalized to Renilla luciferase activity recovered from cell lysate. Data are representative of three independent experiments.
RNA extraction and real-time quantitative RT-PCR
The culture medium was aspirated, and the transfected cells were lysed with RNAiso Plus reagent kit (Takara Bio). Total cellular RNA was extracted, according to the manufacturer’s protocol. First-strand cDNA synthesis was performed by reverse transcription with Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics), according to the manufacturer’s procedures. Quantitative PCR was performed using StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). PCR was run for 40 cycles with an annealing temperature of 60°C. Cycle threshold (CT) values were measured, and relative expression was calculated by ΔCT method normalized with GAPDH as the internal housekeeping gene. Fold expression was calculated by the ΔΔCT method normalized with the mock. The primers were 5′-TTG AAT GGG AGG CTT GAA TA-3′ (forward) and 5′-GCC AGG AGG TTC TCA ACA ATA G-3′ (reverse) for human IFN-β as well as 5′-AAC GTG TCA GTG GTG GAC CTG-3′ (forward) and 5′-AGT GGG TGT CGC TGT TGA AGT-3′ (reverse) for human GAPDH and 5′-CAAAACCTTCCCTAAGAAGGAAAAG-3′ (forward) and 5′-GCTCCTTTGGAGGTTCAGACAT-3′ (reverse) for MERS-N. Data are representative of three independent experiments.
Generation of recombinant virus
Homologous recombination of bacterial artificial chromosome (BAC) was performed to insert a galK expression cassette to the area desired for mutation at first and to replace the galK cassette with the mutant construct subsequently. The galK expression cassette was amplified by PCR with pMOD4-GalK-G kindly provided by Dr. Søren Warming (National Cancer Institute, Bethesda, MD) with primers consisting of homology arms flanking the desired area of the BAC and the galK sequence.
The two primers are 5′-CAA AAT GCT GGG TAT TGG CGG AGA CAG GAC AGA AAA ATT AAT ACC GGG AAC CTG TTG ACA ATT AAT CAT CGG CA-3′ (forward) and 5′-TGG GAG TGC TGC TTC GGG TCC AGT TCC AGT GTA GTA GAA GTA CCA CCT GGT CAG CAC TGT CCT GCT CCT T-3′ (reverse). The sequence that binds to pMOD4-GalK-G was underlined. The amplified products were treated with DpnI restriction enzyme (New England BioLabs, Ipswich, MA) for 1 h at 37°C to remove the pMOD4-GalK-G template. The DpnI-treated PCR product was purified by Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI). Electroporation was performed at 25 μF, 1.75 kV, and 200 Ω to introduce 2.5 μl of purified PCR product into 25 μl of Escherichia coli strain SW102 carrying the pBeloBAC11-EMC-2012 BAC in a 0.1-cm cuvette (Bio-Rad Laboratories, Hercules, CA). Cells were then allowed to recover in 1 ml of Luria–Bertani broth at 32°C for 1 h. The recovered cells were washed with 1× M9 minimal salt solution (MilliporeSigma). M63 minimal agar plate (M63 medium from MilliporeSigma) was used for spreading the washed bacteria and was allowed to incubate for 3 d at 32°C. Bacterial colonies were picked and streaked in MacConkey agar plate for further incubation at 32°C for 3 d. Bright red colonies were picked after 3 d and grown for validation of the introduction of galK expression cassette.
Oligonucleotides designed to carry desired mutations in MERS-CoV ORF8b gene were synthesized by Sigma-Proligo (MilliporeSigma). The two oligonucleotides are 5′-GGT ATT GGC GGA GAC AGG ACA GAA AAA TTA ATA CCG GGA ATG GAA TAA AGC AAC TAG CTC CCA GGT GGT ACT TCT ACT ACA CTG GAA CTG GAC CCG AAG C-3′ (forward) and 5′-GCT TCG GGT CCA GTT CCA GTG TAG TAG AAG TAC CAC CTG GGA GCT AGT TGC TTT ATT CCA TTC CCG GTA TTA ATT TTT CTG TCC TGT CTC CGC CAA TAC C-3′ (reverse). Residues that introduce mutations were underlined. The synthesized oligonucleotides were annealed by incubating at 95°C for 5 min followed by incubation at a successive decrease of 5°C within 5 min until 65°C and then incubation at 60°C for 20 min. Electroporation was performed as above to introduce 1 μl of annealed oligonucleotides into 25 μl of E. coli strain SW102 carrying the pBeloBAC11-EMC-2012 BAC-galK in a 0.1-cm cuvette. The electroporated cells were recovered in 10 ml Luria–Bertani broth at 32°C for 4.5 h. One milliliter of the recovered cells was pelleted and washed with 1× M9 salt solution. DOG-M63 minimal agar plate with 20% 2-deoxygalactose was used for spreading the washed bacteria and was allowed to incubate for 3 d at 32°C. The colonies present were subjected to clone validation by sequencing. The positive clones were recovered as described previously (44).
Virus infection and quantification
Huh-7 cells were seeded 1 d before infection. Cells were washed with serum-free DMEM before infection and infected at the indicated multiplicity of infection (MOI) by diluting viruses in serum-free DMEM. Cells were incubated at 37°C for 1 h. The inoculum was removed, replaced with complete culture medium, and further incubated until harvest. Viral titers of MERS-CoV Δ8b and wild-type (WT) were determined by standard plaque assay or quantitative RT-PCR (RT-qPCR) method as described (46).
Silver staining and mass spectrometry
HEK293 cells were transfected with the pCAGEN empty vector or pCAGEN-ORF8b plasmid in a 100-mm dish. Cells were subjected to immunoprecipitation 48 h posttransfection. The immunoprecipitates were loaded and resolved in 10% denaturing gel. The resolved gel was fixed in 50% methanol and 12% acetic acid in Milli-Q water for at least 2 h at room temperature. The gel was washed in 35% ethanol in Milli-Q water three times each for 20 min at room temperature. The washed gel was sensitized with sensitizing solution prepared by dissolving 0.1 g of sodium thiosulfate (MilliporeSigma) in 400 ml Milli-Q water for 10 min at room temperature. The sensitized gel was washed again with Milli-Q water for three times each for 5 min at room temperature. The gel was stained with silver nitrate solution prepared by dissolving 0.8 g of silver nitrate (MilliporeSigma) and 288 ml of formaldehyde (MilliporeSigma) in 400 ml Milli-Q water in darkness for 20 min at room temperature. The gel was washed with Milli-Q water twice each for 10 s at room temperature. The bands in the gel were developed in developing solution prepared by adding 24 g of sodium carbonate and 189 μl of formaldehyde into 8 ml of sensitizing solution topped up to 400 ml with Milli-Q water for 3 min. The reaction was stopped by adding 50% methanol and 12% acetic acid in Milli-Q water to the gel for 5 min. The bands were cut and kept in 1% acetic acid at 4°C until sent for characterization. Unique major bands were cut and subjected to mass spectrometric analysis using a Thermo Finnigan LTQ Orbitrap mass spectrometer with a dynamic nano-electrospray ionization interface. In brief, the samples were treated with trypsin for the generation of smaller peptides, and the peptides were introduced to the mass spectrometer for analysis. The resulting molecular ion peaks were searched against SwissProt_2015_10 database by mascot search engine.
Confocal microscopy
Huh-7 cells were transfected with the corresponding expression construct and infected as mentioned 24 h posttransfection. Cells were washed once with PBS and fixed with 4% paraformaldehyde in PBS, followed by permeabilization with 0.1% Triton X-100 in PBS. Cells were further washed with PBS for three times before blocking with 3% BSA in PBS for 1 h. Primary Abs anti-FLAG (M2; Invitrogen) and anti-myc (A14; Santa Cruz Biotechnology) as well as the guinea pig antiserum against N protein of MERS-CoV were diluted at 1:1000, 1:100, and 1:200, respectively, in 3% BSA in PBS for incubation at 4°C overnight. Cells were washed three times with PBS the next day before incubation with secondary Abs AP307R (myc; MilliporeSigma), AP124F (FLAG; MilliporeSigma), and Alexa Fluor 647–conjugated goat anti-guinea pig IgG (H chain + L chain) Ab (A-21450, 1:500; Invitrogen) plus DAPI at 1:200 in 3% BSA in PBS for 1 h at room temperature. Cells were washed with PBS for three times and visualized by confocal microscope.
Results
MERS-CoV ORF8b is a type I IFN antagonist
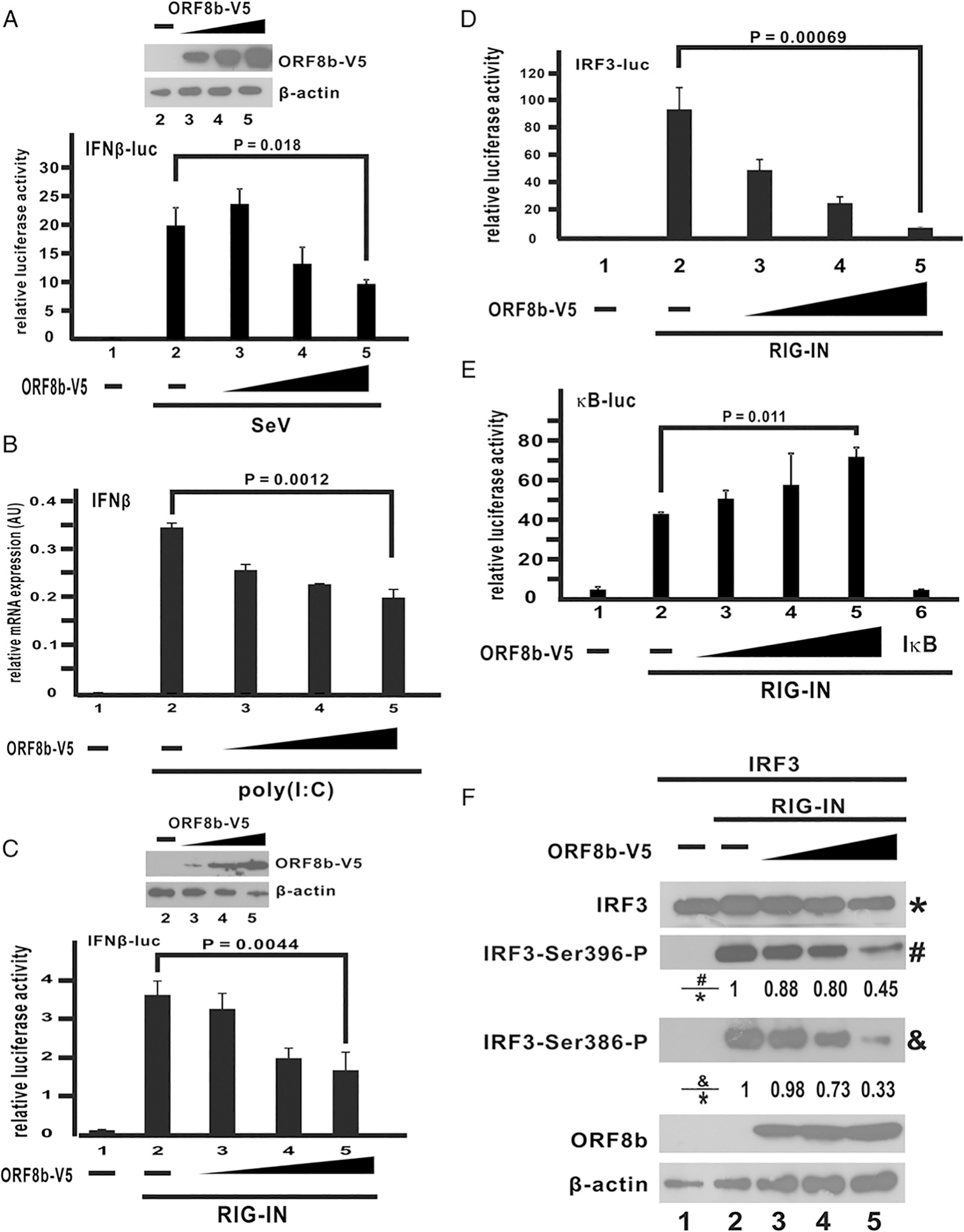
To explore the role of MERS-CoV ORF8b in type I IFN suppression, a luciferase reporter construct under the control of IFN-β promoter was introduced into HEK293 cells to assess IFN-β promoter activity under the challenge with SeV, which is a classic activator of RLR-induced IFN-β pathway. HEK293 cells are competent in type I IFN production and commonly used in the study of IFN response. Ectopic expression of MERS-CoV ORF8b at an increasing dose reduced SeV-induced IFN-β promoter activity (Fig. 1A; bars 3–5 versus 2). To corroborate these findings, IFN-β transcript was quantified by real-time RT-qPCR with synthetic dsRNA analogue poly(I:C) as the inducer of RLR-mediated IFN production. IFN-β expression was potently induced by poly(I:C), and a dose-dependent repression of such induction by ectopically expressed MERS-CoV ORF8b was observed (Fig. 1B; bars 3–5 versus 2). When the truncated version of RIG-I containing only the N-terminal CARD domain (RIG-IN), which serves as a constitutively active form of RIG-I (28), was used as a stimulant in Huh-7 cells, a similar observation was obtained. Ectopic expression of MERS-CoV ORF8b suppressed RIG-IN–induced IFN-β promoter activity in a dose-dependent manner (Fig. 1C; bars 3–5 versus 2). These results supported the IFN antagonism of MERS-CoV ORF8b in a cell line–independent manner with different RNA stimuli.
FIGURE 1.
MERS-CoV ORF8b inhibits IFN-β expression and IRF3 activation. (A) V5-tagged ORF8b expression construct (ORF8b-V5) was transfected into HEK293 cells at an increasing dose (100, 200, and 400 ng per well) together with a firefly luciferase reporter plasmid driven by IFN-β promoter (IFN-β-luc) and a control Renilla luciferase reporter plasmid. Cells were infected with SeV (100 hemagglutinating U/ml) at 24 h posttransfection of ORF8b-V5 for 16 h. At 16 hpi, cells were harvested for dual-luciferase reporter assay. The expression levels of the three doses introduced were detected by Western blotting with anti-V5 Ab (inset). β-Actin was included as an internal loading control. (B) V5-tagged ORF8b expression construct (ORF8b-V5) was transfected into HEK293 cells at an increasing dose (100, 200, and 400 ng per well). IFN-β expression was induced by poly(I:C) (1 μg/ml) at 24 h posttransfection, and cells were harvested for RT-qPCR at 16 hpi. GAPDH mRNA was used as an internal control. Relative expression of IFN-β transcripts was determined by ΔCT against GAPDH. (C) V5-tagged ORF8b expression construct (ORF8b-V5) was transfected into Huh-7 cells at an increasing dose (100, 200, and 400 ng per well) together with an expression vector for RIG-IN, a firefly luciferase reporter plasmid driven by IFN-β promoter (IFN-β-luc), and a control Renilla luciferase reporter plasmid. At 40 h posttransfection, cells were harvested for dual-luciferase reporter assay. The expression levels of the three doses introduced were detected by Western blotting with anti-V5 Ab (inset). β-Actin was included as an internal loading control. (D and E) ORF8b-V5 expression construct was transfected into HEK293 cells at an increasing dose (100, 200, and 400 ng) together with firefly luciferase reporter plasmid driven by either tandem copies of IRF3-binding elements (D) or κB elements (E) and a control Renilla luciferase reporter plasmid. Expression of IFN-β was induced by RIG-IN. HEK293 cells were harvested for dual-luciferase assay at 40 h posttransfection. (F) IRF3 and V5-ORF8b expression constructs were transfected into HEK293 cells at the dose mentioned above. RIG-IN was used to induce IRF3 phosphorylation. Cells were harvested 40 h posttransfection. The protein samples were then resolved with SDS-PAGE, and the indicated proteins were detected by Western blotting. The relative band intensity of phospho-IRF3 at serine 396 versus total IRF3 (#/*) and phospho-IRF3 at serine 386 versus total IRF3 (&/*) were measured using ImageJ software. Three independent experiments were performed. All bars represent the mean (n = 3), and error bars indicate their SD. The statistical significance between selected samples was evaluated by a two-tailed Student t test for unpaired samples with equal variance, and p values (P) were indicated. AU, arbitrary unit.
Expression of IFN-β in cells is known to be subject to tight regulation by long-distance interaction of multiple transcription factors that result in the formation of a giant enhanceosome complex (47). Among the transcription factors that regulate the expression of IFN-β, IRF3 and NF-κB are predominantly activated by RLR signaling (28, 48, 49). It therefore remains unknown whether the immunosuppressive effects of MERS-CoV ORF8b is mediated through IRF3 or NF-κB. To answer this question, RIG-IN, which is known to potently induce IRF3 and NF-κB signaling (28), was used to induce the activity of two different luciferase reporter constructs with tandem copies of either IRF3 or NF-κB binding sites cloned into the promoter region. Consistent with previous reports, RIG-IN significantly induced the activity of both IRF3- and NF-κB–driven luciferase activities (Fig. 1D, 1E; bar 2 versus 1). Ectopic expression of ORF8b effectively blunted RIG-IN–induced IRF3 activity in a dose-dependent manner (Fig. 1D; bars 3–5 versus 2). However, no suppressive effect on RIG-IN–induced NF-κB activity was observed when ORF8b was introduced (Fig. 1E; bars 3–5 versus 2), even though complete ablation of NF-κB activity was seen when the canonical inhibitor IκB-α was expressed (Fig. 1E; bar 6 versus 2). Phosphorylation of IRF3 at several residues is essential for dimerization, activation, and translocation of IRF3 (50). We therefore investigated if the steady-state amounts of phospho-IRF3 would decrease in the presence of ORF8b. IRF3 was overexpressed in cultured cells with RIG-IN to stimulate its phosphorylation. The levels of phosphorylation at two serine residues Ser-386 and Ser-396 of IRF3 were detected with Abs against phospho–serine 386 and 396, respectively. RIG-IN effectively induced IRF3 phosphorylation at Ser-386 and Ser396 (Fig. 1F; lane 2 versus 1). Introduction of ORF8b reduced both the levels of phospho–serine 386 and 396 in a dose-dependent manner (Fig. 1F; lanes 3–5 versus 2), indicating the inhibition of IRF3 activation. From these data, we conclude that MERS-CoV ORF8b exerts its immunosuppressive effect on IRF3 while sparing NF-κB signaling.
Whether ORF8b is expressed and functions to antagonize type I IFNs during MERS-CoVinfection remains elusive. To shed light on this, we raised a rabbit antiserum against ORF8b. The Abs specifically reacted to V5-tagged ORF8b (ORF8b-V5) expressed in HEK293 cells (Fig. 2A, 2B, lane 2 versus lane 1) and could precipitate ORF8b-V5 protein from transfected HEK293 cells (Fig. 2C). In addition, a band with the corresponding size of the ORF8b protein expressed in MERS-CoV–infected Huh-7 cells was detected (Fig. 2D, lane 2 versus 1), hence providing the first evidence, to our knowledge, for abundant expression of ORF8b in infected cells. Huh-7 cells were used in this experiment because they are highly susceptible to MERS-CoV infection (46). To construct a recombinant ORF8b-deficient MERS-CoV (Δ8b), we made use of the molecular clone of MERS-CoV strain EMC-2012 on BAC (44). The design of the Δ8b virus is shown in Fig. 2E. Particularly, two premature stop codons were introduced into the ORF8b, resulting in disruption of ORF8b expression without affecting the amino acid sequence of N protein. Multiple clones of Δ8b virus were obtained as verified by DNA sequencing (Fig. 2F). The anti-ORF8b Abs described above were used to detect ORF8b expression upon MERS-CoV infection. Compared with cells infected with WT virus, in which ORF8b was abundantly expressed, no ORF8b expression was detected in Huh-7 cells infected with the three Δ8b viruses. Meanwhile, N protein expression was unaffected in all viruses (Fig. 2G; lanes 3–5 versus 2). Because no clonal difference was noted in this and all subsequent experiments, results generated from clone 3 only were presented hereafter.
FIGURE 2.
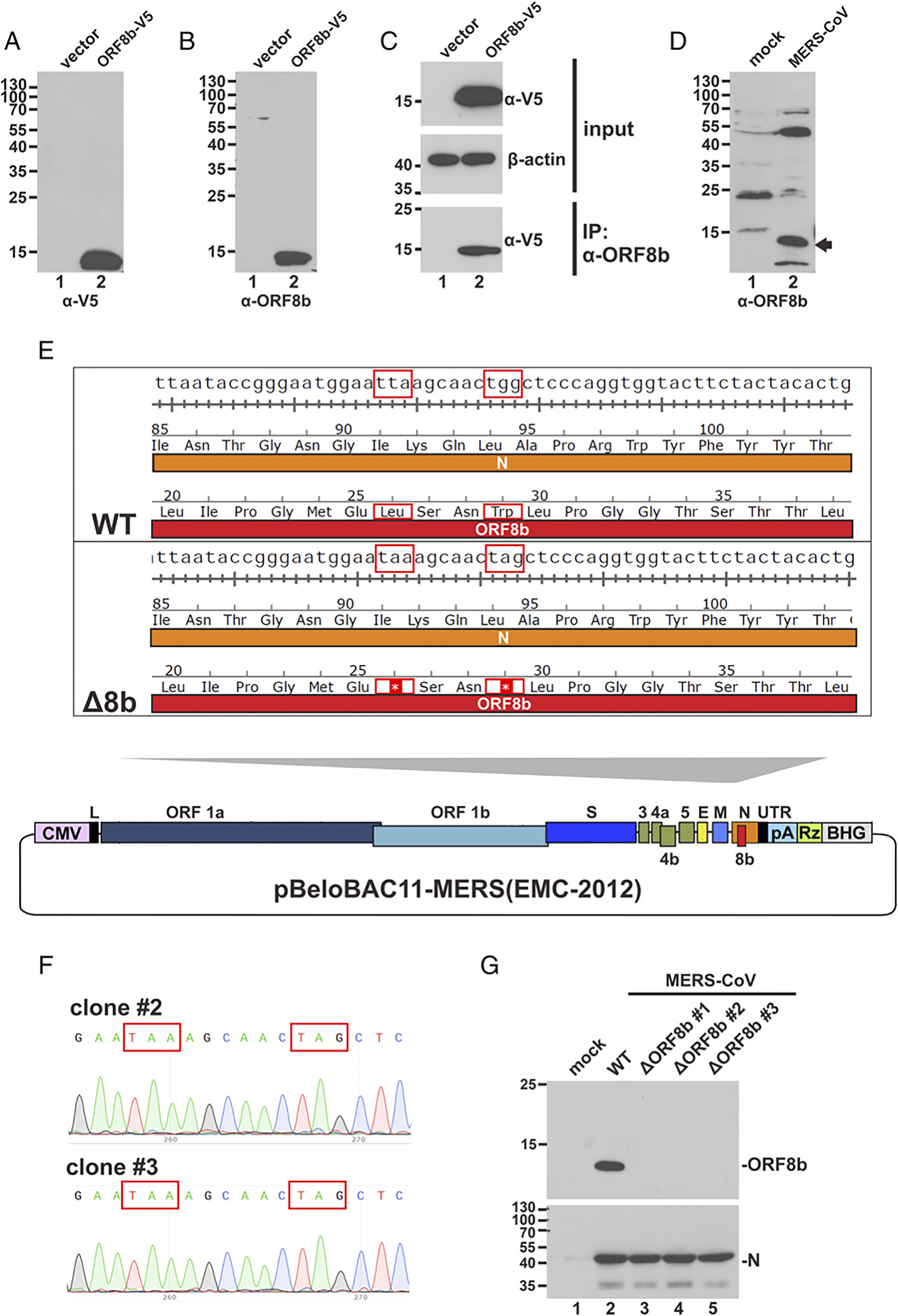
Generation, verification, and rescue of MERS-CoV-Δ8b. (A and B) HEK293 cells were transfected with either empty vector or ORF8b-V5 expression construct. Transfected cells were harvested 40 h posttransfection for Western blot analysis with anti-V5 (α-V5) or anti-ORF8b (α-ORF8b) Abs. (C) HEK293 cells were transfected with either empty vector or ORF8b-V5 plasmid. Transfected cells were harvested 40 h posttransfection for immunoprecipitation (IP) with anti-ORF8b Ab. (D) Huh-7 cells were either mock infected or infected with MERS-CoV at an MOI of 0.01. Infected cells were harvested 24 hpi for Western blot analysis with anti-ORF8b Ab. The band indicated by the arrow corresponds to the size of ORF8b. (E) Schematic diagram showing pBeloBAC11-based molecular clone harboring the complete genome of MERS-CoV strain EMC-2012. Essential (ORF1a, ORF1b, S, E, M, and N) and accessory (3, 4a, 4b, 5, and 8b) genes are indicated. The ORF8b region was zoomed in. Overlapping region of N and ORF8b genes of WT and ORF8b-deficient (Δ8b) viruses was compared side by side, highlighting (red boxes) the point mutations affecting the nucleotide and amino acid sequences. The two premature stop codons introduced to ORF8b sequence are indicated by asterisks (*). The amino acid sequence of N protein remains unchanged. (F) Sequencing result of Δ8b clones. The desired point mutations indicated by red boxes were found in two independent clones. Because no difference in sequence was observed between these two clones, results derived from only one clone (clone 3) were presented hereafter. (G) Huh7 cells were either mock infected or infected with WT or Δ8b MERS-CoV for 48 h before harvested for Western blot analysis with anti-ORF8b. Virus production was confirmed by detecting N protein expression using anti-N Ab.
To characterize WT and Δ8b viruses, Huh-7 and Vero cells were infected at an MOI of 0.01. Both cells are highly susceptible to infection with MERS-CoV (46). In contrast to Huh-7 cells that are IFN proficient, Vero cells are IFN deficient (51). Comparison of these cell lines might therefore be insightful when we studied the role of ORF8a in the induction of type I IFN production. The replication kinetics of WT and Δ8b viruses were compared, and a very minor difference between the two viruses was observed. In Huh-7 cells in which IFN production and signaling pathways are intact, very subtle differences were observed in the viral titers among the two viruses at any given time points (Fig. 3A–C). The overall growth rates of the two viruses were also similar, and viral titers of both WT and Δ8b viruses were essentially identical at any given time points in Vero cells (Fig. 3D–F). The viral titers of both viruses were largely comparable at any time point during the experiment, but they dropped earlier or more sharply in Huh-7 cells than in Vero cells, suggesting a role of IFN in limiting viral replication. Generally consistent with previous findings on other lineage-specific accessory genes of MERS-CoV, our results suggest that ORF8b is not essential for MERS-CoV replication in Huh-7 and Vero cells. Levels of IFN-β mRNA induced by WT and Δ8b viruses were measured at three different time points postinfection of Huh-7 cells at an MOI of 0.01 (Fig. 3G). The expression of IFN-β mRNA in WT-infected cells gradually increased from 0 to 48 h postinfection (hpi) and reached a maximum of around 10-fold induction at 48 hpi. The peak of IFN-β expression did not extend beyond 48 hpi and remained at a similar level until 72 hpi. This was generally consistent with previous reports that MERS-CoV infection failed to induce a strong IFN response (20–25). However, although IFN-β mRNA levels in WT- and Δ8b-infected cells were not much different at 24 hpi, the Δ8b virus induced consistently higher IFN-β mRNA expression at 48 and 72 hpi (Fig. 3G). The amount of IFN-β protein in the supernatant detected in the supernatant was much higher at 48 hpi (Fig. 3H). Consistently, mRNA expression of ISG15, a representative ISG, was more prominent at 48 and 72 hpi (Fig. 3I). The induction of the transcript of IL-8, a representative proinflammatory chemokine that is an atypical ISG (52), was also considerably more pronounced at 72 hpi (Fig. 3J). Expression levels of ORF8b in infected cells were determined. The expression level of ORF8b remained relatively constant from 24 to 48 hpi but diminished significantly at 72 hpi (Fig. 3K, lanes 2–4 versus 1). There was not a significant change in IFN-β mRNA expression from 48 to 72 hpi in both WT- and Δ8b-infected cells despite the change in ORF8b expression level. In other words, WT and Δ8b viruses were of similar replication kinetics and initial titers, but IFN-β and ISG induction by Δ8b virus was generally more robust. Thus, MERS-CoV ORF8b plays a suppressive role in the production of type I IFN when ectopically expressed and during infection.
FIGURE 3.
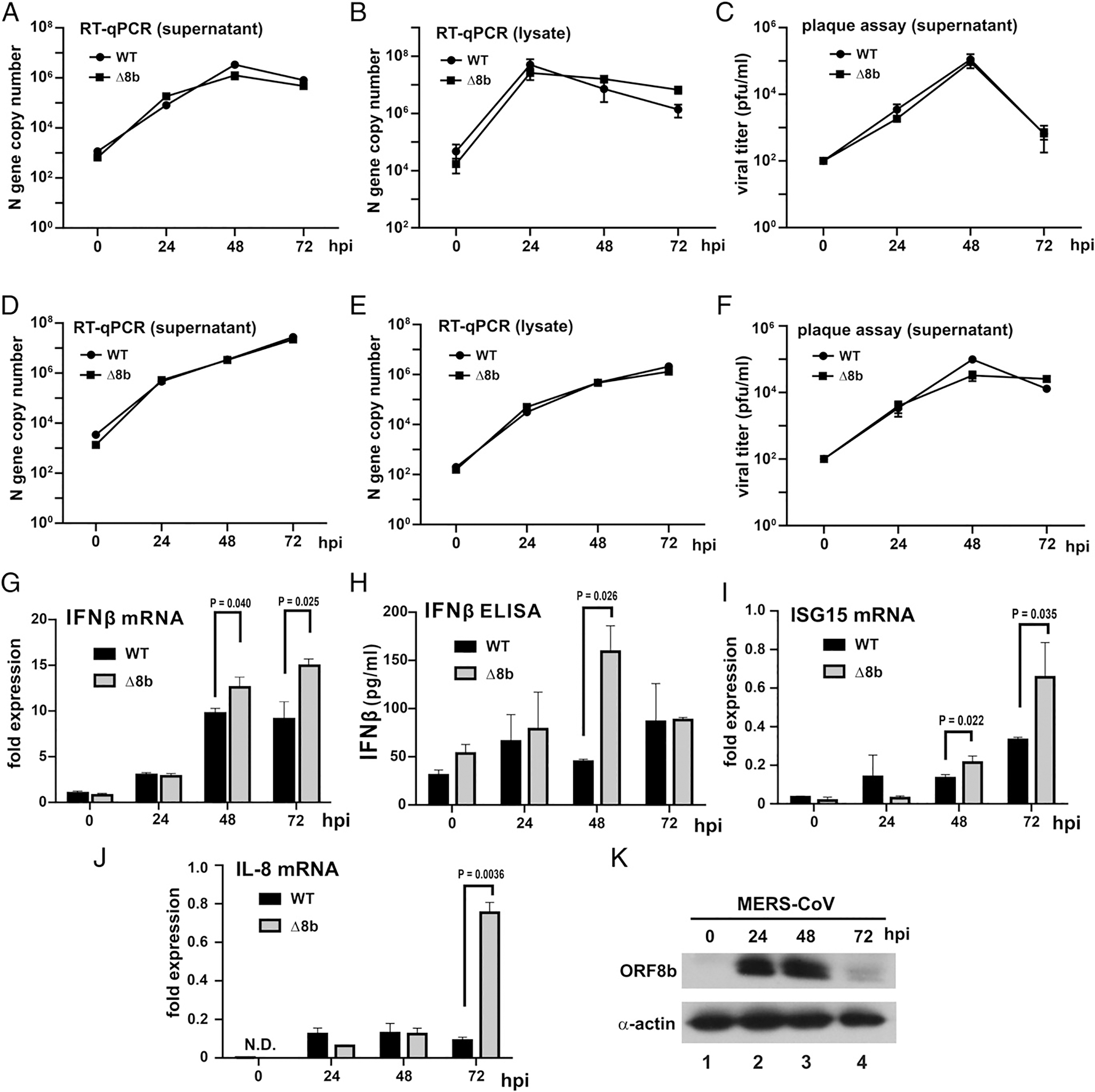
Replication kinetics and IFN-β induction of WT and Δ8b viruses. Huh-7 (A–C) and Vero E6 (D–F) cells were infected by WT and Δ8b viruses at an MOI of 0.01 for the indicated time points. Supernatants (A and D) and cell lysates (B and E) were harvested, and viral titers were quantified by N gene quantification with reference to a standard curve of WT MERS-CoV with the titer previously determined by standard plaque assay. Viral plaque assay from supernatant of Huh-7 (C) and Vero E6 (F) were performed to determine the amount of infectious virus present at the indicated time point. Each data point represents the mean (n = 3), and error bars indicate their SD. (G–K) Enhanced IFN-β induction by Δ8b viruses. Huh-7 cells were infected by WT and Δ8b viruses at an MOI of 0.01 for the indicated time points. Infected cells were harvested for total RNA, and the levels of IFN-β (G), ISG15 (I), and IL-8 (J) transcripts were determined by RT-qPCR. Supernatants of the same experiment were also collected for measuring the abundance of IFN-β secreted by ELISA (H). Fold expression of IFN-β transcripts was determined by ΔΔCT against GAPDH at time 0. Three independent experiments were performed. Bars represent the mean (n = 3), and error bars indicate their SD. The statistical significance between selected samples was evaluated by a two-tailed Student t test for unpaired samples with equal variance, and p values (P) were indicated. (K) Huh-7 cells were infected at an MOI of 0.01 for the indicated time points. Cells were harvested, and total cell lysates were resolved by SDS-PAGE followed by Western blotting. β-Actin was included as an internal loading control. N.D., not detected.
MERS-CoV ORF8b exerts its IFN-suppressive effect on IRF3
The RLR signaling pathway involves several key components that transduce the activation signal from an upstream inducer to a downstream effector with IRF3 activation as the end result (28). Alteration of the function of any of the key components upstream of IRF3 may lead to suppression of IRF3 activation and hence IFN-β production. To shed light on the action point of ORF8b, key components in the RLR signaling pathway were separately overexpressed in cultured HEK293 cells to produce an activation signal to a luciferase reporter construct driven by IFN-β promoter. The activation triggered by enforced expression of a component of the RLR pathway remains mostly unaffected when it is downstream of the action point of ORF8b. In this regard, RIG-IN being a constitutively active form of RIG-I serve as an upstream activation signal in the RLR pathway. ORF8b suppressed RIG-IN–induced IFN-β promoter activity in a dose-dependent manner in HEK293 cells (Fig. 4A; bars 3–5 versus 2). The results were similar when MAVS (Fig. 4B; bars 3–5 versus 2) or IRF kinases TBK1 and IKKε (37) were used as the activation signal (Fig. 4C, 4D; bars 3–5 versus 2). However, the inhibitory effect was not observed when IRF3 and IRF3–5D, which is a constitutively active form of IRF3 (50), served as the activation signal (Fig. 4E, 4F; bars 3–5 versus 2). Hence, the action point of MERS-CoV ORF8b lies upstream of IRF3, and the inhibitory effect of ORF8b results in the suppression of IRF3 activation.
FIGURE 4.
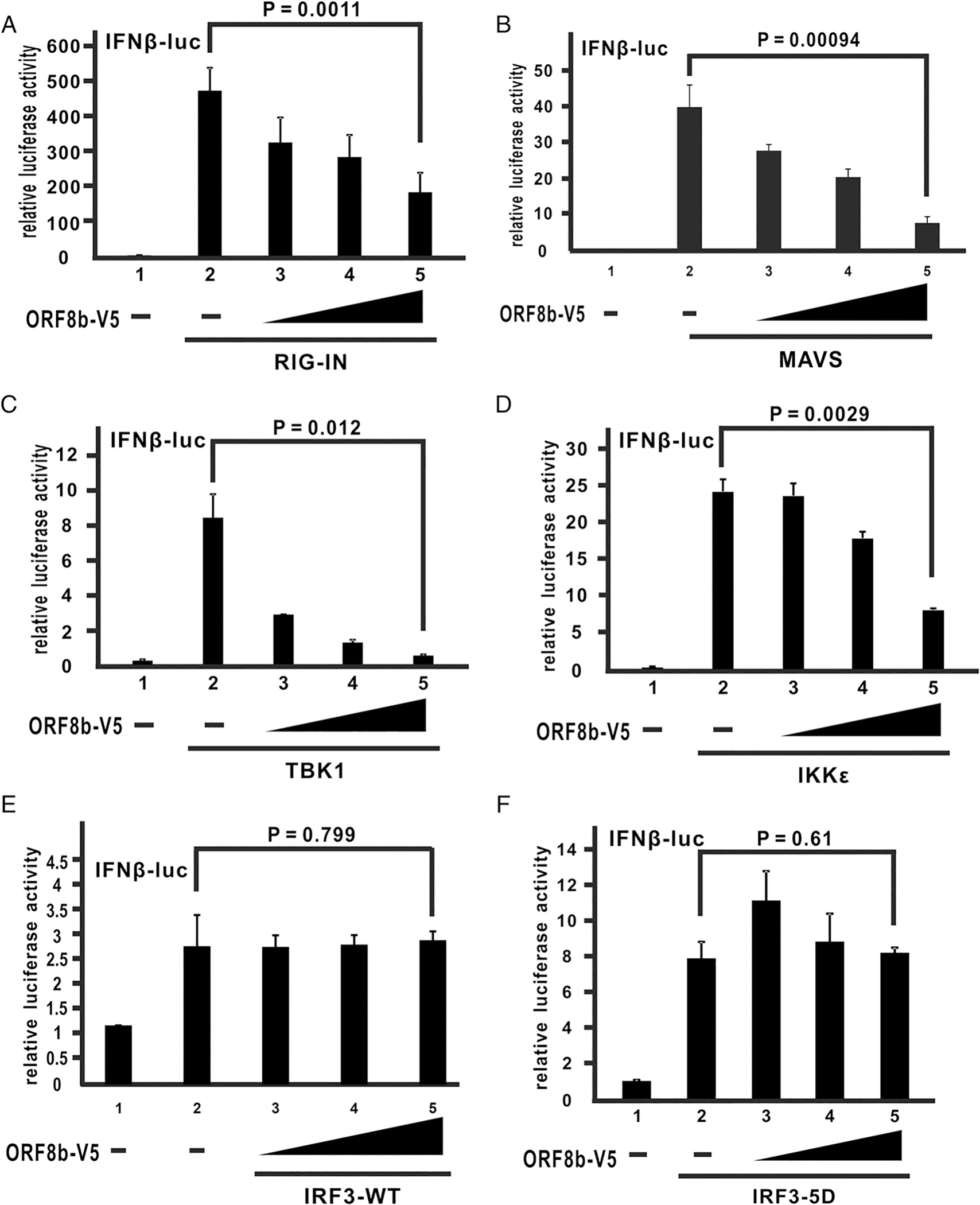
MERS-CoV ORF8b exerts its suppressive effect upstream of IRF3. (A–F) HEK293 cells were transfected with an increasing dose of ORF8b-V5 (100, 200, and 400 ng), a firefly luciferase reporter driven by IFN-β promoter, and a Renilla luciferase reporter as control, together with the expression plasmids of different effectors of the RLR pathway, RIG-IN (A), MAVS (B), TBK1 (C), IKKε (D), IRF3-WT (E), and IRF3–5D (F). HEK293 cells were harvested for dual-luciferase assay at 40 h posttransfection. Lysates were mixed with luciferase substrate and analyzed by luminometer to obtain readings. Relative luciferase activity was obtained by normalizing firefly luciferase to Renilla luciferase activity. Bars represent the mean of three biological replicates (n = 3), and error bars indicate their SD. The statistical significance between selected samples was evaluated by a two-tailed Student t test for unpaired samples with equal variance, and p values (P) were indicated.
MERS-CoV ORF8b targets IKKε through HSP70
Because the action point of ORF8b was shown to be upstream of IRF3, we postulated that ORF8b might function through an interaction with RLR components upstream of IRF3. To address this, coimmunoprecipitation was performed. FLAG-tagged RLR components including RIG-I, MAVS, TRAF3, TBK1, and IKKε were individually overexpressed in cultured cells together with ORF8b-V5. Whole cell lysates were subjected to immunoprecipitation with anti-FLAG Ab. Surprisingly, no V5-specific band was detected in the immunoprecipitates (Fig. 5A), indicating the lack of direct interaction between ORF8b and any RLR component tested. To identify its binding partners, ORF8b-V5 was overexpressed in HEK293 cells for immunoprecipitation. The anti-V5 precipitates were resolved in denaturing SDS-PAGE followed by silver staining (Fig. 5B). Three prominent bands (arrowed with a, b, and c) detected in the ORF8b-V5–expressing cells but not in control cells transfected with the empty vector were sent for mass spectrometric analysis. The result indicated that several HSPs, namely HSP60-D1, HSP70–1A, HSP90α, and HSP90β, were proteins with the highest scores (Fig. 5C). HSP70–1A is a key member in the HSP70 family (53).
FIGURE 5.
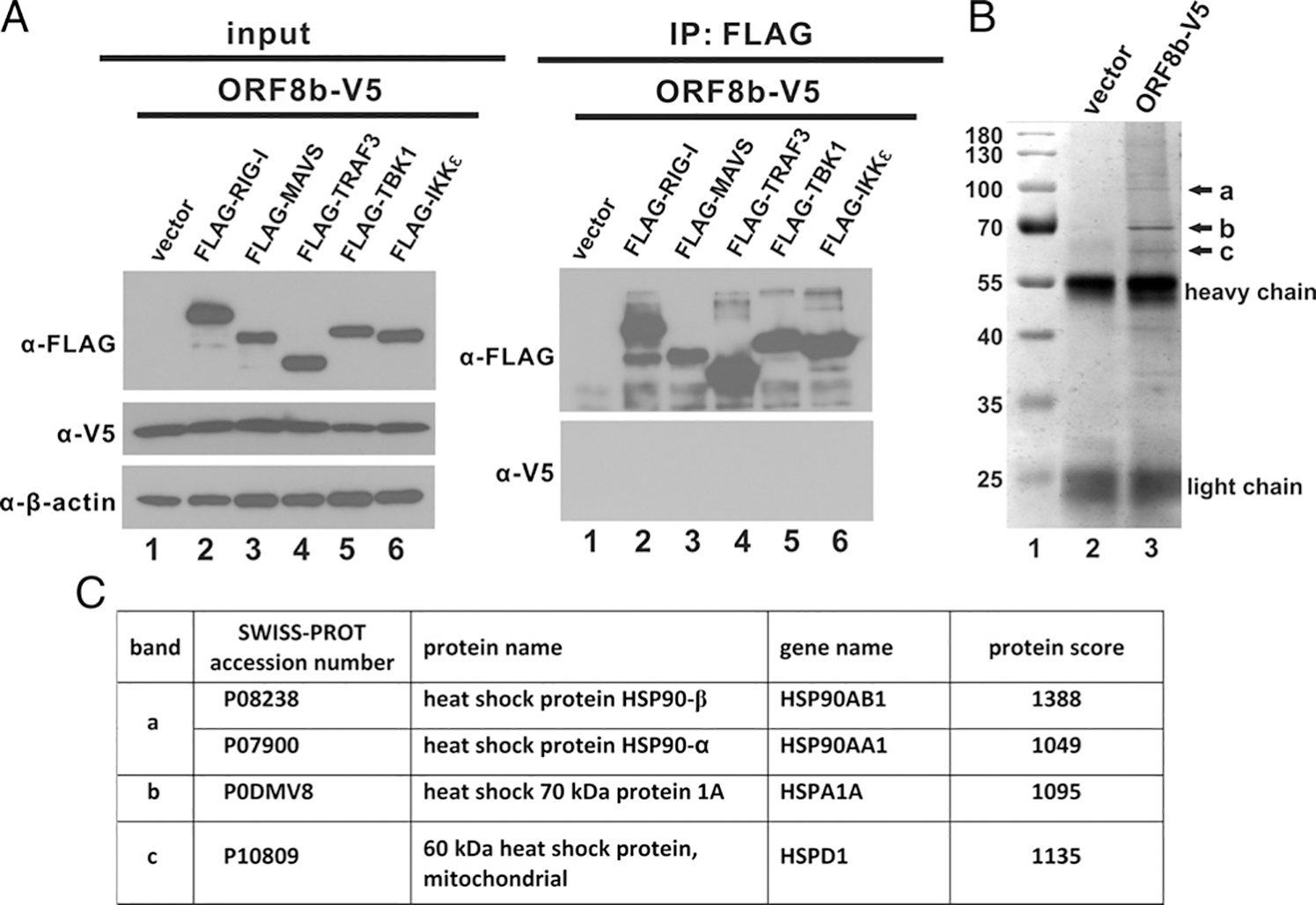
MERS-CoV ORF8b interacts with HSPs as determined by mass spectrometry. (A) Expression constructs of FLAG-tagged RLR pathway effectors were individually cotransfected with ORF8b-V5 plasmid into HEK293 cells as indicated. Transfected cells were harvested 40 h posttransfection and subjected to immunoprecipitation (IP) with anti-FLAG Ab. The total cell lysate and Ab-bound fraction were resolved by SDS-PAGE and analyzed by Western blotting with anti-V5 and anti-FLAG Abs. β-Actin was also detected as an internal loading control. (B) HEK293 cells were transfected with either ORF8b-V5 plasmid or empty vector. Transfected cells were harvested for immunoprecipitation with anti-V5 Ab 40 h posttransfection. Immunoprecipitates were then resolved in denaturing SDS-PAGE gel followed by silver staining for band visualization. Unique major bands (indicated by arrows a, b, and c) in ORF8b-V5 sample were cut and subjected to mass spectrometric analysis. HSPs identified to be interacting with ORF8b were listed in the table. (C) A table listing the proteins identified in bands a, b, and c by mass spectrometry.
The four HSPs identified by mass spectrometry were cloned into expression vector for verification by coimmunoprecipitation. V5-tagged ORF8b and myc-tagged HSPs were cotransfected into cultured cells. Immunoprecipitation with anti-V5 Ab was performed on whole cell lysates. HSP70–1A was the sole candidate among the four that interacted with ORF8b as an myc-specific band was observed in the anti-V5 precipitate (Fig. 6A; lane 5). Consistent with coimmunoprecipitation experiments, the luciferase functional assay showed that expression of HSP70–1A could reverse ORF8b-mediated inhibition of IFN-β promoter activity (Fig. 6B; lane 5 versus 3), suggesting that HSP70–1A interacts with ORF8b and is influential in its innate immunosuppressive effect.
FIGURE 6.
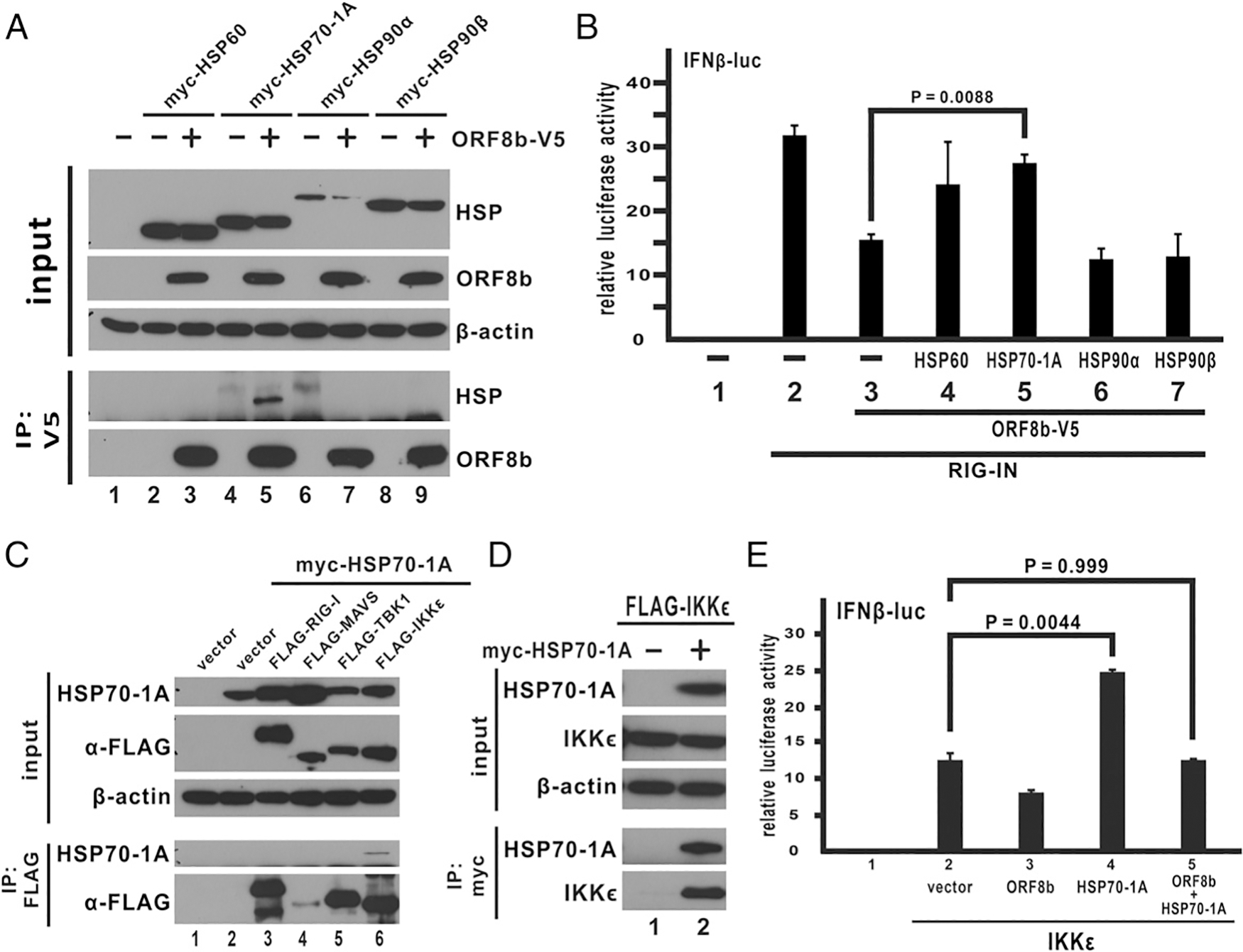
MERS-CoV ORF8b targets IKKε through HSP70. (A) Expression construct of ORF8b-V5 was cotransfected with myc-tagged HSPs into HEK293 cells as indicated. Transfected cells were collected 40 h posttransfection for immunoprecipitation (IP) with anti-V5 Ab. The total cell lysate and the Ab-bound fraction were resolved in denaturing SDS-PAGE and visualized by Western blot with anti-V5 and anti-myc Abs. β-Actin was also detected as an internal loading control. (B) HSP expression constructs were cotransfected into HEK293 cells together with ORF8b and RIG-IN expression plasmids, a firefly luciferase reporter driven by IFN-β promoter, and a control Renilla luciferase plasmid. Transfected cells were harvested for dual-luciferase reporter assay 40 h posttransfection. (C and D) Transfection of HEK293 cells and immunoprecipitation (IP) were performed as indicated. (E) HSP70–1A facilitates activation of IKKε-mediated IFN-β promoter activity. Transfection of HEK293 cells and dual-luciferase assay were carried out as described above with the lowest corresponding dose of ORF8b (100 ng). The bars indicate the mean, and the error bars indicate the SD of the three biological replicates (n = 3). Two-tailed Student t test for unpaired samples with equal variance was used to determine the statistical significance between designated samples with the p value (P) indicated.
HSP70–1A was shown to interact with ORF8b and rescue ORF8b-mediated suppression of IFN-β expression. We therefore hypothesized that HSP70–1A might have a role in regulating the RLR signaling pathway to achieve such an effect (Fig. 6B; bar 5 versus 3). HSP60 was not further pursued as the difference was not statistically significant. To test this hypothesis, we performed reciprocal coimmunoprecipitation experiments between myc-tagged HSP70–1A and FLAG-tagged RLR components (Fig. 6C, 6D). The results suggested that HSP70–1A specifically interacts with IKKε. Because IKKε is a kinase directly phosphorylating IRF3 (37), any inhibitory effects on IKKε could lead to the observations obtained above. We reasoned that HSP70–1A might have a role in IKKε-mediated IFN-β expression as HSP70–1A interacts with both ORF8b and IKKε, although the mode of interaction is not known. To this end, IKKε-induced IFN-β promoter activity was tested with or without HSP70–1A (Fig. 6E). With ectopically expressed HSP70–1A, IKKε-induced IFN-β promoter activity increased by ~2-fold (Fig. 6E, bar 4 versus 2). In addition, HSP70–1A rescued the suppression of IKKε-induced IFN-β promoter activity by ORF8b (Fig. 6E, bar 5 versus 3). All these suggest a specific role of HSP70–1A in enhancing IKKε-mediated IFN-β expression.
To analyze further the role of HSP70 in IFN induction, two short hairpin RNAs (shRNAs) targeting HSP70–1A were individually expressed, followed by transfection of an IKKε expression construct or an empty vector as a control (Fig. 7A). The expression levels of HSP70–1A and knockdown efficiency of the two shRNAs against HSP70–1A in Huh-7 cells are shown in Fig. 7B. shHSP70–2 was more efficient in ablating the expression of endogenous HSP70–1A in Huh-7 cells. mRNA expression driven by IFN-β promoter was detected at basal levels with or without shRNA expression in the control group (Fig. 7A; empty bars). However, despite the elevated level of IFN-β promoter activity induced by IKKε in the absence of shRNAs, IKKε failed to induce such activity in the presence of shRNA against HSP70–1A (Fig. 7A; solid bars in groups 2 and 3 compared with empty bar in group 1). The effect of HSP70–1A depletion on viral induction of IFN-β was assessed by RT-qPCR analysis of IFN-β mRNA (Fig. 7C). Consistent with results presented in Fig. 3G, Δ8b virus induced IFN-β expression more robustly compared with WT virus in the absence of shRNA against HSP70–1A (Fig. 7C, group 1). In the presence of shRNAs targeting HSP70–1A, IFN-β induction by both WT and Δ8b viruses was dampened (Fig. 7C; groups 2 and 3), consistent with the positive role of HSP70–1A in IFN-β expression. Notably, the expression of IFN-β mRNA induced by the Δ8b virus was less prominent in the presence of shHSP70–1 than in its absence (Fig. 7C, group 2 versus 1) but remained more pronounced when compared with the induction by WT virus (Fig. 7C; group 2, gray versus black bar). This was likely ascribed to incomplete knockdown of HSP70–1A by shHSP70–1. The residual HSP70–1A was suppressed by WT virus with intact ORF8b expression but not Δ8b virus. In the presence of shHSP70–2, by which a near-complete knockdown of HSP70–1A was achieved (Fig. 7B), no statistically significant difference in IFN-β mRNA expression was observed between WT and Δ8b viruses (Fig. 7C, group 3, gray versus black bar). Plausibly, the near-complete depletion of HSP70–1A resulted in the reversion of elevated IFN-β mRNA expression attributed to the absence of ORF8b to a level comparable to that induced by WT virus. This is consistent with the notion that the IFN antagonism of ORF8b is mediated primarily through HSP70–1A. Generally, in keeping with the mRNA induction results, the amounts of secreted IFN-β protein in the supernatant induced by the WT and Δ8b viruses were comparable in the presence of shHSP70–1 or shHSP70–2 (Fig. 7E, groups 2 and 3, gray versus black bars). The viral titers of WT and Δ8b viruses were also compared to determine the impact of HSP70–1A depletion on viral replication. WT and Δ8b viruses did not differ significantly in viral replication in the absence of HSP70–1A depletion (Fig. 7E, group 1). The viral titers measured in all settings were highly comparable. The difference in viral titers of the two viruses in HSP70–1A–depleted cells were only marginal despite being statistically significant (Fig. 7E; groups 2 and 3, gray versus black bars). Thus, HSP70–1A upregulates IFN expression when stimulated by ectopic inducer or during viral infection independently of the changes in viral titers.
FIGURE 7.
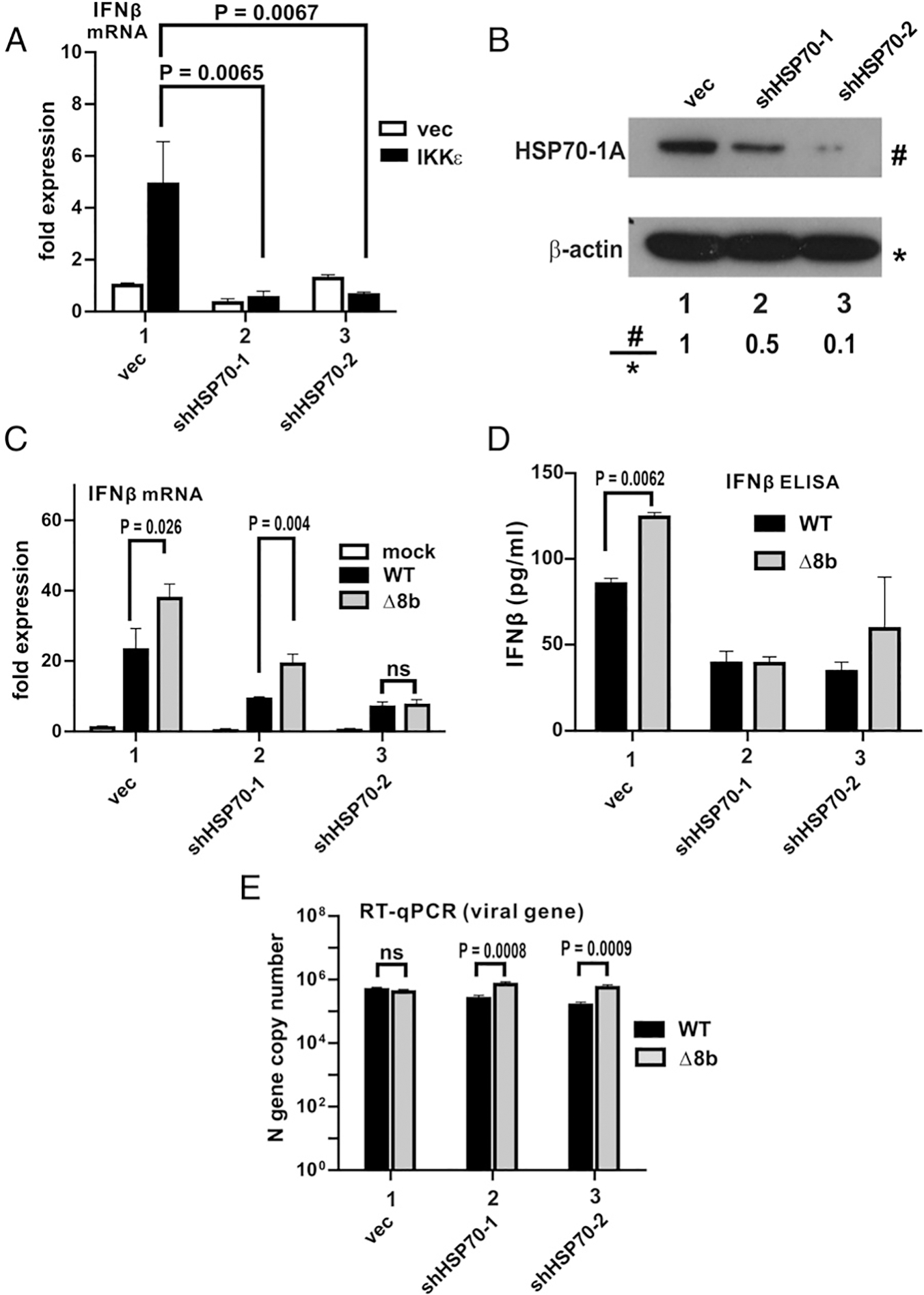
HSP70–1A is required to enhance IFN expression in viral infection. (A) Huh-7 cells were transfected with empty vector (vec) or shRNA targeting HSP70–1A (shHSP70–1 or shHSP70–2). A second transfection was performed at 24 h after the first transfection with empty vector (white panel) or IKKε expression construct (black panel). At 48 h after the first transfection, total cellular RNA was harvested, and IFN-β transcript levels were determined by RT-qPCR. GAPDH mRNA was used as an internal control. Fold of expression of IFN-β transcripts was determined by ΔΔCT against GAPDH of mock. Bars represent the mean from three independent experiments (n = 3), and error bars indicate their SD. The statistical significance between selected samples was evaluated by a two-tailed Student t test for unpaired samples with equal variance, and p values (P) were indicated. (B) Huh-7 cells were transfected with empty vector (vec) or shRNA targeting HSP70–1A. Cells were harvested 48 h posttransfection, and total cell lysates were resolved by SDS-PAGE followed by Western blot analysis with the Abs specified. The relative band intensity (#/*) of HSP70–1A and β-actin was determined using ImageJ software. (C–E) Huh-7 cells were transfected with vec or shRNA targeting HSP70–1A. Cells were infected with WT or Δ8b viruses at an MOI of 0.01 at 24 h posttransfection. IFN-β transcript levels (C) and N gene copy number (E) in cells were determined by real-time RT-qPCR, whereas IFN-β protein levels (D) were detected by ELISA after harvesting at 24 hpi. GAPDH mRNA was used as an internal control, and the fold of expression of IFN-β transcripts was determined by ΔΔCT against GAPDH of mock (C). N gene copy number was measured with reference to a standard curve of WT MERS-CoV with the titer previously determined by standard plaque assay (E). Bars represent the mean of three independent experiments (n = 3), and error bars indicate their SD. The statistical significance between selected samples was evaluated by a two-tailed Student t test for unpaired samples with equal variance, and p values (P) were indicated. ns, not significant.
MERS-CoV ORF8b suppresses IFN-β production by impeding the interaction between HSP70 and IKKε
Site-directed mutagenesis of Ser-172 of IKKε showed that phosphorylation at Ser-172 is required for the kinase activity of IKKε (54). To identify the role of HSP70–1A on IKKε-mediated induction of IFN-β expression, activation of IKKε was measured with phospho–serine 172 of IKKε as a readout. IKKε activation was observed in HEK293 cells transfected with IKKε expression vector with the presence of a prominent band when immunoblotted with anti–phospho-serine 172 of IKKε (Fig. 8A; lane 2 versus 1). This is consistent with previous reports that TBK1 and the structurally related IKKε are activated by autotransphosphorylation at Ser-172 (54, 55). This also accounts for the results that ectopically expressing IKKε induced IFN-β promoter activity as shown above. When HSP70–1A was cotransfected with IKKε, the level of phosphorylated Ser-172 was significantly higher than when IKKε alone was present with comparable levels of total IKKε in all lanes as the presence of HSP70–1A did not have a significant effect on the levels of IKKε expressed (Fig. 8A; lane 4 versus 2). In addition, the introduction of ORF8b inhibited the level of HSP70–1A–mediated IKKε activation (Fig. 8A; lane 5 versus 4). Hence, HSP70–1A enhances IKKε activation, and ORF8b functions to prevent HSP70–1A–mediated activation of IKKε.
FIGURE 8.
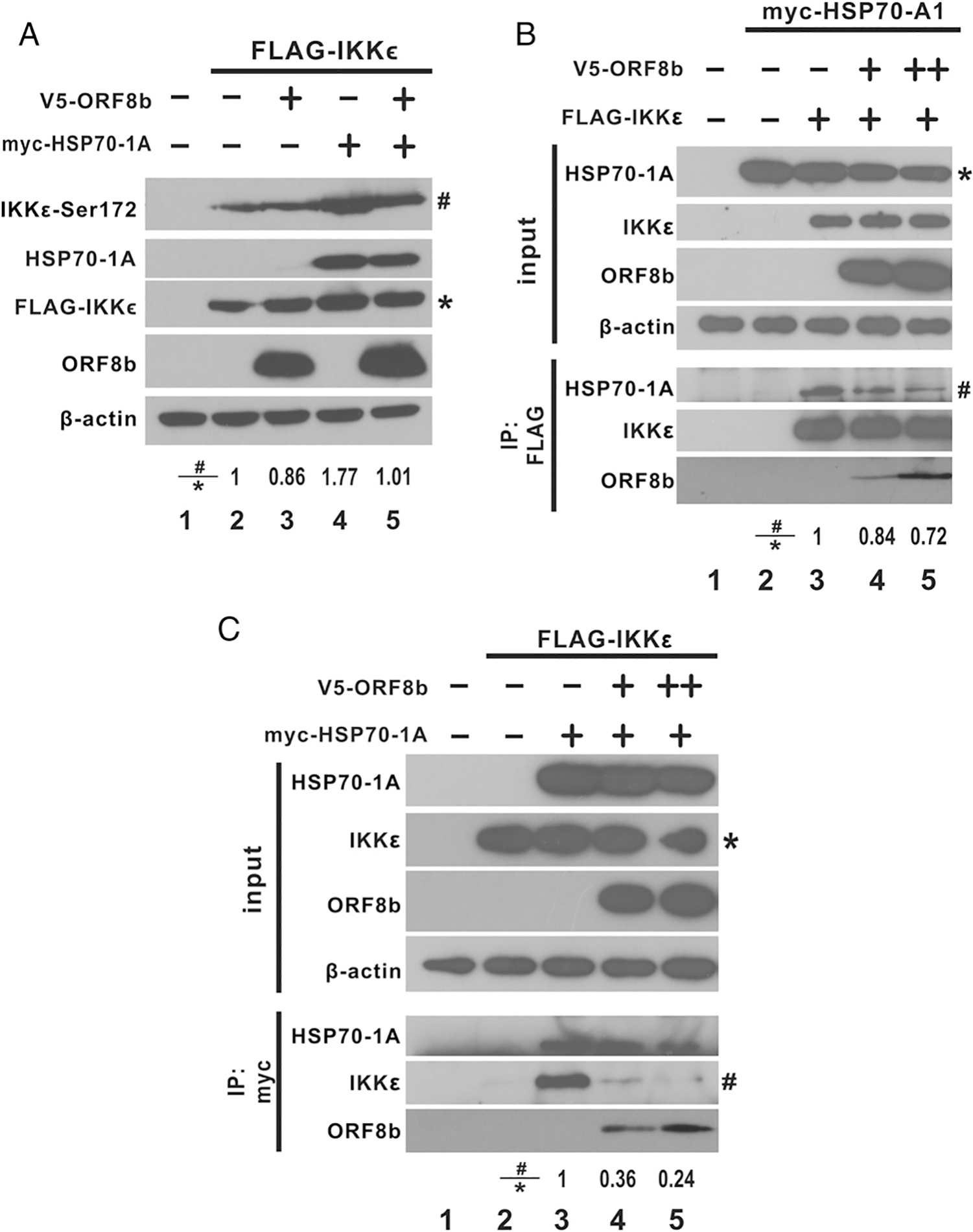
MERS-CoV ORF8b impedes interaction between IKKε and HSP70–1A, thereby suppressing HSP70–1A–dependent IKKε activation. (A) MERS-CoV ORF8b and HSP70–1A plasmids were cotransfected into HEK293 cells as indicated. IKKε phosphorylation at residue serine 172 was induced by expressing IKKε. Transfected cells were harvested 40 h posttransfection and subjected to denaturing SDS-PAGE and Western blot analysis with anti–phospho-IKKε at serine 172 (anti-IKKε-Ser172), anti-myc, anti-V5, and anti-FLAG Abs. β-Actin was also detected as an internal loading control. (B and C) IKKε expression construct was cotransfected into HEK293 cells with HSP70–1A plasmid as indicated. Expression of ORF8b-V5 was introduced as indicated. Transfected cells were collected 40 h posttransfection for immunoprecipitation (IP) with anti-FLAG (B) and anti-myc (C) Abs. The total cell lysate and the Ab-bound fraction were resolved in denaturing SDS-PAGE and visualized by Western blot with anti-V5, anti-FLAG, and anti-myc Abs. The relative band intensity (#/*) of phospho-IKKε at serine 172 (A), coimmunoprecipitated HSP70–1A (B), and coimmunoprecipitated IKKε (C) was measured using ImageJ software.
From the above results, HSP70–1A interacts with IKKε to enhance its activation, although ORF8b also interacts with HSP70–1A. We hypothesized that ORF8b might compete with IKKε for binding with HSP70–1A. To test this possibility, the interaction between IKKε and HSP70–1A was detected by reciprocal coimmunoprecipitation in the presence or absence of ORF8b (Fig. 8B, 8C). When IKKε was pulled down, the level of myc-tagged HSP70–1A in the anti-FLAG precipitate decreased with an increasing level of ORF8b, whereas the level of IKKε remained essentially similar (Fig. 8B; lanes 4 and 5 versus 3). Similar results were obtained when myc-tagged HSP70–1A was pulled down. The level of FLAG-tagged IKKε decreased when the dose of ORF8b was increased, but the amount of HSP70–1A was essentially unchanged (Fig. 8C; lanes 4 and 5 versus 3). Notably, ectopic expression of ORF8b did not significantly alter the level of expression of IKKε or HSP70–1A (Fig. 8B, 8C; lanes 4 and 5 versus 3 in the input), suggesting that ORF8b impedes the interaction between HSP70–1A and IKKε without affecting their expression levels.
To strengthen our results obtained from the ectopic expression model, we also analyzed the interplay of ORF8b, IKKε, and HSP70–1A in infected cells. HSP70–1A and IKKε were ectopically expressed in Huh-7 cells before they were challenged with WT and Δ8b viruses (Fig. 9). In mock-infected cells, extensive colocalization of HSP70–1A and IKKε was observed as expected from the earlier results presented in Figs. 6 and 8. Because the ORF8b antiserum raised was not good for immunofluorescent staining, infected cells were stained with Abs against N protein of MERS-CoV (MERS-N in pink). When challenged with WT viruses, the expression of HSP70–1A and IKKε was not altered (Fig. 9, panels 6 and 7). However, the colocalization between the two proteins was almost completely abolished, suggesting the disruption of their interaction by WT virus (Fig. 9, panel 10). The colocalization between HSP70–1A and IKKε was partially restored when cells were infected with the Δ8b virus (Fig. 9, panel 15), implicating the role of ORF8b in the disruption of HSP70–1A–IKKε interaction. Taken together, our results are compatible with a competition between MERS-CoV ORF8b and IKKε for binding with HSP70–1A, which is required for full activation of IKKε (Fig. 10).
FIGURE 9.
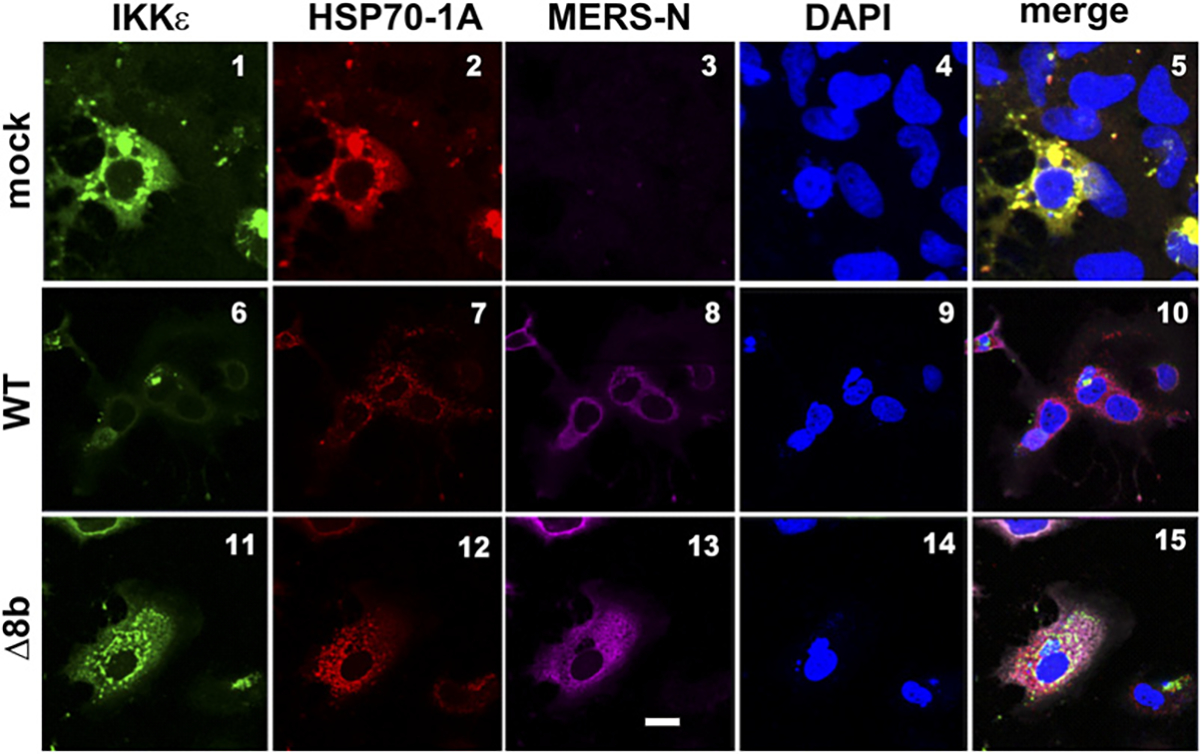
MERS-CoV perturbs colocalization between HSP70–1A and IKKε in infected cells. FLAG-IKKε and myc-HSP70–1A expression constructs were transfected into Huh-7 cells. Cells were either mock infected or infected with WT or Δ8b virus at 24 h posttransfection with an MOI of 0.01. Cells were fixed and labeled with primary Abs against epitope tag and N protein of MERS-CoV at 24 hpi. Cells were incubated with secondary Abs conjugated with fluorophores after primary Ab incubation and subsequently visualized by confocal microscope. Nuclei were costained with DAPI with secondary Abs. DAPI is in blue. IKKε is in green. HSP70–1A is in red. MERS-N is in pink. Scale bar, 20 μm.
FIGURE 10.
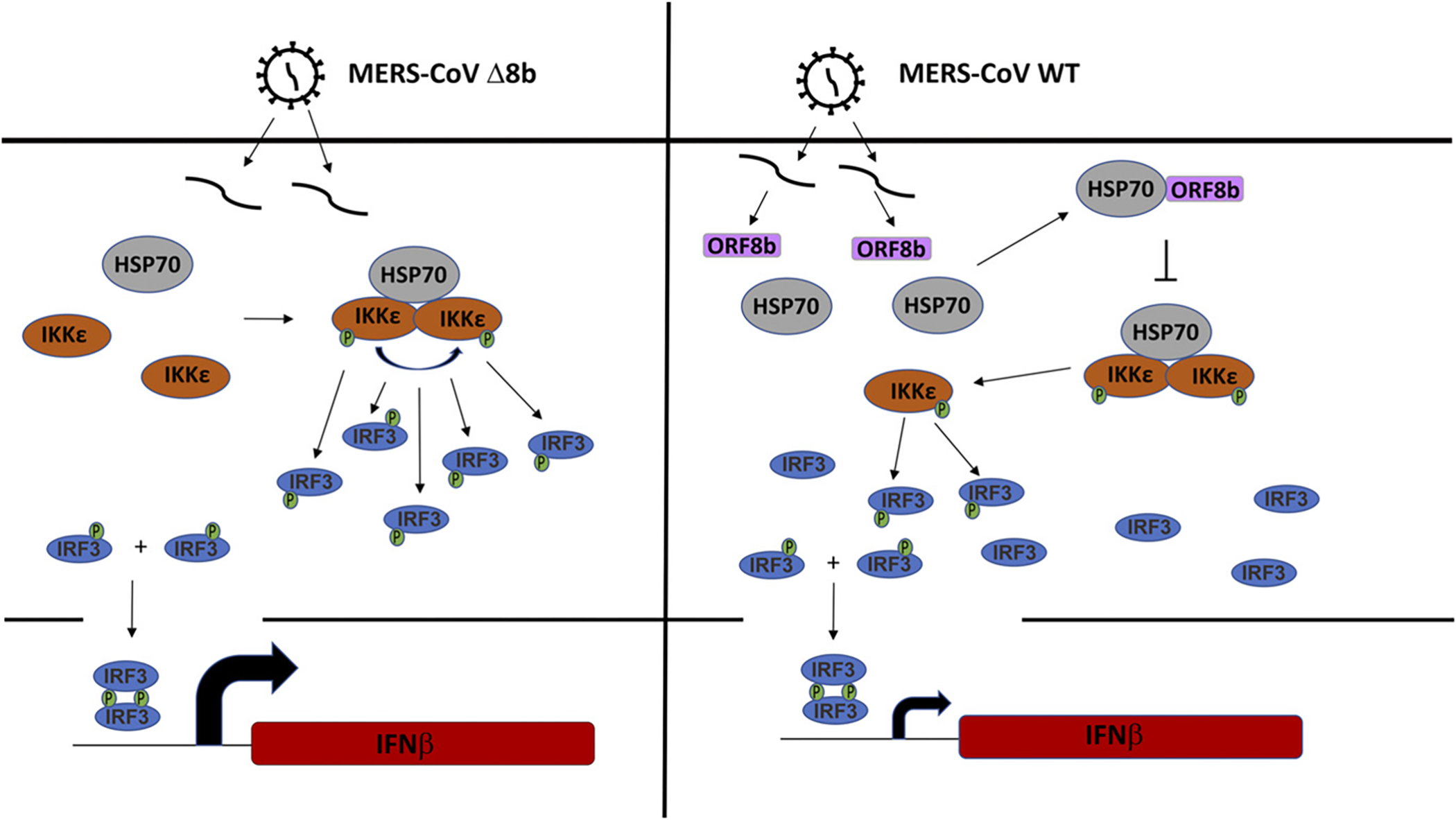
Schematic illustration of the mode of action of MERS-CoV ORF8b. HSP70 interacts with IKKε in the absence of ORF8b when infected with Δ8b virus. HSP70 serves as a bridge to recruit IKKε; as a result, more IKKε becomes phosphorylated, likely by autotransphosphorylation. With more phosphorylated IKKε, more IRF3 become phosphorylated and activated, resulting in higher expression of IFN-β (left panel). When infected with WT virus, ORF8b is expressed and sequesters HSP70 from IKKε. ORF8b also compete with IKKε for HSP70. As a result, the interaction between IKKε and HSP70 is impeded, resulting in less IKKε being phosphorylated, consequently less IRF3 being phosphorylated, and eventually lower expression of IFN-β.
Discussion
MERS-CoV encodes five accessory proteins. Four of the accessory proteins were characterized previously in terms of innate immunosuppressive activities (13, 14, 29, 30). However, the role of ORF8b has not been documented. In this study, we reported for the first time, to our knowledge, that the internal gene encoded by the same mRNA as the N protein named ORF8b (32) is a type I IFN antagonist that suppresses IFN-β expression when both ectopically expressed and during infection. Functional analysis suggested that ORF8b fulfills this role by perturbing IRF3 signaling at a level upstream of IRF3. Moreover, mass spectrometric analysis revealed that cellular factor HSP70–1A interacts specifically with ORF8b. HSP70–1A was further demonstrated to be involved in IKKε-mediated or MERS-CoV–induced IFN-β expression. Mechanistically, ORF8b was shown to compete with IKKε for HSP70–1A binding to achieve its immunosuppressive effects without altering the expression level or localization pattern of IKKε and HSP70–1A. Our findings are summarized in a schematic diagram in Fig. 10. Because suppression of IFN response is also seen in COVID-19 caused by SARS-CoV-2 (1–3), it will be of interest to see whether any SARS-CoV-2 proteins might also target HSP70–1A and IKKε.
ORF8b has also been identified in two recent papers on IFN antagonism of MERS-CoV proteins based completely on luciferase reporter assays conducted in HEK293T cells (55, 56). Generally consistent with our finding, ORF8b suppresses IFN-β promoter activity in those studies. However, ORF8b has no influence on the activation of IFN-β promoter by MAVS, TBK1, IKKε, or IRF3 but exerts an inhibitory effect on TBK1- and MDA5-induced activation of κB-Luc (56, 57). Their results are apparently at odds with our findings. Notably, the activity patterns of ORF4a and ORF4b on IFN-β-Luc and κB-Luc reported in those two papers are also inconsistent with previous finding from us and other groups (13–15, 29, 30, 58). We do not understand whether the expression of SV40 T Ag in the cells used in those studies might make a difference. Clarification of the discrepancies requires analysis in more cell lines, with the same reagents and with more methods.
The presence of an internal gene overlapping with the N segment is a unique feature observed in betacoronaviruses such as MHV, HKU1, SARS-CoV, and MERS-CoV (9, 31). When compared with its counterparts in the most closely related bat coronaviruses HKU4 and HKU5, MERS-CoV ORF8b is apparently a truncated version in which the start codon is shifted downstream with respect to that in ORF8b of HKU4 and HKU5 because of a single nucleotide difference. Sequence conservation immediately downstream of the start codon is observed among the ORF8b proteins of the three viruses (9). However, MERS-CoV ORF8b likely acquires an early stop codon, resulting in a shorter sequence (9). This suggests that MERS-CoV may have evolved to encode an ORF8b with a function beneficial to the virus under selection pressure. The downstream shift of the start codon and the truncation at the 3′-end might alter protein function. MERS-CoV might not require all amino acid residues of the ORF8b proteins of HKU4 and HKU5 for infection of a human host. Further analysis along this line may derive new understanding of how MERS-CoV crossed the species barrier to become a virulent infectious agent of humans. In particular, it will be of great interest to compare the IFN-antagonizing properties of the ORF8b proteins of the three viruses side by side.
MERS-CoV is known to encode multiple proteins to evade host innate immune surveillance (12–15). The reason for MERS-CoV to encode several proteins to achieve the same goal with redundancy is largely unknown. One possible explanation is that MERS-CoV is extremely sensitive to IFN so that multiple viral proteins are required to suppress IFN production and signaling at different steps so that the induction and function of IFN are kept minimal upon infection. We found that the Δ8b virus induces IFN transcription more robustly, but its replication kinetics remained unchanged compared with that of the WT virus. Plausibly, additional IFN-antagonizing proteins in the Δ8b virus could still suppress IFN signaling effectively so that the antiviral activity of ISGs is not seen. The peak of ORF8b expression occurred during 24–48 hpi, and ORF8b almost disappeared at 72 hpi. However, no difference in IFN-β induction by WT and Δ8b viruses was seen at 24 hpi despite the abundance of ORF8b in infected cells. Whether additional viral or host factors, which might not be induced until 48 hpi, are required for optimal IFN antagonism of ORF8b remains to be clarified. Alternatively, the discrepancies between ORF8b and IFN-β expression at 24 and 48 hpi might also be explained by the time lag for the suppressive effect of ORF8b to be seen and to disappear. Whether IFN antagonism of ORF8b might operate within a specific window requires further investigations.
Several studies have suggested the administration of exogenous IFN as a potential treatment for MERS-CoV infection (21, 24). Full evaluation of this idea requires a better understanding of the relative importance of all IFN-antagonizing viral proteins encoded by MERS-CoV. Among all the viral proteins reported to inhibit innate immunity, MERS-CoV ORF4b has been studied most extensively, and it is capable of perturbing multiple innate immune signaling pathways (14, 15, 29, 30). The other IFN-antagonizing viral proteins of MERS-CoV have not been well studied. Further characterization of the IFN antagonism of these proteins as well as clarification of the influence of MERS-CoV ORF8b on other cell signaling pathways and its cooperation with other IFN-antagonizing viral proteins are warranted.
HSPs are molecular chaperones involved in protein quality control. HSPs generally assist the folding of nonnative proteins by preventing aggregation, promoting native state folding, and refolding aggregated proteins by solubilization (53). The expression of HSP70 is regulated by heat shock factor, which is activated at stressed conditions (59). Apart from heat shock factor, HSP expression can also be induced by IL-6 and non-HSP transcription factors such as STAT1 and STAT3 (59, 60). In addition to this link between HSPs and innate immune signaling, HSPs are also involved in other aspects of innate immune regulation (35, 36). In this study, we demonstrated that HSP70–1A is important for IKKε phosphorylation, which is essential for IFN-β production. However, it is not clear exactly how HSP70 achieves this. One postulation is that HSP70–1A serves to recruit IKKε and stabilize its self-association, thus allowing optimal activation by autotransphosphorylation. We noted that only a fraction of HSP70–1A was bound to ORF8b, yet overexpression of HSP70–1A did make a difference in supporting IKKε activation. Plausibly, there is a threshold for IKKε-bound HSP70–1A that governs the activation of IKKε kinase. The addition of extra HSP70–1A to the existing pool might therefore tip the balance toward IKKε activation. Introduction of MERS-CoV ORF8b might sequester HSP70–1A from IKKε. The mode of action of HSP70–1A on IKKε requires further investigation. HSP70 performs its function by working closely with cochaperones J-domain proteins (61), but whether the innate immune function mediated by HSP70 requires cochaperones merits further investigations.
IKKε is structurally related to TBK1. Both IKKε and TBK1 are components of a large protein complex that phosphorylates and activates IRF3 (37, 62, 63). Although IKKε was originally thought to phosphorylate IκB-α (64), IRF3 but not IκB-α is its physiological substrate (54, 65). This might explain the specific suppressive effect of ORF8b on IRF3 but not NF-κB. The inhibition of TBK1-induced activation of IFN-β promoter by ORF8b (Fig. 4) was surprising because no interaction between HSP70–1A and TBK1 was observed (Fig. 5). Whether this might be explained by the cross-talk between IKKε and TBK1 (66) remains to be clarified. Our work reveals a specific requirement of HSP70–1A for full activity of IKKε (Figs. 6–8). In this regard, HSP90 has previously been shown to stabilize TBK1 (35). Plausibly, different HSPs might cooperate to support optimal activation of TBK1 and IKKε. IKKε is a multifunctional protein critically involved in many aspects of innate immune response (63, 67). In particular, IKKε is influential in IFN signaling through its phosphorylation of STAT1 (67). We showed in this study the suppression of IKKε by MERS-CoV ORF8b. It will be of great interest to see whether ORF8b might also exert a suppressive effect on other functions of IKKε and particularly its ability to activate IFN signaling.
Acknowledgments
We thank Connie Cepko, Genhong Cheng, Luis Enjuanes, Takashi Fujita, and Søren Warming for gifts of plasmids and reagents and members of Jin Laboratory for critical reading of the manuscript.
This work was supported by the Hong Kong Health and Medical Research Fund (HKM-15-M01) and Hong Kong Research Grants Council, University Grants Committee (T11-707/15-R). The funders had no role in the design and conduct of the study, in the collection, analysis, and interpretation of the data, or in the preparation, review, or approval of the manuscript.
Abbreviations used in this article:
- Δ8b
ORF8b knockout
- BAC
bacterial artificial chromosome
- COVID-19
coronavirus disease 2019
- CT
cycle threshold
- hpi
hour postinfection
- HSP
heat shock protein
- ISG
IFN-stimulated gene
- MERS-CoV
Middle East respiratory syndrome coronavirus
- MHV
mouse hepatitis virus
- MOI
multiplicity of infection
- ORF
open reading frame
- RLR
RIG-I–like receptor
- RT-qPCR
quantitative RT-PCR
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- SeV
Sendai virus
- shRNA
short hairpin RNA
- WT
wild-type
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, et al. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579: 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan JFW, Yuan S, Kok KH, To KK, Chu H, Yang J, Xing F, Liu J, Yip CC, Poon RW, et al. 2020. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet 395: 514–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fung SY, Yuen KS, Ye ZW, Chan CP, and Jin DY. 2020. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: lessons from other pathogenic viruses. Emerg. Microbes Infect. 9: 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, and Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. [Published erratum appears in 2013 N. Engl. J. Med. 369: 394.] N. Engl. J. Med. 367: 1814–1820. [DOI] [PubMed] [Google Scholar]
- 5.Oboho IK, Tomczyk SM, Al-Asmari AM, Banjar AA, Al-Mugti H, Aloraini MS, Alkhaldi KZ, Almohammadi EL, Alraddadi BM, Gerber SI, et al. 2015. 2014 MERS-CoV outbreak in Jeddah--a link to health care facilities. N. Engl. J. Med. 372: 846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hui DS, Azhar EI, Kim YJ, Memish ZA, Oh MD, and Zumla A. 2018. Middle East respiratory syndrome coronavirus: risk factors and determinants of primary, household, and nosocomial transmission. Lancet Infect. Dis. 18: e217–e227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Groot RJ, Baker SC, Baric RS, Brown CS, Drosten C, Enjuanes L, Fouchier RA, Galiano M, Gorbalenya AE, Memish ZA, et al. 2013. Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J. Virol. 87: 7790–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woo PCY, Lau SKP, Li KS, Tsang AK, and Yuen KY. 2012. Genetic relatedness of the novel human group C betacoronavirus to Tylonycteris bat coronavirus HKU4 and Pipistrellus bat coronavirus HKU5. Emerg. Microbes Infect. 1: e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus ADME, Haagmans BL, Gorbalenya AE, Snijder EJ, and Fouchier RAM. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3: e00473–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durai P, Batool M, Shah M, and Choi S. 2015. Middle East respiratory syndrome coronavirus: transmission, virology and therapeutic targeting to aid in outbreak control. Exp. Mol. Med. 47: e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raj VS, Osterhaus AD, Fouchier RA, and Haagmans BL. 2014. MERS: emergence of a novel human coronavirus. Curr. Opin. Virol. 5: 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lui PY, Wong LYR, Fung CL, Siu KL, Yeung ML, Yuen KS, Chan CP, Woo PCY, Yuen KY, and Jin DY. 2016. Middle East respiratory syndrome coronavirus M protein suppresses type I interferon expression through the inhibition of TBK1-dependent phosphorylation of IRF3. Emerg. Microbes Infect. 5: e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niemeyer D, Zillinger T, Muth D, Zielecki F, Horvath G, Suliman T, Barchet W, Weber F, Drosten C, and Mu MA¨ller. 2013. Middle East respiratory syndrome coronavirus accessory protein 4a is a type I interferon antagonist. J. Virol. 87: 12489–12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Y, Ye F, Zhu N, Wang W, Deng Y, Zhao Z, and Tan W. 2015. Middle East respiratory syndrome coronavirus ORF4b protein inhibits type I interferon production through both cytoplasmic and nuclear targets. Sci. Rep. 5: 17554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Zhang L, Geng H, Deng Y, Huang B, Guo Y, Zhao Z, and Tan W. 2013. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell 4: 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mielech AM, Kilianski A, Baez-Santos YM, Mesecar AD, and Baker SC. 2014. MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology 450–451: 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menachery VD, Mitchell HD, Cockrell AS, Gralinski LE, Yount BL Jr., Graham RL, McAnarney ET, Douglas MG, Scobey T, Beall A, et al. 2017. MERS-CoV accessory ORFs play key role for infection and pathogenesis. mBio 8: e00665–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samuel CE 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14: 778–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stetson DB, and Medzhitov R. 2006. Type I interferons in host defense. Immunity 25: 373–381. [DOI] [PubMed] [Google Scholar]
- 20.Chan RW, Chan MC, Agnihothram S, Chan LL, Kuok DI, Fong JH, Guan Y, Poon LLM, Baric RS, Nicholls JM, and Peiris JSM. 2013. Tropism of and innate immune responses to the novel human betacoronavirus lineage C virus in human ex vivo respiratory organ cultures. J. Virol. 87: 6604–6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan JFW, Yao Y, Yeung ML, Deng W, Bao L, Jia L, Li F, Xiao C, Gao H, Yu P, et al. 2015. Treatment with Lopinavir/Ritonavir or interferon-β1b improves outcome of MERS-CoV infection in a nonhuman primate model of common marmoset. J. Infect. Dis. 212: 1904–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faure E, Poissy J, Goffard A, Fournier C, Kipnis E, Titecat M, Bortolotti P, Martinez L, Dubucquoi S, Dessein R, et al. 2014. Distinct immune response in two MERS-CoV-infected patients: can we go from bench to bedside? PLoS One 9: e88716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kindler E, Jónsdóttir HR, Muth D, Hamming OJ, Hartmann R, Rodriguez R, Geffers R, Fouchier RAM, Drosten C, Müller MA, et al. 2013. Efficient replication of the novel human betacoronavirus EMC on primary human epithelium highlights its zoonotic potential. mBio 4: e00611–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shalhoub S, Farahat F, Al-Jiffri A, Simhairi R, Shamma O, Siddiqi N, and Mushtaq A. 2015. IFN-α2a or IFN-β1a in combination with ribavirin to treat Middle East respiratory syndrome coronavirus pneumonia: a retrospective study. J. Antimicrob. Chemother. 70: 2129–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zielecki F, Weber M, Eickmann M, Spiegelberg L, Zaki AM, Matrosovich M, Becker S, and Weber F. 2013. Human cell tropism and innate immune system interactions of human respiratory coronavirus EMC compared to those of severe acute respiratory syndrome coronavirus. J. Virol. 87: 5300–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raj VS, Mou H, Smits SL, Dekkers DH, Müler MA, Dijkman R, Muth D, Demmers JA, Zaki A, Fouchier RA, et al. 2013. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495: 251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu G, Hu Y, Wang Q, Qi J, Gao F, Li Y, Zhang Y, Zhang W, Yuan Y, Bao J, et al. 2013. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 500: 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, and Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5: 730–737. [DOI] [PubMed] [Google Scholar]
- 29.Canton J, Fehr AR, Fernandez-Delgado R, Gutierrez-Alvarez FJ, Sanchez-Aparicio MT, García-Sastre A, Perlman S, Enjuanes L, and Sola I. 2018. MERS-CoV 4b protein interferes with the NF-κB-dependent innate immune response during infection. PLoS Pathog. 14: e1006838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thornbrough JM, Jha BK, Yount B, Goldstein SA, Li Y, Elliott R, Sims AC, Baric RS, Silverman RH, and Weiss SR. 2016. Middle East respiratory syndrome coronavirus NS4b protein inhibits host RNase L activation. mBio 7: e00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fehr AR, and Perlman S. 2015. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 1282: 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corman VM, Ithete NL, Richards LR, Schoeman MC, Preiser W,Drosten C, and Drexler JF. 2014. Rooting the phylogenetic tree of middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. J. Virol. 88: 11297–11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fischer F, Peng D, Hingley ST, Weiss SR, and Masters PS. 1997. The internal open reading frame within the nucleocapsid gene of mouse hepatitis virus encodes a structural protein that is not essential for viral replication. J. Virol. 71: 996–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi CS, Qi HY, Boularan C, Huang NN, Abu-Asab M, Shelhamer JH,and Kehrl JH. 2014. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 193: 3080–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang K, Shi H, Qi R, Sun S, Tang Y, Zhang B, and Wang C. 2006. Hsp90 regulates activation of interferon regulatory factor 3 and TBK-1 stabilization in Sendai virus-infected cells. Mol. Biol. Cell 17: 1461–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim MY, Shu Y, Carsillo T, Zhang J, Yu L, Peterson C, Longhi S,Girod S, Niewiesk S, and Oglesbee M. 2013. hsp70 and a novel axis of type I interferon-dependent antiviral immunity in the measles virus-infected brain. J. Virol. 87: 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, and Maniatis T. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4: 491–496. [DOI] [PubMed] [Google Scholar]
- 38.Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G,Sun R, Haberland M, Modlin R, and Cheng G. 2002. IRF3 mediates a TLR3/ TLR4-specific antiviral gene program. Immunity 17: 251–263. [DOI] [PubMed] [Google Scholar]
- 39.Guo B, and Cheng G. 2007. Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J. Biol. Chem. 282: 11817–11826. [DOI] [PubMed] [Google Scholar]
- 40.Siu KL, Kok KH, Ng MH, Poon VK, Yuen KY, Zheng BJ, and Jin DY. 2009. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J. Biol. Chem. 284: 16202–16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ng MH, Ho TH, Kok KH, Siu KL, Li J, and Jin DY. 2011. MIP-T3 is a negative regulator of innate type I IFN response. J. Immunol. 187: 6473–6482. [DOI] [PubMed] [Google Scholar]
- 42.Kok KH, Lui PY, Ng MH, Siu KL, Au SWN, and Jin DY. 2011. Thedouble-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 9: 299–309. [DOI] [PubMed] [Google Scholar]
- 43.Chaudhary V, Zhang S, Yuen KS, Li C, Lui PY, Fung SY, Wang PH,Chan CP, Li D, Kok KH, et al. 2015. Suppression of type I and type III IFN signalling by NSs protein of severe fever with thrombocytopenia syndrome virus through inhibition of STAT1 phosphorylation and activation. J. Gen. Virol. 96: 3204–3211. [DOI] [PubMed] [Google Scholar]
- 44.Almazán F, Márquez-Jurado S, Nogales A, and Enjuanes L. 2015. Engineering infectious cDNAs of coronavirus as bacterial artificial chromosomes. Methods Mol. Biol. 1282: 135–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsuda T, and Cepko CL. 2004. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. USA 101: 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan S, Chu H, Chan JFW, Ye ZW, Wen L, Yan B, Lai PM, Tee KM,Huang J, Chen D, et al. 2019. SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat. Commun. 10: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thanos D, and Maniatis T. 1995. Virus induction of human IFN b gene expression requires the assembly of an enhanceosome. Cell 83: 1091–1100. [DOI] [PubMed] [Google Scholar]
- 48.Seth RB, Sun L, Ea CK, and Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122: 669–682. [DOI] [PubMed] [Google Scholar]
- 49.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, and Akira S. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6: 981–988. [DOI] [PubMed] [Google Scholar]
- 50.Panne D, McWhirter SM, Maniatis T, and Harrison SC. 2007. Interferon regulatory factor 3 is regulated by a dual phosphorylation-dependent switch. J. Biol. Chem. 282: 22816–22822. [DOI] [PubMed] [Google Scholar]
- 51.Emeny JM, and Morgan MJ. 1979. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J. Gen. Virol. 43: 247–252. [DOI] [PubMed] [Google Scholar]
- 52.Yamaoka Y, Kudo T, Lu H, Casola A, Brasier AR, and Graham DY. 2004. Role of interferon-stimulated responsive element-like element in interleukin-8 promoter in Helicobacter pylori infection. Gastroenterology 126: 1030–1043. [DOI] [PubMed] [Google Scholar]
- 53.Mayer MP, and Bukau B. 2005. Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62: 670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kishore N, Huynh QK, Mathialagan S, Hall T, Rouw S, Creely D,Lange G, Caroll J, Reitz B, Donnelly A, et al. 2002. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J. Biol. Chem. 277: 13840–13847. [DOI] [PubMed] [Google Scholar]
- 55.Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW,Bowman KK, Starovasnik MA, and Dueber EC. 2012. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc. Natl. Acad. Sci. USA 109: 9378–9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee JY, Bae S, and Myoung J. 2019. Middle East respiratory syndrome coronavirus-encoded ORF8b strongly antagonizes IFN-β promoter activation: its implication for vaccine design. J. Microbiol. 57: 803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee JY, Bae S, and Myoung J. 2019. Middle East respiratory syndrome coronavirus-encoded accessory proteins impair MDA5- and TBK1-mediated activation of NF-kB. J. Microbiol. Biotechnol. 29: 1316–1323. [DOI] [PubMed] [Google Scholar]
- 58.Siu KL, Yeung ML, Kok KH, Yuen KS, Kew C, Lui PY, Chan CP,Tse H, Woo PC, Yuen KY, and Jin DY. 2014. Middle east respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J. Virol. 88: 4866–4876. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Stephanou A, and Latchman DS. 2011. Transcriptional modulation of heatshock protein gene expression. Biochem. Res. Int. 2011: 238601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stephanou A, Isenberg DA, Nakajima K, and Latchman DS. 1999. Signal transducer and activator of transcription-1 and heat shock factor-1 interact and activate the transcription of the Hsp-70 and Hsp-90b gene promoters. J. Biol. Chem. 274: 1723–1728. [DOI] [PubMed] [Google Scholar]
- 61.Sima S, and Richter K. 2018. Regulation of the Hsp90 system. Biochim. Biophys. Acta Mol. Cell Res. 1865: 889–897. [DOI] [PubMed] [Google Scholar]
- 62.Perry AK, Chow EK, Goodnough JB, Yeh WC, and Cheng G. 2004. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J. Exp. Med. 199: 1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang J, Tian M, Xia Z, and Feng P. 2016. Roles of IκB kinase ε in the innate immune defense and beyond. Virol. Sin. 31: 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peters RT, Liao SM, and Maniatis T. 2000. IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol. Cell 5: 513–522. [DOI] [PubMed] [Google Scholar]
- 65.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, and Hiscott J.2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300: 1148–1151. [DOI] [PubMed] [Google Scholar]
- 66.Clark K, Takeuchi O, Akira S, and Cohen P. 2011. The TRAF-associated protein TANK facilitates cross-talk within the IkappaB kinase family during toll-like receptor signaling. Proc. Natl. Acad. Sci. USA 108: 17093–17098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tenoever BR, Ng SL, Chua MA, McWhirter SM, García-Sastre A, and Maniatis T. 2007. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science 315: 1274–1278. [DOI] [PubMed] [Google Scholar]