

ABSTRACT
Escherichia coli is one of the most frequent human pathogens, increasingly exhibits antimicrobial resistance, and has complex interactions with the host immune system. E. coli exposure or infection can result in the generation of antibodies specific for outer membrane protein A (OmpA), a multifunctional porin. We identified four OmpA-specific naturally occurring antibodies from healthy human donor B cells and assessed their interactions with E. coli and OmpA. These antibodies are highly specific for OmpA, exhibiting no cross-reactivity to a strain lacking ompA and retaining binding to both laboratory and clinical isolates of E. coli in enzyme-linked immunosorbent assay (ELISA) and immunofluorescence assays. One monoclonal antibody (Mab), designated ECOL-11, is specific for the extracellular N-terminal porin domain of OmpA and induces growth phase-specific bacterial aggregation. This aggregation is not induced by the fragment antigen binding (Fab) form of the MAb, suggesting the importance of bivalency for this aggregating activity. ECOL-11 decreases adhesion and phagocytosis of E. coli by RAW 264.7 macrophage-like cells, possibly by inhibiting the adhesion functions of OmpA. Despite this in vitro phenotype, organ E. coli burdens were not altered by antibody prophylaxis in a murine model of lethal E. coli septic shock. Our findings support the importance of OmpA at the host-pathogen interface and begin to explore the implications and utility of E. coli-specific antibodies in human hosts.
KEYWORDS: Escherichia coli, aggregation, monoclonal antibodies, outer membrane proteins, phagocytosis
INTRODUCTION
Escherichia coli is a versatile Gram-negative bacterial organism that can be a human enteric commensal, a widely used laboratory reagent, and in some settings a dangerous human pathogen. The latter of these is especially concerning because pathogenic E. coli frequently exhibits antimicrobial resistance (AMR), even to the most advanced antibiotics in clinical use. Carbapenem-resistant or extended-spectrum beta lactamase (ESBL)-producing E. coli causes over 200,000 infections and more than 10,000 deaths annually in the United States (1). These infections lead to extensive morbidity, mortality, and more than 1 billion USD of excess health care costs annually (2). Treating E. coli infections and stopping the spread of AMR bacteria require a multipronged approach involving public health containment, clinical care improvement, and novel therapeutic strategies.
The utility of monoclonal antibodies (MAbs) as an antibacterial strategy has only recently begun to be systematically evaluated, and yet, numerous groups have espoused the promise of this class of therapeutics (3–7). Although interest in antibacterial MAbs has recently increased, several MAbs have been tested in clinical trials with various degrees of success, including MEDI4983 (8), 514G3 (9), and AR301 (10) against Staphylococcus aureus and MEDI3902 (11, 12) against Pseudomonas aeruginosa. MAbs to E. coli have not been evaluated extensively, although previous work identified an outer membrane porin, likely outer membrane protein A (OmpA), as a dominant antibody target following E. coli infection (13–17). More recently, a murine MAb, 49.4-15, was identified that is specific for E. coli OmpA (18). Multiple studies of OmpA protein variants as vaccine candidates have also demonstrated the development of OmpA-specific humoral immunity in immunized animals, although the usefulness of OmpA as a vaccine may be limited by the high level of conservation of ompA across commensal bacteria (19–22). It is evident that antibodies to E. coli are induced in humans after infection, and the murine antibody studies suggest these human antibodies may impact the virulence or pathogenesis of E. coli infection by targeting OmpA.
In this study, we identified and evaluated naturally occurring human MAbs that specifically bind to OmpA. We investigated the binding of these antibodies to protein and intact bacteria under various growth conditions and determined functional properties of MAb-OmpA binding upon E. coli pathogenesis. We assessed the characteristics of this MAb against K-12 MG1655 and the well-characterized urinary tract isolate UTI89. Urinary tract infections represent a large burden of disease caused by E. coli, and OmpA has been reported previously as important for E. coli uropathogenesis (23, 24). This work provides a foundation for experiments to obtain a better understanding of the interaction of E. coli outer membrane proteins and the human immune system.
RESULTS
Isolation of human MAbs that bind to OmpA.
Peripheral blood samples were collected after written informed consent was obtained from healthy laboratory personnel working in microbiology research laboratories. Peripheral blood mononuclear cells (PBMCs) were isolated by gradient fractionation and used to make human B cell hybridoma cell lines secreting naturally occurring MAbs that bound to E. coli outer membrane fractions by ELISA. Most MAbs were found to be of the IgG1 isotype, and the antibody variable gene sequences were obtained and used to produce recombinant IgG1 proteins. If the native isotype was not IgG1, we recombinantly produced and tested both the native isotype IgG as secreted by the hybridoma cell line and the IgG that was isotype switched to IgG1. We also produced recombinant LALA-PG Fc region-variant IgG proteins (which do not bind to mouse nor human FcγR [25]), fragment antigen binding (Fab) variants (which only contain a single binding site instead of the paired binding arms of IgG MAbs), and rSTAU-228 IgG1 (a negative-control antibody specific for Staphylococcus aureus IsdA [26]). To determine the antigen specificity of these MAbs, they were assessed for binding to bacterial lysates of several strains of E. coli, including strains in which the genes encoding the most common outer membrane proteins (ompA, ompC, ompF, and lamB) were inactivated (27). Four MAbs bound to BL21(DE3) wild type (WT) but not to BL21(DE3) ΔompA, indicating specificity for the gene product OmpA (Fig. 1A). Furthermore, when we tested binding to a recombinant partial OmpA protein that includes only the C-terminal periplasmic domain, three MAbs (rECOL-2, rECOL-3, and rECOL-4) bound, but one (rECOL-11) did not. This result suggests that three of four MAbs are specific for the C-terminal periplasmic domain of OmpA (OmpACTerm) and that one of four MAbs is specific for the N-terminal porin domain (OmpANTerm). All MAbs retain binding to lysates from both the laboratory strain E. coli K-12 and the clinical isolate E. coli UTI89 (Fig. 1A). This degree of conserved binding is not surprising, as OmpA is generally highly conserved in sequence. Most E. coli strains exhibit >90% OmpA identity, and the three strains we tested differ only at three amino acid positions (Fig. 1B).
FIG 1.

rECOL MAbs bind to the conserved outer membrane protein (OmpA). (A) Enzyme-linked immunosorbent assays (ELISA) of recombinant monoclonal antibody binding to E. coli lysate [BL21(DE3), K-12, and UTI89] or recombinant protein (rOmpACTerm). Points represent arithmetic mean, error bars represent standard deviation, curves indicate four-parameter variable slope logistic analysis, and data are representative of three independent experiments performed in quadruplicate. (B) Clustal Omega alignment of OmpA from the strains tested by ELISA, namely, an engineered E. coli (BL21), a laboratory or commensal strain (K-12 MG1655), and a clinical uropathogenic strain (UTI89), shows 99% identity (black, conserved; white, variable). Orange indicates the N-terminal porin domain with secondary structure above mapped from PDB 1BXW using ESPript 3; green indicates the C-terminal periplasmic domain with secondary structure below mapped from PDB 2MQE.
OmpANTerm MAb ECOL-11 binds whole E. coli.
Having demonstrated the binding of four MAbs to OmpA in bacterial lysates, we next sought to examine the capacity of these MAbs to bind intact whole E. coli. Using bacteria cultured to mid-exponential phase or late stationary phase, we performed immunofluorescence imaging using human MAbs. OmpACTerm MAbs, such as rECOL-4, did not stain E. coli of any growth phase. However, the OmpANTerm MAb rECOL-11 stained exponential-phase K-12 E. coli (Fig. 2A) and strongly stained stationary-phase K-12 E. coli (Fig. 2B). Binding to uropathogenic isolate UTI89 was also evident, but the level of binding was inconsistent (Fig. 2C). To evaluate potential causes of differential binding, we assessed the expression of OmpA across multiple time points. mRNA transcripts for OmpA were assessed by reverse transcriptase quantitative PCR (RT-qPCR) and found to be more abundant in exponential-phase E. coli than in stationary-phase E. coli, consistent with previously reported differential ompA mRNA stability during growth (Fig. 3A) (28). OmpA protein levels were assessed at multiple time points during growth by immunoblot. While slight differences were noted between stationary-phase E. coli and exponential-phase E. coli, these differences were small, and OmpA levels in both K-12 (Fig. 3B) and UTI89 (Fig. 3C) E. coli were relatively consistent across all time points. We observed two bands stained by rECOL-4 IgG1, and based upon relative molecular weight and previously reported studies of OmpA, we predict the 37-kDa band is consistent with full-length OmpA and the 25-kDa band is consistent with a partially denatured protein or isolated C-terminal domain. Neither ompA mRNA nor OmpA protein levels explained the minimal binding of ECOL-11 to intact UTI89 by immunofluorescence. Previous literature has reported that polysaccharide capsules may impair the binding of antibodies to bacteria, such as Acinetobacter baumannii (29), and the K1 capsule of UTI89 has been found to be an important virulence factor (30, 31). We performed Maneval’s capsule staining of K-12 and UTI89 E. coli to determine whether greater capsule production might explain the decreased binding of ECOL-11 to UTI89. Our data show that UTI89 exhibits a thicker capsular polysaccharide layer than E. coli K-12 (Fig. 3D and E), and we hypothesize that this finding explains the differential ECOL-11 binding observed by immunofluorescence.
FIG 2.
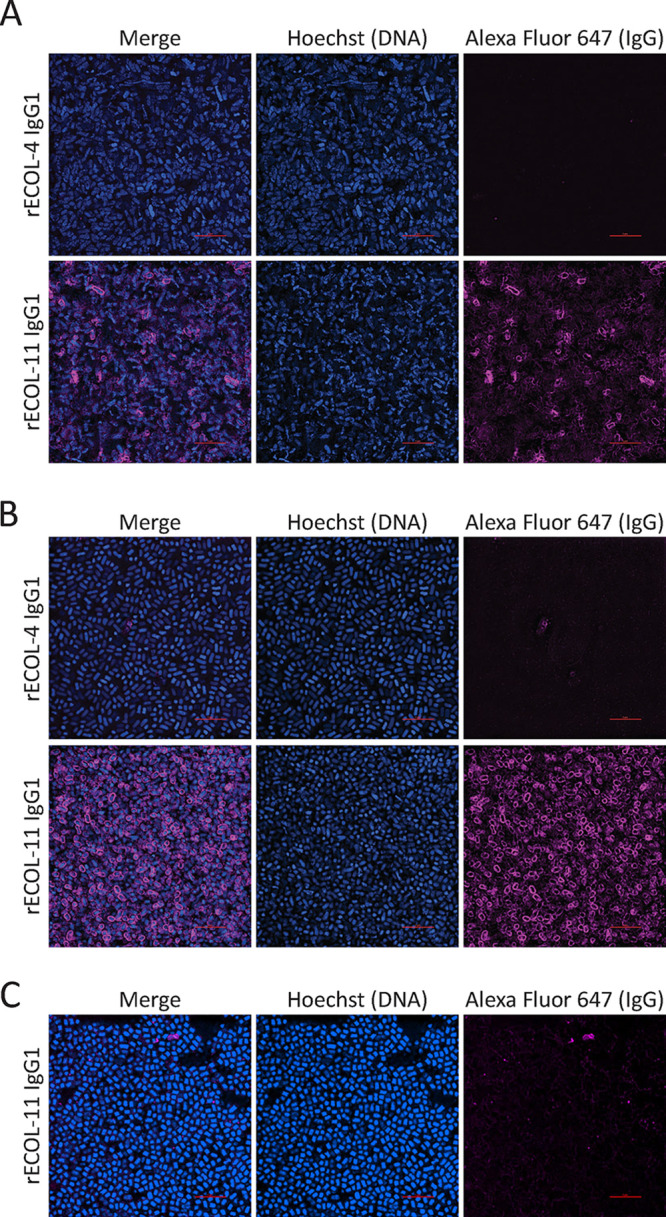
Immunofluorescence staining of exponential (A) or stationary-phase K-12 E. coli (B) or stationary-phase UTI89 E. coli (C) with Hoechst (blue), recombinant MAb, and mouse anti-human IgG Alexa Fluor 647 (magenta). Image locations were randomly selected from slides, gathered with identical image and laser settings using a 100× oil objective, and processed with identical look-up tables. Red scale bars are 5 μm.
FIG 3.
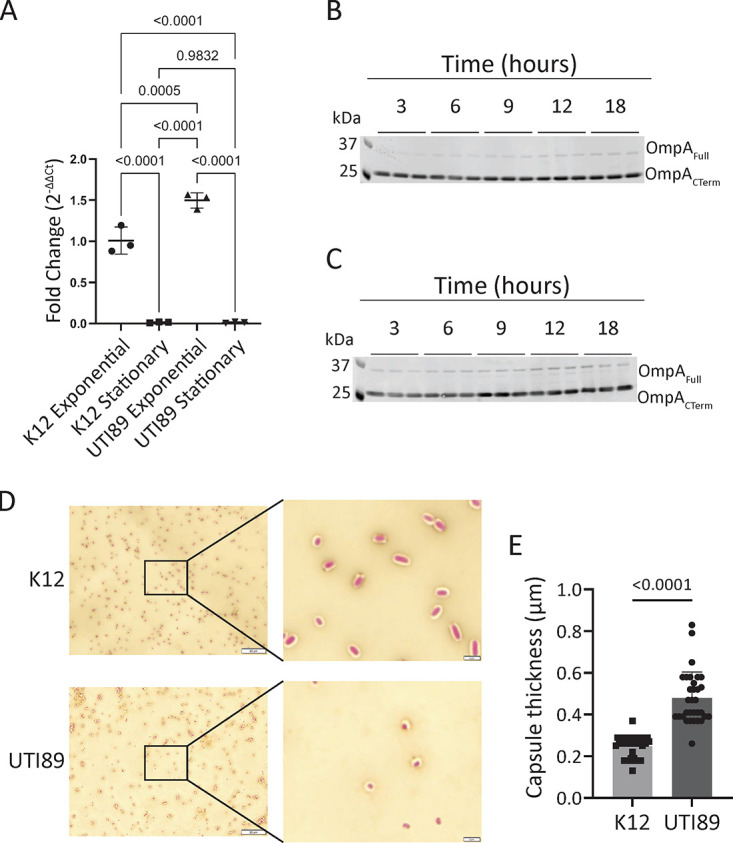
Neither ompA mRNA transcript levels nor OmpA protein expression levels explain binding differences between K-12 and UTI89 E. coli, but UTI89 produces more capsule. (A) Transcript-level expression of ompA by RT-qPCR normalized to 16S housekeeping gene expression relative to exponential-phase K-12 E. coli. RT-qPCR was analyzed by one-way ANOVA. Error bars show standard deviation. Data are representative of two independent experiments. Protein level expression of OmpA by immunoblot to K-12 (B) or UTI89 (C) E. coli. Two bands representing two forms of OmpA are observed; based upon molecular weight, they are predicted to be two forms of OmpA. Data are representative of three independent experiments. (D) Maneval’s capsule staining of K-12 or UTI89. Background is counterstained with Congo red, and cells are stained with Maneval’s solution. Images were taken with a 100× objective, the primary image scale bar is 20 μm, and the inset scale bar is 2 μm. (E) The zone of stain exclusion by capsule was measured from eight cells in three independent replicates. Data were pooled, each data point represents one cell, data were analyzed by Mann-Whitney U test, and error bars show standard deviation.
Bivalent IgG aggregates stationary-phase E. coli.
Murine MAbs to E. coli have been demonstrated previously to induce aggregation or clumping of E. coli (32). We incubated IgG1 or fragment antigen binding (Fab) forms of MAbs with exponential- or stationary-phase K-12 or UTI89 E. coli and imaged bacterial pellet morphology after allowing the bacteria to settle. Aggregation, or cross-linking, of bacteria prevents the cells from settling. The images shown in Fig. 4A reveal that rECOL-11 IgG1 causes stationary-phase K-12 E. coli to aggregate, but the rECOL-11 Fab does not, indicating that bivalency is essential for aggregation to occur. rECOL-11 IgG1 does not cause aggregation of UTI89 E. coli, likely due to decreased MAb binding to this strain as described in Fig. 2. Furthermore, when bacteria are imaged immediately after the addition of antibody, rECOL-11 IgG1 causes the formation of visible clumps that are absent in bacteria treated with rECOL-4 IgG1 or other OmpACTerm-specific MAbs (Fig. 4B and 4C). This observation shows that aggregation depends upon the accessibility of the antigen on the bacterial surface and could be used as an assay to quickly assess antibodies for binding to clinical isolates in an antigen-independent screen.
FIG 4.
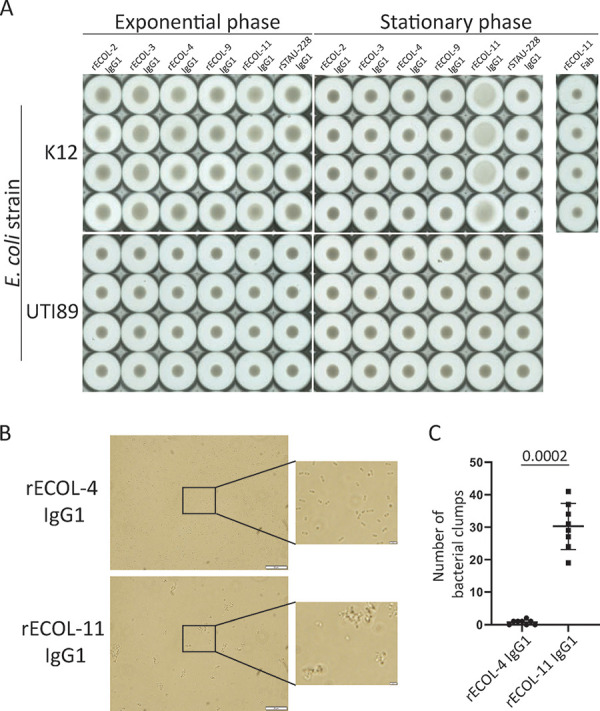
Aggregation of exponential or stationary-phase K-12 E. coli by IgG1 or Fab. (A) Aggregation was imaged on a Cellular Technology Limited (CTL) Immunospot device to visualize bacterial pellets. Aggregation of bacteria prevents the formation of a denser pellet. (B) Aggregation of stationary-phase K-12 E. coli by brightfield microscopy. (C) Number of clumps (five or more bacteria in direct contact) per 100× objective field from brightfield microscopy (n = 8 random images per condition) were analyzed by Mann-Whitney U test. Data are representative of two independent experiments, and error bars show standard deviation.
MAb treatment of E. coli alters macrophage phagocytosis in vitro.
Aggregation has been associated with increased opsonophagocytosis (33). We used a temperature-dependent model with RAW264.7 macrophage-like cells to test opsonophagocytosis in vitro. Opsonized K-12 E. coli was incubated with cells at 4°C to allow adhesion but prevent phagocytosis or at 37°C to allow adhesion and phagocytosis. Cells were then rinsed, lysed, and plated to count adhered or phagocytosed bacteria. Opsonization of E. coli with rECOL-11 expressed in the format of an IgG1, IgG3, IgG1-LALA-PG Fc variant, or Fab resulted in decreased adhesion to and phagocytosis by RAW 264.7 cells (Fig. 5). Interestingly, this decrease persisted despite eliminating Fcγ receptor (FcγR) interactions with a LALA-PG variant IgG and persisted despite eliminating aggregatory activity with a Fab. To explain this phenotype, we propose that under the tested conditions, OmpA is an important adhesion factor and rECOL-11 binding inhibits this adhesion. This finding is consistent with prior work indicating that OmpA is important for adhesion or entry to eukaryotic cells (18, 23, 34). This interaction complicates an in vitro assessment of opsonophagocytosis.
FIG 5.
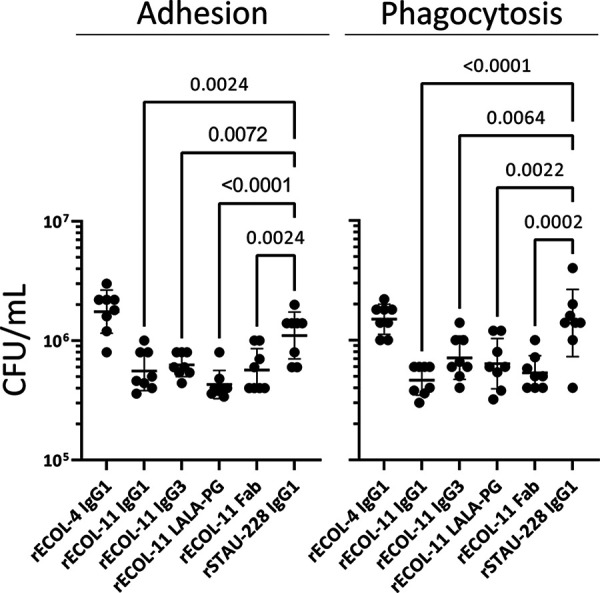
rECOL-11 opsonization alters K-12 E. coli adhesion to and phagocytosis by RAW264.7 macrophage-like cells. Adhesion is the bacterial count after coincubating opsonized bacteria and RAW 264.7 cells at 4°C, and phagocytosis is the bacterial count after coincubating at 37°C. Data representative of three independent experiments were analyzed in comparison to rSTAU-228 IgG1 by one-way ANOVA. Comparisons where P values of <0.01 are shown, and error bars show standard deviation.
MAb treatment does not reduce bacterial burdens in vivo.
To better understand the interactions of OmpA MAbs, E. coli, and the mammalian immune system, we adapted a systemic infection model of E. coli in mice. Mice were pretreated with MAb and inoculated with UTI89 E. coli, and then organ bacterial burdens were enumerated at 24 h postinoculation. If the disease was allowed to progress, mice reached terminal endpoints due to septic shock at around 36 h postinoculation. When pretreated with 10 mg of MAb per kg of body weight, organ bacterial burdens at 24 h postinoculation did not differ significantly between treatments using any of the OmpA-specific MAbs compared with a control antibody (the similarly prepared human IgG rSTAU-228, directed to a Staphylococcus aureus protein [26]) (Fig. 6A). Increasing the dose of MAb to 25 mg of MAb per kg of body weight also did not have an impact upon bacterial burdens in this stringent model of E. coli disease (Fig. 6B). Thus, we concluded that despite altering phagocytosis, MAb treatment was not sufficient to alter acutely lethal systemic E. coli disease in mice.
FIG 6.

In vivo assessment of MAb prophylaxis in a lethal systemic model of E. coli disease. (A and B) Six- to 8-week-old female BALB/cJ mice were pretreated with 10 mg/kg (A) or 25 mg/kg (B) MAb, then inoculated with ~6.5 × 106 CFU of UTI89 E. coli, and monitored for 24 h before euthanasia and enumeration of organ bacterial burdens. Limit of detection (LoD) was 200 CFU/mL. All groups began with 5 mice, and mice that did not recover from anesthesia were excluded and account for any groups with <5 data points. Data were compared to control MAb (rSTAU-228) by Kruskal-Wallis testing with Dunn’s correction for multiple comparisons. No conditions exhibited statistically significant differences. Error bars show standard deviation.
DISCUSSION
E. coli, particularly multi-drug resistant strains, is a common human pathogen that can be challenging to prevent or treat. While a variety of factors contribute to these difficulties, one reason is the lack of the development of new antibacterial strategies, and another reason is the minimal progress toward identifying promising vaccine candidates. One possible antigen that might serve as a target of protective immunity is OmpA, a multifunctional virulence factor found almost universally in pathogenic E. coli strains and exhibiting remarkable conservation across widely divergent strains. OmpA is composed of an N-terminal porin domain that resides within the bacterial outer membrane joined by a trypsin-digestible region to a C-terminal domain that is found in the periplasmic space (35–37). Although OmpA has been studied previously as a vaccine candidate and found to elicit some degree of immunity, little has been done to study the functions and mechanisms of OmpA-specific immunity in humans. In this study, we sought to isolate and study naturally occurring antibodies obtained from healthy human donors and identified four OmpA-targeting MAbs. Most interestingly, one of these MAbs, ECOL-11, was specific for the N-terminal porin domain of OmpA, a region of the protein that is essential for a variety of OmpA-dependent effector functions (34, 38, 39). Manipulating the interactions of OmpA with host immune effectors provides insight into host-pathogen relationships and may prove to be a potent strategy to combat E. coli pathogenesis.
The utility of this strategy has already been demonstrated by using cyclic peptides to inhibit OmpA (40). We took a similar approach to assess the ability of naturally occurring MAbs binding to OmpA to alter E. coli biology. It is important to note that the MAbs we evaluated came from healthy laboratory workers with no specific history of E. coli infection, and yet, we were still able to isolate OmpA-specific, affinity-matured, class-switched antibodies. This finding may be due to the ubiquity of E. coli as a commensal organism in the intestine or could be a result of prior infection with E. coli that was not reported by the donors upon sample collection. We assessed the impact of these MAbs upon a subset of antigen-agnostic and OmpA-specific possible effector functions. Aggregation, which we observed with rECOL-11 treatment of E. coli, is an antigen-agnostic MAb function that has been reported sporadically after exposing bacteria to an antibody (32). Aggregation has various effects upon interactions between bacteria and host phagocytes. It may impede phagocytosis due to large aggregate size or irregular shape, or it may enhance phagocytosis due to increased complement fixation (41, 42). Coupling this complex effect with the known role of OmpA as an adhesin and the interactions of MAb with Fc receptors suggests that assessing phagocytosis in vitro is challenging. We observed decreased adhesion to and decreased phagocytosis of rECOL-11-treated E. coli by RAW 264.7 macrophage-like cells. This phenotype persists with a LALA-PG Fc variant IgG, suggesting it is independent of FcγR interactions, and with Fab, indicating it is not a result of aggregation as the Fab does not induce aggregate formation. Although we have not directly shown that ECOL-11 blocks the adhesin activity of OmpA, by eliminating other possible causes, our data support this model as the best explanation of this observed phenotype.
Regardless of the underlying cause, when tested for effects upon bacterial burdens in a lethal model of E. coli septic shock, these MAbs did not alter organ bacterial counts. Our study design euthanized mice well before they began to exhibit significant signs of disease and was not designed to assess survival. The statistical power of our experiments was limited by stochastic anesthesia-related mortality that reduced the sample size of some groups; however, it is unlikely we missed a significant phenotype in this severe infection model. The systemic model of E. coli septic shock that we used is highly stringent and may detect only extreme phenotypes. Other research has demonstrated the functional utility of MAbs to E. coli in highly nuanced in vivo models of microbiome interactions (32). Overall, our in vivo data did not show reduced bacterial burdens in MAb-prophylaxed septic mice. However, further evaluation of these MAbs using additional E. coli strains, greater numbers of replicates, milder disease models, or microbiome interaction models such as competitive intestinal infections are likely necessary to fully understand the interactions of antibodies with E. coli within a mammalian host.
In addition to the antibody-mediated effector functions we analyzed, OmpA has been implicated in many different aspects of E. coli biology and pathogenesis, including cell adhesion, immunomodulation, membrane stability, bacteriophage binding, and biofilm formation (34, 43–48). The extracellular loops of OmpA have been implicated directly in meningeal invasion of E. coli by association with GlcNAcβ1-4GlcNAc on endothelial cells (34, 44). OmpA also triggers dendritic cell activation in intestinal cell coculture and is responsible for binding to polarized intestinal epithelial cells (46). Taken together, these studies support the further exploration of OmpA as a potent immunogen and potential therapeutic target. Importantly, although E. coli is frequently a pathogen, it is even more commonly a commensal (49–51). In this work, we assessed some of the roles MAbs may have during pathogenesis, but the roles that human MAbs exhibit in E. coli commensalism remain largely unknown. The data presented here indicate that antibodies naturally arise in humans against OmpA and different clones bind to multiple antigenic sites on the surface of OmpA. Furthermore, MAbs that bind extracellular portions of outer membrane proteins may have effects that impact host-pathogen interactions, such as aggregation or altering phagocytosis, that are worth studying as tools to understand membrane protein biology and may yet be found to have impacts upon E. coli pathogenesis or commensalism.
Our work has also recapitulated some of the challenges of understanding membrane protein biology and antibody interactions with bacteria. We noted growth phase-dependent and strain-dependent differences in antibody binding to E. coli. We found minor differences in OmpA expression between growth phases that could explain variable binding during growth. Strain-dependent binding differences are critically important to understand in the context of vaccination and therapeutic strategy development. Our antibodies bound identically to protein isolated from disparate E. coli strains, such as UTI89 and K-12, but exhibited significantly less binding to UTI89 E. coli by immunofluorescence. OmpA was expressed in UTI89 to similar levels as that in K-12, so we concluded that additional factors must be contributing to this strain-dependent difference. We observed an increased capsule thickness of UTI89, which could explain this differential binding between clinical and laboratory isolates, or it may be due to different lipopolysaccharide structures, as has been reported previously for an antibody targeting E. coli BamA (52, 53). Elucidating the biological processes that explain sequence-independent strain-to-strain MAb binding variability will be valuable follow-up studies. rECOL-11 may provide a critical tool to enable an increased understanding of the mechanisms for strain-dependent binding of antibacterial MAbs that will guide future vaccine and therapeutic development.
MATERIALS AND METHODS
Human subjects.
Peripheral venous blood was collected at Vanderbilt University Medical Center (VUMC) with informed written consent from healthy individuals with exposure to Gram-negative pathogens through laboratory work but no known history of infection with E. coli. Heparinized peripheral blood was processed to isolate peripheral blood mononuclear cells (PMBCs) using SepMate-50 tubes (Stemcell Technologies; 85450), and PBMCs were cryopreserved and stored in the vapor phase of liquid nitrogen until used. The studies were approved by the VUMC Institutional Review Board.
Generation of human MAbs.
Human PBMCs were thawed and transformed with Epstein-Barr virus; plated in 384-well plates in the presence of CpG (5′-C-phosphate-G-3′) DNA, Chk2 inhibitor, and cyclosporine as described previously (54); and expanded into 96-well plates containing a feeder layer of irradiated human PBMCs from an unrelated donor. Then supernatants were screened by ELISA for binding to E. coli outer membrane proteins. Cells from reactive wells were electrofused with HMMA2.5 myeloma cells to generate oligoclonal hybridoma cell lines. These cell lines were cultured, stained with propidium iodide, and single-cell sorted using fluorescence-activated cell sorting with an SH800S cytometer (Sony Biotechnology). Monoclonal hybridomas were cultured, and the MAb IgG protein was purified from cell supernatants with MabSelect or Protein G affinity columns (GE Healthcare; 11003493) using fast protein liquid chromatography (FPLC) on an ÄKTA pure chromatography system (GE Healthcare). RNA was extracted from monoclonal hybridoma cells, and the antibody transcript was sequenced using 5′ rapid amplification of cDNA ends and cloned into a monocistronic expression vector by Twist Biosciences. Antibodies were expressed by transfection of these constructs into ExpiCHO cells and purified from supernatants using affinity chromatography as above for hybridoma MAbs. All data presented were generated using recombinant MAb proteins.
Bacterial strains.
E. coli strains BL21(DE3), BL21(DE3) ΔA, and BL21(DE3) ΔABCF were gifts from Jack Leo (Addgene; 102256 and 102270) (27). Strain UTI89 was a gift from Maria Hadjifrangiskou, MicroVU, and the VUMC Center for Personalized Microbiology. All wild-type bacterial strains were grown for 14 to 16 h on Miller LB agar (LBA) plates or in Miller LB broth at 37°C. Mutant bacterial strains were cultured for 24 h at 30°C in the same medium. Bacterial lysates were prepared using bacterial protein extraction reagent (BPER) with DNase and lysozyme (Thermo Fisher Scientific; 78248).
E. coli outer membrane protein isolation.
Outer membranes from E. coli were purified using previously described techniques (55, 56). Briefly, 1 L of cell pellets was lysed in buffer containing lysozyme and EDTA followed by buffer with magnesium, RNase/DNase, and EDTA-free protease inhibitor and homogenized using a Microfluidics LM20 instrument. Unlysed cells were removed by centrifugation followed by ultracentrifugation at 100,000 × g to pellet inner and outer membranes. Inner membranes were solubilized with 1% N-lauroylsarcosine (Millipore Sigma; L9150) and 10% glycerol, and afterward, outer membranes were pelleted at 100,000 × g before being solubilized in β-octyl glucoside (Anatrace; O311) or Fos-choline-12 (Anatrace; F308S). Membrane separation was verified with SDS-PAGE.
Enzyme linked immunosorbent assay (ELISA).
ELISA plates (Thermo Fisher Scientific; 3455 or 265203) were coated with 1 to 10 μg/mL E. coli outer membrane proteins or crude lysate in carbonate buffer and then blocked with nonfat milk (Bio-Rad; 1706404). Antigens were probed with cell supernatants or purified MAb followed by horseradish peroxidase (HRP)-conjugated secondary antibodies, developed with 3,3′,5,5′-tetramethylbenzidine (Thermo Fisher Scientific; 34029), and read at a 450-nm absorbance on a Biotek Synergy H1 or Powerwave HT microplate reader.
Multiple sequence alignment.
OmpA amino acid sequences were extracted from or translated from deposited genomes (BL21, GenBank accession no. ACT29660.1; K-12, NP_415477.1; UTI89, ABE06507.1). Sequences were aligned using Clustal Omega (57). Multiple sequence alignment graphics were developed using ESPript 3.0 (58) and PDB 1BXW for the N-terminal porin domain (35) and PDB 2MQE for the C-terminal periplasmic domain (36).
Immunofluorescence.
Bacterial cultures were grown overnight or subcultured and then rinsed with phosphate-buffered saline (PBS) and normalized to an optical density at 600 nm (OD600) of 1.0. Cells were stained with Hoechst 33342 (Thermo Fisher Scientific; 62249) at 5 μL/mL for 2 min, and excess was removed with a PBS wash. Bacteria then were incubated with MAb for 30 min, rinsed, and then incubated with mouse anti-human IgG Alexa Fluor 647 (SouthernBiotech; 9040-31) for 30 min. Stained bacteria were dried overnight on a number 1.5 cover glass, then fixed to glass slides with Prolong Gold Antifade (Thermo Fisher Scientific; P36930), imaged on a Nikon structured illumination microscope with an Andor iXon Ultra DU-897 EMCCD monochrome camera, and reconstructed in NIS-Elements.
Reverse transcriptase quantitative PCR.
Bacterial RNA was extracted in triplicate from exponential or stationary-phase E. coli cultured in LB using lysing matrix B tubes (MP; 6911100) and RNeasy kits (Qiagen; 74104). Carryover DNA was removed by DNase digestion (Thermo Fisher; AM1907), and 1 μg of RNA was used to synthesize cDNA (Bio-Rad; 1708891). Synthesized cDNA was diluted in nuclease-free water, and qPCR was performed on a Bio-Rad CFX96 instrument using SYBR green chemistry (Bio-Rad; 1708882) and primers as shown in Table 1. Target gene expression was normalized to 16S housekeeping gene expression by using the threshold cycle (ΔΔCT) method.
TABLE 1.
RT-qPCR primers used in this study
| Primer target | Primer sequence | Source |
|---|---|---|
| ompA | CTGGGTGGTATGGTATG | This publication |
| ompA | TAGCGATTTCAGGAGTG | This publication |
| 16S | TGATCATGGCTCAGATT | This publication |
| 16S | CAGTTTCCCAGACATTAC | This publication |
Western blot.
Overnight or subcultured bacteria were rinsed with PBS, and pellets were lysed with BPER + DNase + lysozyme. Protein content was measured using the bicinchoninic acid assay, and 2 μg of lysate was prepared with NuPAGE sample buffer and reducing agent and then heated at 70°C for 10 min before loading on 4% to 12% Bis-Tris protein gels. SDS-PAGE was run in morpholinepropanesulfonic acid (MOPS) buffer for 60 min at a constant 200 V. Gels were transferred to polyvinylidene fluoride (PVDF), then stained for total protein with Revert 700 stain (Li-Cor; 926-11015), and imaged with a Li-Cor Odyssey CLx system. Membranes were then blocked with Intercept blocker (Li-Cor; 924-70001) and probed with rECOL-4 IgG1 followed by IRDye800CW goat anti-human IgG (Li-Cor; 926-32232), and then membranes were imaged again.
Capsule staining.
A 10- to 20-μL volume of bacterial culture was added to 20 μL of 1% Congo red aqueous solution on a glass slide and allowed to air dry. The slide was then flooded with Maneval’s solution (acid fuchsin, ferric chloride, acetic acid, and phenol) for 5 min and then gently rinsed with distilled water. Slides were air-dried and then imaged with an Olympus brightfield microscope and camera. Four randomly selected regions of each slide were photographed, and the capsule thicknesses of eight individual bacteria were measured for each replicate and condition.
Aggregation assays.
Overnight or subcultured bacteria were rinsed with and resuspended in PBS to a OD600 of 1.0, and then 50 μL was transferred to U-bottom 96-well plates. MAb or Fab was added to each well and gently mixed before the plates were allowed to sit at room temperature for 16 to 24 h. Plates were then imaged on an ImmunoSpot plate reader (Cellular Technology Limited). Alternatively, 15 μL of rinsed bacteria was applied to glass slides, treated with MAb for 15 to 30 min, protected with a cover glass, and imaged with an Olympus brightfield microscope and camera. Eight randomly selected regions of each slide were photographed, and aggregated clumps of bacteria were counted.
Adhesion assays.
RAW 264.7 cells were maintained in Dulbecco’s modified Eagle medium (DMEM) with 10% heat-inactivated fetal bovine serum and 10 mM sodium pyruvate at low passages (<15 passages). Cells were cultured in 24-well plates until confluent. Overnight or subcultured bacteria were rinsed with PBS, resuspended to an OD600 of ~0.150 (~1.0 × 108 CFU/mL), and opsonized with MAb for 60 min. Cell culture medium was refreshed with medium as above except with ultra-low IgG heat-inactivated fetal bovine serum (Thermo Fisher; 16250078), and 50 μL of opsonized bacteria was added and incubated for 15 min at either 37°C to measure adhesion and phagocytosis or 4°C to measure adhesion but prevent phagocytosis. Cell layers were rinsed four times with PBS to remove nonadhered bacteria and then lysed with 0.25% sodium deoxycholate in PBS supplemented with Benzonase nuclease (Millipore Sigma; 71205-3). Cell lysates were serially diluted and plated on LB agar to enable counting of CFU.
Mouse experiments.
All animal studies were approved by and performed according to the guidelines of the VUMC Institutional Animal Care and Use Committee. A mouse septic model of E. coli disease was developed. After a 16- to 18-h overnight culture, E. coli UTI89 was subcultured 1:100 and grown to mid-exponential phase for 3 h at 37°C and 225 rpm before being pelleted and rinsed with PBS. Bacteria were normalized in PBS using OD600 to approximately 5.0 × 107 CFU (CFU)/mL and stored on ice until inoculation. Female, 6- to 8-week-old, BALB/cJ mice (The Jackson Laboratory; 000651) were weighed, and filter-sterilized MAb in PBS was administered intraperitoneally 90 min prior to inoculation at 10 mg/kg or 25 mg/kg. After treatment, mice were anesthetized by intraperitoneal injection of avertin (~400 mg/kg) and then inoculated retro-orbitally with ~5.0 × 106 CFU of E. coli UTI89. Mice were monitored for 24 h and then euthanized by CO2 inhalation. Blood was collected by cardiac puncture, and organs were collected by dissection in a sterile environment. Organs were homogenized in sterile PBS using a bead beater and then homogenates were serially diluted and spot plated in technical duplicates on LBA plates for the enumeration of CFUs.
ACKNOWLEDGMENTS
We acknowledge Maria Hadjifrangiskou, Connor Beebout, and Grace Morales for providing key reagents and advice. Additionally, Erica Armstrong, Christopher Gainza, Ryan Irving, Summer Morales, Stacey Seeback, and Andrew Trivette of the Vanderbilt Vaccine Center assisted with antibody preparation. Jessica R. Sheldon provided critical training.
B.D.F. was supported by T32GM007347, UL1 TR002243, and the Vanderbilt Initiative for Personalized Microbial Discovery and Innovation (IPMDI).
J.E.C. has served as a consultant for Luna Innovations, Merck, and GlaxoSmithKline; is a member of the Scientific Advisory Board of Meissa Vaccines; and is Founder of IDBiologics. The Crowe laboratory at Vanderbilt University Medical Center has received sponsored research agreements from Takeda Pharmaceuticals, IDBiologics, and AstraZeneca.
Footnotes
This article is a direct contribution from Eric P. Skaar, a member of the Infection and Immunity Editorial Board, who arranged for and secured reviews by Alfredo Torres, UTMB, and Matthew Mulvey, University of Utah School of Medicine.
[This article was published on 18 May 2022 with an error in Fig. 1A. Figure 1 was updated in the current version, posted on 24 May 2022.]
Contributor Information
Eric P. Skaar, Email: eric.skaar@vumc.org.
James E. Crowe, Jr., Email: james.crowe@vumc.org.
Andreas J. Bäumler, University of California, Davis
REFERENCES
- 1.CDC. 2019. Antibiotic resistance threats in the United States, 2019. US Department of Health and Human Services, Atlanta, GA. [Google Scholar]
- 2.Antimicrobial Resistance Collaborators. 2022. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399:629–655. 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DiGiandomenico A, Sellman BR. 2015. Antibacterial monoclonal antibodies: the next generation? Curr Opin Microbiol 27:78–85. 10.1016/j.mib.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 4.Grunenwald CM, Bennett MR, Skaar EP. 2018. Nonconventional therapeutics against Staphylococcus aureus. Microbiol Spectr 6:6.6.07. 10.1128/microbiolspec.GPP3-0047-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma YX, Wang CY, Li YY, Li J, Wan QQ, Chen JH, Tay FR, Niu LN. 2020. Considerations and caveats in combating ESKAPE pathogens against nosocomial infections. Adv Sci (Weinh) 7:1901872. 10.1002/advs.201901872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oleksiewicz MB, Nagy G, Nagy E. 2012. Anti-bacterial monoclonal antibodies: back to the future? Arch Biochem Biophys 526:124–131. 10.1016/j.abb.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 7.Motley MP, Fries BC. 2017. A new take on an old remedy: generating antibodies against multidrug-resistant Gram-negative bacteria in a postantibiotic world. mSphere 2:e00397-17. 10.1128/mSphere.00397-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruzin A, Wu Y, Yu L, Yu XQ, Tabor DE, Mok H, Tkaczyk C, Jensen K, Bellamy T, Roskos L, Esser MT, Jafri HS. 2018. Characterisation of anti-alpha toxin antibody levels and colonisation status after administration of an investigational human monoclonal antibody, MEDI4893, against Staphylococcus aureus alpha toxin. Clin Transl Immunology 7:e1009. 10.1002/cti2.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varshney AK, Kuzmicheva GA, Lin J, Sunley KM, Bowling RA, Jr, Kwan TY, Mays HR, Rambhadran A, Zhang Y, Martin RL, Cavalier MC, Simard J, Shivaswamy S. 2018. A natural human monoclonal antibody targeting Staphylococcus Protein A protects against Staphylococcus aureus bacteremia. PLoS One 13:e0190537. 10.1371/journal.pone.0190537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Francois B, Mercier E, Gonzalez C, Asehnoune K, Nseir S, Fiancette M, Desachy A, Plantefeve G, Meziani F, de Lame PA, Laterre PF, Pierre-François Laterre for the MASTER 1 study group . 2018. Safety and tolerability of a single administration of AR-301, a human monoclonal antibody, in ICU patients with severe pneumonia caused by Staphylococcus aureus: first-in-human trial. Intensive Care Med 44:1787–1796. 10.1007/s00134-018-5229-2. [DOI] [PubMed] [Google Scholar]
- 11.Ali SO, Yu XQ, Robbie GJ, Wu Y, Shoemaker K, Yu L, DiGiandomenico A, Keller AE, Anude C, Hernandez-Illas M, Bellamy T, Falloon J, Dubovsky F, Jafri HS. 2019. Phase 1 study of MEDI3902, an investigational anti-Pseudomonas aeruginosa PcrV and Psl bispecific human monoclonal antibody, in healthy adults. Clin Microbiol Infect 25:629.e1–629.e6. 10.1016/j.cmi.2018.08.004. [DOI] [PubMed] [Google Scholar]
- 12.Hebert W, DiGiandomenico A, Zegans M. 2020. Multifunctional monoclonal antibody targeting Pseudomonas aeruginosa keratitis in mice. Vaccines (Basel) 8:638. 10.3390/vaccines8040638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henriksen AZ, Maeland JA. 1987. Serum antibodies to outer membrane proteins of Escherichia coli in healthy persons and patients with bacteremia. J Clin Microbiol 25:2181–2188. 10.1128/jcm.25.11.2181-2188.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henriksen AZ, Maeland JA, Brakstad OG. 1989. Monoclonal antibodies against three different enterobacterial outer membrane proteins. Characterization, cross-reactivity, and binding to bacteria. APMIS 97:559–568. 10.1111/j.1699-0463.1989.tb00831.x. [DOI] [PubMed] [Google Scholar]
- 15.Henriksen AZ, Maeland JA. 1990. Antibody response to defined domains on enterobacterial outer membrane proteins in healthy persons and patients with bacteraemia. APMIS 98:163–172. 10.1111/j.1699-0463.1990.tb01017.x. [DOI] [PubMed] [Google Scholar]
- 16.Henriksen AZ, Maeland JA. 1991. A conserved domain on enterobacterial porin protein analysed by monoclonal antibody. APMIS 99:49–57. 10.1111/j.1699-0463.1991.tb05117.x. [DOI] [PubMed] [Google Scholar]
- 17.Henriksen AZ, Maeland JA, Wetzler LM. 1998. An epitope shared by enterobacterial and neisserial porin proteins. APMIS 106:818–824. 10.1111/j.1699-0463.1998.tb00228.x. [DOI] [PubMed] [Google Scholar]
- 18.Abe Y, Haruta I, Yanagisawa N, Yagi J. 2013. Mouse monoclonal antibody specific for outer membrane protein A of Escherichia coli. Monoclon Antib Immunodiagn Immunother 32:32–35. 10.1089/mab.2012.0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puohiniemi R, Karvonen M, Vuopio-Varkila J, Muotiala A, Helander IM, Sarvas M. 1990. A strong antibody response to the periplasmic C-terminal domain of the OmpA protein of Escherichia coli is produced by immunization with purified OmpA or with whole E. coli or Salmonella typhimurium bacteria. Infect Immun 58:1691–1696. 10.1128/iai.58.6.1691-1696.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rainard P, Reperant-Ferter M, Gitton C, Gilbert FB, Germon P. 2017. Cellular and humoral immune response to recombinant Escherichia coli OmpA in cows. PLoS One 12:e0187369. 10.1371/journal.pone.0187369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu H, Liao Y, Zhang J, Wang Y, Liu Z, Cheng P, Wang X, Zou Q, Gu J. 2018. Rational design and evaluation of an artificial Escherichia coli K1 protein vaccine candidate based on the structure of OmpA. Front Cell Infect Microbiol 8:172. 10.3389/fcimb.2018.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jeannin P, Magistrelli G, Goetsch L, Haeuw JF, Thieblemont N, Bonnefoy JY, Delneste Y. 2002. Outer membrane protein A (OmpA): a new pathogen-associated molecular pattern that interacts with antigen presenting cells-impact on vaccine strategies. Vaccine 20:A23–A27. 10.1016/S0264-410X(02)00383-3. [DOI] [PubMed] [Google Scholar]
- 23.Nicholson TF, Watts KM, Hunstad DA. 2009. OmpA of uropathogenic Escherichia coli promotes postinvasion pathogenesis of cystitis. Infect Immun 77:5245–5251. 10.1128/IAI.00670-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. 2015. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol 13:269–284. 10.1038/nrmicro3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes JM, Zhang Y, Lin YL, Chung S, Dennis MS, Zuchero YJ, Watts RJ, Couch JA, Meng YG, Atwal JK, Brezski RJ, Spiess C, Ernst JA. 2017. Effector-attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J Biol Chem 292:3900–3908. 10.1074/jbc.M116.767749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett MR, Bombardi RG, Kose N, Parrish EH, Nagel MB, Petit RA, Read TD, Schey KL, Thomsen IP, Skaar EP, Crowe JE. 2019. Human mAbs to Staphylococcus aureus IsdA provide protection through both heme-blocking and Fc-mediated mechanisms. J Infect Dis 219:1264–1273. 10.1093/infdis/jiy635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meuskens I, Michalik M, Chauhan N, Linke D, Leo JC. 2017. A new strain collection for improved expression of outer membrane proteins. Front Cell Infect Microbiol 7:464. 10.3389/fcimb.2017.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nilsson G, Belasco JG, Cohen SN, von Gabain A. 1984. Growth-rate dependent regulation of mRNA stability in Escherichia coli. Nature 312:75–77. 10.1038/312075a0. [DOI] [PubMed] [Google Scholar]
- 29.Wang-Lin SX, Olson R, Beanan JM, MacDonald U, Balthasar JP, Russo TA. 2017. The capsular polysaccharide of Acinetobacter baumannii is an obstacle for therapeutic passive immunization strategies. Infect Immun 85:e00591-17. 10.1128/IAI.00591-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goh KGK, Phan MD, Forde BM, Chong TM, Yin WF, Chan KG, Ulett GC, Sweet MJ, Beatson SA, Schembri MA. 2017. Genome-wide discovery of genes required for capsule production by uropathogenic Escherichia coli. mBio 8:e01558-17. 10.1128/mBio.01558-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.King JE, Aal Owaif HA, Jia J, Roberts IS. 2015. Phenotypic heterogeneity in expression of the K1 polysaccharide capsule of uropathogenic Escherichia coli and downregulation of the capsule genes during growth in urine. Infect Immun 83:2605–2613. 10.1128/IAI.00188-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rollenske T, Burkhalter S, Muerner L, von Gunten S, Lukasiewicz J, Wardemann H, Macpherson AJ. 2021. Parallelism of intestinal secretory IgA shapes functional microbial fitness. Nature 598:657–661. 10.1038/s41586-021-03973-7. [DOI] [PubMed] [Google Scholar]
- 33.Lu T, Porter AR, Kennedy AD, Kobayashi SD, DeLeo FR. 2014. Phagocytosis and killing of Staphylococcus aureus by human neutrophils. J Innate Immun 6:639–649. 10.1159/000360478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shin S, Lu G, Cai M, Kim KS. 2005. Escherichia coli outer membrane protein A adheres to human brain microvascular endothelial cells. Biochem Biophys Res Commun 330:1199–1204. 10.1016/j.bbrc.2005.03.097. [DOI] [PubMed] [Google Scholar]
- 35.Pautsch A, Schulz GE. 2000. High-resolution structure of the OmpA membrane domain. J Mol Biol 298:273–282. 10.1006/jmbi.2000.3671. [DOI] [PubMed] [Google Scholar]
- 36.Ishida H, Garcia-Herrero A, Vogel HJ. 2014. The periplasmic domain of Escherichia coli outer membrane protein A can undergo a localized temperature dependent structural transition. Biochim Biophys Acta 1838:3014–3024. 10.1016/j.bbamem.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 37.Reusch RN. 2012. Insights into the structure and assembly of Escherichia coli outer membrane protein A. FEBS J 279:894–909. 10.1111/j.1742-4658.2012.08484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maruvada R, Kim KS. 2011. Extracellular loops of the Escherichia coli outer membrane protein A contribute to the pathogenesis of meningitis. J Infect Dis 203:131–140. 10.1093/infdis/jiq009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mittal R, Krishnan S, Gonzalez-Gomez I, Prasadarao NV. 2011. Deciphering the roles of outer membrane protein A extracellular loops in the pathogenesis of Escherichia coli K1 meningitis. J Biol Chem 286:2183–2193. 10.1074/jbc.M110.178236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vila-Farres X, Parra-Millan R, Sanchez-Encinales V, Varese M, Ayerbe-Algaba R, Bayo N, Guardiola S, Pachon-Ibanez ME, Kotev M, Garcia J, Teixido M, Vila J, Pachon J, Giralt E, Smani Y. 2017. Combating virulence of Gram-negative bacilli by OmpA inhibition. Sci Rep 7:14683. 10.1038/s41598-017-14972-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kapral FA. 1966. Clumping of Staphylococcus aureus in the peritoneal cavity of mice. J Bacteriol 92:1188–1195. 10.1128/jb.92.4.1188-1195.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dalia AB, Weiser JN. 2011. Minimization of bacterial size allows for complement evasion and is overcome by the agglutinating effect of antibody. Cell Host Microbe 10:486–496. 10.1016/j.chom.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim KS. 2001. Escherichia coli translocation at the blood-brain barrier. Infect Immun 69:5217–5222. 10.1128/IAI.69.9.5217-5222.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prasadarao NV, Wass CA, Kim KS. 1996. Endothelial cell GlcNAc beta 1-4GlcNAc epitopes for outer membrane protein a enhance traversal of Escherichia coli across the blood-brain barrier. Infect Immun 64:154–160. 10.1128/iai.64.1.154-160.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Torres AG, Kaper JB. 2003. Multiple elements controlling adherence of enterohemorrhagic Escherichia coli O157: H7 to HeLa cells. Infect Immun 71:4985–4995. 10.1128/IAI.71.9.4985-4995.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Torres AG, Li YG, Tutt CB, Xin LJ, Eaves-Pyles T, Soong L. 2006. Outer membrane protein A of Escherichia coli O157: H7 stimulates dendritic cell activation. Infect Immun 74:2676–2685. 10.1128/IAI.74.5.2676-2685.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krishnan S, Prasadarao NV. 2012. Outer membrane protein A and OprF: versatile roles in Gram-negative bacterial infections. FEBS J 279:919–931. 10.1111/j.1742-4658.2012.08482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prasadarao NV, Blom AM, Villoutreix BO, Linsangan LC. 2002. A novel interaction of outer membrane protein A with C4b binding protein mediates serum resistance of Escherichia coli K1. J Immunol 169:6352–6360. 10.4049/jimmunol.169.11.6352. [DOI] [PubMed] [Google Scholar]
- 49.Leimbach A, Hacker J, Dobrindt U. 2013. E. coli as an all-rounder: the thin line between commensalism and pathogenicity. Curr Top Microbiol Immunol 358:3–32. 10.1007/82_2012_303. [DOI] [PubMed] [Google Scholar]
- 50.Tenaillon O, Skurnik D, Picard B, Denamur E. 2010. The population genetics of commensal Escherichia coli. Nat Rev Microbiol 8:207–217. 10.1038/nrmicro2298. [DOI] [PubMed] [Google Scholar]
- 51.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. 2007. Development of the human infant intestinal microbiota. PLoS Biol 5:e177. 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Storek KM, Auerbach MR, Shi HD, Garcia NK, Sun DW, Nickerson NN, Vij R, Lin ZH, Chiang N, Schneider K, Wecksler AT, Skippington E, Nakamura G, Seshasayee D, Koerber JT, Payandeh J, Smith PA, Rutherford ST. 2018. Monoclonal antibody targeting the ss-barrel assembly machine of Escherichia coli is bactericidal. Proc Natl Acad Sci USA 115:3692–3697. 10.1073/pnas.1800043115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vij R, Lin ZH, Chiang N, Vernes JM, Storek KM, Park S, Chan J, Meng YG, Comps-Agrar L, Luan P, Lee S, Schneider K, Bevers J, Zilberleyb I, Tam C, Koth CM, Xu M, Gill A, Auerbach MR, Smith PA, Rutherford ST, Nakamura G, Seshasayee D, Payandeh J, Koerber JT. 2018. A targeted boost-and-sort immunization strategy using Escherichia coli BamA identifies rare growth inhibitory antibodies. Sci Rep 8:7136. 10.1038/s41598-018-25609-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith SA, Crowe JE. 2015. Use of human hybridoma technology to isolate human monoclonal antibodies. Microbiol Spectr 3:AID-0027-2014. 10.1128/microbiolspec.AID-0027-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cian MB, Giordano NP, Mettlach JA, Minor KE, Dalebroux ZD. 2020. Separation of the cell envelope for Gram-negative bacteria into inner and outer membrane fractions with technical adjustments for Acinetobacter baumannii. JoVE 158:e60517. 10.3791/60517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moynie L, Serra I, Scorciapino MA, Oueis E, Page MGP, Ceccarelli M, Naismith JH. 2018. Preacinetobactin not acinetobactin is essential for iron uptake by the BauA transporter of the pathogen Acinetobacter baumannii. Elife 7:e42270. 10.7554/eLife.42270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]