

Abstract
Impaired cortical maturation is a postulated mechanism in the etiology of neurodevelopmental disorders, including schizophrenia. In sensory cortex, activity relayed by the thalamus during a postnatal sensitive period is essential for proper cortical maturation. Whether thalamic activity also shapes prefrontal cortical maturation is unknown. We show that inhibiting the mediodorsal and midline thalamus in mice during adolescence leads to a long-lasting decrease in thalamo-prefrontal projection density and reduced excitatory drive to prefrontal neurons. It also caused prefrontal-dependent cognitive deficits during adulthood, associated with disrupted prefrontal cross-correlations and task-outcome encoding. Thalamic inhibition during adulthood had no long-lasting consequences. Exciting the thalamus in adulthood during a cognitive task rescued prefrontal cross-correlations, task outcome encoding, and cognitive deficits. These data point to adolescence as a sensitive window of thalamo-cortical circuit maturation. Furthermore, by supporting prefrontal network activity, boosting thalamic activity provides a potential therapeutic strategy for rescuing cognitive deficits in neurodevelopmental disorders.
Introduction
Sensitive periods denote developmental time windows of heightened plasticity during which alterations in experience can lead to long-lasting changes in the anatomy and function of the nervous system1. A classic example is in the visual system, where transient developmental monocular deprivation permanently impairs acuity in the deprived eye in an activity-dependent manner2. This impairment in function persists even after the deprivation in visual input is reversed, as the thalamo-cortical inputs representing the closed eye are permanently disrupted. While sensitive periods for the circuit refinement of sensory cortices have been well-documented2–5, similar transient changes in activity during postnatal development may also have lasting changes in the prefrontal cortex (PFC), an associative cortical area supporting higher cognitive functioning6–10.
Disturbances in PFC function are believed to underlie the cognitive symptoms found in psychiatric disorders, such as schizophrenia. Schizophrenia is thought to have a developmental origin11,12, and one prominent hypothesis is that during adolescence, a vulnerable period for the development of this disorder, the maturation of the PFC is disrupted13. Recent studies have identified decreased correlated activity between the thalamus and the dorsolateral PFC under resting conditions and during cognitive testing, a finding which may have a structural basis14–17. Strikingly, decreased thalamo-prefrontal connectivity was also seen in younger adolescents at high risk for psychosis, and it predicted later illness conversion14,18,19, raising the intriguing possibility that decreased input from the thalamus could contribute to the developmental etiology of PFC dysfunction in the disorder18–20.
Here, we directly test the hypothesis that input activity from the thalamus during adolescence is important for PFC circuit maturation and that decreasing this input during adolescence will lead to long-lasting impairments in the functioning of the PFC. To address this question, we used the designer receptor, hM4DGi, to selectively reduce adolescent activity of the midline thalamus, including the mediodorsal thalamus, which projects to the medial PFC (mPFC) in the mouse. We found that transient thalamic inhibition during adolescence led to several persistent changes in adulthood, including (1) deficits in two mPFC-dependent cognitive tasks, (2) decreased excitatory drive onto mPFC pyramidal cells, (3) decreased anatomical thalamo-mPFC input, (4) reduced mPFC neuron cross-correlations, and (5) impaired mPFC neuron encoding of extra-dimensional set shifting task outcomes. In contrast, inhibiting the thalamus for a comparable period during adulthood had no long-lasting effects. These data point to adolescence as a sensitive time window of thalamo-cortical circuit maturation. Strikingly, enhancing thalamic excitability during adulthood rescued the behavioral deficits and restored the ability of mPFC neurons to encode task outcome after adolescent thalamic inhibition. Prior studies have suggested that the thalamic inputs act as a non-specific amplifier supporting prefrontal activity during the delay periods of a working memory task and a contextual switching task21–24. Our data suggest that the thalamus plays a broader function in facilitating mPFC activity that is not restricted to delay-containing tasks. Thus, this study identifies neuronal mechanisms of prefrontal circuit maturation and offers therapeutic insights into how to reverse cognitive deficits arising from a developmentally altered brain.
Results
Thalamic inhibition during adolescence or adulthood
To inhibit thalamic activity during adolescence and adulthood, we injected an adeno-associated virus (AAV) carrying a Cre-dependent version of the inhibitory designer receptor, hM4DGi (hereafter referred to as hM4D), into the thalamus of GBX2-CreERT mice. Viral injections were performed at postnatal day P13 and Cre-mediated recombination was induced by tamoxifen injection at P15–16, at a time when GBX2 expression is restricted to the midline thalamus, thereby limiting viral spread (Extended Data 1). To determine the efficacy of hM4D-mediated inhibition, we performed whole-cell patch clamp recordings from thalamic neurons in both adolescent and adult brain slices. Application of the DREADD ligand, clozapine-n-oxide (CNO), hyperpolarized thalamic neurons consistent with activation of G-protein coupled inward rectifying potassium (GIRK) channels (Extended Data 1d). Thalamic neurons in control animals did not respond to CNO. CNO-application led to comparable effects sizes in adolescent and adult brain slices that were consistent with published results in adult thalamic neurons25. Crucially, CNO-application hyperpolarized thalamic neurons in animals that had been exposed to twice daily intraperitoneal (i.p.) CNO injections for 15 days (P20–35, P90–105) or 30 days (P20–50), suggesting that chronic hM4D activation does not lead to receptor de-sensitization (Extended Data 1d). These data indicate that repeated hM4D activation inhibits thalamic neuron activity during adolescence and adulthood.
Adolescent thalamic activity is required for adult cognition
We then tested the long-term effects of transient thalamic inhibition during adolescence (P20–50) on prefrontal-dependent cognitive task performance. To this end, CNO (1 mg/kg) was injected twice daily in hM4D and control GFP mice from days P20–50, and the animals were tested forty days later, at P90 (Fig. 1a). To assess cognition during adulthood, we chose an operant-based Non-Match to Sample (NMS) working memory task (Fig. 1b), whose acquisition is delayed after a lesion of the mPFC26 and an odor- and texture- based attentional set shifting task (ASST), in which the extra-dimensional set shifting component of the task (EDSS) is sensitive to mPFC lesions (Fig. 1c)27. Following adolescent thalamic inhibition from P20–50, we found that the acquisition of the NMS task was impaired in animals expressing hM4D compared with controls (Fig. 1d, Extended Data 2a). No changes were seen in any other task variables, such as trial length, task latencies, or rewards consumed (Extended Data 2b–e), excluding deficits in motivation or mobility.
Figure 1. Adolescent, but not adult, thalamic inhibition leads to persistent cognitive impairments in adulthood.

Schematics of (a) the Non-Match to Sample (NMS) task and (b) the attentional set-shifting task (ASST). (c) Adolescent experimental timeline. CNO was administered from P20–50 to mice expressing hM4D or GFP in the thalamus, and behavioral testing was conducted 40 days later, at P90. Created with Biorender.com. (d) Adolescent-inhibited hM4D animals take significantly longer to acquire the NMS task (left), taking significantly more days to reach criterion (right). Control: n=17 animals; hM4D: n=21 animals; learning curve: 2-way repeated measures (rm) ANOVA, effect of time F(4.201,151.2)=102.0, p<0.0001, effect of group F(1,36)=3.143, p=0.0847, effect of group × time F(13,468)=2.088, *p=0.0137; days to criterion (3 consecutive days above 70%): Control: (mean ± standard error of the mean, SEM) 5.35±0.27 days, hM4D: 7.05±0.51 days; two-sided unpaired t-test: t=2.746, df=36, **p=0.0094. (e) Adolescent-inhibited hM4D animals are no different than controls in the initial acquisition (IA) of the ASST (left, Control: n=14 animals, 15.71±1.88 trials, hM4D: n=16 animals, hM4D: 11.81±1.50 trials; two-sided unpaired t-test, t=1.639, df=28, p=0.1125) but take significantly more trials in the extra-dimensional set shift (EDSS) than controls (right, Control: n=14 animals, 10.57±0.42 trials, hM4D: n=15 animals, hM4D: 15.07±1.79 trials; unpaired t-test, t=2.372, df=27, *p=0.0251). (f) Adult experimental timeline, with CNO administered P90–120 and testing at P160. There were no differences in either (g) the acquisition of the NMS task (Control: n=6 animals, hM4D: n=10 animals; learning curve: 2-way rmANOVA, effect of time F(5.501,77.01)=40.21, p<0.0001, effect of group F(1,14)=1.462, p=0.2467, effect of group × time F(17,238)=0.8680, p=0.6126; days to criterion: Control: 7.33±0.67 days, hM4D: 7.40±1.02 days; two-sided unpaired t-test, t=0.04654, df=14, p=0.9635) or (h) the IA (Control: n=20 animals, 10.60±0.59 days, hM4D: n=18 animals, 11.39±0.76 trials; two-sided unpaired t-test, t=0.8260, df=36, p=0.4142) and EDSS (Control: 12.40±0.89 trials, hM4D: 10.76±0.64 days; unpaired t-test, t=1.442, df=35, p=0.1583) portions of the ASST between adult-inhibited hM4D animals and controls. Learning curves depict mean performance ± SEM for each day. For other plots, dots represent individual animals; lines represent mean ± SEM. *p<0.05, **p<0.01
Similarly, in the ASST, we found that the mPFC-dependent EDSS was impaired in animals expressing hM4D compared with controls (Fig. 1e). Meanwhile, behavior in the non-mPFC-dependent initial acquisition portion of the set shifting task (IA) was unchanged. No changes were seen in any other task variables, including IA or EDSS task latencies (Extended Data 2f–h).
To address whether the primary contribution of this behavioral deficit came from projections to the mPFC, as opposed to other thalamic projections, we selectively inhibited thalamo-mPFC projections during adolescence. We used a dual virus approach, injecting a retrogradely-transported AAV expressing Cre recombinase into the mPFC and an AAV expressing Cre-dependent hM4D into the thalamus (Extended Data 3a). Using this method, hM4D expression was restricted to thalamic cells projecting to the mPFC (Extended Data 3b, c). As in Fig. 1d, we observed intact IA and impaired EDSS after projection-specific inhibition (Extended Data 3d, e).
To determine whether adolescence is a sensitive period, or whether the circuit is sensitive to transient changes at any age, we also inhibited the thalamus for a comparable time window during adulthood, P90–120, and tested the mice forty days later, at P160 (Figure 1f).
While there was an effect of age on performance in the NMS task, with the older P160 animals performing worse than the P90 animals, adult thalamic inhibition affected neither acquisition of the NMS task (Fig. 1g) nor trials to criterion in the EDSS task (Fig. 1h), supporting the hypothesis that adolescence is a sensitive period in which changes in thalamic activity influence the development of thalamo-mPFC circuit maturation.
Adolescent thalamic activity regulates adult mPFC excitation
To determine whether thalamic inhibition during adolescence leads to long-lasting changes in mPFC circuit function, we used slice physiology to measure spontaneous excitatory and inhibitory activity in mPFC layer II/III pyramidal cells, which receive projections from the thalamus (Fig. 2a). Following adolescent thalamic inhibition, the frequency of spontaneous excitatory post-synaptic currents (sEPSCs) was reduced, while the sEPSC amplitude was unchanged (Fig. 2b, c). This change in frequency, but not amplitude, suggests a decrease in pre-synaptic excitatory inputs. In contrast, we found no changes in frequency or amplitude of spontaneous inhibitory post-synaptic currents (sIPSCs) (Fig. 2b, d).
Figure 2. Adolescent, but not adult, thalamic inhibition leads to a persistent reduction in excitatory drive onto mPFC pyramidal neurons.

(a) Adolescent experimental timeline and schematic. Whole cell patch clamp recordings were made from pyramidal cells in layer II/III of the mPFC from hM4D and control mice. These pyramidal cells receive excitatory inputs from the thalamus as well as inhibitory inputs from local interneurons. Created with Biorender.com. (b) Representative traces showing spontaneous excitatory post-synaptic currents (sEPSCs, left) and spontaneous inhibitory post-synaptic currents (sIPSCs, right). (c) sEPSC frequency is significantly reduced following adolescent thalamic inhibition relative to control mice, but sEPSC amplitude is unchanged. Control: n=20 cells, 5 animals; hM4D: n=24 cells, 7 animals; frequency: Control: 4.438±0.429 Hz, hM4D: 3.202±0.325 Hz; two-sided unpaired t-test, t=2.337, df=42, *p=0.0243; amplitude: Control: 28.71±2.57 pA, hM4D: 29.82±1.47 pA; unpaired t-test, t=0.3881, df=42, p=0.6999. (d) sIPSC frequency and amplitude are also unchanged. Control: n=20 cells, 5 animals; hM4D: n=21 cells, 7 animals; frequency: Control: 3.421±0.376 Hz, hM4D: 2.627±0.323 Hz; two-sided unpaired t-test, t=1.606, df=39, p=0.1163; amplitude: Control: 32.29±2.08 pA, hM4D: 31.03±1.84 pA; unpaired t-test, t=0.4450, df=34, p=0.6592. (e) Adult experimental timeline. (f) sEPSC and (g) sIPSC frequency and amplitude are unchanged following adult thalamic inhibition. Control: n=12 cells, 3 animals; hM4D: n=12 cells, 3 animals; sEPSC: n=12 Control cells, n=12 hM4D cells; frequency: Control: 4.674±0.448 Hz, hM4D: 4.675±0.561 Hz; two-sided unpaired t-test, t=0.001936, df=22, p=0.9985; amplitude: Control: 27.78±1.68 pA, hM4D: 29.75±1.78 pA; unpaired t-test, t=0.8048, df=22, p=0.4296; sIPSC: n=12 Control cells, n=12 hM4D cells; frequency: Control: 3.775±0.506 Hz, hM4D: 2.825±0.625 Hz; unpaired t-test, t=1.181, df=22, p=0.2501; amplitude: Control: 25.49±0.82 pA, hM4D: 23.69±1.82 pA; two-sided unpaired t-test, t=0.9030, df=22, p=0.3763. Dots represent individual animals; lines represent mean ± SEM. *p<0.05
These effects were again selective to thalamic inhibition during adolescence as we found no changes in excitatory or inhibitory inputs following chronic thalamic inhibition in adulthood (Fig. 2f, g). Consistent with the behavioral results, these results point to adolescence as a sensitive time-period during which thalamic activity regulates the development of the mPFC. Note that adolescent thalamic inhibition did not alter the intrinsic properties and excitatory inputs to MD neurons (Extended Fig. 4).
Adolescent activity regulates adult thalamo-mPFC projections
We next determined whether decreased subcortical anatomical inputs may contribute to the decrease in sEPSC frequency. To address this question, we injected a retrogradely transported fluorescent protein, GFP, into the mPFC of adult mice that had experienced adolescent thalamic inhibition. Three weeks later, we calculated the density of retrogradely labelled neurons in the thalamus and basolateral amygdala (BLA, Fig. 3a), which both project to layer II/III of the mPFC. We observed a decrease in the density of cells projecting from the thalamus to the mPFC in adult mice (Fig. 3c), which already emerges at P35 (Extended Fig. 5). In contrast, we found no change in the density of cells projecting from the BLA (Fig. 3d, e), suggesting that there is no global competition between subcortical mPFC projections, as has previously been observed after early postnatal lesion studies28. We measured no change in overall cell density within the thalamus based on DAPI-positive cell counts (two-sided unpaired t-test, t=0.6050, df=11, p=0.5575), suggesting a loss of thalamic inputs to the mPFC rather than thalamic cells.
Figure 3. Adolescent thalamic inhibition leads to a persistent reduction in the density of thalamo-prefrontal, but not BLA-prefrontal, projections.

(a) Experimental timeline and schematic. At P70, a retrograde tracer, GFP, was injected into the mPFC, before perfusion 3 weeks later. Created with Biorender.com. (b) Representative confocal images illustrating GFP staining in the mPFC (left), thalamus (middle), and basolateral amygdala (BLA, right) in control (top) and hM4D (bottom) animals. Outlines were determined using DAPI staining. For each animal, 6 slices were used for each of the 3 regions. Control: n=6 animals, hM4D: n=7 animals. (c) Stereology was conducted using DAPI staining for outlines of regions and GFP staining for cell counting. Quantification of GFP-positive cell density showed a significant decrease in thalamo-mPFC projecting cells in adolescent-inhibited hM4D animals compared to controls (top, Control: n=6 animals, 453.2±61.3 cells/mm2, hM4D: n=7 animals, 260.3±56.1 cells/mm2; two-sided unpaired t-test, t=2.326, df=11, *p=0.0401). Stereological estimates showed no difference in overall thalamic area (bottom, Control: 5.539±0.232 mm2, hM4D: 5.429±0.178 mm2; two-sided unpaired t-test, t=0.3834, df=11, p=0.7087). (d) Stereology in the BLA showed no differences in either GFP-positive cell density (top, Control: 602.4±61.1 cells/mm2, hM4D: 578.5±61.0 cells/mm2; two-sided unpaired t-test, t=0.2749, df=11, p=0.7885) or BLA area (bottom, Control: 5.687±0.211 mm2, hM4D: 5.432±0.163 mm2; two-sided unpaired t-test, t=0.9713, df=11, p=0.3523). (e) The ratio of thalamic to BLA projection cell densities showed a significant reduction in adolescent-inhibited hM4D animals compared to controls (top, Control: 0.742±0.065, hM4D: 0.467±0.091; two-sided unpaired t-test, t=2.376, df=11, *p=0.0368) but no change in region area (bottom, Control: 0.981±0.055, hM4D: 1.006±0.049; two-sided unpaired t-test, t=0.3471, df=11, p=0.7351). Dots represent individual animals; lines represent mean ± SEM. *p<0.05
Adult thalamic excitation rescues the cognitive deficit
The anatomical changes suggest that the circuit alterations are persistent. Previous work has shown that exciting the thalamus enhances performance in prefrontal-dependent working memory and a 2-alternative forced choice tasks21,24.
Therefore, we excited the thalamus during the set shifting task using a stabilized step-function opsin (SSFO, Fig. 4a). We activated the SSFO with a 5 second pulse (473 nm, 4 mW) before the start of the EDSS portion of the task (Fig. 4b). Because the SSFO will slowly inactivate over time, we repeated the 5 s pulse every 30 minutes during the intertrial interval (ITI) of the task.
Figure 4. Acute thalamic activity enhancement rescues the ASST behavioral deficit following adolescent thalamic inhibition.

(a) Experimental timeline. At P70, a stabilized step-function opsin (SSFO) was injected into the thalamus along with optrodes. Behavioral testing was done at P90 and P100. (b) Schematic for behavior. For the light ON animals, the SSFO was stimulated before the EDSS and again every 30 min during the inter-trial interval (ITI) until completion of the task. Animals were randomly assigned to two groups: (1) light ON (SSFO activation) on Day 1 at P90 and light OFF on Day 2 at P100; (2) light OFF on Day 1 at P90 and light ON on Day 2 at P100. Created with Biorender.com. (c) There is no significant difference in IA performance between the control or hM4D light OFF groups (2-way rmANOVA; effect of group F(1,23)=2.407, p=0.1344, effect of light F(1,23)=0.3319, p=0.5702, effect of group × light F(1,23)=0.001148, p=0.9733; Holm-Sidak post-hoc: Control Light OFF vs. hM4D Light OFF p=0.4425). Further, all groups showed equivalent trials to criterion during the IA (Holm-Sidak post-hoc: Control Light OFF vs. ON p=0.8925, hM4D Light OFF vs. ON p=0.8925). (d) hM4D light OFF animals take significantly more trials to reach criterion during EDSS compared with control light OFF animals. Acute SSFO stimulation (light ON) during the EDSS rescued the behavior in the adolescent-inhibited hM4D animals but had no effect on control animals (2-way rmANOVA; effect of group F(1,23)=5.407, p=0.0292, effect of light F(1,23)=5.002, p=0.0353, effect of group × light F(1,23)=5.002, p=0.0353; Holm-Sidak post-hoc: Control Light OFF vs. ON p>0.9999, hM4D Light OFF vs. ON **p=0.0035; Light OFF Control vs. hM4D **p=0.0046, Light ON Control vs. hM4D p=0.8197). Dots represent individual animals, lines connecting performance with light OFF and light ON. Control: n=10; hM4D: n=15. **p<0.01
We performed a crossover experiment where each animal performed the ASST twice, with and without SSFO activation, ten days apart. We replicated the behavioral deficit described in Fig. 1e and found that increasing thalamic excitability via SSFO activation during EDSS rescued the deficit in EDSS (Fig. 4d). The effects of SSFO activation did not persist from the first day of testing to the second testing day, and repeating the experiment did not influence behavior (2-way rmANOVA; effect of light F(1,46)=6.302, p=0.0156, effect of run day F(1,46)=2.512, p=0.1199, effect of light × day run F(1,46)=1.364, p=0.2488). Thus, even though the sensitive period of circuit maturation occurs in adolescence, activating thalamo-mPFC circuitry can still rescue the behavioral deficits in adulthood.
Oscillatory activity does not explain the cognitive deficits
To better understand the network mechanisms driving the behavioral deficits, we examined several metrics of mPFC activity during the behavior: local field potential (LFP) activity, single unit cellular activity, and neural ensemble activity.
Prior work, using the same set shifting task, identified an increase in the power of gamma frequency (40–90 Hz) oscillations in the mPFC before correct, but not incorrect, choices during the EDSS behavior6.
Consistent with these prior results6, we found that mPFC gamma power was increased in correct versus incorrect trials before the mouse made a decision (Fig. 5a, d, e). However, this difference was observed in both controls and after adolescent inhibition of the MD, albeit with a smaller-appearing effect size (Fig. 5b, d). Moreover, thalamic SSFO activation had no significant effect on mPFC gamma power (Fig. 5c–e). These results suggest that changes in gamma power do not explain the deficit in EDSS.
Figure 5. Adolescent thalamic inhibition does not affect mPFC gamma power in adult mice performing the EDSS task.

(a) Control animal without SSFO activation (Light OFF) mPFC normalized power (artificial units, A.U.) as a function of frequency during the 6 seconds preceding the decision point during the EDSS during correct trials (green), and incorrect trials (orange). Red shading indicates 40–90 Hz (gamma). Lines and shading indicate mean ± SEM. (b) As in (a), but for adolescent-inhibited hM4D Light OFF animals. (c) As in (a), but for adolescent-inhibited hM4D animals that have acute SSFO activation during EDSS (Light ON). (d) Mean gamma power (40–90 Hz) is significantly increased in correct vs incorrect trials for all three groups, and this pattern is not significantly different across groups. Control Light OFF: left, blue; hM4D Light OFF: center, pink; hM4D Light ON: right, purple. Control Light OFF: n=9 animals, 88 correct trials, 23 incorrect trials, Correct: 0.1774±0.0064; Incorrect: 0.1625±0.0066; linear mixed effects model (power~trial type+(1|animal)): fixed effect (Trial Type), ****p=5.1208e-05; hM4D Light OFF: n=14 animals, 177 correct trials, 89 incorrect trials, Correct: 0.1813±0.0048; Incorrect: 0.1765±0.0068; linear mixed effects model (power~trial type+(1|animal)): fixed effect (Trial Type), **p=0.0014916; hM4D Light ON: n=15 animals, 137 correct trials, 41 incorrect trials, Correct: 0.1697±0.0043; Incorrect: 0.1652±0.0099; linear mixed effects model (power~trial type+(1|animal)): fixed effect (Trial Type), *p=0.015341. Linear hypothesis two-sided F-test to compare differences: Control vs. hM4D: p=0.3092; hM4D vs. hM4D Light ON: p=0.7607. Lines and error represent mean ± SEM. Dots represent individual trials for each animal (colors of the dots). (e) Mean difference in gamma power between correct and incorrect trials by animal shows no differences across groups, all groups having an increased gamma power for correct over incorrect trials. Control Light OFF: n=9 animals, 0.0134±0.0043; hM4D Light OFF: n=14 animals, 0.0093±0.0025; hM4D Light ON: n=15 animals, 0.0115±0.0045; 1-way ANOVA, F(2,35)=0.2329, p=0.7935. Lines and error represent mean ± SEM. Dots represent individual animal mean difference. *p<0.05, **p<0.01, ****p<0.0001
Other cognitive tasks are known to generate thalamo-cortical oscillations in the beta frequency range (12–30 Hz)21,22,25. In the ASST, we recorded an increase in beta power during the trial compared to the inter-trial interval (ITI, Extended Data 6a). This beta activation was equivalent across both trial types and was not affected by the developmental manipulation (Extended Data 6b, e). In addition, we found no changes between trial types in thalamo-mPFC coherence (Extended Data 6c, d, f) or phase-locking in the beta frequency range (Extended Data 6g, h).
Altogether, these data show that changes in oscillatory activity cannot explain the behavioral deficit in adolescent-inhibited mice.
Adolescent thalamic activity regulates adult mPFC encoding
To determine whether thalamic inhibition alters encoding of information within the mPFC, we analyzed the firing rates of single units in the mPFC (Fig. 6a). Most mPFC units showed task-modulated activity with cells showing either enhanced or decreased activity during the EDSS task trials compared with the ITI (Extended Data 7a, b, c). However, overall single unit firing rates (FR) were not altered by either the developmental manipulation or the SSFO rescue (Fig. 6b). This was consistent during the ITI, over the course of the trial, in the pre-decision, and in the post-decision periods, when looking at either raw FR (Extended Data 7d–g) or FR that were normalized to ITI activity (Extended Data 8). Furthermore, FR did not significantly vary between different trial types, such as correct trials and incorrect trials (Fig. 6c). Again, this was found throughout the different epochs of the trial (Extended Data 7, 8). Thus, individual FR do not predict trial outcomes in control animals, and this metric was not affected by either developmental thalamic inhibition or acute thalamic activation. These findings were consistent, even when selecting only the task-modulated cells. Note that overall multiunit firing rates were also not changed in the MD thalamus during EDSS trials (Extended Data 9).
Figure 6. Adolescent thalamic inhibition leads to loss of mPFC cellular encoding of ASST trial outcome in adult mice.

(a) Control Light OFF mean firing rate (FR) during EDSS, normalized to ITI, before/after the decision point (dashed line). Color scale represents z-scored FR. (b) No differences in FR during the trial. Control OFF: n=8 animals, 75 cells, 3.650±0.639Hz; hM4D OFF: n=12 animals, 55 cells, 3.487±0.777Hz; hM4D ON: n=13 animals, 71 cells, 3.058±0.516Hz; 1-way ANOVA, F(2,194)=0.2493, p=0.7796. Dots represent cells; bars represent mean±SEM. (c) No differences in FR during correct and incorrect trials. Control OFF: FR Correct:3.570±0.647Hz, Incorrect:3.744±0.630Hz; two-sided paired t-test: t=0.6546, df=71, p=0.5148; hM4D OFF: Correct:3.607±0.859Hz, Incorrect:3.676±0.873Hz; two-sided paired t-test: t=0.3174, df=48, p=0.7523; hM4D ON: Correct:3.058±0.519Hz, Incorrect:3.023±0.550Hz; two-sided paired t-test: t=0.1539, df=70, p=0.8781. Dots represent individual cells, lines connecting FR for correct and incorrect trials. (d) Peak cross-correlations for cellpairs during correct (green) and incorrect (orange) trials show decrease in hM4D OFF animals compared to Control OFF and hM4D ON (graph truncated along y-axis; untruncated in Extended Data 8g). Control OFF: n=6 animals, 73 cells, 507 cellpairs, 0.0177±0.0430; hM4D OFF: n=9 animals, 52 cells, 181 cellpairs, 0.0124±0.0212; hM4D ON: n=11 animals, 69 cells, 327 cellpairs, 0.0201±0.0414; linear mixed effects model (peak cross-correlation~group+trial type+(1|cellpair)+(1|animal)+(1|cell1)+(1|cell2)), fixed effect of group: Control OFF vs. hM4D OFF: *p=0.041622; hM4D OFF vs. ON: **p=0.0090838. Bars with error represent mean±SEM; dots represent cellpairs. (e) Schematic of linear decoder. Neither hypothetical cell’s firing rate can distinguish between correct and incorrect trials (dots along axes). When plotted together, a linear decoder (red line) can discriminate between outcomes in additional trials (light circles). (f) Decoding trial outcome using EDSS FR. Decoder performance is significantly above chance for Control OFF (blue), at chance for hM4D OFF (red), and rescued by thalamic activation (purple). Actual performance in colored diamonds. Shuffled trial outcomes show chance performance, mean±standard deviation (black circles and lines) and individual shuffles (1000, grey circles), two-sided z-score to calculate p-values. Control OFF: n=4 animals, 60 cells, actual:74.71%, shuffled:49.95±3.75%, ****p=3.9604e-11; hM4D OFF: n=7 animals, 45 cells, actual:43.25%, shuffled:51.13±7.49%, p=0.2926; hM4D ON: n=9 animals, 61 cells, actual:69.41%, shuffled:50.15±3.08%, ****p=3.9472e-10. *p<0.05, **p<0.01, ****p<0.0001
However, previous studies have highlighted the benefits of analyzing firing rates across populations of neurons to better elucidate task behaviors, contexts, and outcomes29–31.
First, we analyzed the correlation between the firing of cells. We correlated peak cross-correlation values for each cell pair for each trial × to the outcome of trial x+1. This analysis showed that trial x+1 outcome is positively correlated with trial × peak cross-correlation (p=0.025). This was true even when taking trial × outcome into account (i.e., this was true across correct and incorrect trials), with no significant interaction of the effect of trial × cross-correlation and trial × outcome (p=0.165). This analysis indicates that higher peak cross-correlation values for trial × are associated with improved performance in trial x+1.
Next, we explored the effects of thalamic inhibition on peak cross-correlations. Using a previous data set21, we found that acute inhibition of thalamo-mPFC projections in adult mice decreases peak cross-correlation values for pairs of mPFC single units during the delay of a T-maze working memory task (Extended Data 10a).
We then analyzed cross-correlations for each cell pair during the EDSS trials. Adolescent thalamic inhibition reduced peak cross-correlations, and they were recovered following acute thalamic SSFO activation (Fig. 6d). This analysis supports the hypothesis that higher peak cross-correlations lead to improved subsequent learning and behavioral outcomes, which are compromised following adolescent thalamic inhibition.
We also employed a linear decoder to elucidate differences in population activity for trials that have different outcomes (Fig. 6e). Taking the firing rates for all cells in an experimental group across all trials, we trained a linear decoding algorithm using 50% of all trials for each cell to predict whether the behavioral outcome would yield a correct or incorrect trial. We then tested the decoder on the other 50% of trials to determine whether we could predict trial outcome based on cell firing rates. To determine chance performance, we employed the same decoding algorithm using randomly shuffled trial outcomes, repeated 1000 times32. Employing this decoder on the control group showed a resulting performance that was significantly better than chance, at 74.71% accuracy (Fig. 6f). This finding is eliminated following adolescent thalamic inhibition, where the decoder was no better than chance at 43.25% accuracy. Crucially, acute thalamic enhancement rescued the decoder performance to 69.41% accuracy.
Of note, no subset of neurons contributed more to the decoder performance, with an even distribution across the populations in all three groups (Extended Data 10b, c). Similarly, the decoding performance discrepancies across groups are visible with randomly selected subgroups of neurons. The pattern can be seen with as few as 5 neurons (Extended Data 10d). Moreover, the control decoding performance was not observed when applied to trials in the IA portion of the ASST (Extended Data 10e), indicating the specificity of the role of the mPFC during the EDSS.
Together, these findings show that adolescent thalamic inhibition disrupts prefrontal encoding of EDSS task outcome in adulthood while decreasing correlated activity between prefrontal neurons. This disruption can be rescued by acute thalamic activation during adulthood.
Discussion
An adolescent sensitive period for thalamo-mPFC development
Thalamic input activity has been shown to be important for sensory cortex maturation, including the visual cortex1–4. More recent studies have also begun to explore how neuronal activity shapes the development of higher cognitive structures, such as the mPFC6–8. Primarily, these studies have focused on changes to intrinsic components of mPFC circuitry, such as excitation and inhibition or layer II/III pyramidal neuron activity6,9,10,33. Some have also highlighted similarities between mechanisms found in sensory sensitive periods and mPFC adolescent development, including BDNF expression, NMDA receptor changes, and the formation of perineuronal nets8,34–36. This paper is the first to explore whether afferent input from the thalamus shapes cortical maturation and whether inhibition of thalamic activity leads to long-lasting changes in mPFC function and behavior.
We found that thalamic inhibition during adolescence leads to persistent impairments in mPFC circuit function and cognitive behaviors in adulthood. Specifically, we observed impairments in two mPFC-dependent tasks assessing the acquisition of a non-match to sample rule and attentional set shifting. These deficits were associated with a decrease in excitatory drive and anatomical projections to mPFC neurons. We determined that adolescence is a sensitive period because the impairments in behavior and excitatory drive were not observed following a comparable thalamic inhibition during adulthood. These results indicate that excitatory activity from the thalamus during adolescence is essential for thalamo-mPFC circuit development. This mirrors the findings in sensory sensitive periods, where thalamo-cortical inputs are compromised following sensory deprivation, ultimately leading to cortical restructuring2,37.
Adolescence is a key period of cortical maturation
We found that adolescence is a sensitive period for mPFC circuit development. Adolescence is known to be a period of vulnerability in the development of psychiatric disorders, such as schizophrenia, in humans11,12,38. Moreover, functional imaging studies have shown that thalamo-prefrontal hypoconnectivity, a finding in patients with schizophrenia, is already present in young adolescents at clinical high risk for the disorder14–19. We chose to inhibit thalamic activity in mice during the P20–50 window because it is known that the mPFC is maturing during this time38–45. In rodents, the volume of the mPFC peaks around P24 after which point it decreases, reflecting a period of dendritic pruning in mPFC pyramidal neurons, which peaks around P3046–48. It has been postulated that this volumetric change and pruning could result in part from refinement of thalamo-cortical synaptic contacts during this period49. Furthermore, one classic hypothesis originally presented by Feinberg states that in schizophrenia aberrant activity-dependent pruning during adolescence may lead to persistent changes in prefrontal circuit function13.
Although the literature describing the density of thalamo-mPFC during postnatal development is scarce, published data suggest that projections are already fully established at P10, followed by a decrease later on50. This suggests that adolescent inhibition mostly affects the stability of thalamo-mPFC projections and that projections that are not used during adolescence are pruned away. Moreover, the relative change in projection density between the control and developmentally manipulated animals is similar at P35 (39.4% reduction) and P90 (42.6% reduction), indicating that the difference in projection density between the groups comes primarily from thalamic inhibition early on, between P20–35.
Whether this reduction in thalamic input also leads to long-lasting changes in intra-cortical connectivity remains unclear. During the same time window, the mPFC undergoes changes in myelination and interneuron development, which together promote emergent changes in network activity and behavioral functionality6,38,39,51. Future studies will determine whether the decrease in excitatory events is fully explained by a decrease in thalamic input or whether there are changes within the mPFC at the level of intra-cortical synaptic connectivity. Our finding that the frequency and amplitude of inhibitory events is unaltered in the adult mPFC offers evidence against a strong involvement of interneurons in this process, although we did not determine whether cortical inhibition may have been altered during adolescence at the time of thalamic inhibition.
While we provide strong evidence that adolescence is a sensitive time window for thalamo-mPFC circuit maturation, the underlying mechanisms by which this occurs are unclear. The observation that activity is important for the development of thalamo-cortical projections suggests a Hebbian rule where projections that are not used are not stable, as discussed above.
Recent studies also point to intra-cortical mechanisms in the maturation of cortical circuitry. DREADD-mediated inhibition of anterior cingulate (ACC) to visual cortex projection neurons during adolescence has been shown to disrupt the maintenance of local connectivity within the ACC10. By analogy, if inhibition of thalamic inputs decreases activity of mPFC-thalamic projection neurons during adolescence, this may lead to a disruption in the maintenance of local excitation in the mPFC. A different mechanism has also been described within the visual cortex. DREADD-mediated inhibition of layer II/III neurons during the critical period of primary visual cortex development (P24–29) led to excitatory synaptic scaling and increased intrinsic excitability suggesting homeostatic plasticity as a mechanism affecting visual cortex maturation52. Future studies will be able to identify the exact plasticity mechanisms in mPFC circuitry that are induced by thalamic inhibition during adolescence.
Adolescent thalamic activity regulates adult mPFC circuitry
Thalamic projections to the mPFC are a crucial source of excitatory input to mPFC pyramidal cells. Following adolescent thalamic inhibition, we found reduced excitatory drive to mPFC pyramidal cells in adulthood. Due to the reduction in sEPSC frequency, but not amplitude, we hypothesized that this change was primarily driven by a reduction in pre-synaptic inputs and that decreased inputs from the thalamus may contribute to this change.
We confirmed this hypothesis through retrograde labelling. We believe this thalamo-mPFC projection reduction is due to a decrease in axonal arborization rather than thalamic cell numbers because DAPI staining in the thalamus was unchanged. This is consistent with the observation that the thalamus has already undergone a period of heightened apoptosis around P13, which is before the time window of our inhibition experiments49,50.
Adolescent thalamic inhibition had no effect on mPFC-projecting cells from another subcortical region, the BLA, indicating specificity to thalamo-cortical projections. This result is distinct from what has been observed after early developmental subcortical lesions, which showed a compensatory increase in BLA-mPFC projections following early postnatal (P7) ventral hippocampal lesions28. Since we used bilateral injections of retrograde virus to investigate the impact of developmental thalamic inhibition on cortical projections, we were unable to determine whether there were any changes to cortico-cortical contralateral projections. As already discussed above, future studies should examine whether intra-cortical connectivity is also affected.
Oscillatory activity cannot explain the behavioral outcomes
Previous studies have shown the importance of task-induced gamma for predicting behavioral performance during EDSS and that this signal is persistently disrupted following adolescent inhibition of mPFC PV interneurons6. Here, we also found that mPFC gamma power was correlated with behavioral performance in control animals, with elevated gamma in correct trials compared with incorrect trials, but this pattern was not affected by adolescent thalamic inhibition. Consistent with unchanged gamma power after developmental thalamic inhibition, we did not find any deficits in cortical inhibition in adult mice (assessed by recording sIPSCs in cortical pyramidal cells). This is different from what had been observed following adolescent PV inhibition6 and suggests that the long-term consequences of adolescent thalamic inhibition may not involve cortical PV interneurons.
Beta oscillatory activity has also previously been identified in thalamo-prefrontal manipulations, often in the context of working memory behaviors21,25. While we did find task-induced beta oscillations, these were not correlated with behavioral outcome in controls and were not affected by adolescent thalamic inhibition.
Altogether, these data suggest that, despite reduced thalamic inputs to the mPFC, oscillatory measures of the thalamo-mPFC circuitry cannot explain the deficits observed during the EDSS behavior. Thus, while these oscillations may be necessary for the proper execution of this task, they are not the only mechanism at play.
The thalamus supports mPFC outcome encoding
To determine whether adolescent thalamic inhibition disrupts single unit activity in the adult animal, we examined mPFC cell firing rates during EDSS trials. We found no changes in individual neuron firing rates across different trial types or throughout the trial. However, recent theories suggest that multiple neurons form ensembles that determine functional properties and outcomes, in ways beyond single neuron firing29,30.
When we studied the cross-correlations between cell pairs, we found that cross-correlations were disrupted following adolescent thalamic inhibition, which is reminiscent of what we measured after acute thalamo-mPFC inhibition in the adult21. This disruption was rescued by acute thalamic activation. Together, these results point to a role of thalamic inputs in enhancing mPFC cellular communication. The decrease in cross-correlations following adolescent thalamic inhibition is found in both correct and incorrect trials, which likely reflects the fact that the animals are learning throughout the task, receiving feedback during both types of trials. Given the positive correlation between peak cross-correlation in trial × and trial x+1 outcome regardless of trial × outcome, we further hypothesize that these cross-correlations allow the animals to incorporate feedback during both correct and incorrect trials. By contrast, following adolescent thalamic inhibition, the decreased cross-correlations across both trial types speaks to the animals’ inability to incorporate information during both correct and incorrect trials.
To further explore the effects on population encoding, we trained a linear decoding algorithm using a subset of trials to predict the EDSS trial outcome based on mPFC neuronal ensemble activity. Using this decoder, we were able to accurately predict trial outcome in control animals, but the decoding ability was down to chance levels following adolescent thalamic inhibition. Of note, this inability to decode does not necessarily imply that there is no information present in the activity. For example, there are several technical reasons why we may see a chance level decoder performance, including a high level of noise. Alternatively, hM4D animals with impaired thalamo-mPFC circuitry may be using a different circuit to perform the task, which could explain why activity of mPFC neurons no longer helps to predict trial outcome. Importantly, mPFC neurons regained the ability to encode task outcome after thalamic stimulation, suggesting that thalamic excitation rescues outcome encoding and task performance. Other thalamo-cortical circuits, namely in motor circuitry, have shown task-specific roles for both thalamic and cortical activity53. In addition, modifying activity of different mPFC cell types have also demonstrated task-specific roles for mPFC cellular subpopulations54. Meanwhile, thalamic input to the mPFC has been hypothesized to non-specifically amplify or sustain local mPFC connectivity and encoding23,24. This study supports the hypothesized role of the thalamus as a non-specific amplifier of mPFC cellular encoding during this cognitive flexibility task in two major ways. First, adolescent thalamic inhibition disrupted thalamo-mPFC projections in adulthood, which coincides with both reduced mPFC cellular cross-correlations and disrupted mPFC task outcome encoding. Second, non-specific thalamic activation, even in the context of fewer thalamic projections, during the EDSS was sufficient to restore cross-correlations and outcome encoding.
Prior studies have found that hyper-activation of mPFC neuronal populations can have detrimental effects, reflecting an inverted U-shaped pattern of mPFC neuronal activity and functional outcomes55,56. We found improved behavioral performance in the adolescent-inhibited animals and no change in performance in the control animals during thalamic excitation. By facilitating activity of thalamic neurons that are engaged in the task, thalamic SSFO activation, may not lead to the over-stimulation of mPFC neurons. This is consistent with our finding that thalamic SSFO activation does not increase overall firing rates in the mPFC. Moreover, this finding further supports the theory of the thalamus as a facilitator of mPFC ensemble activity as this manipulation may modulate activity of a subset of mPFC neurons without increasing overall mPFC activity.
The mPFC itself has been intensively studied in cognitive flexibility tasks, such as the ASST. Some studies have pointed to the post-decision period as a crucial point for the mPFC during the EDSS57. While we found mPFC encoding throughout the trial, the decoder performed better when using post-decision versus pre-decision period firing activity within a trial, indicating that the mPFC may indeed be particularly important in the period following the choice.
The mPFC and the thalamus are interconnected in cognition
Many of the analyses and interpretations in this study have focused on the impact of adolescent thalamic inhibition on adult mPFC functioning. However, separating mPFC function from thalamic function in the context of cognition is almost impossible as the two regions are reciprocally connected. Thus, changing activity in one part of the circuit will change activity in the other part of the circuit. In agreement with this tenet, we found that cognitive deficits were associated with reduced thalamo-mPFC projections.
In addition, an analysis of intrinsic thalamic cellular properties revealed no changes in ex vivo slice sEPSC frequency and excitability, nor did we observe changes in the in vivo power in the epsilon band (>100 Hz, a metric for multi-unit activity58). Together, these findings support a model in which the impact of transient thalamic inhibition during adolescence selectively disrupts thalamo-mPFC projections and prefrontal physiology, while the physiological properties of thalamic neurons are largely spared.
Concurrent thalamic excitation as a therapeutic intervention
Following thalamic inhibition during adolescence, we found persistent anatomical changes in thalamic projections to the mPFC. Nevertheless, we rescued the behavioral deficits by acutely exciting the thalamic in the adult mouse, even though this manipulation is unlikely to reverse the anatomical changes. It has previously been shown that exciting the thalamus during the delay of a prefrontal-dependent working memory and a 2AFC task enhanced performance in both tasks21,24, while inhibiting the thalamus impaired mPFC-dependent contextual switching23. Our data suggest that the thalamus plays a broader function in amplifying mPFC activity that is not restricted to delay-containing cognitive processes. This result offers a major insight into potential therapeutic interventions in this circuit, as it indicates that even with persistent changes in circuit anatomy, a relatively non-specific thalamic excitation may still be able to improve behavior.
Relevance for Human Disorders
Human imaging studies have pointed to the importance of the thalamo-prefrontal connectivity in cognitive functioning15–17. In patients with schizophrenia, deficits in cognition have been related to hypo-connectivity between the thalamus and PFC, which is already seen in young adolescents before their diagnosis14,18,19. Indeed, given our findings, these early changes in thalamo-cortical functioning observed in adolescent patients at clinical high risk or with early onset schizophrenia may lead to long-lasting consequences on PFC function, which may increase the likelihood of severe cognitive deficits. Given the relevance of thalamo-prefrontal circuitry in psychiatric disorders like schizophrenia, this study offers key mechanistic insights into the etiology of, and potential therapies for, these disorders.
Methods
Animal Husbandry.
All procedures were done in accordance with guidelines approved by the Institutional Animal Care and Use Committees at Columbia University and the New York State Psychiatric Institute (Protocol NYSPI 1499). Animals were housed under a 12h light-dark cycle in a temperature-controlled environment (72°F, humidity 30%−70%) with food and water available ad libitum, unless otherwise noted. Offspring of heterozygous GBX2-CreERT (Jackson Labs, #022135; back-crossed to C57/Bl6) and C57/Bl6 females (Jackson Labs, #000664) were used for most experiments. At postnatal day 10 (P10), tail samples were collected for genotyping (Transnetyx, Inc). At P13, GBX2-CreERT heterozygous mice were used for viral injections. Littermates were randomly assigned to each group, with random and equal distribution across males and females. Mice were housed together with dams and littermates. At P15 and P16, all offspring were given intraperitoneal (i.p.) injections of tamoxifen (Sigma-Aldrich, T5648), dissolved in corn oil, at 75mg/kg to induce Cre recombination. Offspring were weaned at P28 and group-housed with same-sex littermates (5 mice/cage).
For thalamic inhibition, mice were given i.p. injections of clozapine-N-oxide (CNO), dissolved in 0.9% saline, at 1mg/kg, twice per day. All mice were given CNO, regardless of viral vector or group. Throughout data collection and analysis, experimenters were blinded to the group of the animal. These injections took place every day P20–50 for adolescent inhibition and P90–120 for adult inhibition.
At P70, mice used for cell density studies were injected with virus, and mice used for in vivo optogenetic neurophysiology recordings during behavioral experiments were virally injected and implanted with optrodes. Implanted mice were subsequently housed in cages of 2–3 mice/cage.
All behavioral testing and in vivo recordings were done 40 days after the last CNO injection in adult mice. During behavioral training and testing, mice were food-restricted and maintained at 85% of their initial weight.
For the dual virus approach, C57/Bl6 males and females were bred, and all pups were used for the experiment. Surgeries were conducted at P13, and all mice injected from P20–50 i.p. with JHU37160 (a CNO analog59; 0.9% saline, 0.01mg/kg, twice/day). We based sample sizes on previous experiments and no statistical methods were used to calculate sample sizes6, 26, 60–61.
Surgical procedures.
For viral injections at P13, mice were anesthetized with ketamine (4mg/ml) and xylazine (0.6mg/ml) and head-fixed in a stereotactic apparatus (Kopf). Mice were injected bilaterally in the midline thalamus with AAV5-hSyn-DIO-hM4D-mCherry (Addgene #44362) or a control virus, either AAV5-hSyn-DIO-EGFP (Addgene #50457) or AAV5-hSyn-DIO-mCherry (Addgene #50459) at a volume of 0.25 μl (0.1μl/min).
For the dual virus approach surgeries at P13, mice were injected bilaterally in the midline thalamus, with AAV5-hSyn-DIO-hM4D-mCherry or the control, AAV5-hSyn-DIO-EGFP. They were also injected bilaterally in the mPFC with rgAAV-hSyn-Cre-WPRE-hGH (Addgene #105553) at a volume of 0.25 μl (0.1μl/min).
P13 coordinates: thalamus: −1.0 AP, ±0.25 ML, −3.0 DV (skull at bregma), and mPFC: −0.92 AP, ±0.13 ML, −1.45 DV (skull at bregma). For surgeries at P70, mice were anesthetized with ketamine (10 mg/ml) and xylazine (1mg/ml). Mice were injected bilaterally into the mPFC ay +1.8 AP, ±0.35 ML, −2.5 DV (bregma) with retrograde AAV-CAG-GFP (Addgene #37825) at a volume of 0.25μl (0.1μl/min).
For in vivo optogenetic neurophysiology experiments, mice were anesthetized with isoflurane. All mice were injected bilaterally into the midline thalamus at −1.2 AP, ±0.35 ML, −3.2 DV (skull at bregma) with AAV5-CaMKII-hChR2(C128S/D156A)-EYFP (University of North Carolina Vector Core; 0.4μl, 0.1μl/min). During the same surgery mice were implanted with an optrode, consisting of a 36-channel narrow electronic interface board (Neuralynx, Bozeman, MT), a single stereotrode bundle, additional local field potential (LFP) wires, and 2 flat tipped, ferrule-coupled optical fibers (0.22 NA, 200μm diameter). Stereotrodes for recording spikes were made from 13μM tungsten fine wire (California Fine Wire, Grover Beach, CA) and were coupled to one 50μM tungsten wire for recording LFPs. This stereotrode bundle was then unilaterally targeted to the left mPFC (+1.85 AP, −0.35 ML, −1.4 DV (brain). Another 50μM tungsten wire was glued to the left optical fiber in the midline thalamus, extending 450μm below the tip of the fibers. Skull screws placed over the cerebellum and olfactory bulb served as ground and reference, respectively, while spikes were referenced to a local mPFC stereotrode wire.
Behavioral procedures.
All behavioral tasks were conducted during the light cycle. At P90, mice were gradually restricted to 85% of their body weight.
Non-Match to Sample working memory task:
Eight operant-conditioning chambers (ENV-307A; Med Associates, Georgia, VT) were used (15.24cm long × 13.34cm wide × 12.7cm high). Each chamber was housed in a sound-attenuated box and equipped with two retractable levers (ENV-312-3M) on the front wall, with one milk dipper between them (ENV-302RM-S). The back wall contained one noseport (ENV-313M) directly opposite to the milk dipper, which delivers 1 drop of evaporated milk (0.01ml). A 1.0A house light was positioned directly above the noseport. A computer (COM-106-NV, Intel i5-7400) controlled and recorded all experimental events and responses via an interface (MED-SYST-16e-V). Med-PC V programs were used to administer and record the task. Mice were first given 2 days of habituation to the milk dipper, followed by 7 days of training to associate a lever press with a milk reward. Lastly, they were given 5 days of noseport training before beginning the acquisition stage.
During acquisition, each trial began with the house light being turned on and an illuminated noseport to signal an initial noseport entry. Noseport entry triggered the start of the sample phase with sample lever presentation in a pseudo-random order. After sample lever press, the noseport was re-illuminated (following a 0-second delay) signaling a second noseport entry. Following the second noseport entry, the choice phase began, and both levers were presented. If the animal pressed the opposite lever to the sample lever of that trial (non-match), the trial was recorded as “correct,” and a milk reward was given. If the animal pressed the same lever as the sample, the trial was recorded as “incorrect,” and the dipper was not presented. This final step was followed by a 10-second inter-trial interval (ITI) during which the house light was turned off. Acquisition was repeated every day with 120 trials per day. For the final 3 days, the total number of trials was increased to 160 trials. Throughout the experiment, mice were given unlimited time to complete the required trials. All mice achieved a criterion level of performance, defined as 3 consecutive days above 70% correct.
Attentional Set Shifting cognitive flexibility task:
Mice were habituated to the testing arena on day 1. On days 2–3, they were trained to dig in both bedding media (corn cob and paper pellet, both unscented) to obtain a food reward. Once mice dug reliably, testing began. For each trial, mice were placed at the opposite end from 2 terra cotta bowls containing different odor/medium combinations. For initial acquisition (IA), mice needed to learn that the cinnamon scent, not the paprika scent, predicted a Honey Nut Cheerio reward, irrespective of the bedding media. For the first 5 trials, mice could explore both bowls until they found the reward, but the trial was only scored as correct if the animal initially chose the correct bowl. From the 6th trial onward, once the mouse began digging in a bowl, the entrance to the other bowl was closed off. Criterion was reached when the mouse made eight of ten consecutive correct choices. If the mouse did not meet criterion in 30 trials, the animal did not advance to the next stage (one animal from the adolescent manipulation hM4D group did not meet the IA criterion). If the mouse did reach criterion, extra-dimensional set shifting (EDSS) began. In EDSS, the animal needed to learn that the type of bedding medium (paper pellets, not corn cobs) predicted the Honey Nut Cheerio reward, irrespective of odor. Criterion was reached with eight of ten consecutive correct choices.
For optogenetic experiments, mice completed the task twice, 10 days apart. Animals were randomized to receive the light ON or OFF on Run Day 1 or Run Day 2 during EDSS. For Run Day 1, the rules in IA and EDSS were cinnamon rewarded in IA and paper rewarded in EDSS. For Run Day 2, the rule in IA was the same as EDSS for Run Day 1, with paper rewarded. The rule in EDSS for Run Day 2 was odor (paprika) predicting the reward. For EDSS on the second run, mice that previously had the light ON for Run Day 1 had the light OFF and vice versa. There was no effect of Run Day on overall performance; therefore, light conditions were pooled across runs for analysis.
Optogenetic Parameters.
In optogenetic stabilized step-function opsin (SSFO) experiments, for the light ON run, a 5s blue light pulse (473nm, 4mW) was used for opsin activation prior to the first EDSS trial. Light was delivered via flat tipped 200μm diameter, 0.22 NA optical fibers. To ensure continued opsin activation throughout EDSS, the 5s pulse was repeated between trials every 30min.
Slice Electrophysiology.
Whole-cell current and voltage clamp recordings were performed in layer II/III mPFC pyramidal cells and MD. Recordings were obtained with a Multiclamp 700B amplifier (Molecular Devices) and digitized using a Digidata 1440A acquisition system (Molecular Devices) with Clampex 10 (Molecular Devices) and analyzed with pClamp 10 (Molecular Devices). Following decapitation, 300μM slices containing mPFC or MD were incubated in artificial cerebral spinal fluid (ACSF) containing (in mM) 126 NaCl, 2.5 KCl, 2.0 MgCl2, 1.25 NaH2PO4, 2.0 CaCl2, 26.2 NaHCO3, and 10.0 D-Glucose, bubbled with oxygen, at 32°C for 30min before being returned to room temperature for at least 30min prior to use. During recording, slices were perfused in ACSF at a rate of 5mL/min. Electrodes were pulled from 1.5mm borosilicate-glass pipettes on a P-97 puller (Sutter Instruments). Electrode resistance was typically 3–5 MΩ when filled with internal solution consisting of (in mM): 130 K-Gluconate, 5 NaCl, 10 HEPES, 0.5 EGTA, 2 Mg-ATP, and 0.3 Na-GTP (for thalamic recordings; pH 7.3, 280mOsm) or 130mM Cs-Gluconate, 10 HEPES, 2 MgCl2, 0.2 EGTA, 2.5 MgATP, 0.3 NaGTP, and 5 Lidocaine N-ethyl bromide (for pyramidal cell recordings; pH 7.3, 280mOsm).
MD recordings:
Animals were sacrificed at P35 or P105 after either receiving CNO for 2 weeks or not. hM4D (mCherry-tagged) or GFP-infected thalamic cells were identified by their fluorescence at 40x magnification under infrared and diffusion interference contrast microscopy using an inverted Olympus BX51W1 microscope coupled to a Hamamatsu C8484 camera. Intrinsic and active membrane properties (resting membrane potential, input-output firing frequency curve) were recorded in current clamp using the K-Gluconate intracellular solution detailed above before and after 10μM CNO was bath applied to the slice.
mPFC recordings:
Animals were sacrificed for recordings at P90 for the adolescent and P160 for the adult manipulation. mPFC pyramidal cells were visually identified based on their shape and prominent apical dendrite at 40x magnification under infrared and diffusion interference contrast microscopy. Spontaneous excitatory post-synaptic currents (sEPSCs) were recorded in voltage clamp at a holding potential of −55mV and spontaneous inhibitory post-synaptic currents (sIPSCs) were recorded in voltage clamp at a holding potential of +10mV. 60s of the current recording for each condition was analyzed. Recordings were filtered with an eight-pole low-pass Bessel filter, and sEPSCs and sIPSCs were detected using MiniAnalysis (Synaptosoft). All event data was averaged by cell.
In vivo electrophysiology.
In vivo electrophysiology recordings were performed while the animals were performing the ASST. Field potential signals from the mPFC and MD were referenced against a screw implanted in the anterior portion of the skull above the olfactory bulb. Recordings were amplified, band-pass filtered (1–1000Hz LFPs; 600–6000Hz spikes) and digitized using a Digital Lynx system (Neuralynx). LFPs were collected at 2kHz, while spikes were detected by online thresholding, collected at 32kHz, and sorted off-line. TTLs were manually inserted to record the timing of relevant events (e.g., trial start, decision point, trial end).
Histology.
Adult mice were anesthetized with 100mg/kg ketamine and 5mg/kg xylazine (i.p.). For in vivo electrophysiology experiments, electrolytic lesions were induced at each recording site by passing current (50μA, 30s) through electrodes prior to perfusion. All animals were perfused with 4% paraformaldehyde in PBS. Brains were post-fixed in 4% PBS overnight before being transferred to 1% PBS for long-term storage. Brains were sectioned serially at 50μm on a vibratome (Leica, Buffalo Grove, IL, USA). The following primary antibodies were used: mCherry (rabbit-anti-dsred; Takara Bio, Mountainview, CA, USA; 632496, 1:250) or green fluorescent protein (GFP; Abcam, Cambridge, UK, ab13970, 1:1000). Primary antibody incubation was 48h at 4°C. Alexa Fluor-conjugated secondary antibodies (donkey anti-rabbit Alexa Fluor-546 and goat anti-chicken Alexa Fluor-488, Invitrogen, 1:1000) were used for secondary detection. Stained tissue slices were then mounted on slides with Vectashield containing DAPI (Vector Labs). Viral expression was confirmed from mCherry or GFP staining, and locations of recording site lesions were confirmed under DAPI.
Stereology was used to assess retrogradely-labeled cell numbers in the MD and BLA in adult developmental manipulation and control animals using StereoInvestigator software (MBF Biosciences, Williston, VT, USA). Every 3rd slice was used, and regions were traced using DAPI staining. During image acquisition and quantification, the investigator was blind to the treatment group.
LFP and single-unit analysis.
Neuralynx files containing LFP, and spike data were imported into Matlab with Neuralynx MATLAB import/export package v4.10.
LFP samples were notch filtered using the MATLAB Chronux package to remove 60-cycle noise (http://chronux.org/; rmlinesmovingwinc.m). Mechanical artifacts were eliminated by removing samples whose voltage was more than 3 standard deviations from the entire signal mean. The cleaned signal was then root-mean-squared. Power and coherence were calculated using the wavelet transformation package in MATLAB. These values were averaged over the relevant time windows (e.g., 6s before the decision point). Frequency ranges were defined as 40–90Hz for gamma and 12–30Hz for beta.
Single units were clustered using Klustakwik (Ken Harris) based on spike sorting of the first two principal components, peak voltage and energy from each stereotrode channel. Clusters were then accepted, merged or removed based on isolation distance, visual inspection of feature segregation, inter-spike interval distribution, cross-correlation in spike timing for simultaneously recorded units, and stability across the recording session.
To analyze the phase-locking of single cells in the mPFC with the LFP in the thalamus in the beta range, we calculated the pairwise phase comparison (PPC)62 of mPFC spikes to thalamic LFP. The LFP signal was first digitally bandpass-filtered (12–30Hz) using a zero-phase-delay filter (filter0, K. Harris and G. Buzsaki), and the Hilbert transform of the bandpass-filtered signal was calculated to obtain the oscillatory phase. The magnitude of the phase-nonuniformity of spike times relative to the filtered LFP oscillation was then calculated for the 6s before the decision point in correct and incorrect trials. The 6s period was chosen based on previous findings in that window4. However, we found similar results when looking in the 6s period after the decision point or the full 12s window. To avoid spuriously high or low PPC values, only units that fired at least 50 spikes in each condition were used.
Statistics.
Statistical analysis and graph preparations were done using Prism 9 software (Graphpad Software, San Diego, CA, USA) or custom scripts in MATLAB (Mathworks, Natick, MA, USA) and Python. One-way ANOVA, two-way repeated measures ANOVA, and unpaired or paired two-tailed t-tests were used to analyze slice physiology, behavior, cell density, and single unit firing rates, with equal variances and normal distributions found for each analysis. For the slice physiology acute CNO experiment, Holm-Sidak post-hoc analyses were used to compare the hyperpolarization upon bath application of CNO for all hM4D groups to the control. For the optogenetic behavior, Holm-Sidak post-hoc analyses were used to compare light off vs. light on outcomes.
To analyze differences in gamma power, we fit linear mixed models with gamma power as outcome. The random effect was animal, and the fixed effect was either trial (ITI vs. trial) or trial outcome type (correct vs. incorrect). Power as a function of frequency was plotted by averaging the gamma power across the 6s before the decision point. Mean power or coherence was calculated for those 6s for the range of 40–90Hz for gamma, or 12–30Hz for beta.
For analyzing firing rates data were binned into 50ms windows. Firing rates were smoothed for analysis (where indicated) by taking the average firing rate of the surrounding 5 bins (i.e., 250ms).
To represent z-scored firing rates, the mean and standard deviation was calculated for the firing rate for all EDSS ITI time bins. Smoothed firing rates for each time bin for the 12s surrounding the decision in each trial were calculated using the ITI mean and standard deviation. The mean z-score was then taken across all trials for each time bin. Mean firing rates were taken for each cell across the 12s surrounding the decision of each trial. Mean firing rates were calculated first for all trials. Then, the mean firing rate was taken for each trial outcome type (correct vs. incorrect). Paired t-tests were used to compare the firing rates across trial types.
For cross-correlations, firing rates were binned into 50μs windows. For each trial, the 12s surrounding the decision point was taken, and the spike train for each trial was concatenated with the trains for that cell and trial outcome type. The firing for each spike train was normalized to overall firing rate, and the Matlab function, xcorr, was applied to all pairs of cells within each animal, using a maximum lag time of ±80ms. The peak cross-correlation value for each cell pair was used in the analysis, with each cell pair having a peak cross-correlation during correct and incorrect trials. We then fit a linear mixed model with peak cross-correlation as outcome, fixed effects of group (control, hM4D, hM4D Light ON) and trial outcome type (correct vs. incorrect), and random effects of animal and cell. Since the analysis requires cell pairs, certain animals were removed from the analysis if they had only one isolated cell (Control: 2 animals out of 8; hM4D: 3/12; hM4D Light ON: 2/13).
Decoder.
The linear decoder is custom-written in Python. Smoothed firing rates for the 12 s around the decision (described above) and the trial outcome (i.e., correct or incorrect) were used for each trial for the decoder. The analysis was done during IA and EDSS of the ASST task. Certain animals were removed from the analysis if they had fewer than 2 neurons or fewer than 2 of each trial outcome (EDSS: Control: 4; hM4D: 5; hM4D Light ON: 4; IA: Control: 5). The decoder algorithm was based on linear classifiers trained on pseudo-simultaneous population activity created by combining 50ms-binned neural patterns recorded from different animals performing the same behavioral task. The decoding algorithm was cross-validated and tested against a null model with shuffled trial condition labels.
Cross-validation:
We computed the decoding performance using a 20-fold cross validation (CV) scheme. For each CV fold, we randomly selected half of the trials of each condition and used them to build pseudo-simultaneous (PS) activity (see below) which was used to train a Support Vector Machine (SVM) with a linear kernel to classify PS patterns into one of the two conditions. Similarly, the remaining half of the trials were used to build PS activity that was used to test the trained SVM. The decoding performance was then assessed as the mean accuracy on the test set over the CV folds.
Pseudo-population:
To build pseudo-populations, we randomly selected 50ms binned neural patterns from training and testing trials of all animals and concatenated them to form a larger pseudo-simultaneous neural pattern. To obtain training and testing data sets used in the cross-validation scheme, this procedure was repeated 10xN times per condition, where N is the total number of neurons.
n-time-bins decoding:
To increase the signal to noise ratio of the decoder, we used a procedure where the decoder is trained to classify groups of n time bins sampled from the two conditions (n=1 corresponding to standard single time-bin decoding). In practice, this was done when building pseudo-population activity by randomly sampling n different time bins for each individual animal to build a single pseudo-simultaneous time bin. Unless specified otherwise, we used n=5.
Null model and p-value:
All decoding performance values were tested against M repetitions of a null model by shuffling the condition labels of individual trials. After each shuffle of the labels, the exact same decoding procedure described above was repeated on the shuffled data. Unless specified otherwise, we used M=1000. The p-value associated to the decoding performance was computed by comparing the performance of the shuffled model to the performance of the data.
Implementation:
The analysis was performed in Python3, using a linear classifier based on a support vector machine with custom-written Python scripts based on the scikit-learn SVC package63.
Data availability.
The source data that support the findings of this study are available on figshare: https://figshare.com/projects/Benoit_Kellendonk_NN-A76458A/135581 or from the corresponding author upon reasonable request.
Code availability.
Med-PC V, MATLAB, and Python code used for administering the behavior and analysis of the data that support the findings of this study is available on figshare: https://figshare.com/projects/Benoit_Kellendonk_NN-A76458A/135581 or from the corresponding author upon reasonable request.
Extended Data
Extended Data Fig. 1. Extended Data 1. A chemogenetic approach to reversibly and chronically inhibit thalamic cells during development or adulthood.
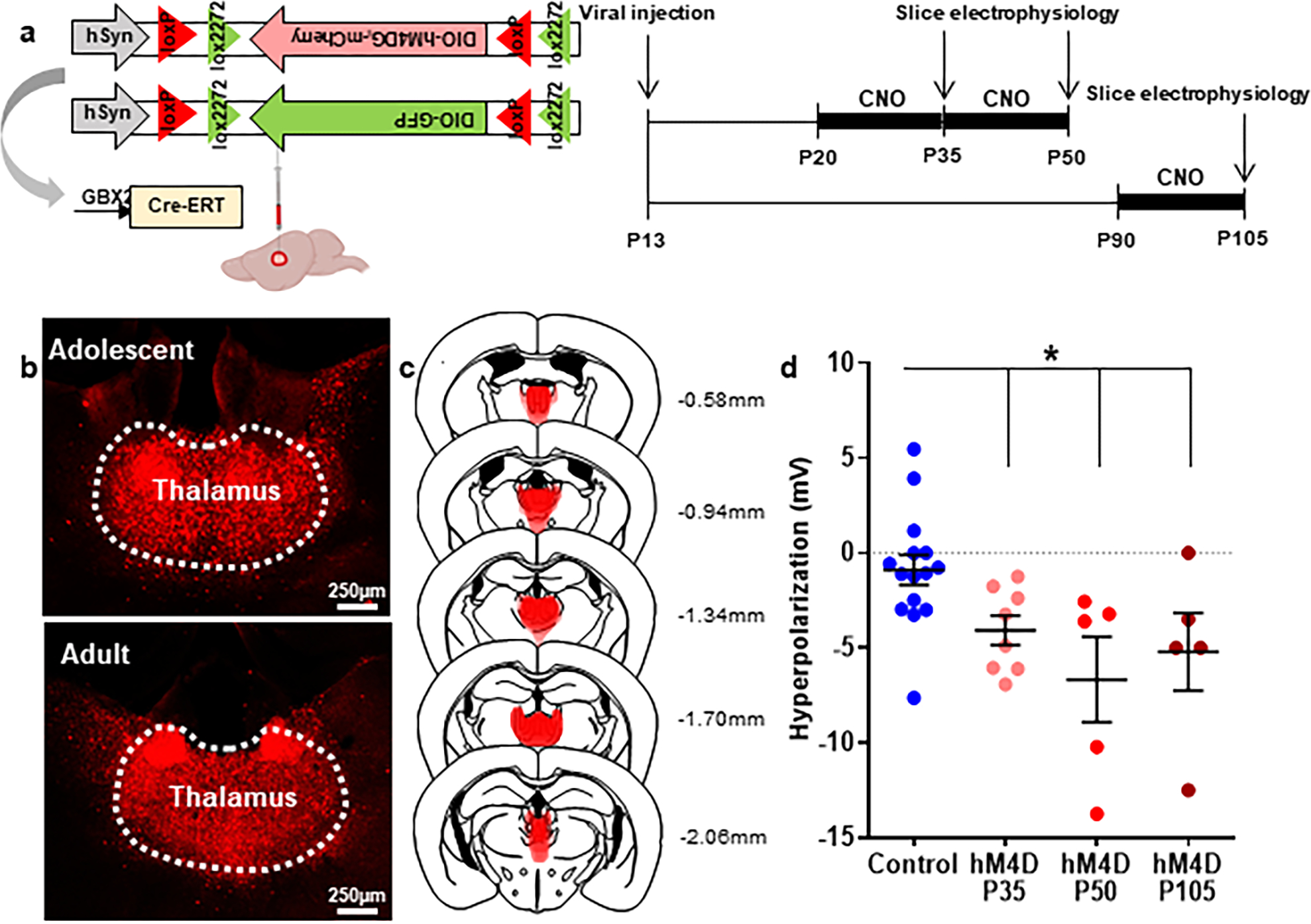
(a) Experimental design and timeline. Mice were injected with virus at P13, and whole cell patch clamp recordings were made at P35, P50, or P105 in cells expressing hM4D-mCherry or control cells at baseline and in response to bath application of 10 μM. Animals expressing hM4D were given twice daily 1 mg/kg CNO i.p. injections for 15 days (P20–35 and P90–105) or 30 days (P20–50). Created with Biorender.com. (b) Example images illustrating hM4D-mCherry expression in the midline thalamus in adolescent and adult animals. Histology images were collected from each animal for each cohort, with at least 6 slices taken for each animal and at least 8 adolescent and 5 adult animals. (c) Superimposed traces of hM4D-mCherry viral spread (red shading) relative to mediodorsal and midline thalamic nuclei (dashed black lines) in coronal slices. Distance from bregma listed beside each coronal slice. (d) Quantification of CNO-induced hyperpolarization. Control cells at P35 and P105 were pooled because CNO did not show an effect at either age. CNO induced a significant hyperpolarization in P35, P50, and P105 cells expressing hM4D relative to control cells. Dots indicate individual cell responses and bars indicate mean ± SEM. Control: n=15 cells, 5 animalshM4D, P35 (CNO P20–35, 15 days): n=8 cells, 3 animals; hM4D, P50 (CNO P20–50, 30 days): n=5 cells, 2 animals; hM4D, P105 (CNO P90–105, 15 days): n=5 cells, 3 animals; 1-way ANOVA, effect of treatment F(3, 29)=4.573, p=0.0097; Holm-Sidak post-hoc, Control vs. hM4D P35 *p=0.0460, Control vs. hM4D P50 **p=0.0095, Control vs. hM4D P105 *p=0.0459. *p<0.05.
Extended Data Fig. 2. Extended Data 2.

a) Adolescent-inhibited hM4D animals have a significantly worse performance during the first 8 days of the NMS task, particularly at day 4. Control: n=17 animals; hM4D=21 animals. 2-way rmANOVA, effect of time F(3.129,112.7)=87.66, p<0.0001, effect of group F(1,36)=4.575, *p=0.0358, effect of group × time F(7,252)=1.546, p=0.1523; Holm-Sidak post-hoc analysis day 4, *p=0.0456. Learning curves depict mean performance ± SEM each day. b) In the NMS task, mobility and motivation were unaffected by adolescent (top) or adult (bottom) thalamic inhibition: mean trial length, adolescent: Control: n=17 animals, 18.94±1.61s, hM4D: n=21 animals, 19.03±1.26s; two-sided unpaired t-test: t=0.04228, df=36, p=0.9665; adult: Control: n=6 animals, 19.41±4.19s, hM4D: n=10 animals, 19.05±1.50s; two-sided unpaired t-test: t=0.09631, df=14, p=0.9246, (c) sample lever-choice lever press latency, adolescent: Control: 4.932±0.199s, hM4D: 5.372±0.252s; two-sided unpaired t-test: t=1.321, df=36, p=0.1949; adult: Control: 5.304±0.416s, hM4D: 4.999±0.102s; two-sided unpaired t-test: t=0.8949, df=14, p=0.3875, (d) latency to collect reward, adolescent: Control: 0.5781±0.0143s, hM4D: 0.5801±0.0125s; two-sided unpaired t-test: t=0.1086, df=36, p=0.9141; adult: Control: 0.6091±0.0396s, hM4D: 0.6137±0.0233s; two-sided unpaired t-test: t=0.1064, df=14, p=0.9168, and (e) percentage of rewards retrieved, adolescent: Control: 99.78±0.05%, hM4D: 99.71±0.07%; two-sided unpaired t-test: t=0.7668, df=36, p=0.4482; adult: Control: 99.36±0.21s, hM4D: 99.62±0.10s; two-sided unpaired t-test: t=1.199, df=14, p=0.2503. f) In the ASST, mobility and motivation were unaffected by adolescent (top) or adult (bottom) thalamic inhibition: median latency to dig during IA (adolescent: Control: n=14 animals, 24.29±5.84s, hM4D: n=16 animals, 32.47±8.91s; two-sided unpaired t-test: t=0.7448, df=28, p=0.4626; adult: Control: n=20 animals, 53.50±9.41s, hM4D: n=17 animals, 45.44±6.95s; two-sided unpaired t-test: t=0.6682, df=35, p=0.5084) or (g) SS (adolescent: Control: n=14 animals, 34.57±8.39s, hM4D: n=15 animals, 32.07±4.32s; two-sided unpaired t-test: t=0.2708, df=27, p=0.7886; adult: Control: n=20 animals, 90.20±12.58s, hM4D: n=17 animals, 78.44±13.70s; two-sided unpaired t-test: t=0.6323, df=35, p=0.5313). h) EDSS error type breakdown, perseverative (P) and random (R), was unaffected. Adolescent inhibition (top) caused increased P and R errors (Control: n=14 animals, hM4D: n=15 animals; 2-way rmANOVA, effect of group F(1,27)=4.215, *p=0.0499). Adult inhibition (bottom) caused no change in error type (Control: n=20 animals, hM4D: n=17 animals; 2-way rmANOVA, effect of group F(1,35)=1.369, p=0.2499). Dots represent individual animals; lines represent mean ± SEM. *p<0.05
Extended Data Fig. 3. Extended Data 3. Thalamo-mPFC projection activity during adolescence is required for adult cognitive flexibility.
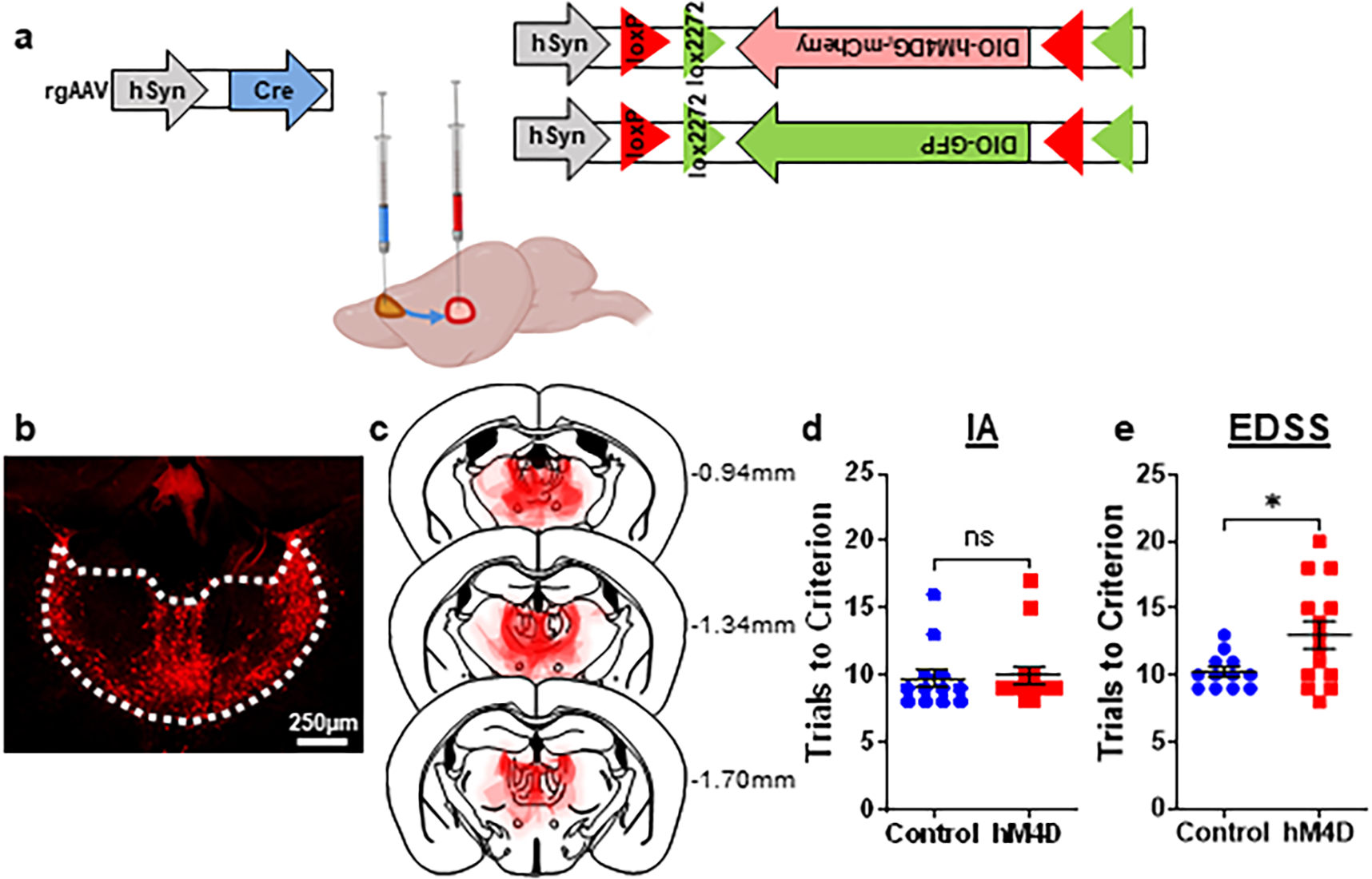
(a) Schematic for viral injections to target thalamo-mPFC projections. At P13, one virus was injected into the mPFC containing a retrogradely transported Cre driver, and another virus was injected into the thalamus containing floxed, Cre-dependent hM4D or the control GFP. Created with Biorender.com. (b) Example image illustrating hM4D-mCherry expression in the thalamo-mPFC projections. Histology images were collected from each animal, with at least 6 slices taken for each animal (n=14 animals). (c) Superimposed traces of hM4D-mCherry viral spread (red shading) relative to mediodorsal and midline thalamic nuclei (dashed black lines) in coronal slices. Distance from bregma listed beside each coronal slice. (d) Adolescent-inhibited hM4D animals are no different than controls in the IA portion of the ASST. Control: n=12 animals, 9.75±0.70 trials; hM4D: n=14 animals, 10.00±0.70 trials; two-sided unpaired t-test, t=0.2507, df=24, p=0.8042. Dots represent individual animals; lines represent mean ± SEM. (e) Adolescent-inhibited hM4D animals take significantly more trials in the EDSS to reach criterion than controls. Control: n=12 animals, 10.25±0.37 trials; hM4D: n=14 animals, 13.00±1.02 trials; two-sided unpaired t-test, t=2.385, df=24, *p=0.0254. Dots represent individual animals; lines represent mean ± SEM. *p<0.05.
Extended Data Fig. 4. Extended Data 4. Thalamic activity in adolescence is not required for thalamic cell activity in adulthood.
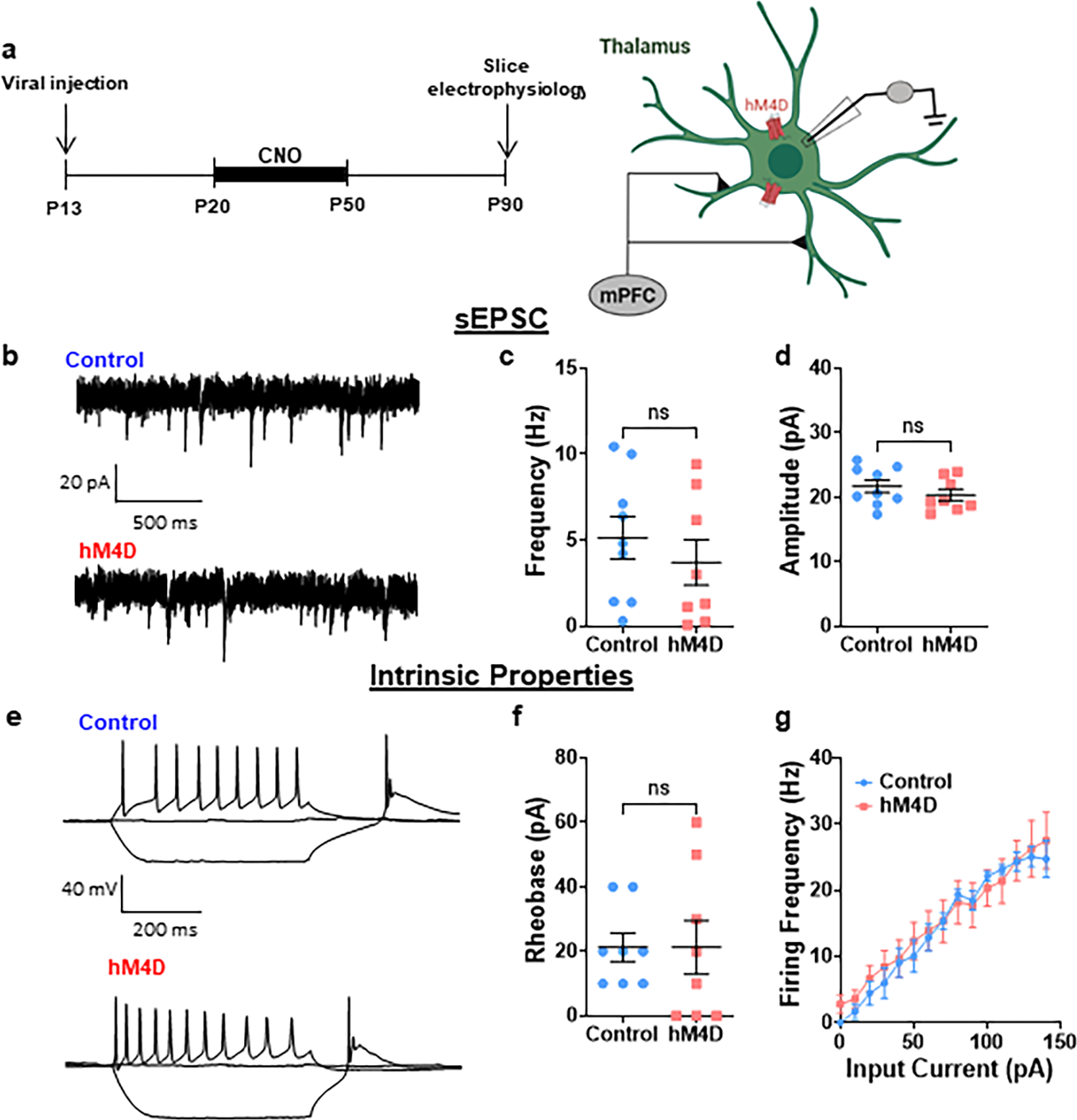
(a) Adolescent experimental timeline and schematic. Whole cell patch clamp recordings were made from thalamic cells from control and hM4D mice. These cells receive inputs from the mPFC and express the control or hM4D virus. Created with Biorender.com. (b) Representative traces showing spontaneous excitatory post-synaptic currents (sEPSCs). sEPSC (c) frequency and (d) amplitude are unchanged following adolescent thalamic inhibition relative to control mice. Control: n=9 cells, 3 animals; hM4D: n=8 cells, 2 animals; frequency: Control: 5.131±1.234 Hz, hM4D: 3.710±1.318 Hz; two-sided unpaired t-test, t=0.7874, df=15, p=0.4433; amplitude: Control: 21.70±0.98 pA, hM4D: 20.32±0.89 pA; two-sided unpaired t-test, t=1.023, df=15, p=0.3227. (e) Representative traces showing events in current clamp recordings. (f) Rheobase is unchanged following adolescent thalamic inhibition, as is (g) firing frequency in response to input currents. Control: n=8 cells, 3 animals; hM4D: n=8 cells, 2 animals; rheobase: Control: 21.25±4.41 pA, hM4D: 21.25±8.33 pA; two-sided unpaired t-test, t=0.000, df=14, p>0.9999; input current vs. firing frequency: 2-way rmANOVA, effect of input current F(2.484,34.59)=48.33, p<0.0001, effect of group F(1,14)=0.08940, p=0.7693, effect of group × input current F(14,195)=0.4870, p=0.9383. Curves depict mean firing frequency ± SEM for each input current. For all other plots, dots represent individual animals; lines represent mean ± SEM.
Extended Data Fig. 5. Extended Data 5. Density of thalamic projections to the mPFC was already decreased at P35.
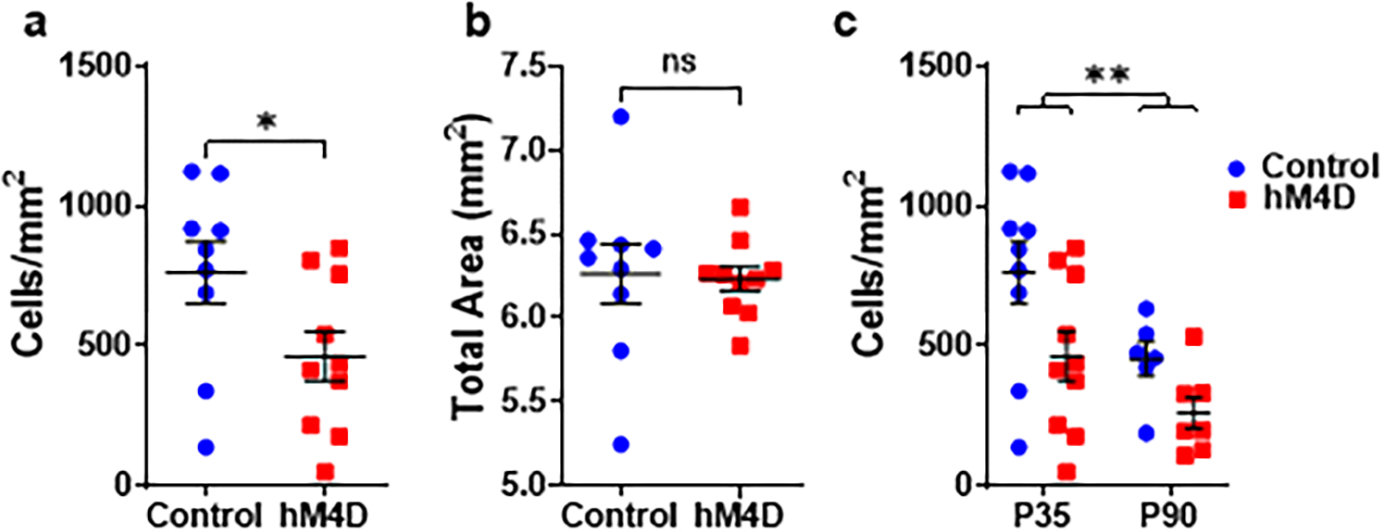
(a) After twice daily CNO injections from P20–35, stereological estimates of thalamo-mPFC projections at P35 showed a significant decrease in density in adolescent-inhibited hM4D animals compared to controls (Control: n=9 animals, 762.3±110.9 cells/mm2, hM4D: n=10 animals, 462.1±87.21 cells/mm2; two-sided unpaired t-test, t=2.149, df=17, *p=0.0463). (b) Meanwhile, there were no differences in overall thalamic area (Control: n=9 animals, 6.262±0.176 mm2, hM4D: n=10 animals, 6.231±0.072 mm2; two-sided unpaired t-test, t=0.1712, df=17, p=0.8661). (c) Moreover, there is a higher density of thalamo-mPFC projections at P35 than at P90 (P35: Control: n=9 animals, hM4D: n=10 animals; P90: Control: n=6 animals, hM4D: n=7 animals; 2-way rmANOVA, effect of age F(1,28)=7.731, **p=0.0096, effect of group F(1,28)=7.205, *p=0.0121, effect of age × group F(1,28)=0.3405, p=0.5642). Dots represent individual animals; lines represent mean ± SEM. *p<0.05, **p<0.01
Extended Data Fig. 6. Extended Data 6.
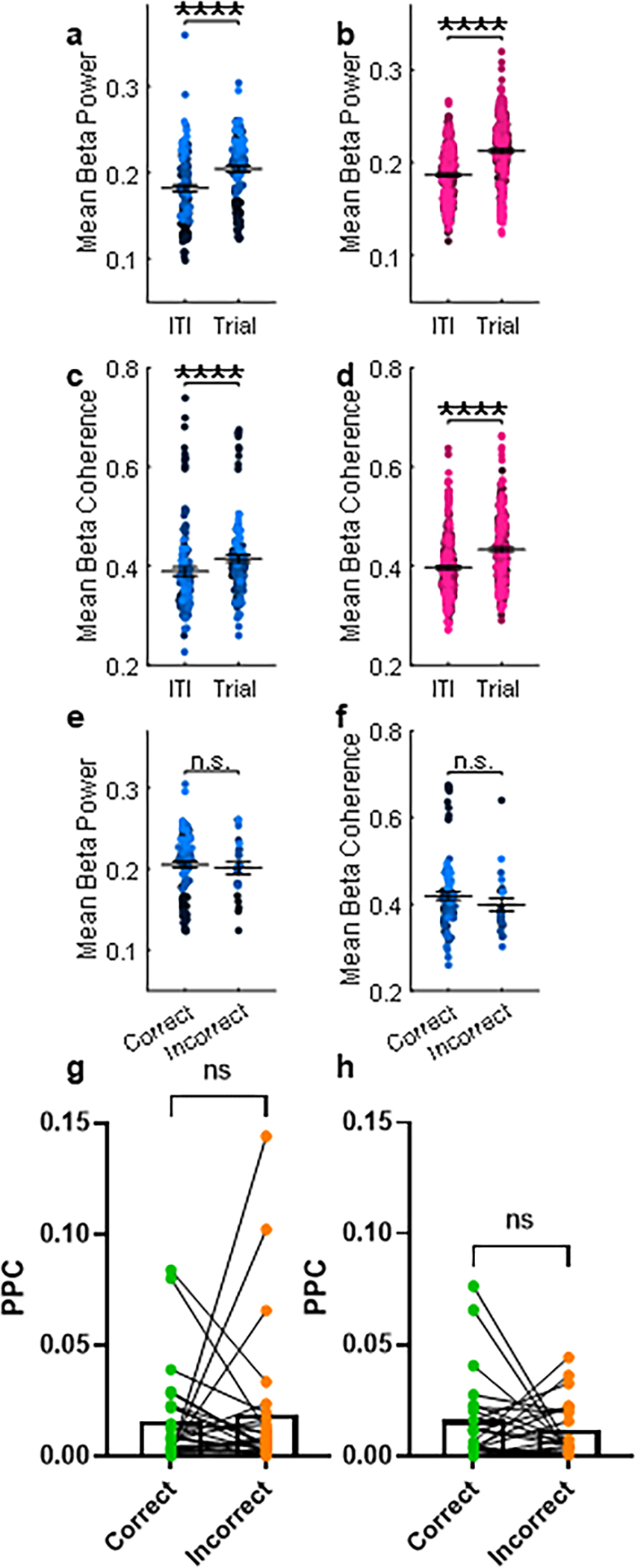
Mean thalamic beta (12–30 Hz) power is specifically enhanced during the EDSS trials compared to the ITI for (a) control (n=10 animals, ITI: 0.1822±0.0042; Trial: 0.2044±0.0036; linear mixed effects model (power~trial+(1|animal)): fixed effect (Trial), ****p=3.2642e-16) and (b) hM4D (n=15 animals, ITI: 0.1866±0.0018; Trial: 0.2124±0.0022; linear mixed effects model (power~trial+(1|animal)): fixed effect (Trial), ****p=2.0872e-41) animals. (c) As in (a) except for mean beta (12–30 Hz) mPFC-thalamic coherence (n= 10 animals, ITI: 0.3890±0.0090; Trial: 0.4146±0.0080; linear mixed effects model (coherence~trial+(1|animal)): fixed effect (Trial), ****p=2.0137e-07). (d) As in (b) except for mean beta mPFC-thalamic coherence (n=15 animals, ITI: 0.3968±0.0041; Trial: 0.4331±0.0044; linear mixed effects model (coherence~trial+(1|animal)): fixed effect (Trial), ****p=6.7099e-19). (e) Mean thalamic beta power (n=10 animals, 88 correct trials, 23 incorrect trials, Correct: 0.2052±0.0041; Incorrect: 0.2011±0.0075; linear mixed effects model (power~trial type+(1|animal)): fixed effect (Trial Type), p=0.18827) and (f) beta mPFC-thalamic coherence (n=10 animals, Correct: 0.4188±0.0092; Incorrect: 0.3984±0.0152; linear mixed effects model (coherence~trial type+(1|animal)): fixed effect (Trial Type), p=0.72808) are unchanged across trial types in controls. Dots represent individual trials for each animal (colors of the dots). Lines and error represent mean ± SEM. g) Pairwise phase consistency (PPC) values show no differences between phase-locking of mPFC cell firing and thalamic beta oscillatory activity in control (n=6 animals, 27 cells, Correct PPC: 0.01575±0.00420; Incorrect: 0.01904±0.00642; two-sided paired t-test: t=0.4114, df=26, p=0.6841) or (h) hM4D (n=7 animals, 22 cells, Correct: 0.01623±0.00441; Incorrect: 0.01205±0.00561; two-sided paired t-test: t=0.7443, df=21, p=0.4649) animals. Dots represent individual cells, with lines connecting each cell’s correct and incorrect PPC value. ****p<0.0001
Extended Data Fig. 7. Extended Data 7.

Breakdown of cells by behavior during the trial compared to the inter-trial interval (ITI), with cells that have a significantly increased firing rate during the trial (increasers), decreased firing rate (decreasers), or unchanged firing rate compared to the ITI (non-changers). This shows a majority of cells modulated during EDSS trials, with (a) 80% modulated in control animals, (b) 81.82% modulated in adolescent-inhibited hM4D animals, and (c) 71.83% modulated in hM4D animals during EDSS thalamic activation. d) Raw firing rates show no differences between groups across different epochs of the EDSS: during the ITI (Control Light OFF: n=8 animals, 75 cells, 3.503±0.561 Hz; hM4D Light OFF: n=12 animals, 55 cells, 2.619±0.645 Hz; hM4D Light ON: n=13 animals, 71 cells, 2.877±0.570 Hz; 1-way ANOVA, F(2,194)=0.5002, p=0.6072, dots represent individual cells; lines represent mean ± SEM), (e) over the course of the trial (overlapping 2s bins, with x-axis labels depicting the middle of each bin; dots represent mean FR for each bin, error bars represent SEM); Control Light OFF: n=8 animals, 75 cells; hM4D Light OFF: n=12 animals, 55 cells; hM4D Light ON: n=13 animals, 71 cells; 2-way rmANOVA, effect of group, F(2,194)=0.2743, p=0.7604), during the (f) pre-decision (all trials: Control Light OFF: n=8 animals, 75 cells, 3.717±0.634 Hz; hM4D Light OFF: n=12 animals, 55 cells, 3.482±0.765 Hz; hM4D Light ON: n=13 animals, 71 cells, 3.097±0.533 Hz; 1-way ANOVA, F(2,194)=0.2492, p=0.7797; correct vs. incorrect: 2-way rmANOVA, Holm-Sidak post-hoc, Control Light OFF: p=0.9962, hM4D Light OFF: p>0.9999, hM4D Light ON: p=0.9707) and (g) post-decision (all trials: Control Light OFF: n=8 animals, 75 cells, 3.585±0.650 Hz; hM4D Light OFF: n=12 animals, 55 cells, 3.493±0.795 Hz; hM4D Light ON: n=13 animals, 71 cells, 3.014±0.500 Hz; 1-way ANOVA, F(2,194)=0.2693, p=0.7642; correct vs. incorrect: 2-way rmANOVA, Holm-Sidak post-hoc, Control Light OFF: p=0.6988, hM4D Light OFF: p=0.9761, hM4D Light ON: p=0.9475) periods, both across trial types (left; dots represent individual cells; lines represent mean ± SEM) and between correct and incorrect trials (right; dots represent individual cells, lines connecting FR for correct and incorrect trials).
Extended Data Fig. 8. Extended Data 8.
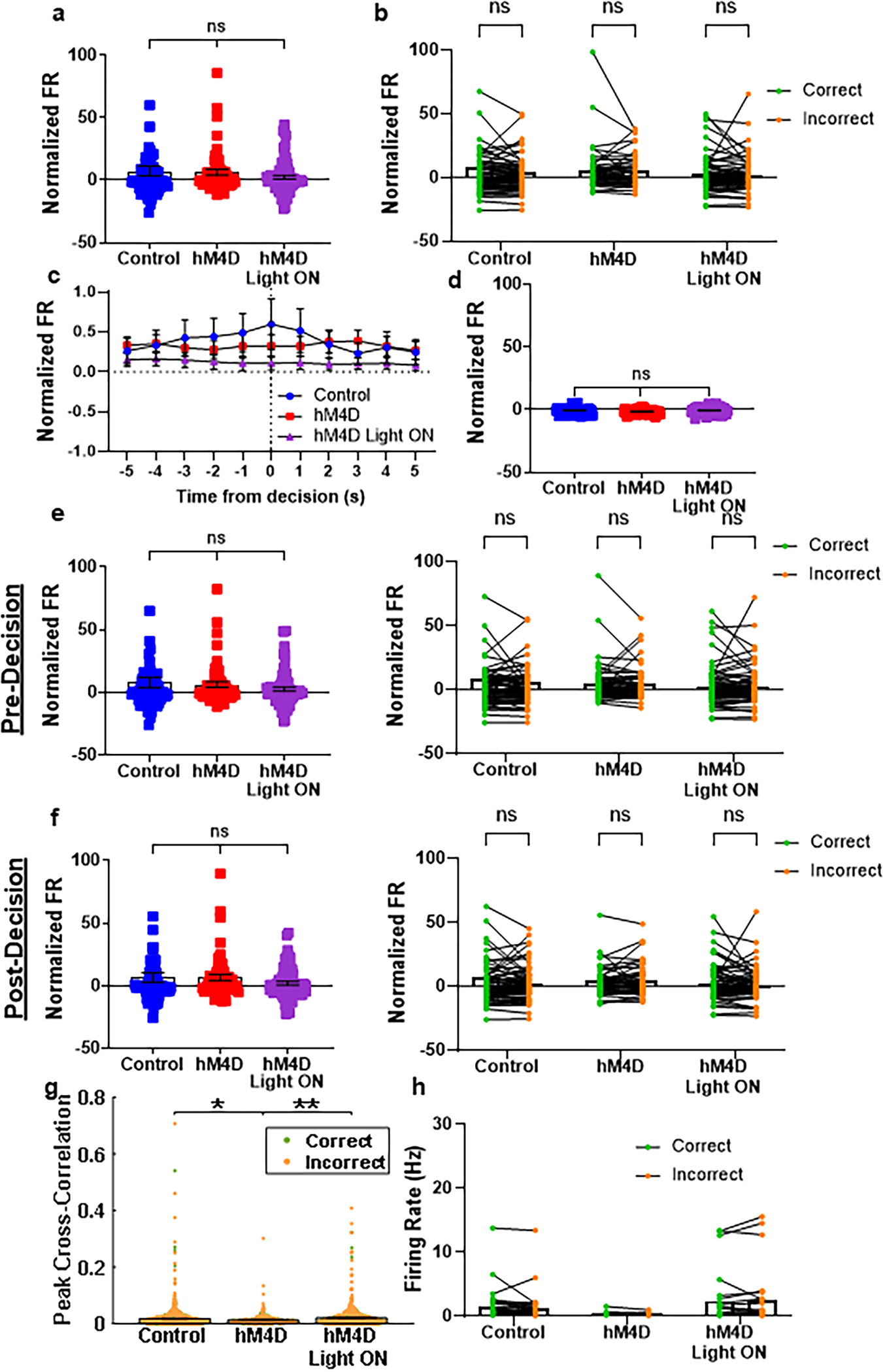
Firing rates normalized to the ITI show no differences between groups across different EDSS epochs (for a-f: Control OFF: n=8 animals, 75 cells; hM4D OFF: n=12 animals, 55 cells; hM4D ON: n=13 animals, 71 cells): (a) during the trial (Control OFF:7.250±3.907; hM4D OFF:6.442±2.351; hM4D ON:2.353±1.556; 1-way ANOVA, F(2,194)=1.346, p=0.2628; dots represent individual cells; lines represent mean ± SEM), (b) between trial types (2-way rmANOVA, Holm-Sidak post-hoc, Control OFF: p=0.2067, hM4D OFF: p=0.9981, hM4D ON: p=0.9848; dots represent individual cells, lines connecting FR for correct and incorrect trials), (c) over the course of the trial (overlapping 2s bins, with x-axis labels depicting the middle of each bin; dots represent mean FR, error bars represent SEM; 2-way rmANOVA, effect of group, F(2,194)=0.9097, p=0.4044), (d) during the ITI (Control OFF:−0.7287±0.2749; hM4D OFF:−1.508±0.2542; hM4D ON:−0.7053±0.2727; 1-way ANOVA, F(2,194)=0.5465, p=0.5798; dots represent individual cells; lines represent mean ± SEM), (e) pre-decision (all trials: Control OFF:7.898±4.166; hM4D OFF:6.356±2.273; hM4D ON:2.716±1.722; 1-way ANOVA, F(2,194)=1.241, p=0.2914; correct vs. incorrect: 2-way rmANOVA, Holm-Sidak post-hoc, Control OFF:p=0.5151, hM4D OFF:p=0.9983, hM4D ON:p>0.9999) and (f) post-decision (all trials: Control OFF:6.608±3.720; hM4D OFF:6.521±2.443; hM4D ON:1.993±1.440; 1-way ANOVA, F(2,194)=1.441, p=0.2391; correct vs. incorrect: 2-way rmANOVA, Holm-Sidak post-hoc, Control OFF:p=0.2171, hM4D OFF:p=0.9994, hM4D ON:p=0.9347), across trial types (left; dots represent individual cells; lines represent mean ± SEM) and between trial types (right; dots represent individual cells, lines connecting FR for correct and incorrect trials). (g) Un-truncated plot from Figure 6d. Control OFF: n=6 animals, 73 cells, 507 cellpairs, 0.0177±0.0430; hM4D OFF: n=9 animals, 52 cells, 181 cellpairs, 0.0124 ±0.0212; hM4D ON: n=11 animals, 69 cells, 327 cellpairs, 0.0201±0.0414; linear mixed effects model (peak cross-correlation~group+outcome+(1|cellpair)+(1|animal)+(1|cell1)+(1|cell2)), fixed effect of group: Control OFF vs. hM4D OFF:*p=0.041622; hM4D OFF vs. hM4D ON:**p=0.0090838. Bars with error represent mean ± SEM. Individual dots represent cell pair correlations for each trial type. (h) FRs of cells with peak cross-correlation above 0.08 show no FR pattern. Dots represent cells, lines connecting each cell’s FR for correct and incorrect trials. Control OFF: n=6 animals, 33 cells; hM4D OFF: n=3 animals, 6 cells; hM4D ON: n=5 animals, 26 cells.
Extended Data Fig. 9. Extended Data 9.
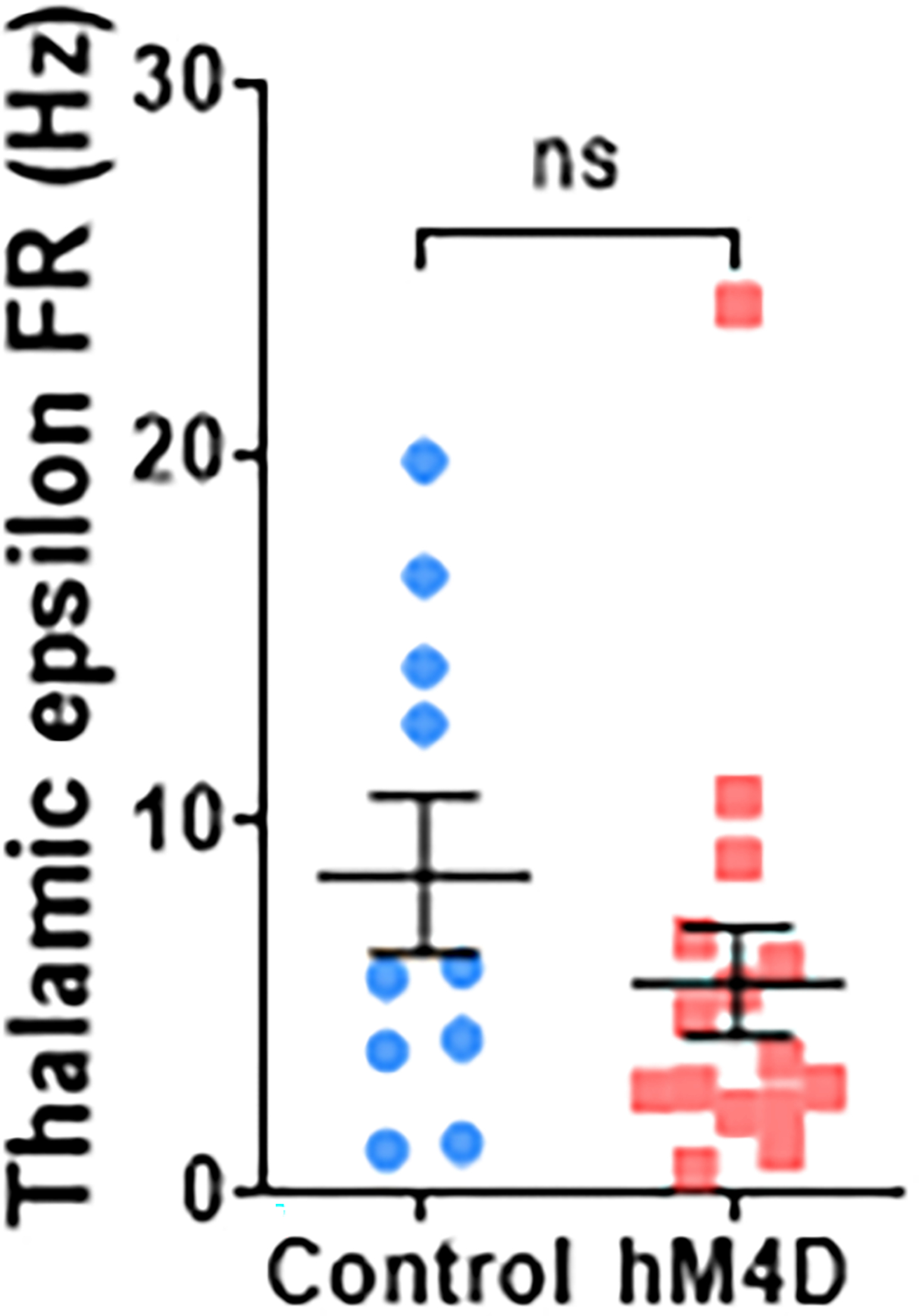
Mean thalamic epsilon firing rates during EDSS trials for each animal show no significant differences in thalamic activity for control or adolescent-inhibited hM4D animals. Control: n=10 animals, 8.606±2.114 Hz; hM4D: n=15 animals, 5.726±1.499 Hz; two-sided unpaired t-test, t=1.144, df=23, p=0.2643. Dots represent individual animals; lines represent mean ± SEM.
Extended Data Fig. 10. Extended Data 10.
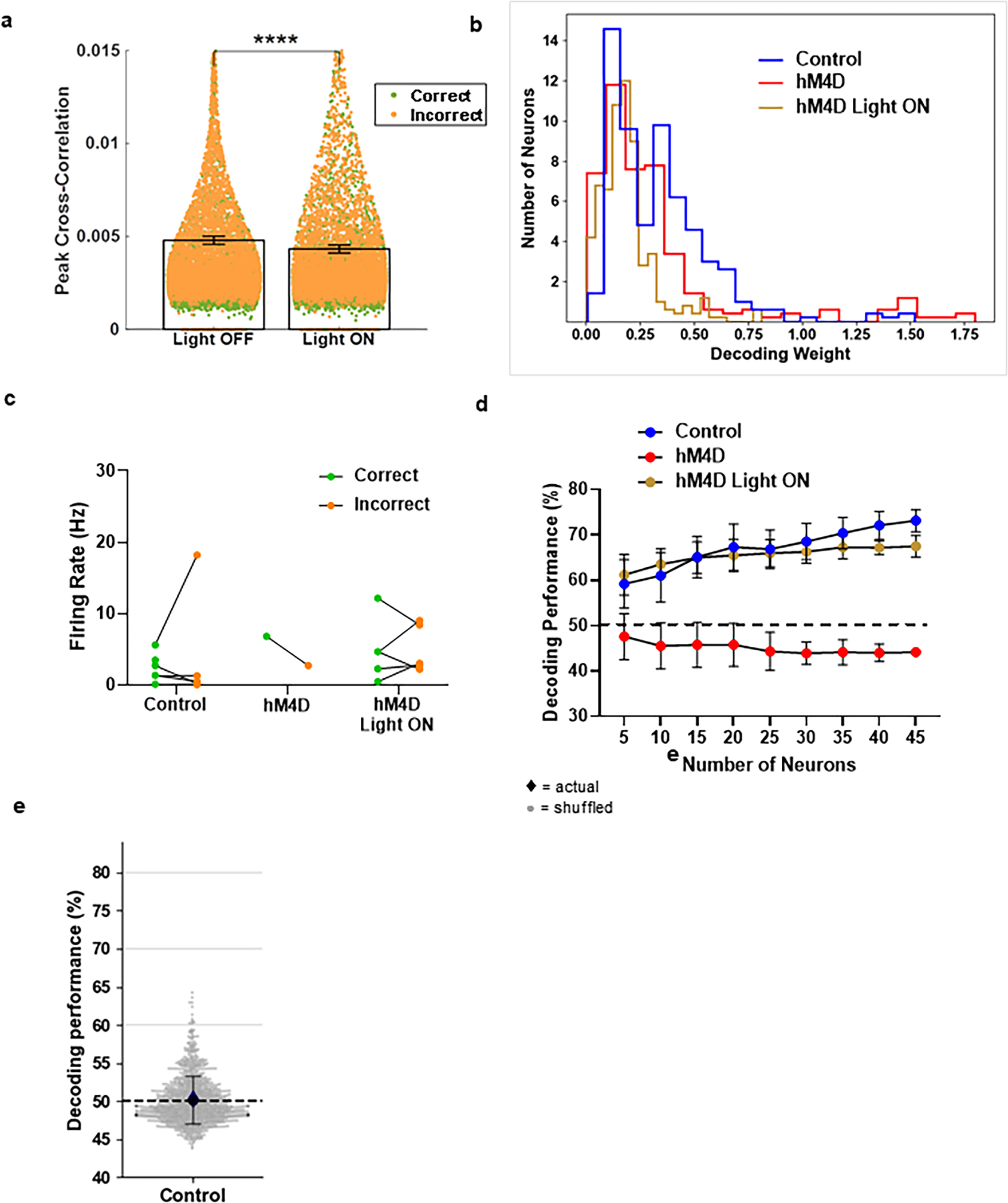
(a) Peak cross-correlation values for pairs of mPFC single units during the delay of a working memory T-maze task for correct (green) and incorrect (orange) trials, as described in Bolkan et al 201721. Acute thalamo-mPFC inhibition (Light ON) during the delay shows decreased cross-correlations compared with baseline (Light OFF). n=9 animals, 891 cells, 5254 cell pairs; Light OFF: 0.0048±0.0002; Light ON: 0.0043±0.0002; linear mixed effects model (peak cross-correlation~group+trial type+(1|cellpair)+(1|animal)+(1|cell1)+(1|cell2)), fixed effect of group: Light OFF vs. ON: ****p=3.587e-17. Bars with error represent mean ± SEM. Individual dots represent cell pairs. This graph has been truncated along the y-axis.
(b) Histogram of control (blue), hM4D (red), and hM4D Light ON (gold) cell decoding weights show the distribution of contributions across cells is unchanged across groups.
(c) Firing rates of cells with a significantly elevated decoding weight relative to shuffled data. Very few cells (<10% for each group) contribute significantly more than when shuffled, and these cells do not have different FRs across correct and incorrect trials. Dots represent cell FRs for each trial type, lines connecting each cell’s correct and incorrect trials. Control: n=3 animals, 7 cells; hM4D: n=1 animal, 1 cell; hM4D Light ON: n=2 animals, 5 cells.
(d) Decoder performance was calculated using random subgroups of neurons, repeated 25 times for each multiple of 5 neurons. Significant separation between hM4D and both Control and hM4D Light ON groups is seen with 5 neurons. Dashed line represents chance performance (50%). Dots represent mean performance for the repetitions, error bars represent standard deviation. Control Light OFF: n=4 animals, 60 cells; hM4D Light OFF: n=7 animals, 45 cells; hM4D Light ON: n=9 animals, 61 cells; 2-way rmANOVA Holm-Sidak post-hoc analysis, with 5 neurons: Control vs. hM4D: p<0.0001, Control vs. hM4D Light ON: p=0.1557, hM4D vs. hM4D Light ON: p<0.0001.
(e) Decoding performance (blue diamond) is no better than chance for control animals during IA. Shuffled trial outcomes show chance decoder performance, mean ± standard deviation (black circles and error bars) and individual shuffles (grey circles). n=3 animals, 47 cells, 1000 shuffles; actual: 50.35%, shuffled: 50.13±3.14%, p=0.9438.
Acknowledgments
We thank members of the Kellendonk, Canetta, and Harris labs for technical assistance and discussions. This work was supported by grants from the NIMH (K01MH107760 to S.C, R21 MH121334 and R21 MH117454 to C.K., F31 MH119691 to L.J.B., R01MH124998 to A.Z.H., NSF Neuronex to L.P. and S.F.).
Footnotes
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Takesian AE & Hensch TK Balancing plasticity/stability across brain development. Prog Brain Res 207, 3–34, doi: 10.1016/B978-0-444-63327-9.00001-1 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Wiesel TN & Hubel DH Single-Cell Responses in Striate Cortex of Kittens Deprived of Vision in One Eye. J Neurophysiol 26, 1003–1017, doi: 10.1152/jn.1963.26.6.1003 (1963). [DOI] [PubMed] [Google Scholar]
- 3.de Villers-Sidani E, Chang EF, Bao S & Merzenich MM Critical period window for spectral tuning defined in the primary auditory cortex (A1) in the rat. J Neurosci 27, 180–189, doi: 10.1523/JNEUROSCI.3227-06.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caras ML & Sanes DH Sustained Perceptual Deficits from Transient Sensory Deprivation. J Neurosci 35, 10831–10842, doi: 10.1523/JNEUROSCI.0837-15.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun YJ, Espinosa JS, Hoseini MS & Stryker MP Experience-dependent structural plasticity at pre- and postsynaptic sites of layer 2/3 cells in developing visual cortex. Proc Natl Acad Sci U S A 116, 21812–21820, doi: 10.1073/pnas.1914661116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canetta SE et al. Mature parvalbumin interneuron function in prefrontal cortex requires activity during a postnatal sensitive period. bioRxiv, 2021.2003.2004.433943, doi: 10.1101/2021.03.04.433943 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bitzenhofer SH, Pöpplau JA, Chini M, Marquardt A & Hanganu-Opatz IL A transient developmental increase in prefrontal activity alters network maturation and causes cognitive dysfunction in adult mice. Neuron 109, 1350–1364.e1356, doi: 10.1016/j.neuron.2021.02.011 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larsen B & Luna B Adolescence as a neurobiological critical period for the development of higher-order cognition. Neuroscience & Biobehavioral Reviews 94, 179–195, doi: 10.1016/j.neubiorev.2018.09.005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bicks LK et al. Prefrontal parvalbumin interneurons require juvenile social experience to establish adult social behavior. Nature communications 11, 1003, doi: 10.1038/s41467-020-14740-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nabel EM et al. Adolescent frontal top-down neurons receive heightened local drive to establish adult attentional behavior in mice. Nat Commun 11, 3983, doi: 10.1038/s41467-020-17787-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinberger DR & Berman KF Prefrontal function in schizophrenia: confounds and controversies. Philos Trans R Soc Lond B Biol Sci 351, 1495–1503, doi: 10.1098/rstb.1996.0135 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Sakurai T & Gamo NJ Cognitive functions associated with developing prefrontal cortex during adolescence and developmental neuropsychiatric disorders. Neurobiology of Disease 131, 104322, doi: 10.1016/j.nbd.2018.11.007 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Feinberg I Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res 17, 319–334, doi: 10.1016/0022-3956(82)90038-3 (1982). [DOI] [PubMed] [Google Scholar]
- 14.Marenco S et al. Investigation of anatomical thalamo-cortical connectivity and FMRI activation in schizophrenia. Neuropsychopharmacology 37, 499–507, doi: 10.1038/npp.2011.215 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woodward ND, Karbasforoushan H & Heckers S Thalamocortical dysconnectivity in schizophrenia. Am J Psychiatry 169, 1092–1099, doi: 10.1176/appi.ajp.2012.12010056 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitelman SA, Byne W, Kemether EM, Hazlett EA & Buchsbaum MS Metabolic disconnection between the mediodorsal nucleus of the thalamus and cortical Brodmann’s areas of the left hemisphere in schizophrenia. Am J Psychiatry 162, 1733–1735, doi:162/9/1733 [pii] 10.1176/appi.ajp.162.9.1733 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Giraldo-Chica M, Rogers BP, Damon SM, Landman BA & Woodward ND Prefrontal-Thalamic Anatomical Connectivity and Executive Cognitive Function in Schizophrenia. Biol Psychiatry, doi: 10.1016/j.biopsych.2017.09.022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodward ND & Heckers S Mapping Thalamocortical Functional Connectivity in Chronic and Early Stages of Psychotic Disorders. Biol Psychiatry 79, 1016–1025, doi: 10.1016/j.biopsych.2015.06.026 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anticevic A et al. Association of Thalamic Dysconnectivity and Conversion to Psychosis in Youth and Young Adults at Elevated Clinical Risk. JAMA Psychiatry 72, 882–891, doi: 10.1001/jamapsychiatry.2015.0566 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho KI et al. Altered Thalamo-Cortical White Matter Connectivity: Probabilistic Tractography Study in Clinical-High Risk for Psychosis and First-Episode Psychosis. Schizophrenia bulletin 42, 723–731, doi: 10.1093/schbul/sbv169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolkan SS et al. Thalamic projections sustain prefrontal activity during working memory maintenance. Nature neuroscience 20, 987–996, doi: 10.1038/nn.4568 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parnaudeau S, Bolkan SS & Kellendonk C The Mediodorsal Thalamus: An Essential Partner of the Prefrontal Cortex for Cognition. Biological psychiatry 83, 648–656, doi: 10.1016/j.biopsych.2017.11.008 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rikhye RV, Gilra A & Halassa MM Thalamic regulation of switching between cortical representations enables cognitive flexibility. Nature neuroscience 21, 1753–1763, doi: 10.1038/s41593-018-0269-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmitt LI et al. Thalamic amplification of cortical connectivity sustains attentional control. Nature 545, 219–223, doi: 10.1038/nature22073 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parnaudeau S et al. Inhibition of mediodorsal thalamus disrupts thalamofrontal connectivity and cognition. Neuron 77, 1151–1162, doi:S0896–6273(13)00139–6 [pii] 10.1016/j.neuron.2013.01.038 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benoit LJ et al. Medial prefrontal lesions impair performance in an operant delayed nonmatch to sample working memory task. Behavioral neuroscience, doi: 10.1037/bne0000357 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bissonette GB et al. Double dissociation of the effects of medial and orbital prefrontal cortical lesions on attentional and affective shifts in mice. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 11124–11130, doi:28/44/11124 [pii] 10.1523/JNEUROSCI.2820-08.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guirado R, Umemori J, Sipila P & Castren E Evidence for Competition for Target Innervation in the Medial Prefrontal Cortex. Cereb Cortex 26, 1287–1294, doi: 10.1093/cercor/bhv280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stefanini F et al. A Distributed Neural Code in the Dentate Gyrus and in CA1. Neuron 107, 703–716 e704, doi: 10.1016/j.neuron.2020.05.022 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuste R From the neuron doctrine to neural networks. Nature reviews. Neuroscience 16, 487–497, doi: 10.1038/nrn3962 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Narayanan NS & Laubach M Methods for studying functional interactions among neuronal populations. Methods Mol Biol 489, 135–165, doi: 10.1007/978-1-59745-543-5_7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernardi S et al. The Geometry of Abstraction in the Hippocampus and Prefrontal Cortex. Cell 183, 954–967 e921, doi: 10.1016/j.cell.2020.09.031 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bitzenhofer SH, Pöpplau JA, Chini M, Marquardt A & Hanganu-Opatz IL A transient developmental increase in prefrontal activity alters network maturation and causes cognitive dysfunction in adult mice. Neuron, doi: 10.1016/j.neuron.2021.02.011 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hill RA, Wu YW, Kwek P & van den Buuse M Modulatory effects of sex steroid hormones on brain-derived neurotrophic factor-tyrosine kinase B expression during adolescent development in C57Bl/6 mice. J Neuroendocrinol 24, 774–788, doi: 10.1111/j.1365-2826.2012.02277.x (2012). [DOI] [PubMed] [Google Scholar]
- 35.Flores-Barrera E et al. Late adolescent expression of GluN2B transmission in the prefrontal cortex is input-specific and requires postsynaptic protein kinase A and D1 dopamine receptor signaling. Biol Psychiatry 75, 508–516, doi: 10.1016/j.biopsych.2013.07.033 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker KD, Gray AR & Richardson R The development of perineuronal nets around parvalbumin gabaergic neurons in the medial prefrontal cortex and basolateral amygdala of rats. Behav Neurosci 131, 289–303, doi: 10.1037/bne0000203 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Fagiolini M & Hensch TK Inhibitory threshold for critical-period activation in primary visual cortex. Nature 404, 183–186, doi: 10.1038/35004582 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Paus T, Keshavan M & Giedd JN Why do many psychiatric disorders emerge during adolescence? Nature reviews. Neuroscience 9, 947–957, doi:nrn2513 [pii] 10.1038/nrn2513 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caballero A, Flores-Barrera E, Cass DK & Tseng KY Differential regulation of parvalbumin and calretinin interneurons in the prefrontal cortex during adolescence. Brain Struct Funct 219, 395–406, doi: 10.1007/s00429-013-0508-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chini M & Hanganu-Opatz IL Prefrontal Cortex Development in Health and Disease: Lessons from Rodents and Humans. Trends Neurosci 44, 227–240, doi: 10.1016/j.tins.2020.10.017 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Konstantoudaki X et al. Prefrontal cortical-specific differences in behavior and synaptic plasticity between adolescent and adult mice. J Neurophysiol 119, 822–833, doi: 10.1152/jn.00189.2017 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Miyamae T, Chen K, Lewis DA & Gonzalez-Burgos G Distinct Physiological Maturation of Parvalbumin-Positive Neuron Subtypes in Mouse Prefrontal Cortex. J Neurosci 37, 4883–4902, doi: 10.1523/JNEUROSCI.3325-16.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang JM, Zhang J, Yu YQ, Duan S & Li XM Postnatal development of 2 microcircuits involving fast-spiking interneurons in the mouse prefrontal cortex. Cereb Cortex 24, 98–109, doi: 10.1093/cercor/bhs291 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Bitzenhofer SH, Popplau JA & Hanganu-Opatz I Gamma activity accelerates during prefrontal development. eLife 9, doi: 10.7554/eLife.56795 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delevich K, Klinger M, Okada NJ & Wilbrecht L Coming of age in the frontal cortex: The role of puberty in cortical maturation. Seminars in Cell & Developmental Biology, doi: 10.1016/j.semcdb.2021.04.021 (2021). [DOI] [PubMed] [Google Scholar]
- 46.Zuo Y, Lin A, Chang P & Gan W-B Development of Long-Term Dendritic Spine Stability in Diverse Regions of Cerebral Cortex. Neuron 46, 181–189, doi: 10.1016/j.neuron.2005.04.001 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Pattwell SS et al. Dynamic changes in neural circuitry during adolescence are associated with persistent attenuation of fear memories. Nat Commun 7, 11475, doi: 10.1038/ncomms11475 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Eden CG & Uylings HB Postnatal volumetric development of the prefrontal cortex in the rat. The Journal of comparative neurology 241, 268–274, doi: 10.1002/cne.902410303 (1985). [DOI] [PubMed] [Google Scholar]
- 49.Ferguson BR & Gao WJ Development of thalamocortical connections between the mediodorsal thalamus and the prefrontal cortex and its implication in cognition. Frontiers in human neuroscience 8, 1027, doi: 10.3389/fnhum.2014.01027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rios O & Villalobos J Postnatal development of the afferent projections from the dorsomedial thalamic nucleus to the frontal cortex in mice. Brain Res Dev Brain Res 150, 47–50, doi: 10.1016/j.devbrainres.2004.02.005 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Uhlhaas PJ & Singer W The development of neural synchrony and large-scale cortical networks during adolescence: relevance for the pathophysiology of schizophrenia and neurodevelopmental hypothesis. Schizophrenia bulletin 37, 514–523, doi: 10.1093/schbul/sbr034 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wen W & Turrigiano GG Developmental Regulation of Homeostatic Plasticity in Mouse Primary Visual Cortex. The Journal of Neuroscience 41, 9891–9905, doi: 10.1523/jneurosci.1200-21.2021 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo ZV et al. Maintenance of persistent activity in a frontal thalamocortical loop. Nature 545, 181–186, doi: 10.1038/nature22324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamigaki T & Dan Y Delay activity of specific prefrontal interneuron subtypes modulates memory-guided behavior. Nature neuroscience 20, 854–863, doi: 10.1038/nn.4554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanaka S Dysfunctional GABAergic inhibition in the prefrontal cortex leading to “psychotic” hyperactivation. BMC Neurosci 9, 41, doi: 10.1186/1471-2202-9-41 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taylor SF, Welsh RC, Chen AC, Velander AJ & Liberzon I Medial frontal hyperactivity in reality distortion. Biol Psychiatry 61, 1171–1178, doi: 10.1016/j.biopsych.2006.11.029 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Spellman T, Svei M, Kaminsky J, Manzano-Nieves G & Liston C Prefrontal deep projection neurons enable cognitive flexibility via persistent feedback monitoring. Cell 184, 2750–2766 e2717, doi: 10.1016/j.cell.2021.03.047 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park AJ et al. Reset of hippocampal-prefrontal circuitry facilitates learning. Nature 591, 615–619, doi: 10.1038/s41586-021-03272-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonaventura J et al. High-potency ligands for DREADD imaging and activation in rodents and monkeys. Nature Communications 10, 4627, doi: 10.1038/s41467-019-12236-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Canetta S et al. Maternal immune activation leads to selective functional deficits in offspring parvalbumin interneurons. Mol Psychiatry 21, 956–968, doi: 10.1038/mp.2015.222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guirado R, Umemori J, Sipila P & Castren E Evidence for Competition for Target Innervation in the Medial Prefrontal Cortex. Cereb Cortex 26, 1287–1294, doi: 10.1093/cercor/bhv280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vinck M, van Wingerden M, Womelsdorf T, Fries P & Pennartz CM The pairwise phase consistency: a bias-free measure of rhythmic neuronal synchronization. Neuroimage 51, 112–122, doi: 10.1016/j.neuroimage.2010.01.073 (2010). [DOI] [PubMed] [Google Scholar]
- 63.Pedregosa F et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The source data that support the findings of this study are available on figshare: https://figshare.com/projects/Benoit_Kellendonk_NN-A76458A/135581 or from the corresponding author upon reasonable request.