

Abstract
The APOBEC3 (A3) family of single-stranded DNA cytidine deaminases are host restriction factors that inhibit lentiviruses, such as HIV-1, in the absence of the Vif protein that causes their degradation. Deamination of cytidine in HIV-1 (−)DNA forms uracil that causes inactivating mutations when uracil is used as a template for (+)DNA synthesis. For APOBEC3C (A3C), the chimpanzee and gorilla orthologues are more active than human A3C, and we determined that Old World Monkey A3C from rhesus macaque (rh) is not active against HIV-1. Biochemical, virological, and coevolutionary analyses combined with molecular dynamics simulations showed that the key amino acids needed to promote rhA3C antiviral activity, 44, 45, and 144, also promoted dimerization and changes to the dynamics of loop 1, near the enzyme active site. Although forced evolution of rhA3C resulted in a similar dimer interface with hominid A3C, the key amino acid contacts were different. Overall, our results determine the basis for why rhA3C is less active than human A3C and establish the amino acid network for dimerization and increased activity. Based on identification of the key amino acids determining Old World Monkey antiviral activity we predict that other Old World Monkey A3Cs did not impart anti-lentiviral activity, despite fixation of a key residue needed for hominid A3C activity. Overall, the coevolutionary analysis of the A3C dimerization interface presented also provides a basis from which to analyze dimerization interfaces of other A3 family members.
Keywords: APOBEC3, coevolutionary analysis, molecular dynamics simulations, dimerization, HIV
Introduction
Host restriction factors act as a cross-species transmission barrier for the Simian and Human Immunodeficiency viruses, SIV and HIV1. Overcoming these barriers by evolution of virally-encoded ‘accessory proteins’ which antagonize these restriction factors has characterized successful adaptation of the primate lentiviruses to new hosts2. In some cases, such as the infection of chimpanzees with SIV from Old World Monkeys (OWM) these adaptions to a new host was accompanied by major changes in the viral genome, which also facilitated the transmission to humans as HIV-13. One of the major barriers to cross-species transmission of SIVs to a new host is the family of APOBEC3 (A3) host restriction factors that in most primates constitutes a minimum of seven enzymes, named A3A through A3H, excluding E4. The A3 family are single-stranded (ss) DNA cytidine deaminases that can inhibit retroelements, such as LINE-1, retroviruses, such as HIV and SIV, and some other viruses5–9. They belong to a larger family of APOBEC enzymes that have diverse roles in immunity and metabolism10.
For restricting HIV-1 replication, A3 enzymes first need to become packaged in the budding virion6,9. When these newly formed virions infect the next target cell, the packaged A3 enzymes deaminate cytidines on single-stranded regions of the (−) DNA to uridines during reverse transcription6,9. Uracil is promutagenic in DNA since it templates the addition of adenosine and results in G to A hypermutations on the (+)DNA when it is synthesized using a deaminated (−)DNA as a template6,9. This resulting double-stranded (ds) DNA provirus can become integrated but is non-functional6,9. Some A3 enzymes can also physically inhibit HIV reverse transcriptase activity11,12. To counteract the actions of A3 enzymes, HIV encodes a protein Vif that becomes the substrate receptor of an E3 ubiquitin ligase with host proteins CBF-β, Cul5, EloB, EloC and Rbx2 to cause polyubiquitination and degradation of A3 enzymes via the 26S proteasome13. Members of the human A3 family exhibit considerable variation in their antiviral activity; A3G can restrict HIV-1 ΔVif replication the most, with A3H, A3F, and A3D having decreasing abilities to restrict HIV-1 ΔVif14–17. A3A and A3B can potently restrict endogenous retroelements and some DNA viruses, respectively, but not HIV-17,18,19.
A3C is the least active member of the human A3 family when it comes to restricting HIV-1 and HIV-220,21 because of its reduced ability to significantly deaminate (−)DNA due to a lack of processivity15. Since A3 enzymes deaminate only ssDNA and the availability of ssDNA during reverse transcription is transient, the enzymes must have an efficient way to search for cytidines in the correct deamination motif, e.g., 5′TTC for A3C, in order to maximize deaminations in the short time that the ssDNA is available6. However, a polymorphism in A3C that exists in about 10% of African individuals causes a change of S188 to I188, and this increases its HIV ΔVif restriction ability 5- to 10- fold, but it does not reach the level of restriction that A3G can achieve22,23. The increased restrictive activity of A3C S188I is due to acquiring the ability to form a homodimer, which enables processive deamination of HIV (−)DNA. Notably, the A3C S188I does not appear to be directly involved in the dimer interface, but instead induces conformational changes conducive to dimerization15. In addition, for the common form of human (h)A3C there is also a contribution of a specific loop near the active site of A3C (loop 1) to catalytic activity24. Interestingly, chimpanzee (c)A3C and gorilla (g)A3C despite having a S188 have equivalent HIV restriction activity to hA3C S188I15. This restriction activity of cA3C and gA3C is also attributed to their ability to form dimers, but through a different amino acid at position 11515.
In contrast to hominid A3C, the rhesus macaque (rh)A3C has been found to not be able to restrict replication of HIV-1, HIV-2, SIVmac (rhesus macaque), or SIVagm (African green monkey) in the majority of studies20,25,26. Paradoxically, the rhA3C contains the I188 that enables activity of hA3C and enhances activity of cA3C, and gA3C15,22. Through direct coupling analysis (DCA)/coevolutionary analysis and subsequent Molecular Dynamics (MD), virological, and biochemical experiments, we uncovered a series of amino acid replacements required to promote dimerization in rhA3C and therefore promote HIV-1 restriction ability. Analysis of several OWM A3C sequences showed that they did not contain the “right” combination of amino acids for activity against HIV-1 suggesting that the evolutionary pressures that formed OWM A3C were different from hominid A3Cs that are active against lentiviruses. Overall, we form a model for the determinants of A3C antiviral activity and estimate its loss and gain throughout primate evolution.
Results
rhA3C is a monomer, rather than a dimer
Dimerization of hA3C, cA3C, and gA3C has previously been shown to correlate with HIV-1 restriction efficiency15. Since rhA3C has been reported as not being active against HIV20,25,26, we hypothesized that this could be due to a lack of dimerization. To determine the overall amino acid similarity, particularly at the dimer interface residues, the amino acid sequences for hA3C, cA3C, gA3C, and rhA3C were aligned (Figure 1(A)). The rhA3C does contain an I188, which stabilizes dimerization in hA3C, cA3C, and gA3C (Figure 1(A))15. However, the other determinant identified for cA3C and gA3C that promotes activity against HIV-1 is K115. The rhA3C has a different amino acid at this position, M115, which is also different from hA3C that has an N115 (Figure 1(A–B)). Furthermore, amino acid R44 (and possibly R45) was predicted to interact with K115 in cA3C15. but in rhA3C these amino acids are Q44 and H45 (Figure 1 (A–B)). The role of I188 in dimerization is indirect and the proposed mechanism for human A3C is that I188 causes a steric clash with F126 and N132 causing a repositioning of helix 6, enabling dimerization [15]. Due to the indirect nature of the role of I188 and that there are several key amino acid differences between hA3C and rhA3C, we purified the rhA3C wild type (WT) to determine its oligomerization state (Figure 1(A–B)).
Figure 1. Amino acid differences of rhA3C in comparison to hA3C, cA3C, and gA3C.
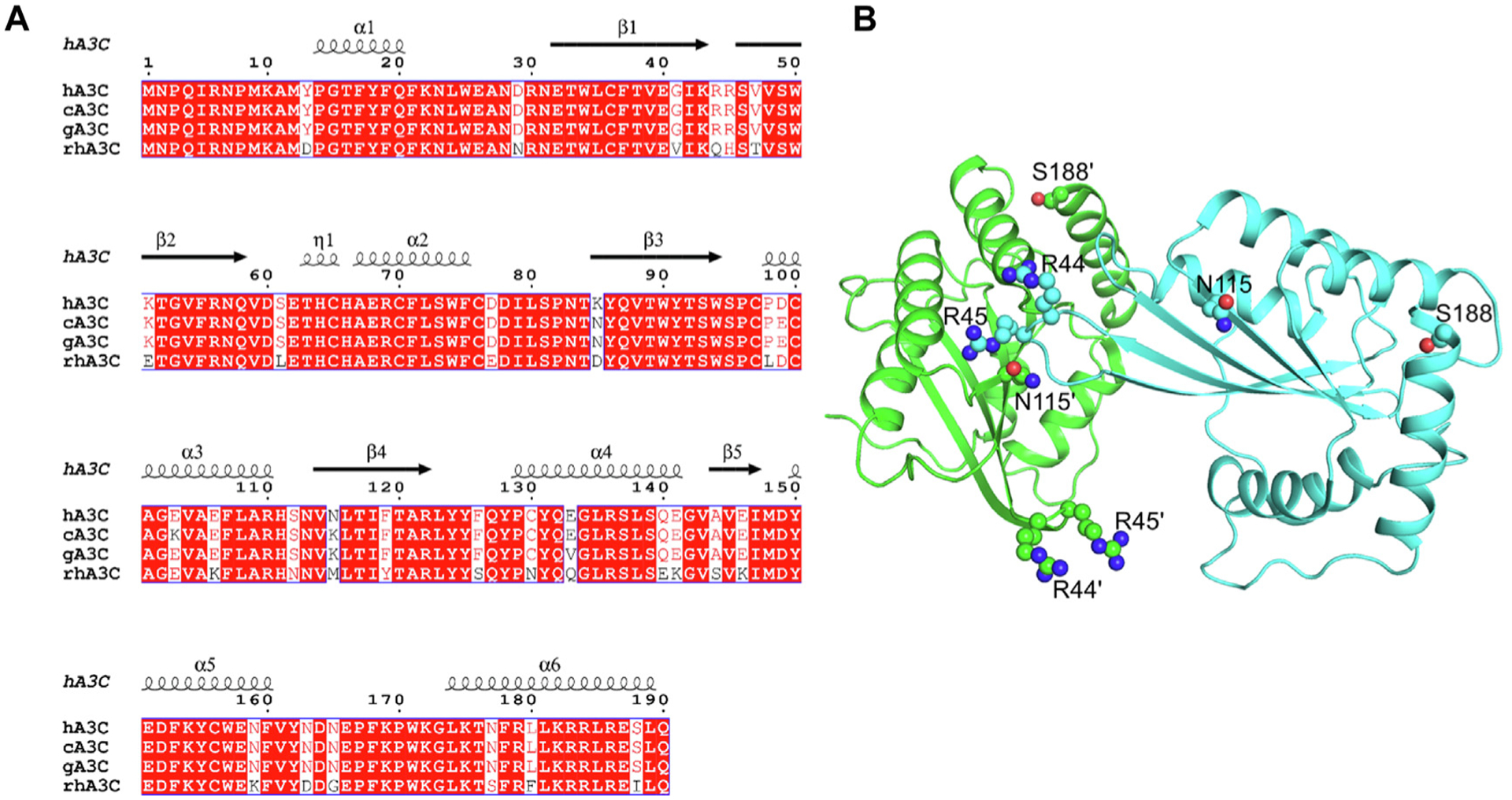
Sequence alignment and structural analysis of A3C. (A) Sequence alignment of hA3C, cA3C, gA3C, and rhA3C with amino acid differences shown in white. The sequence alignment was performed by a Clustal Omega multiple sequence alignment [50] and plotted using the program ESPript [78]. The α-helices and β-strands are shown above the alignment and are based on the hA3C 3VOW structure [35]. (B) The hA3C 3VOW structure is shown with amino acids important for hominid A3C dimerization shown on each monomer. The monomer shown in green has amino acids labeled with a prime symbol to differentiate them from amino acids belonging to the cyan monomer.
The rhA3C WT was purified from Sf9 cells in the presence of RNase A and we used size exclusion chromatography (SEC) to determine the oligomerization state. We found that the rhA3C WT had only one peak in the chromatogram that was a monomer (M, Figure 2(A–B)), suggesting that the amino acid differences identified prevent dimerization. We then created rhA3C mutants to make it more hominid-like. We purified rhA3C mutants Q44R/H45R (hA3C-, cA3C-, and gA3C- like), Q44R/H45R/M115K (cA3C- and gA3C- like), and Q44R/H45R/M115N (hA3C-like). The rhA3C Q44R/H45R and Q44R/H45R/M115K mutants also only had one peak in the chromatogram that was a monomer (M, Figure 2(A–B)). However, the rhA3C Q44R/H45R/M115N had two peaks demonstrating formation of a dimer (D), but the monomer peak was more predominant (Figure 2 (A–B)).
Figure 2. Mutation of rhA3C amino acids to hA3C amino acids enables dimerization.
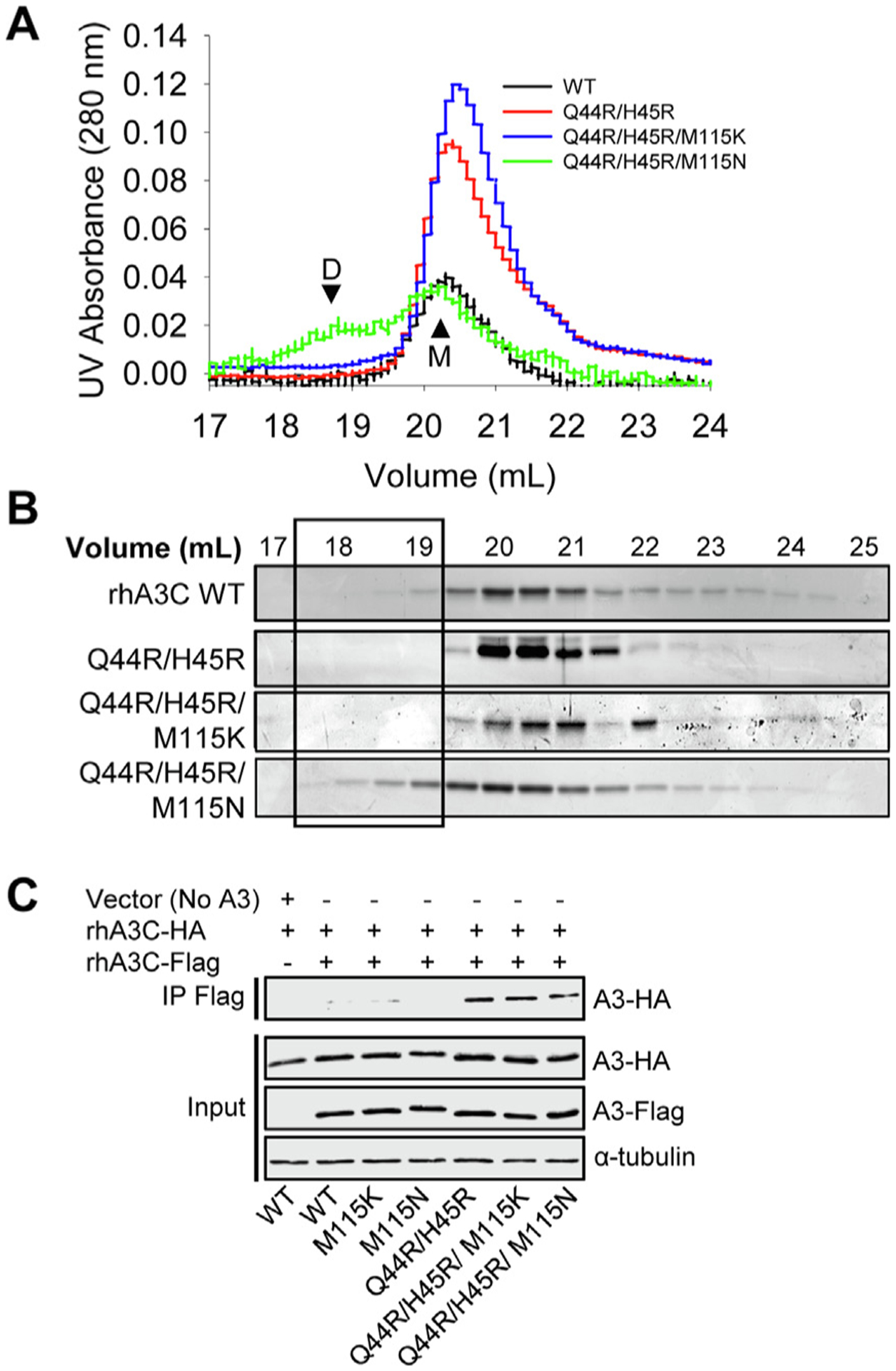
The oligomerization of rhA3C was studied (A–B) in vitro and (C) in cell lysates. (A–B) SEC profile for rhA3C WT and rhA3C mutants Q44R/H45R, Q44R/H45R/M115K, and Q44R/H45R/M115N. Elution profiles for all enzymes was composed of a monomer peak (M, ~20 ml elution volume). Only for the rhA3C Q44R/H45R/M115N there was a larger dimer peak (D, ~19 mL elution volume). The elution profiles are shown as the (A) UV absorbance during SEC elution and (B) coomassie stained protein fractions resolved by SDS-PAGE. Box shows where the dimeric fractions eluted. During the determination of the apparent molecular weights in comparison to a series of standards, we found that the A3C retention time in the SEC column did not reflect the actual molecular weight (Figure S1). However, comparison to cA3C confirmed our assignment of monomer and dimer forms and use of Multi-angle light scattering to empirically determine the molecular mass of rhA3C Q44R/H45R/M115K confirmed that all A3C forms were monomeric except Q44R/H45R/M115N (Figure S1). (C) Co-immunoprecipitation of rhA3C-3xHA with rhA3C-3xFlag. The A3C-3xHA and A3C-3xFlag were transfected in combination and immunoprecipitation of the cell lysates used magnetic anti-Flag beads. Immunoblotting was conducted with antibodies against α-tubulin, HA and Flag. Cell lysate (input) shows the expression of α-tubulin, HA and Flag.
To determine if the rhA3C Q44R/H45R/M115N could also form dimers in cells we used co-immunoprecipitation (co-IP). Due to the propensity of A3s to bind RNA in cells and cell lysates27, the co-IP was carried out in the presence of RNaseA to ensure that interactions were due to protein–protein contacts and not mediated by an RNA bridge. Using two rhA3C constructs with either a 3xFlag- or 3xHA- tag we carried out a co-IP and found that the rhA3C Q44R/H45R/M115N was able to self-associate in cells. As a control, we also tested the single mutant M115N and this could not immunoprecipitate, suggesting that the Q44R/H45R change enabled dimer formation (Figure 2(C)). Consistent with this, the rhA3C self-interaction was also found with Q44R/H45R and Q44R/H45R/M115K, but not M115K (Figure 2(C)). Thus, although in cells all three mutants with the Q44R/H45R change can form dimers, they do not all appear to be stable in vitro (Figure 2(A–B)). Collectively, the data suggested that amino acids 44 and 45 are key for rhA3C dimerization, but there may be different amino acids required to stabilize rhA3C dimerization than for hA3C.
rhA3C deaminase activity is increased with hominid-like amino acids at positions 44 and 45
Since the rhA3C appeared to have different determinants for stable dimerization than hA3C, we wanted to test that dimerization did indeed correlate with increased deamination activity in rhA3C. We examined the specific activity of rhA3C by conducting a uracil DNA glycosylase assay with a 118 nt substrate containing two 5′TTC motifs (Figure 3(A–B)). We used a time course of deamination to determine the linear range of activity (Figure 3(A–B)) and then calculated the specific activity of the rhA3C in that region (i.e., 5 to 15 min) (Figure 3(C)). We found that the rhA3C WT was approximately 2-fold less active than the three rhA3C mutants containing the Q44R/H45R change, confirming that residues that promote dimerization are of primary importance to rhA3C activity (Figure 3(C)). However, since the rhA3C needs to search the DNA for the 5′TTC motifs among the non-motif containing DNA, the specific activity is made up of both the chemistry in the catalytic site once reaching the substrate C and the time it took to search for the 5′TTC motif.6 The search occurs by facilitated diffusion and results in the enzyme being processive, i.e., deaminating more than one 5′TTC motif in a single enzyme substrate encounter. The processivity was calculated by analyzing the individual bands from the uracil DNA glycosylase assay (Figure 3(B and D)). The processivity requires single-hit conditions in which an enzyme would have acted only on one DNA (see Materials and Methods).
Figure 3. Arginines at positions 44 and 45 in rhA3C increase enzyme activity.
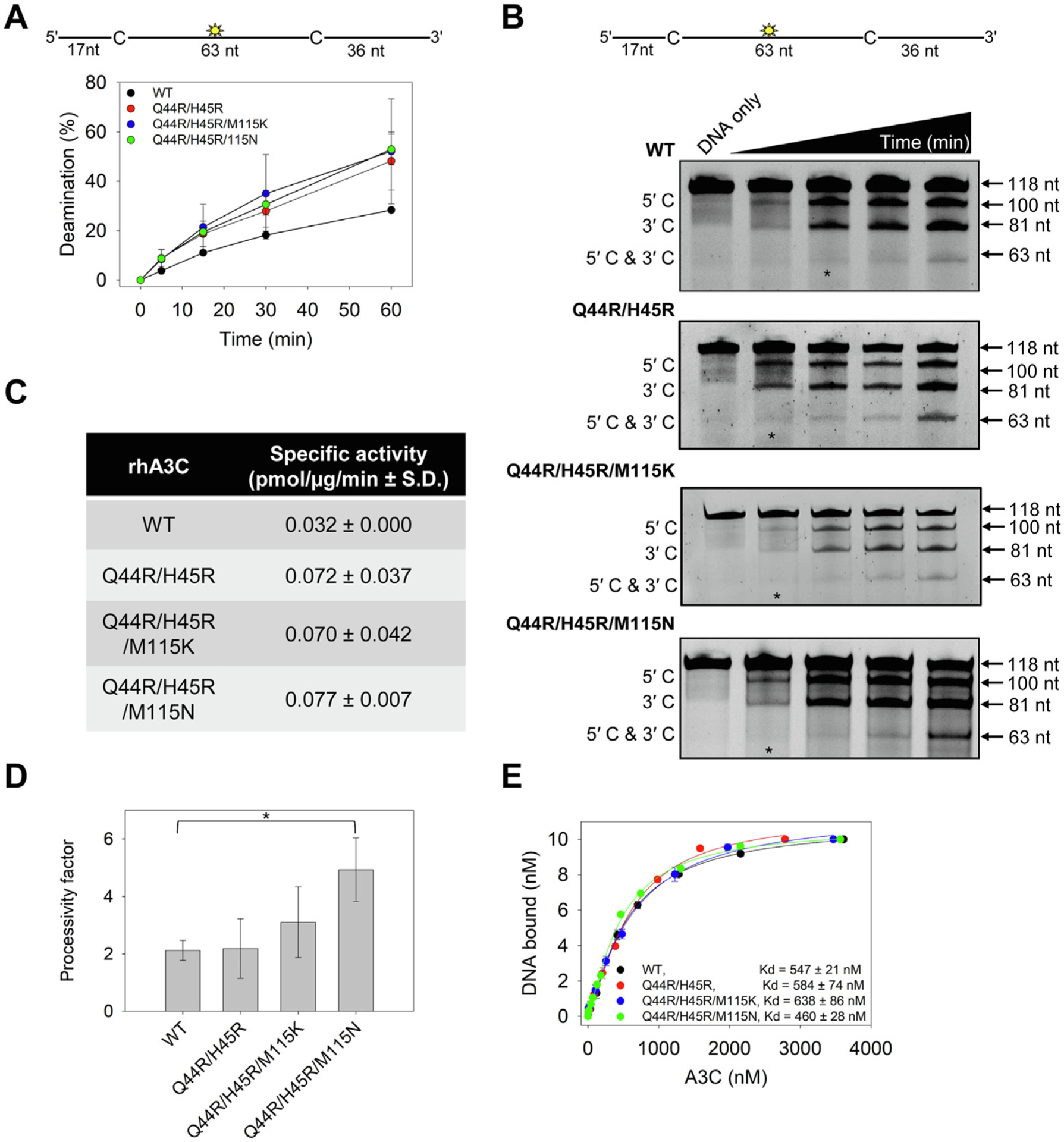
(A–B) Time course of rhA3C WT, Q44R/H45R, Q44R/H45R/M115K, and Q44R/H45R/M115N on a 118 nt fluorescently labeled ssDNA with two 5′TTC deamination motifs spaced 63 nt apart. Reactions were performed with 100 nM substrate DNA and 1000 nM rhA3C for the indicated amount of time (5–60 min). (B) Representative gel from three independent deamination assay experiments. Lanes with an asterisk were used for processivity calculations shown in panel (D). (C) Specific activity as calculated from the time course in (A–B) and showed increased activity if the rhA3C had the Q44R/H45R mutation. (D) Processivity of rhA3C WT, Q44R/H45R, Q44R/H45R/M115K, and Q44R/H45R/M115N calculated from the time course in (A–B). The processivity factor measures the likelihood of a processive deamination over a nonprocessive deamination and is described further in the ‘Materials and Methods‘ section. Designations for significant difference of values were ***P ≤ 0.001, **P ≤ 0.01 or *P ≤ 0.05. (E) The apparent Kd of rhA3C enzymes from a 118 nt fluorescently labeled ssDNA was analyzed by steady-state rotational anisotropy for rhA3C WT, Q44R/H45R, Q44R/H45R/M115K, and Q44R/H45R/M115N. Apparent Kd values are shown in the figure with the standard deviation. (A, D, E) On the graphs, the standard deviation for three independent experiments is shown as error bars.
Using these conditions, we determined the number of deaminations at both 5′TTC motifs (5′C & 3′C) compared to the single deaminations at either the 5′TTC motif closest to the 5′-end (5′C) or 3′-end (3′C) (Figure 3(B)). From these values a processivity factor can be calculated which is the fold likelihood that a processive deamination would take place. We found that the processivity of rhA3C WT was 2.1 (Figure 3(D)). In comparison, a nonprocessive enzyme has a processivity factor of 1. Thus, the rhA3C WT is not very processive, consistent with it being a monomer. The mutants had small but consistent increases in processivity where the Q44R/H45R processivity factor was 2.5, the Q44R/H45R/M115K slightly more processive (processivity factor of 3.2) and the Q44R/H45R/M115N that could partially dimerize most stably had a significant 2-fold higher processivity factor than the WT (processivity factor of 4.9, Figure 3(D)). However, dimerization did not increase the affinity of the A3C for the 118 nt ssDNA, as measured by steady-state rotational anisotropy (Figure 3(E)).
Increase in rhA3C specific activity correlates with increase in HIV-1 restriction activity
To determine if the in vitro differences of rhA3C from hA3C affect HIV-1 ΔVif ΔEnv (referred to as HIV) restriction ability we used a single-cycle infectivity assay. We compared hA3C S188I that is restrictive to HIV and rhA3C. We found that hA3C S188I could restrict HIV, but rhA3C could not, despite naturally having amino acid I188 (Figure 1(A) and Figure 4(A)). We also checked rhA3C activity against the OWM SIV from sooty mangabey monkey (smm). The rhA3C only restricted SIVsmm ΔVif 2-fold, in contrast to hA3C S188I (13-fold), hA3C (6-fold) and cA3C (4-fold) that were more restrictive (Figure S2). Notably, the hA3C S188I was able to restrict SIV ΔVif better than HIV, suggesting that SIV is restricted more easily (Figure 4(A) and Figure S2)15. Thus, overall, the data show that rhA3C is not active against HIV or SIVsmm.
Figure 4. Amino acids 44 and 45 in rhA3C are key determinants for HIV restriction ability.
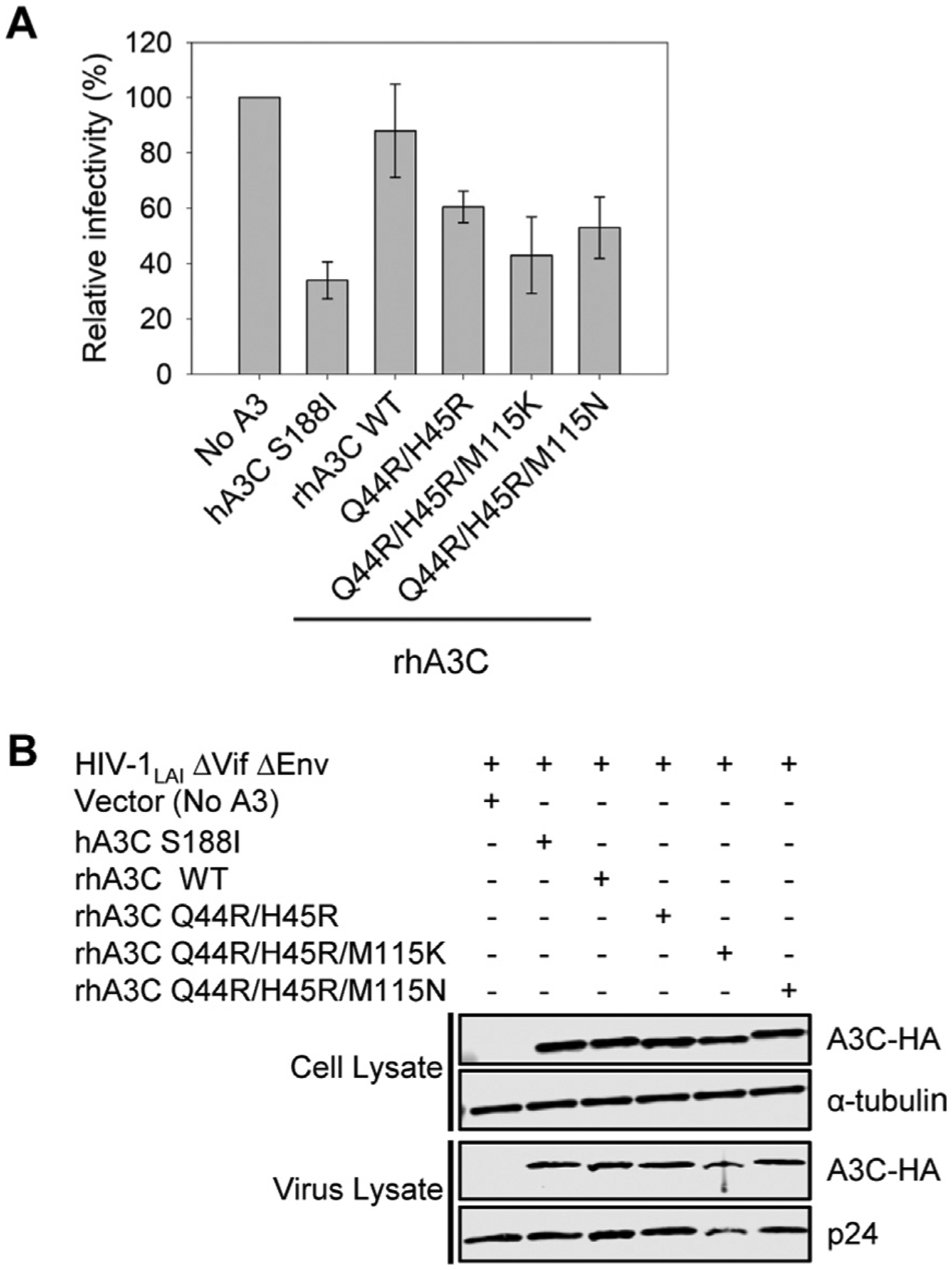
(A) Infectivity was measured by β-galactosidase expression driven by the HIV-1 5΄LTR from TZM-bl cells infected with VSV-G pseudotyped HIV ΔVif ΔEnv that was produced in the absence or presence of 3xHA tagged hA3C S188I, rhA3C WT, and rhA3C mutants Q44R/H45R, Q44R/H45R/M115K, and Q44R/H45R/M115N. Results normalized to the no A3 condition are shown with error bars representing the Standard Deviation of the mean calculated from three independent experiments. (B) Immunoblotting for the HA tag was used to detect A3C enzymes expressed in cells and encapsidated into HIV ΔVif ΔEnv pseudotyped virions. The cell lysate and virion loading controls were α-tubulin and p24, respectively.
To determine if the lack of rhA3C restriction was due to it being a monomer, we tested the rhA3C mutants. We began by converting only the rhA3C Q44/H45 to R44/R45 (as in hA3C). We found that this mutation alone resulted in a 1.5-fold increase in restriction from rhA3C wild type (WT) (Figure 4 (A)). The WT and mutant were expressed and encapsidated into virions similarly (Figure 4(B)). Upon the addition of M115K and M115N mutations in a Q44R/H45R background, we found that there was a small increase in restriction. These triple mutants had a 1.7–2-fold increase in restriction activity against HIV in comparison to rhA3C WT, which was not due to increased encapsidation into virions (Figure 4(B)). Importantly, just the changes at amino acids 44 and 45 increased HIV restriction similarly to changes at position 115 (Figure 4(A–B)). Altogether, the data showed that the 2-fold increase in specific activity (Figure 3(C)) corresponded well with an approximate 2-fold increase in virus restriction (Figure 4(A)) and suggested that processivity may not be essential to HIV restriction by rhA3C. Further, these data demonstrated that rhA3C restriction activity was improved by converting dimer interface amino acids to the hA3C or cA3C amino acids, demonstrating that the rhA3C WT as a monomer is less able to restrict HIV. However, the restrictive activity of the triple mutants was still 2-fold lower than that of hA3C S188I (Figure 4(A)) which suggested that there were likely additional determinants for dimerization in rhA3C.
A key residue for rhA3C dimerization is uncovered via direct coupling analysis
Since we could not achieve stable dimerization or HIV restriction activity equivalent to hA3C S188I from mutation of rhA3C amino acids 44/45/115 we turned to direct coupling analysis (DCA), also termed coevolutionary analysis28. Since dimerization is relevant for catalytic activity, we hypothesized that amino acid interactions that maintain function must be encoded in the evolutionary history of the A3 family of sequences (Pfam PF18771). The sequences included any organism and homologs that have a sequence that is classified as a member of the A3 family. A hidden Markov Model profile (HMMER) was used to search the NCBI database, including non-curated sequences, to find sequences whose statistics look like members of the family (Figure 5). This allowed inclusion of more distant sequences as opposed to direct matches to A3C. A resulting multiple sequence alignment (MSA) with ~2500 sequences was compiled (see Materials and Methods and Supplementary File 1). Methods like coevolutionary analysis have been useful to identify amino acid coevolution in sequence alignments and to predict residue-residue contacts in the 3D structure of proteins28,29 and to predict 3D folds30.
Figure 5. Coevolutionary analysis to identify relevant interaction residues involved in dimerization.
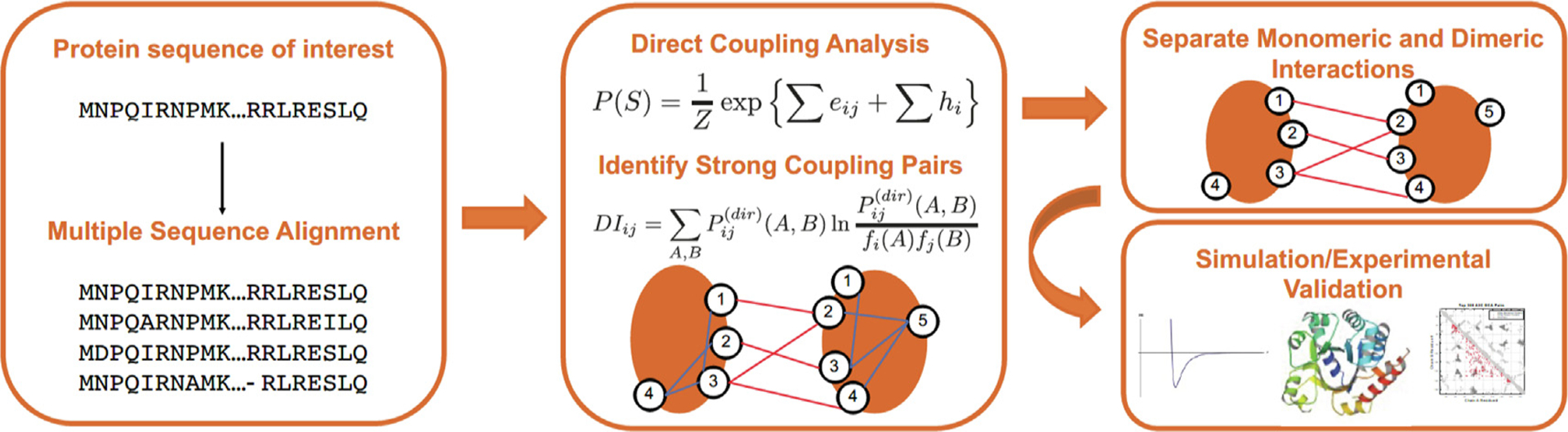
First, a multiple sequence alignment (MSA) is created to identify members of the A3C family. The MSA is then processed using Direct Coupling Analysis (DCA) and a metric of coupling strength called Direct Information (DI). Once the top DI pairs have been identified, the monomeric crystal structure is used to distinguish monomeric from dimeric interactions. The resulting interacting residues are used to drive a simulation that predicts dimeric complexes. The residues involved in more coupled interactions are then proposed for experimental validation.
Interestingly, coupled amino acid changes that preserve dimeric interactions at the interface between oligomers can also be inferred and used to predict complex formation in dimers31–33. Therefore, we analyzed the alignment of approximately 2500 sequences (Figure 5 and Materials and Methods) to identify the most important dimeric coevolved interactions for the A3 family. Using DCA we identified a set of residue-residue pairs that are important for dimerization (Figure 6 and Materials and Methods). Figure 6(A) shows, in a contact map, the residue-residue contacts found in the crystal structure of hA3C (PDB 3VOW) for monomeric interactions (light gray) and the dimeric contacts (blue). In the same map, red dots indicate the top 300 coevolving interactions found by DCA. We notice that monomeric contacts can be predicted from the analysis of sequences, but more relevant to our study, we uncover a region (Figure 6(B)) that coincides with homodimeric contacts in the crystal structure (Figure 6(C)).
Figure 6. Directly coupled residue pairs identify relevant dimeric interacting residues preserved through evolution.
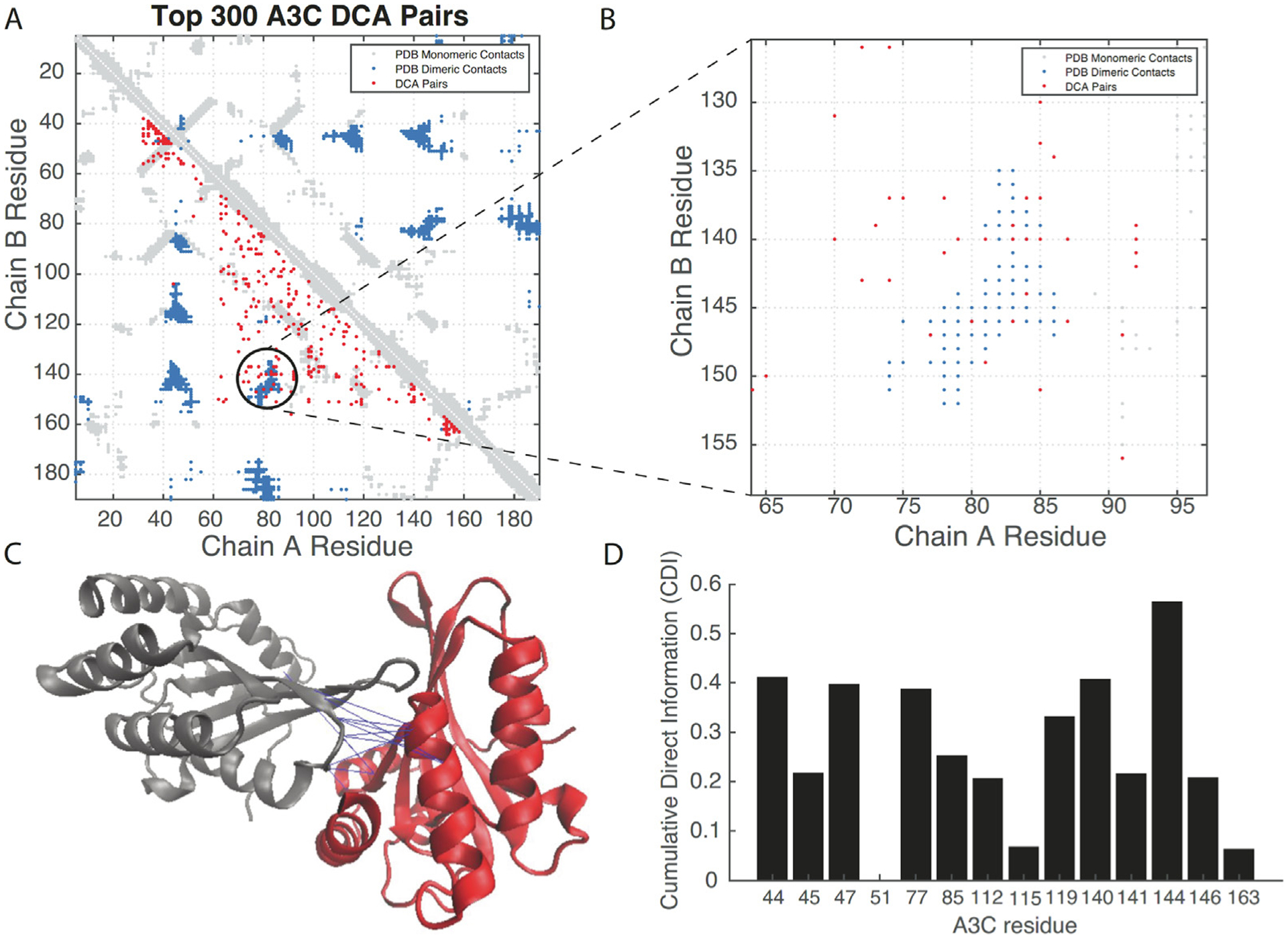
(A) A contact map comparing the residue contacts in the x-ray crystal structure (3VOW) of hA3C (light gray for monomeric, dark blue for dimeric) and those interactions found by DCA (red). (B) A region of dimeric interactions shared by the crystal and coevolutionary analysis, highlighting the importance of those residues for dimer formation. (C) Overlay of the coevolved pairs on the 3VOW structure, depicting pairs inferred by the coevolutionary analysis. (D) A Cumulative Direct Information (CDI) metric identifies relevant residues at the interface. Of note, is the residue 144 that appears to have strong interactions with several residues and was therefore a candidate for further analysis.
To further validate the relevance of those contacts for homodimeric interactions, we ran a coarse-grained MD simulation that uses such coupled coevolved pairs to drive a dimerization process (see Materials and Methods). The outcome of such simulation is a homodimer predicted completely from coevolutionary signatures. We notice that this predicted complex deviates from the x-ray structure, but it does share a large portion of the homodimeric interface (Figure S3).
Having identified a relevant coevolving interface for dimerization, we proposed a metric to identify key residues for dimer formation and that at the same time are part of the set of amino acid differences between hA3C and rhA3C. Our metric, called Cumulative Direct Information (CDI) quantifies how much a given residue is involved in coevolving interactions from the most important ranked residue pairs. We reasoned that if a residue is involved in several coevolving interactions, then they could be ideal candidates for mutational studies. This quantification for the top 200 dimeric interactions in the A3 family and for the residues that differ among the hA3C and rhA3C is shown in Figure 6(D). We first noticed that the area between residues 44–47 was important according to this quantification, which was consistent with the experimental data (Figure 2). This analysis showed that residue 115 does not have a high CDI score, which agrees with our results of a limited impact of residue 115 on activity beyond residues 44 and 45 (Figures 3 and 4). More importantly, Figure 6(D) shows a dominant role of residue 144 in dimerization and predicts residue 144 as a potentially important candidate for further mutational analysis.
MD simulation predicts an important role for amino acid 144 in dimerization
To test the DCA and coarse-grained MD simulation we performed classical MD simulations using the dimeric hA3C crystal structure. MD simulations of hA3C tested the effect of converting the hA3C A144 to the rhA3C amino acid S144 in the context of changes at amino acids 44 and 45, forming the single amino acid hA3C mutants and the hA3C R44Q/R45H/A144S triple mutant (rhA3C-like). If amino acid 144 is involved in dimerization then we would expect introduction of the rhA3C amino acid to disrupt the existing dimerization of hA3C. Consistent with this hypothesis, this mutant exhibited greater than 0.5 Å change in RMSF on 119 amino acids, and 9 changing by more than 1.0 Å with respect to the hA3C WT (Figure 7(A)). This corresponded to larger regions of change on the protein near the dimer interface, most particularly on loop 1, which is located between α-helix 1 and β-strand 1 (Figures 1A and 7(A)). Due to the asymmetric nature of the dimer, only the loop 1 on the monomer subunit closest to the dimer interface was affected, not the loop 1 on the other monomer distal to the dimer interface. Normal mode analysis based on the trajectories indicated that the essential dynamics of all the systems are captured by the first two modes (Figures S4 and S5). Principal component analysis (PCA) revealed a clear change in the dynamic motion especially on the first mode (Figures S6). These larger changes are observed alongside a noticeable reduction of average hydrogen bonding interactions at the dimer interface throughout the simulation (Figure 7(B)). The hydrogen bonds at the three mutation sites all showed considerable change, with position 144 increasing by more than 10% and positions 44 and 45 having six interactions decreasing by more than 10% (Figure 7(B)). The correlated motion of the variant is noticeably different compared with the hA3C, with a general trend of loss of correlated motion between the two monomers, consistent with a loss of dimer character (Figures 7(C–D) and S7). These changes in correlated motion are also consistent with the observed changes in root mean squared fluctuation (RMSF) (Figure S8). Energy decomposition analysis indicates the dimer interaction is destabilized by approximately 770 kcal/mol (Table S1). This destabilization is greater than the sum of the individual variants, consistent with what has been previously observed with multiple mutants34.
Figure 7. Model analysis of hA3C R44Q/R45H/A144S shows large changes in specific conformations.
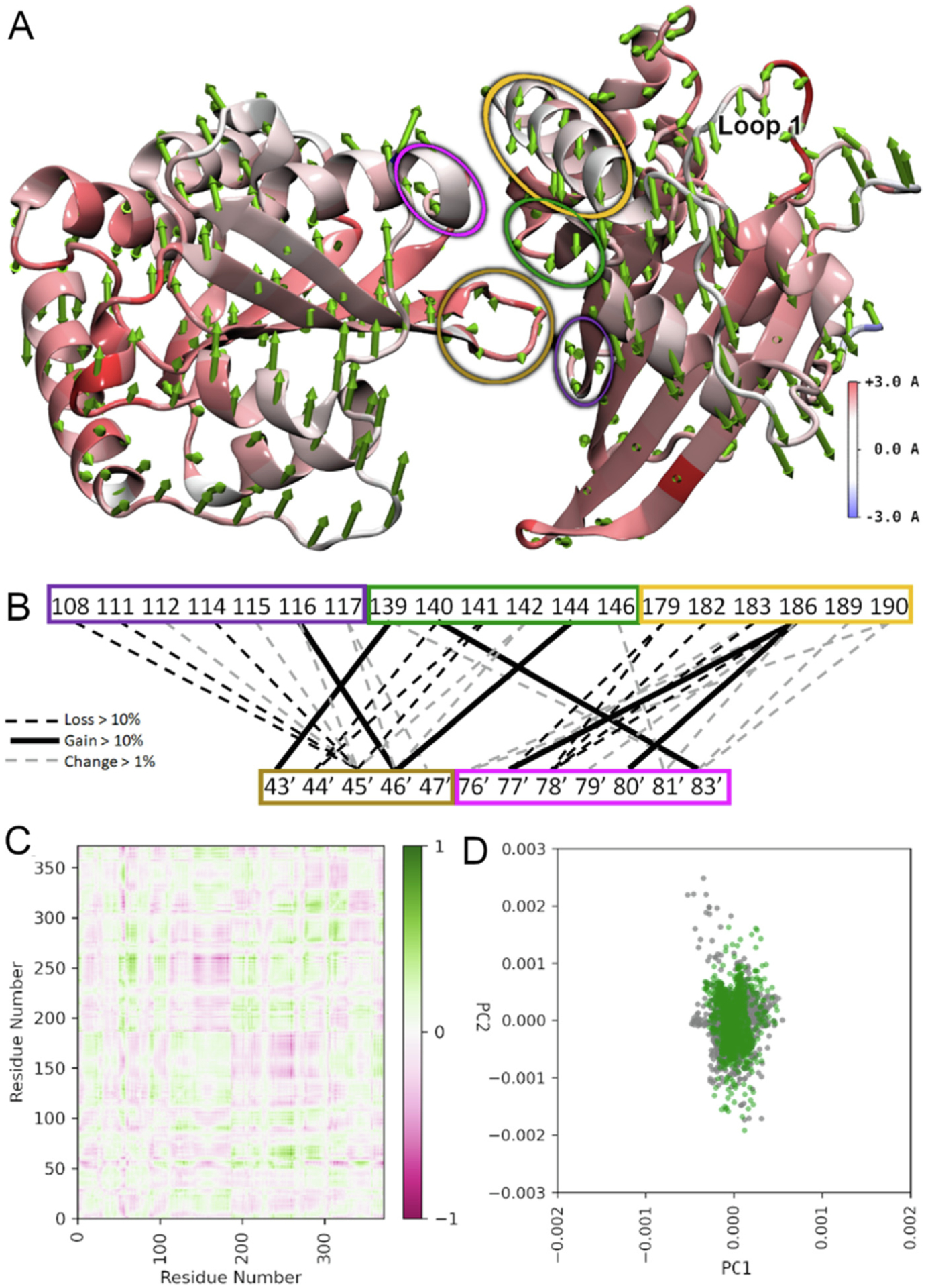
The hA3C R44Q/R45H/A144S variant compared to hA3C WT. (A) RMSF heatmapped to the protein structure with first normal mode shown as arrows from each amino acid in sequence. Normal modes were calculated based on the motion of the α-carbon to eliminate disproportionate contributions from amino acid side chains. Arrow direction shows the motion of each residue as a portion of the total largest contributor to essential motion. Arrow size denotes magnitude of motion. The RMSF heatmapping shows the total fluctuation of each residue from an average position over the trajectory. Higher values of RMSF indicate greater movement from this average position. Loop 1 that is important for activity is labeled. (B) Dimer interface hydrogen bonding changes. Colored blocks of residues correspond to regions encircled in panel (A) with same color. Black hatched lines mean a loss >10%, black lines mean a gain of >10%, and grey hatched lines mean a change of >1%. (C) Difference correlation plot between hA3C R44Q/R45H/A144A and hA3C WT. (D) PCA showing first two modes of hA3C R44Q/R45H/A144A (green) against hA3C WT (grey).
A rhA3C S144A mutant in combination with Q44R/H45R enables stable dimerization and increased catalytic activity by a unique mechanism
Based on the predictions from coevolutionary analysis and MD simulations, we produced from Sf9 cells a rhA3C S144A mutant alone and in combination with changes at residues 44 and 45 to create a rhA3C Q44R/H45R/S144A mutant (hA3C-like). Consistent with the coevolutionary analysis, the S144A mutation enabled rhA3C to interact with itself (Figure 8(A–B)). However, a prominent dimer peak was only formed in the presence of a Q44R/H45R background (Figure 8 (A–B)). Based on the hA3C crystal structure, we hypothesized that the rhA3C S144′ pushes S46 away due to unfavorable non-bonded interactions (Figure 8(C)).35 In contrast, the A144′ mutant enables the S46 to come closer to the dimer interface, allowing the loop to properly orient and stabilize the dimer in conjunction with R44/R45 interactions (Figure 8(C)).
Figure 8. Mutation of rhA3C amino acids 44, 45, and 144 to hA3C amino acids enables dimerization.
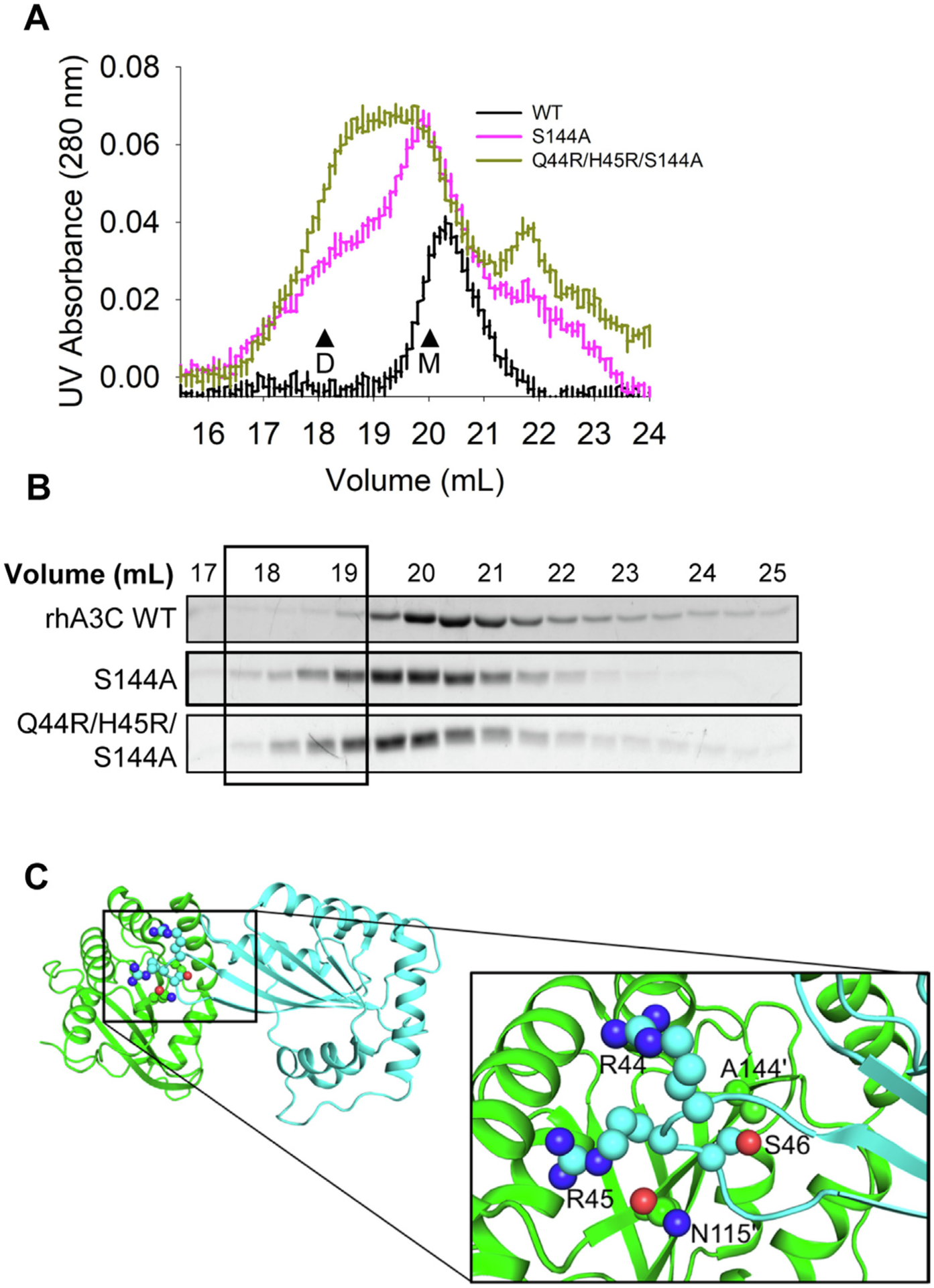
(A) SEC profile for rhA3C WT and rhA3C mutants S144A, and Q44R/H45R/S144A. Elution profiles for rhA3C WT was composed of a monomer peak (M, ~20 ml elution volume). The rhA3C S144A showed a monomer and dimer peak (D, ~19 mL elution volume). Only for the rhA3C Q44R/H45R/S144A there was a more prominent dimer peak. The elution profiles are shown as the UV absorbance during SEC elution. (B) Coomassie stained protein fractions resolved by SDS-PAGE that correspond to the eluted fractions in (A). Box shows where the dimeric fractions eluted. (C) The hA3C 3VOW structure is shown with amino acids important for rhA3C dimerization shown on each monomer. The monomer shown in green has amino acids labeled with a prime symbol to differentiate them from amino acids belonging to the cyan monomer.
Consistent with the contribution to activity of amino acids 44 and 45 the rhA3C Q44R/H45R/S144A mutant, but not the rhA3C S144A mutant, increased activity 2-fold compared to rhA3C WT (Figure 9(A–C)). In addition, the rhA3C S144A, but not Q44R/H45R/S144A mutant had a 1.5-fold increase in its Kd for ssDNA compared to rhA3C WT, suggesting that ssDNA binding is negatively affected when dimerization occurs in the absence of Q44R/H45R (Figure 9(D)). Surprisingly, for rhA3C Q44R/H45R/S144A, the increase in specific activity and maintenance of WT ssDNA binding affinity did not result in an increase in processivity (Figure 9(E)). Rather, the reason for this increased dimerization and catalytic activity without the expected increase in processivity appears to be due to changes in loop 1 (Figure 7). In multiple A3s, including A3C, loop 1 has been found to mediate accessibility of the ssDNA substrate to the active site [24,36,37]. The MD simulations revealed effects of changes at residue 144 on loop 1 that were unique from changes at residue 115 (Figures 7, S9, and S10). In the hA3C R44Q/R45H/A144S mutant, the increased RMSF on loop 1 corresponds to a reduction in dynamic motion compared with hA3C WT (Figures 7(A) and S9). The helices around the active site maintain similar RMSF across the hA3C WT and hA3C R44Q/R45H/A144S mutant consistent with a specific change in loop 1 mediating the changes in specific activity (Figure 7). Namely, loop 1 in hA3C is in an open/transition/closed conformation for 2%/80%/19% of the simulation time, while for R44Q/R45H/N115M this changes to 13%/23%/64%, and in R44Q/R45H/A144S this changes to 3%/36%/61%. In the hA3C WT and R44Q/R45H/N115M variant, there is little to no correlated motion between loops 1 and 7. However, in the hA3C R44Q/R45H/A144S variant, there is a region of increased correlation between these two regions, indicating that the A144S mutation affects the motion of nearby loop 7, which in turn is more correlated with the motion of loop 1 (Figure S11). In the rhA3C, the opposite effect is expected, where the open conformation time of rhA3C Q44R/H45R/S144A loop 1 is increased compared to rhA3C WT. This is consistent with previous observations that more time in an open loop 1 state correlates with increases in deamination activity and is consistent with our observation of increased specific activity for rhA3C Q44R/H45R/S144A (Figures 7, 9(C), and S9)24.
Figure 9. An S144A amino acid change in rhA3C increases deamination activity, but not processivity.
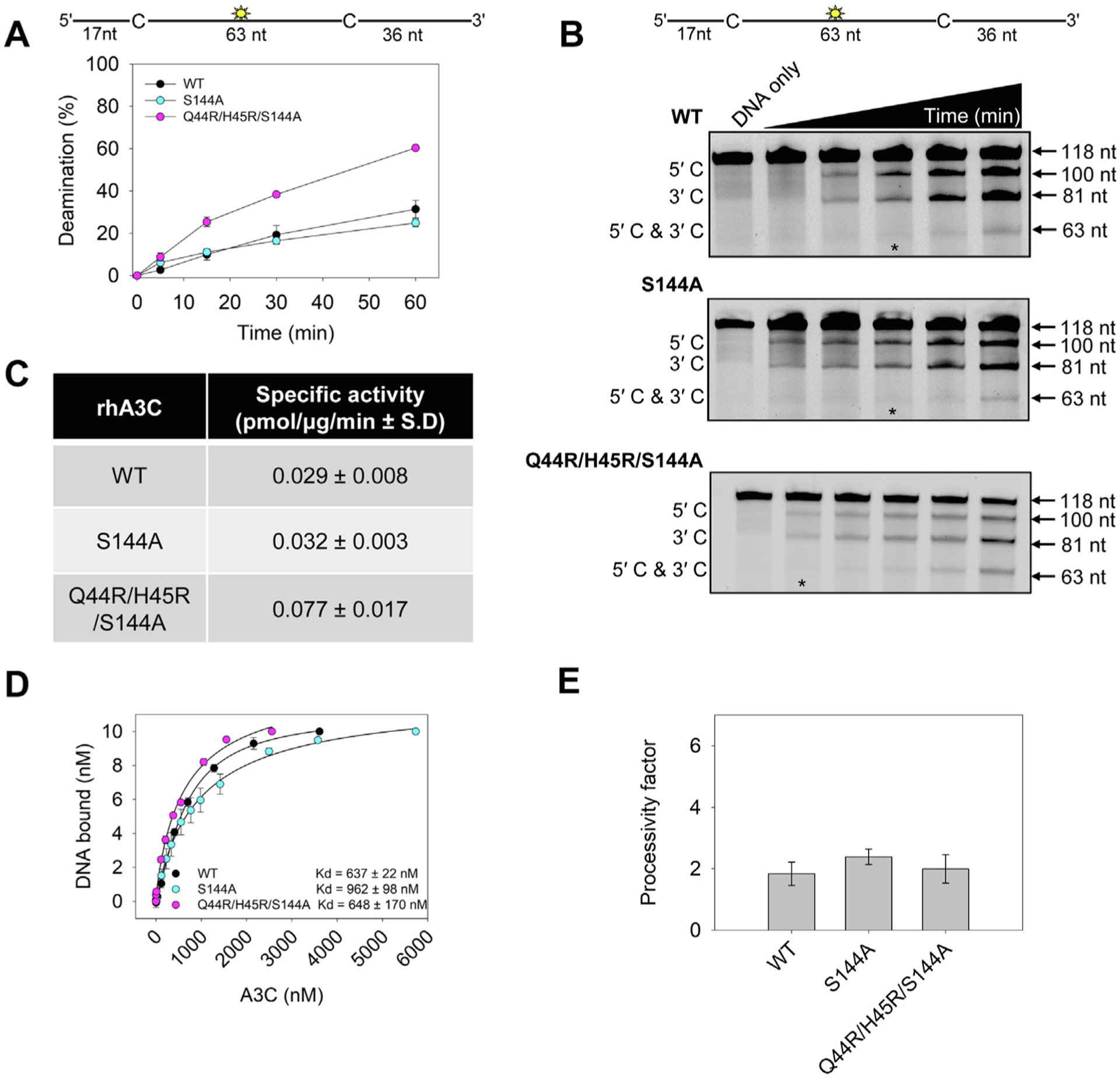
(A–B) Time course of rhA3C WT, S144A, and Q44R/H45R/S144A on a 118 nt fluorescently labeled ssDNA with two 5′TTC deamination motifs spaced 63 nt apart. Reactions were performed with 100 nM substrate DNA and 1000 nM rhA3C for the indicated amount of time (5–60 min). (B) Representative gel from three independent deamination assay experiments. Lanes with an asterisk were used for processivity calculations shown in panel (E). (C) Specific activity was calculated from the time course in (A) and showed increased activity if the rhA3C had the Q44R/H45R/S144A mutation. (D) The apparent Kd of rhA3C enzymes from a 118 nt fluorescently labeled ssDNA was analyzed by steady-state rotational anisotropy for rhA3C WT, S144A, and Q44R/H45R/S144A. Apparent Kd values are shown in the figure with the standard deviation. (E) Processivity of rhA3C S144A and Q44R/H45R/S144A calculated from the time course in (A–B) demonstrated that it was not increased above WT rhA3C. The processivity factor measures the likelihood of a processive deamination over a nonprocessive deamination and is described further in the ‘Materials and Methods‘ section. (A, D, E) On the graphs, the standard deviation for three independent experiments is shown as error bars.
Efficient HIV restriction by rhA3C when dimerization is mediated by amino acid 144
To test if the rhA3C S144A-mediated dimer form enabled HIV restriction, we conducted a single-cycle infectivity assay. We found that the rhA3C Q44R/H45R/S144A (Figure 10(A), 33% infectivity) could restrict HIV at an equivalent level to hA3C S188I (Figure 10(A), 24% infectivity). This was not due to increased encapsidation as it was encapsidated into HIV virions at an equivalent or slightly lesser amount that hA3C S188I (Figure 10 (B)). Although the rhA3C S144A could dimerize, it could not restrict HIV, consistent with no increase in catalytic activity (Figures 8, 9(C), and 10(A)). This result was interesting since both the S144A and M115N induced dimerization in combination with the Q44R/H45R mutations, but the rhA3C Q44R/H45R/S144A had a higher level of HIV restriction activity (Figure 10(A), 33% infectivity and Figure 4(A), 53% infectivity). We hypothesized that this simply could be because the Q44R/H45R/S144A had a more dominant dimer peak than Q44R/H45R/M115N (compare Figures 2(A–B) and 8(A–B)). Alternatively, since the in vitro specific activities were similar, the difference could be due to a larger change of loop 1 for rhA3C Q44R/H45R/S144A compared to rhA3C Q44R/H45R/M115N (Figures 7 and S9). Since the rhA3C Q44R/H45R/S144A processivity was less than rhA3C Q44R/H45R/M115N (compare Figures 3(D) and 9(E)) this led us to hypothesize that the rhA3C Q44R/H45R/S144A restricts HIV by a predominantly deamination-independent mode (mediated by nucleic acid binding) and rhA3C Q44R/H45R/M115N a deamination-dependent mode (mediated by deamination).
Figure 10. Amino acids 144 in combination with 44 and 45 in rhA3C enable HIV restriction ability.
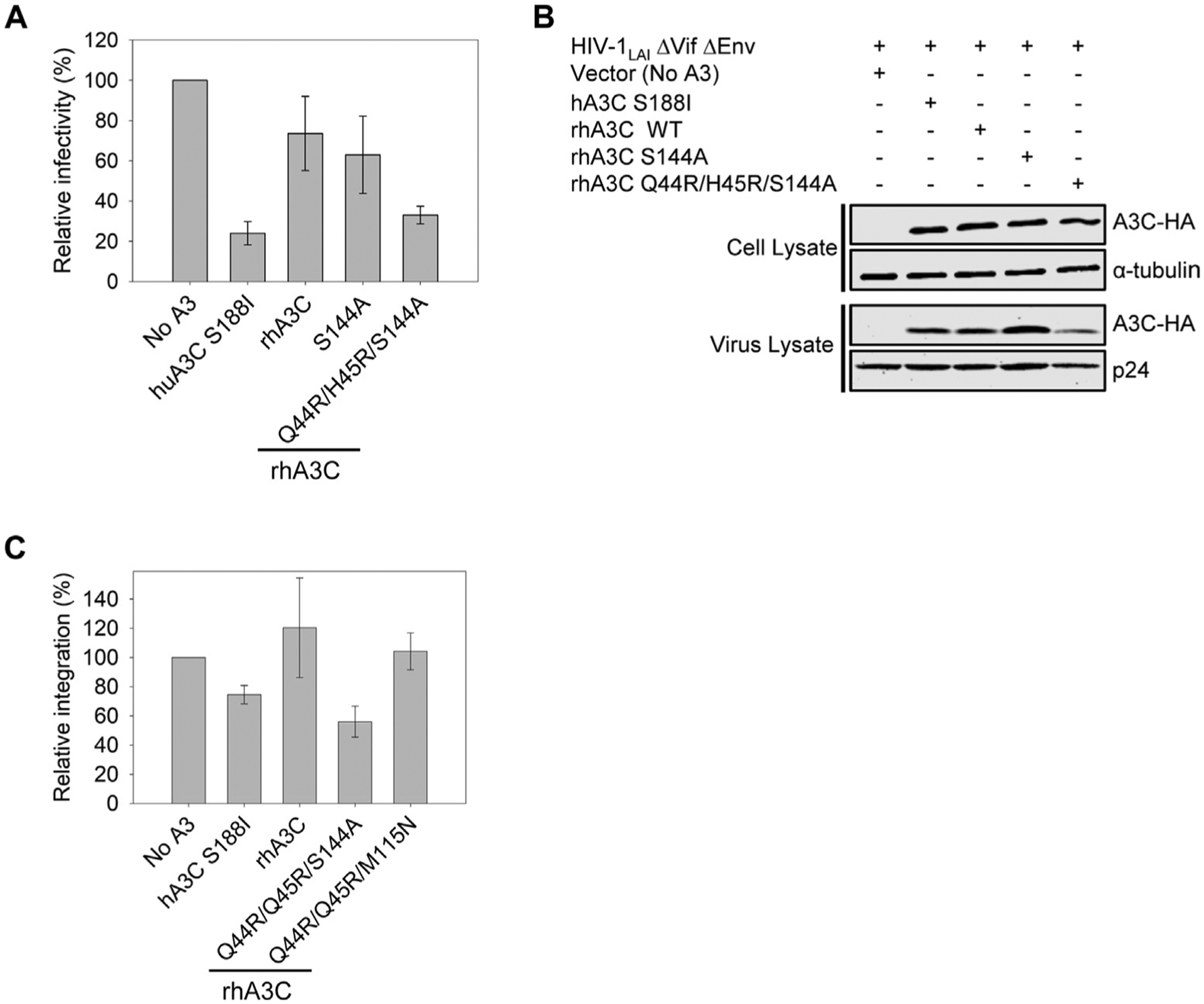
(A) Infectivity was measured by β-galactosidase expression driven by the HIV-1 5΄LTR from TZM-bl cells infected with VSV-G pseudotyped HIV ΔVif ΔEnv that was produced in the absence or presence of 3xHA tagged hA3C S188I, rhA3C WT, and rhA3C mutants S144A and Q44R/H45R/S144A. Results normalized to the no A3 condition are shown with error bars representing the Standard Deviation of the mean calculated from three independent experiments. (B) Immunoblotting for the HA tag was used to detect A3C enzymes expressed in cells and encapsidated into HIV ΔVif ΔEnv pseudotyped virions. The cell lysate and virion loading controls were α-tubulin and p24, respectively. (C) The relative amount of proviral DNA integration in infected HEK293T cells in the presence of hA3C S188I, rhA3C WT, and rhA3C mutants Q44R/H45R/S144A and Q44R/H45R/M115N in comparison to the No A3 condition was determined by qPCR. Error bars represent the standard deviation of the mean calculated from two independent experiments.
We tested the deamination-dependent mode by determining the level of G → A mutations in the coding strand of the integrated proviral DNA. We tested hA3C S188I, rhA3C WT and the two rhA3C mutants, Q44R/H45R/S144A and Q44R/H45R/M115N. The hA3C S188I had the highest level of G → A mutations (Table 1, 6.54 G → A mutations/kb). The majority mutations were in the GA → AA context (5′TC on the (−)DNA), which is the commonly preferred context for hA3C (Table 1). Consistent with lower activity against HIV (Figure 10(A)), the rhA3C WT had the lowest level of G → A mutations (Table 1, 2.15 mutations/kb). For rhA3C WT there was an approximately equal amount of mutations with a GG → AG context (5′CC on the (−)DNA) and GA → AA context, indicating that the rhA3C active site has a more relaxed sequence preference. The GG → AG context is primarily associated with A3G38. The rhA3C Q44R/H45R/S144A induced 1.5-fold less mutations than the rhA3C Q44R/H45R/M115N (Table 1, 3.20 and 4.67 G → A mutations/kb, respectively) consistent with rhA3C Q44R/H45R/S144A being 2-fold less processive than rhA3C Q44R/H45R/M115N.
Table 1.
Analysis of A3-induced mutagenesis of pol gene from integrated HIVΔVif.
| A3 enzyme | Base Pairs Sequenced | Total G → A mutations | Total GG → AG mutations | Total GA → AA mutations | G → A Mutations per kb | GG → AG mutations per kb | GA → AA mutations per kb |
|---|---|---|---|---|---|---|---|
| No A3 | 8715 | 4 | 2 | 2 | 0.46 | 0.23 | 0.23 |
| hA3C S188I | 8715 | 57 | 15 | 41 | 6.54 | 1.72 | 4.70 |
| rhA3C WT | 6972 | 15 | 6 | 7 | 2.15 | 0.86 | 1.00 |
| rhA3C Q44R/H45R/S144A | 8134 | 26 | 12 | 11 | 3.20 | 1.48 | 1.35 |
| rhA3C Q44R/H45R/M115N | 8134 | 38 | 20 | 15 | 4.67 | 2.46 | 1.84 |
To further explore how the rhA3C Q44R/H45R/S144A could restrict HIV more than rhA3C Q44R/H45R/M115N we also determined the level of proviral DNA integration. A3s can inhibit reverse transcriptase activity, which results in less completed proviral DNA synthesis, and less integration11,12. The rhA3C WT and rhA3C Q44R/H45R/M115N did not decrease proviral DNA integration (Figure 10(C)). However, hA3C S188I and rhA3C Q44R/H45R/S144A did decrease proviral DNA integration (Figure 10(C)). The rhA3C Q44R/H45R/S144A allowed only 56% of the total proviral DNA to integrate, relative to the no A3 condition (Figure 10(C)). These data show that rhA3C Q44R/H45R/S144A restricts HIV using both deamination -dependent and -independent modes of restriction which are more effective than only the deamination-dependent mode used by rhA3C Q44R/H45R/M115N. Since the ssDNA binding affinities for rhA3C Q44R/H45R/S144A and Q44R/H45R/M115N were similar (Figures 3(E) and 9 (D)), these data suggest that the rhA3C Q44R/H45R/S144A changes to loop 1 dynamics, increased dimerization, or both features enabled more deamination-independent restriction than rhA3C Q44R/H45R/M115N.
Evolutionary dynamics of key residues involved in A3C dimerization
Since amino acids at residues 44, 45, 115, and 144 were all found to be important in the gain of dimerization and restriction activity of rhA3C, we examined the evolution of these residues over primate evolution. None of these residues corresponds to those that were previously reported to be under positive selection22 Nonetheless, they do vary in Old World Primates and in Hominoids. While both the rhesus and the crab-eating macaque encode the QHMS at residues 44, 45, 115, and 144, respectively (Figure 11), the Northern pig tailed macaque encodes a cysteine (C) at residue 45 while the nearest outgroup, the Baboon, encodes an arginine (R) at residue 45, which is the residue found in Gorilla, Chimpanzee, and Humans at that position (Figure 11). However, the glutamine (Q) at amino acid 44 which is unfavorable for dimerization of rhA3C is also found in Baboons as well as the Collard Mangabey (Figure 11). Furthermore, the African Green Monkeys (Sabaeus, Vervet, Grivet, Tantalus) all have a histidine (H) at position 44. There is also variation at position 115 and 144 among primates. These results suggest that loss of dimerization of rhA3C is not a unique event, but rather may have occurred by a different mechanism in multiple primate lineages.
Figure 11. Phylogeny of A3C shows variation in primates at key residues important for dimerization.
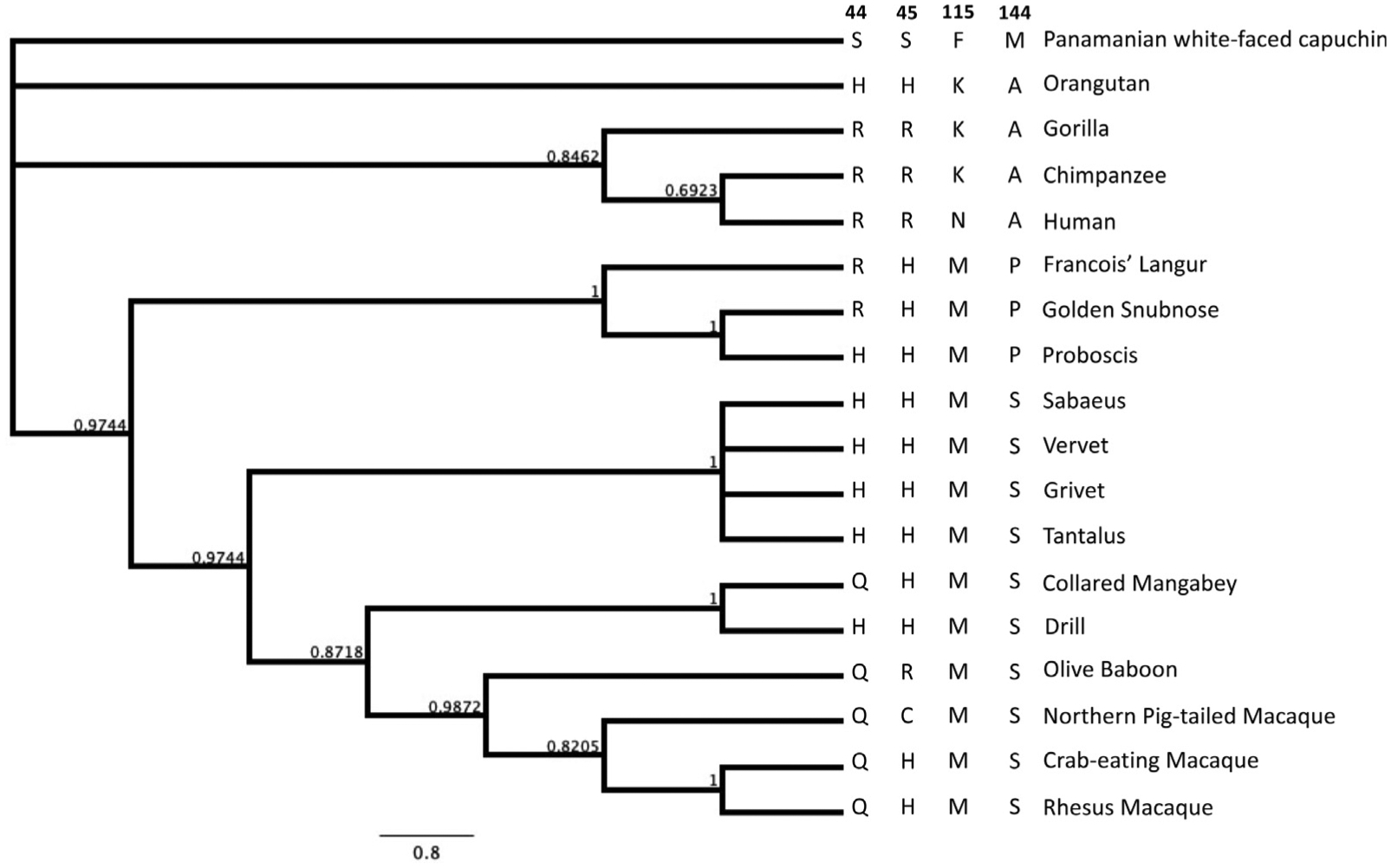
A phylogeny depicting residues crucial for APOBEC3C dimerization, including values indicating posterior probability of branch lengths. The species included are on the right of the phylogenetic tree of A3C sequences as well as the four amino acids at residues 44, 45, 115, and 144, respectively.
Discussion
Throughout the evolution of a host restriction factor, the protein must be able to retain activity against different viral pathogens, which may require compensatory mutations over time to either keep up with evolution of the initial virus or to counter-act new viral transmissions within the species39 In this study, we investigated the impact of evolutionary changes on a specific A3 enzyme, rhA3C. Our work highlights the importance of an integrated strategy to identify relevant functional interactions in enzymes. In particular, it describes a cycle of experiment and theory that allowed us to efficiently identify key positions from a combinatorial web of potential interactions. This methodology is transferrable and warrants its application to study other members of the APOBEC family as our analysis showed that the evolutionary signals for dimerization seem to be preserved across multiple organisms and family members with distinct functions. This methodology would also be useful in other protein systems.
Determination of activity against lentiviruses for A3 enzymes is multifactorial. First, it requires viral encapsidation, which occurs for all A3C orthologues tested thus far (Figure 4)15,22 After that, there is a requirement to deaminate cytidines in lentiviral (−)DNA or inhibit reverse transcriptase.40 Despite rhA3C inducing 2 mutations/kb, approximately 20 mutations in the total genome, the infectivity of the HIV was not significantly decreased (Figure 10 and Table 1). Since the mutations are stochastic, there may be some genomes with many mutations, some with none, and some with mutations that are not inactivating. Therefore, the more mutations, the better for ensuring inactivation. Increased processivity can achieve this, as evidenced by the 2-fold increase in mutations by the more processive rhA3C Q44R/H45R/M115N mutant, but this was still not enough for robust HIV restriction (Figure 4 and Table 1). What was needed was a deamination independent restriction of reverse transcriptase in combination with inducing mutagenesis to enable more robust restriction as for rhA3C Q44R/H45R/S144A (Figure 10 and Table 1).
However, the rhA3C Q44R/H45R/S144A results seemly go against numerous studies showing a direct correlation between processivity and mutation frequency in multiple A3 enzymes, including hominid A3Cs14,15,41,42 Using an in vitro system to study the effect of processivity on mutations during reverse transcription, it was shown that A3A, a non-processive enzyme, could introduce a similar number of mutations to A3G, a highly processive enzyme, but not at the same locations along the ssDNA43. The processivity was needed for mutations to accumulate in ssDNA regions rapidly lost to replication and dsDNA formation. The deaminations of A3A were achieved by a quasi-processive search involving multiple on and off interactions with the ssDNA. In this process time is lost, but deaminations still occur. This may be occurring with rhA3C Q44R/H45R/S144A.
Also interesting was that for rhA3C, unlike hominid A3Cs, the dimer did not always result in a processive enzyme. The rhA3C Q44R/H45R/S144A had equal specific activity to rhA3C Q44R/H45R/M115N but by a seemingly different mechanism than processivity (Figures 3 and 9). In addition, the rhA3C S144A single mutant could dimerize, but had no increase in activity (Figures 8 and 9). The rhA3C Q44R/H45R/S144A is predicted to have an altered loop 1 that is able to increase the deamination activity (Figures 7 and S9). Loop 1 has been identified to be a gate-like structure that controls access of the ssDNA to the active site36,37. A3A that has an open loop 1 conformation and displays high specific activity with low processivity44. In contrast, A3B and a related family member Activation Induced Cytidine deaminase (AID) have closed loop structures36,37. However, A3B and AID are processive and still achieve similar activity to A3A but are more selective for which cytidines are deaminated or under which types of conditions, e.g., A3B is more active when ssDNA is in excess to the enzyme45 Our data suggest that the rhA3C Q44R/H45R/S144A has increased activity in an A3A-like manner (Figures 9 and 10). Further, the rhA3C S144A mutant demonstrates that dimerization was necessary, but not sufficient, since arginines at residues 44 and 45 were also required for the increase in specific activity and restriction activity (Figures 9 and 10). The reason for why the processive rhA3C Q44R/H45R/M115N does not have an increase in anti-viral activity similar to hA3C S188I may simply be due to the lack of a stable dimer, which would be needed in the absence of the loop 1 alteration (Figure 2). Loop 1 has previously been shown to regulate activity of hA3C24. The hA3C was shown to have an amino acid pair 25 W/26E that decreased activity compared to a hA3C 25R/26 K mutant24. Our MD simulations data suggest that alterations in the protein structure from a 144A substitution changed the dynamics of loop 1 indirectly, which also resulted in increased specific activity, in a 44R/45R background (Figures 7, 9, and S9).
Interestingly, the majority of positive selection in A3C has taken place outside of the interaction motif with Vif, which suggests an evolution of enzyme activity, rather than avoidance of the lentiviral antagonist, providing a unique model to study restriction factor evolution22. Here we examined the residues needed for activity against HIV in rhA3C in their evolutionary context in other OWMs. We found that none of the analyzed OWM A3Cs contained the right amino acid combinations for robust activity, although there were considerable changes. The rhA3C sequences at the four key amino acids is not unique in OWM, but other OWMs also had other changes to one or two sites. This perhaps indicates that different selective pressures were on OWM A3C from a non-lentivirus pathogen. This may be a retroelement, such as LINE-1, since hA3C can restrict LINE-1 similarly to hA3C S188I, indicating that there is no requirement for dimerization22. Alternatively, A3C may act in concert with other A3s. For example, in humans, A3G and A3F have been found to hetero-oligomerize and this increases the activity of both enzymes46. This hetero-oligomerization has been understudied and perhaps A3C acts with another A3 and has greater activity. This would perhaps explain the different requirements for dimerization in comparison to hA3C although rhA3C and hA3C use the same general interface. Finally, it is possible that A3C, like A3H, has lost activity during evolution in primate47, perhaps because of selection against the deleterious effects of the enzyme, or because its activity has been usurped by other A3 enzymes.
The data show that rhA3C activity can be enhanced through dimerization that either increases processivity or more robustly through dimerization that causes an alteration of loop 1 conformation and dynamics. This is important for understanding the biochemical basis of activity of A3C and other A3 family members and for using it as a tool to predict anti-lentiviral activity in other OWMs. The OWM A3C has evidence of positive selection both within and outside the Vif binding region, suggesting that it has antiviral activity22. However, the fixation of the I188 in rhA3C and likely other OWMs did not impart anti-lentiviral activity, perhaps due to other compensatory mutations being made at the sites identified here, which were needed to combat other pathogens. Altogether, the data provide an in-depth structure–function analysis of rhA3C and suggest that the rhA3C viral targets of restriction have yet to be thoroughly identified.
Materials and methods
Plasmid constructs
The rhA3C DNA (GenBank: EU381233.1) was synthesized by GeneArt DNA synthesis and then subcloned into the appropriate vector. The rhA3C was cloned into the baculovirus transfer vector pFAST-bac1 containing an N-terminal GST tag as described previously using SmaI and NotI restriction sites42, pcDNA3.1 vector with a C-terminal 3xHA tag using a XbaI restriction site, or pCMV vector with a N-terminal 3xFlag using NotI and SalI restriction sites. Site directed mutagenesis of rhA3C WT was used to create rhA3C M115K, rhA3C M115N, rhA3C Q44R/H45R, rhA3C Q44R/H45R/M115K, rhA3C Q44R/H45R/M115N, rhA3C S144A and rhA3C Q44R/H45R/S144A mutants. The hA3C, hA3C S188I, and cA3C have been previously described15. All constructed plasmids were verified by DNA sequencing.
Single-cycle infectivity assay
HEK293T cells (1 × 105 cells per well) in 12 well plates were co-transfected with 500 ng of pHIV-1 LAI ΔVif ΔEnv, 180 ng of pMDG, which expresses VSV-G, and 100 ng of pcDNA A3C-3xHA expression plasmid using GeneJuice transfection reagent (EMD Millipore). The SIVsmm lineage 5 (L5) infectious molecular clone48 was obtained from Dr. Frank Kirchhoff with an inactivated Vif (SIVsmm ΔVif)21. After 24 h post transfection the media was changed. After 44 h post transfection, culture supernatants containing the virus were harvested, filtered through 0.45 μm polyvinylidene difluoride (PVDF) syringe filters and used to infect TZM-bl cells. For infection of TZM-bl cells 1 × 104 cells per well of a 96-well plate were infected with a serial dilution of virus in the presence of 8 μg/mL polybrene. Forty-eight hours after infection the cells were washed with PBS and infectivity was measured through colorimetric detection using a β-galactosidase assay reagent and spectrophotometer. Infectivity of each virus was compared by setting the infectivity of the “No A3” condition as 100%.
Immunoblotting viral and cell lysates
A portion of the viral supernatant collected for infectivity assays was concentrated using the Retro-X (Clontech) following the manufacturer’s protocol. For immunoblotting 8 μL of concentrated virus was used. Producer cells were washed with PBS and lysed using 2 × Laemmli buffer. Total protein in the cell lysate was estimated using the Lowry assay and 30 μg total protein from each cell lysate was used for Western blotting. A3C was detected in cell lysates and virions using anti-HA antibody (Mouse monoclonal, Cat# H9658 (Sigma) for cell lysate, Rabbit polyclonal, Cat# H6908 (Sigma) for virus). Loading controls for cell lysates (α-tubulin, rabbit polyclonal, Cat# PA1–20988, Invitrogen) and virus (p24, mouse monoclonal, Cat# 3537, NIH HIV Reagent Program) were detected using specific antibodies. Secondary detection was performed using Licor IRDye antibodies produced in goat (IRDye 680-labeled anti-rabbit 1: 10,000 Cat# 926–68071 and IRDye 800-labeled anti mouse 1:10000 Cat# 926–32210).
Proviral DNA sequencing
For proviral sequencing, 1 × 105 HEK293T cells per well of a 24-well plate were infected with supernatant containing virus in the presence of 8 μg/mL polybrene. The plates were spinoculated at 800g for 1 h. Cells were harvested after 48 h by removing the media, washing with PBS, and lysing the cells and extracting DNA with DNAzol (Invitrogen). The PCR amplification of a pol region of HIV-1 (581 bp) and treatment of DNA with DpnI was carried out as previously described14. Primers have been previously described49. Sequences were analyzed with Clustal Omega50 and Hypermut51.
Proviral DNA integration
Methods to quantify the integrated proviral DNA were adapted from52. For infections, 1 × 105 HEK293T cells per well of a 12-well plate were infected by spinoculation (1 h at 800g) and in the presence of polybrene (8 μg/mL) with HIV produced from the single-cycle replication assays. DNA was extracted after 24 h using DNAzol (Invitrogen) according to manufacturer’s instructions. The DNA was then treated with DpnI and 50 ng was used in a PCR as previously described52. The PCR was then diluted 40-fold and used as the template in a qPCR as previously described52.
Co-immunoprecipitation
For co-immunoprecipitation HEK293T cells in T75 cm2 flask were transfected with 1 μg of each plasmid DNA using GeneJuice transfection reagent (EMD Millipore) as per manufacturer’s instructions. At 48 h post transfection, the cells were washed with PBS and lysed using IP buffer (50 mM Tris-Cl pH 7.4, 1% Nonidet-P40, 10% glycerol, 150 mM NaCl) supplemented with EDTA-free protease inhibitor (Roche). The protein concentration of cell lysates was measured using Bradford assay and equal amount of protein was added to anti-Flag M2 magnetic beads (Sigma) in the presence of RNaseA (50 μg/ml; Roche) and incubated for 2 h with gentle rocking at 4 °C. The beads were subsequently washed 5 times with the Tris-Buffered Saline (TBS), and the immunoprecipitated proteins were subjected to SDS-PAGE and immunoblotting with anti-Flag antibody (Cat# F1804, Sigma), anti-HA antibody (Cat# H6908, Sigma). Secondary detection was performed using Licor IRDye antibodies produced in goat (IRDye 680-labeled anti-rabbit 1: 10,000 Cat# 926–68071 and IRDye 800-labeled anti mouse 1:10000 Cat# 926–32210).
Protein purification
The pFAST-bac1 vectors were used to produce recombinant baculovirus according to the protocol for the Bac-to-Bac system (Life Technologies), except using Insect GeneJuice (EMD Millipore) for bacmid transfection into Sf9 cells. The rhA3C WT and mutants were expressed in Sf9 cells following infection with recombinant baculovirus with a multiplicity of infection of 2.5 and harvested after 72 h. The purification of cA3C has been previously described15.
Pellets were stored at −80 °C until use. Thawed cell pellets from one litre cultures were resuspended in 35 ml of lysis buffer containing (20 mM HEPES (pH 7.5), 150 mM NaCl, 1% (v/v) Triton X-100, 10 mM NaF, 10 mM sodium phosphate, 10 mM sodium pyrophosphate, 100 μM ZnCl2, 1 mM EDTA, 10 mM DTT, 10% (v/v) glycerol and one Complete protease inhibitor tablet (Roche)). Following sonication and centrifugation, cleared lysate was incubated with glutathione-Sepharose resin (GE Healthcare) for several hours before washing the resin first with PBS buffer containing 1% (v/v) Triton X-100 and 500 mM NaCl followed by subsequent washes with PBS NaCl (250 mM). Finally, the resin was washed with digestion buffer containing 50 mM HEPES, pH 7.5, 250 mM NaCl, 1 mM DTT, and 10 % glycerol (v/v). The GST-A3C fusion enzyme containing slurry was treated with 0.02 units/μl thrombin (GE Healthcare) for a minimum of 4 hours at room temperature to release A3C from the GST tag. Purified proteins resolved by SDS-PAGE are shown in Figure S12.
Size exclusion chromatography (SEC) and multi angle light scattering (MALS)
The SEC was performed using the Superdex 200 Increase 10/300 (GE Healthcare). Purified protein (230 ± 25 μg) was applied to the column equilibrated with 50 mM Tris (pH 8), 200 mM NaCl, 1 mM DTT. The flow rate was set to 0.6 ml/min and the eluate was monitored by the UV absorbance at 280 nm. Fractions were collected and further assessed using Coomassie stained SDS-PAGE. The same protocol was used to measure the light scattering and refractive index using a Wyatt Technology Multi-Angle Light Scattering (MALS) DAWN HELEOS II and Refractive Index (RI) OPTILAB T-rex, connected in tandem to a Bio-Rad FPLC system.
Deamination assay
Reactions were conducted with 100 nM of a fluorescein labeled 118 nt DNA substrate (Tri-Link Biotechnologies) with two 5′TTC motifs at 37 °C in RT buffer (50 mM Tris, pH 7.5, 40 mM KCl, 10 mM MgCl2 and 1 mM DTT). The substrate was designed to not contain secondary structure and the sequence has been previously described14. Reactions used 1000 nM A3C and were incubated for 0 to 60 min. The reaction was initiated by the addition of ssDNA. Deamination reactions were stopped using phenol:chloroform extraction followed by two additional chloroform extractions. The deaminations were detected by treating the substrates with Uracil DNA Glycosylase (New England Biolabs) and heating under alkaline conditions. The ssDNA fragments were resolved on 10% (v/v) denaturing polyacrylamide gel. Gel photos were obtained using a Chemidoc-MP imaging system (Bio-Rad) and integrated gel band intensities were analyzed using ImageQuant (GE Healthcare).
Processivity reactions were carried out under single-hit conditions (<15% substrate usage) to ensure a single enzyme-substrate encounter53. A processivity factor can be calculated under these conditions by comparing the quantified total amount of deaminations occurring at the two sites on the same ssDNA with a calculated theoretical value of deaminations assuming they were different deamination events54,55. To define these conditions, we determined the deamination over time and chose a time point with less than 15% substrate usage (see Materials and Methods). A processivity factor greater than 1.0 means the majority of double deaminations are catalyzed by a single enzyme, and therefore x-fold more likely to deaminate processively. A non-processive enzyme has a processivity factor of 1.0 or more commonly, does not have a visible amount of deamination at two sites under the single-hit conditions of the reaction. The specific activity was calculated under single-hit conditions by determining the picomoles of substrate used per minute for a microgram of enzyme.
Steady-state rotational anisotropy
Steady-state rotational anisotropy reactions (60 μL) were conducted in buffer containing 50 mM Tris, pH 7.5, 40 mM KCl, 10 mM MgCl2, and 1 mM DTT and contained 10 nM of the fluorescein-labeled 118 nt ssDNA used for deamination assays. Increasing amounts of A3C was added (0 to −4500 nM). A QuantaMaster QM-4 spectrofluorometer (Photon Technology International) with a dual emission channel was used to collect data and calculate anisotropy. Measurements were performed at 21 °C. Samples were excited with vertically polarized light at 495 nm (8-nm band pass), and vertical and horizontal emissions were measured at 520 nm (8-nm band pass). The apparent Kd was obtained by fitting to a single rectangular hyperbola equation using SigmaPlot version 11.2 software.
Classical molecular dynamics
The initial structure of the human hA3C dimer was taken from the Protein Data Bank (PDB accession: 3VOW) and the peptide sequence was confirmed using Uniprot. The rhesus model (rhA3C) and all human-background variants were generated from the human dimer using Chimera to modify the peptide sequence56. Four single mutants (R44Q, R45H, N115M, A144S), a double mutant (R44Q/R45H), and two triple mutants (R44Q/R45H/N115M, R44Q/R45H/A144S) were all generated from the human wildtype (WT) background. MolProbity and H++ were used for all systems to determine protonation states of amino acids at pH of 7.057–62. All models were neutralized to zero net charge with Cl− and K+ counterions and solvated using TIP3P water with a 12 Å minimum distance from protein surface to the edge of the solvent box63. This resulted in a solvated rectangular box unit cell measuring 82 Å × 84 Å × 108 Å with 90° angles between all adjacent edges. The AMBER FF14SB forcefield was used for the protein and TIP3P for water, Zn2+, and counterions63,64.
Molecular dynamics simulations were performed using OpenMM on XSEDE’s Comet HPC cluster65,66. Each system was equilibrated with iteratively reduced restraints on the protein to ensure stable simulation environment. The restraints began at 1000 kcal/mol and were reduced by half after every completed stage of equilibration until the restraint fell to below 1 kcal/mol. Each completed stage was run for a minimum of 1.0 ns (106 timesteps) and checked for convergence to ensure stability of temperature (300 K), density (1.0 g/mL), periodic box volume (approx 600 Å2), and total (potential and kinetic) energies of the system, resulting in a total equilibration time of 11.0 ns. To maintain active site geometry, harmonic restraints of 20 kcal/mol Å2 were applied between the active site zinc and the carboxylate carbon of E54 of each monomer subunit with an equilibrium distance of 5.0 Å. These restraints were maintained throughout the simulations. After equilibration, production dynamics were run for 100 ns using a 1 fs timestep and coordinates saved every 10 ps. A Langevin integrator was used as the thermostat with a Monte Carlo barostat in an NPT ensemble. The nonbonded cutoff distance was set to 10.0 Å, and the Ewald error tolerance was set to 10−3. All models were simulated in triplicate for a total of 300 ns of production.
Analysis of MD trajectories was performed using cpptraj (correlated motion, normal mode analysis, root mean squared deviation (RMSD) RMSF, hydrogen bond interaction analysis)67. Data plots were generated with python, and 3D structure visualization was done using Chimera56. The first 100 normal modes were calculated and their relative contribution to the total motion was examined. Dimer interaction energies were calculated using the AMBER-EDA program (available at https://github.com/CisnerosResearch/AMBER-EDA) by calculating the ensemble average Coulomb and Van der Waals interactions between each amino acid on one monomer with each amino acid on the other.
Direct coupling analysis (DCA)
DCA is a global statistical inference model that is used to study coevolution in protein sequences28. Through DCA, an approximation of the global probability distribution for a multiple-sequence alignment creating a large quantity of homologous sequences can be modeled for a set of residual positions in the sequence. The DCA model can accurately estimate the direct covariations between any two variables while excluding secondary correlations between dependent variables. Therefore, DCA can be used to study molecular connectivity in various biological situations, such as the functionality and specificity for interacting proteins. DCA can also be used on non-sequence data, such as pharmacogenomic data68.
In the DCA model, naturally occurring and modified protein sequences are assumed to be sampled from a Boltzmann distribution. Multiple sequences are grouped in a manner such that the homologous positions are aligned, forming a large sample space. The Boltzmann distribution is the most general and least-constrained model derived from maximum entropy modeling. DCA infers in an efficient manner the parameters of a large joint-probability distribution and uses these inferred parameters to determine estimates of the coupling between pairs of variables in such distribution. A metric to quantify such coupling between two positions in the alignment is called Direct Information (DI)28 which is zero if the two positions are uncoupled and positive otherwise. Higher DI values indicate a stronger dependency or functional relevance is present for the two residue sites. In this study, we utilize a metric called Cumulative Direct Information CDIi, which is the sum of the DI values for all the residue-residue pairs that a given residue i participates in a list of top x ranked pairs.
Sequence datasets
An NCBI multiple sequence alignment (MSA) was downloaded from the Pfam database for the APOBEC3 (PF18771) family69. Sequences with more than 27 (20% of the aligned sequence length) consecutive mispairing positions were removed. The resulting MSA was composed of 2504 sequences (Supplementary file 1). The MSA was processed using mean field DCA (mfDCA). The resulting DI pairs were then filtered for monomeric interactions and ranked from greatest to least DI value. A domain matching script using the FASTA sequence for hA3C (Q9NRW3) was used to match the calculated DI pairs to the original hA3C sequence.
Molecular simulation of homodimeric complexes driven by evolutionary couplings
To estimate a complex of the hA3C dimer that is consistent with the relevant evolutionary couplings for dimerization uncovered by DCA, we utilized a coarse-grained simulation methodology that uses the coordinates of low energy states of individual monomers and represents them as a bonded chain of residues, as opposed to an all-atom representation of the side chains. These Structure Based Models (SBMs)70 have been used extensively to study the dynamics of protein folding and molecular interactions and recently we have shown that they can be enhanced with the addition of evolutionary residue-residue interactions to study folding and complex formation71. In the SBM formulation, there is no implicit solvation and molecular interactions are modelled though a Hamiltonian composed of bonded (VB) and non-bonded interactions (VNB):
| (1) |
where rij is the distance between two residues i and j in the amino acid chain. The bonded interactions VB have, in turn, a distance potential determined by bonds, angles and dihedrals parametrized from the native structures. The non-covalent interactions have components for tertiary contacts as well as repulsive interactions. We employ a specific Gaussian potential developed by Lammert et al. that allows non-bonded interactions to be parametrized to allow the introduction of dimeric contacts predicted from evolutionary constraints72. We have shown that these coarse-grained models are predictive of homodimerization with highly accurate RMSDs31 and we have employed such methodology to predict dimerization states of hA3C monomers. This methodology complements well the full-atomic analysis described in the “Classical Molecular Dynamics” methodology since it provides an accelerated way to estimate complexes that agree with the evolutionary interactions predicted by Direct Coupling Analysis.
The protein data bank (PDB) for hA3C (3VOW) was accessed to obtain all-atom coordinates of the protein crystal structure of hA3C35. The structure was used to compute the solvent accessible surface area (SASA) of individual residues using getarea program73. From the list of top coevolving DCA pairs, only those whose SASA summations were greater than 100 were taken for modeling studies to prioritize interactions at the protein surface. Using the PDB structure, all DCA pairs that were monomeric interactions, i.e., that contribute to the monomeric fold, were filtered out. From the remaining ranked DCA pairs, the top 25 were selected as dimeric interactions in a coarse-grain molecular simulation of the two interacting monomers.
The top 25 DCA pairs were incorporated as Gaussian potentials to drive formation of the hA3C dimer complex in a SBM-MD simulation31. The simulation process iteratively reduced the equilibrium distance between the monomer units to allow exploration of the dimer interface. A PDB file containing the two monomer subunits at 50 Å was used as input in the SMOG server to generate parameter and topology files70,74. SBM potentials generated from the top 25 DCA pairs were inserted into the SMOG files. A version of GROMACS with support for SBM Gaussian potentials was used to run the molecular dynamics simulation72. The final coordinates of the model were then compared to the initial PDB dimer structure.
Contact maps
The x-ray coordinates for hA3C (3VOW) were used to identify residue-residue physical contacts. A monomeric contact map was then created based on the proximity between amino acids of the same subunit, with a distance cutoff at 8 Å between α carbon atoms. A dimeric contact map was generated based on the proximity between residues of different subunit, with a distance cutoff at 10 Å between α carbon atoms. The two contact maps were then overlayed on top of each other to show all the residue-residue interactions in the PDB structure as well as the top 300 DI pairs calculated using DCA to give the final map.
Phylogenetic analysis
Sequences of 18 primates were obtained from NCBI and were also described in22. Sequence alignments were created using the MUSCLE 3.8.425 function75. We used a HKY85 substitution model, a gamma rate variation, unconstrained branch lengths and estimated base frequencies. Sequences were analyzed phylogenetically using a Bayesian Monte Carlo Markov (MCMC) approach within MrBayes 3.2.676 in Geneious Prime 2021.2.2 (Biomatters Ltd). Nucleotide alignments were translated into codons to extract information about the amino acid identities at positions 44, 45, 115, and 144.
Data availability
All data is within the manuscript or available upon request.
Supplementary Material
Acknowledgements
We thank Ossama Ibrahim for assistance with site directed mutagenesis. Research described in this paper was performed in part at the Protein Characterization and Crystallization Facility (PCCF), which is supported by the College of Medicine, University of Saskatchewan, Saskatoon, Canada. Reagents obtained through the NIH HIV Reagent Program, Division of AIDS, NIAID, NIH and Centre for AIDS Reagents, NIBSC, UK, were supported by EURIPRED (EC FP7 INFRASTRUCTURES-2012 – INFRA-2012–1.1.5.: Grant Number 31266). This work was supported by a Canadian Institutes of Health Research (CIHR) Grant PJT-162407 (L.C.), NIH R01GM108583 and NSF CHE-1856162 (G.A.C.), support to B.B. via NSF CHE-1757946, computing time from XSEDE TG-CHE160044 (G.A.C.), CASCaM with partial support from NSF CHE-1531468 (G.A.C.), NIH R35GM133631 and NSF MCB-1943442 (F.M.), NIH R01AI030927 (M.E.), D.W. was in the UW Post-Baccalaureate Research Education Program (PREP), and A.G. was supported by a Saskatchewan Health Research Foundation (SHRF) postdoctoral fellowship.
Footnotes
CRediT authorship contribution statement
Amit Gaba: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Writing – original draft, Visualization. Mark A. Hix: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data curation, Writing – original draft, Visualization. Sana Suhail: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data curation, Writing – review & editing, Visualization. Ben Flath: Methodology, Validation, Formal analysis, Investigation, Writing – review & editing, Visualization. Brock Boysan: Investigation. Danielle R. Williams: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data curation, Writing – review & editing, Visualization. Tomas Pelletier: Investigation. Michael Emerman: Validation, Supervision, Funding acquisition. Faruck Morcos: Conceptualization, Methodology, Validation, Formal analysis, Writing – original draft, Visualization, Supervision, Funding acquisition. G. Andrés Cisneros: Conceptualization, Methodology, Validation, Formal analysis, Writing – original draft, Visualization, Supervision, Funding acquisition. Linda Chelico: Conceptualization, Methodology, Validation, Formal analysis, Writing – original draft, Visualization, Supervision, Funding acquisition, Project administration.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2021.167306.
References
- 1.Gaba A, Flath B, Chelico L, (2021). Examination of the APOBEC3 Barrier to Cross Species Transmission of Primate Lentiviruses. Viruses 13, 1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris RS, Hultquist JF, Evans DT, (2012). The restriction factors of human immunodeficiency virus. J. Biol. Chem 287, 40875–40883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Etienne L, Hahn BH, Sharp PM, Matsen FA, Emerman M, (2013). Gene loss and adaptation to hominids underlie the ancient origin of HIV-1. Cell Host Microbe 14, 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uriu K, Kosugi Y, Ito J, Sato K, (2021). The Battle between Retroviruses and APOBEC3 Genes: Its Past and Present. Viruses 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng AZ, Moraes SN, Shaban NM, Fanunza E, Bierle CJ, Southern PJ, et al. , (2021). APOBECs and Herpesviruses. Viruses 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adolph MB, Love RP, Chelico L, (2018). Biochemical Basis of APOBEC3 Deoxycytidine Deaminase Activity on Diverse DNA Substrates. ACS Infect. Dis 4, 224–238. [DOI] [PubMed] [Google Scholar]
- 7.Arias JF, Koyama T, Kinomoto M, Tokunaga K, (2012). Retroelements versus APOBEC3 family members: No great escape from the magnificent seven. Front. Microbiol 3, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delviks-Frankenberry KA, Desimmie BA, Pathak VK, (2020). Structural Insights into APOBEC3-Mediated Lentiviral Restriction. Viruses 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu WK, Byun H, Dudley JP, (2020). The Role of APOBECs in Viral Replication. Microorganisms 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salter JD, Bennett RP, Smith HC, (2016). The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem Sci. 41, 578–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pollpeter D, Parsons M, Sobala AE, Coxhead S, Lang RD, Bruns AM, et al. , (2018). Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nature Microbiol. 3, 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwatani Y, Chan DS, Wang F, Maynard KS, Sugiura W, Gronenborn AM, et al. , (2007). Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 35, 7096–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu Y, Knecht KM, Shen Q, Xiong Y, (2020). Multifaceted HIV-1 Vif interactions with human E3 ubiquitin ligase and APOBEC3s. FEBS J.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ara A, Love RP, Chelico L, (2014). Different mutagenic potential of HIV-1 restriction factors APOBEC3G and APOBEC3F is determined by distinct single-stranded DNA scanning mechanisms. PLoS Pathog. 10, e1004024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adolph MB, Ara A, Feng Y, Wittkopp CJ, Emerman M, Fraser JS, et al. , (2017). Cytidine deaminase efficiency of the lentiviral viral restriction factor APOBEC3C correlates with dimerization. Nucleic Acids Res. 45, 3378–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaipan C, Smith JL, Hu WS, Pathak VK, (2013). APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J. Virol 87, 444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.OhAinle M, Kerns JA, Li MM, Malik HS, Emerman M, (2008). Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 4, 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richardson SR, Narvaiza I, Planegger RA, Weitzman MD, Moran JV, (2014). APOBEC3A deaminates transiently exposed single-strand DNA during LINE-1 retrotransposition. Elife 3, e02008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng AZ, Yockteng-Melgar J, Jarvis MC, Malik-Soni N, Borozan I, Carpenter MA, et al. , (2019). Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nature Microbiol. 4, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, et al. , (2011). Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol 85, 11220–11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nchioua R, Kmiec D, Gaba A, Stürzel CM, Follack T, Patrick S, et al. , (2021). APOBEC3F constitutes a barrier to successful cross-species transmission of SIVsmm to humans. J Virol.. Jvi0080821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wittkopp CJ, Adolph MB, Wu LI, Chelico L, Emerman M, (2016). A Single Nucleotide Polymorphism in Human APOBEC3C Enhances Restriction of Lentiviruses. PLoS Pathog. 12, e1005865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson BD, Ikeda T, Moghadasi SA, Martin AS, Brown WL, Harris RS, (2018). Natural APOBEC3C variants can elicit differential HIV-1 restriction activity. Retrovirology 15, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaguva Vasudevan AA, Balakrishnan K, Gertzen CGW, Borveto F, Zhang Z, Sangwiman A, et al. , (2020). Loop 1 of APOBEC3C Regulates its Antiviral Activity against HIV-1. J. Mol. Biol 432, 6200–6227. [DOI] [PubMed] [Google Scholar]
- 25.Virgen CA, Hatziioannou T, (2007). Antiretroviral activity and Vif sensitivity of rhesus macaque APOBEC3 proteins. J. Virol 81, 13932–13937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, Gu Q, Jaguva Vasudevan AA, Jeyaraj M, Schmidt S, Zielonka J, et al. , (2016). Vif Proteins from Diverse Human Immunodeficiency Virus/Simian Immunodeficiency Virus Lineages Have Distinct Binding Sites in A3C. J. Virol 90, 10193–10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith HC, (2016). RNA binding to APOBEC deaminases; Not simply a substrate for C to U editing. RNA Biol., 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, et al. , (2011). Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci. U. S. A 108, E1293–E1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anishchenko I, Ovchinnikov S, Kamisetty H, Baker D, (2017). Origins of coevolution between residues distant in protein 3D structures. Proc. Natl. Acad. Sci 114, 9122–9127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marks DS, Colwell LJ, Sheridan R, Hopf TA, Pagnani A, Zecchina R, et al. , (2011). Protein 3D Structure Computed from Evolutionary Sequence Variation. PLoS ONE 6, e28766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.dos Santos RN, Morcos F, Jana B, Andricopulo AD, Onuchic JN, (2015). Dimeric interactions and complex formation using direct coevolutionary couplings. Sci. Rep 5, 13652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ovchinnikov S, Kamisetty H, Baker D, (2014). Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. Elife 3, e02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malinverni D, Jost Lopez A, De Los RP, Hummer G, Barducci A, (2017). Modeling Hsp70/Hsp40 interaction by multi-scale molecular simulations and coevolutionary sequence analysis. Elife 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Werner M, Gapsys V, de Groot BL, (2021). One Plus One Makes Three: Triangular Coupling of Correlated Amino Acid Mutations. J. Phys. Chem. Lett 12, 3195–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kitamura S, Ode H, Nakashima M, Imahashi M, Naganawa Y, Kurosawa T, et al. , (2012). The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nature Struct. Mol. Biol 19, 1005–1010. [DOI] [PubMed] [Google Scholar]
- 36.Shi K, Demir Ö, Carpenter MA, Wagner J, Kurahashi K, Harris RS, et al. , (2017). Conformational Switch Regulates the DNA Cytosine Deaminase Activity of Human APOBEC3B. Sci. Rep 7, 17415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.King JJ, Larijani M, (2017). A Novel Regulator of Activation-Induced Cytidine Deaminase/APOBECs in Immunity and Cancer: Schrödinger’s CATalytic Pocket. Front. Immunol 8, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, et al. , (2004). Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nature Struct. Mol. Biol 11, 435–442. [DOI] [PubMed] [Google Scholar]
- 39.Duggal NK, Emerman M, (2012). Evolutionary conflicts between viruses and restriction factors shape immunity. Nature Rev. Immunol 12, 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harris RS, Dudley JP, (2015). APOBECs and virus restriction. Virology 479–480, 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feng Y, Chelico L, (2011). Intensity of deoxycytidine deamination of HIV-1 proviral DNA by the retroviral restriction factor APOBEC3G is mediated by the noncatalytic domain. J. Biol. Chem 286, 11415–11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng Y, Love RP, Ara A, Baig TT, Adolph MB, Chelico L, (2015). Natural Polymorphisms and Oligomerization of Human APOBEC3H Contribute to Single-stranded DNA Scanning Ability. J. Biol. Chem 290, 27188–27203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Love RP, Xu H, Chelico L, (2012). Biochemical analysis of hypermutation by the deoxycytidine deaminase APOBEC3A. J. Biol. Chem 287, 30812–30822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi K, Carpenter MA, Banerjee S, Shaban NM, Kurahashi K, Salamango DJ, et al. , (2017). Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nature Struct. Mol. Biol 24, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adolph MB, Love RP, Feng Y, Chelico L, (2017). Enzyme cycling contributes to efficient induction of genome mutagenesis by the cytidine deaminase APOBEC3B. Nucleic Acids Res. 45, 11925–11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ara A, Love RP, Follack TB, Ahmed KA, Adolph MB, Chelico L, (2017). Mechanism of Enhanced HIV Restriction by Virion Coencapsidated Cytidine Deaminases APOBEC3F and APOBEC3G. J. Virol 91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia EI, Emerman M, (2018). Recurrent Loss of APOBEC3H Activity during Primate Evolution. J. Virol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fischer W, Apetrei C, Santiago ML, Li Y, Gautam R, Pandrea I, et al. , (2012). Distinct evolutionary pressures underlie diversity in simian immunodeficiency virus and human immunodeficiency virus lineages. J. Virol 86, 13217–13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mohammadzadeh N, Love RP, Gibson R, Arts EJ, Poon AFY, Chelico L, (2019). Role of co-expressed APOBEC3F and APOBEC3G in inducing HIV-1 drug resistance. Heliyon 5, e01498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. , (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rose PP, Korber BT, (2000). Detecting hypermutations in viral sequences with an emphasis on G → A hypermutation. Bioinformatics 16, 400–401. [DOI] [PubMed] [Google Scholar]
- 52.Belanger K, Savoie M, Rosales Gerpe MC, Couture JF, Langlois MA, (2013). Binding of RNA by APOBEC3G controls deamination-independent restriction of retroviruses. Nucleic Acids Res. 41, 7438–7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Creighton S, Bloom LB, Goodman MF, (1995). Gel fidelity assay measuring nucleotide misinsertion, exonucleolytic proofreading, and lesion bypass efficiencies. Methods Enzymol. 262, 232–256. [DOI] [PubMed] [Google Scholar]
- 54.Chelico L, Pham P, Calabrese P, Goodman MF, (2006). APOBEC3G DNA deaminase acts processively 3’ → 5’ on single-stranded DNA. Nature Struct. Mol. Biol 13, 392–399. [DOI] [PubMed] [Google Scholar]
- 55.Pham P, Chelico L, Goodman MF, (2007). DNA deaminases AID and APOBEC3G act processively on single-stranded DNA. DNA Repair (Amst) 6, 689–692. author reply 93–4. [DOI] [PubMed] [Google Scholar]
- 56.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. , (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 57.Anandakrishnan R, Aguilar B, Onufriev AV, (2012). H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40, W537–W541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Myers J, Grothaus G, Narayanan S, Onufriev A, (2006). A simple clustering algorithm can be accurate enough for use in calculations of pKs in macromolecules. Proteins Struct. Funct. Bioinf 63, 928–938. [DOI] [PubMed] [Google Scholar]
- 59.Chen VB, Bryan AW, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson, (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D, Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gordon J, Myers J, Folta T, Shoja V, Heath L, Onufriev A, (2005). H++: a server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 33, 368–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, et al. , (2007). MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davis IW, Murray LW, Richardson JS, Richardson DC, (2004). MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32, W615–W619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926–935. [Google Scholar]
- 64.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C, (2015). ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Towns J, Cockerill T, Dahan M, Foster I, Gaither K, et al. , (2014). XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng 16, 62–74. [Google Scholar]
- 66.Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, et al. , (2017). OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol 13, e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Case DA, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham ITE, Cruzeiro VWD, et al. , (2018). AMBER 2018. University of California, San Francisco. [Google Scholar]
- 68.Jiang XL, Martinez-Ledesma E, Morcos F, (2017). Revealing protein networks and gene-drug connectivity in cancer from direct information. Sci. Rep 7, 3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, et al. , (2019). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clementi C, Nymeyer H, Onuchic JN, (2000). Topological and energetic factors: what determines the structural details of the transition state ensemble and “en-route” intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol 298, 937–953. [DOI] [PubMed] [Google Scholar]
- 71.Noel JK, Morcos F, Onuchic JN, (2016). Sequence co-evolutionary information is a natural partner to minimally-frustrated models of biomolecular dynamics. F1000Res 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lammert H, Schug A, Onuchic JN, (2009). Robustness and generalization of structure-based models for protein folding and function. Proteins 77, 881–891. [DOI] [PubMed] [Google Scholar]
- 73.Fraczkiewicz R, Braun W, (1998). Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem 19, 319–333. [Google Scholar]
- 74.Noel JK, Whitford PC, Sanbonmatsu KY, Onuchic JN, (2010). SMOG@ctbp: simplified deployment of structure-based models in GROMACS. Nucleic Acids Res. 38, W657–W661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Edgar RC, (2004). MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 5, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huelsenbeck JP, Ronquist F, (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. [DOI] [PubMed] [Google Scholar]
- 77.Robert X, Gouet P, (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is within the manuscript or available upon request.