

Abstract
In eukaryotic cells, intron lariats produced by the spliceosome contain a 2′5′ phosphodiester linkage. The RNA lariat debranching enzyme, Dbr1, is the only enzyme known to hydrolyze this bond. Dbr1 is a member of the metallophosphoesterase (MPE) family of enzymes, and recent X-ray crystal structures and biochemistry data demonstrate that Dbr1 from Entamoeba histolytica uses combinations of Mn2+, Zn2+, and Fe2+ as enzymatic cofactors. Here, we examine the kinetic properties and metal dependence of the Dbr1 homolog from Saccharomyces cerevisiae (yDbr1). Elemental analysis measured stoichiometric quantities of Fe and Zn in yDbr1 purified following heterologous expression E. coli. We analyzed the ability of Fe2+, Zn2+, and Mn2+ to reconstitute activity in metal-free apoenzyme. Purified yDbr1 was highly active, turning over substrate at 5.6 sec−1, and apo-yDbr1 reconstituted with Fe2+ was the most active species, turning over at 9.2 sec−1. We treated human lymphoblastoid cells with the iron-chelator deferoxamine and measured a twofold increase in cellular lariats. These data suggest that Fe is an important biological cofactor for Dbr1 enzymes.
Keywords: RNA, lariats, introns, debranching enzyme, metalloenzymes, metallophosphoesterase
INTRODUCTION
The presence of noncoding introns in protein coding genes is a hallmark of eukaryotic cells. These introns are removed by the spliceosome, a large riboprotein complex that recognizes the splice-sites and the branchpoint sequence. The 2′-OH of the branchpoint nucleotide nucleophilically attacks the first 5′–3′ phosphodiester bond of the intron leaving a “lariat” topology where the 3′ end of the intron is the “tail” and the 5′ end forms the loop at the branchpoint. After a second nucleophilic attack, the excised lariat is liberated (Wilkinson et al. 2020). Although one-third of the human genome “codes” for these introns, their specific biological roles remain unclear beyond the observation that some snoRNA and miRNA are located within introns (Ooi et al. 1998; Ladewig et al. 2012). Thus, the utility of introns remains a mystery. Significant cellular resources are used to maintain and transcribe intronic sequences, yet the products are largely degraded following splicing.
Recently, hints as to the roles introns play in biology have come from systems where lariat metabolism is defective due to the loss of Dbr1. When humans are homozygous for loss-of-function mutations in the DBR1 loci, they are susceptible to severe juvenile brainstem encephalitis from viruses that are normally benign (Zhang et al. 2018). These mutations reduce Dbr1 protein levels and enzymatic activity, while increasing the levels of lariat RNA. However, the specific molecular interactions that link Dbr1, lariats, and innate immunity remain unknown.
Intron lariats modulate the proteotoxicity of TDP-43 protein aggregates in a yeast model of amyotrophic lateral sclerosis (ALS) (Armakola et al. 2012). TDP-43 aggregates are found in the spinal cords of the majority of nonfamilial amyotrophic lateral sclerosis patients (Neumann et al. 2006), as well as in frontotemporal dementia (Goossens et al. 2015), and chronic traumatic encephalopathy (McKee et al. 2010). This result suggests that Dbr1 inhibition, with concomitant increase in lariat RNA, may protect from TDP-43 toxicity. Also, a recent study showed that down-regulation of Dbr1 can induce oncogenesis in cellular models by disrupting splicing and impacting snRNP recycling (Han et al. 2017).
Debranching activity was first isolated and characterized from HeLa cells in 1985 (Ruskin and Green 1985). The Dbr1 gene was identified in yeast in 1991 as a factor required for the retrovirus-like Ty-1 retrotransposon (Chapman and Boeke 1991), and several groups have explored potential mechanisms of how Dbr1 affects Ty-1 retrotransposition (Cheng and Menees 2004, 2011; Pratico and Silverman 2007). Dbr1 knockdown also impairs replication of the HIV retrovirus (Ye et al. 2005; Galvis et al. 2017) and is required for class-switch recombination of antibodies (Zheng et al. 2015).
The metals Mn2+, Fe2+, and Zn2+ support Dbr1 catalysis in biochemical assays (Khalid et al. 2005; Clark et al. 2016). The first X-ray crystal structure of Dbr1, from the amoeba Entamoeba histolytica (EhDbr1), contained Mn2+, and only a single metal ion was bound in the β-pocket of the active site. This was a surprising result, because all related MPEs have two metal ion cofactors (Matange et al. 2015), and Dbr1 was predicted to have a di-Mn active site (Khalid et al. 2005; Schwer et al. 2016). Subsequent structures from two different groups resolved this issue by observing a second metal, a Zn ion, bound in the α-pocket where it was coordinated by a cysteine (Cys) residue that is unique to Dbr1 enzymes (Clark et al. 2016; Ransey et al. 2017). This residue is an aspartic acid (Asp) in other MPEs. Therefore, it appears that the Cys in Dbr1 enzymes provides for stable Zn binding. EhDbr1 is the only Dbr1 protein with a reported crystal structure, and based on the 33% sequence identity between E. histolytica and yeast Saccharomyces cerevisiae Dbr1 (yDbr1), we hypothesized that yDbr1 would also use a hetero-metal active site with Zn and either Mn or Fe. To test this hypothesis, we measured the metal content and kinetic properties of purified yDbr1, and performed metal reconstitution assays with metal-free apo yDbr1. We also found that the iron chelator deferoximine (DFO) can increase lariat levels in a human cell line, suggesting that Fe is an important cofactor for yDbr1.
RESULTS AND DISCUSSION
Homology modeling of yDbr1
A homology model of yDbr1 was calculated based on the structure of EhDbr1 with Fe/Zn metals and no active site ligands (PDB 5k73) using SWISS-MODEL (Waterhouse et al. 2018). yDbr1 is 33% identical to EhDbr1. The seven metal-ligating residues and the catalytic acid (histidine [His] 86) are all identical between EhDbr1 and yDbr1 (Fig. 1A). Of the nine Dbr1 structures in the protein data bank, the beta site is occupied by a Mn, Fe, or Ni. The alpha site is either populated with a Zn, or it can be unoccupied (Fig. 1B). Only in the apo structure (PDB 5k71) are both sites unoccupied.
FIGURE 1.

Homology model of yeast Dbr1. (A) The yDbr1 homology model was calculated from EhDbr1 PDB 5k73. The α-pocket contains a Zn2+ ion, and the β-pocket contains an Fe2+ ion. All seven metal ligands are identical between EhDbr1(gray backbone) and yDbr1 (tan backbone). The OH− nucleophile is the red sphere. (B) Summary of the metal content of all EhDbr1 structures in the PDB. The metals found in the α and β pockets are indicated, along with PDB ID, and a description of the unique ligand if present.
Elemental analysis of purified yeast Dbr1
The yeast Dbr1 gene was expressed in BL 21 E. coli in Terrific Broth media made with distilled water. No metals were added at any point in the culture or purification. The purification scheme consisted of initial purification with a Ni-affinity column, overnight dialysis with TEV protease to remove the His-tag, followed by purification on a Heparin-SO4 column. For three independent experiments, the metal content of the purified proteins was analyzed with inductively coupled plasma mass spectrometry (ICP-MS). We observed a mean of 2.7 equivalents of Zn, and 0.8 equivalents of Fe per yDbr1 monomer (Fig. 2A, blue points). Ni and Cu were present at ∼0.02 equivalents, and Mn and Co were <0.001 equivalents. In comparison, EhDbr1 purified with 0.7 equivalents of Zn and 0.7 equivalents of Fe (Clark et al. 2016). This additional Zn binding we observed for yDbr1 could represent adventitious surface binding, or structural Zn binding sites that are present in yDbr1 but not EhDbr1. Without a crystal structure of yDbr1, we cannot distinguish between these possibilities or hypothesize what, if any, role these additional Zn ions may have. We also prepared and analyzed a sample of apo-yDbr1, and this contained <0.02 eq. Fe, Ni, and Cu, and <0.00001 eq. Zn, Mn, Co (Fig. 2A, red points).
FIGURE 2.
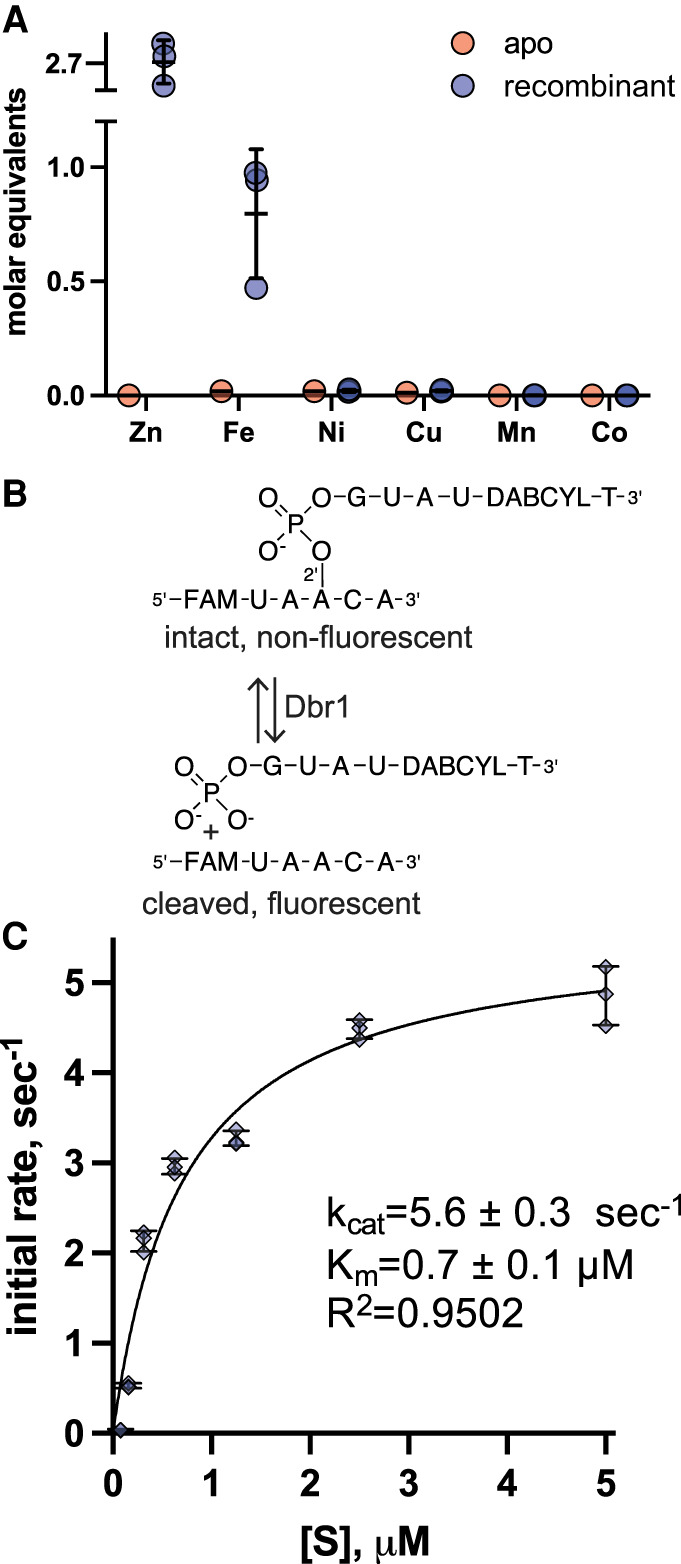
Metal content and steady state kinetics of recombinant yDbr1. (A) Molar equivalents of metals ions for recombinant and apo yDbr1 measured with ICP-MS. Zn and Fe were present at 2.7 and 0.7 molar equivalents, while Ni, Cu, Mn, and Co are <0.02 equivalents. Apo-yDbr1 has <0.02 equivalents of all metals. (B) Schematic of the fluorescent AK88 bRNA substrate. When intact, the “black-hole” quencher DABCYL prevents the fluorescein (FAM) from emitting light at 488 nm. Following Dbr1 cleavage, the quencher is no longer proximal to the FAM which becomes highly fluorescent. (C) Steady-state kinetic analysis of recombinant yDbr1. The AK88 bRNA was prepared at concentrations from 0.08–5 µM and initial rates of product formation were measured. The resulting data were fit with the Michaelis–Menten equation to obtain estimates of kcat = 5.6 sec−1, Km = 0.7 µM. No metal ions were added to any component of these reactions; the only metal ions present are those that copurified with the enzyme or those present as trace metal contaminants.
To understand the relation between the metal content of the purified enzyme and the growth media and water used to prepare it, we measured the metal content of deionized (DI) water, ultrapure water, TB media, and insect culture media by ICP-MS. Both DI and ultrapure water had 0.01 µM Mn, and <0.01 µM Fe, Cu, Co, Ni, Zn. Terrific Broth had 24 µM Fe, 56 µM Zn, 1 µM Mn, 2 µM Co, 1.3 µM Cu, and 0.1 µM Ni (Supplemental Table 1). We analyzed a eukaryotic growth media, serum-free insect media, and found similar proportions of metals at reduced concentrations; 8 µM Fe, 11 mM Zn, and 0.3 µM Mn; 0.6 µM Co, 1.1 µM Cu, and 0.1 µM Ni. We previously observed that adding 1–10 mM MnSO4 into the culture media was ineffective at increasing the Mn content of purified EhDbr1 (Clark et al. 2016). In human tissue, Zn and Fe are present at ∼300 µM, and Mn is ∼6 µM (cervix, Supplemental Table 1; Cunzhi et al. 2003).
Kinetic analysis of recombinant yDbr1
To analyze the steady-state kinetic parameters of recombinant yDbr1 (which copurified with Fe and Zn), we used a fluorogenic branched RNA (bRNA) substrate (Katolik et al. 2017). This bRNA substrate increases in fluorescence after Dbr1-hydrolysis, enabling real-time kinetic measurements of Dbr1 enzymes (Fig. 2B). We assayed this substrate at seven concentrations between 0.08–5 µM, and fit the resulting data with the Michaelis–Menten equation (Fig. 2C). No exogenous metal ions were added to any of the buffers or growth media used to produce the yDbr1 sample, or to any component of the kinetic assay. We measured a kcat of 5.6 sec−1 and Km of 0.7 µM for recombinant yDbr1 (Fig. 2C). We ascribe this activity to the Fe and Zn cofactors which purified with yDbr1, as no metals were added at any point. In comparison, when EhDbr1 protein was prepared with similar methods, a kcat of 2.0 sec−1, and Km of 0.2 µM were reported (Clark et al. 2016). Based on this turnover rate, we concluded that yDbr1 is highly active when purified from E. coli with Zn and Fe and requires no exogenous metals for catalysis.
Metal reconstitution assay
To determine how the activity of recombinant yDbr1 with Fe2+/Zn2+ compares to Fe2+, Mn2+, or Zn2+ reconstituted yDbr1, we prepared metal-free apoenzyme by stripping bound metal ions with high concentrations of metal chelators. We verified that the apoenzyme was metal free with ICP-MS (Fig. 2A), and inactive (Fig. 3A, metal = 0 datapoint). Trace metal ions are present in water (and buffers) in the low nanomolar range (Supplemental Table 1). We used a high concentration of apoenzyme (1 µM) in our metal-dependence assays to prevent trace ion interference. To prevent oxidation of the Fe2+, the experiments were performed in an anaerobic chamber equipped with a fluorescent plate reader. Metals (0.1–100 µM) and apoenzyme (1 µM) were combined in the anaerobic environment and incubated for 10 min in a multiwell plate. An equal volume of substrate (20 µM) was injected to initiate the reaction, and product development was followed using the rapid-kinetic settings. Fe2+ reconstitution produced the highest rates: 9.2 sec−1. Mn2+ reconstitution gave a rate of 1.1 sec−1, and Zn2+ gave a rate 0.2 sec−1 (Fig. 3A,B). Unlike Fe2+ and Mn2+, the velocity of Zn2+ reconstitutions decreased at higher concentrations. Zn may be ineffective in these reconstitutions because it may be unable to occupy the β-pocket of yDbr1 and occupies only the α-pocket. Alternatively, it is possible that a di-Zn2+ enzyme cannot support catalysis. The latter explanation would be consistent with the finding that Zn2+ is inhibitory in the presence of Mn2+ (Khalid et al. 2005; Clark et al. 2016).
FIGURE 3.
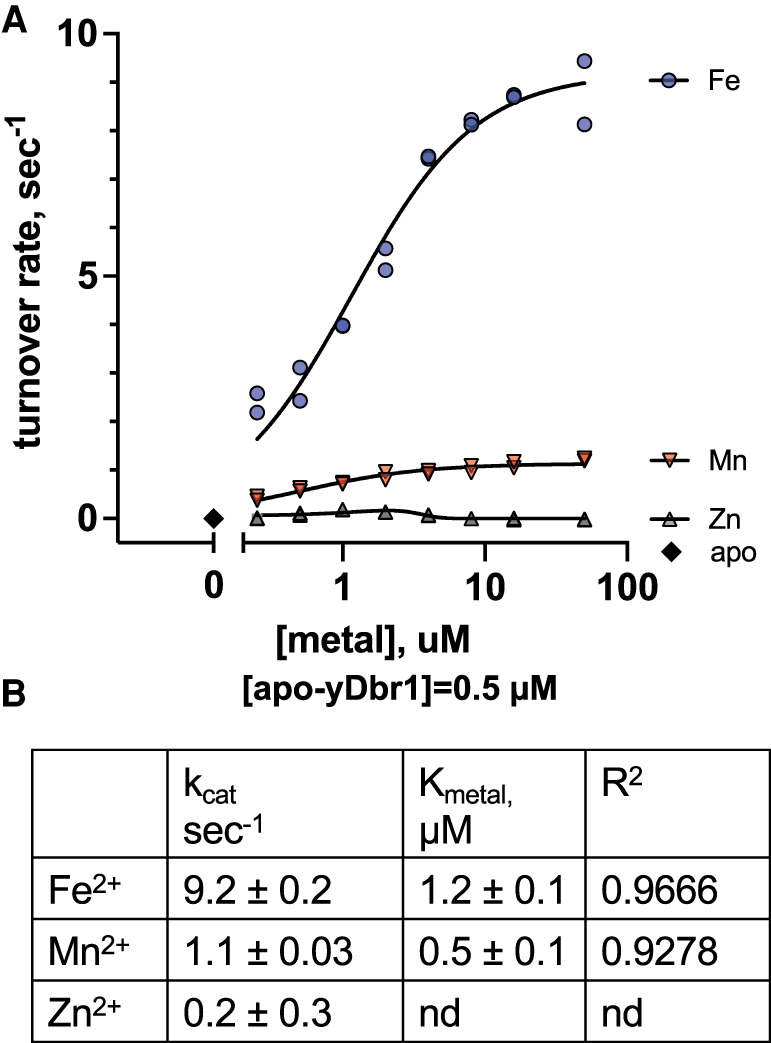
Reconstitution of apo-yDbr1 with Fe2+, Zn2+, and Mn2+. (A) Apo-yDbr1 (1 µM) was reconstituted with 0–100 µM of the indicated metals under anaerobic conditions. Apo-yDbr1, black diamonds at metal = 0, had no detectable activity without exogenous metals. (B) Results of curve fitting to obtain estimates of kcat and Kmetal.
Compared to Dbr1 with Fe2+/Zn2+ (as-purified Dbr1) or Fe2+/Fe2+ (Fe2+ reconstituted Dbr1), other metalation states result in bRNA turnover rates that are orders of magnitudes slower. For example, a preparation of EhDbr1 was exposed to 1 mM MnSO4 during cell resuspension and lysis (Ransey et al. 2017). The authors measured 1.25 eq Zn, 0.75 eq Mn, and 0.21 eq Fe in the final purified protein using ICP-AES. This Mn2+/Zn2+ containing sample debranched a synthetic fluorescent 19-mer branched RNA at a rate of 0.040 sec−1. -Adding 1 mM MnSO4 to the debranching reaction increased the rate to 0.044 sec−1. In a 2005 study, after purifying yDbr1 in the presence of EDTA to remove any metal cofactors, the authors added 2.5 mM MnSO4 to debranching reactions with a 32P-labeled 18-mer synthetic branched RNAs (Khalid et al. 2005). The authors did not report turnover rates, but based on the concentrations and incubation times reported, we estimated a turnover rate of approximately 0.004 sec−1.
Steady-state turnover rates are determined by combinations of enzyme, cofactors, and substrates. When assayed against a bis-p-nitrophenylphosphate substrate (15 mM), yDbr1 with 5 mM MnCl2 turned over substrate at 4.2 sec−1 (Schwer et al. 2016). The authors point out that the pKa of the nitrophenylphospate leaving group is 5–6 pH units lower than the pKa of a 2′-OH of a branched RNA leaving group, and the hydrolysis of bis-p-nitrophenylphosphate does not require the catalytic acid H86 to protonate the 2′-OH leaving group of bRNA. yDbr1 preparations with the substitutions H86A, H86N, and H86Q retained 20%–40% activity against bis-p-nitrophenylphosphate (Schwer et al. 2016), but substitution of this histidine residue completely abolishes bRNA hydrolysis (Khalid et al. 2005; Clark et al. 2016; Schwer et al. 2016). The large difference in the size of the products of bis-p-nitrophenylphosphate and AK88 bRNA hydrolysis (122 Da vs. 2070 Da, respectively) could also affect the rate of product release. Therefore, in addition to metal cofactor identity, substrate chemistry affects Dbr1 kinetics.
Iron chelation of lymphoblastoid cells
To determine if the dependence on Fe2+ for efficient debranching is observed in vivo, we incubated a human lymphoblastoid cell line with the iron-chelator deferoxamine (DFO) at 200 µM for 60 h. DFO is used to treat iron overdose and iron accumulation disorders, and aluminum toxicity in dialysis patients (Yawalkar 1993). We hypothesized that if Dbr1 depends on Fe for in vivo lariat debranching, treating cells with an iron-specific chelator may elevate cellular lariat levels. RNA was extracted from duplicates of DFO and untreated cells, and total RNA-sequencing was performed. The data are available in GEO (GSE199077) and are also available as UCSC Genome Browser tracks: https://genome.ucsc.edu/s/allisontaggart/DFO. An example of an elevated lariat, intron 6 from the SUDS3 gene, is shown in Figure 4A. The intron has approximately fivefold more raw counts in the DFO-treated samples, and has the characteristic read density profile of a lariat, with a dip in intensity at the 5′ and 3′ ends of the intron (Taggart and Fairbrother 2018). Three additional examples of elevated introns are presented in Supplemental Figure 1. If the intron reads were from pre-mRNA, splicing intermediates, or retained introns, these dips in read density would not be present (Fig. 4A; Supplemental Fig. 1). DFO does not appear to increase the transcription rates of the genes with elevated lariats, as read counts for the neighboring exons are identical in DFO-treated versus control cells (Fig. 4A; Supplemental Fig. 1)
FIGURE 4.

The iron-chelator DFO increases lariat levels in lymphoblastoid cells. (A) Genome browser screen capture showing raw read counts for the SUDS3 intron 6, an example of a lariat that is elevated in DFO-treated cells. The read density fall-off at the 3′ and 5′ splice sites is characteristic of intron lariats. (B) Lariat enrichment, calculated from branchpoint-traversing reads, is shown for duplicate samples of DFO-treated lymphoblastoid cells and controls. DFO treatment elevates lariats approximately twofold. (C) Plot of log2 fold change in lariat abundance of DFO-treated versus untreated controls cells. (D) DFO treatment does not significantly change DBR1 expression level, FDR-adjusted P-value = 0.18. (E) DFO and EDTA inhibit Dbr1 in a time-dependent manner, with 67% and 38% inhibition, respectively, after 3 h.
We quantified lariats in DFO-treated cells using two different computational approaches. The lariat read method (Fig. 4B) searches for gapped, inverted reads that link a 5′ splice site with a downstream intronic branchpoint (Taggart et al. 2012, 2017; Taggart and Fairbrother 2018). However, reverse transcriptase (RT) reads through the 2′5′ linkage with very low frequency, in a context-dependent manner. Many introns are measured only once, making relative quantification difficult using this method. The ShapeShifter (Fig. 4C) method is more quantitative as it uses the characteristic shape of read density profiles to differentiate lariats from linear introns and partially processed pre-mRNA (Taggart and Fairbrother 2018), and is not dependent on the infrequent RT branchpoint readthrough. In both cases, lariat levels are normalized to total reads to control for possible changes in transcription levels. Intron lariats are rapidly degraded by Dbr1 under normal conditions, thus the pool of lariats in untreated control cells is very sparsely populated and the same lariats are not recovered in control vs. DFO-treated cells. Both approaches (Fig. 4B,C) find elevated lariats in DFO-treated samples. We observed an approximately twofold increase in branchpoint-traversing reads following DFO treatment (Fig. 4B). A log2 fold change plot of lariat abundance (Fig. 4C, calculated with ShapeShifter) shows that ∼150 introns are elevated twofold or greater with DFO treatment.
To examine if the increase in lariats was due to a DFO-mediated decrease in DBR1 expression, we compared the normalized DBR1 read counts for control and DFO samples (Fig. 4D). We did not observe a significant decrease in DBR1 expression (FDR-adjusted P-value of 0.18). Prior works suggest that an 85% decrease in Dbr1 is necessary to result in a twofold increase in lariats (Zhang et al. 2018). Therefore, we concluded that DFO mediated depletion of Fe2+ was responsible for the loss of activity. We combined recombinant yDbr1 with 100 µM DFO, or 100 µM EDTA (as a control), incubated the mixtures for 30 min or 3 h at room temperature, and measured the debranching activity (Fig. 4E). Both chelators showed a time-dependent inhibition of debranching activity. DFO inhibited 67%, and EDTA inhibited 39% after 3 h of coincubation.
In light of these observations (Fig. 4A–E), we propose that the increase in lariat levels following DFO treatment is due to inhibition of Dbr1. DFO might directly strip Fe from Dbr1 as we observed in vitro (Fig. 4E), or DFO could chelate cellular stores and reduce Fe available for loading into enzymes such as Dbr1 (Dayani et al. 2004; Umemura et al. 2017).
Conclusions
Here we demonstrate that yDbr1 contains Fe and Zn when purified following recombinant expression in E. coli. This enzyme preparation has a relatively high kcat of 5.6 sec−1. In comparison, EhDbr1 has a kcat of 2.0 sec−1 against the same bRNA substrate. We report that apo-yDbr1 reconstituted with Fe2+ debranches at 9.2 sec−1, and 1.1 sec−1 when reconstituted with Mn2+. Working with redox-sensitive Fe2+ under anaerobic conditions is technically difficult and requires specialized equipment. Mn2+ can be substituted for Fe2+ in the β-pocket of Dbr1 (Figs. 1A,B; 3A) when anaerobic experiments are not feasible or justified. In general, Mn2+ is a good analog for Fe2+ as it can accommodate similar coordination states and metal-ligand distances (Kuppuraj et al. 2009). Considering how similar the two metal ions are, it is surprising that the catalytic activity is approximately ninefold higher with Fe2+ compared to Mn2+. While we did not examine combinations of Fe2+ and Zn2+ in this study, we previously showed that combinations of Fe2+ and Zn2+ reconstituted full activity to apo-EhDbr1 (Clark et al. 2016).
We believe that the difficulty of working with ferrous iron in vitro formed a barrier to recognizing the ability of Fe2+ to support debranching by Dbr1. In vivo, cells are able to maintain a reducing environment and actively manage the pools of Fe2+ (Andrews and Schmidt 2007). When cells are lysed in a normal laboratory environment, oxygen can diffuse into the buffers and oxidize Fe2+ to the insoluble ferric form (Fe3+). To minimize oxidation, we de-gas our buffers, use reducing agents (TCEP and DTT), and snap-freeze our enzyme preparations.
One of the earliest yeast Dbr1 publications assayed the enzyme without the addition of any exogenous metal ions and found a robust activity (Nam et al. 1994). We also observe no requirement of exogenous metal ions for robust activity if the purification scheme does not include EDTA or other metal chelators, and some care is taken to minimize oxidation. The Cys residue that distinguishes Dbr1 from other MPEs binds a Zn ion (Fig. 1A; Clark et al. 2016; Ransey et al. 2017). We propose that the Zn-Cys bond stabilizes the α-pocket Zn and protects the β-pocket Fe2+ from oxidation. This enables purification of highly active Fe2+/Zn2+ recombinant yDbr1 and EhDbr1 under normal aerobic laboratory conditions (Clark et al. 2016).
Fe and Zn are the most abundant metals in culture media and human tissue (Supplemental Table 1). Highly active yDbr1 retains stoichiometric levels of Fe and Zn throughout a 72 h purification scheme consisting of two affinity columns and overnight dialysis, suggesting that the Fe and Zn ions are tightly bound and slow to exchange. We hypothesize that Fe and Zn are the biologically relevant metals for Dbr1. This hypothesis is supported by our observation that the iron chelator DFO raises the lariat levels in a human cell line. In light of this apparent Fe-dependence, we believe that future studies could link oxidative stress to Dbr1 activity and lariat levels. Such studies could provide further insights into the roles of lariat RNA in viral immunity, antibody generation, neurodegeneration, and retrovirus replication (Ye et al. 2005; Armakola et al. 2012; Zheng et al. 2015; Galvis et al. 2017; Zhang et al. 2018).
MATERIALS AND METHODS
Homology modeling
The yDbr1 homology model was generated with SWISS-MODEL (Waterhouse et al. 2018), aligned with EhDbr1 PDB 5k73 in Coot (Emsley et al. 2010) using the SSM function, and the figure was prepared in PyMol (Schrodinger). yDbr1 is 33% identical to EhDbr1, and the average model confidence (QMEANDisCo) is 0.63 ± 0.05 (Benkert et al. 2011).
Protein expression and purification
The yeast Dbr1 gene was PCR amplified from S. cerevisiae genomic DNA, cloned into a modified pET 32 vector, and sequenced to confirm integrity of the open reading frame. The sequence verified expression vector was transformed into BL21 cells, and 6 L of transformed cells were cultured in Terrific Broth with ampicillin at 37°C. When the culture reached an absorbance of 3 OD (λ = 600 nm), the temperature was reduced to 18°C, 1 mM IPTG was added, and the cells were cultured overnight. The following day, cells were harvested by centrifugation, resuspended in 300 mL of Ni-A buffer (500 mM NaCl, 50 mM Tris pH 8, 20 mM imidazole, 1 mM TCEP, and 1 mM DTT) supplemented with protease inhibitor cocktail (Sigma P8849). All buffers were treated with Chelex 100 resin (Bio-Rad) to minimize trace metal contamination. Cells were lysed with a high intensity 1” sonicator horn (Qsonica) with cooling provided by an ice water bath, then clarified by centrifugation at 15,000g for 30 min at 4°C. Clarified lysate was filtered with a 0.45 µm filter, and then loaded onto a 20 mL Ni column (Ni-Sepharose FF, GE Healthcare), and eluted with a shallow gradient of Ni-A supplemented with 400 mM imidazole. Fractions containing yDbr1 were pooled in a dialysis bag with TEV protease at a 1:50 ratio, and dialyzed overnight in Heparin-A buffer (50 mM HEPES pH 7, 50 mM NaCl, 1 mM TCEP, and 1 mM DTT). Dialyzed fractions were applied to a 5 mL HiTrap Heparin column (GE Healthcare), washed, and eluted with a shallow gradient of Heparin-A buffer supplemented with 1 M NaCl. The major yDbr1 peak eluted at ∼45 mS/cm conductivity.
Preparation of apo-yDbr1
To prepare metal-free apo-enzyme, heparin-column fractions of yDbr1 were pooled and diluted in stripping buffer to a volume of 14 mL (10 mM EDTA, 10 mM NTA, 50 mM HEPES pH 7, 100 mM NaCl, 1 mM TCEP, 1 mM DTT). The protein was concentrated in an Amicon 30,000 mwco spin concentrator (Millipore) to a volume of 1 mL, then diluted again to 14 mL with stripping buffer, and this was repeated a total of six times with stripping buffer, followed by four cycles of dilution and concentration with Chelex-treated assay buffer (100 mM NaCl, 50 mM HEPES pH 7, 1 mM TCEP, 1 mM DTT). The concentrated protein (∼1 mL) was then dialyzed against 4 L of assay buffer for 24 h at 4°C to remove any remaining metal chelators.
Inductively coupled plasma mass spectrometry
Protein samples of recombinant and apo yDbr1 at 10 mg/mL were diluted 50-fold in 5 mL of 2% nitric acid. Ultrapure water was from a Milli-Q Academic system (Millipore), and deionized water was from a lab faucet. Terrific Broth was from RPI, made according to manufacturer's protocols with deionized water. Insect media was serum-free SFX media from Expression Systems.
The concentrations of Mn, Fe, Co, Ni, Cu, and Zn were measured with an Agilent 7500ce ICP-MS at the University of Texas at Austin (Dept. of Geological Sciences). The instrument was optimized for sensitivity across the AMU range, while minimizing oxide production (<1.5%). The analytical method employed an octopole reaction system (ORS) operated in hydrogen (reaction-mode) for removal of polyatomic interferences, and no gas mode for interference-free analytes. Internal standards, mixed into unknowns via in-run pumping, were used to compensate for instrumental drift and internal standard intensity variations were well within QA tolerances (±50%). Limits of detection, based upon the population of blank (2% HNO3) analyses interspersed throughout the analytical sequence, were below 0.2 ppb. Analyte recoveries obtained for 2–4 replicates of two independent quality control standards were typically within 5% of certified values. Relative precisions obtained for these quality control standards were typically within 0.8 to 1.6% of replicate averages. Analyte concentrations in diluted sample fractions were typically 10× higher than detection limits for Fe, Ni, Cu, and Zn, 1–5× higher for Mn, and <1 for Co. ICP-MS analytical parameters and quality control metrics are shown in Supplemental Tables 1, 2. Molar equivalents of transition and alkali metals were calculated by dividing the metal concentration measured with ICP-MS by the protein concentration measured with UV280 absorbance.
Kinetic analysis of recombinant yDbr1
To determine the Km and kcat of recombinant yDbr1, we measured the hydrolysis rate of the synthetic branched RNA substrate AK88 at different concentrations. AK88 was prepared at 10, 5, 2.5, 1.25, 0.63, 0.31, and 0.16 µM. Recombinant yDbr1 was prepared at 5 nM. Protein and substrate were prepared in assay buffer (100 mM NaCl, 50 mM HEPES pH 7, 1 mM TCEP, 1 mM DTT). No exogenous metal ions were added to any component of the reaction. 20 µL of substrate was dispensed into a multiwell plate (Costar 3694, ½ area black 96-well plate), then 20 µL of enzyme was dispensed immediately before reading in a BioTek Synergy H1 plate reader. Each substrate concentration was assayed in triplicate. Product development was measured by exciting at 485 nM, and measuring emission at 528 nm with 20 nm bandwidth filters. The maximum velocity of the reactions (in RFU/sec) was calculated using the BioTek Gen5 software (Version 3.08.01). To convert from RFU/sec to µM/sec, the difference between fluorescence of substrate-only and completed reactions was divided by the substrate concentrations, and the µM/sec rates were then divided by the enzyme concentration (2.5 nM final) to obtain initial turnover rates (sec−1). The resulting curves were fit to the Michaelis–Menten equation in Prism (GraphPad) to obtain estimates of Km and kcat.
Metal-dependence assays
To measure the ability of Zn2+, Mn2+, and Fe2+ to reconstitute debranching activity to metal-free apo-yDbr1, we utilized a BMG Omega plate reader (BMG Labtech) housed in an anaerobic chamber (Coy labs) maintained with a mixture of 10% hydrogen/90% nitrogen. 1 M solutions of Zn2+SO4, Mn2+SO4, and Fe2+ (NH4)2(SO4)2 were prepared fresh in 2 mM HCl (inside the chamber) prior to the assays. Buffers were sparged with nitrogen prior to entrance into the anaerobic chamber, and all solutions (metals, apo-enzyme) were subjected to five vacuum/nitrogen purge cycles in the chamber interlock prior to entry into the chamber. Inside the chamber, 9 μL of 1.1 µM apo-yDbr1 (in assay buffer) was mixed with 1 μL of metal stocks in black nonbinding 384-well plates (Corning 4514). The final concentrations were 0.5–100 µM metals, and 1 µM apo-yDbr1. To ensure that apo-yDbr1 was devoid of activity, mock reconstitutions with 2 mM HCL were performed in parallel. Apoenzyme and metals were equilibrated for 10 min prior to substrate addition. The reactions were initiated by injecting 10 μL of 20 µM AK88 bRNA substrate in assay buffer using the on-board reagent injectors. Final concentrations were 10 µM AK88 bRNA, and 0.5 µM yDbr1. Fluorescence intensity (488 ex/520 em) was measured every 0.08 sec for 20 sec using “well-mode” for rapid-kinetics. The slope of the linear portion of the reactions (RFU/sec) was calculated in Excel, and otherwise data analysis was explained in the previous section.
Lymphoblastoid culture, DFO treatment, and sequencing
Lymphoblast cells were cultured in RPMI 1640 with 15% fetal bovine serum and 1% penicillin/streptomycin. 200 µM DFO was added to two wells of a six-well plate, and two control wells were maintained in parallel. After 60 h, RNA was extracted with TRIzol, and libraries were prepared with a TruSeq Stranded Total RNA Human/Mouse/Rat kit (Illumina). Sequencing libraries (25 pM) were chemically denatured and applied to an Illumina HiSeq v4 paired-end flow cell using an Illumina cBot. Hybridized molecules were clonally amplified and annealed to sequencing primers with reagents from an Illumina HiSeq PE Cluster Kit v4-cBot (PE-401-4001). Following transfer of the flow cell to an Illumina HiSeq 2500 instrument (HCS v2.2.38 and RTA v1.18.61), a 125 cycle paired-end sequence run was performed using HiSeq SBS Kit v4 sequencing reagents (FC-401-4003). Following sequencing, lariats were counted using gapped, inverted branchpoint alignments (Taggart et al. 2017) and the ShapeShifter method (Taggart and Fairbrother 2018). To calculate gene expression changes, transcripts were quantified with Salmon (Patro et al. 2017), and differential expression was analyzed with DESeq2 (Love et al. 2014), and adjusted P-values used the false discovery rate correction (Benjamini and Hochberg 1995). The RNA-seq data are available as UCSC Genome Brower tracks: https://genome.ucsc.edu/s/allisontaggart/DFO and in GEO under accession numbers (GSE199077).
DFO and EDTA inhibition assay
Recombinant yDbr1 was diluted to 50 nM in assay buffer (100 mM NaCl, 50 mM HEPES pH 7, 1 mM TCEP, 1 mM DTT). 3 × 100 µL aliquots were mixed with either 1 µL of water, 10 mM EDTA, or 10 mM DFO at time = 0, and the mixtures were incubated at room temperature. Final concentrations were 100 µM EDTA or DFO. At 30 min and 3 h, 20 µL of the enzyme-chelator samples were mixed with 20 µL of 5 µM AK88 substrate and activity was measured as described in “Kinetic analysis of recombinant yDbr1.” Duplicate reactions were measured for each sample at each timepoint. The debranching rates (RFU/sec) were calculated using Gen5 software (BioTek), normalized to the mean of water-only control reactions with Excel, and plotted with GraphPad (Prism).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant numbers RO1GM105681 and RO1GM127472.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079159.122.
MEET THE FIRST AUTHOR
Nathaniel Clark.

Meet the First Author(s) is a new editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Nathaniel Clark is the first author of this paper, “Metal content and kinetic properties of yeast RNA lariat debranching enzyme Dbr1.” Nathaniel is a research faculty member in the laboratory of Dr. William G. Fairbrother in the Department of Molecular Biology, Cell Biology, and Biochemistry at Brown University in Providence, Rhode Island.
What are the major results described in your paper and how do they impact this branch of the field?
The RNA lariat debranching enzyme Dbr1 is the only enzyme known to hydrolyze the 2′5′ bond found in spliceosomal intron lariats. The gene for yeast Dbr1 was isolated in 1991, but the enzyme has resisted crystallization, and there is a lack of consensus in the field as to which metal ions yeast Dbr1 requires. Through elemental analysis of yeast Dbr1 protein samples, enzyme kinetics, and reconstitution of metal-free Dbr1 under anaerobic conditions, we discovered that Dbr1 has a strong preference for iron. This implies that lariat metabolism is dependent on iron availability. Because iron is a redox-sensitive metal, the ability of yeast cells (and other cells) to metabolize intron lariats may be linked to the redox status of the cell, and Dbr1 may shut down with oxidative stress. This finding offers some clues to the unresolved question of why cells devote significant resources to maintaining and transcribing intronic sequences when the products (intron lariats) are rapidly degraded and to date, have few well-defined biological roles.
What led you to study RNA or this aspect of RNA science?
I began to study RNA as a postdoc, when I started to work on structural biology and enzymology of Dbr1 enzymes. Dbr1 enzymes are fascinating, and Dbr1 is implicated in human diseases such as amyotrophic lateral sclerosis, HIV, and innate immunity to viruses. However, many of the mechanistic details as to precisely how Dbr1 activity affects these processes remain obscure, and there are many open questions in the field.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
When I started working on the Dbr1 enzyme, the consensus in the field was that the enzyme required manganese. Through a collaboration with Drs. Masad J. Damha and Adam Katolik, we acquired a truly novel reagent—a fluorogenic Dbr1 substrate amenable to real-time kinetics. All prior Dbr1 enzymology relied on gel-based autoradiographic assays. As I began to work out assay conditions, I was surprised to find that the Dbr1 enzymes were highly active without added metals, and this observation lead to an unexpected new focus for my research. Initially, I analyzed the metal dependence of E. histolytica Dbr1 (a Dbr1 homolog for which we determined the crystal structure) and found that the enzyme was iron-dependent. In this manuscript, we extend our analysis and demonstrate that yeast Dbr1 is an iron-dependent enzyme. This surprising result suggests that lariat metabolism is intrinsically linked to iron homeostasis and redox state (ferrous/2 + vs ferric/3 + oxidation states).
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
Solving my first crystal structure as a graduate student was an important moment in my development as a scientist. It was a long battle to crystallize this human glycoprotein (human α-NAGAL), and I had to develop an insect-cell expression system, purify the protein from buckets of spent culture media, and then mutate specific glycosylation sites to obtain high quality crystals. Another landmark moment was determining the metal dependence of Dbr1, because everyone (including my advisor, Dr. P. John Hart) thought I was wrong (and possibly crazy) when I floated the idea that Dbr1 required iron (and not manganese). In the end, they were convinced by the data I obtained with the help of excellent collaborators and colleagues.
REFERENCES
- Andrews NC, Schmidt PJ. 2007. Iron homeostasis. Annu Rev Physiol 69: 69–85. 10.1146/annurev.physiol.69.031905.164337 [DOI] [PubMed] [Google Scholar]
- Armakola M, Higgins MJ, Figley MD, Barmada SJ, Scarborough EA, Diaz Z, Fang X, Shorter J, Krogan NJ, Finkbeiner S, et al. 2012. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet 44: 1302–1309. 10.1038/ng.2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Benkert P, Biasini M, Schwede T. 2011. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27: 343–350. 10.1093/bioinformatics/btq662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman KB, Boeke JD. 1991. Isolation and characterization of the gene encoding yeast debranching enzyme. Cell 65: 483–492. 10.1016/0092-8674(91)90466-C [DOI] [PubMed] [Google Scholar]
- Cheng Z, Menees TM. 2004. RNA branching and debranching in the yeast retrovirus-like element Ty1. Science 303: 240–243. 10.1126/science.1087023 [DOI] [PubMed] [Google Scholar]
- Cheng Z, Menees TM. 2011. RNA splicing and debranching viewed through analysis of RNA lariats. Mol Genet Genomics 286: 395–410. 10.1007/s00438-011-0635-y [DOI] [PubMed] [Google Scholar]
- Clark NE, Katolik A, Roberts KM, Taylor AB, Holloway SP, Schuermann JP, Montemayor EJ, Stevens SW, Fitzpatrick PF, Damha MJ, et al. 2016. Metal dependence and branched RNA cocrystal structures of the RNA lariat debranching enzyme Dbr1. Proc Natl Acad Sci 113: 14727–14732. 10.1073/pnas.1612729114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunzhi H, Jiexian J, Xianwen Z, Jingang G, Shumin Z, Lili D. 2003. Serum and tissue levels of six trace elements and copper/zinc ratio in patients with cervical cancer and uterine myoma. Biol Trace Elem Res 94: 113–122. 10.1385/BTER:94:2:113 [DOI] [PubMed] [Google Scholar]
- Dayani PN, Bishop MC, Black K, Zeltzer PM. 2004. Desferoxamine (DFO)–mediated iron chelation: rationale for a novel approach to therapy for brain cancer. J Neurooncol 67: 367–377. 10.1023/B:NEON.0000024238.21349.37 [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvis AE, Fisher HE, Fan H, Camerini D. 2017. Conformational changes in the 5′ end of the HIV-1 genome dependent on the debranching enzyme DBR1 during early stages of infection. J Virol 91: e01377-17. 10.1128/JVI.01377-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossens J, Vanmechelen E, Trojanowski JQ, Lee VMY, Van Broeckhoven C, van der Zee J, Engelborghs S. 2015. TDP-43 as a possible biomarker for frontotemporal lobar degeneration: a systematic review of existing antibodies. Acta Neuropathol Commun 3: 15. 10.1186/s40478-015-0195-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Park HK, Ching T, Panneerselvam J, Wang H, Shen Y, Zhang J, Li L, Che R, Garmire L, et al. 2017. Human DBR1 modulates the recycling of snRNPs to affect alternative RNA splicing and contributes to the suppression of cancer development. Oncogene 36: 5382–5391. 10.1038/onc.2017.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katolik A, Clark NE, Tago N, Montemayor EJ, Hart PJ, Damha MJ. 2017. Fluorescent branched RNAs for high-throughput analysis of Dbr1 enzyme kinetics and inhibition. ACS Chem Biol 12: 622–627. 10.1021/acschembio.6b00971 [DOI] [PubMed] [Google Scholar]
- Khalid MF, Damha MJ, Shuman S, Schwer B. 2005. Structure-function analysis of yeast RNA debranching enzyme (Dbr1), a manganese-dependent phosphodiesterase. Nucleic Acids Res 33: 6349–6360. 10.1093/nar/gki934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppuraj G, Dudev M, Lim C. 2009. Factors governing metal-ligand distances and coordination geometries of metal complexes. J Phys Chem B 113: 2952–2960. 10.1021/jp807972e [DOI] [PubMed] [Google Scholar]
- Ladewig E, Okamura K, Flynt AS, Westholm JO, Lai EC. 2012. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res 22: 1634–1645. 10.1101/gr.133553.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matange N, Podobnik M, Visweswariah SS. 2015. Metallophosphoesterases: structural fidelity with functional promiscuity. Biochem J 467: 201–216. 10.1042/BJ20150028 [DOI] [PubMed] [Google Scholar]
- McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW, Perl DP, Hedley-Whyte ET, Price B, Sullivan C, et al. 2010. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 69: 918–929. 10.1097/NEN.0b013e3181ee7d85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam K, Hudson RH, Chapman KB, Ganeshan K, Damha MJ, Boeke JD. 1994. Yeast lariat debranching enzyme. Substrate and sequence specificity. J Biol Chem 269: 20613–20621. 10.1016/S0021-9258(17)32037-9 [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133. 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- Ooi SL, Samarsky DA, Fournier MJ, Boeke JD. 1998. Intronic snoRNA biosynthesis in Saccharomyces cerevisiae depends on the lariat-debranching enzyme: intron length effects and activity of a precursor snoRNA. RNA 4: 1096–1110. 10.1017/S1355838298980785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14: 417–419. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratico ED, Silverman SK. 2007. Ty1 reverse transcriptase does not read through the proposed 2′,5′-branched retrotransposition intermediate in vitro. RNA 13: 1528–1536. 10.1261/rna.629607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransey E, Paredes E, Dey SK, Das SR, Heroux A, Macbeth MR. 2017. Crystal structure of the Entamoeba histolytica RNA lariat debranching enzyme EhDbr1 reveals a catalytic Zn2+/Mn2+ heterobinucleation. FEBS Lett 591: 2003–2010. 10.1002/1873-3468.12677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin B, Green MR. 1985. An RNA processing activity that debranches RNA lariats. Science 229: 135–140. 10.1126/science.2990042 [DOI] [PubMed] [Google Scholar]
- Schwer B, Khalid F, Shuman S. 2016. Mechanistic insights into the manganese-dependent phosphodiesterase activity of yeast Dbr1 with bis-p-nitrophenylphosphate and branched RNA substrates. RNA 22: 1819–1827. 10.1261/rna.058552.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart AJ, Fairbrother WG. 2018. ShapeShifter: a novel approach for identifying and quantifying stable lariat intronic species in RNAseq data. Quant Biol 6: 267–274. 10.1007/s40484-018-0141-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart AJ, DeSimone AM, Shih JS, Filloux ME, Fairbrother WG. 2012. Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat Struct Mol Biol 19: 719–721. 10.1038/nsmb.2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart AJ, Lin CL, Shrestha B, Heintzelman C, Kim S, Fairbrother WG. 2017. Large-scale analysis of branchpoint usage across species and cell lines. Genome Res 27: 639–649. 10.1101/gr.202820.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemura M, Kim JH, Aoyama H, Hoshino Y, Fukumura H, Nakakaji R, Sato I, Ohtake M, Akimoto T, Narikawa M, et al. 2017. The iron chelating agent, deferoxamine detoxifies Fe(Salen)-induced cytotoxicity. J Pharmacol Sci 134: 203–210. 10.1016/j.jphs.2017.07.002 [DOI] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46: W296–W303. 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson ME, Charenton C, Nagai K. 2020. RNA splicing by the spliceosome. Annu Rev Biochem 89: 359–388. 10.1146/annurev-biochem-091719-064225 [DOI] [PubMed] [Google Scholar]
- Yawalkar SJ. 1993. Milestones in the research and development of desferrioxamine. Nephrol Dial Transplant 8: 40–42. 10.1093/ndt/8.supp1.40 [DOI] [PubMed] [Google Scholar]
- Ye Y, De Leon J, Yokoyama N, Naidu Y, Camerini D. 2005. DBR1 siRNA inhibition of HIV-1 replication. Retrovirology 2: 63. 10.1186/1742-4690-2-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SY, Clark NE, Freije CA, Pauwels E, Taggart AJ, Okada S, Mandel H, Garcia P, Ciancanelli MJ, Biran A, et al. 2018. Inborn errors of RNA lariat metabolism in humans with brainstem viral infection. Cell 172: 952–965.e918. 10.1016/j.cell.2018.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Vuong BQ, Vaidyanathan B, Lin JY, Huang FT, Chaudhuri J. 2015. Non-coding RNA generated following lariat debranching mediates targeting of AID to DNA. Cell 161: 762–773. 10.1016/j.cell.2015.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.