

Abstract
N6-isopentenyladenosine (i6A), a modified adenosine monomer, is known to induce cell death upon its addition to the culture medium. However, the molecular fate of extracellularly added i6A has yet to be identified. Here we show that i6A addition to cell culture medium results in i6A incorporation into cellular RNA in several cell lines, including the 5-fluorouracil (5-FU)-resistant human oral squamous cell carcinoma cell line FR2-SAS and its parental 5-FU-sensitive cell line SAS. i6A was predominantly incorporated into 18S and 28S rRNAs, and i6A incorporation into total RNA was mostly suppressed by treating these cell lines with an RNA polymerase I (Pol I) inhibitor. i6A was incorporated into RNA even upon inactivation of TRIT1, the only cellular i6A-modifying enzyme. These results indicate that upon cellular uptake of i6A, it is anabolized to be used for Pol I transcription. Interestingly, at lower i6A concentrations, the cytotoxic effect of i6A was substantially more pronounced in FR2-SAS cells than in SAS cells. Moreover, in FR2-SAS cells, i6A treatment decreased the rate of cellular protein synthesis and increased intracellular protein aggregation, and these effects were more pronounced than in SAS cells. Our work provides insights into the molecular fate of extracellularly applied i6A in the context of intracellular nucleic acid anabolism and suggests investigation of i6A as a candidate for a chemotherapy agent against 5-FU-resistant cancer cells.
Keywords: RNA modification, N6-isopentenyladenosine, 5-fluorouracil, ribosomal RNA, oral squamous cell carcinoma
INTRODUCTION
Post-transcriptional RNA modifications exist in most RNA species and play pivotal roles in maintaining RNA structural integrity, function, and metabolism (Frye et al. 2016; Agris et al. 2018; Suzuki 2021). To date, more than 150 RNA modifications have been identified in the three domains of life (Boccaletto et al. 2018). RNA modifications are pivotal for life, and aberrations in over 60 human RNA modification enzymes are associated with diseases that frequently manifest as brain dysfunction, mitochondrial diseases, diabetes, or cancer (Wei et al. 2011; Chujo and Tomizawa 2021; Suzuki 2021).
N6-isopentenyladenosine (i6A) is an RNA modification that exists in tRNAs. The adenosine at tRNA position 37 of specific human tRNA species are post-transcriptionally modified to i6A by tRNA isopentenyltransferase 1 (TRIT1) via recognition of specific anticodon loop sequences (Lamichhane et al. 2011, 2013; Khalique et al. 2020).The TRIT1 gene has been proposed as a cancer suppressor gene candidate because TRIT1 expression is low in lung cancer cells and TRIT1 overexpression suppresses lung cancer cell growth (Spinola et al. 2005). Addition of i6A to cell culture media induces cell-cycle-arrest and cell death in cancer cell lines, including colon cancer, bladder cancer, and glioma cell lines (Laezza et al. 2009; Castiglioni et al. 2013; Ciaglia et al. 2017). Also, an increase of endogenous i6A monomer level has been shown to exert an antitumor effect by inducing excessive autophagy in a glioma cell line (Yamamoto et al. 2019). Despite these reports on cytotoxicity of i6A, the molecular fate and molecular function of extracellularly added i6A monomer has yet to be identified.
At the end of its life, RNA is degraded to nucleoside monomers. In human cells, in comparison to unmodified nucleosides that are salvaged and reused, modified nucleosides are resistant to further degradation and many are exported out of cells through nucleoside transporters called ENT1 and ENT2 (Shi et al. 2021). The modified nucleosides are then circulated in the blood and finally discarded into urine (Mandel et al. 1966; Pane et al. 1992). Recently, a physiological function of a modified nucleoside in the extracellular space has been reported in human and higher mammals. The extracellular N6-methyladenosine (m6A) monomer was found to function as a ligand that strongly binds to human A3 adenosine receptor and stimulates intracellular signaling pathways to play physiological roles such as type I allergy (Ogawa et al. 2021).
Various artificial analogs of DNA and RNA nucleosides and nucleobases are used for chemotherapy to cure cancer and viral infection. Generally, these artificial nucleosides and nucleobases inhibit the function of nucleoside biosynthetic enzymes and/or are anabolized where their products are used for nucleotide polymerization by DNA/RNA polymerase (Longley et al. 2003; Eyer et al. 2018). For example, the deoxycytidine analog cytarabine is used in cancer chemotherapy, and adenosine analog remdesivir is used for COVID-19 treatment (Braess et al. 1998; Beigel et al. 2020).
5-fluorouracil (5-FU, Fig. 1A) is a uracil base analog and is commonly used for chemotherapy against various tumors including colon, breast, stomach, and oral cancers (Longley et al. 2003). After 5-FU enters cells, 5-FU is converted to various derivatives including 5-fluorodeoxyuridine monophosphate (5-FdUMP) and 5-fluorouridine triphosphate (5-FUTP). These analogs perturb DNA and RNA synthesis and inhibit specific RNA modification. The fluorine atom at uracil C5-position causes 5-FdUMP to covalently bind to thymidylate synthase to inhibit dTMP synthesis, thereby blocking DNA nucleotide synthesis (Santi et al. 1974). FdUTP can be misincorporated into DNA (Longley et al. 2003) and 5-FUTP used for RNA transcription, whereby it is incorporated into RNA polynucleotides and exists as 5-fluorouridine (5-FUrd, Fig. 1B) within RNA (Longley et al. 2003). Similarly to the case of thymidylate synthase inhibition by 5-FdUMP, 5-fluorouridine taken into tRNAs inhibits the tRNA modification enzymes responsible for 5-methyluridine and pseudouridine by tightly binding to the enzymes, although cytotoxicity by loss of RNA modification may be limited (Spenkuch et al. 2014; Carter et al. 2019). In the human breast cancer cell line MCF-7, 5-FU incorporation into RNA correlated with cytotoxicity, whereas the simultaneous addition of thymidine to bypass the thymidylate synthase block does not cancel this effect. This result suggests that 5-FU incorporation into RNA is the major mechanism of cytotoxic action in MCF-7 cells (Kufe and Major 1981).
FIGURE 1.

Incorporation of 5-FU into 5-FU-resistant cell line RNA. (A) Chemical structure of 5-fluorouracil (5-FU). The position numbers are indicated in blue. (B) Chemical structure of 5-fluorouridine (5-FUrd). (C) Cell growth of SAS (left) and FR2-SAS cells (right) cultured in the medium containing 0, 5, or 15 µM 5-FU. Cell growth was quantified by performing WST assay and measuring absorbance at 450 nm. The values are shown as the relative levels versus the mean of 0 h cells. Mean ± standard error of the mean (SEM) from n = 8 biological replicates. (D) 5-FUrd level in cellular RNA from SAS cells and FR2-SAS cells upon addition of 5-FU into cell culture medium. After 48 h of 0 or 10 µM 5-FU addition in the medium, total RNA was collected and digested into nucleosides for LC-MS analysis. Peak areas were normalized by uridine peak areas of the same samples and are shown as the relative levels versus SAS 5-FU 10 µM samples. (E) DPYD mRNA levels in SAS cells and FR2-SAS cells. DPYD mRNA levels were quantified by RT-qPCR and normalized by GAPDH mRNA levels, and are shown as the relative levels versus SAS cells. (F,G) 5-methyluridine (m5U) and pseudouridine levels in cellular RNA of SAS cells and FR2-SAS cells upon addition of 5-FU into cell culture medium. After 48 h of 0 or 10 µM 5-FU addition in the medium, total RNA was collected and digested into nucleosides for LC-MS analysis. m5U (F) or pseudouridine (G) peak area was normalized by uridine peak area of the same sample and are shown as the relative levels versus SAS 0 µM samples. Mean ± SEM from n = 4 biological replicates in D–G. (*) P < 0.05 by Mann–Whitney test.
5-FU is one of the most effective and commonly used chemotherapeutic agents for various cancers (Longley et al. 2003). However, its clinical applications have been limited by acquired drug resistance of the cancer cells. For example, 5-FU inactivation is initiated by dihydropyridine dehydrogenase (DPYD), an enzyme that incorporates two hydrogen atoms to 5-FU (Lu et al. 1992), and high DPYD expression is observed in some cases of 5-FU-resistant tumors (Salonga et al. 2000). However, despite some advances in the mechanistic understanding of 5-FU resistance, acquired resistance remains a significant limitation to the clinical use of 5-FU. New strategies for overcoming 5-FU-resistant cells are urgently required.
The most common head and neck cancer is oral cancer, especially oral squamous cell carcinoma (OSCC). In the present study, we searched for a means to remove a 5-FU-resistant OSCC cell line and found that i6A was able to induce cell death in a 5-FU-resistant OSCC cell line FR2-SAS. Interestingly, i6A was substantially more cytotoxic to FR2-SAS cells than the parental 5-FU-sensitive SAS cells. We also revealed that i6A addition to the medium induces incorporation of i6A into cellular 18S and 28S ribosomal RNAs in an RNA polymerase I transcription-coupled manner. Our work provides insights into the molecular fate of extracellularly added i6A and suggests that i6A should be considered as a candidate chemotherapy agent for fighting 5-FU-resistant cancer cells.
RESULTS
Incorporation of 5-FU into cellular RNA is not decreased in 5-FU-resistant FR2-SAS cells
In search of clues toward eliminating 5-FU resistant cancer cells, we started by comparing our 5-FU-resistant oral squamous cell carcinoma (OSCC) cell line FR2-SAS cells to SAS cells, from which FR2-SAS cells were previously generated (Nagata et al. 2011). In SAS cells, a marked decrease of growth speed was observed when the cells were incubated in the medium containing 15 µM 5-FU (Fig. 1C). In contrast, although 5-FU mildly suppressed growth of FR2-SAS cells, the FR2-SAS cells continued to grow even when incubated in the medium containing 15 µM 5-FU (Fig. 1C), in accordance with a previous study (Nagata et al. 2011).
Inside cells, 5-FU is converted to 5-fluorouridine (5-FUrd), which is used for RNA transcription (Longley et al. 2003), and 5-FUrd incorporation into RNA correlates with 5-FU cytotoxicity in certain cancer cells (Kufe and Major 1981). Thus, to compare the amount of 5-FUrd intake into RNA of SAS and FR2-SAS cells, the cells were incubated in 5-FU-containing medium and total cellular RNA was degraded to nucleosides for liquid chromatography mass spectrometry (LC-MS) analysis. Upon addition of 5-FU to cell culture medium, total RNA from SAS cells and FR2-SAS cells showed equivalent 5-FUrd levels (Fig. 1D; Supplemental Fig. 1A), suggesting that 5-FU anabolism into RNA did not substantially differ between 5-FU-resitant FR2-SAS cells and the parental SAS cells. This result was supported by the equivalent DYPD mRNA steady-state levels in SAS and FR2-SAS cells (Fig. 1E), the translated protein of which plays a major role in inactivation of 5-FU (Lu et al. 1992).
5-FU is known to suppress tRNA 5-methyluridine (m5U) modification and pseudouridine modification. Decrease of these modifications may or may not affect cell viability depending on cell lines (Spenkuch et al. 2014; Carter et al. 2019). To investigate the effect of 5-FU on m5U and pseudouridine modification in SAS and FR2-SAS cells, we checked the steady-state levels of m5U and pseudouridine modifications in cellular RNA after 5-FU addition. Upon incubation of cells with 5-FU, while both m5U and pseudouridine modification levels showed some tendency to decrease in both SAS and FR2-SAS cells, the degrees of changes did not show significant differences between SAS and FR2-SAS cells (Fig. 1F,G; Supplemental Fig. 1B,C). Collectively, these results imply that the 5-FU resistance of OSCC-derived FR2-SAS cells was not acquired through alteration of 5-FU incorporation into cellular RNA or changes in RNA modification levels. Therefore, to induce cell growth arrest or cell death of 5-FU-resistant cells, rather than targeting 5-FU metabolism or transport pathways, a different approach may be more effective.
i6A suppresses cell growth of SAS cells
Several groups previously showed that N6-isopentenyladenosine (i6A, Fig. 2A) added to cell culture medium is cytotoxic to various cancer cell lines, such as colon, bladder, and glioma (Laezza et al. 2009; Castiglioni et al. 2013; Ciaglia et al. 2017), prompting us to investigate if i6A is cytotoxic to the OSCC cell lines SAS and FR2-SAS. The addition of 2 µM i6A into the cell culture medium decreased SAS cell growth, and addition of 5 or 10 µM i6A further suppressed SAS cell growth (Fig. 2B,C). In SAS cells, using either 5 µM i6A alone, 10 µM 5-FU alone, or both 5 µM i6A and 10 µM 5-FU, caused similar levels of cell growth arrest (Fig. 2D,E), showing that the use of i6A alone or 5-FU alone is sufficient to exert a similar effect on cell growth suppression in the OSCC cell line SAS.
FIGURE 2.

i6A-induced cell growth suppression of oral cancer cell line SAS. (A) Chemical structure of N6-isopentenyladenosine (i6A). The position numbers are indicated in blue. (B) Cell growth time course experiment upon addition of 0, 2, 5, or 10 µM i6A in cell culture medium for indicated hours. Cell growth was quantified by performing WST assay and measuring absorbance at 450 nm. The values are shown as the relative levels versus the mean of 0 µM i6A 0 h cells. Mean ± SEM from n = 8 biological replicates. (C) Microscopic images of SAS cells after 48 h incubation with indicated concentration of i6A in the medium. Scale bar, 50 µm. (D) Microscopic images of SAS cells after 48 h incubation with indicated concentration of 5-FU and/or i6A in the medium. Scale bar, 50 µm. (E) Cell numbers of alive (blue) and dead (red) cells after 48 h incubation with indicated concentrations of 5-FU and/or i6A in the medium. Alive and dead cells were counted using trypan blue and a cell counting device. Mean ± SEM from n = 4 biological replicates. (***) P < 0.001, (*) P < 0.05 by Mann–Whitney test.
Susceptibility of FR2-SAS cells to cell death upon i6A addition compared to SAS cells
To assess the effects of i6A on 5-FU-resistant FR2-SAS cells, i6A was added to the cell culture medium of FR2-SAS cells and cell viability monitored (Fig. 3A). Surprisingly, the growth of FR2-SAS was completely stopped by the addition of 2 µM i6A (Fig. 3A). In comparison, SAS cells to which 2 µM i6A was added only partially suppressed cell growth (Fig. 2B). Addition of both 5-FU and i6A showed a limited degree of synergistic effect on FR2-SAS cell growth (Fig. 3B,C), showing that use of i6A alone is sufficient for cytotoxicity against FR2-SAS cells.
FIGURE 3.

i6A-induced cell death of 5-FU-resistant oral cancer cell line FR2-SAS. (A) Cell growth time course experiment upon addition of 0, 2, 5, or 10 µM i6A in cell culture medium for indicated number of hours. Cell growth was quantified by performing WST assay and measuring absorbance at 450 nm. The values shown as the relative levels versus the mean of 0 µM i6A 0 h cells. Mean ± SEM from n = 8 biological replicates. (B,C) Cell growth of SAS cells (B) and FR2-SAS cells (C) after 48 h incubation with indicated concentrations of 5-FU and/or i6A in the medium, quantified by performing WST assay and measuring absorbance at 450 nm, and the values shown as the relative levels versus the mean of 0 h, 0 µM cells. Mean ± s.e.m. from n = 8 biological replicates. (D) Microscopic images of SAS cells and FR2-SAS cells after 48 h incubation with indicated concentration of i6A in the medium. SAS cells were seeded at two densities, that is, at the same density as FR2-SAS cells (top panels) and at lower density (middle panels). Scale bar, 50 µm. (E,F) Cell numbers (left) and percentages (right) of alive (blue) and dead (red) SAS cells (E) and FR2-SAS cells (F) after 48 h incubation with indicated concentrations of i6A in the medium. SAS cells were seeded at two densities, that is, at the same density as FR2-SAS cells (E, upper graphs) and at lower density (E, lower graphs). Alive and dead cells were counted using trypan blue and a cell counting device. Mean ± SEM from n = 4 biological replicates. (***) P < 0.001, (**) P < 0.01, (*) P < 0.05 by Mann–Whitney test.
Since FR2-SAS cells were more sensitive to i6A than SAS cells, we monitored the effect of i6A from smaller concentrations by incubating SAS cells and FR2-SAS cells in medium containing 0, 1, 2, and 4 µM i6A. We then collected nonadherent and adherent cells and distinguished between and counted alive and dead cells using trypan blue staining. SAS cells to which 0, 1, 2, and 4 µM i6A were applied started to die only upon 4 µM i6A addition and only when they were seeded at lower cell density (Fig. 3D, upper and middle panels and Fig. 3E). Conversely, 1 µM i6A significantly decreased FR2-SAS cell number, and 2 or 4 µM i6A caused further cell death in FR2-SAS cells (Fig. 3D, lower panels and Fig. 3F). Even when FR2-SAS cells were cultured at a higher cell density than SAS cells, FR2-SAS cells were more prone to cell death than SAS cells (Fig. 3E, lower graph and Fig. 3F). Therefore, although FR2-SAS cells are resistant to 5-FU, i6A effectively causes cell death in FR2-SAS cells.
i6A addition to the medium causes incorporation of i6A into cellular RNAs
Although i6A has been reported to be cytotoxic to several cancer cell lines, the molecular fate and direct molecular function(s) of i6A have been unknown. Considering that 5-FU in the medium is taken into the cells, anabolized and incorporated into RNA, we hypothesized that i6A in the medium might incorporate into RNA. To investigate this idea, we incubated SAS and FR2-SAS cells in i6A-containing medium, collected total cellular RNA, and subjected the samples to RNA digestion and LC-MS analysis. In accordance with our hypothesis, upon i6A addition to the medium, the amount of i6A contained in the total cellular RNA increased four- to fivefold in both SAS and FR2-SAS cells (“Total RNA” in Fig. 4A; Supplemental Fig. 2).
FIGURE 4.

Incorporation of i6A into RNAs upon i6A addition to the medium. (A–F) Modified nucleoside levels in RNAs after incubation of SAS or FR2-SAS cells in medium containing no i6A (blue), 1 µM i6A (green), 2 µM i6A (yellow), or 4 µM i6A (red) for 24 h. After total RNA extraction, 18S rRNA, 28S rRNA, and tRNA were each fractioned by gel electrophoresis and gel excision, and subjected to digestion to nucleosides and LC-MS analysis. i6A (A), t6A (B), ms2i6A (C), m3C (D), m62A (E), and Um (F) peak areas were normalized by uridine peak area of the same sample, and the values are shown as the relative levels versus the mean of 0 µM i6A SAS cell total RNA samples. Mean ± SEM from n = 4 biological replicates. (*) P < 0.05 by Mann–Whitney test.
To investigate whether i6A is widely incorporated into various RNA species or specifically incorporated into specific RNA species, we used gel fractionation to collect fractions of tRNA, 18S ribosomal RNA (rRNA), and 28S rRNA. N6-threonylcarbamoyladenosine (t6A), 2-methylthio-i6A (ms2i6A) and 3-methylcytidine (m3C), which are considered to exist exclusively in tRNAs, were enriched in the tRNA fraction and barely detected in rRNA fractions, confirming that the tRNA fractionation was successful (Fig. 4B–D). Similarly, N6,N6-dimethyladenosine (m62A), which is considered to exist only in 18S rRNA, was enriched in the 18S rRNA fraction, confirming 18S rRNA fractionation was successful (Fig. 4E). In this situation, we unexpectedly observed substantial increases of i6A incorporation into the18S rRNA and 28S rRNA fractions in both SAS cells and FR2-SAS cells (Fig. 4A; Supplemental Fig. 2). We also attempted to fractionate poly(A)+ RNA, which is primarily composed of mRNAs, but could not completely remove contaminations of abundant tRNAs and rRNAs. Thus, poly(A)+ RNAs could not be analyzed. Considering that rRNAs comprise 80%–90% of cellular RNA, tRNAs comprise about 10% of cellular RNA, and mRNAs and other noncoding RNAs comprise the remaining few percent, our results indicate that i6A addition to the cell culture medium predominantly induced i6A incorporation into 18S and 28S rRNAs.
Apart from the endogenous i6A at tRNA position 37 incorporated by the TRIT1 enzyme, i6A addition to the medium slightly increased the i6A level in the SAS cell tRNA fraction (Fig. 4A). The i6A level in the FR2-SAS cell tRNA fraction repeatedly fluctuated (Fig. 4A), which may or may not be due to vulnerability of FR2-SAS cells to i6A. In SAS cell tRNA, if i6A increase is due to an increased activity of TRIT1-mediated i6A modification at tRNA position 37, ms2i6A modification in mitochondrial tRNA position 37 might also increase, because i6A modification within ms2i6A is performed by TRIT1 (Schweizer et al. 2017; Takenouchi et al. 2019). However, upon i6A addition to the medium, the ms2i6A level did not increase in SAS cells and FR2-SAS cells (Fig. 4C), implying that TRIT1 is irrelevant in this process. In addition, endogenous i6A at tRNA position 37 is known to promote m3C modification at tRNA position 32 in human mitochondria and fission yeast and 2′-O-methyuridine (Um) modification at tRNA position 34 in E. coli (Zhou et al. 2015; Arimbasseri et al. 2016; Scholler et al. 2021). However, m3C and Um levels did not increase upon i6A addition to the medium (Fig. 4D,F), further supporting the irrelevancy of TRIT1.
TRIT1-catalyzed post-transcriptional i6A modification is not responsible for i6A incorporation into cellular RNA
Within the cell, the artificial nucleobase analog 5-FU is converted to 5-FUTP and incorporated into RNA, presumably during RNA transcription (Longley et al. 2003). However, to the best of our knowledge, all endogenous RNA modifications are incorporated post-transcriptionally by specific RNA modifying enzymes that act on RNA polynucleotides, with the exception of the inosine monomer, which can be made from the adenosine monomer. We set out to differentiate between whether adding i6A to the medium induces cotranscriptional or post-transcriptional i6A incorporation into RNA by RNA-modifying enzymes. We first investigated if inactivation of the only known human endogenous i6A-modifying enzyme TRIT1 prevents i6A incorporation upon i6A addition to the medium. Our rationale was based on observations that various cellular stresses induce recruitment of diverse proteins into the nucleolus (Audas et al. 2012). Thus, TRIT1 could be recruited to the nucleolus upon i6A-induced cellular stress to promiscuously incorporate i6A into rRNAs.
To investigate the role of TRIT1 in i6A incorporation into RNA, we mutated the TRIT1 gene in HEK293FT cells using the CRISPR/Cas9 system. We obtained TRIT1 mutant cells that have a 1 nt insertion on one allele and a 14 nt deletion on the other, each of which causes a frameshift that induces a premature termination codon after translation of several amino acids (Fig. 5A; Supplemental Fig. 3). Using western blot, we confirmed that mature TRIT1 protein was present in the control cell lysate and barely detected in the TRIT1 mutant cell lysate (Fig. 5B). Because the mutation site and the premature termination codon are near the 3′ end of the TRIT1 open reading frame, a small amount of near-full-length TRIT1 may be synthesized by the pioneer-round translation prior to the nonsense-mediated mRNA decay. However, we confirmed that TRIT1 was dysfunctional in TRIT1 mutant cells by showing that i6A contained in the total cellular RNA was reduced to less than one percent compared to the control cells (Fig. 5C).
FIGURE 5.
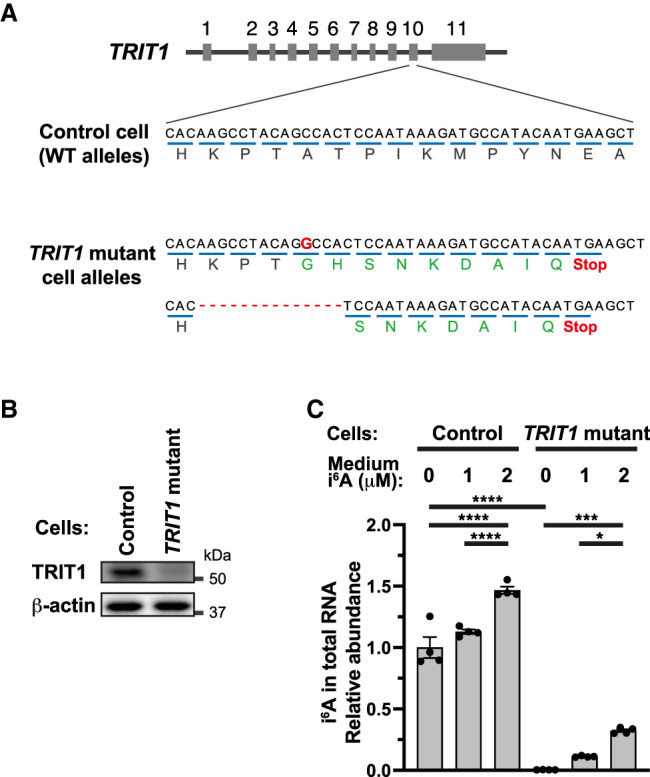
TRIT1-independent i6A incorporation into cellular RNA upon i6A addition to the medium. (A) TRIT1 alleles in TRIT1 mutant HEK293FT cells and control HEK293FT cells. Control cells were generated by the same CRISPR/Cas9 cloning procedures using nonhuman DNA targeting guide sequence. Encoded amino acids are indicated under codons. The red G in the mutated allele indicates an extra base insertion. Red lines indicate deletions, and red “Stop” indicates premature termination codon made by frameshift mutations. (B) Western blot of TRIT1 protein in the cells. β-actin is shown as a loading control. (C) i6A incorporation into control HEK293FT cells or TRIT1 mutant HEK293FT cells. Cells were incubated in medium containing 0, 1 or 2 µM i6A for 24 h, and total RNA was extracted and digested to nucleosides for LC-MS analysis. i6A peak area was normalized by uridine peak area of the same sample, and the values are shown as the relative levels versus the mean of 0 µM i6A control cell samples. Mean ± SEM from n = 4 biological replicates. (****) P < 0.0001, (***) P < 0.001, (*) P < 0.05 by two-way ANOVA followed by Tukey's multiple comparison test.
We then performed LC-MS on total cellular RNA nucleosides and showed that i6A addition to the medium induced i6A incorporation into total cellular RNA for both control cells and TRIT1 mutant cells (Fig. 5C). Therefore, TRIT1, which is responsible for post-transcriptional i6A incorporation at tRNA position 37, does not mediate incorporation of i6A into RNA upon i6A addition to the medium. Notably, i6A addition to the medium caused i6A incorporation into RNA not only in SAS and FR2-SAS cells (Fig. 4A), but also in HEK293FT cells (Fig. 5C), suggesting that i6A incorporation into cellular RNA upon i6A addition to the medium could be a general phenomenon in different types of human cells.
i6A incorporation into rRNAs is coupled with Pol I transcription
We next investigated the extent to which RNA polymerase I (Pol I)-dependent rRNA transcription is involved in incorporation of i6A, available from the growth medium, into rRNA. For these experiments, we used CX-5461, a potent inhibitor of Pol I (Drygin et al. 2011; Harold et al. 2021). Pol I transcribes 18S, 5.8S and 28S rRNAs in a single precursor RNA containing spacer sequences around the rRNAs. Subsequently, the RNA precursor is processed by specific ribonucleases to produce mature 18S, 5.8S, and 28S rRNAs (Fig. 6A; Aubert et al. 2018). In the Pol I inhibition experiments, we monitored Pol I transcriptional inhibition by detecting the ribosomal RNA precursors using RT-qPCR with primers designed across the spacer RNA and rRNA (Fig. 6A). Upon incubation of SAS or FR2-SAS cells in the cell culture medium containing Pol I inhibitor for 24 h, we detected a reduction of 18S and 28S rRNA precursors (Fig. 6B, blue bars of “Pre-18S” and “Pre-28S”) at a comparable level with the previous study (Drygin et al. 2011). We also performed RT-qPCR using primers designed within 18S or 28S rRNA regions. 18S and 28S rRNA steady-state levels, normalized by Pol II-transcribed GAPDH mRNA, showed a tendency to decrease to about 75% after 24 h of Pol I inhibitor treatment (Fig. 6B, blue bars of “18S” and “28S”). These decreases also imply that Pol I transcription was largely inhibited. About 75% of 18S and 28S rRNAs remained after 24 h of Pol I inhibitor treatment, presumably due to the long half-lives of cytoplasmic rRNAs.
FIGURE 6.

Pol I transcription-coupled i6A incorporation into ribosomal RNAs. (A) Schematic of rRNA transcription, processing, and RT-qPCR primers. 18S, 5.8S, and 28S rRNAs are transcribed by Pol I as a polycistronic 47S precursor RNA followed by processing by specific ribonucleases. Pre-18S rRNA primers and pre-28S primers are designed across the boundary of 18S or 28S rRNAs and their adjacent spacer RNAs. (B) RT-qPCR of 18S and 28S rRNAs and their precursors upon addition of 2 µM i6A and/or 2 µM Pol I inhibitor, CX-5461 into the medium for 24 h. RNA levels were normalized by Pol II-transcribed GAPDH mRNA levels. (C) i6A level in total cellular RNA, 18S rRNA and 28S rRNA in SAS cells or FR2-SAS cells upon addition of 2 µM i6A and/or 2 µM RNA polymerase I inhibitor, CX-5461 into medium for 24 h. Total RNA samples were used to fractionate 18S and 28S rRNAs using agarose gel electrophoresis and gel excision. The RNA samples were digested to nucleosides and subjected to LC-MS analysis. i6A peak area was normalized by uridine peak area of the same sample. Mean ± SEM from n = 4 samples of which RNA samples were independently collected on different days and measured with LC-MS on the same day. (*) P < 0.05 by Mann–Whitney test.
i6A addition to the medium resulted in i6A incorporation into total RNA of SAS and FR2-SAS cells; however, this was inhibited by the further addition of CX-5461 (Fig. 6C). Some increase of i6A levels in total RNA upon only Pol I inhibitor treatment (blue bars in Fig. 6C) might be due to the relative increase of naturally i6A-containing tRNA due to a decrease of rRNAs.
Moreover, we showed by the fractionating of 18S and 28S rRNAs isolated by gel excision that i6A incorporation into 18S and 28S rRNAs upon i6A addition to the medium was inhibited by the further addition of CX-5461 (Fig. 6C). Collectively, these data show that i6A incorporation into 18S and 28S rRNAs did not occur after Pol I transcription, and that i6A incorporation into 18S and 28S rRNAs is coupled with Pol I transcription of ribosomal RNAs.
i6A induces decrease of cellular protein synthesis and increase of protein aggregation at stronger magnitudes in FR2-SAS cells than in SAS cells
Because i6A has a bulky isopentenyl residue (Fig. 2A), incorporation of i6A into 18S and 28S rRNAs may alter the 40S and 60S ribosome subunit structure. The ribosome is a ribozyme for protein synthesis (Cech 2000). Thus, ribosomal structural alterations might interfere with protein synthesis, leading to (1) decreased translation rate, and/or (2) decreased translational fidelity, inducing misincorporation of amino acids and misfolding of proteins.
To investigate whether i6A incorporation leads to a decreased translation rate, we observed nascent protein synthesis after adding 35S-labeled methionine to the cell culture medium for 35 min, followed by total protein electrophoresis and radio imaging (Fig. 7A). Considering that SAS cells and FR2-SAS cells showed similar levels of i6A incorporation into rRNAs (Figs. 4, 6), we initially expected that SAS cells and FR2-SAS cells would show a similar trend. In SAS cells, incubating the cells in the medium containing 0 to 4 µM i6A for 24 h did not change the detectable nascent protein level (Fig. 7A,B). In contrast, to our surprise, in FR2-SAS cells, increasing the medium i6A concentration from 0 to 4 µM reduced the global protein synthesis rate in a i6A dose-dependent manner (Fig. 7A,B).
FIGURE 7.

i6A-induced translational decrease and protein aggregation in FR2-SAS cells. (A) Nascent cellular protein synthesis observed by 35S-methionine pulse-labeling. Radiation images of the electrophoresed gel is shown on the left, and Coomassie brilliant blue (CBB)-staining of the same gel on the right as a loading control. n = 3 independent experiments were performed, and representative images are shown. (B) Quantification of the nascent protein signal lanes in A. The relative signal intensities of the lanes versus SAS 0 µM i6A lane were quantified. Mean ± SEM from n = 3 independent experiments. (C) Quantification of endoplasmic reticulum (ER) stress marker mRNAs by RT-qPCR. Mean ± SEM from n = 4 biological replicates. RNA levels were normalized by GAPDH mRNA levels. (D) Detection of protein aggregation in SAS and FR2-SAS cells. After 24 h of 0 or 4 µM i6A treatment, the cells were fixed and subjected to ProteoStat assay and confocal microscopy. Protein aggregates are shown in green, and Hoechst stained nuclei are shown in blue. Scale bar, 20 µm. (****) P < 0.0001, (***) P < 0.001, (**) P < 0.01, (*) P < 0.05 by two-way ANOVA followed by Tukey's multiple comparison test.
Next, to determine whether i6A incorporation compromises protein quality, we monitored endoplasmic reticulum (ER) stress markers. The extremely high concentration of proteins within the ER makes this organelle susceptible to protein misfolding and aggregation. Accumulation of misfolded or aggregated proteins causes ER stress, which can induce cell death (Hetz et al. 2011). ER stress can be monitored by up-regulation of BIP, CHOP and XBP1 mRNAs, as well as by splicing of XBP1 mRNA (Hetz et al. 2011; Fakruddin et al. 2018). Thus, we determined levels of mRNA for these ER stress markers in SAS cells and FR2-SAS cells after incubation in the medium containing 0, 2, or 4 µM of i6A for 24 h (Fig. 7C). In SAS cells, ER stress marker mRNA levels were not up-regulated by 2 or 4 µM i6A addition. In contrast, in FR2-SAS cells, we observed clear up-regulations of ER stress marker mRNAs upon 2 or 4 µM i6A addition, suggesting that i6A-mediated impaired ER protein quality.
Decreased translational fidelity can increase protein misfolding and aggregation. Thus, to investigate if i6A treatment compromises protein quality in FR2-SAS cells by a different method, we monitored protein aggregation using the ProteoStat assay, with i6A addition to the medium for 24 h. Although we observed some increase in protein aggregation in SAS cells, protein aggregation was much more prominent in FR2-SAS cells (Fig. 7D). Collectively, the results shown in Figure 7C and D strongly suggest that i6A induces more pronounced levels of protein aggregation in FR2-SAS cells than in SAS cells.
DISCUSSION
5-fluorouracil (5-FU) is commonly used for cancer therapy. However, its clinical applications have often been hampered by acquired drug resistance of cancer cells. Our study revealed that use of another nucleic acid analog, i6A, effectively induced cell death of a 5-FU-resistant cell line, FR2-SAS, whereas the parental oral squamous cell carcinoma cell line SAS showed only a mild reduction in cell growth at the same i6A concentrations. Future studies are needed to investigate whether this effect of i6A is generalizable to cancer cells resistant to 5-FU and other cancer chemotherapeutic drugs. Also, since artificial nucleoside analogs are used to treat RNA virus infections (Eyer et al. 2018), it may be of importance to test if i6A is effective against RNA viruses including SARS-CoV-2.
We also discovered that upon i6A addition to cell growth media, i6A is incorporated into cellular RNA, especially into 18S and 28S rRNAs. i6A incorporation occurred in a Pol I-transcription coupled manner and even in cells lacking TRIT1, the cell's sole post-transcriptional i6A modifying enzyme. Our data strongly suggest that i6A incorporation into rRNAs occurs at the level of transcription, implying that extracellular i6A is imported into cells and phosphorylated to produce i6ATP, which is then used for transcription. Considering that 5-FU addition to the medium is known to induce 5-FUTP production and 5-FUrd incorporation into cellular RNA (Kufe and Major 1981; Longley et al. 2003), we propose that intracellular conversion of i6A into i6ATP is the most reasonable hypothesis to explain our results. To investigate this hypothesis, chemical synthesis of authentic i6ATP and development of a detection method for i6ATP will be required.
A limitation to our study is that we could not establish causation of i6A incorporation into RNA and cell death by experiments such as the use of CX-5461 to inhibit Pol I, which would test if i6A-mediated cell death could be suppressed through a Pol I-mediated mechanism. CX-5461 has itself proven to be cytotoxic, as demonstrated by its use in a phase I clinical trial against hematologic cancers (Khot et al. 2019; Harold et al. 2021). It should be noted that even without i6A addition, FR2-SAS cells showed more protein aggregation than SAS cells (Fig. 7D, 0 µM i6A SAS and FR2-SAS cells). In addition, the CHOP mRNA level was higher in FR2-SAS cells than in SAS cells even without i6A addition (Fig. 7C, CHOP mRNA, blue bars). Thus, FR2-SAS cells might inherently show slightly compromised protein quality, and upon i6A incorporation into rRNAs, compromised protein quality may further deteriorate and cause ER stress, the excess of which can cause cell death (Hetz et al. 2011). Also, although the majority of i6A incorporation into RNA depended on Pol I (Fig. 6), it is possible that i6A also incorporates based on Pol II-mediated mRNA transcription. Such potential i6A incorporation into mRNA coding sequences might also interfere with mRNA-tRNA interaction in the ribosomes, causing decreased translational speed and decreased translational fidelity.
Generally, artificial analogs of DNA and RNA nucleosides and nucleobases (1) inhibit the function of nucleoside biosynthetic enzymes and/or (2) are used for nucleotide polymerization by DNA/RNA polymerase (Longley et al. 2003; Eyer et al. 2018). 5-FU meets both criteria. Our study strongly suggests that i6A fits in the latter case and is used in transcription mediated by Pol I. We do not currently know if i6A inhibits RNA modification enzymes, but our RNA LC-MS experiments, whereby we simultaneously monitored more than 50 modified nucleosides in total RNA, failed to detect observable differences in modification levels of other modified nucleosides, including other N6-modified adenosines such as t6A, ms2i6A, and m62A (Fig. 4B,C,E).
Extracellular 5-FU has been shown to be imported into cells, converted to 5-FdUTP, and used for DNA synthesis (Longley et al. 2003). We wondered if i6A can also be converted to deoxy-i6ATP and used for DNA synthesis. Although our LC-MS cannot detect deoxy-i6ATP, it can be used to detect the mass equivalent of dephosphorylated deoxy-i6A. Upon incubation of cells in a medium containing i6A, we detected a small amount of the mass equivalent to deoxy-i6A (MH+ m/z = 320) from nuclease-digested cellular genomic DNA (Supplemental Fig. 4A). Moreover, the detection occurred in an i6A concentration-dependent manner and an i6A addition time-dependent manner (Supplemental Fig. 4B). The detected peak equivalent to the mass of deoxy-i6A (MH+ m/z = 320) showed a LC retention time 0.4 min after i6A (Supplemental Fig. 4A). This delay may be due to increased hydrophobicity of deoxy-i6A compared to i6A, owing to the lack of the 2′ hydroxyl group of ribose, thus making a stronger interaction of deoxy-i6A with the hydrophobic column in the LC system. In addition, a collision-induced dissociation analysis of this peak detected the mass equivalent to the N6-isopentenyladenine base (MH+ m/z = 204) (Supplemental Fig. 4A). Thus, our preliminary results are in accordance with the presumed conversion of medium-derived i6A into deoxy-i6ATP and incorporation into DNA. A conclusive investigation will require chemical synthesis of authentic deoxy-i6A and deoxy-i6ATP.
In addition to aberrant translation-mediated effects, considering that the mass equivalent to deoxy-i6A was detected from the genomic DNA (Supplemental Fig. 4A,B), we should not exclude the possibility of bulky deoxy-i6A within genomic DNA sterically blocking specific proteins that function on DNA. Moreover, since incorporation of i6A into rRNAs implies the putative presence of i6ATP in cells, we cannot exclude the possibility of i6ATP blocking various enzymatic processes that use ATP, such as phosphorylation, thiolation, and ADP-ribosylation. Therefore, a comprehensive analysis of the cytotoxic mechanisms of i6A is needed. Such studies may be initiated by performing transcriptome and/or proteome analyses of SAS and FR2-SAS cells that are either treated or not treated with i6A.
In summary, we searched for means to cause cell death of 5-FU-resitant oral squamous cell carcinoma cell line FR2-SAS and found that i6A can induce cell death of FR2-SAS cells. i6A was more effective in inducing cell death in 5-FU-resistant FR2-SAS cells than 5-FU-sensitive parental SAS cells. We also revealed that i6A addition to the medium induced incorporation of i6A into cellular 18S and 28S ribosomal RNAs in a Pol I-coupled manner, providing insights into the molecular fate of extracellularly added i6A in the context of cellular nucleic acid anabolism.
MATERIALS AND METHODS
Cell culture
Oral squamous cell carcinoma cell line SAS was acquired from the Cell Resource Center for Biomedical Research in Tohoku University. FR2-SAS cells (originally named SAS/FR2 cells in Nagata et al. 2011) were previously generated from SAS cells (Nagata et al. 2011), and 5-FU-resistance was maintained by culturing the cells in a medium containing 10 µM 5-FU. SAS and FR2-SAS cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, in a humidified, 37°C atmosphere with 5% CO2. 5-FU (Wako), i6A (Cayman Chemical Company), CX-5461 (Millipore) or dimethyl sulfoxide (DMSO) vehicle (Nacalai Tesque) were used in experiments.
Cell number quantification
Both nonadherent cells and adherent cells were collected, mixed and stained with trypan blue (Fujifilm Wako), and alive and dead cells were distinguished and counted using a cell counting device (Olympus). Cells cultured in 96-well plates were quantified using Cell Counting Kit-8 (DOJINBO) and a 96-well plate reader (TECAN).
Construction of TRIT1 mutant cells and control cells
Human TRIT1 mutant cells and control cells were generated using the CRISPR/Cas9 system essentially as described previously (Shalem et al. 2014). Briefly, sense and antisense oligonucleotides encoding a single guide RNA (sgRNA) against TRIT1 gene or a control sgRNA (not targeting human genome) (Supplemental Table 1; Shalem et al. 2014) were cloned into the BsmBI sites of lentiCRISPR v2 Blast plasmid (Addgene #83480). Lentiviruses were generated in HEK293FT cells by transfecting the sgRNA sequence-containing lentiCRISPR v2 Blast plasmid, psPAX2 plasmid (Addgene #12260) and pMD2.G plasmid (Addgene #12259) with Lipofectamine 3000 (Invitrogen). Fresh HEK293FT cells were transduced with the generated viruses, followed by blasticidin selection of the transduced cells. Subsequently, single clones were acquired by diluting the cells in 96-well plates followed by expansion of the clones. The target region of the genome in each clone was PCR-amplified and sequenced with conventional Sanger sequencing, using the primers listed in Supplemental Table 1.
RNA extraction
Cells were rinsed with phosphate-buffered saline (PBS) at least three times, and total cellular RNA was prepared using TRI Reagent (MRC), according to the manufacturer's protocol.
tRNA and rRNA fractionation
Total tRNA fraction was collected from total RNA by electrophoresis of total RNA in denaturing 7 M urea/tris-borate-EDTA (TBE) buffer/10% polyacrylamide gel electrophoresis, staining using SYBR Gold (Invitrogen), gel excision of the tRNA band, and elution of tRNA using 60°C water. 18S and 28S rRNAs were collected by agarose gel electrophoresis of formamide-denatured total RNA, gel excision of rRNA band, and RNA extraction using Zymoclean Gel RNA Recovery Kit (Zymo Research).
Quantitative reverse-transcription real-time PCR (RT-qPCR)
RT-qPCR was performed as previously described (Takesue et al. 2019). The primer sequences are listed in Supplemental Table 1. For RT-qPCR to detect spliced XBP1 mRNA, pre-18S rRNA, and pre-28S rRNA, we used previously reported qPCR primers (Chujo et al. 2017; Yoon et al. 2019).
Genomic DNA extraction
After incubating cells in medium containing 0, 1, or 2 µM i6A for 24 or 48 h, cells were rinsed with PBS three times, trypsinized, and collected. The cell pellets were lysed in 1 ml of Proteinase K buffer (20 mM Tris-HCl [pH 8.0], 5 mM EDTA, 400 mM NaCl, 0.3% sodium dodecyl sulfate) and sonicated for a total of 24 sec using Sonifier 250 (Branson) at power 4. Then, 10 µL of Proteinase K (NEB) was added to the solution, and the samples were incubated at 55°C for 1 h for protein digestion, followed by Proteinase K inactivation using phenol-chloroform-isoamylalcohol (Invitrogen). Nucleic acids were collected by isopropanol precipitation, cleaned by 75% ethanol washing, and eluted into 10 mM Tris-HCl (pH 8.0)/1 mM EDTA (pH 8.0). RNA in the solution was digested using RNaseA (Qiagen), and RNaseA was inactivated using phenol-chloroform-isoamylalcohol (Invitrogen). The remaining DNA was isopropanol-precipitated, 75% ethanol-washed, and dissolved into MilliQ water.
Nucleoside mass spectrometry
RNA or genomic DNA was digested to nucleosides, which were subjected to liquid chromatography triple quadrupole mass spectrometry (LC-MS) using LCMS-8050 (Shimadzu), as previously described (Hirayama et al. 2020; Nagayoshi et al. 2021). A program to detect 5-fluorouridine (molecular weight: 262.2) was added by setting the Q1 m/z to 263.2 Da and Q3 m/z to 131.2 Da with the same collision energy as uridine. The LC elution time and mass spectrum were confirmed by using authentic 5-fluorouridine (Sigma). Programs to detect the DNA-digested nucleosides with the molecular weights equivalent to deoxy-i6A (Q1 m/z = 320.0 Da, Q3 m/z = 204.0 Da) and deoxycytidine (Q1 m/z = 228.2 Da, Q3 m/z = 112.1 Da) were added with the same collision energy values as i6A and cytidine, respectively. The LC elution time and mass spectrum of deoxycytidine were confirmed by using authentic deoxycytidine prepared by dephosphorylating dNTP (TOYOBO) with bacterial alkaline phosphatase (TAKARA).
Western blotting
Western blotting was performed essentially as described previously (Takesue et al. 2019). The antibodies and their conditions for use are listed in Supplemental Table 2.
35S-methionine labeling of nascent proteins
Pulse-labeling of nascent proteins was performed essentially as previously described (Fukuda et al. 2021), with some modifications. Briefly, cells grown in 6 cm dishes were incubated in the medium containing 0, 2, or 4 µM i6A for 24 h, and briefly washed using 37°C DMEM without Met, Cys, and Gln (Gibco). A total of 3 mL of 37°C preincubation medium (DMEM without Met, Cys, and Gln, supplemented with 2% FBS, 2 mM Gln, 0.2 mM Cys, with 0, 2, or 4 µM i6A) was added to the cells, and the cells were incubated in 37°C CO2 incubator for 15 min. The medium was then exchanged with 2 mL of incubation medium (2 mL of preincubation medium containing 5.92 MBq of 35S-labeled methionine and 0, 2, or 4 µM i6A), and the cells were incubated in 37°C CO2 incubator for 35 min. Subsequently, the medium was removed and the cells were washed with PBS and collected using trypsin and DMEM containing 10% FBS. Cells were lysed and protein concentration was measured using the BCA Protein Assay Kit (Pierce). A total of 20 µg of total proteins were run on Tricine PAGE gel (NOVEX). The gel was stained using Coomassie brilliant blue (CBB) staining solution (Bio-Rad), dried on a gel dryer, photographed, and the radiation image was acquired using an imaging plate and imager (Fujifilm).
ProteoStat staining assay
A ProteoStat Aggresome Detection Kit (Enzo Life Sciences) was used to monitor protein aggregation. A total of 4.5 × 105 SAS or FR2-SAS cells were seeded on 35 mm glass-bottom cell culture dishes (IWAKI), cultured for 48 h, and incubated in the medium containing 0 or 4 µM i6A for 24 h. Subsequently, the ProteoStat staining assay was performed according to the manufacturer's instructions, followed by image acquisition using FLUOVIEW FV3000 confocal microscope (Olympus).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Tomizawa laboratory for fruitful discussions, Yuka Tashiro for technical assistance, and Natalie D. DeWitt for critical reading and language editing. We also thank the Kumamoto University Radioisotope Center at which the 35S-methionine pulse-labeling experiment was performed. This work was supported by Japan Society for the Promotion of Science KAKENHI grants to K.T. and T.C. (18H02865 and 17KT0054 to K.T., and 20H03187 to T.C.), a Japan Science and Technology Agency FOREST grant to T.C. (JPMJFR204Z), and a Japan Agency for Medical Research and Development grant to K.T. (20gm4010003h0002).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079176.122.
MEET THE FIRST AUTHOR
Maya Yakita.

Meet the First Author(s) is a new editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Maya Yakita is the first author of this paper, “Extracellular N6-isopentenyladenosine (i6A) addition induces cotranscriptional i6A incorporation into ribosomal RNAs.” Maya is a dentist and a graduate student in the Department of Molecular Physiology in Kumamoto University, Japan.
What are the major results described in your paper and how do they impact this branch of the field?
N6-isopentenyladenosine (i6A) is an RNA modification that is post-transcriptionally added to a tRNA anticodon loop by a specific tRNA modification enzyme. Using a human oral squamous cell carcinoma (OSCC) cell line, we found that addition of i6A monomer into the cell culture medium causes the RNA polymerase I to cotranscriptionally incorporate i6A into ribosomal RNA, accompanied by protein aggregation. We were able to reveal an unexpected molecular fate of extracellularly added modified RNA nucleoside monomer. In addition, we found that i6A is cytotoxic especially to an OSCC line resistant to the chemotherapeutic drug 5-fluorouracil (5-FU) in a more prominent manner than to its parental 5-FU-sensitive OSCC line.
What led you to study RNA or this aspect of RNA science?
I am a dentist. I face patients with oral cancers, as well as cancers that have become resistant to chemotherapy. Some chemotherapeutic drugs such as 5-FU are nucleobase/nucleoside analogs, making me interested in RNA modifications and in entering graduate school to study RNA modifications.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
i6A monomer has been known to be cytotoxic, but no one knew its molecular function. After incubating the cells in culture medium containing i6A, I extracted cellular RNA and checked the RNA modification, just as a control. Then, we were totally surprised and puzzled to detect a lot of i6A incorporated into total RNA. We could not believe it, so we fractionated total RNA to tRNA and rRNA, and we were further surprised to see that i6A was incorporated mostly into rRNAs, although endogenous i6A is a tRNA modification.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
As a graduate student, through studying and struggling with my mentors, I gained confidence on which way to proceed and started to make findings. The more I proceeded, the more I wanted to deepen understanding of my RNA molecules.
If you were able to give one piece of advice to your younger self, what would that be?
To find and reach the truth, there is no short cut, but also no waste. If you are having a hard time, it means that you are sincerely doing science, so do not give up, but keep going. At the same time, it is also important to discuss often with the people around you to get good advice.
REFERENCES
- Agris PF, Eruysal ER, Narendran A, Vare VYP, Vangaveti S, Ranganathan SV. 2018. Celebrating wobble decoding: half a century and still much is new. RNA Biol 15: 537–553. 10.1080/15476286.2017.1356562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimbasseri AG, Iben J, Wei FY, Rijal K, Tomizawa K, Hafner M, Maraia RJ. 2016. Evolving specificity of tRNA 3-methyl-cytidine-32 (m3C32) modification: a subset of tRNAsSer requires N6-isopentenylation of A37. RNA 22: 1400–1410. 10.1261/rna.056259.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert M, O'Donohue MF, Lebaron S, Gleizes PE. 2018. Pre-ribosomal RNA processing in human cells: from mechanisms to congenital diseases. Biomolecules 8: 123. 10.3390/biom8040123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audas TE, Jacob MD, Lee S. 2012. Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol Cell 45: 147–157. 10.1016/j.molcel.2011.12.012 [DOI] [PubMed] [Google Scholar]
- Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, Hohmann E, Chu HY, Luetkemeyer A, Kline S, et al. 2020. Remdesivir for the treatment of Covid-19—final report. N Engl J Med 383: 1813–1826. 10.1056/NEJMoa2007764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, de Crecy-Lagard V, Ross R, Limbach PA, Kotter A, et al. 2018. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res 46: D303–D307. 10.1093/nar/gkx1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braess J, Freund M, Hanauske A, Heil G, Kaufmann C, Kern W, Schussler M, Hiddemann W, Schleyer E. 1998. Oral cytarabine ocfosfate in acute myeloid leukemia and non-Hodgkin's lymphoma–phase I/II studies and pharmacokinetics. Leukemia 12: 1618–1626. 10.1038/sj.leu.2401152 [DOI] [PubMed] [Google Scholar]
- Carter JM, Emmett W, Mozos IR, Kotter A, Helm M, Ule J, Hussain S. 2019. FICC-Seq: a method for enzyme-specified profiling of methyl-5-uridine in cellular RNA. Nucleic Acids Res 47: e113. 10.1093/nar/gkz658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castiglioni S, Casati S, Ottria R, Ciuffreda P, Maier JA. 2013. N6-isopentenyladenosine and its analogue N6-benzyladenosine induce cell cycle arrest and apoptosis in bladder carcinoma T24 cells. Anticancer Agents Med Chem 13: 672–678. 10.2174/1871520611313040016 [DOI] [PubMed] [Google Scholar]
- Cech TR. 2000. Structural biology. The ribosome is a ribozyme. Science 289: 878–879. 10.1126/science.289.5481.878 [DOI] [PubMed] [Google Scholar]
- Chujo T, Tomizawa K. 2021. Human transfer RNA modopathies: diseases caused by aberrations in transfer RNA modifications. FEBS J 288: 7096–7122. 10.1111/febs.15736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chujo T, Yamazaki T, Kawaguchi T, Kurosaka S, Takumi T, Nakagawa S, Hirose T. 2017. Unusual semi-extractability as a hallmark of nuclear body-associated architectural noncoding RNAs. EMBO J 36: 1447–1462. 10.15252/embj.201695848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciaglia E, Abate M, Laezza C, Pisanti S, Vitale M, Seneca V, Torelli G, Franceschelli S, Catapano G, Gazzerro P, et al. 2017. Antiglioma effects of N6-isopentenyladenosine, an endogenous isoprenoid end product, through the downregulation of epidermal growth factor receptor. Int J Cancer 140: 959–972. 10.1002/ijc.30505 [DOI] [PubMed] [Google Scholar]
- Drygin D, Lin A, Bliesath J, Ho CB, O'Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, et al. 2011. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res 71: 1418–1430. 10.1158/0008-5472.CAN-10-1728 [DOI] [PubMed] [Google Scholar]
- Eyer L, Nencka R, de Clercq E, Seley-Radtke K, Ruzek D. 2018. Nucleoside analogs as a rich source of antiviral agents active against arthropod-borne flaviviruses. Antivir Chem Chemother 26: 2040206618761299. 10.1177/2040206618761299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakruddin M, Wei FY, Suzuki T, Asano K, Kaieda T, Omori A, Izumi R, Fujimura A, Kaitsuka T, Miyata K, et al. 2018. Defective mitochondrial tRNA taurine modification activates global proteostress and leads to mitochondrial disease. Cell Rep 22: 482–496. 10.1016/j.celrep.2017.12.051 [DOI] [PubMed] [Google Scholar]
- Frye M, Jaffrey SR, Pan T, Rechavi G, Suzuki T. 2016. RNA modifications: what have we learned and where are we headed? Nat Rev Genet 17: 365–372. 10.1038/nrg.2016.47 [DOI] [PubMed] [Google Scholar]
- Fukuda H, Chujo T, Wei FY, Shi SL, Hirayama M, Kaitsuka T, Yamamoto T, Oshiumi H, Tomizawa K. 2021. Cooperative methylation of human tRNA3Lys at positions A58 and U54 drives the early and late steps of HIV-1 replication. Nucleic Acids Res 49: 11855–11867. 10.1093/nar/gkab879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold CM, Buhagiar AF, Cheng Y, Baserga SJ. 2021. Ribosomal RNA transcription regulation in breast cancer. Genes (Basel) 12: 502. 10.3390/genes12040502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Martinon F, Rodriguez D, Glimcher LH. 2011. The unfolded protein response: integrating stress signals through the stress sensor IRE1α. Physiol Rev 91: 1219–1243. 10.1152/physrev.00001.2011 [DOI] [PubMed] [Google Scholar]
- Hirayama M, Wei FY, Chujo T, Oki S, Yakita M, Kobayashi D, Araki N, Takahashi N, Yoshida R, Nakayama H, et al. 2020. FTO demethylates cyclin D1 mRNA and controls cell-cycle progression. Cell Rep 31: 107464. 10.1016/j.celrep.2020.03.028 [DOI] [PubMed] [Google Scholar]
- Khalique A, Mattijssen S, Haddad AF, Chaudhry S, Maraia RJ. 2020. Targeting mitochondrial and cytosolic substrates of TRIT1 isopentenyltransferase: specificity determinants and tRNA-i6A37 profiles. PLoS Genet 16: e1008330. 10.1371/journal.pgen.1008330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khot A, Brajanovski N, Cameron DP, Hein N, Maclachlan KH, Sanij E, Lim J, Soong J, Link E, Blombery P, et al. 2019. First-in-human RNA polymerase I transcription inhibitor CX-5461 in patients with advanced hematologic cancers: results of a phase I dose-escalation study. Cancer Discov 9: 1036–1049. 10.1158/2159-8290.CD-18-1455 [DOI] [PubMed] [Google Scholar]
- Kufe DW, Major PP. 1981. 5-Fluorouracil incorporation into human breast carcinoma RNA correlates with cytotoxicity. J Biol Chem 256: 9802–9805. 10.1016/S0021-9258(19)68695-3 [DOI] [PubMed] [Google Scholar]
- Laezza C, Caruso MG, Gentile T, Notarnicola M, Malfitano AM, Di Matola T, Messa C, Gazzerro P, Bifulco M. 2009. N6-isopentenyladenosine inhibits cell proliferation and induces apoptosis in a human colon cancer cell line DLD1. Int J Cancer 124: 1322–1329. 10.1002/ijc.24056 [DOI] [PubMed] [Google Scholar]
- Lamichhane TN, Blewett NH, Maraia RJ. 2011. Plasticity and diversity of tRNA anticodon determinants of substrate recognition by eukaryotic A37 isopentenyltransferases. RNA 17: 1846–1857. 10.1261/rna.2628611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane TN, Mattijssen S, Maraia RJ. 2013. Human cells have a limited set of tRNA anticodon loop substrates of the tRNA isopentenyltransferase TRIT1 tumor suppressor. Mol Cell Biol 33: 4900–4908. 10.1128/MCB.01041-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longley DB, Harkin DP, Johnston PG. 2003. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 3: 330–338. 10.1038/nrc1074 [DOI] [PubMed] [Google Scholar]
- Lu ZH, Zhang R, Diasio RB. 1992. Purification and characterization of dihydropyrimidine dehydrogenase from human liver. J Biol Chem 267: 17102–17109. 10.1016/S0021-9258(18)41899-6 [DOI] [PubMed] [Google Scholar]
- Mandel LR, Srinivasan PR, Borek E. 1966. Origin of urinary methylated purines. Nature 209: 586–588. 10.1038/209586a0 [DOI] [PubMed] [Google Scholar]
- Nagata M, Nakayama H, Tanaka T, Yoshida R, Yoshitake Y, Fukuma D, Kawahara K, Nakagawa Y, Ota K, Hiraki A, et al. 2011. Overexpression of cIAP2 contributes to 5-FU resistance and a poor prognosis in oral squamous cell carcinoma. Br J Cancer 105: 1322–1330. 10.1038/bjc.2011.387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayoshi Y, Chujo T, Hirata S, Nakatsuka H, Chen CW, Takakura M, Miyauchi K, Ikeuchi Y, Carlyle BC, Kitchen RR, et al. 2021. Loss of Ftsj1 perturbs codon-specific translation in the brain and is associated with X-linked intellectual disability. Sci Adv 7: eabf3072. 10.1126/sciadv.abf3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa A, Nagiri C, Shihoya W, Inoue A, Kawakami K, Hiratsuka S, Aoki J, Ito Y, Suzuki T, Suzuki T, et al. 2021. N6-methyladenosine (m6A) is an endogenous A3 adenosine receptor ligand. Mol Cell 81: 659–674. 10.1016/j.molcel.2020.12.038 [DOI] [PubMed] [Google Scholar]
- Pane F, Oriani G, Kuo KC, Gehrke CW, Salvatore F, Sacchetti L. 1992. Reference intervals for eight modified nucleosides in serum in a healthy population from Italy and the United States. Clin Chem 38: 671–677. 10.1093/clinchem/38.5.671 [DOI] [PubMed] [Google Scholar]
- Salonga D, Danenberg D, Johnson M, Metzger R, Groshen S, Tsao-Wei D, Lenz H, Leichman C, Leichman L, Diasio R, et al. 2000. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res 6: 1322–1327. [PubMed] [Google Scholar]
- Santi DV, McHenry CS, Sommer H. 1974. Mechanism of interaction of thymidylate synthetase with 5-fluorodeoxyuridylate. Biochemistry 13: 471–481. 10.1021/bi00700a012 [DOI] [PubMed] [Google Scholar]
- Scholler E, Marks J, Marchand V, Bruckmann A, Powell CA, Reichold M, Mutti CD, Dettmer K, Feederle R, Huttelmaier S, et al. 2021. Balancing of mitochondrial translation through METTL8-mediated m3C modification of mitochondrial tRNAs. Mol Cell 81: 4810–4825.e4812. 10.1016/j.molcel.2021.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer U, Bohleber S, Fradejas-Villar N. 2017. The modified base isopentenyladenosine and its derivatives in tRNA. RNA Biol 14: 1197–1208. 10.1080/15476286.2017.1294309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem S, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343: 84–87. 10.1126/science.1247005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Fukuda H, Chujo T, Kouwaki T, Oshiumi H, Tomizawa K, Wei FY. 2021. Export of RNA-derived modified nucleosides by equilibrative nucleoside transporters defines the magnitude of autophagy response and Zika virus replication. RNA Biol 18: 478–495. 10.1080/15476286.2021.1960689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spenkuch F, Motorin Y, Helm M. 2014. Pseudouridine: still mysterious, but never a fake (uridine)! RNA Biol 11: 1540–1554. 10.4161/15476286.2014.992278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinola M, Galvan A, Pignatiello C, Conti B, Pastorino U, Nicander B, Paroni R, Dragani TA. 2005. Identification and functional characterization of the candidate tumor suppressor gene TRIT1 in human lung cancer. Oncogene 24: 5502–5509. 10.1038/sj.onc.1208687 [DOI] [PubMed] [Google Scholar]
- Suzuki T. 2021. The expanding world of tRNA modifications and their disease relevance. Nat Rev Mol Cell Biol 22: 375–392. 10.1038/s41580-021-00342-0 [DOI] [PubMed] [Google Scholar]
- Takenouchi T, Wei FY, Suzuki H, Uehara T, Takahashi T, Okazaki Y, Kosaki K, Tomizawa K. 2019. Noninvasive diagnosis of TRIT1-related mitochondrial disorder by measuring i6A37 and ms2i6A37 modifications in tRNAs from blood and urine samples. Am J Med Genet A 179: 1609–1614. 10.1002/ajmg.a.61211 [DOI] [PubMed] [Google Scholar]
- Takesue Y, Wei FY, Fukuda H, Tanoue Y, Yamamoto T, Chujo T, Shinojima N, Yano S, Morioka M, Mukasa A, et al. 2019. Regulation of growth hormone biosynthesis by Cdk5 regulatory subunit associated protein 1-like 1 (CDKAL1) in pituitary adenomas. Endocr J 66: 807–816. 10.1507/endocrj.EJ18-0536 [DOI] [PubMed] [Google Scholar]
- Wei FY, Suzuki T, Watanabe S, Kimura S, Kaitsuka T, Fujimura A, Matsui H, Atta M, Michiue H, Fontecave M, et al. 2011. Deficit of tRNALys modification by Cdkal1 causes the development of type 2 diabetes in mice. J Clin Invest 121: 3598–3608. 10.1172/JCI58056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Fujimura A, Wei FY, Shinojima N, Kuroda JI, Mukasa A, Tomizawa K. 2019. 2-Methylthio conversion of N6-isopentenyladenosine in mitochondrial tRNAs by CDK5RAP1 promotes the maintenance of glioma-initiating cells. iScience 21: 42–56. 10.1016/j.isci.2019.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon SB, Park YH, Choi SA, Yang HJ, Jeong PS, Cha JJ, Lee S, Lee SH, Lee JH, Sim BW, et al. 2019. Real-time PCR quantification of spliced X-box binding protein 1 (XBP1) using a universal primer method. PLoS One 14: e0219978. 10.1371/journal.pone.0219978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Long T, Fang ZP, Zhou XL, Liu RJ, Wang ED. 2015. Identification of determinants for tRNA substrate recognition by Escherichia coli C/U34 2′-O-methyltransferase. RNA Biol 12: 900–911. 10.1080/15476286.2015.1050576 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.