

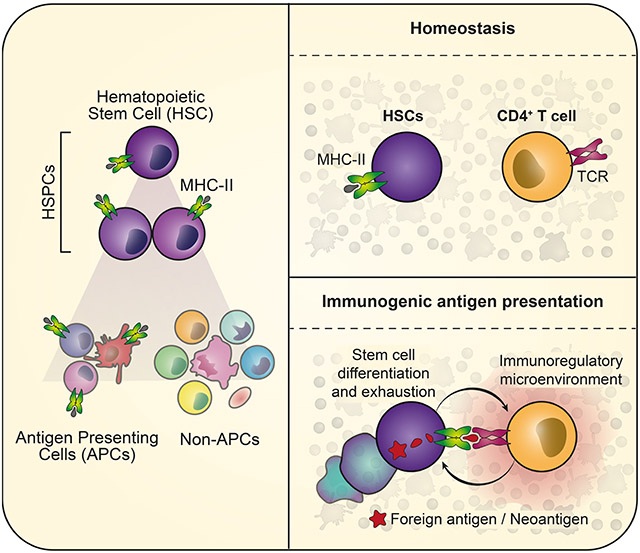
Summary
Hematopoietic stem and progenitor cells (HSPCs) are responsible for the production of blood and immune cells. Throughout life, HSPCs acquire oncogenic aberrations that can cause hematological cancers. While molecular programs maintaining stem cell integrity have been identified, safety mechanisms eliminating malignant HSPCs from the stem cell pool remain poorly characterized. Here we show that HSPCs constitutively present antigens via major histocompatibility complex class II. The presentation of immunogenic antigens, as occurring during malignant transformation, triggers bidirectional interactions between HSPCs and antigen-specific CD4+ T cells, causing stem cell proliferation, differentiation and specific exhaustion of aberrant HSPCs. This immunosurveillance mechanism effectively eliminates transformed HSPCs from the hematopoietic system, thereby preventing leukemia onset. Together, our data reveal a bidirectional interaction between HSPCs and CD4+ T cells, demonstrating that HSPCs are not only passive receivers of immunological signals, but actively engage in adaptive immune responses to safeguard the integrity of the stem cell pool.
Graphical Abstract
eTOC blurb:
Haas and colleagues show that hematopoietic stem cells (HSCs) act as antigen presenting cells. The presentation of immunogenic antigens triggers a bidirectional interaction with antigen-specific CD4+ T cells, resulting in the rapid exhaustion of diseased HSCs, thereby safeguarding the integrity of the stem cell pool.
Introduction
Hematopoietic stem and progenitor cells (HSPCs) are the ultimate source of blood and immune cells, including antigen presenting cells (APCs) and T cells (Doulatov et al., 2012; Eaves, 2015). In contrast to mature cell types, HSPCs are multipotent, long-lived and self-renew. The acquisition of genomic aberrations in HSPCs constitutes a major threat to the hematopoietic system since genomic errors are passed on to daughter stem cells and eventually to the entire hematopoietic system, where they are maintained throughout life. In the elderly, the establishment of such clonally expanded populations carrying pre-leukemic mutations is a frequent event and associated with a high risk of malignant transformation to hematological cancers (Genovese et al., 2014; Jaiswal et al., 2014). To protect stem cells from damage induced by replicative stress and reactive oxygen species, HSPCs are maintained in a long-term quiescent and low metabolic state (van Galen et al., 2014; Walter et al., 2015; Ho et al., 2017). While inflammatory signals released during infections activate HSPCs to propel blood production, excessive exposure to inflammation induces replicative stress causing DNA damage and stem cell exhaustion (Essers et al., 2009; Sato et al., 2009; Walter et al., 2015; Zhang et al., 2016; Takizawa et al., 2017). CD4+ regulatory T cells (Tregs) have been suggested to establish an immune privileged niche in the bone marrow maintaining stem cell quiescence, presumably by protecting stem cells from replicative stress induced by inflammatory insults (Fujisaki et al., 2011; Hirata et al., 2018). While several mechansisms have been described how stem cells are passively protected by their microenvironment to prevent the acquisition of damage, active safety mechanisms that specifically eliminate malignant HSPCs from the system remain unknown.
Professional APCs, such as B cells or mature dendritic cells (DCs), induce adaptive immune responses by presenting antigens via the major histocompatibility complex class II (MHC-II) to the T cell receptor of CD4+ T cells (Neefjes et al., 2011; Roche and Furuta, 2015). Microenvironmental factors, the maturation state of APCs and the expression of co-stimulatory molecules on APCs have been implicated in balancing immunogenic versus tolerogenic T cell responses (Wakkach et al., 2003; Goodnow et al., 2005; Jurewicz and Stern, 2019). Professional APCs constitutively express high levels of MHC-II (Steinman, 2007; Merad et al., 2013; Roche and Furuta, 2015), whereas immature or non-professional APCs acquire antigen presentation activity only upon exposure to inflammatory signals associated with MHC-II up-regulation (Kambayashi and Laufer, 2014; Jakubzick, Randolph and Henson, 2017). The majority of other cell types are typically devoid of MHC-II expression and are not capable of priming CD4+ T cells. Despite several studies reporting that MHC-II might be expressed on immature cells of the hematopoietic system (Russell and Engh, 1979; Fitchen, Foon and Cline, 1981; Sieff et al., 1982; Szer et al., 1985), HSPCs have not been considered capable of actively interacting with the adaptive immune system. Moreover, a systematic understanding of the MHC-II expression patterns is lacking and the functionality as well as the role of MHC-II-mediated antigen presentation in HSPCs during health and disease remains unexplored.
Here, we demonstrate that HSPCs constitutively present antigens via MHC-II. Upon presentation of immunogenic antigens, HSPCs directly interact with antigen-specific CD4+ T cells, driving HSPC differentiation and extinction from the system. On the other hand, CD4+ T cells are activated and subsequently adopt an immunoregulatory state preventing harmful pro-inflammatory bone marrow responses. This immunosurveillance mechanism effectively suppresses leukemia onset upon malignant transformation of HSPCs.
Results
Mouse HSPCs express the MHC-II antigen presenting machinery
To systematically explore the expression patterns of the MHC-II antigen presentation machinery in the hematopoietic system, we performed a series of analyses. First, global transcriptome datasets of mouse multipotent HSPCs (Lin−Sca1+cKit+ cells, LSKs) revealed high expression of genes encoding MHC-II molecules (H2-Aa, H2-Ab1, H2-Eb1), the related antigen loading machinery (H2-Dma, H2-Dmb2, H2-Oa, H2-Ob, Cd74) and Ciita, the master regulator of MHC-II gene expression (Steimle et al., 1993, 1994) (Figure 1A). Targeted transcriptional profiling confirmed that MHC-II genes were highly expressed in mouse hematopoietic stem cells (HSCs) and multipotent progenitors (MPPs) 1-4 (Cabezas-Wallscheid et al., 2014), together comprising the LSK compartment (Figure 1B). These genes were gradually downregulated upon transition to committed progenitors that comprise the Lineage−Sca-1−cKit+ (LS−K) compartment. To analyze MHC-II protein expression, we measured MHC-II surface expression levels across all major cell populations present in the mouse bone marrow and spleen by flow cytometry (Figures 1C, 1D, S1A and S1B). As expected, professional APCs expressed consistently high levels of MHC-II, non-professional APCs expressed MHC-II at heterogeneous levels, and non-APCs did not express MHC-II. Importantly, HSCs and MPPs showed prominent surface expression levels of MHC-II, which were gradually downregulated upon transition to committed progenitors of the LS−K compartment, in line with our transcriptomic data. Notably, homeostatic levels of MHC-II molecules in HSCs and MPPs were only slightly lower as compared to professional APCs, but significantly higher when compared to any other population examined, including macrophages (Figures 1C, 1D, S1A and S1B). Transcript and protein levels of MHC-II genes were efficiently up-regulated in HSCs in vivo upon administration of bacterial lipopolysaccharide (LPS), recombinant type-I interferon, the viral mimetic polyinosinic:polycytidylic acid (pI:C) or following viral infection with MCMV (Figures 1C, 1D and S1A-S1C). Stimulation by LPS or pI:C treatment of mice enhanced expression of MHC-II surface levels in HSCs comparable to those observed in professional APCs, but had only an negligible impact on non-APCs.
Figure 1. Mouse HSPCs express the MHC-II antigen presentation machinery.

See also Figure S1.
(A) z-scores of MHC-II antigen presentation machinery genes in mouse HSCs and MPPs (LSK) and progenitors (LS−K) derived from RNA-Seq data (Klimmeck et al., 2014), n=3.
(B) Relative gene expression of MHC-II genes across bone marrow (BM) populations measured by qPCR, n=2-3.
(C) Heatmap summarizing MHC-II surface measurements for BM and spleen (Sp) populations by flow cytometry at homeostasis or 24h post pI:C or LPS treatment.
(D) Representative histograms (left) and quantification (right) of MHC-II surface expression as in (C), n=4-5.
(E and F) Transplantation experiments of MHC-II+/− BM populations, n=4-6.
(E) Peripheral blood (PB) chimerism, n=4-6.
(F) BM chimerism at the endpoint of primary (left) and secondary (right) transplantation, n=4-6.
Individual values are shown in A and C, means and SEM are depicted otherwise. No significance = ns, P<0.05 *, P<0.01 **, P<0.001 ***, P<0.0001 ****. Two-way ANOVA was performed in D as discovery test, followed by a paired two-tailed t-test. If not stated otherwise, unpaired two-tailed t-tests were performed as post-hoc tests. Two-way ANOVA was performed in E.
To unambiguously determine whether MHC-II expression marks HSCs with long-term self-renewal capacity, we separated lineage-depleted bone marrow solely based on MHC-II surface expression, followed by transplantation into lethally irradiated mice (Figures 1E, 1F, S1D and S1E). While MHC-II-negative bone marrow cells were not capable of repopulating all hematopoietic lineages efficiently, MHC-II-positive bone marrow cells reconstituted hematopoiesis long-term, demonstrating that MHC-II surface expression is an explicit feature of self-renewal capacity and therefore marks all functional HSCs.
Mouse HSPCs present antigens via MHC-II
To determine whether mouse HSPCs are capable of presenting antigens via MHC-II, we made use of the Y-Ae antibody that recognizes the MHC-II-derived Eα peptide52-68 when presented in the context of MHC-II I-Ab haplotype (Murphy et al., 1989; Rudensky et al., 1991). Accordingly, in C57BL/6 mice, that display the I-Ab haplotype but lack expression of Eα, exogenous Eα peptide can be used as foreign antigen to characterize antigen presentation capacities of cell populations ex vivo. While professional APCs efficiently presented the Eα peptide via MHC-II and non-APCs failed to do so, HSPCs presented MHC-II-restricted peptides efficiently, suggesting that HSPCs can present exogenous peptides ex vivo (Figure 2A). In support of this, HSPCs efficiently incorporated and processed exogenously administered BODIPY-conjugated DQ-ovalbumin (DQ-OVA), a self-quenched conjugate that exhibits fluorescence upon cleavage, ex vivo and in vivo (Figure S1F-S1H).
Figure 2. Mouse HSPCs present self-antigens via MHC-II.

See also Figure S1.
(A) Ex vivo antigen presentation assay. Representative histograms (left) and quantification (right), n=4.
(B and C) In vivo antigen presentation assay, n=6. (B) Heatmap summarizing the percentage of Eα presenting cells in C57BL/6xBALB/c mice and control C57BL/6 mice. (C) Quantification of selected populations in C57BL/6xBALB/c.
(D-F) Mass spectrometry analyses of peptides recovered from MHC-II of indicated populations. (D) MHC-II-eluted peptide size distribution. (E) Gene set enrichment analysis (GSEA) of presented peptides related to their gene expression in HSPCs. (F) MHC-II-derived peptides in HSPCs that are transcribed (endogenous) or not transcribed (exogenous) within HSPCs based on a threshold of 100 RPKM.
Individual values are shown in B and D, means and SEM are depicted otherwise. No significance = ns, P<0.05 *, P<0.01 **, P<0.001 ***, P<0.0001 ****. One- (A) or two-way ANOVA (C) were performed as discovery tests. Paired two-tailed t-test was performed in C. If not stated otherwise, unpaired two-tailed t-tests were performed as post-hoc tests.
To investigate whether HSPCs of naïve mice present self-antigens via MHC-II in vivo, we crossed BALB/C mice, which express Eα but exhibit the I-Ad haplotype, to C57BL/6 mice (I-Ab, Eα-negative). In mice of the F1 generation, MHC-II mediated self-antigen presentation can be assessed by the Y-Ae antibody, due to the expression of Eα in the presence of MHC-II molecules with I-Ab haplotype (Henri et al., 2010) (Figure S1I). In line with previous reports, professional APCs displayed efficient MHC-II mediated presentation of Eα during homeostasis and upon LPS treatment in vivo, macrophages did not present Eα at homeostasis, but acquired strong antigen presentation capacity upon LPS treatment, and non-APCs showed no or highly restricted antigen presenting activity (Kambayashi and Laufer, 2014; Jakubzick, Randolph and Henson, 2017) (Figure 2B, 2C and S1J). Importantly, HSCs and MPPs exhibited significant antigen presentation of Eα at homeostasis, and efficiently increased antigen presenting capacity upon LPS treatment in an MHC-II restricted manner, suggesting that HSPCs constantly present self-peptides via MHC-II in naïve mice (Figure 2B, 2C and S1J).
To identify antigens presented by HSPCs, we performed immunoprecipitation of MHC-II molecules from HSPCs of naïve mice, followed by peptide elution and mass spectrometry (Figure 2D and 2E). We also included T cells and splenocytes, serving as negative and positive control of APCs, respectively. MHC-II eluted peptides from HSPCs resembled those from splenocytes in number and length distribution, and considerably outnumbered peptides eluted from non-APCs (Figure 2D). The evaluation of detected peptides confirmed that predominantly self-peptides are presented by HSPCs in naïve mice (Figure 2E and F, Supplementary Table 1). Together, these data demonstrate that HSPCs constitutively present self-antigens via MHC-II at homeostasis and further increase antigen presentation upon inflammation.
Antigen presenting HSPCs engage in bidirectional interactions with antigen-specific CD4+ T cells
The main feature of APCs is the antigen-specific activation of CD4+ T cells. To study whether HSPCs can interact with CD4+ T cells in an antigen-specific manner, we made use of OT-II and 2D2 mice that express transgenic T cell receptors specifically recognizing the chicken ovalbumin (OVA323-339) or myelin oligodendrocyte glycoprotein (MOG35-55) peptides, respectively, when presented by MHC-II (Barnden et al., 1998; Bettelli et al., 2003). In co-cultures with multipotent HSPCs (LSKs), naïve antigen-specific CD4+ T cells were efficiently activated and proliferated specifically in the presence of the respective peptides (Figures 3A, 3B, S2A-S2D). Notably, all populations of the LSK compartment, including HSCs and MPPs1-4 induced antigen-specific CD4+ T cell responses (Figure 3C). Since these populations also express similar levels of MHC-II and exhibit comparable presentation of endogenous antigens in vivo (see above), we used LSK cells in the majority of functional experiments that characterize antigen presentation of mouse multipotent HSPCs. Importantly, blocking MHC-II abrogated HSPC-mediated activation of CD4+ T cells, demonstrating that antigen-specific CD4+ T cell activation is MHC-II dependent (Figure S2E). While HSPCs efficiently activated CD4+ and CD8+ T cells in the presence of processed peptides, they were also able to present antigens derived from OVA protein, albeit to a lesser extent if compared to DCs (Figure S2F and S2G). However, LPS inflammatory signals significantly enhanced the HSPC-mediated antigen-specific activation of T cells.
Figure 3. MHC-II mediates an antigen-specific bidirectional interaction between HSPCs and CD4+ T cells.

See also Figure S2.
(A and B) HSPCs activate naïve OT-II CD4+ T cells in co-culture assays, n=8. (A) Representative histograms of CD44 expression and cell trace violet (CTV). (B) Quantification of T cell activation.
(C) T cell activation assays for different HSPC subpopulations (2.5x103 cells) as in 3A, n=4.
(D) In vivo antigen presentation assay for exogenous antigens. Experimental approach (left), quantification of T cell activation (right), n=8.
(E) In vivo antigen presentation assay for endogenous antigens. Experimental approach (left), quantification of T cell activation (right), n=4.
(F) Antigen presentation impacts on HSPC proliferation in co-culture assays with naïve OT-II CD4+ T cells (see methods). Representative plots (left) and quantification (right), n=4.
(G and H) In vivo antigen-specific HSPC-T cell interaction promotes HSPC cell cycle entry in a Scl-CreERT2 H2-Ab floxed YFP-stop floxed mouse model. (G) Experimental scheme (left), and cell cycle analyses (right). (H) Representative plots (left) and cell cycle analysis (right) of YFP+MHC-II− or YFP−MHC-II+ HSPCs from Cre+ mice, n=5.
Means and SEM are depicted. No significance = ns, P<0.05 *, P<0.01 **, P<0.001 ***, P<0.0001 ****. One- (B, C) or two-way ANOVA (D, E, G and H) were performed as discovery tests. Paired two-tailed t-test was performed in G. If not stated otherwise, unpaired two-tailed t-tests were performed as post-hoc tests.
To determine whether HSPCs are capable of incorporating, processing and presenting exogenous antigens in vivo, we administered OVA protein to mice. Indeed, HSPCs isolated from OVA-injected mice were able to activate antigen-specific OT-II CD4+ T cells ex vivo (Figure 3D), indicating their capability to process and present exogenous antigens in vivo. To confirm whether HSPCs were also able to present endogenous antigens, we co-cultured HSPCs from wild type or ovalbumin-expressing mice (CAG-OVA) with OT-II T cells. Indeed, OT-II T cells were specifically activated in the presence of HSPCs expressing OVA endogenously, albeit to a lesser extent if compared to mature DCs (Figure 3E). Together these experiments suggest that HSPCs are capable of activating CD4+ T cells upon presentation of both, endogenous and exogenous antigens via MHC-II.
Next, we investigated the impact of MHC-II mediated antigen presentation on HSPCs. In co-cultures, antigen-specific interactions with naïve CD4+ T cells resulted in substantial proliferation of HSPCs (Figure 3F and S2H). Moreover, transwell assays demonstrated that direct contact between HSPCs and CD4+ T cell cells is required for full cell cycle activation of HSPCs ex vivo (Figure S2I and S2J). To evaluate the mechanistic role of MHC-II in HSPC-T cell interactions in vivo, we generated mice carrying a tamoxifen-inducible recombinase CreERT2 under the control of the HSPC-specific SCL promoter, a loxP-flanked MHC-II allele (H2-Ab) and a loxP-flanked STOP sequence followed by the Enhanced Yellow Fluorescent Protein (YFP) (Figure 3G, see Methods). This enabled an efficient conditional deletion of MHC-II in HSPCs and their progeny (Figures S2K and S2L). Co-transfer of OVA-specific OT-II cells into tamoxifen-treated mice, followed by OVA immunization resulted in specific cell cycle induction of HSPCs that maintained physiological MHC-II levels, while MHC-II deficient HSPCs from the same mice did not respond to OVA treatment (Figures 3G and 3H). Together, these observations demonstrate that transient presentation of immunogenic antigens via MHC-II by HSPCs mediates bidirectional interactions with antigen-specific CD4+ T cells, resulting in simultaneous activation of stem and T cells.
Sustained antigen presentation drives differentiation and elimination of HSPCs from the stem cell pool
To investigate the physiological relevance of our findings, we modelled the long-term consequences of sustained presentation of immunogenic antigens by HSPCs as occurring during chronic infections or malignant transformation. For this purpose, we generated mice with chimeric hematopoietic systems by co-transplantation of equal numbers of wild type HSPCs and CAG-OVA HSPCs, constitutively presenting OVA, into lethally irradiated congenic mice (Figure 4A). In the absence of antigen-specific CD4+ T cells, this resulted in a stable 50:50 chimerism of the two hematopoietic systems throughout primary and secondary transplantation, suggesting that the presentation of antigens in the absence of antigen-specific CD4+ T cells does not affect hematopoiesis (Figure 4B). In contrast, upon co-transfer of OVA-specific OT-II CD4+ T cells at the beginning of the primary transplantation (d0), OVA-expressing HSPCs were immediately removed from the system, resulting in a complete and specific engraftment failure of stem cells presenting the T cell-recognized antigen (Figure 4B and 4C). If OT-II cells were co-transferred after stable engraftment of the two hematopoietic systems (d60 post transplantation), the chimerism was kept stable initially, but started dropping upon secondary transplantation. Importantly, also in this setting, antigen presenting HSPCs were efficiently decreased and eliminated after primary and secondary transplantation, respectively.
Figure 4. Sustained presentation of immunogenic antigens drives differentiation and exhaustion of HSPCs.

See also Figure S2.
(A-E) Sustained in vivo interactions of antigen presenting HSPCs and antigen-specific CD4+ T cells trigger HSPC differentiation and exhaustion.
(A) Experimental scheme: Co-transplantations of CAG-OVA and wt HSPCs with or without OT-II CD4+ T cells.
(B) Percentage of CAG-OVA progeny in the blood of recipient mice, n=4-6.
(C) BM chimerism at the endpoints of primary (left) and secondary (right) transplantations, n=4-6.
(D) Percentage of OT-II T cells of total CD4+ T cells in recipient mice (week 20), n=6.
(E) Lineage-output upon HSPC-T cell interactions in vivo. Percentage of CAG-OVA HSPC derived progeny 20 weeks after transplantation, n=4.
(F-I) Impact of antigen presentation on HSPC differentiation.
(F) Experimental scheme. Co-cultures between HSPCs and OT-II T cells were analyzed by flow cytometry (G) or transplanted into lethally irradiated mice (H and I).
(G) Indicated populations derived from transplanted HSPCs were quantified, n=4.
(H) PB engraftment, n=6.
(I) BM engraftment at week 16, n=6.
(J and K) In vivo impact of antigen presentation on HSPCs.
(J) Experimental scheme. OVA loaded HSPCs were co-transferred with naïve OT-II CD4+ T cells.
(K) Indicated populations were quantified 3 days post transfer.
Means and SEM are depicted. No significance = ns, P<0.05 *, P<0.01 **, P<0.001 ***, P<0.0001 ****. One-way (C, D, E) and two-way (B, H) ANOVA was performed. If not stated otherwise, unpaired two-tailed t-tests were performed as post-hoc tests.
While antigen-specific CD4+ T cells strongly expanded and accumulated in the bone marrow during stem cell exhaustion, antigen-specific CD8+ T cells were not detected (Figures 4D, S2M and S2N), suggesting that the elimination of antigen presenting HSPCs was mediated by direct CD4+ T cell interactions and not by secondary activation of cytotoxic CD8+ T cells. Of note, loss of OVA-presenting HSPCs was associated with an increased myeloid-biased differentiation (Figure 4E). To determine whether differentiation is the main cause of elimination of antigen presenting stem cells, we first investigated the impact of antigen presentation on HSPCs differentiation. In co-cultures, antigen-specific interactions with CD4+ T cells induced rapid differentiation of HSPCs into the myeloid lineage, associated with loss of in vivo stem cell potential as measured by bone marrow transplantations (Figures 4F-4I and S2O). Gene expression analyses confirmed the upregulation of differentiation programs in HSPCs and their progeny (Figure S2P). In line with this, co-transfer of OVA-loaded HSPCs with OVA-specific CD4+ T cells into mice resulted in rapid differentiation of HSPCs in vivo (Figure 4J and 4K). Finally, transwell assays demonstrated that direct HSPC-CD4+ T cell interactions are required to effectively drive HSPC differentiation, while additional indirect effects mediated by the establishment of cytokine gradients are likely to contribute to the observed effect as well (Figure S2Q). Together, these data suggest that direct interactions with antigen-specific CD4+ T cells drive differentiation and exhaustion of HSPCs that present the cognate immunogenic antigens via MHC-II, thereby irreversibly removing them from the system, while leaving unrecognized self-antigen presenting HSPCs unaffected.
Antigen-specific HSPC-CD4+ T cell interactions promote an immune-regulatory state
Inflammatory signals, such as those released during pro-inflammatory T cell responses, induce systemic HSPC proliferation (Essers et al., 2009; Baldridge et al., 2010; Walter et al., 2015). However, antigen presentation by HSPCs resulted in the specific activation and exhaustion of stem cells that actively present immunogenic antigens, while leaving self-antigen presenting HSPCs unaffected (see above), suggesting that HSPC-mediated T cell activation occurs in the absence of global pro-inflammatory bone marrow responses. Since naïve CD4+ T cells can be polarized into pro-inflammatory or immunosuppressive T helper subsets depending on the properties of the APC and environmental factors (Zhu and Paul, 2010), we investigated the exact nature of HSPC-induced T cell polarization.
First, we characterized the APC properties of HSPCs. Gene expression analyses of HSPCs revealed low to intermediate expression of classical co-stimulatory molecules, but high surface presentation of the co-inhibitory molecule PD-L1 (Figure S3A and S3B). Moreover, the most highly expressed cytokine genes in HSPCs are Ebi3 and Il12a (Figure S3C), forming together the suppressive cytokine IL-35 (Collison et al., 2007, 2010). Upon engagement with antigen-specific CD4+ T cells, HSPCs further upregulated PD-L1, acquired features of myeloid-derived suppressor cells and expressed high levels of the immunoregulatory cytokines IL-10 and IL-27 (Figure S3D-S3H). Since high expression of immunoregulatory cytokines and co-inhibitory receptors by APCs are associated with the promotion of anti-inflammatory or regulatory T cell responses (Ness, Lin and Gordon, 2021), we investigated whether antigen presentation by HSPCs might polarize CD4+ T cells to an immunoregulatory state. Indeed, in contrast to CD4+ T cells activated by other APCs, CD4+ T cells activated by HSPCs acquired a unique state, characterized by high surface expression of co-inhibitory molecules, such as PD-L1 (Figures S4A and S4B). This was also the case for CD4+ T cells activated by highly purified HSCs and MPP populations (Figure S4C). Global transcriptomic comparisons of CD4+ T cells activated by HSPCs, in the following termed THSCs, with CD4+ T cells activated by conventional DCs (TDCs) confirmed that they acquired fundamentally distinct transcriptomic states, with THSCs adopting an immunoregulatory and anti-inflammatory phenotype (Figure 5A and 5B). Of note, the expression of the signature transcription factor of regulatory T cells (Tregs), Foxp3, remained absent upon HSPC-mediated T cell activation (Figure S4D). In contrast, an upregulation of the transcription factors c-Maf and Prdm1 was observed, which act as master regulators of type 1 regulatory T (Tr1) cell differentiation and mediate the transcriptional induction of co-inhibitory gene modules in T cells (Chihara et al., 2018; Zhang et al., 2020) (Figures 5B and S4D). In line with this, THSCs up-regulated robust and sustained expression of the immune suppressive cytokine IL-10 and a co-inhibitory gene module comprising the co-inhibitory molecules PD-1 (Pdcd1), PD-L1 (Cd274), LAG3 (Lag3) and TIM3 (Havcr2) on the mRNA and protein level (Figures 5C, S4D-S4F). The expression of co-inhibitory molecules and signature Tr1 transcription factors in THSCs remained elevated upon resting, antigen-dependent or -independent re-stimulation and exposure to inflammatory molecules, suggesting that the regulatory phenotype is not due to a transient activation state, but rather reflects a stable state linked to differentiation (Figures S4G-S4J).
Figure 5. HSPC-mediated antigen presentation induces a suppressive phenotype in CD4+ T cells.

(A) Principle component analyses (PCA) of Nanostring (left) and RNA-Seq (right) gene expression of OT-II CD4+ T cells activated by HSPCs (THSCs) or dendritic cells (TDCs), n=3-4.
(B) Top THSC-enriched gene sets of gene set enrichment analyses (GSEA) of RNA-Seq data from (A), comparing THSCs and TDCs.
(C) Heatmap representing z-scored expressions of co-inhibitory module genes (Chihara et al., 2018).
(D-F and K) Ex vivo CD4+ T cell suppression assays. (D) Representative plots for the 1:2 suppressive/bystander naïve CD4+ T cell condition. (E) Suppression index for different bystander/suppressive ratios, n=4. (F) Proliferation index of responder CD4+ T cells for the 1:2 ratio, n=4.
(G) Ex vivo CD8+ T cell suppression assay, n=4.
(H and I) Ex vivo CD8+ T cell activation (H) and annexin V cytotoxicity assay (I), n=4. OVA1: ovalbumin 257-264, OVA2: ovalbumin 323-339.
(J) Ex vivo macrophage polarization assay, n=4.
(K) Role of IL-10 in THSC-mediated suppression. Activation of WT (left) or Il10rb−/− (right) bystander T cells in the presence of THSCs or TDCs in a 1:2 suppressive:bystander ratio., n=4.
(L) In vivo suppression assay. Representative plots (left) and quantification of bystander T cell proliferation (right), n=3.
Individual values are shown in A and C, means and SEM are depicted otherwise. No significance = ns, P<0.05 *, P<0.01 **, P<0.001 ***, P<0.0001 ****. One- (F, G, H, I, L) or two-way ANOVA (K) were performed as discovery test. Two-way ANOVA was performed in E and J. If not stated otherwise, unpaired two-tailed t-tests were performed as post-hoc tests.
To evaluate whether THSCs acquired a functionally suppressive phenotype ex vivo, we performed suppression assays using canonical Tregs as control (Figure 5D-5G). In contrast to TDCs, THSCs efficiently suppressed CD4+ and CD8+ T cell responses in an antigen-dependent and -independent manner (Figure 5D-5H). Moreover, THSCs reduced the cytotoxic activity of CD8+ T cells and supported macrophage polarization to an anti-inflammatory M2 state (Figure 5I and 5J). Mechanistically, both the capacity of THSCs to suppress bystander T cells and to polarize macrophages to an M2 state was, at least partly, driven by IL-10 (Figure 5J and 5K), which is upregulated both in HSPCs and CD4+ T cells upon bidirectional interactions (see above). Adoptive transfers of THSCs into mice effectively suppressed the response of naïve OT-II T cells to OVA immunizations, demonstrating the in vivo capacity of THSCs to dampen the immune system (Figure 5L).
In line with our ex vivo results, upon sustained interactions with CAG-OVA HSPCs in vivo, antigen-specific CD4+ T cells acquired a PD-L1 high phenotype (Figure S4K). Both antigen-specific CD4+ T cells and the CAG-OVA HSPC-derived bone marrow cells of mice in which HSPC-mediated antigen presentation occurred, adopted a functionally suppressive phenotype, confirming that sustained antigen presentation by HSPCs causes an overall immunoregulatory bone marrow response in vivo (Figures S4L and S4M).
Together, these findings demonstrate that antigen presentation by HSPCs to CD4+ T cells triggers an immunoregulatory T cell state, causing HSPC and T cell activation while promoting an immune-regulatory environment to avoid harmful pro-inflammatory responses in the bone marrow.
Human HSPCs are antigen presenting cells
In order to investigate whether our findings in the mouse system can be translated to humans, we first analyzed bulk and single-cell transcriptome datasets of human HSPCs (Novershtern et al., 2011; Hay et al., 2018; Pellin et al., 2019). These analyses revealed high expression of genes encoding MHC-II (e.g. HLA-DRA, HLA-DRB) and the machinery related to antigen presentation via MHC-II (e.g. HLA-DMA, HLA-DMB, CD74) in HSCs and MPPs (Figure 6A, S5A and S5B). While the expression of MHC-II and its antigen presenting machinery was maintained throughout commitment of HSCs to lineages with APC function (DC, B cell and monocyte/macrophage lineages), it was gradually downregulated upon commitment to all other lineages (neutrophil, eosinophil/basophil/mast cell, erythroid, megakaryocytic lineages). We next performed a flow cytometric characterization of the cell surface expression of the MHC-II molecule HLA-DR across major hematopoietic compartments of the bone marrow from healthy donors. The results accurately recapitulated our findings from the mouse system, with no expression of HLA-DR in non-APCs, high expression in professional APCs, and robust, albeit slightly lower expression in HSPCs and early progenitors of the CD34+ compartment (Figures 6B and 6C and S5C). To confirm that MHC-II marks human HSCs with long-term self-renewal capacity, we transplanted human bone marrow, separated based solely on HLA-DR expression, into sublethally irradiated immunodeficient mice (Figure 6D). 20 weeks post-transplant, HLA-DR-positive bone marrow cells gave rise to significantly higher levels of human multilineage engraftment compared to HLA-DR-negative bone marrow, suggesting that functional HSC activity is associated with MHC-II expression (Figure 6E).
Figure 6. Human HSPCs act as antigen presenting cells.

See also Figure S5.
(A) SPRING plots of human HSPC differentiation trajectories from scRNA-RNAseq data (Pellin et al., 2019). Lineage annotation (left) and MHC-II gene expression (right).
(B) Representative plots of HLA-DR expression in human BM aspirates, n=6.
(C) Quantification of HLA-DR+ expression from 6B.
(D) Experimental scheme for xenotransplantations.
(E) Quantification of human CD45+ cells in the BM (left) and multilineage engraftment (right) in xenotransplantations. Donut plots depict myeloid, lymphoid and HSPC percentages for every donor, n=3.
(F-H) Human CD4+ T cell activation assays using CytoStim (CS, F and G) or an MHC-II-restricted peptide pool (PP, H). Representative plots (F) and quantification of T cell activation (G and H), n=3-4.
(I) qPCR analyses of CD4+ T cells activated by HSPCs (THSCs) or dendritic cells (DCs) (TDCs) in the presence of CytoStim as in 6F, n=4.
(J and K) Surface protein expression in THSCs and TDCs, n=4.
Means and SEM are depicted in all bar-plots. No significance = ns, P<0.05 *, P<0.01 **, P<0.001 ***, P<0.0001 ****. One-way ANOVA was performed in C, G, H, I and J as discovery test. Unpaired two-tailed t-tests were performed as post-hoc tests.
To investigate whether human HSPCs are capable of presenting antigens via MHC-II, we made use of CytoStim, an antibody-based reagent that cross-links MHC-II of APCs with the T cell receptor of CD4+ T cells, resulting in T cell activation. As expected, addition of CytoStim resulted in efficient activation of CD4+ T cells in co-cultures with professional APCs, but had little or no effect in co-cultures with non-APCs (Figure 6F and 6G). In contrast, addition of CytoStim to co-cultures of CD34+ HSPCs and CD4+ T cells resulted in efficient T cell activation, which was comparable to DC-mediated T cell activation. To determine whether human HSPCs can activate CD4+ T cells in an antigen-dependent manner, we made use of a pool of peptides frequently recognized by a small subset of antigen-experienced CD4+ T cells (Figure 6H). Notably, the CD34+ cells that were used for functional assays have been purely sorted, displaying an neglectable amount of immature DCs and B cells (Figure S5D).According to our previous observations, APCs and HSPCs comparably activated CD4+ T cells reactive to the peptides. Of note, similar to their mouse counterparts, human CD4+ T cells activated by human CD34+ HSPCs acquired an immunoregulatory phenotype associated with upregulation of co-inhibitory molecules, such as LAG3, PD-L1 and TIM3, as well as increased expression of the IL10 gene and Tr1 associated transcription factors, suggesting a conserved mechanism from mouse to human (Figure 6I-6K). Collectively, these data suggest that human HSPCs, similar to their mouse counterparts, act as antigen presenting cells capable of interacting with CD4+ T cells via MHC-II.
MHC-II mediated antigen presentation is associated with a stem-like state in AML
Acute myeloid leukemia (AML) is an aggressive hematological cancer characterized by the accumulation of immature blasts that originate from HSCs or myeloid progenitors. MHC-II has been described to be expressed in a heterogeneous manner in AML (Miale et al., 1982; Newman et al., 1983), and its deregulation has been linked to relapse after allogeneic stem cell transplantation (Christopher et al., 2018; Toffalori et al., 2019). However, neither a rationale for MHC-II expression heterogeneity, nor a link to APC capacity and clinical or biological features of AMLs have been established (Miale et al., 1982; Mutis et al., 1997, 1998; Costello et al., 1999; Berlin et al., 2015). In line with our previous findings in the healthy hematopoietic system, transcriptomic analyses of 523 leukemia samples of AML patients revealed that high expression of the MHC-II antigen presentation machinery is associated with a transcriptomic state of stemness (Pölönen et al., 2019) (Figure 7A). In accordance with this, flow cytometric analyses of 63 AML patients confirmed that high HLA-DR (MHC-II) surface expression identifies patients with stem-like or monocyte-like AMLs and marks immature stem-like populations within the leukemic blast hierarchy of individual patients (Figure 7B, 7C and S5E-S5G). To determine whether a stem-like state in human AML is indeed associated with functional APC capacities, we screened 23 human AML cell lines and categorized them based on their immunophenotype into stem- or mature-like (Figure S5H and S5I). In line with our observations in primary AMLs, stem-like AML cell lines expressed higher HLA-DR levels, displayed higher CD4+ T cell activation and immunosuppressive polarization capacities, and underwent myeloid differentiation upon antigen presentation, suggesting that the above-described bidirectional interaction might also be operational in stem-like AML cells (Figure 7D-F and S5I).
Figure 7. MHC-II-mediated neoantigen presentation of HSPCs protects from leukemia onset.

See also Figure S5.
(A) Stemness correlates with MHC-II expression in AML. Sum of scaled HLA-DR (MHC-II) gene expression and stem cell scores for AMLs from (Pölönen et al., 2019), n=523.
(B and C) AML patient samples were analyzed by flow cytometry and stratified into indicated groups.
(B) HLA-DR surface expression in the different AML groups, n=63.
(C) HLA-DR geometric mean fluorescence intensity within AML blasts positive or negative for representative stem or mature markers (left), n=63. Representative flow cytometry histograms (right).
(D) T cell activation capability in AML cell lines stratified as stem-like or mature-like, n=23.
(E) Immunosuppressive score in activated CD4+ T cells in the presence of the AML cell lines from 7D based on the z-scored expression of LAG3, PD-L1 and TIM3, n=23.
(F) CD11b (left) and CD64 (right) expression in AML cell lines after co-culture with CD4+ T cell in the presence or absence of CS, n=23.
(G) Sum of scaled MHC-II related genes (left) or stem cell scores (Ng et al., 2016) (right) in AML patients segregated based on NPM1 and FLT3 mutational state (Kohlmann et al., 2010), n=78.
(H) Antigen presentation assays of HSC- and GMP-derived MLL-AF9 leukemias, n=4.
(I) Proportion of NPM1wt or NPM1mut co-occurrence with immunogenic IDH1R132H (n=33), non-immunogenic IDH1R132C (n=31) and FLT3wt AMLs (n=144) (Ley et al., 2013; Falini et al., 2019).
(J-P) Stem cell-derived leukemia antigen presentation impacts on disease onset.
(J) Experimental scheme. CAG-OVA HSPCs were transformed with MLL-AF9 and co-transplanted with or without OT-II T cells at day 0 (K-O) or 2 weeks post transplantation (P).
(K and P) AML cells over time in the peripheral blood, n=5-8.
(L) AML cells in the BM at the endpoint, n=8.
(M) OT-II CD4+ T cells in the BM at the endpoint, n=8.
(N) PD-L1 expression in BM CD4+ T cells, n=8.
(O) Relative frequencies of host CD4+ (left) and CD8+ (right) naïve, effector memory (EM) and central memory (CM) T cells in presence or absence of OT-II CD4+ T cells, n=8.
Individual values are shown in A. Minimum to maximum are depicted in E and G, means and SEM are depicted otherwise. No significance = ns, P<0.05 *, P<0.01 **, P<0.001 ***, P<0.0001 ****. Two-way ANOVA (H) or Kruskal-Wallis (B and F) were performed as discovery tests. Linear regression analysis (A), Chi-squared test (I), two-way ANOVA (K, O, P), paired (C,F) unpaired Mann-Whitney test (B, D, E, G, L) was performed. If not stated otherwise, unpaired two-tailed t-tests were performed as post-hoc tests.
Interestingly, human AMLs with genomic alterations capable of transforming only HSCs, such as FLT3-ITD AMLs (without NPM1 alterations), displayed a transcriptomic state of stemness and expressed consistently high levels of the MHC-II machinery (Figure 7G). In contrast, AMLs with NPM1 mutations (without FLT3 alterations), capable of also transforming differentiated progenitors, displayed a more differentiated phenotype and lower expression of the MHC-II machinery. These data suggest that the leukemic cell origin might determine the APC capacity of the AML. To experimentally test this, we generated stem cell-derived AMLs and mature granulocyte progenitor-derived AMLs by transducing either mouse LSK or GMP populations with the oncogene MLL-AF9, followed by transplantation into recipient mice (Krivtsov et al., 2006, 2013). In line with our hypothesis, stem cell-derived AMLs expressed more MHC-II and were significantly more efficient in inducing MHC-II-dependent, antigen-specific CD4+ T cell responses, if compared to myeloid progenitor-derived AMLs (Figure 7H, S5J and S5K). Together, these data demonstrate that the state of differentiation, linked to the cellular origin of AML, impacts on the capability of the disease to interact with the adaptive immune system. Moreover, similar to their healthy counterparts, stem cell-like leukemia cells display most efficient APC function, which is lost during granulocytic differentiation.
MHC-II mediated interactions between transformed stem cells and antigen-specific CD4+ T cells prevent leukemia onset
Since healthy and malignant stem cells displayed APC capacities, we investigated whether the above described mechanism driving differentiation and depletion of immunogenic antigen-presenting stem cells could serve as an immunosurveillance mechanism to prevent leukemia onset by eliminating transformed HSPCs. According to our hypothesis, mutations generating MHC-II restricted neoantigens in humans should be efficiently out-selected in stem-like AMLs, but not in differentiated leukemias that express low levels of MHC-II, such as NPM1mut AMLs. In line with this, the IDH1(R132H) mutation, generating a well-established MHC-II restricted neoepitope (Schumacher et al., 2014), occurred almost exclusively in differentiated NPM1mut AML but not in more immature NPM1wt AMLs (Figure 7I). In contrast, AMLs with a non-immunogenic IDH1(R132C) mutation displayed a comparable proportion of NPM1mut to NPM1wt AMLs to a general AML cohort, supporting the hypothesis that immature HSPCs acquiring immunogenic aberrations presented via MHC-II are efficiently removed from the hematopoietic system in humans.
To experimentally validate this hypothesis, we mimicked a malignant transformation resulting in immunogenic neoantigen presentation by transforming OVA-expressing HSPCs with the oncogene MLL-AF9, followed by transplantation into mice in the presence or absence of OVA-specific OT-II CD4+ T cells (Figure 7J). While mice rapidly developed leukemias in the absence of CD4+ T cells that specifically recognize the malignant leukemia stem cells, in the presence of OT-II T cells, transformed HSPCs were efficiently removed, preventing leukemia formation and accumulation of leukemia cells in the bone marrow (Figure 7K and 7L). Similar to our observations in the healthy system, upon bidirectional interactions with leukemia stem cells in vivo, antigen-specific CD4+ T cells expanded in the bone marrow and acquired a PD-L1 high phenotype resembling THSCs capable of preventing harmful pro-inflammatory bone marrow reactions (Figure 7M and 7N). In line with this, bystander bone marrow T cells remained in a homeostatic state in the presence of PD-L1 high antigen-specific CD4+ T cells, but were highly activated in the absence of antigen-specific CD4+ T cells (Figure 7O). Of note, while newly transformed stem cells were efficiently eliminated by interactions with CD4+ T cells before disease onset, addition of antigen-specific CD4+ T cells after the establishment of the disease, did not rescue the animals (Figure 7P, see discussion). Together, these data suggest that presentation of immunogenic antigens by transformed stem cells act as surveillance mechanism to remove malignant cells from the hematopoietic system thereby preventing leukemia onset.
Discussion
Here we demonstrate that mouse and human HSPCs present antigens via MHC-II, and induce T cell responses. Interestingly, helper T cells have been described to regulate tissue homeostasis and stem cells in different tissues (Fujisaki et al., 2011; Burzyn et al., 2013; Arpaia et al., 2015; Ali et al., 2017; Biton et al., 2018; Hirata et al., 2018; Naik et al., 2018). In the intestine, MHC-II has been suggested to act as a scaffold for the recruitment of T helper subsets that modulate stem cell differentiation and tumorigenesis (Biton et al., 2018; Beyaz et al., 2021).
The acquisition of genomic aberrations in HSPCs is the main cause for the development of hematological malignancies. Accordingly, several passive protection mechanisms reduce the exposure of HSPCs to molecular, cellular and inflammatory stress, minimizing the risk for a malignant transformation (Essers et al., 2009; Sato et al., 2009; Fujisaki et al., 2011; van Galen et al., 2014; Walter et al., 2015; Zhang et al., 2016; Ho et al., 2017). In addition, regulatory T cells have been implicated in maintaining HSC quiescence and establishing an immune privileged niche to further protect HSPC integrity (Zou et al., 2004; Urbieta et al., 2010; Fujisaki et al., 2011; Pierini et al., 2017; Hirata et al., 2018). However, active mechanisms that specifically eliminate abberant HSPCs from the stem cell pool have not been described to date. Here, we demonstrate that mouse and human HSPCs continuously present antigens via MHC-II repurposing this process as an immunosurveillance mechanism. While the presentation of self-antigens during homeostasis is immunologically ignored, the presentation of immunogenic antigens results in a bidirectional interaction between antigen presenting HSPCs and antigen-specific CD4+ T cells. Within the HSPC pool, antigen-specific interactions with CD4+ T cells trigger a rapid cycle entry and differentiation specifically of those HSPCs that present immunogenic antigens, thereby effectively eliminating potentially (pre)malignant HSPCs. Simultaneously, this bidirectional interaction results in the activation CD4+ T cells. However, in contrast to pro-inflammatory antigen-specific interactions, CD4+ T cells are polarized to an immune regulatory state, thereby preventing excessive inflammatory responses in the bone marrow that would endanger the remaining healthy HSPCs (Sato et al., 2009; Walter et al., 2015).
In our study we have investigated the effect of the presentation of immunogenic antigens derived from endogenous sources on HSPCs, reflecting the presentation of neoepitopes during leukemogenesis, which trigger the effective elimination of (pre)malignant HSPCs. However, similar safeguarding mechanisms might be in place, in which the presentation of exogenous pathogen-derived peptides could drive the rapid elimination of infected HSPCs.
All in all, our data demonstrate that MHC-II based antigen presentation by HSPCs acts an immunosurveillance mechanism operational both in mouse and human, providing a mechanistic understanding for the recent clinical findings that relapse after allogeneic stem cell transplantation is tightly associated with loss of MHC-II in AML (Christopher et al., 2018; Toffalori et al., 2019). These findings may also provide a potential explanation for the heterogeneous response of AMLs to immunotherapies (Liao et al., 2019; Barrett, 2020; Vago and Gojo, 2020). Boosting or restoring MHC-II mediated antigen presentation might serve as a future therapeutic avenue to prevent AML relapse. Lastly, a deregulation of this immunoregulatory MHC-II-T cell axis might also result in loss of stem cell function as observed in acquired idiopathic aplastic anemia, an autoimmune disease caused by the immune-mediated destruction of HSCs. In this line, particular MHC-II haplotypes and loss of heterozygosity are associated with aplastic anemia susceptibility and response to immunosuppressive therapy (Nakao et al., 1994; Nimer et al., 1994; Saunthararajah et al., 2002; Rehman et al., 2009; Dhaliwal et al., 2011; Liu et al., 2016; Young, 2018).
Together, our study reveals so far unrecognized insights into antigen-specific bidirectional interactions between HSPCs and CD4+ T cells, demonstrating that HSPCs are not only passive receivers of immunological signals, but actively engage in adaptive immune responses to safeguard the integrity of the stem cell pool.
Limitations of the study
Our study has uncovered a novel immunosurveillance that is driven by a direct, MHC-II dependent interaction between antigen-specific CD4+ T cells and antigen presenting HSPCs. Even though there is no evidence for a direct participation of CD8+ T cells in the elimination of malignant HSPCs, we cannot completely rule out that secondary activation of CD8+ T cells may play a role in some experimental settings. While our data demonstrate that leukemic HSPCs are rapidly cleared upon presentation of immunogenic neoantigens via MHC-II during disease onset, the functional role of MHC-II in fully established leukemias remains more elusive. Since antigen presentation by HSPCs polarizes CD4+ T cells to an immunoregulatory state, it is conceivable that fully established leukemias may hijack this mechanism to create an immune suppressive environment and evade immune clearance. In line with this, immune suppression is frequently observed in AML (Vago and Gojo, 2020).
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Simon Haas (simon.haas@bih-charite.de).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
RNA-seq count and NanoString data have been deposited at Figshare and and are publicly available as of the date of publication. DOIs are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Mouse Antibodies | ||
| Anti-mouse B220 FITC | ThermoFisher | RRID:AB_2621690 |
| Anti-mouse B220 AF700 | ThermoFisher | RRID: AB_891458 |
| Anti-mouse B220 APC-efluor780 | ThermoFisher | RRID:AB_2866434 |
| Anti-mouse CD105 efluor 450 | ThermoFisher | RRID:AB_10548959 |
| Anti-mouse CD117 BV711 | BioLegend | RRID:AB_2565956 |
| Anti-mouse CD117 PE | ThermoFisher | RRID:AB_469643 |
| Anti-mouse CD117 PE-Cy5 | BioLegend | RRID:AB_468786 |
| Anti-mouse CD11b FITC | ThermoFisher | RRID:AB_11152193 |
| Anti-mouse CD11b AF700 | ThermoFisher | RRID:AB_657586 |
| Anti-mouse CD127 PE | BioLegend | RRID:AB_953562 |
| Anti-mouse CD150 PE-Cy5 | ThermoFisher | RRID:AB_493598 |
| Anti-mouse CD16/32 AF700 | ThermoFisher | RRID:AB_493995 |
| Anti-mouse CD16/32 APC | ThermoFisher | RRID:AB_469356 |
| Anti-mouse CD19 APC | BioLegend | RRID:AB_313646 |
| Anti-mouse CD206 FITC | BioLegend | RRID:AB_10901166 |
| Anti-mouse CD25 BV785 | BioLegend | RRID:AB_2564131 |
| Anti-mouse CD25 APC | BioLegend | RRID:AB_2280288 |
| Anti-mouse CD274 (PD-L1) BV711 | BioLegend | RRID:AB_2563619 |
| Anti-mouse CD274 (PD-L1) PE | BioLegend | RRID:AB_2073556 |
| Anti-mouse CD279 (PD-1) APC | ThermoFisher | RRID:AB_11149358 |
| Anti-mouse CD34 PE | ThermoFisher | RRID:AB_467210 |
| Anti-mouse CD3e FITC | ThermoFisher | RRID:AB_2572431 |
| Anti-mouse CD3e | BioXCell | RRID:AB_1107632 |
| Anti-mouse CD4 BUV805 | BD | RRID:AB_2827960 |
| Anti-mouse CD4 FITC | ThermoFisher | RRID:AB_464892 |
| Anti-mouse CD4 AF700 | ThermoFisher | RRID:AB_493999 |
| Anti-mouse CD4 APC-Cy7 | BD | RRID:AB_394331 |
| Anti-mouse CD41 APC | BioLegend | RRID:AB_11126751 |
| Anti-mouse CD41 FITC | BD | RRID:AB_10892804 |
| Anti-mouse CD44 FITC | BioLegend | RRID:AB_312957 |
| Anti-mouse CD45 Pacific Blue | BioLegend | RRID:AB_493536 |
| Anti-mouse CD45.1 BUV395 | BD | RRID:AB_2722493 |
| Anti-mouse CD45.1 BV605 | BioLegend | RRID:AB_11204076 |
| Anti-mouse CD45.1 PE | ThermoFisher | RRID:AB_465675 |
| Anti-mouse CD45.1 PE-Cy5 | ThermoFisher | RRID:AB_468759 |
| Anti-mouse CD45.2 FITC | ThermoFisher | RRID:AB_465061 |
| Anti-mouse CD45.2 APC-efluor780 | ThermoFisher | RRID:AB_1272211 |
| Anti-mouse CD48 BUV395 | BD | RRID:AB_2739984 |
| Anti-mouse CD48 APC | ThermoFisher | RRID:AB_469408 |
| Anti-mouse CD48 BV421 | BioLegend | RRID:AB_10895922 |
| Anti-mouse CD69 PE-Cy5 | BioLegend | RRID:AB_313112 |
| Anti-mouse CD8 BUV395 | BD | RRID:AB_2739421 |
| Anti-mouse CD8 FITC | ThermoFisher | RRID:AB_464915 |
| Anti-mouse CD8 AF700 | ThermoFisher | RRID:AB_494005 |
| Anti-mouse CD84 PE | BioLegend | RRID:AB_2074756 |
| Anti-mouse CD90.1 FITC | BioLegend | RRID:AB_314014 |
| Anti-mouse F4/80 Pacific Blue | ThermoFisher | RRID:AB_10373419 |
| Anti-mouse Gr-1 FITC | ThermoFisher | RRID:AB_11152477 |
| Anti-mouse Gr-1 AF700 | ThermoFisher | RRID:AB_494007 |
| Anti-mouse Ki67 PE-Cy7 | BD | RRID:AB_10716060 |
| Anti-mouse Lag3 APC-Cy7 | ThermoFisher | RRID:AB_2637323 |
| Anti-mouse MHC-II (I-A/I-E) | BioXCell | RRID:AB_10949298 |
| Anti-mouse MHC-II (I-A/I-E) BV785 | BioLegend | RRID:AB_2565977 |
| Anti-mouse MHC-II (I-A/I-E) PE | BioLegend | RRID:AB_313323 |
| Anti-mouse IL-10 PE | BioLegend | RRID:AB_466176 |
| Anti-mouse Sca-1 APC-Cy7 | BD | RRID:AB_1727552 |
| Anti-mouse SiglecF BV421 | BD | RRID:AB_2739398 |
| Anti-mouse SiglecH PE | ThermoFisher | RRID:AB_10597139 |
| Anti-mouse TCRb PE-Cy7 | Biolegend | RRID:AB_893627 |
| Anti-mouse Ter119 FITC | ThermoFisher | RRID:AB_465312 |
| Anti-mouse Ter119 AF700 | BioLegend | RRID:AB_528963 |
| Anti-mouse Tim3 APC | BioLegend | Clone:5D12 (custom) |
| Anti-mouse I-Ab-Ea FITC | ThermoFisher | RRID:AB_996692 |
| Anti-mouse I-Ab-Ea Biotin | ThermoFisher | RRID:AB_657823 |
| Human Antibodies | ||
| Anti-human CD3 BUV395 | BD | RRID:AB_2744387 |
| Anti-human CD4 APC | BD | RRID:AB_11153855 |
| Anti-human CD4 BUV805 | ThermoFisher | RRID:AB_2870176 |
| Anti-human CD8 APC | BD | RRID:AB_398595 |
| Anti-human CD11b APC | BD | RRID:AB_10561676 |
| Anti-human CD11c BV605 | BD | RRID:AB_2744276 |
| Anti-human CD11c Pe-Cy7 | BioLegend | RRID:AB_389351 |
| Anti-human CD19 APC | ThermoFisher | RRID:AB_10804519 |
| Anti-human CD19 APC-Cy7 | BioLegend | RRID:AB_2564193 |
| Anti-human CD19 BV786 | BioLegend | RRID:AB_2563442 |
| Anti-human CD20 APC | BD | RRID:AB_398670 |
| Anti-human CD25 PE-Cy7 | BioLegend | RRID:AB_314282 |
| Anti-human CD33 BV421 | BioLegend | RRID:AB_2561690 |
| Anti-human CD34 APC-Cy7 | ThermoFisher | RRID:AB_2573956 |
| Anti-human CD38 A700 | ThermoFisher | RRID:AB_10852837 |
| Anti-human CD41a APC | BioLegend | RRID:AB_2129464 |
| Anti-human CD45 APC | ThermoFisher | RRID:AB_10667894 |
| Anti-human CD45 PE | ThermoFisher | RRID:AB_1724079 |
| Anti-human CD45RA FITC | BioLegend | RRID:AB_2650650 |
| Anti-human CD45RO FITC | BioLegend | RRID:AB_2564159 |
| Anti-human CD49b FITC | BioLegend | RRID:AB_2562531 |
| Anti-human CD49f PE-Cy7 | ThermoFisher | RRID:AB_10804881 |
| Anti-human CD56 APC | BD | RRID:AB_398601 |
| Anti-human CD56 Alexa Fluor 488 | BD | RRID:AB_396808 |
| Anti-human CD56 BV711 | BioLegend | RRID:AB_2562417 |
| Anti-human CD69 BUV395 | BD | RRID:AB_2738770 |
| Anti-human CD90 PE-Cy5 | BD | RRID:AB_395971 |
| Anti-human HLA-DR PE | ThermoFisher | RRID:AB_10698015 |
| Anti-human CD154 PE-Cy5 | BioLegend | RRID:AB_314831 |
| Anti-human CD197 (CCR7) Pacific Blue | BioLegend | RRID:AB_10918984 |
| Anti-human CD223 (LAG3) BV711 | BioLegend | RRID:AB_2716125 |
| Anti-human CD235 APC | ThermoFisher | RRID:AB_2043823 |
| Anti-human CD274 (PD-L1) BV785 | BioLegend | RRID:AB_2629582 |
| Anti-human CD279 (PD1) APC | BioLegend | RRID:AB_940473 |
| Anti-human CD366 (TIM3) BV605 | BioLegend | RRID:AB_2562194 |
| Bacterial and Virus Strains | ||
| MCMV-Δm157 (MCMV) | Hirche et al., 2017 | N/A |
| Biological Samples | ||
| Human Healthy Bone Marrow Aspirates | Heidelberg University Hospital | N/A |
| Human Peripheral Blood | University Hospital Mannheim | N/A |
| Human AML Bone Marrow Aspirates | AML-SG and SAL biorepositories | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| pI:C | Invivogen | Cat#tlrl-pic |
| LPS | ThermoFisher | Cat#00-4976-03 |
| IFNα | Miltenyi | Cat#130-093-131 |
| Ovalbumin | Invivogen | Cat#vac-stova |
| DQ Ovalbumin | Invitrogen | Cat#D12053 |
| Ovalbumin 323-339 peptide | Invivogen | Cat#vac-isq |
| Ovalbumin 257-264 peptide | Invivogen | Cat#vac-sin |
| MOG peptide | Genemed Sythesis | Cat#MOG3555-P2-1 |
| Eα peptide (52-68) | Mimotopes | Cat#68827-005 |
| ACK Buffer | Lonza | Cat#10-548E |
| Sodium pyruvate | Gibco | Cat#11360039 |
| L-Glutamine | Gibco | Cat#25030081 |
| L-arginine | Sigma | Cat#A5006-100G |
| L-asparagine | Sigma | Cat#A0884-100G |
| Penicillin/Streptomycin | Sigma | Cat#P4458-100ml |
| Folic acid | Sigma | Cat#F7876-10G |
| MEM non-essential amino acids | ThermoFisher | Cat#11140050 |
| MEM vitamin solution | ThermoFisher | Cat#11120052 |
| β-mercaptoethanol | Sigma | Cat# M3148 |
| Cell Trace Violet | ThermoFisher | Cat#C34557 |
| Dynabeads Mouse T-Activator | ThermoFisher | Cat#11452D |
| CytoStim | Miltenyi | Cat#130-092-172 |
| PepMix CEFX Ultra SuperStim MHC-II Subset Pool | JPT | Cat#PM-CEFX-3 |
| Mouse TPO | PreproTech | Cat#315-14 |
| Mouse SCF | PreproTech | Cat#250-03 |
| CNBr-activated Sepharose | GE Healthcare | Cat#17-0430-01 |
| Trifluoroacetic acid | Merck | Cat#108262 |
| DNAseI | Roche | Cat#4716728001 |
| 2′,7′-Dichlorofluorescin diacetate | Sigma | Cat#D6883-50MG |
| DAPI | ThermoFisher | RRID:AB_2629482 |
| Sunflower oil | Sigma | S5007-250ML |
| Tamoxifen | Sigma | T5648-1G |
| RNAsin+ | Promega | N2611 |
| Triton X-100 | Sigma | 9002-93-1 |
| Smart-seq2 Oligo-dT primer | Sigma | N/A |
| dNTP mix | NEB | N0447S |
| SmartScribe | Takara | 639538 |
| Smart-seq2 TSO | IDT | N/A |
| Smart-seq2 ISPCR primer | IDT | N/A |
| Ctrl IgG2b | ThermoFisher | RRID:AB_470099 |
| Streptavidin PE | BioLegend | Cat#12-4317-82 |
| Critical Commercial Assays | ||
| Dynabeads Untouched Mouse CD4 Cells Kit | Invitrogen | Cat#11416D |
| Cell Stimulation Cocktail (plus protein transport inhibitors) | eBioscience | Cat#00-4975-93 |
| Fixation/Permeabilization Solution Kit | BD | Cat#554714 |
| Arcturus PicoPure RNA Isolation Kit | Invitrogen | Cat#KIT0204 |
| SuperScript VILO cDNA synthesis Kit | Invitrogen | Cat#11754050 |
| PowerUP SybrGreen Mastermix | ThermoFisher | Cat#A25741 |
| RNA 6000 Pico Kit | Agilent | Cat#5067-1513 |
| SMARTer Ultra Low Input RNA Kit | Takara | Cat# 634940 |
| NEBNext ChIP-seq Library Prep Kit for Illumina | NEB | Cat# E6240 |
| Qubit™ dsDNA HS Assay Kit | Invitrogen | Cat# Q32851 |
| SureSelect HS XT Target Enrichment System v6 | Agilent | N/A |
| KAPA HiFi HS Mastermix | Roche | Cat#07958935001 |
| Experimental Models: Mice | ||
| BALB/c | Harlan / Jackson / Taconic | JAX:000651 |
| C57BL/6J | Harlan/Taconic/Jackso n Laboratory | JAX:000664 |
| B6.SJL-Ptprca Pepcb/BoyJ | Harlan/Taconic/Jackso n Laboratory | JAX:002014 |
| NOD.Cg-PrkdcscidIL2rgtmWjl/SzJ | Jackson | JAX:005557 |
| C57BL/6-Tg(CAG-OVA)916Jen/J | Jackson | JAX:005145 |
| C57BL/6-Tg(Tcra2D2,Tcrb2D2)1Kuch/J | Jackson | JAX:006912 |
| B6.Cg-Tg(TcraTcrb)425Cbn/J | Jackson | JAX:004194 |
| C57BL/6-Tg(TcraTcrb)1100Mjb/J | Jackson | JAX:003831 |
| B6.129S2-Il10rbtmlAgt/J | Jackson | JAX:005027 |
| B6.129S(Cg)-Stat1tm1Dlv/J | Durbin et al., 1996 | JAX:012606 |
| BALB/c x C57BL/6J | N/A | N/A |
| B6-Tg(Tal1-cre)42-056Jrg H2-Ab1tm1KoniGt(ROSA)26Sortm1(EYFP)Cos/Atp | N/A | N/A |
| H2-Ab1tm1Koni Gt(ROSA)26Sortm1(EYFP)Cos/Atp | N/A | N/A |
| Experimental Models: Cell Lines | ||
| CTV-1 | Leibniz Institute DSMZ | ACC 40 |
| GDM-1 | Leibniz Institute DSMZ | ACC 87 |
| HL-60 | Cell Lines Service (CLS) | 300209 |
| Kasumi-1 | Leibniz Institute DSMZ | ACC 220 |
| Kasumi-3 | Leibniz Institute DSMZ | 16469 |
| Kasumi-6 | Leibniz Institute DSMZ | 15974 |
| KG-1 | Leibniz Institute DSMZ | ACC 14 |
| KG-1a | Leibniz Institute DSMZ | ACC 421 |
| ME-1 | Leibniz Institute DSMZ | ACC 537 |
| ML-1 | Leibniz Institute DSMZ | ACC 464 |
| ML-2 | Leibniz Institute DSMZ | ACC 15 |
| MOLM-14 | Leibniz Institute DSMZ | ACC 777 |
| MONO-MAC-6 | Leibniz Institute DSMZ | ACC 124 |
| MV4-11 | American Type Culture Collection (ATCC) | ATCC-CRL-9591 |
| NB-4 | Leibniz Institute DSMZ | ACC 207 |
| OCI-AML2 | Leibniz Institute DSMZ | ACC 99 |
| OCI-AML3 | Leibniz Institute DSMZ | ACC 582 |
| OCI-M1 | Leibniz Institute DSMZ | ACC 529 |
| PL-21 | Leibniz Institute DSMZ | ACC 536 |
| SET-2 | Leibniz Institute DSMZ | ACC 608 |
| SKM-1 | Leibniz Institute DSMZ | ACC 547 |
| THP-1 | Leibniz Institute DSMZ | ACC 16 |
| U-937 | Leibniz Institute DSMZ | ACC 5 |
| YNH-1 | Leibniz Institute DSMZ | ACC 692 |
| Oligonucleotides | ||
| Mouse primers for qPCR, see Table S2 | Sigma | |
| Human primers for qPCR, see Table S3 | Sigma | |
| Deposited data | ||
| Nanostring targeted gene expression data | This study | https://doi.org/10.6084/m9.figshare.19337429 |
| RNA sequencing count data | This study | https://doi.org/10.6084/m9.figshare.19425302 |
| Software and Algorithms | ||
| Quant StudioTM Real-Time PCR Software v1.3 | Applied Biosystems | https://www.thermofisher.com/de/de/home/global/forms/life-science/quantstudio-6-7-flex-software.html |
| FACSDIVA v8.0 | BD | https://www.bdbiosciences.com/en-eu/products/software/instrument-software/bd-facsdiva-software |
| Flowjo v10 | BD | https://www.flowjo.com/solutions/flowjo |
| Proteome Discoverer v1.3 | ThermoFisher | https://www.thermofisher.com/de/de/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
| Sequest search engine | ThermoFisher | https://proteomicsresource.washington.edu/protocols06/sequest.php |
| nSolver Analysis Software | Nanostring | https://nanostring.com/products/analysis-solutions/ncounter-analysis-solutions/nsolver-data-analysis-support/ |
| cluster v2.1.0 | Maechler et al., 2019 | |
| NbClust v3.0 | Charrad et al., 2014 | |
| ComplexHeatmap v.2.0.0 | Gu, Eils and Schlesner, 2016 | |
| DESeq2 | Love, Huber and Anders, 2014 | |
| ClusterProfiler | Yu et al., 2012 | |
| FactoMinR | Lê et al., 2008 | |
| GraphPad Prism v8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Others | ||
| Analysis code RNA-seq | This study | https://doi.org/10.6084/m9.figshare.19437236 |
| Immunopeptidomics sequences, see Table S1 | This study | |
All original code has been deposited at Figshare and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this work is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experiments were approved by the Animal Care and Use Committees of the German Regierungspräsidium Karlsruhe für Tierschutz und Arzneimittelüberwachung (Karlsruhe, Germany), the Harvard Medical Area Standing Committee on Animals, the Brigham and Women’s Hospital Institutional Animal Care and Use Committee (Boston, USA) or the Institutional Animal Care and Use Committees (IACUC) of the Dana-Farber Cancer Institute (Boston, USA). All mice were maintained in individually ventilated cages under SPF conditions in the animal facility of the DKFZ (Heidelberg, Germany), the Hale Building for Transformative Medicine of the Brigham and Women's Hospital (Boston, USA) or Dana-Farber Cancer Institute (Boston, USA). Wild type mice (BALB/c, C57BL/6J (CD45.2) and B6.SJL-Ptprca Pepcb/BoyJ (CD45.1)) were purchased from Harlan Laboratories, Taconic or the Jackson Laboratories. NOD.Cg-PrkdcscidIL2rgtmWjl/SzJ (NSG), C57BL/6-Tg(CAG-OVA)916Jen/J (CAG-OVA), C57BL/6-Tg(Tcra2D2,Tcrb2D2)1Kuch/J (2D2) and B6.Cg-Tg(TcraTcrb)425Cbn/J (OT-II) mice were purchased from the Jackson Laboratories. B6.129S(Cg)-Stat1tm1Dlv/J (Stat1−/−) and B6.129S2-Il10rbtm1Agt/J (Il10rb−/−) have been described before (Durbin et al., 1996; Spencer et al., 1998). B6.129S2-Il10rbtm1Agt/J mice were kindly provided by Dr. Laura Llaó-Cid. C57BL/6-FLT3wt/ITD/Mx1-Cre mice were kindly provided by the group of Prof. Dr. Carsten Müller Tidow. C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I) mice were kindly provided by Stephanie Lindner from the group of Prof. Dr. Rienk Offringa. BALB/cxC57BL/6J F1 and B6-Tg(Tal1-cre)42-056Jrg H2-Ab1tm1Koni Gt(ROSA)26Sortm1(EYFP)Cos/Atp (SclCreERT2 x MHC-II-flox x Rosa26-EYFP-flox) mice were generated in house.
To induce inflammatory conditions, mice were injected intraperitoneally with a single dose of 5 mg/kg pI:C (Invivogen), 0.25 mg/kg LPS (ThermoFisher), 500U/g IFNα (Miltenyi) and MCMV (Hirche et al., 2017). For administration of ovalbumin, a single dose of 500mg/kg of full ovalbumin protein (Invivogen), 500mg/kg of DQ-OVA (Invitrogen), or 12.5mg/kg of ovalbumin 323-339 peptide (Invivogen) was administered. For knock-out induction, 100mg/kg of tamoxifen were resuspended in sunflower oil with ethanol (10%) and injected intraperitoneally once a day for five consecutive days.
Human samples
Peripheral blood and bone marrow samples from healthy donors were obtained from the University Hospital Mannheim and Heidelberg University Hospital after informed written consent using ethic application number S480/2011. Mononuclear cells were isolated by density gradient centrifugation and stored in liquid nitrogen until further use. All experiments involving human samples were conducted in compliance with the Declaration of Helsinki and approved by and in accordance with regulations and guidelines by the ethics committee of the medical faculty of the University of Heidelberg.
METHOD DETAILS
Preparation of mouse bone marrow, spleen and lymph nodes
Mouse bone marrow was prepared by crushing femur, tibia, humerus, ilium, sternum and columna vertebralis in PBS (Sigma) supplemented with 2% heat-inactivated FCS (Gibco). Subsequently, cells were filtered through 40μm cell strainers (Falcon) and erythrocyte lysis was performed for 5 min using ACK buffer (Lonza), followed by washing and centrifugation for 5 min at 250 x g. For isolation of HSPCs, cells were incubated in PBS 2% FCS for 15 minutes with antibodies against the lineage markers CD11b (M1/70), Gr-1 (RB6.8C5), CD4 (GK1.5), CD8a (53.6.7), Ter119 (Ter119) and B220 (RA3-6B2) at 4°C. Subsequently, cells were washed and incubated for 15 minutes with pre-washed anti-rat IgG-coated Dynabeads 4,5μm magnetic polystyrene beads (Invitrogen) in the ratio of 1mL of beads/mouse. Cells expressing lineage markers were depleted using a separation magnet (Invitrogen), followed by staining the remaining lineage-negative cells described below.
Spleen and lymph nodes (inguinal, axial, submandibular, mesenteric) were dissected and homogenized through a 40μm filter into PBS 2% FCS using the plunger of a syringe. Erythrocyte lysis was performed for 5 min using ACK buffer (Lonza). For CD4+ T cell sorts, the Dynabeads Untouched Mouse CD4 Cells Kit (Invitrogen) was used according to the manufacturer’s instructions. Enriched cells were stained and isolated by FACS sorting as described below.
Flow cytometry staining, acquisition and FACS sorting
For flow cytometric analyses and FACS sorts, lineage-depleted, CD4+ T cell enriched or unfractionated cells were stained in PBS 2% FCS for 20 min with corresponding antibodies and washed. For Y-Ae antibody conjugated with biotin, cells were washed and incubated for another 20 minutes with Streptavidin-PE (ThermoFisher). For intracellular cytokine staining, cells were stimulated for 4h at 37°C with the Cell Stimulation Cocktail (plus protein transport inhibitors) (eBioscience). After surface staining, cells were fixed, permeabilized and stained using the BD Fixation/Permeabilization Solution Kit (BD Biosciences) according to manufacturer’s instructions. Finally, cells were filtered through a 35-40μM filter and acquired by a flow cytometer (LSR II or LSRFortessa, Becton Dickinson) or cell sorter (FACSAria II or FACSAria Fusion, Becton Dickinson) for analysis or sort, respectively. Common gating strategies used in this study to define populations are depicted in Figure S6 and S7.
Quantitative Polymerase Chain Reaction (qPCR)
For qPCR analyses, cells were directly sorted into RNA lysis buffer (Arcturus PicoPure RNA Isolation Kit (Invitrogen), incubated for 30 min at 42°C and processed for cDNA synthesis using SuperScript VILO cDNA synthesis kit (Invitrogen) according to manufacturer’s instructions. The newly synthesized cDNA was diluted 1:10 in RNase free H2O and 6 μL were mixed in technical triplicates in 384-well plates with 0.5 μl of forward and reverse primer (10 μM) (Table S2 and S3) and 7 μl PowerUP SybrGreen Mastermix (ThermoFisher). Program: 50°C for 2 minutes, 95°C for 10 minutes and 40 cycles of 95°C for 15 seconds, 60°C 1 minute. Primers were designed to be intron spanning whenever possible using the Universal ProbeLibrary Assay Design Center (Roche) and purchased from Sigma Aldrich. Experiments were performed on the ViiA7 System (ThermoFisher) and analysis of gene amplification curves was performed using the Quant StudioTM Real-Time PCR Software v1.3 (Applied Biosystems). RNA expression was normalized to the housekeepers Gapdh/Actb for murine and B2M/ACTB for human gene expression analysis. Relative expression levels are depicted in 2−ΔCt values, ΔCt = (geoMean Housekeeper Ct) - (gene of interest Ct).
Murine ex vivo cultures
Cells were cultured at 37°C and 5% CO2 in U-bottom plates in a total volume of 200μL of Dulbecco’s Modified Eagle’s Medium GlutaMAX (DMEM GlutaMAX, Gibco) supplemented with 10% heat-inactivated Fetal Calf Serum (FCS, Gibco), sodium pyruvate (1.5mM, Gibco), L-glutamine (2mM, Gibco), L-arginine (1x, Sigma), L-asparagine (1x, Sigma), penicillin/streptomycin (100 U/mL, Sigma), folic acid (14μM, Sigma), MEM non-essential amino acids (1x, ThermoFisher), MEM vitamin solution (1x, ThermoFisher) and β-mercaptoethanol (57.2μM, Sigma). Cells were sorted and, when mentioned, labelled with cell trace violet (ThermoFisher) according to manufacturer’s instructions. 5x104 naïve CD4+ T cells were cultured with 2x104 HSPCs, DCs or CD8+ T cells, unless stated otherwise. When stated, ovalbumin peptides (323-339 or 257-264) (both 25μg/mL, Invivogen), full ovalbumin protein (10 mg/mL, Invivogen), DQ-OVA (100μg/mL, Invitrogen), MOG peptide (50μg/mL, Genemed Sythesis), Eα peptide (52-68) (100μg/mL, Mimotopes), LPS (100 ng/mL, ThermoFisher), αMHC-II blocking antibody (10μg/mL, M5/114.15.2, BioXCell) or a control IgG2b antibody (10μg/mL, eB149/10H5, ThermoFisher) were added to the cultures. For transwell experiments, cells were plated as described with additional 2x104 HSPCs plated on 96-well plate inserts with polyester membrane and 1 μm pore size (Corning). For resting of T cells, culture medium was replaced by fresh culture medium in the absence of ovalbumin peptide, followed by culturing for two days. Re-stimulation was performed by addition of Dynabeads Mouse T-Activator (ThermoFisher) according to manufacturer’s instructions.
Human ex vivo cultures
Human cells were cultured under the same conditions as murine cells. For T cell activation assays, 5x104 naïve CD4+ T cells were cultured with 5x103 antigen presenting cells (either HLA-DR+ CD11c+ DCs, CD34+ HSPCs or additional CD3+ T cells) from an unrelated donor in the presence or absence of CytoStim (Miltenyi) or an MHC-II-restricted peptide pool (JPT) according to manufacturer’s instructions. All analyses were performed after three days of co-culture using flow cytometry. For AML cell line experiments, 23 AML cell lines were characterized as stem-like (CD34, CD117, HLA-DR expression) or mature-like (CD14, CD15, CD16, CD64 expression) and co-cultured with human PBMC naive CD4+ T cells in the presence or absence of CytoStim (CS) for 72h.
BM transplantation
For mouse stem cell transplantation experiments, HSPCs were transplanted intravenously into lethally irradiated (2x500rad) recipient mice together with 1x105 rescue bone marrow cells. For testing of stem cell potential of MHC+ populations, lineage-negative, MHC+ or MHC− BM cells were transplanted as described above. Four months post transplantation, total BM cells were transplanted into secondary recipients.
Mice were bled periodically and cells were stained as described above to assess engraftment. After 4 months, mice were sacrificed, analyzed for engraftment and 1x106 bone marrow cells were intravenously transplanted into secondary lethally irradiated recipients. For xenotransplantation assays, HLA-DR+ and HLA-DR− cells from three healthy donor bone marrow aspirates were sorted, and 1x105 cells were transplanted intrafemorally into sublethal irradiated (175x1rad) NSG mice. Engraftment of human cells was measured 4 months later by flow cytometry.
Adoptive co-transfer of OVA-loaded HSCs and antigen-specific T cells
1.5x105 BM OT-II CD4+ T cells were sorted and intravenously transferred into Ly5.1 mice. LSK cells were isolated as described above and cultured for 12 hours in presence or absence of ovalbumin peptide (50μg/mL) in culture medium supplemented with TPO (50 ng/mL, PreproTech) and SCF (50 ng/mL, PreproTech) at 37°C, 5% CO2 levels. Subsequently, cells were washed and (1x105 cells per mouse) adoptively transferred into the recipient mice from above. After three days, mice were sacrificed and the BM was isolated for flow cytometric analysis of HSPC-derived cells.
In vivo antigen presentation assays
For analysis of presentation of exogenous antigens on HSPCs, ovalbumin or LPS were administered to mice as described above and 1h later 4x104 CD8+ T cells, DCs or LSKs were isolated from mice and co-cultured with naïve OT-II CD4+ T cells in the absence of exogenous ovalbumin peptide. For analysis of presentation of endogenous antigens, CD8+ T cells, DCs or LSKs populations were isolated from CAG-OVA and control mice. Antigen presentation capacity was read out by co-culture with OT-II CD4+ T cells in the absence of exogenous ovalbumin peptide. In a transplantation setup, WT or CAG-OVA HSPCs were co-transplanted in equal ratios into irradiated WT recipients with or without OT-II T cells at day 0 or day 60 post BM reconstitution. 20 weeks post transplantation, the BM was analyzed and total BM was re-transplanted into secondary recipients. In another setup, HSPCs were cultured for 12h with our without ovalbumin peptide and then adoptively co-transferred with freshly isolated naïve OT-II CD4+ T cells, and HSPC derived progeny was analyzed after three days by flow cytometry.
MLL-AF9 experiments
LSK or GMP cells were sorted and transduced with an MLL-AF9 construct and transplanted into C57BL/6J mice (Taconic) as previously described (Krivtsov et al., 2006, 2013). In brief, LSK and GMPs were isolated from BM of C57BL/6J (wt) or C57BL/6-Tg(CAG-OVA)916Jen/J (CAG-OVA) mice cultured with retroviral supernatant for 48h. GFP+ cells were isolated via FACS and transplanted in sublethally irradiated wt recipient mice. One month post-transplant, mice were sacrificed and leukemic GFP+ cells were sorted and co-cultured with naïve OT-II T cells as described above and T cell activation was analyzed via flow cytometry after 72h. When indicated, 1x106 naïve OT-II T cells were co-transplanted at d0 or transplanted at d15 after initial transplantation of transduced LSKs/GMPs and the disease growth in blood was measured weekly. Bone marrow and spleen of recipient mice were analyzed at the endpoint via flow cytometry.
Immunopeptidomics
Isolation of MHC ligands
2.5x107-5x107 splenocytes (CD3−), T cells (CD3+) or HSPCs (Lineage-cKit+) were sorted and snap frozen. The MHC class II molecules were isolated using standard immunoaffinity purification (Falk et al., 1991; Kowalewski and Stevanović, 2013). In brief, snap-frozen primary samples were lysed in 10 mM CHAPS/PBS (AppliChem) with 1× protease inhibitor (Roche). For the immunoprecipitation of MHC class II–peptide complexes the monoclonal antibody M5/114.15.2 (eBioscience) covalently linked to CNBr-activated Sepharose were used (GE Healthcare). MHC–peptide complexes were eluted by repeated addition of 0.2% TFA (trifluoroacetic acid, Merck). Eluted MHC ligands were purified by ultrafiltration using centrifugal filter units (Amicon). Peptides were desalted using ZipTip C18 pipette tips (Millipore), eluted in 35 μl 80% acetonitrile (Merck)/0.2% TFA, vacuum-centrifuged and resuspended in 25 μl of 1% acetonitrile/0.05% TFA and samples stored at −20 °C until LC–MS/MS analysis.
Analysis of MHC ligands by LC–MS/MS
Isolated peptides were separated by reversed-phase liquid chromatography (nano-UHPLC, UltiMate 3000 RSLCnano; ThermoFisher) and analyzed in an online-coupled Orbitrap Fusion Lumos mass spectrometer (ThermoFisher). Samples were analyzed in three technical replicates and sample shares of 33% trapped on a 75 μm × 2 cm trapping column (Acclaim PepMap RSLC; Thermo Fisher) at 4 μl/min for 5.75 min. Peptide separation was performed at 50 °C and a flow rate of 175 nl/min on a 50 μm × 25 cm separation column (Acclaim PepMap RSLC; Thermo Fisher) applying a gradient ranging from 2.4 to 32.0% of acetonitrile over the course of 90 min. Samples were analyzed on the Orbitrap Fusion Lumos implementing a top-speed CID method with survey scans at 120k resolution and fragment detection in the Orbitrap (OTMS2) at 60 k resolution. A mass range of 300–1500 m/z was analyzed with charge states ≥ 2 selected for fragmentation.
Database search and spectral annotation
LC-MS/MS results were processed using Proteome Discoverer (v.1.3; ThermoFisher) to perform database search using the Sequest search engine (ThermoFisher) and the murine proteome as reference database annotated by the UniProtKB/Swiss-Prot (http://www.uniprot.org), status February 2014 containing 20,270 ORFs. The search combined data of three technical replicates, was not restricted by enzymatic specificity and oxidation of methionine residues was allowed as dynamic modification. Precursor mass tolerance was set to 5 ppm, and fragment mass tolerance to 0.02 Da. False discovery rate was estimated using the Percolator node (Käll et al., 2007) and was limited to 5%. Peptide length was limited to 12–25 AA of length.
NanoString and RNA-Seq gene expression analysis
After 3 days of co-culture with 2.5x103 (NanoString) or 2x104 (RNA-Seq) HSPCs or 2x104 DCs, CD4+ T cells were FACS-sorted and lysed in RLT Buffer (Qiagen) with 1% β-mercaptoethanol (Sigma). For NanoString, RNA was hybridized with the PanCancer Mouse Immune Profiling CodeSet provided by NanoString Technologies. The barcodes were counted on an nCounter Digital Analyzer. The obtained raw data was analyzed using the nSolver Analysis Software. For RNA-Seq, 5x103 THSC or TDC were sorted and RNA was extracted using Arcturus PicoPure Kit, and reverse transcribed, amplified and tagmented using the SmartSeq2 protocol (Picelli et al., 2013, 2014) and using a homemade Tn5 enzyme and sequenced on an Illumina NextSeq 550 (75bp high-output). Reads were aligned to the murine reference genome (Ensembl GRCm38) using STAR v2.5.2b. Gene count tables were generated using Gencode M12 annotations. Differential expression between samples was tested using the R/Bioconductor package DESeq2 (Love, Huber and Anders, 2014). GSEA was run with the R/Bioconductor package clusterProfiler (Yu et al., 2012) and PCA was performed with FactoMineR (LÊ et al., 2008).
In vitro suppression assay
TDCs and THSCs were generated by 3 days of culture as described above, rested in the absence of ovalbumin peptide for 2 days and FACS-sorted. Subsequently, 105 CTV-labelled naïve bystander CD4+ or CD8+ T cells were cultured with 105 CD19−CD3− splenocytes and different ratios of in vitro-generated THSCs or TDCs, or freshly purified CD4+ Tregs relative to the amount of naïve bystander CD4+ T cells, and soluble anti-CD3 antibody (1 μg/mL,145-2C11, BioXCell). Cells were analyzed by flow cytometry and proliferation of bystander cells was assessed.
i = Number of cell divisions, seen by CTV dilution
In vitro CD8+ T cell cytotoxicity assay
5x104 CTV-labelled naïve OT-I CD8+ T cells were co-cultured with 5x104 naïve OT-II CD4+ T cells and 2x104 CD19−CD3− splenocytes or HSPCs in the presence or absence of the MHC-I- and/or MHC-II-restricted OVA peptides. T cells and APCs were analyzed after 3 days via flow cytometry. Cytotoxicity was measured by annexin V positivity in the APCs.
In vitro macrophage polarization assay
TDCs and THSCs were generated by 3 days of culture as described above, rested in the absence of ovalbumin peptide for 2 days and FACS-sorted. Subsequently, 5x104 freshly sorted TDCs or THSCs were cultured with 1x105 CD19−CD3−CD11b+SSClow bone marrow monocytes and macrophages and anti-CD3/anti-CD28 activating beads according to manufacturer’s instructions. Cells were analyzed after 24 hours by flow cytometry.
In vivo suppression assay
For the in vivo suppression assay, TDCs and THSCs were generated as described above and FACS sorted at day 3 of the co-culture. Subsequently, 1.5x105 cells were adoptively transferred intravenously together with 106 CTV-labelled naïve OT-II CD4+ T cells into naive mice. One day post transfer, mice were injected with ovalbumin peptide and LPS as described above, and splenic T cells were analyzed after 3 days via flow cytometry. The proliferation index was calculated as described above.
EuroFlow analysis of diagnostic AML samples
Diagnostic bone marrow aspirates of AML patients were analyzed using the EuroFlow panels (van Dongen et al., 2012) at the University Hospital Heidelberg, Germany. AML blast cells were gated in FlowJo as CD45+ excluding CD45highSSClow healthy lymphoid cells, and geometric mean fluorescence intensities (gMFIs) for all FACS markers were exported. Before z-score scaling the data, values larger than the 95 percentile and smaller than the 5th percentile were considered to be outliers and adjusted to the 95th or 5th percentile, respectively. The data was partitioned into 4 clusters by PAM (partitioning around medoids) clustering using the R package cluster v2.1.0 (Maechler et al., 2019), after determining the best number of clusters using NbClust v3.0 (Charrad et al., 2014). Heatmap visualizations of the data were done using the R/Bioconductor package ComplexHeatmap v.2.0.0 (Gu, Eils and Schlesner, 2016). Stem-, Mono-, and Granulo-indices were calculated by adding the scaled gMFIs of the respective signature for each patient and min-max feature scaling each index between patients: Stem-index = CD34 and CD117, Mono-index = CD14, CD64, CD300e and CD45, Granulo-index = CD35, CD15, CD16 and SSC-A.
Quantification and statistical analysis
Flow cytometric analyses were performed in FlowJo (BD). Bioinformatic analyses were performed in R, and visualized or further analyzed in R or GraphPad Prism (v8.4.2, GraphPad Software). The vast majority of ex vivo experiments have been performed multiple times. Most experiments for large-scale gene and protein expression analyses and in vivo experiments, have been performed once. The number of biological replicates per experiment are indicated in the figure legends. Statistical tests used in every figure legend. In short, one- or two-way ANOVA, or Kruskal-Wallis tests were performed as discovery tests wherever necessary. Only when the discovery test was significant, post-hoc two-tailed t-tests or Mann-Whitney tests were performed based on normality of the data. In case of multiple comparisons, p-values were corrected by the Benjamini-Hochberg false discovery rate of 5% and q-values were subsequently used to indicate significance. Significance is depicted as: no significance = ns, P<0.05 *, P<0.01 **, P<0.001 ***, P<0.0001 **** according to statistical tests indicated in each figure legend.
Supplementary Material
Highlights:
HSPCs constitutively present antigens via major histocompatibility complex class II
Presentation of immunogenic antigens results in the activation of CD4+ T cells
Antigen presentation causes differentiation and depletion of immunogenic HSPCs
This prohibits the onset of HSC-derived leukemias presenting neoantigens via MHC-II
Acknowledgements
We would like to thank all members from the DKFZ flow cytometry, the DKFZ animal facility, D. Kozoriz and J. Panten for their assistance. We thank L. Bunse and M. Platten for providing the MHC-II conditional knockout mouse model. This work was supported by a German Academic Scholarship Foundation PhD fellowship (to A.S.), the Emmy Noether Fellowship RA3166/1-1 funded by the Deutsche Forschungsgemeinschaft (DFG) (to S.RA), the P01AI073748 and R01NS030843, funded by the National Institute of Health (NIH) (to V.K.K.), the SFB873 (to A.T.), FOR2674 (to A.T. and L.B.) and FOR2033 (to A.T.), funded by the DFG, the SyTASC consortium (Deutsche Krebshilfe) (all to A.T. and L.B.), the Dietmar Hopp Foundation and the José Carreras Foundation for Leukemia Research (grant no. DCJLS 20R/2017) (both to A.T. and S.H.) and the LeukoSyStem consortium (BMBF) (to S.RA and S.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
V.K.K. is a co-founder, has ownership interest, and is on the SAB of Celsius Therapeutics and Tizona Therapeutics.
References
- Ali N et al. (2017) ‘Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation’, Cell. Elsevier Inc., 169(6), pp. 1119–1129.e11. doi: 10.1016/j.cell.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshetaiwi H et al. (2020) ‘Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics’, Science Immunology, 5(44), p. eaay6017. doi: 10.1126/sciimmunol.aay6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N et al. (2015) ‘A Distinct Function of Regulatory T Cells in Tissue Protection’, Cell. Elsevier Inc., 162(5), pp. 1078–1089. doi: 10.1016/j.cell.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldridge MT et al. (2010) ‘Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection’, Nature. Nature Publishing Group, 465(7299), pp. 793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnden MJ et al. (1998) ‘Defective TCR expression in transgenic mice constructed using cDNA-based α- and β-chain genes under the control of heterologous regulatory elements’, Immunology and Cell Biology, 76(1), pp. 34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- Barrett AJ (2020) ‘Acute myeloid leukaemia and the immune system: implications for immunotherapy’, British Journal of Haematology, 188(1), pp. 147–158. doi: 10.1111/bjh.16310. [DOI] [PubMed] [Google Scholar]
- Beyaz S et al. (2021) ‘Dietary suppression of MHC class II expression in intestinal epithelial cells enhances intestinal tumorigenesis’, Cell Stem Cell, 28(11), pp. 1922–1935.e5. doi: 10.1016/j.stem.2021.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin C et al. (2015) ‘Mapping the HLA ligandome landscape of acute myeloid leukemia: a targeted approach toward peptide-based immunotherapy’, Leukemia, 29(3), pp. 647–659. doi: 10.1038/leu.2014.233. [DOI] [PubMed] [Google Scholar]
- Bettelli E et al. (2003) ‘Myelin Oligodendrocyte Glycoprotein–specific T Cell Receptor Transgenic Mice Develop Spontaneous Autoimmune Optic Neuritis’, Journal of Experimental Medicine, 197(9), pp. 1073–1081. doi: 10.1084/jem.20021603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biton M et al. (2018) ‘T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation’, Cell. Elsevier Inc., 175(5), pp. 1307–1320.e22. doi: 10.1016/j.cell.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzyn D et al. (2013) ‘Special Population of Regulatory T Cells Potentiates Muscle Repair’, Cell. Elsevier Inc., 155(6), pp. 1282–1295. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas-Wallscheid N et al. (2014) ‘Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis’, Cell Stem Cell, 15(4), pp. 507–522. doi: 10.1016/j.stem.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Charrad M et al. (2014) ‘Nbclust: An R package for determining the relevant number of clusters in a data set’, Journal of Statistical Software. American Statistical Association, 61(6), pp. 1–36. doi: 10.18637/jss.v061.i06. [DOI] [Google Scholar]
- Chihara N et al. (2018) ‘Induction and transcriptional regulation of the co-inhibitory gene module in T cells’, Nature, 558(7710), pp. 454–459. doi: 10.1038/s41586-018-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher MJ et al. (2018) ‘Immune Escape of Relapsed AML Cells after Allogeneic Transplantation’, New England Journal of Medicine, 379(24), pp. 2330–2341. doi: 10.1056/NEJMoa1808777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison LW et al. (2007) ‘The inhibitory cytokine IL-35 contributes to regulatory T-cell function’, Nature, 450(7169), pp. 566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- Collison LW et al. (2010) ‘IL-35-mediated induction of a potent regulatory T cell population’, Nature Immunology. Nature Publishing Group, 11(12), pp. 1093–1101. doi: 10.1038/ni.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello R et al. (1999) ‘The immunophenotype of minimally differentiated acute myeloid leukemia (AML-M0): reduced immunogenicity and high frequency of CD34+/CD38− leukemic progenitors’, Leukemia, 13(10), pp. 1513–1518. doi: 10.1038/sj.leu.2401519. [DOI] [PubMed] [Google Scholar]
- Dhaliwal JS et al. (2011) ‘Susceptibility to aplastic anemia is associated with HLA-DRB1*1501 in an aboriginal population in Sabah, Malaysia’, Human Immunology. Elsevier Inc., 72(10), pp. 889–892. doi: 10.1016/j.humimm.2011.06.013. [DOI] [PubMed] [Google Scholar]
- van Dongen JJM et al. (2012) ‘EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes’, Leukemia, 26(9), pp. 1908–1975. doi: 10.1038/leu.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov S et al. (2012) ‘Hematopoiesis: A Human Perspective’, Cell Stem Cell. Elsevier Inc., 10(2), pp. 120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Durbin JE et al. (1996) ‘Targeted Disruption of the Mouse Stat1 Gene Results in Compromised Innate Immunity to Viral Disease’, Cell, 84(3), pp. 443–450. doi: 10.1016/S0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- Eaves C (2015) ‘Hematopoietic stem cells: concepts, definitions, and the new reality’, Blood, 125(17), pp. 2605–2614. doi: 10.1182/blood-2014-12-570200.Lessons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers MAG et al. (2009) ‘IFNα activates dormant haematopoietic stem cells in vivo’, Nature. Nature Publishing Group, 458(7240), pp. 904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- Falini B et al. (2019) ‘IDH1-R132 changes vary according to NPM1 and other mutations status in AML.’, Leukemia, 33(4), pp. 1043–1047. doi: 10.1038/s41375-018-0299-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk K et al. (1991) ‘Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules’, Nature, 351(6324), pp. 290–296. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- Fitchen JH, Foon KA and Cline MJ (1981) ‘The Expression of Histocompatibility-2 Antigens on Hemopoietic Stem Cells’, New England Journal of Medicine, 305(1). doi: 10.1056/NEJM198107023050104. [DOI] [Google Scholar]
- Fujisaki J et al. (2011) ‘In vivo imaging of T reg cells providing immune privilege to the haematopoietic stem-cell niche’, Nature, 474(7350), pp. 216–220. doi: 10.1038/nature10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Galen P et al. (2014) ‘The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress’, Nature, 510(7504), pp. 268–272. doi: 10.1038/nature13228. [DOI] [PubMed] [Google Scholar]
- Genovese G et al. (2014) ‘Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence’, New England Journal of Medicine, 371(26), pp. 2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow CC et al. (2005) ‘Cellular and genetic mechanisms of self tolerance and autoimmunity’, Nature, 435(7042), pp. 590–597. doi: 10.1038/nature03724. [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R and Schlesner M (2016) ‘Complex heatmaps reveal patterns and correlations in multidimensional genomic data’, Bioinformatics, 32(18), pp. 2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- Haas S et al. (2015) ‘Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors’, Cell Stem Cell, 17(4), pp. 422–434. doi: 10.1016/j.stem.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Hay SB et al. (2018) ‘The Human Cell Atlas bone marrow single-cell interactive web portal’, Experimental Hematology. Elsevier Inc., 68, pp. 51–61. doi: 10.1016/j.exphem.2018.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henri S et al. (2010) ‘CD207+ CD103+ dermal dendritic cells cross-present keratinocyte-derived antigens irrespective of the presence of Langerhans cells’, The Journal of Experimental Medicine, 207(1), pp. 189–206. doi: 10.1084/jem.20091964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata Y et al. (2018) ‘CD150highBone Marrow Tregs Maintain Hematopoietic Stem Cell Quiescence and Immune Privilege via Adenosine’, Cell Stem Cell. Elsevier Inc., 22(3), pp. 445–453.e5. doi: 10.1016/j.stem.2018.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirche C et al. (2017) ‘Systemic Virus Infections Differentially Modulate Cell Cycle State and Functionality of Long-Term Hematopoietic Stem Cells In Vivo’, Cell Reports, 19(11), pp. 2345–2356. doi: 10.1016/j.celrep.2017.05.063. [DOI] [PubMed] [Google Scholar]
- Ho TT et al. (2017) ‘Autophagy maintains the metabolism and function of young and old stem cells’, Nature, 543(7644), pp. 205–210. doi: 10.1038/nature21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S et al. (2014) ‘Age-related clonal hematopoiesis associated with adverse outcomes.’, The New England journal of medicine, 371(26), pp. 2488–98. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubzick CV, Randolph GJ and Henson PM (2017) ‘Monocyte differentiation and antigen-presenting functions’, Nature Reviews Immunology. Nature Publishing Group, 17(6), pp. 349–362. doi: 10.1038/nri.2017.28. [DOI] [PubMed] [Google Scholar]
- Jurewicz MM and Stern LJ (2019) ‘Class II MHC antigen processing in immune tolerance and inflammation’, Immunogenetics. Immunogenetics, 71(3), pp. 171–187. doi: 10.1007/s00251-018-1095-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käil L et al. (2007) ‘Semi-supervised learning for peptide identification from shotgun proteomics datasets’, Nature Methods, 4(11), pp. 923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- Kambayashi T and Laufer TM (2014) ‘Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell?’, Nature Reviews Immunology, 14(11), pp. 719–730. doi: 10.1038/nri3754. [DOI] [PubMed] [Google Scholar]
- Klimmeck D et al. (2014) ‘Transcriptome-wide Profiling and Posttranscriptional Analysis of Hematopoietic Stem/Progenitor Cell Differentiation toward Myeloid Commitment’, Stem Cell Reports. The Authors, 3(5), pp. 858–875. doi: 10.1016/j.stemcr.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmann A et al. (2010) ‘Gene expression profiling in AML with normal karyotype can predict mutations for molecular markers and allows novel insights into perturbed biological pathways’, Leukemia, 24(6), pp. 1216–1220. doi: 10.1038/leu.2010.73. [DOI] [PubMed] [Google Scholar]
- Kowalewski DJ and Stevanović S (2013) ‘Biochemical Large-Scale Identification of MHC Class I Ligands’, in Methods Mol Biol, pp. 145–157. doi: 10.1007/978-1-62703-218-6_12. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV et al. (2006) ‘Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9’, Nature, 442(7104), pp. 818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV et al. (2013) ‘Cell of origin determines clinically relevant subtypes of MLL-rearranged AML.’, Leukemia, 27(4), pp. 852–60. doi: 10.1038/leu.2012.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê S, Josse J, and Husson F (2008). FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw 25, 1–18. [Google Scholar]
- Ley TJ et al. (2013) ‘Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia’, New England Journal of Medicine, 368(22), pp. 2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D et al. (2019) ‘review of efficacy and safety of checkpoint inhibitor for the treatment of acute myeloid leukemia’, Frontiers in Pharmacology, 10(JUN), pp. 1–11. doi: 10.3389/fphar.2019.00609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S et al. (2016) ‘Association of Human Leukocyte Antigen DRB1*15 and DRB1*15:01 Polymorphisms with Response to Immunosuppressive Therapy in Patients with Aplastic Anemia: A Meta-Analysis’, PLOS ONE. Edited by Devaney J, 11(9), p. e0162382. doi: 10.1371/journal.pone.0162382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W and Anders S (2014) ‘Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2’, Genome Biology, 15(12), p. 550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maechler M et al. (2019) ‘cluster: Cluster Analysis Basics and Extensions. R package version 2.1.0.’ [Google Scholar]
- Merad M, Sathe P, Helft J, Miller J & Mortha A The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Re’, Annual Review of Immunology, 31(1), pp. 563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miale TD et al. (1982) ‘Surface la-Like Expression and MLR-Stimulating Capacity of Human Leukemic Myeloblasts: Implications for Immunotherapy and Prognosis’, Acta Haematologica, 68(1), pp. 3–13. doi: 10.1159/000206941. [DOI] [PubMed] [Google Scholar]
- Murphy DB et al. (1989) ‘A novel MHC class II epitope expressed in thymic medulla but not cortex’, Nature, 338(6218), pp. 765–768. doi: 10.1038/338765a0. [DOI] [PubMed] [Google Scholar]
- Mutis T et al. (1997) ‘HLA Class II Restricted T-Cell Reactivity to a Developmentally Regulated Antigen Shared by Leukemic Cells and CD34+ Early Progenitor Cells’, Blood, 90(3), pp. 1083–1090. doi: 10.1182/blood.V90.3.1083.1083_1083_1090. [DOI] [PubMed] [Google Scholar]
- Mutis T et al. (1998) ‘CD80-Transfected Acute Myeloid Leukemia Cells Induce Primary Allogeneic T-Cell Responses Directed at Patient Specific Minor Histocompatibility Antigens and Leukemia-Associated Antigens’, Blood, 92(5), pp. 1677–1684. doi: 10.1182/blood.V92.5.1677.417k14_1677_1684. [DOI] [PubMed] [Google Scholar]
- Naik S et al. (2018) ‘Two to Tango: Dialog between Immunity and Stem Cells in Health and Disease’, Cell. Elsevier Inc., 175(4), pp. 908–920. doi: 10.1016/j.cell.2018.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao S et al. (1994) ‘Identification of a specific HLA class II haplotype strongly associated with susceptibility to cyclosporine-dependent aplastic anemia’, Blood, 84(12), pp. 4257–4261. doi: 10.1182/blood.V84.12.4257.bloodjournal84124257. [DOI] [PubMed] [Google Scholar]
- Neefjes J et al. (2011) ‘Towards a systems understanding of MHC class I and MHC class II antigen presentation’, Nature Reviews Immunology. Nature Publishing Group, 11(12), pp. 823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- Ness S, Lin S and Gordon JR (2021) ‘Regulatory Dendritic Cells, T Cell Tolerance, and Dendritic Cell Therapy for Immunologic Disease’, Frontiers in Immunology, 12. doi: 10.3389/fimmu.2021.633436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman RA et al. (1983) ‘Differential expression of HLA-DR and DR-linked determinants on human leukemias and lymphoid cells’, European Journal of Immunology, 13(2), pp. 172–176. doi: 10.1002/eji.1830130215. [DOI] [PubMed] [Google Scholar]
- Ng SWK et al. (2016) ‘A 17-gene stemness score for rapid determination of risk in acute leukaemia’, Nature. Nature Publishing Group, 540(7633), pp. 433–437. doi: 10.1038/nature20598. [DOI] [PubMed] [Google Scholar]
- Nimer SD et al. (1994) ‘An increased HLA DR2 frequency is seen in aplastic anemia patients.’, Blood, 84(3), pp. 923–7. [PubMed] [Google Scholar]
- Novershtern N et al. (2011) ‘Densely Interconnected Transcriptional Circuits Control Cell States in Human Hematopoiesis’, Cell. Elsevier Inc., 144(2), pp. 296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellin D et al. (2019) ‘A comprehensive single cell transcriptional landscape of human hematopoietic progenitors’, Nature Communications, 10(1), p. 2395. doi: 10.1038/s41467-019-10291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S et al. (2013) ‘Smart-seq2 for sensitive full-length transcriptome profiling in single cells’, Nature Methods, 10(11), pp. 1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- Picelli S et al. (2014) ‘Full-length RNA-seq from single cells using Smart-seq2’, Nature Protocols, 9(1), pp. 171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Pierini A et al. (2017) ‘Foxp3+ regulatory T cells maintain the bone marrow microenvironment for B cell lymphopoiesis’, Nature Communications. Nature Publishing Group, 8(1), p. 15068. doi: 10.1038/ncomms15068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pölönen P et al. (2019) ‘Hemap: An Interactive Online Resource for Characterizing Molecular Phenotypes across Hematologic Malignancies’, Cancer Research, 79(10), pp. 2466–2479. doi: 10.1158/0008-5472.CAN-18-2970. [DOI] [PubMed] [Google Scholar]
- Rehman S et al. (2009) ‘The Frequency of HLA Class I and II Alleles in Pakistani Patients with Aplastic Anemia’, Immunological Investigations, 38(8), pp. 812–819. doi: 10.3109/08820130903271415. [DOI] [PubMed] [Google Scholar]
- Roche PA and Furuta K (2015) ‘The ins and outs of MHC class II-mediated antigen processing and presentation’, Nature Reviews Immunology. Nature Publishing Group, 15(4), pp. 203–216. doi: 10.1038/nri3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudensky AY et al. (1991) ‘On the complexity of self’, Nature, 353(6345), pp. 660–662. doi: 10.1038/353660a0. [DOI] [PubMed] [Google Scholar]
- Russell JL and Engh GJ (1979) ‘The Expression of Histocompatibility-2 Antigens on Hemopoietic Stem Cells’, Tissue Antigens, 13. doi: http://doi.wiley.com/10.1111/j.1399-0039.1979.tb01135.x. [DOI] [PubMed] [Google Scholar]
- Sato T et al. (2009) ‘Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon–dependent exhaustion’, Nature Medicine, 15(6), pp. 696–700. doi: 10.1038/nm.1973. [DOI] [PubMed] [Google Scholar]
- Saunthararajah Y et al. (2002) ‘HLA-DR15 (DR2) is overrepresented in myelodysplastic syndrome and aplastic anemia and predicts a response to immunosuppression in myelodysplastic syndrome’, Blood, 100(5), pp. 1570–1574. doi: 10.1182/blood.V100.5.1570.h81702001570_1570_1574. [DOI] [PubMed] [Google Scholar]
- Schumacher T et al. (2014) ‘A vaccine targeting mutant IDH1 induces antitumour immunity’, Nature, 512(7514), pp. 324–327. doi: 10.1038/nature13387. [DOI] [PubMed] [Google Scholar]
- Spencer SD et al. (1998) ‘The Orphan Receptor CRF2–4 Is an Essential Subunit of the Interleukin 10 Receptor’, Journal of Experimental Medicine, 187(4), pp. 571–578. doi: 10.1084/jem.187.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieff C et al. (1982) ‘Changes in Cell Surface Antigen Expression During Hemopoietic Differentiation’, Blood, 60(3). [PubMed] [Google Scholar]
- Steimle V et al. (1993) ‘Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome)’, Cell, 75(1), pp. 135–146. doi: 10.1016/S0092-8674(05)80090-X. [DOI] [PubMed] [Google Scholar]
- Steimle V et al. (1994) ‘Regulation of MHC class II expression by interferon-γ mediated by the transactivator gene CIITA’, Science, 265(5168), pp. 106–109. doi: 10.1126/science.8016643. [DOI] [PubMed] [Google Scholar]
- Steinman RM (2007) ‘Dendritic cells: Understanding immunogenicity’, European Journal of Immunology, 37(SUPPL. 1), pp. 53–60. doi: 10.1002/eji.200737400. [DOI] [PubMed] [Google Scholar]
- Szer J et al. (1985) ‘Failure of autologous marrow reconstitution after cytolytic treatment of marrow with anti-Ia monoclonal antibody’, Blood, 65(4), pp. 819–822. doi: 10.1182/blood.v65.4.819.bloodjournal654819. [DOI] [PubMed] [Google Scholar]
- Takizawa H et al. (2017) ‘Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness’, Cell Stem Cell, 21(2), pp. 225–240.e5. doi: 10.1016/j.stem.2017.06.013. [DOI] [PubMed] [Google Scholar]
- Toffalori C et al. (2019) ‘Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation’, Nature Medicine. Springer US, 25(4), pp. 603–611. doi: 10.1038/s41591-019-0400-z. [DOI] [PubMed] [Google Scholar]
- Urbieta M et al. (2010) ‘Hematopoietic progenitor cell regulation by CD4+CD25+ T cells’, Blood, 115(23), pp. 4934–4943. doi: 10.1182/blood-2009-04-218826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vago L and Gojo I (2020) ‘Immune escape and immunotherapy of acute myeloid leukemia’, Journal of Clinical Investigation, 130(4), pp. 1552–1564. doi: 10.1172/JCI129204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakkach A et al. (2003) ‘Characterization of Dendritic Cells that Induce Tolerance and T Regulatory 1 Cell Differentiation In Vivo’, Immunity, 18(5), pp. 605–617. doi: 10.1016/S1074-7613(03)00113-4. [DOI] [PubMed] [Google Scholar]
- Walter D et al. (2015) ‘Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells’, Nature, 520(7548), pp. 549–552. doi: 10.1038/nature14131. [DOI] [PubMed] [Google Scholar]
- Young NS (2018) ‘Aplastic Anemia’, New England Journal of Medicine, 379(17), pp. 1643–1656. doi: 10.1056/NEJMra1413485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G et al. (2012) ‘clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters’, OMICS: A Journal of Integrative Biology, 16(5), pp. 284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H et al. (2016) ‘Sepsis Induces Hematopoietic Stem Cell Exhaustion and Myelosuppression through Distinct Contributions of TRIF and MYD88’, Stem Cell Reports, 6(6), pp. 940–956. doi: 10.1016/j.stemcr.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H et al. (2020) ‘An IL-27-Driven Transcriptional Network Identifies Regulators of IL-10 Expression across T Helper Cell Subsets’, Cell Reports, 33(8), p. 108433. doi: 10.1016/j.celrep.2020.108433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J and Paul WE (2010) ‘Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors’, Immunological Reviews, 238(1), pp. 247–262. doi: 10.1111/j.1600-065X.2010.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L et al. (2004) ‘Bone Marrow Is a Reservoir for CD4 + CD25 + Regulatory T Cells that Traffic through CXCL12/CXCR4 Signals’, Cancer Research, 64(22), pp. 8451–8455. doi: 10.1158/0008-5472.CAN-04-1987. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq count and NanoString data have been deposited at Figshare and and are publicly available as of the date of publication. DOIs are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Mouse Antibodies | ||
| Anti-mouse B220 FITC | ThermoFisher | RRID:AB_2621690 |
| Anti-mouse B220 AF700 | ThermoFisher | RRID: AB_891458 |
| Anti-mouse B220 APC-efluor780 | ThermoFisher | RRID:AB_2866434 |
| Anti-mouse CD105 efluor 450 | ThermoFisher | RRID:AB_10548959 |
| Anti-mouse CD117 BV711 | BioLegend | RRID:AB_2565956 |
| Anti-mouse CD117 PE | ThermoFisher | RRID:AB_469643 |
| Anti-mouse CD117 PE-Cy5 | BioLegend | RRID:AB_468786 |
| Anti-mouse CD11b FITC | ThermoFisher | RRID:AB_11152193 |
| Anti-mouse CD11b AF700 | ThermoFisher | RRID:AB_657586 |
| Anti-mouse CD127 PE | BioLegend | RRID:AB_953562 |
| Anti-mouse CD150 PE-Cy5 | ThermoFisher | RRID:AB_493598 |
| Anti-mouse CD16/32 AF700 | ThermoFisher | RRID:AB_493995 |
| Anti-mouse CD16/32 APC | ThermoFisher | RRID:AB_469356 |
| Anti-mouse CD19 APC | BioLegend | RRID:AB_313646 |
| Anti-mouse CD206 FITC | BioLegend | RRID:AB_10901166 |
| Anti-mouse CD25 BV785 | BioLegend | RRID:AB_2564131 |
| Anti-mouse CD25 APC | BioLegend | RRID:AB_2280288 |
| Anti-mouse CD274 (PD-L1) BV711 | BioLegend | RRID:AB_2563619 |
| Anti-mouse CD274 (PD-L1) PE | BioLegend | RRID:AB_2073556 |
| Anti-mouse CD279 (PD-1) APC | ThermoFisher | RRID:AB_11149358 |
| Anti-mouse CD34 PE | ThermoFisher | RRID:AB_467210 |
| Anti-mouse CD3e FITC | ThermoFisher | RRID:AB_2572431 |
| Anti-mouse CD3e | BioXCell | RRID:AB_1107632 |
| Anti-mouse CD4 BUV805 | BD | RRID:AB_2827960 |
| Anti-mouse CD4 FITC | ThermoFisher | RRID:AB_464892 |
| Anti-mouse CD4 AF700 | ThermoFisher | RRID:AB_493999 |
| Anti-mouse CD4 APC-Cy7 | BD | RRID:AB_394331 |
| Anti-mouse CD41 APC | BioLegend | RRID:AB_11126751 |
| Anti-mouse CD41 FITC | BD | RRID:AB_10892804 |
| Anti-mouse CD44 FITC | BioLegend | RRID:AB_312957 |
| Anti-mouse CD45 Pacific Blue | BioLegend | RRID:AB_493536 |
| Anti-mouse CD45.1 BUV395 | BD | RRID:AB_2722493 |
| Anti-mouse CD45.1 BV605 | BioLegend | RRID:AB_11204076 |
| Anti-mouse CD45.1 PE | ThermoFisher | RRID:AB_465675 |
| Anti-mouse CD45.1 PE-Cy5 | ThermoFisher | RRID:AB_468759 |
| Anti-mouse CD45.2 FITC | ThermoFisher | RRID:AB_465061 |
| Anti-mouse CD45.2 APC-efluor780 | ThermoFisher | RRID:AB_1272211 |
| Anti-mouse CD48 BUV395 | BD | RRID:AB_2739984 |
| Anti-mouse CD48 APC | ThermoFisher | RRID:AB_469408 |
| Anti-mouse CD48 BV421 | BioLegend | RRID:AB_10895922 |
| Anti-mouse CD69 PE-Cy5 | BioLegend | RRID:AB_313112 |
| Anti-mouse CD8 BUV395 | BD | RRID:AB_2739421 |
| Anti-mouse CD8 FITC | ThermoFisher | RRID:AB_464915 |
| Anti-mouse CD8 AF700 | ThermoFisher | RRID:AB_494005 |
| Anti-mouse CD84 PE | BioLegend | RRID:AB_2074756 |
| Anti-mouse CD90.1 FITC | BioLegend | RRID:AB_314014 |
| Anti-mouse F4/80 Pacific Blue | ThermoFisher | RRID:AB_10373419 |
| Anti-mouse Gr-1 FITC | ThermoFisher | RRID:AB_11152477 |
| Anti-mouse Gr-1 AF700 | ThermoFisher | RRID:AB_494007 |
| Anti-mouse Ki67 PE-Cy7 | BD | RRID:AB_10716060 |
| Anti-mouse Lag3 APC-Cy7 | ThermoFisher | RRID:AB_2637323 |
| Anti-mouse MHC-II (I-A/I-E) | BioXCell | RRID:AB_10949298 |
| Anti-mouse MHC-II (I-A/I-E) BV785 | BioLegend | RRID:AB_2565977 |
| Anti-mouse MHC-II (I-A/I-E) PE | BioLegend | RRID:AB_313323 |
| Anti-mouse IL-10 PE | BioLegend | RRID:AB_466176 |
| Anti-mouse Sca-1 APC-Cy7 | BD | RRID:AB_1727552 |
| Anti-mouse SiglecF BV421 | BD | RRID:AB_2739398 |
| Anti-mouse SiglecH PE | ThermoFisher | RRID:AB_10597139 |
| Anti-mouse TCRb PE-Cy7 | Biolegend | RRID:AB_893627 |
| Anti-mouse Ter119 FITC | ThermoFisher | RRID:AB_465312 |
| Anti-mouse Ter119 AF700 | BioLegend | RRID:AB_528963 |
| Anti-mouse Tim3 APC | BioLegend | Clone:5D12 (custom) |
| Anti-mouse I-Ab-Ea FITC | ThermoFisher | RRID:AB_996692 |
| Anti-mouse I-Ab-Ea Biotin | ThermoFisher | RRID:AB_657823 |
| Human Antibodies | ||
| Anti-human CD3 BUV395 | BD | RRID:AB_2744387 |
| Anti-human CD4 APC | BD | RRID:AB_11153855 |
| Anti-human CD4 BUV805 | ThermoFisher | RRID:AB_2870176 |
| Anti-human CD8 APC | BD | RRID:AB_398595 |
| Anti-human CD11b APC | BD | RRID:AB_10561676 |
| Anti-human CD11c BV605 | BD | RRID:AB_2744276 |
| Anti-human CD11c Pe-Cy7 | BioLegend | RRID:AB_389351 |
| Anti-human CD19 APC | ThermoFisher | RRID:AB_10804519 |
| Anti-human CD19 APC-Cy7 | BioLegend | RRID:AB_2564193 |
| Anti-human CD19 BV786 | BioLegend | RRID:AB_2563442 |
| Anti-human CD20 APC | BD | RRID:AB_398670 |
| Anti-human CD25 PE-Cy7 | BioLegend | RRID:AB_314282 |
| Anti-human CD33 BV421 | BioLegend | RRID:AB_2561690 |
| Anti-human CD34 APC-Cy7 | ThermoFisher | RRID:AB_2573956 |
| Anti-human CD38 A700 | ThermoFisher | RRID:AB_10852837 |
| Anti-human CD41a APC | BioLegend | RRID:AB_2129464 |
| Anti-human CD45 APC | ThermoFisher | RRID:AB_10667894 |
| Anti-human CD45 PE | ThermoFisher | RRID:AB_1724079 |
| Anti-human CD45RA FITC | BioLegend | RRID:AB_2650650 |
| Anti-human CD45RO FITC | BioLegend | RRID:AB_2564159 |
| Anti-human CD49b FITC | BioLegend | RRID:AB_2562531 |
| Anti-human CD49f PE-Cy7 | ThermoFisher | RRID:AB_10804881 |
| Anti-human CD56 APC | BD | RRID:AB_398601 |
| Anti-human CD56 Alexa Fluor 488 | BD | RRID:AB_396808 |
| Anti-human CD56 BV711 | BioLegend | RRID:AB_2562417 |
| Anti-human CD69 BUV395 | BD | RRID:AB_2738770 |
| Anti-human CD90 PE-Cy5 | BD | RRID:AB_395971 |
| Anti-human HLA-DR PE | ThermoFisher | RRID:AB_10698015 |
| Anti-human CD154 PE-Cy5 | BioLegend | RRID:AB_314831 |
| Anti-human CD197 (CCR7) Pacific Blue | BioLegend | RRID:AB_10918984 |
| Anti-human CD223 (LAG3) BV711 | BioLegend | RRID:AB_2716125 |
| Anti-human CD235 APC | ThermoFisher | RRID:AB_2043823 |
| Anti-human CD274 (PD-L1) BV785 | BioLegend | RRID:AB_2629582 |
| Anti-human CD279 (PD1) APC | BioLegend | RRID:AB_940473 |
| Anti-human CD366 (TIM3) BV605 | BioLegend | RRID:AB_2562194 |
| Bacterial and Virus Strains | ||
| MCMV-Δm157 (MCMV) | Hirche et al., 2017 | N/A |
| Biological Samples | ||
| Human Healthy Bone Marrow Aspirates | Heidelberg University Hospital | N/A |
| Human Peripheral Blood | University Hospital Mannheim | N/A |
| Human AML Bone Marrow Aspirates | AML-SG and SAL biorepositories | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| pI:C | Invivogen | Cat#tlrl-pic |
| LPS | ThermoFisher | Cat#00-4976-03 |
| IFNα | Miltenyi | Cat#130-093-131 |
| Ovalbumin | Invivogen | Cat#vac-stova |
| DQ Ovalbumin | Invitrogen | Cat#D12053 |
| Ovalbumin 323-339 peptide | Invivogen | Cat#vac-isq |
| Ovalbumin 257-264 peptide | Invivogen | Cat#vac-sin |
| MOG peptide | Genemed Sythesis | Cat#MOG3555-P2-1 |
| Eα peptide (52-68) | Mimotopes | Cat#68827-005 |
| ACK Buffer | Lonza | Cat#10-548E |
| Sodium pyruvate | Gibco | Cat#11360039 |
| L-Glutamine | Gibco | Cat#25030081 |
| L-arginine | Sigma | Cat#A5006-100G |
| L-asparagine | Sigma | Cat#A0884-100G |
| Penicillin/Streptomycin | Sigma | Cat#P4458-100ml |
| Folic acid | Sigma | Cat#F7876-10G |
| MEM non-essential amino acids | ThermoFisher | Cat#11140050 |
| MEM vitamin solution | ThermoFisher | Cat#11120052 |
| β-mercaptoethanol | Sigma | Cat# M3148 |
| Cell Trace Violet | ThermoFisher | Cat#C34557 |
| Dynabeads Mouse T-Activator | ThermoFisher | Cat#11452D |
| CytoStim | Miltenyi | Cat#130-092-172 |
| PepMix CEFX Ultra SuperStim MHC-II Subset Pool | JPT | Cat#PM-CEFX-3 |
| Mouse TPO | PreproTech | Cat#315-14 |
| Mouse SCF | PreproTech | Cat#250-03 |
| CNBr-activated Sepharose | GE Healthcare | Cat#17-0430-01 |
| Trifluoroacetic acid | Merck | Cat#108262 |
| DNAseI | Roche | Cat#4716728001 |
| 2′,7′-Dichlorofluorescin diacetate | Sigma | Cat#D6883-50MG |
| DAPI | ThermoFisher | RRID:AB_2629482 |
| Sunflower oil | Sigma | S5007-250ML |
| Tamoxifen | Sigma | T5648-1G |
| RNAsin+ | Promega | N2611 |
| Triton X-100 | Sigma | 9002-93-1 |
| Smart-seq2 Oligo-dT primer | Sigma | N/A |
| dNTP mix | NEB | N0447S |
| SmartScribe | Takara | 639538 |
| Smart-seq2 TSO | IDT | N/A |
| Smart-seq2 ISPCR primer | IDT | N/A |
| Ctrl IgG2b | ThermoFisher | RRID:AB_470099 |
| Streptavidin PE | BioLegend | Cat#12-4317-82 |
| Critical Commercial Assays | ||
| Dynabeads Untouched Mouse CD4 Cells Kit | Invitrogen | Cat#11416D |
| Cell Stimulation Cocktail (plus protein transport inhibitors) | eBioscience | Cat#00-4975-93 |
| Fixation/Permeabilization Solution Kit | BD | Cat#554714 |
| Arcturus PicoPure RNA Isolation Kit | Invitrogen | Cat#KIT0204 |
| SuperScript VILO cDNA synthesis Kit | Invitrogen | Cat#11754050 |
| PowerUP SybrGreen Mastermix | ThermoFisher | Cat#A25741 |
| RNA 6000 Pico Kit | Agilent | Cat#5067-1513 |
| SMARTer Ultra Low Input RNA Kit | Takara | Cat# 634940 |
| NEBNext ChIP-seq Library Prep Kit for Illumina | NEB | Cat# E6240 |
| Qubit™ dsDNA HS Assay Kit | Invitrogen | Cat# Q32851 |
| SureSelect HS XT Target Enrichment System v6 | Agilent | N/A |
| KAPA HiFi HS Mastermix | Roche | Cat#07958935001 |
| Experimental Models: Mice | ||
| BALB/c | Harlan / Jackson / Taconic | JAX:000651 |
| C57BL/6J | Harlan/Taconic/Jackso n Laboratory | JAX:000664 |
| B6.SJL-Ptprca Pepcb/BoyJ | Harlan/Taconic/Jackso n Laboratory | JAX:002014 |
| NOD.Cg-PrkdcscidIL2rgtmWjl/SzJ | Jackson | JAX:005557 |
| C57BL/6-Tg(CAG-OVA)916Jen/J | Jackson | JAX:005145 |
| C57BL/6-Tg(Tcra2D2,Tcrb2D2)1Kuch/J | Jackson | JAX:006912 |
| B6.Cg-Tg(TcraTcrb)425Cbn/J | Jackson | JAX:004194 |
| C57BL/6-Tg(TcraTcrb)1100Mjb/J | Jackson | JAX:003831 |
| B6.129S2-Il10rbtmlAgt/J | Jackson | JAX:005027 |
| B6.129S(Cg)-Stat1tm1Dlv/J | Durbin et al., 1996 | JAX:012606 |
| BALB/c x C57BL/6J | N/A | N/A |
| B6-Tg(Tal1-cre)42-056Jrg H2-Ab1tm1KoniGt(ROSA)26Sortm1(EYFP)Cos/Atp | N/A | N/A |
| H2-Ab1tm1Koni Gt(ROSA)26Sortm1(EYFP)Cos/Atp | N/A | N/A |
| Experimental Models: Cell Lines | ||
| CTV-1 | Leibniz Institute DSMZ | ACC 40 |
| GDM-1 | Leibniz Institute DSMZ | ACC 87 |
| HL-60 | Cell Lines Service (CLS) | 300209 |
| Kasumi-1 | Leibniz Institute DSMZ | ACC 220 |
| Kasumi-3 | Leibniz Institute DSMZ | 16469 |
| Kasumi-6 | Leibniz Institute DSMZ | 15974 |
| KG-1 | Leibniz Institute DSMZ | ACC 14 |
| KG-1a | Leibniz Institute DSMZ | ACC 421 |
| ME-1 | Leibniz Institute DSMZ | ACC 537 |
| ML-1 | Leibniz Institute DSMZ | ACC 464 |
| ML-2 | Leibniz Institute DSMZ | ACC 15 |
| MOLM-14 | Leibniz Institute DSMZ | ACC 777 |
| MONO-MAC-6 | Leibniz Institute DSMZ | ACC 124 |
| MV4-11 | American Type Culture Collection (ATCC) | ATCC-CRL-9591 |
| NB-4 | Leibniz Institute DSMZ | ACC 207 |
| OCI-AML2 | Leibniz Institute DSMZ | ACC 99 |
| OCI-AML3 | Leibniz Institute DSMZ | ACC 582 |
| OCI-M1 | Leibniz Institute DSMZ | ACC 529 |
| PL-21 | Leibniz Institute DSMZ | ACC 536 |
| SET-2 | Leibniz Institute DSMZ | ACC 608 |
| SKM-1 | Leibniz Institute DSMZ | ACC 547 |
| THP-1 | Leibniz Institute DSMZ | ACC 16 |
| U-937 | Leibniz Institute DSMZ | ACC 5 |
| YNH-1 | Leibniz Institute DSMZ | ACC 692 |
| Oligonucleotides | ||
| Mouse primers for qPCR, see Table S2 | Sigma | |
| Human primers for qPCR, see Table S3 | Sigma | |
| Deposited data | ||
| Nanostring targeted gene expression data | This study | https://doi.org/10.6084/m9.figshare.19337429 |
| RNA sequencing count data | This study | https://doi.org/10.6084/m9.figshare.19425302 |
| Software and Algorithms | ||
| Quant StudioTM Real-Time PCR Software v1.3 | Applied Biosystems | https://www.thermofisher.com/de/de/home/global/forms/life-science/quantstudio-6-7-flex-software.html |
| FACSDIVA v8.0 | BD | https://www.bdbiosciences.com/en-eu/products/software/instrument-software/bd-facsdiva-software |
| Flowjo v10 | BD | https://www.flowjo.com/solutions/flowjo |
| Proteome Discoverer v1.3 | ThermoFisher | https://www.thermofisher.com/de/de/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/multi-omics-data-analysis/proteome-discoverer-software.html |
| Sequest search engine | ThermoFisher | https://proteomicsresource.washington.edu/protocols06/sequest.php |
| nSolver Analysis Software | Nanostring | https://nanostring.com/products/analysis-solutions/ncounter-analysis-solutions/nsolver-data-analysis-support/ |
| cluster v2.1.0 | Maechler et al., 2019 | |
| NbClust v3.0 | Charrad et al., 2014 | |
| ComplexHeatmap v.2.0.0 | Gu, Eils and Schlesner, 2016 | |
| DESeq2 | Love, Huber and Anders, 2014 | |
| ClusterProfiler | Yu et al., 2012 | |
| FactoMinR | Lê et al., 2008 | |
| GraphPad Prism v8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Others | ||
| Analysis code RNA-seq | This study | https://doi.org/10.6084/m9.figshare.19437236 |
| Immunopeptidomics sequences, see Table S1 | This study | |
All original code has been deposited at Figshare and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this work is available from the Lead Contact upon request.