

Abstract
Nature has developed complexity–generating reactions within natural product biosynthetic pathways. However, direct utilization of these pathways to prepare compound libraries remains challenging due to limited substrate scopes, involvement of multiple-step reactions, and moderate robustness of these sophisticated enzymatic transformations. Synthetic chemistry, on the other hand, offers an alternative approach to prepare natural product analogs. However, owing to complex and diverse functional groups appended on the targeted molecules, dedicated design and development of synthetic strategies are typically required. Herein, by leveraging the power of chemo-enzymatic synthesis, we report an approach to bridge the gap between biological and synthetic strategies in the preparation of quinolone alkaloid analogs. Leading by in silico analysis, the predicted substrate analogs were chemically synthesized. The AsqJ-catalyzed asymmetric epoxidation of these substrate analogues was followed by an Lewis Acid-triggered ring contraction to complete the viridicatin formation. We evaluated the robustness of this method in gram-scale reactions. Lastly, through chemoenzymatic cascades, a library of quinolone alkaloids is effectively prepared.
Keywords: Fe/2OG enzyme, AsqJ, Chemo-enzymatic synthesis, rearrangement, Quinolone
Quinoline and quinolone alkaloids are secondary metabolites produced by several organisms with broad biological properties including antiallergic, antibacterial, antitumor and antiviral activities.1–4 In both chemical and biological syntheses, quinoline and quinolone core serve as key intermediates in the preparation of these bioactive molecules.5 Among them, 3-hydroxyquinolin-2(1H)-one, viridicatin, skeleton has been recognized as a valuable scaffold in numerous natural products as well as synthetic compounds.3, 6–7 Quite a few synthetic strategies have been developed for the construction of this skeleton including ring expansion of isatin and aryldiazomethane, Knoevenagel reaction followed by cyclization, condensation using 2-aminobenzyldehyde with chloroacetic anhydride or N-phenylacetoacetamide derivative, transition metal-catalyzed cross-coupling, and ring contraction of cyclopenin.8–14 In nature, viridicatin is biosynthesized from O-methyl-l-tyrosine/phenylalanine and anthranilic acid through a series of enzymatic transformations involving condensation, methylation, desaturation, epoxidation and rearrangement.15 Although the aforementioned strategies have their merits, the limitations of these methods to generate viridicatin analogs led us to consider developing an efficient method combining the tools employed by chemistry and nature.
AsqK, a methyl transferase domain-containing NRPS (nonribosomal peptide synthetase) catalyzes the cyclization and methylation to form (4’-methoxy)cyclopeptin. An iron and 2-oxoglutatre (Fe/2OG) dependent oxygenase, AsqJ, catalyzes sequential desaturation and asymmetric epoxidation to furnish (4’-methoxy)cyclopenin (2) via (4’methoxy)dehydrocyclopeptin (1).15 Finally, cyclopenase, AsqI, triggers ring contraction to complete (4’-methoxy)viridicatin (3) biosynthesis (Figure 1A).16 In comparison with the enzymatic synthesis, 1 and 5 can be chemically prepared through Perkin condensation (Figure 1B).9 However, chemical asymmetric epoxidation of 1 remains a synthetically challenging task. Herein, we demonstrated that utilization of AsqJ catalyzed epoxidation represents an attractive approach to prepare cyclopenin analogs (6), particularly as the epoxidation requires only molecular oxygen and 2-oxoglutarate to enable the catalysis under mild conditions and generates succinate and CO2 as coproducts. Analogous to the cyclopenase catalyzed reaction where a zinc ion located at the active site triggers the rearrangement, leading by in silico analysis, we demonstrated that an Lewis Acid can effectively convert cyclopenin to viridicatin (2 →3 and 6 → 7). Lastly, a cross-coupling reaction using halogenated viridicatins (7a-7d) yields a quinolone library (Figure 1B).
Figure 1.
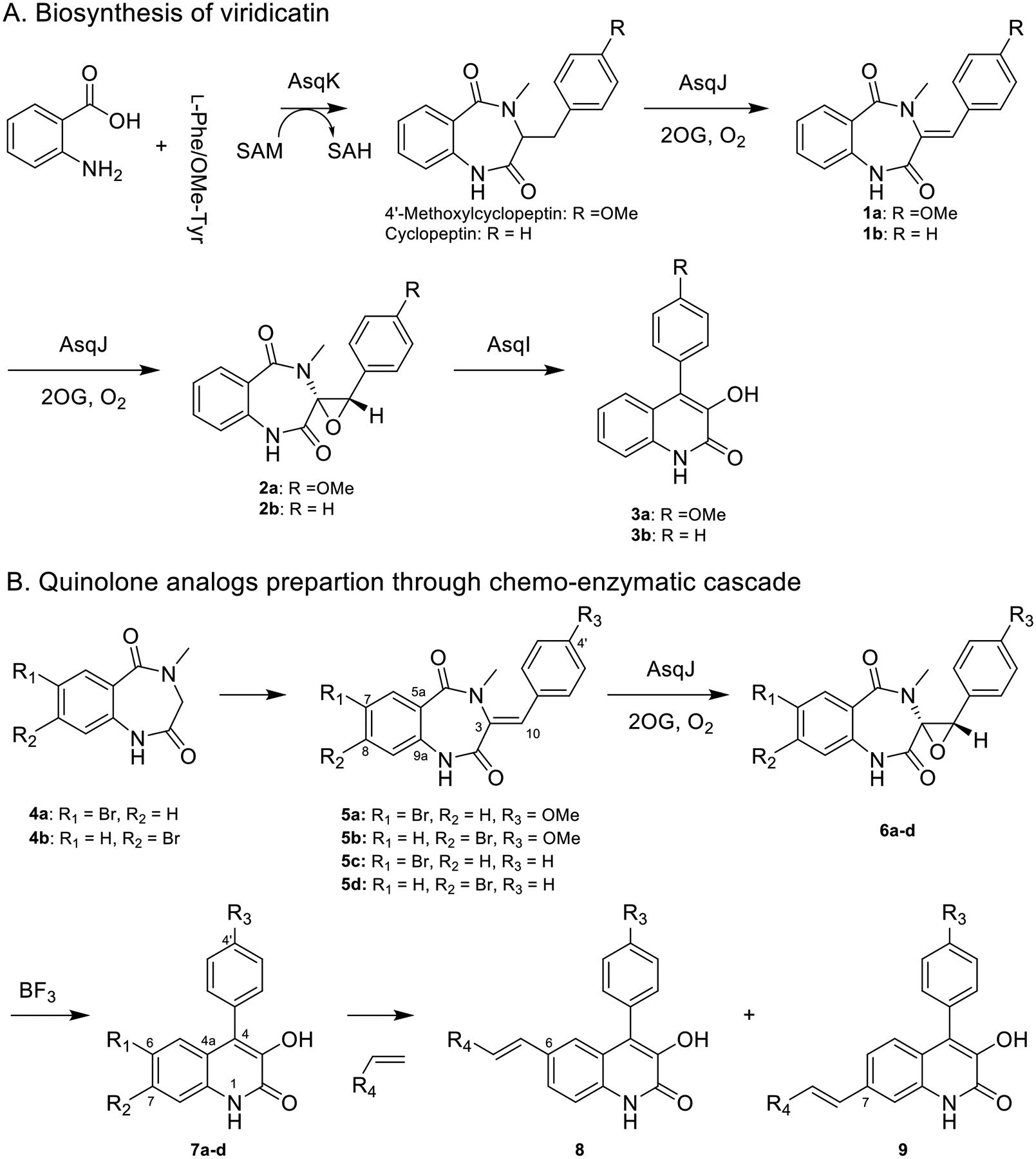
(A) Biosynthetic pathway of viridicatin and (B) preparation of quinolone analogs through chemo-enzymatic transformations.
To harness the advantages provided by AsqJ, challenges including substrate scope, selectivity and effectiveness towards substrate analogs have to be addressed. By inspecting the previously published crystallographic data on AsqJ,17–20 we found that the binding of 1 and analogs (5) are highly conserved. The two carbonyl oxygens from the diazepine-2,5-dione moiety of the substrate participate in hydrogen bonding interactions with the N-atom of Asn70 and the backbone N-atom of Met137, respectively. The aryl ring of diazepine-2,5-dione has hydrophobic interactions with Leu79, Met118, Met122, and Thr227. These four residues are part of the double stranded β-helix structure commonly present in Fe/2OG enzymes. In addition, the phenyl moiety has a π-π interaction with His134, and is also stabilized by hydrophobic interactions with Val72, Phe139 and Pro132 (Figure 2). To efficiently build quinolone library through cross-coupling reactions, we consider brominated viridicatins (7) as our targeted molecules. We performed molecular dynamics (MD) simulations to assess the binding and potential steric effects. As revealed by 10 ns MD trajectory simulations carried out using 1a, 1b and 5a-5d, all compounds exhibit a similar binding configuration in the MD equilibrated structures, which closely resembles those in the AsqJ crystal structures (Figures 2 and S83–S86). In addition, calculations using alchemical ligand transformation21–22 and the Bennett Acceptance Ratio method23–24 suggest that the differences between the binding free energies of all compounds are relatively small. Compared to the native substrates (1a and 1b), the binding energies for 5b and 5d are 4.7 and 2.3 kcal/mol higher. On the other hand, the binding energies for 5a and 5c are lowered by 1.4 kcal/mol. More importantly, the distances between the iron center and the C10 of all compounds are kept in a narrow range of ~ 4.0 – 5.5 Å (Figure 2). According to the recent study of the AsqJ catalyzed epoxidation,20 the iron-oxo of the ferryl intermediate reacts with the olefin moiety at the C10 position of the substrate. Thus, the Fe-C10 distances predicted by the MD simulations strongly suggest that 5a-5d would be suitable for AsqJ catalysis.
Figure 2.
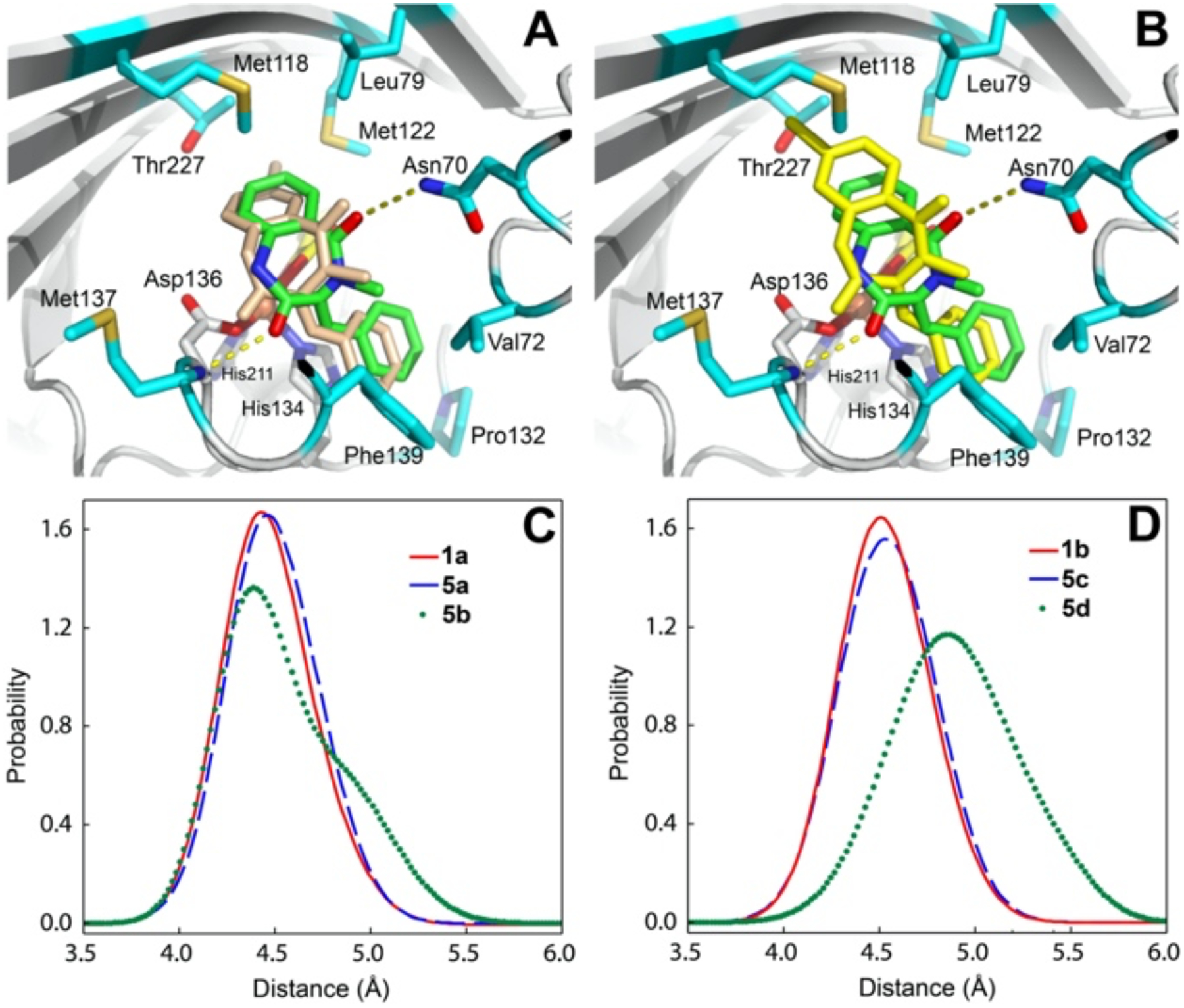
Top: 1b (green) in the AsqJ active site (PDB:6K0E) overlapped with 5c (wheat) (Panel A), or with 5d (yellow) (Panel B) obtained from the MD equilibrated structures. The protein residues surrounding 1b are highlighted. Bottom: The Fe-C10 distance distribution among 1a, 5a, and 5b (Panel C), and among 1b, 5c, and 5d (Panel D) based on MD simulations.
Given the potential substrate promiscuity assessed through in silico analysis, we began by synthesizing the halogenated dehydrocyclopeptin analogs (5a-5d, see Supporting Information (SI) for details). In parallel, AsqJ was heterologously expressed in Escherichia coli and purified using metal-affinity chromatography. Analytical-scale reactions with each substrate were carried out. Analogous to the native substrates, complete substrate consumption of 5c and 5d were detected in 4 hrs (Table1, entry 5 and 6). Moderate conversions (~ 71–75 %) were observed in the reactions using 5a and 5b (entry 3 and 4). For all substrates, longer reaction time led to almost complete substrate consumption.
Table 1.
Substrate consumption monitored by LC-MS.
| Entry | substrate | 4hrs (%) | 20hrs (%) |
|---|---|---|---|
| 1 | 1a | 90 | 100 |
| 2 | 1b | 100 | 100 |
| 3 | 5a | 75 | 93 |
| 4 | 5b | 71 | 92 |
| 5 | 5c | 100 | 100 |
| 6 | 5d | 96 | 97 |
Next, we assessed the reaction outcome and scalability of this biotransformation. Although AsqJ delivers 2a and 2b as the sole products, it is possible that non-native substrates may tilt the reaction outcomes. Fe/2OG enzymes have been reported to produce alternative products when substrate analogs are used. For example, in NapI and SyrB2, the reaction outcomes change from native desaturation and halogenation to hydroxylation.25–26 Therefore, we optimize the reaction conditions to achieve ~1000 turnovers for 5a, 5c and 5d and 250 turnovers for 5b. Currently, the reason for the lower turnover using 5b is unknown. In the scaled-up reactions, the loading of enzyme is at 0.1 – 0.4 % molar ratio to the substrate. All reaction products are purified and characterized by detailed NMR analysis (Figure 3 and SI for details). As revealed by 1H and 13C NMR, 6a-6d display similar resonances as those of 2a and 2b. The up-shifting of the 13C signal (C7 or C8) is due to the introduction of the bromine. Based on HMBC and 1H-1H COSY correlations, the structures of 6a-6d are determined. Furthermore, on the basis of the X-ray structure of 6c, the newly installed chiral centers at C3 and C10 have R and S configurations (Figure 3). These results are consistent with the substrate binding configuration observed in the AsqJ structures and the aforementioned MD simulation predictions where the oxygen atom transfer by the ferryl intermediate is stereo-specific.
Figure 3.
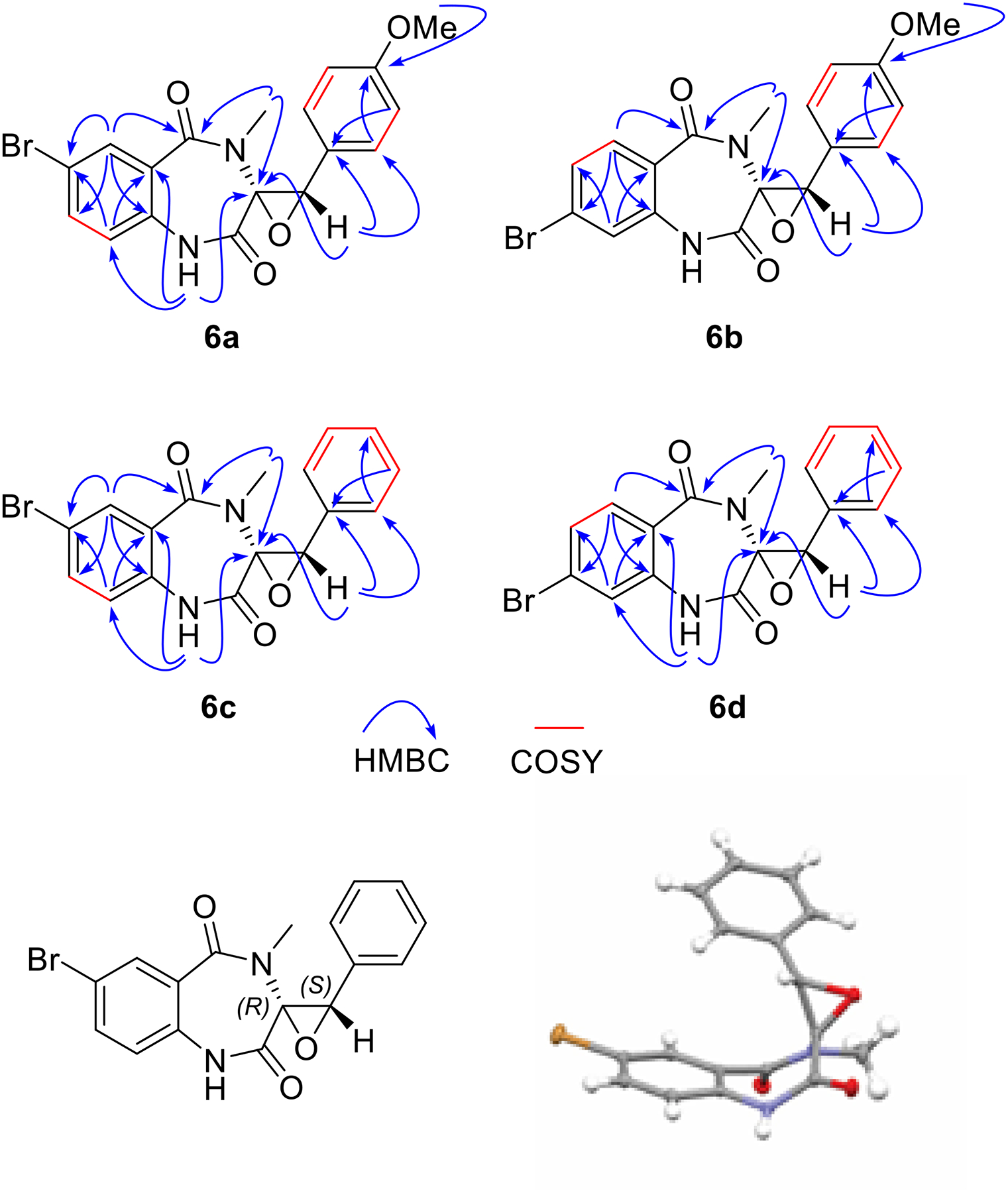
Structural characterization by selective correlations of HMBC and 1H-1H COSY and the x-ray structure of 6c.
Additionally, we determined the steady-state kinetics of the AsqJ catalyzed reactions using 1a, 1b, 5a-5d (Figure S24 and Table S2). Comparing the steady-state kinetics of the two native substrates, 1b has higher catalytic efficiency (kcat/Km of 1b/1a is ~ 5). When analogs (5a-5d) are used, Km values are increased by 510 times. Among 5a–5d, the Km values of 7-brominated analogs (5a and 5c) are ~ 2 times smaller than 8-brominated analogs (5b and 5d). The kcat values of 5a and 5c are similar or even slightly higher than those of the native substrates. Thus, the catalytic efficiency is not perturbed. On the other hand, for 5b and 5d, the overall enzyme efficiency is compromised. Thus, Br-substituent at 7 position (5a and 5c) has minimal influence on AsqJ efficiency. While C8-bromination (5b and 5d) reduces the efficiency by ~10–100 fold. These results are consistent with the MD results where the binding affinity of 5b and 5d are ~ 4.7 and 2.3 kcal/mol higher than native substrates. Taken together, these observations demonstrate that AsqJ is flexible toward the 7- and 8-substitutions and is capable of catalyzing effective asymmetric epoxidation, which is compared favorably to the reported chemical epoxidation of dehydrocyclopeptin.10
Toward preparation of quinolone library, our next goal is to develop an efficient method to convert cyclopenin to viridicatin (2 → 3 and 6 → 7). In the reported AsqI catalysis, the active site Zn ion serves as an Lewis Acid to activate the epoxide and to facilitate an anti-Baldwin-type epoxide-opening 6-endo-tet cyclization.16 Followed by the elimination of methyl isocyanate, a keto-enol tautomerization leads to viridicatin formation (Figure S93). On the other hand, Groll, et al. and Gulder et al. reported that the rearrangement is spontaneous once cyclopenin (2) is released from the active site of AsqJ.17, 27 We first assess the plausible rearrangement pathways through in silico analysis. The DFT calculated reaction coordinate using 2a and 2b largely follows the above-mentioned mechanism (see the SI for the details). The C-C bond formation between C10 and C5a accompanied by epoxide ring opening generates a cyclized intermediate. Subsequently, elimination of a methylisocyanate and keto-enol tautomerization complete the reaction. Interestingly, in both aqueous and organic conditions (calculated by using the Polarizable Continuum Model),28 2a and 2b alone are unlikely to rearrange to 3a and 3b spontaneously. The calculated free energy of the transition state connecting 2a/2b to the cyclized intermediate is ~ 25–30 kcal/mol (ΔG‡) higher than the substrate state (Figure S87, S88). This result is consistent with the DFT calculations reported by Borowski, et al29 and is less consistent with the reported spontaneous rearrangement phenomenon. To explore the possibility of using an Lewis acid as a potential catalyst, we calculated the effect of Lewis acids on the rearrangement reaction. The DFT calculations predict that an Lewis acid, such as BF3, would strongly interact with the epoxide oxygen of cyclopenin to enable epoxide ring opening in organic solvent, such as dichloromethane (Figure 4). Release of the ring strain of the epoxide leads to a better π-π stacking interaction between the two aryl rings of the substrate, which in turn significantly reduces the distance between C10 and C5a from ~ 3.3 to ~ 2.7 Å. As a consequence, the BF3 bound reactant is structurally similar to the transition state structure (the C10 and C5a distance is ~ 2.0 Å), and dramatically reduces the barrier (ΔG‡) of the C10-C5a bond formation from ~ 25–30 to ~ 5 kcal/mol for 2b (Figure 4). For the subsequent methylisocyanate elimination step, the reaction barrier is ~ 6 kcal/mol. Compared to 2b, the methoxy group of 2a seems to raise the transition state barriers for both steps to ~ 8–9 kcal/mol. Besides, other commonly used Lewis acids, such as AlCl3, show a similar effect (Figure S89) and brominated analogs (6a-6d) do not affect the overall reaction coordinate and energetics (Figure S90, S91). It is worthy to mention that 2b obtained in the AsqJ crystal structure and 6c have very different 3-dimentional conformations (Figure S92). Due to the geometric constraints enforced by AsqJ, only the flat, or a chair-like, form retains in the active site. The folded, or a boat-like, form becomes a dominant species once the epoxide is released from the active site of AsqJ. Our DFT calculations suggest that the folded conformation is energetically favorable by ~ 2 kcal/mol for all the compounds used in this study (Table S3).
Figure 4.
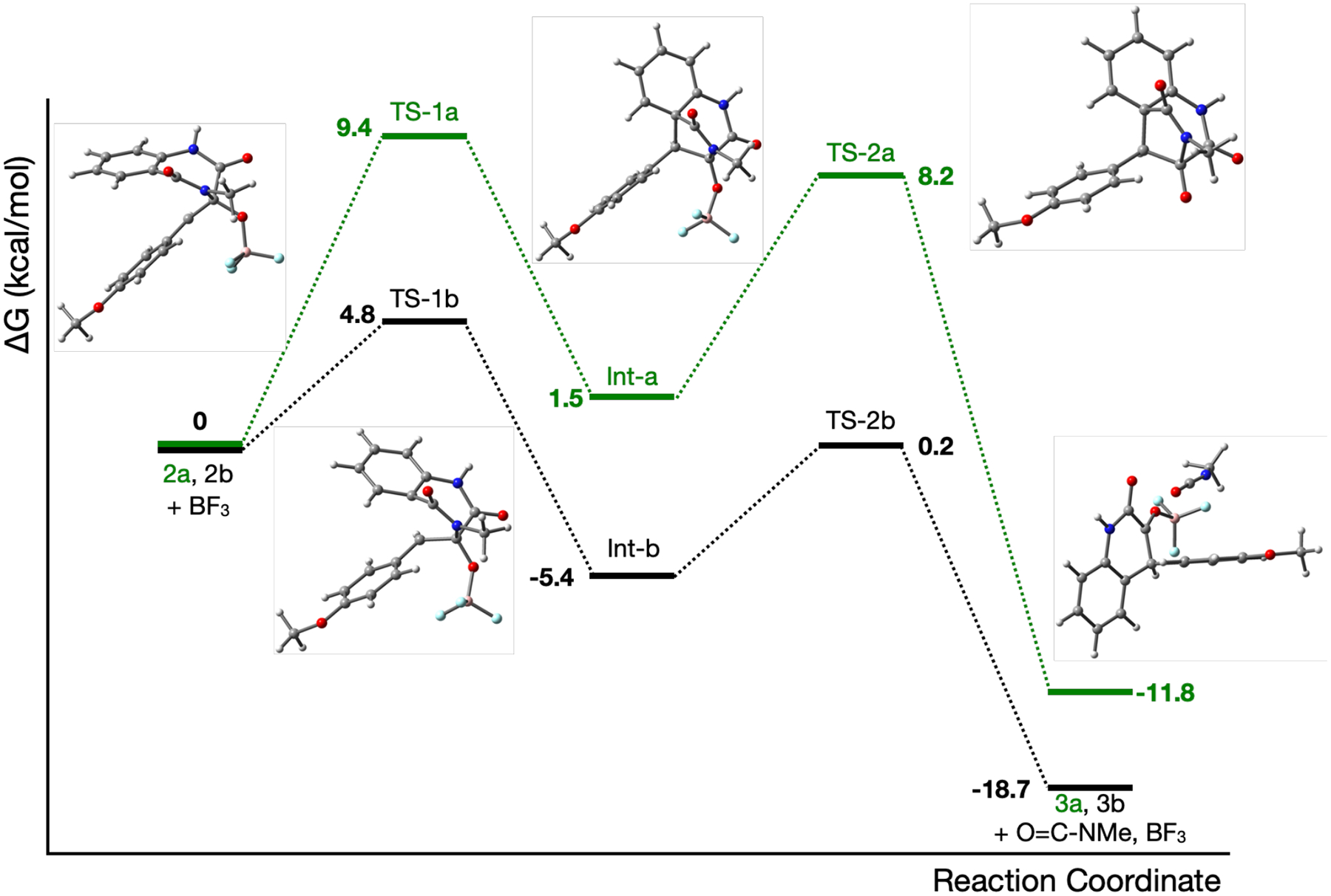
Reaction coordinate for the conversion of 2a (green) and 2b (black) to 3a and 3b in the presence of BF3. The numerical values indicate the calculated free energy differences of various species. The molecular structures of reactant, intermediate, product, and transition states of 2a → 3a are shown.
We then set out to test the rearrangement reaction by monitoring the conversion of 2a and 2b using 1H-NMR. Within 120 mins, no obvious substrate consumption can be observed under pH 5–8.5 (Figure S25, S26). These observations indicate the need of an acid for efficient rearrangement. To our delight, when using BF3 in dichloromethane, through a thin-layer-chromatographic analysis, we observed a complete consumption of the substrates (2a and 2b) and the formation of (4’-methoxy)viridicatin (3a and 3b). Applying this method also affords brominated viridicatins analogues (7a-7d). Although the origin of anti-Baldwin rearrangement remains to be carefully investigated, these results highlight the advantage of our approach which allows for the effective preparation of viridicatins through enzymatic and chemical reactions.
While enzymatic reactions are typically carried out on submilligram to miligram quantities to identify and characterize the reaction products, a potential challenge of preparative-scale reactions can serve as a bottleneck to the enzymatic transformations being embraced for synthetic applications. We envisioned transitioning from sequential reactions/purification to a scalable platform could benefit synthetic usefulness. Toward this goal, reactions using AsqJ-containing lysate were investigated. Although complete consumption of the substrate was achieved, the components originated from the crude lysate severely affect the rearrangement efficiency. Taking advantage of robust expression and stability of AsqJ (~ 38 mg protein per liter of culture), the scale-up reactions were conducted using the purified enzyme. After AsqJ completed epoxidation, the crude product obtained via a simple ethyl acetate extraction was subjected to BF3 catalyzed rearrangement to afford 7a-7d in gram scale with an overall yield of 57–91%.
To build quinolone library, we sought to leverage the reactivity of brominated viridicatins via cross-coupling reactions. We anticipated that a series of compounds can be accessed in one-step by reacting with cross-coupling partners. The 6-substituted vinyl quinolone skeleton is found in aflaquilonones, aspoquinolones, 4-phenyl-3,4-dihydroquinolones and yaequilonones.30–33 On the other hand, the naturally occurring 7-vinyl substituted quinolones are rare. Herein, we explored the synthesis of a library of 6- and 7-vinyl substituted quinolones. To access the 6-substituted vinyl quinolones, 7a and 7c were reacted with substituted alkenes under Heck reaction conditions34 to afford the desired product in reasonable yields (Figure 5 and Figure S94, 8a-(I-XI), 8c-(I-X)). Analogously, the 7-substituted vinyl quinolones were prepared using 7b and 7d as the substrates (9b-(I-XI), 9d-(I-X)). It is worthy to mention that using 1-vinylpyrrolidin-2-one and d-limonene as the coupling partners (X and XI) produces the anticipated products as well as the terminal olefin products (10a-(X-XI), 10b-(X-XI), 10c-X and 10d-X). Production of 10a-d-X likely originates from regio-selectivity at the migratory insertion. On the other hand, formation of 10a-XI and 10b-XI is due to the regio-selectivity of the beta-hydride elimination.
Figure 5.
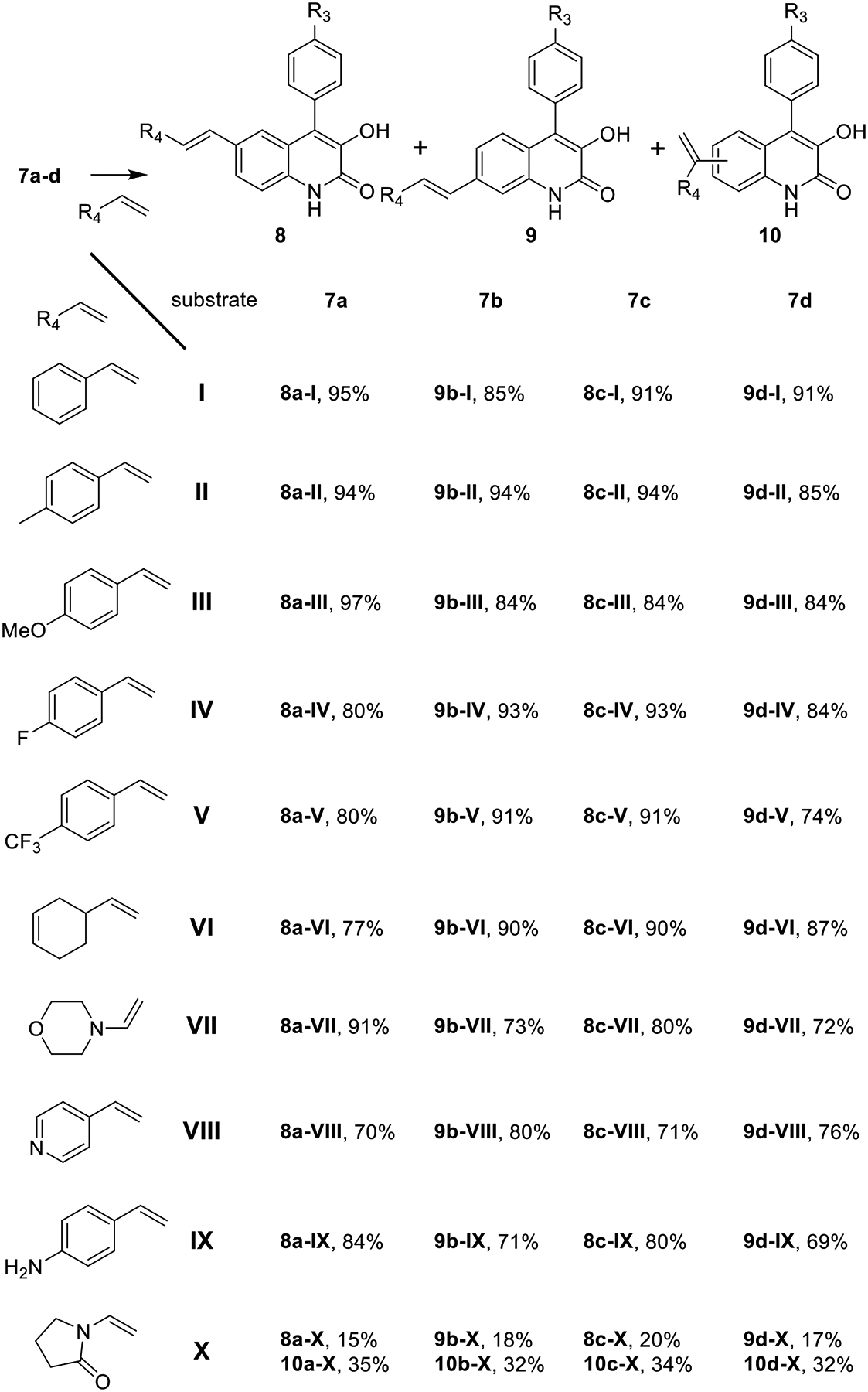
A library of quinolone analogs is prepared via chemo-enzymatic reactions. The products synthesized using XI are shown in the SI (Figure S94).
In summary, the results described herein demonstrated the potential to combine the current tools employed by chemistry and nature for the preparation of important molecules. The substrate diversity available from chemical synthesis, the robustness of AsqJ catalyzed asymmetric epoxidation, a simple Lewis Acid-catalyzed rearrangement, and the cross-coupling reactions have provided a platform for developing a suit of quinolone analogs. Leading by mechanistic and in silico analyses, the catalytic efficiency of AsqJ paired with the mild and environmentally friendly reaction conditions provide an opportunity to apply this biocatalyst in chemo-enzymatic reaction cascades. Along with the development of a biomimetic Lewis acid method of ring contraction, viridicatins can be prepared in gram scale quantity. Overall, this work sets an example to use tools employed by synthetic chemistry and nature for the effective preparation of quinolone alkaloids. It would be interesting to test the biological activities of newly synthesized compounds in future studies.
Supplementary Material
Acknowledgements
This work was supported by the grant from the National Institutes of Health (GM125924 to Y.G., W.-c. C, and M. K.). The authors thank Dr. Roger Sommer for helpful discussion. Crystallography was performed by the Molecular Education, Technology and Research Innovation Center at NC State University Funding for D8 VENTURE acquisition was provided by North Carolina Biotechnology Center (2019-IDG-1010).
Footnotes
Supporting Information
Experimental details, Figure S1–94, and Table S1–3
References
- 1.Heguy A; Cai P; Meyn P; Houck D; Russo S; Michitsch R; Pearce C; Katz B; Bringmann G; Feineis D; Taylor DL; Tyms AS, Isolation and characterization of the fungal metabolite 3-O-methylviridicatin as an inhibitor of tumour necrosis factor alpha-induced human immunodeficiency virus replication. Antivir. Chem. Chemother 1998, 9 (2), 149–55. [DOI] [PubMed] [Google Scholar]
- 2.Mizutani N; Aoki Y; Nabe T; Ishiwara M; Yoshino S; Takagaki H; Kohno S, Effect of TA-270, a novel quinolinone derivative, on antigen-induced nasal blockage in a guinea pig model of allergic rhinitis. Eur. J. Pharmacol 2009, 602 (1), 138–42. [DOI] [PubMed] [Google Scholar]
- 3.Duplantier AJ; Becker SL; Bohanon MJ; Borzilleri KA; Chrunyk BA; Downs JT; Hu LY; El-Kattan A; James LC; Liu S; Lu J; Maklad N; Mansour MN; Mente S; Piotrowski MA; Sakya SM; Sheehan S; Steyn SJ; Strick CA; Williams VA; Zhang L, Discovery, SAR, and pharmacokinetics of a novel 3hydroxyquinolin-2(1H)-one series of potent D-amino acid oxidase (DAAO) inhibitors. J. Med. Chem 2009, 52 (11), 3576–85. [DOI] [PubMed] [Google Scholar]
- 4.Suchaud V; Bailly F; Lion C; Tramontano E; Esposito F; Corona A; Christ F; Debyser Z; Cotelle P, Development of a series of 3-hydroxyquinolin-2(1H)-ones as selective inhibitors of HIV-1 reverse transcriptase associated RNase H activity. Bioorg. Med. Chem. Lett 2012, 22 (12), 3988–92. [DOI] [PubMed] [Google Scholar]
- 5.Muthukrishnan I; Sridharan V; Menendez JC, Progress in the Chemistry of Tetrahydroquinolines. Chem. Rev 2019, 119 (8), 5057–91. [DOI] [PubMed] [Google Scholar]
- 6.Sit SY; Ehrgott FJ; Gao JN; Meanwell NA, 3hydroxy-quinolin-2-ones: Inhibitors of [H-3]-glycine binding to the site associated with the NMDA receptor. Bioorganic. Med, Chem. Lett 1996, 6 (5), 499–504. [Google Scholar]
- 7.Chen MH; Fitzgerald P; Singh SB; O’Neill EA; Schwartz CD; Thompson CM; O’Keefe SJ; Zaller DM; Doherty JB, Synthesis and biological activity of quinolinone and dihydroquinolinone p38 MAP kinase inhibitors. Bioorg. Med. Chem. Lett 2008, 18 (6), 2222–6. [DOI] [PubMed] [Google Scholar]
- 8.Huntress EH; Bornstein J; Hearon WM, An Extension of the Diels-Reese Reaction. J. Am. Chem. Soc 1956, 78 (10), 2225–8. [Google Scholar]
- 9.Martin PK; Rapoport H; Smith HW; Wong JL, Synthesis of Cyclopenin and Isocyclopenin. J. Org. Chem 1969, 34 (5), 1359–63. [Google Scholar]
- 10.Smith HW; Rapoport H, Mechanism of Transformation of Cyclopenin to Viridicatin. J. Am. Chem. Soc 1969, 91 (22), 6083–9. [Google Scholar]
- 11.Kobayashi Y; Harayama T, A concise and versatile synthesis of viridicatin alkaloids from cyanoacetanilides. Org. Lett 2009, 11 (7), 1603–6. [DOI] [PubMed] [Google Scholar]
- 12.Paterna R; Andre V; Duarte MT; Veiros LF; Candeias NR; Gois PMP, Ring-Expansion Reaction of Isatins with Ethyl Diazoacetate Catalyzed by Dirhodium(II)/DBU MetalOrganic System: En Route to Viridicatin Alkaloids. Eur. J. Org. Chem 2013, 2013 (28), 6280–90. [Google Scholar]
- 13.Yuan Y; Yang R; Zhang-Negrerie D; Wang J; Du Y; Zhao K, One-pot synthesis of 3-hydroxyquinolin-2(1H)-ones from N-phenylacetoacetamide via PhI(OCOCF3)2-mediated alphahydroxylation and H2SO4-promoted intramolecular cyclization. J. Org. Chem 2013, 78 (11), 5385–92. [DOI] [PubMed] [Google Scholar]
- 14.Tangella Y; Manasa KL; Krishna NH; Sridhar B; Kamal A; Nagendra Babu B, Regioselective Ring Expansion of Isatins with In Situ Generated alpha-Aryldiazomethanes: Direct Access to Viridicatin Alkaloids. Org. Lett 2018, 20 (12), 3639–42. [DOI] [PubMed] [Google Scholar]
- 15.Ishikawa N; Tanaka H; Koyama F; Noguchi H; Wang CC; Hotta K; Watanabe K, Non-heme dioxygenase catalyzes atypical oxidations of 6,7-bicyclic systems to form the 6,6quinolone core of viridicatin-type fungal alkaloids. Angew. Chem. Int. Ed 2014, 53 (47), 12880–4. [DOI] [PubMed] [Google Scholar]
- 16.Kishimoto S; Hara K; Hashimoto H; Hirayama Y; Champagne PA; Houk KN; Tang Y; Watanabe K, Enzymatic one-step ring contraction for quinolone biosynthesis. Nat. Commun 2018, 9 (1), 2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brauer A; Beck P; Hintermann L; Groll M, Structure of the Dioxygenase AsqJ: Mechanistic Insights into a One-Pot Multistep Quinolone Antibiotic Biosynthesis. Angew. Chem. Int. Ed 2016, 55 (1), 422–6. [DOI] [PubMed] [Google Scholar]
- 18.Liao HJ; Li J; Huang JL; Davidson M; Kurnikov I; Lin TS; Lee JL; Kurnikova M; Guo Y; Chan NL; Chang W. c., Insights into the Desaturation of Cyclopeptin and its C3 Epimer Catalyzed by a non-Heme Iron Enzyme: Structural Characterization and Mechanism Elucidation. Angew. Chem. Int. Ed 2018, 57 (7), 1831–5. [DOI] [PubMed] [Google Scholar]
- 19.Mader SL; Brauer A; Groll M; Kaila VRI, Catalytic mechanism and molecular engineering of quinolone biosynthesis in dioxygenase AsqJ. Nat. Commun 2018, 9 (1), 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J; Liao HJ; Tang Y; Huang JL; Cha L; Lin TS; Lee JL; Kurnikov IV; Kurnikova MG; Chang W.-c.; Chan NL; Guo Y, Epoxidation Catalyzed by the Nonheme Iron(II)- and 2Oxoglutarate-Dependent Oxygenase, AsqJ: Mechanistic Elucidation of Oxygen Atom Transfer by a Ferryl Intermediate. J. Am. Chem. Soc 2020, 142 (13), 6268–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Straatsma TP; Mccammon JA, Computational Alchemy. Annu. Rev. Phys. Chem 1992, 43, 407–35. [Google Scholar]
- 22.Mobley DL; Klimovich PV, Perspective: Alchemical free energy calculations for drug discovery. J. Chem. Phys 2012, 137 (23), 230901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett CH, Efficient Estimation of Free-Energy Differences from Monte-Carlo Data. J. Comput. Phys 1976, 22 (2), 245–68. [Google Scholar]
- 24.Shirts MR; Pande VS, Comparison of efficiency and bias of free energies computed by exponential averaging, the Bennett acceptance ratio, and thermodynamic integration. J. Chem. Phys 2005, 122 (14), 144107. [DOI] [PubMed] [Google Scholar]
- 25.Matthews ML; Neumann CS; Miles LA; Grove TL; Booker SJ; Krebs C; Walsh CT; Bollinger JM Jr., Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc. Natl. Acad. Sci. U.S.A 2009, 106 (42), 17723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunham NP; Chang W.-c.; Mitchell AJ; Martinie RJ; Zhang B; Bergman JA; Rajakovich LJ; Wang B; Silakov A; Krebs C; Boal AK; Bollinger JM Jr., Two Distinct Mechanisms for C-C Desaturation by Iron(II)- and 2-(Oxo)glutarate-Dependent Oxygenases: Importance of alpha-Heteroatom Assistance. J. Am. Chem. Soc 2018, 140 (23), 7116–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Einsiedler M; Jamieson CS; Maskeri M; Houk KN; Gulder TAM, Fungal Dioxygenase AsqJ Is Promiscuous and Bimodal: Substrate-Directed Formation of Quinolones versus Quinazolinones. Angew. Chem. Int. Ed 2021, 60, 8297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomasi J; Mennucci B; Cammi R, Quantum mechanical continuum solvation models. Chem. Rev 2005, 105 (8), 2999–3093. [DOI] [PubMed] [Google Scholar]
- 29.Wojdyla Z; Borowski T, On how the binding cavity of AsqJ dioxygenase controls the desaturation reaction regioselectivity: a QM/MM study. J. Biol. Inorg. Chem 2018, 23 (5), 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uchida R; Imasato R; Tomoda H; Omura S, Yaequinolones, new insecticidal antibiotics produced by Penicillium sp. FKI-2140. II. Structural elucidation. J. Antibiot 2006, 59 (10), 652–8. [DOI] [PubMed] [Google Scholar]
- 31.Scherlach K; Hertweck C, Discovery of aspoquinolones A-D, prenylated quinoline-2-one alkaloids from Aspergillus nidulans, motivated by genome mining. Org. Biomol. Chem 2006, 4 (18), 3517–20. [DOI] [PubMed] [Google Scholar]
- 32.Neff SA; Lee SU; Asami Y; Ahn JS; Oh H; Baltrusaitis J; Gloer JB; Wicklow DT, Aflaquinolones A-G: secondary metabolites from marine and fungicolous isolates of Aspergillus spp. J. Nat. Prod 2012, 75 (3), 464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.An CY; Li XM; Luo H; Li CS; Wang MH; Xu GM; Wang BG, 4-Phenyl-3,4-dihydroquinolone derivatives from Aspergillus nidulans MA-143, an endophytic fungus isolated from the mangrove plant Rhizophora stylosa. J. Nat. Prod 2013, 76 (10), 1896–901. [DOI] [PubMed] [Google Scholar]
- 34.Heck RF; Nolley JP, Palladium-Catalyzed Vinylic Hydrogen Substitution Reactions with Aryl, Benzyl, and Styryl Halides. J. Org. Chem 1972, 37 (14), 2320–2. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.