

Abstract
Although phylogeny estimation is notoriously difficult in radiations that occurred several hundred million years ago, phylogenomic approaches offer new ways to examine relationships among ancient lineages and evaluate hypotheses that are key to evolutionary biology. Here, we reconstruct the deep-rooted relationships of one of the oldest living arthropod clades, the branchiopod crustaceans, using a kaleidoscopic approach. We use concatenation and coalescent tree-building methods to analyze a large multigene data set at the nucleotide and amino acid level and examine gene tree versus species tree discordance. We unequivocally resolve long-debated relationships among extant orders of the Cladocera, the waterfleas, an ecologically relevant zooplankton group in global aquatic and marine ecosystems that is famous for its model systems in ecology and evolution. To build the data set, we assembled eight de novo genomes of key taxa including representatives of all extant cladoceran orders and suborders. Our phylogenetic analysis focused on a BUSCO-based set of 823 conserved single-copy orthologs shared among 23 representative taxa spanning all living branchiopod orders, including 11 cladoceran families. Our analysis supports the monophyly of the Cladocera and reveals remarkable homoplasy in their body plans. We found large phylogenetic distances between lineages with similar ecological specializations, indicating independent evolution in major body plans, such as in the pelagic predatory orders Haplopoda and Onychopoda (the “Gymnomera”). In addition, we assessed rapid cladogenesis by estimating relative timings of divergence in major lineages using reliable fossil-calibrated priors on eight nodes in the branchiopod tree, suggesting a Paleozoic origin around 325 Ma for the cladoceran ancestor and an ancient rapid radiation around 252 Ma at the Perm/Triassic boundary. These findings raise new questions about the roles of homoplasy and rapid radiation in the diversification of the cladocerans and help examine trait evolution from a genomic perspective in a functionally well understood, ancient arthropod group. [Cladocera; Daphnia; evolution; homoplasy; molecular clock; phylogenomics; systematics; waterfleas.]
Reconstructing the evolutionary history of ancient clades is notoriously difficult. The older the clade, the harder it becomes to establish the firm phylogenetic foundations needed to test evolutionary theories. Relationships among deeply divergent lineages are blurred behind a multilayered curtain of hundreds of millions of years of evolution. Phylogenomic approaches are often expected to remove this veil, yet a significant degree of uncertainty is inevitable due to extinctions, the erosion of the phylogenetic signal over time, and analytical artifacts (Delsuc et al. 2005; Whitfield and Lockheart 2007; Telford et al. 2015; Arcila et al. 2017). Enhancing the phylogenetic resolution among highly divergent lineages remains a major challenge in systematic biology.
Interpreting deep evolutionary histories is complicated by the phenomenon of homoplasy, that is, traits that may have evolved multiple times independently in different parts of the tree. Homoplasy requires looking behind the curtain of time from different angles and exploring different data sets using independent, reliable methods (Wake et al. 2011). Phylogenomic approaches, which have recently been used to suggest or confirm phenotypic homoplasy in several deeply divergent lineages (Kusy et al. 2019; Ramírez et al. 2021), have been highly informative when applied critically (Meusemann et al. 2010). A kaleidoscopic or multifacetted analysis of large multilocus data sets can assess the strengths of evolutionary inference (Arcila et al. 2017; Alda et al. 2019; Morales-Briones et al. 2021), as it uses both coalescent and concatenation tree-building methods (Xi et al. 2014), identifies conflicts between species trees and gene trees (Roycroft et al. 2020; Hime et al. 2021), and examines the effects of gene filtering, alignments, partitioning, model selection, and data type (Kainer and Lanfear 2015; Molloy and Warnow 2018; Abadi et al. 2019; Widhelm et al. 2019; Noah et al. 2020). Here, we present a case study of an ancient aquatic arthropod group using a composite of approaches to assess the potential of homoplasy. In addition, we include divergence time estimates to examine rapid cladogenesis.
Arthropods represent the most speciose and diversified animal phylum, occupying vital roles in a wide range of niches in major ecosystems, accompanied by key evolutionary innovations over a vast geological time span. Their morphological diversity can be traced back to conserved arthropod body plans, whose alterations over time are recorded in the genome. These phenotypical changes and the underlying evolutionary mechanisms are best unraveled in a phylogenetic context (Giribet and Edgecombe 2019; Thomas et al. 2020). Among major arthropod lineages, Crustacea exhibit arguably the widest diversity of functional traits in marine and freshwater habitats; however, despite their potential as a model group for understanding the genomic basis of trait evolution over time, study of their genomes has lagged behind similar studies in insects and vertebrates (Meusemann et al. 2010; Havird and Santos 2016; Rotllant et al. 2018).
The Branchiopoda, often represented in comparative arthropod genomic studies by the cladoceran Daphnia O. F. Müller, 1785, is a key lineage for dissecting genome evolution in the Pancrustacea tree of life. They are among the most primitive of extant crustaceans (Fryer 1987a). One of the best-studied arthropod taxa in the world, the model organism Daphnia is the first crustacean for which an annotated genome was available (Colbourne et al. 2011; Lampert 2011; Miner et al. 2012). The highly diverse Cladocera clade, to which Daphnia belongs, is an ecologically and functionally well-understood lineage within the Branchiopoda, constituting an excellent group to examine patterns of arthropod evolution through phylogenetic analysis. Here, we use a multilocus phylogenomic approach to understand the drivers of biological diversity and specifically the role of homoplasy by focusing on the Cladocera as a model taxon.
The Study System
The Cladocera (Crustacea: Branchiopoda), commonly called waterfleas, have successfully colonized a wide range of niches in freshwater, brackish, and marine environments occupying benthic, littoral, and pelagic habitats (Dumont and Negrea 2002). These small animals constitute an old group, with the first unambiguous fossils dating back to the Mesozoic. Their evolutionary history is closely linked to the evolution of aquatic ecosystems over hundreds of millions of years (Van Damme and Kotov 2016). The antiquity of the group is confirmed by fossils and numerous phylogeographic studies (Van Damme and Kotov 2016). Several cladoceran orders have gone extinct since the Jurassic, with the current diversity likely representing only a fraction of what it once was (Korovchinsky 2006; Van Damme and Kotov 2016). The Branchiopoda, of which the Cladocera are the most speciose clade, are considered to have originated in the Cambrian and were suggested to be among the first arthropod lineages to have colonized inland waters from the sea (Harvey et al. 2012; Van Damme and Kotov 2016).
Cladocerans exhibit a wide diversity of body types with elaborate morphologies characterized by bizarre forms and some of the most complex limb arrangements known in crustaceans (Fryer 1968, 1991). Deriving from a basic cladoceran body plan, morphological diversity in waterfleas is strongly linked to aquatic niche and feeding method, which include suspension feeding in the pelagic (Daphnia, Moina Baird, 1850, Bosmina Baird, 1845) or hyponeustonic (Scapholeberis Schoedler, 1858), suspension/deposit-feeding in benthic-littoral environments (Chydorus Leach, 1816; Macrothrix Baird, 1843; Eurycercus Baird, 1843), pure benthic feeding (Ilyocryptus Sars, 1862) and predation in the pelagic using special raptorial limbs (Leptodora Lilljeborg, 1861; Cercopagis Sars, 1897) (e.g., Fryer 1968, 1995; Dumont and Negrea 2002). Functional traits linked to the major feeding modes govern the deeper systematics of the cladocerans (Dumont and Negrea 2002; Kotov 2013), which have not been resolved by molecular phylogenetics.
Cladoceran systematics based on both morphological and molecular data have been widely discussed (Negrea et al. 1999; Dumont and Negrea 2002; Kotov 2013), yet the relationships between cladoceran lineages and the derivations of body types remain elusive (Fig. S1 of the Supplementary material available on Dryad at https://doi.org/10.5061/dryad.mkkwh711b; Table S1 of the Supplementary material available on Dryad). The relationships are debated ever since Georg Ossian Sars described the four extant cladoceran orders in the 19th century (Sars 1865; Forró et al. 2008), largely because the ancient nature and morphological disparity of the Branchiopoda combine to obscure their ancestry and evolutionary history (Fryer 1987a). Molecular phylogenies based on a few genes and morphological data generally support the idea that the Cladocera are monophyletic, with Cyclestheria Sars, 1887 (Cyclestherida) commonly accepted as the sister lineage (e.g., de Waard et al. 2006; Richter et al. 2007; Fig. S1 of the Supplementary material available on Dryad); however, the monophyly of the Cladocera has been challenged, partly due to the lack of a comprehensive morphological analysis (Fryer 1987b, 1999).
An example of phylogenetic uncertainty among cladoceran orders is the debated relationship between the Anomopoda and the Ctenopoda, sometimes grouped together under the Calyptomera (Fig. S1 of the Supplementary material available on Dryad; Table S1 of the Supplementary material available on Dryad). Except for the generally assumed Gymnomera-concept that groups the predatory orders Onychopoda and Haplopoda, there is no consensus (Fig. S1c of the Supplementary material available on Dryad; Swain and Taylor 2003; Forró et al. 2008; Olesen 2009). Within the Anomopoda, hypotheses on how the families relate are based on morphological data, mainly linked to feeding modes (Dumont and Silva-Briano 1998; Kotov 2013). The first studies on (mito)genomes and transcriptomes in the context of cladoceran phylogeny have not examined phylogenetic uncertainties in the group at the deeper levels (Schwentner et al. 2018; Cornetti et al. 2019).
Understanding deep evolutionary relationships in cladocerans is further complicated by their relatively sparse fossil record and the extinctions of higher lineages (Korovchinsky 2006; Kotov 2009; Van Damme and Kotov 2016). For many extant cladoceran lineages, such as the predatory Haplopoda and Onychopoda and most of the Anomopoda families, ages of divergence are undefined, as no pre-Quaternary fossils have been recorded (Van Damme and Kotov 2016). To fill these gaps, divergence times within the Cladocera need to be assessed in a wider paleontological context that the extensive fossil record of larger branchiopods and recently revised cladoceran records now provide (Van Damme and Kotov 2016; Wolfe et al. 2016).
Here, we present a first exploration of the evolutionary history in the Cladocera using a multilocus phylogenomic approach, including representatives of all four extant cladoceran orders that we analyzed in a phylogenomic and fossil-calibrated context against all five extant noncladoceran branchiopod orders. We examine the relationships of the deep lineages, assess the timing of their diversification, and assess the potential of independent evolution and ancient rapid radiation as evolutionary mechanisms.
Materials and Methods
In addition to eight de novo cladoceran genomes, we included representative assemblies of each genus and subgenus in the Daphniidae, one species of the Moinidae and the Bosminidae assembled by Cornetti et al. (2019), three transcriptomes from Schwentner et al. (2018), and other genomes from available databases; in total, we included 23 branchiopod taxa to provide the first phylogenomic analysis of all extant branchiopod orders (Table S2 of the Supplementary material available on Dryad). Our study includes the first de novo high-quality genome assemblies for the families Sididae, Ilyocryptidae, Macrothricidae, Chydoridae, Eurycercidae, Podonidae, and Leptodoridae, and the first genomes of three of the four cladoceran orders (Onychopoda, Ctenopoda, and Haplopoda). Our analysis covers all orders, all suborders and 11 of the 20 extant families in the Cladocera of which 7 (of 11) are in the Anomopoda sensuKotov (2013). The families included in our study harbor a total diversity of at least 90% of the known species in the Cladocera, which are mainly found in the Daphniidae and the Chydoridae (Forró et al. 2008; Kotov et al. 2013). The taxon sampling is therefore comprehensive, aimed at examining relationships between the major cladoceran families and (sub)orders. All data matrices are submitted to GenBank (ENA study number ERZ1964691, project ID: PRJEB44293, accession numbers: OC977944-OC995744).
All nuclear assemblies were assessed for biological completeness using BUSCOv3 (Benchmarking Universal Single-Copy Orthologs) which contains a set of conserved single-copy genes that are present across the arthropod Tree of Life (Simão et al. 2015; Waterhouse et al. 2018, 2019). A total of 1066 single-copy arthropod genes were searched against each individual assembly. We performed multiple phylogenomic analyses using concatenation and coalescent tree-building methods (Lartillot et al. 2013; Zhang et al. 2018; Kozlov et al. 2019; Kapli et al. 2020), taking into account individual substitution models per gene and the effects of saturation (Figs. S2 and S3 of the Supplementary material available on Dryad). We used partitioned data sets of the 823 BUSCO genes as input for the analysis, identified by PartitionFinder v2.1.1 (Lanfear et al. 2017). Molecular clock and species divergence estimates were analyzed in BEAST 2.4.5 (Bouckaert et al. 2014) using the first and second codon position, after examining saturation plots for each codon position and 4-fold degenerate sites (Fig. S2 of the Supplementary material available on Dryad) and taking into account the best substitution models (Fig. S3 of the Supplementary material available on Dryad); we applied eight fossil-calibrated nodes to the branchiopod tree after Van Damme and Kotov (2016). Gene tree–species tree discordance was tested using a multidimensional scaling (MDS) approach (Duchêne et al. 2018; Roycroft et al. 2020). For both the nuclear and amino acid data sets, we calculated the pairwise Robinson–Foulds phylogenetic distances (Robinson and Foulds 1981) of all gene trees, including the best ML species trees obtained by RAxML-NG. Details on genomic DNA extraction, transcriptome and genome assembly, ortholog identification, phylogenomic analyses, data partitioning, gene saturation, model selection, gene tree-species tree discordance (between the nucleotide and amino acid data sets), and fossil-calibrated species divergence estimates, are available as Text ST1 of the Supplementary material available on Dryad.
Results
Phylogenomic Trees
We obtained good quality drafts with an average of 86% complete single-copy arthropod orthologs for the eight newly assembled nuclear genomes and 89% complete single-copy arthropod orthologs for all taxa. Using these genes, together with the complete BUSCOs retrieved for the other Branchiopoda, we compiled a matrix including 823 orthologs containing 10.49% missing data and an average of 2.41 taxa missing per gene (Table S3 of the Supplementary material available on Dryad).
Following the PartitionFinder results, we merged the 823 proteins shared among all branchiopod taxa in our matrix into 319 partitions, which were used to generate the amino acid ML tree with RAxML-NG (Fig. 1; Fig. S4 of the Supplementary material available on Dryad). This tree showed maximal bootstrap support (100) for all nodes except for one internal node in the Daphniidae (88) and the Bosmina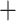 Radopoda node (98) (Fig. S4 of the Supplementary material available on Dryad). When we scored the latter species tree using ASTRAL-III, we obtained maximal (1) or very high (
Radopoda node (98) (Fig. S4 of the Supplementary material available on Dryad). When we scored the latter species tree using ASTRAL-III, we obtained maximal (1) or very high (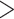 0.9) support for all nodes, except for the Moina
0.9) support for all nodes, except for the Moina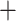 Macrothrix (0.29) node (Fig. 1; Fig. S4 of the Supplementary material available on Dryad). The ASTRAL amino acid tree topology (not shown) was identical to that of the RAxML-NG amino acid tree except for one grouping: Macrothrix formed a sister group to the Daphniidae but remained in one branch with the Aradopoda, supported by posterior probability values of 1 (Moina
Macrothrix (0.29) node (Fig. 1; Fig. S4 of the Supplementary material available on Dryad). The ASTRAL amino acid tree topology (not shown) was identical to that of the RAxML-NG amino acid tree except for one grouping: Macrothrix formed a sister group to the Daphniidae but remained in one branch with the Aradopoda, supported by posterior probability values of 1 (Moina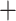 (Macrothrix
(Macrothrix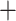 Daphniidae)) and 0.56 (Macrothrix
Daphniidae)) and 0.56 (Macrothrix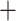 Daphniidae), respectively. Branch lengths for the RAxML-NG protein tree are shown in Fig. S4 of the Supplementary material available on Dryad (Fig. 1 is simplified to focus on the lineage relationships) and those for the RAxML-NG nucleotide tree are shown in Fig. S5 of the Supplementary material available on Dryad. The PhyloBayes amino acid tree (Fig. S6 of the Supplementary material available on Dryad) showed maximal (1) support for all nodes (one of 0.99) and the same topology as the RAxML-NG amino acid tree (Fig. S4 of the Supplementary material available on Dryad).
Daphniidae), respectively. Branch lengths for the RAxML-NG protein tree are shown in Fig. S4 of the Supplementary material available on Dryad (Fig. 1 is simplified to focus on the lineage relationships) and those for the RAxML-NG nucleotide tree are shown in Fig. S5 of the Supplementary material available on Dryad. The PhyloBayes amino acid tree (Fig. S6 of the Supplementary material available on Dryad) showed maximal (1) support for all nodes (one of 0.99) and the same topology as the RAxML-NG amino acid tree (Fig. S4 of the Supplementary material available on Dryad).
Figure 1.

RAxML-NG amino acid tree hypothesis of all extant branchiopod orders based on the BUSCO-matrix of 823 single-copy orthologous genes, merged into 319 partitions. Bootstrap values of the ML analysis and posterior probability values of the ASTRAL-III analysis are shown on the nodes. Inset above left: simplified topology of the extant Cladoceromorpha (four extant cladoceran orders and Cyclestherida) according to the phylogenomic analysis, showing polyphyly of the predatory orders (Gymnomera) and two new superorders, Oligopoda and Magipoda. Predatory body plans in red, suspension- and deposit-feeding body plans in blue, outgroup taxa in grey. For branch lengths, see original tree (Fig. S4 of the Supplementary material available on Dryad).
The topologies of the RAxML-NG, PhyloBayes, and ASTRAL-III amino acid trees all supported the Cladoceromorpha (Cyclestherida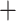 Cladocera) and the four orders of the Cladocera as monophyletic (Table S4 of the Supplementary material available on Dryad); none of the analyses supported the Gymnomera (Onychopoda
Cladocera) and the four orders of the Cladocera as monophyletic (Table S4 of the Supplementary material available on Dryad); none of the analyses supported the Gymnomera (Onychopoda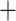 Haplopoda) or the Calyptomera (Anomopoda
Haplopoda) or the Calyptomera (Anomopoda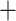 Ctenopoda). The Haplopoda and the Ctenopoda formed a single clade and a sister group to the Onychopoda and Anomopoda, which grouped together as well (Fig. 1). Support for these superorder groupings was maximal in all analyses (bootstrap 100 or posterior probability values 1; Table S4 of the Supplementary material available on Dryad). In the Anomopoda, the suborders Aradopoda and Radopoda and the superfamily Macrothricoidea sensuDumont and Silva-Briano (1998) and Kotov (2013) both appeared polyphyletic in our amino acid and nucleotide RAxML-NG trees and in the PhyloBayes tree (Fig. 1; Figs. S4–S6 of the Supplementary material available on Dryad); the radopod taxon Macrothrix (Macrothricidae) grouped with the aradopod Moina (Moinidae) in one clade, jointly forming a sister group to the Daphniidae. The genera in the latter family related to each other as in the amino acid tree in Cornetti et al. (2019). Our PhyloBayes tree provided an additional test for heterogeneity of the protein data for the unexpected clade Macrothrix
Ctenopoda). The Haplopoda and the Ctenopoda formed a single clade and a sister group to the Onychopoda and Anomopoda, which grouped together as well (Fig. 1). Support for these superorder groupings was maximal in all analyses (bootstrap 100 or posterior probability values 1; Table S4 of the Supplementary material available on Dryad). In the Anomopoda, the suborders Aradopoda and Radopoda and the superfamily Macrothricoidea sensuDumont and Silva-Briano (1998) and Kotov (2013) both appeared polyphyletic in our amino acid and nucleotide RAxML-NG trees and in the PhyloBayes tree (Fig. 1; Figs. S4–S6 of the Supplementary material available on Dryad); the radopod taxon Macrothrix (Macrothricidae) grouped with the aradopod Moina (Moinidae) in one clade, jointly forming a sister group to the Daphniidae. The genera in the latter family related to each other as in the amino acid tree in Cornetti et al. (2019). Our PhyloBayes tree provided an additional test for heterogeneity of the protein data for the unexpected clade Macrothrix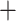 Moina. Showing the same tree topology with maximal support (1) for this group in the PhyloBayes analysis (Fig. S6 of the Supplementary material available on Dryad), Macrothrix placement separate from the other Radopoda in our study probably does not stem from long-branch attraction. The remaining Radopoda clustered together in a highly supported clade (Fig. 1; Figs. S4–S6 of the Supplementary material available on Dryad), with Bosmina as a sister group to the other radopod families except for the Macrothricidae, followed by the monophyletic Ilyocryptidae and finally the superfamily Eurycercoidea (Eurycercidae
Moina. Showing the same tree topology with maximal support (1) for this group in the PhyloBayes analysis (Fig. S6 of the Supplementary material available on Dryad), Macrothrix placement separate from the other Radopoda in our study probably does not stem from long-branch attraction. The remaining Radopoda clustered together in a highly supported clade (Fig. 1; Figs. S4–S6 of the Supplementary material available on Dryad), with Bosmina as a sister group to the other radopod families except for the Macrothricidae, followed by the monophyletic Ilyocryptidae and finally the superfamily Eurycercoidea (Eurycercidae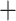 Chydoridae) as defined in Dumont and Silva-Briano (1998).
Chydoridae) as defined in Dumont and Silva-Briano (1998).
The topology of the partitioned nucleotide ML tree (RAxML-NG), based on the BUSCO-matrix of 823 single-copy orthologs merged into 995 partitions following the PartitionFinder results (Fig. S5 of the Supplementary material available on Dryad), was identical to the amino acid tree (Fig. 1; Fig. S4 of the Supplementary material available on Dryad). When scoring the nucleotide RAxML-NG tree with ASTRAL-III, we found maximal (1) or almost maximal (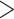 0.9) support for all nodes except for the grouping of Bosmina
0.9) support for all nodes except for the grouping of Bosmina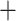 Radopoda (0.52) and Moina
Radopoda (0.52) and Moina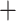 Macrothrix (0.01) (Fig. S5 and Table S4 of the Supplementary material available on Dryad). The ASTRAL nucleotide tree obtained from individual gene trees (not shown) was identical in topology to the RAxML-NG nucleotide tree, except that Bosmina appeared as a sister taxon to Moina
Macrothrix (0.01) (Fig. S5 and Table S4 of the Supplementary material available on Dryad). The ASTRAL nucleotide tree obtained from individual gene trees (not shown) was identical in topology to the RAxML-NG nucleotide tree, except that Bosmina appeared as a sister taxon to Moina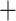 Macrothrix
Macrothrix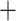 Daphniidae with low support (0.46) and Moina as a sister lineage to Macrothrix
Daphniidae with low support (0.46) and Moina as a sister lineage to Macrothrix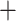 Daphniidae with high support (1) (Table S4 of the Supplementary material available on Dryad).
Daphniidae with high support (1) (Table S4 of the Supplementary material available on Dryad).
In all the above trees, both at the nucleotide and the amino acid level, the groupings of all branchiopod orders were maximally supported, showing a relation- ship in the cladoceran orders between Onychopoda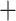 Anomopoda and Haplopoda
Anomopoda and Haplopoda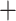 Ctenopoda. In addition, Macrothrix consistently clustered with the suborder Aradopoda (not in the Radopoda) with maximal confidence (bootstrap 100 or posterior probability value 1) (Table S4 of the Supplementary material available on Dryad). The mitogenome RAxML tree was not informative for this type of deep phylogeny (Fig. S7 of the Supplementary material available on Dryad).
Ctenopoda. In addition, Macrothrix consistently clustered with the suborder Aradopoda (not in the Radopoda) with maximal confidence (bootstrap 100 or posterior probability value 1) (Table S4 of the Supplementary material available on Dryad). The mitogenome RAxML tree was not informative for this type of deep phylogeny (Fig. S7 of the Supplementary material available on Dryad).
We chose the amino acid tree, which is more conservative and more informative for the deeper relationships among the Cladocera, as being the most reliable and, therefore, most representative tree (Fig. 1). There is a higher probability of saturation in the nucleotide data set and there is relatively less conflict of gene trees versus species trees in the amino acid data set (Fig. 2; Fig. S2 of the Supplementary material available on Dryad).
Figure 2.

Multidimensional Scaling (MDS) of the pairwise Robinson-Foulds phylogenetic distance among all the amino acid (a) and nucleotide gene trees (c). Each dot represents a gene tree and is colored based on the average bootstrap support calculated among all nodes in its topology. In the pairwise comparisons, the relative data set (species tree) is included as a blue dot indicated by a black arrow, obtained using RAxML-NG from the concatenated partitioned matrices. Average bootstrap values of both data sets are shown in (b) where the black dots inside the violin plots represent the mean values (for the amino acid and nucleotide data sets), while the black whiskers indicate the standard deviations.
Gene versus Species Trees
For both nucleotide and amino matrices, we observed a substantial degree of discordance between individual gene trees and the species tree, as well as wide variation in the phylogenetic distance among gene trees and tree support. The distribution is scattered for both data sets (Fig. 2). None of the gene trees completely matched the topology of the inferred species trees (Fig. 2a,c). In the MDS plot, the gene trees with lower average bootstraps, which contain less phylogenetic information, tend to show greater distance to the species tree compared to gene trees with higher average bootstraps, which have higher phylogenetic information content (Fig. 2a,c). The amino acid gene trees showed significantly higher (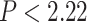 e-16) average bootstrap values than the nucleotide gene trees (Fig. 2b), indicating that our amino acid data set contains more robust phylogenetic content, making it preferable to the nucleotide data set for a clade as ancient as the Branchiopoda.
e-16) average bootstrap values than the nucleotide gene trees (Fig. 2b), indicating that our amino acid data set contains more robust phylogenetic content, making it preferable to the nucleotide data set for a clade as ancient as the Branchiopoda.
Divergence Time Estimates
Our molecular clock estimates were based on the combined first and second codon positions and showed the same topology as the RAxML-NG amino acid (Fig. 1; Fig. S4 of the Supplementary material available on Dryad) and nucleotide analysis (Fig. S5 of the Supplementary material available on Dryad) except for Bosmina (Fig. 3). All nodes in the molecular clock tree were maximally supported except for the node in the Chydoridae between the subfamilies Aloninae (Alona Baird, 1843) and the Chydorinae (Chydorus) (0.96); confidence intervals were relatively small (Fig. 3; Table S5 of the Supplementary material available on Dryad).
Figure 3.

Fossil-calibrated molecular phylogeny of the Branchiopoda. The BEAST tree is based on the combined first and second codon positions of the BUSCO nucleotide matrix containing 823 single-copy orthologs, constrained with eight fossil-calibrated uniform priors (red stars). Posterior probability values for all nodes are 1 except for the internal Chydoridae node (0.96). Blue bars indicate the 95% confidence intervals of the node ages. Black arrow below indicates the Permian/Triassic (P/T) boundary. Differences in topology from the RaxML-NG amino acid tree are indicated with asterisk.
According to the divergence time estimates of key nodes (Fig. 3; Table S5 of the Supplementary material available on Dryad), the most recent common ancestor (MRCA) of the Diplostraca dates from around 455 Ma (Ordovician), while the MRCA of the Onychocaudata (Spinicaudata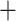 Cladoceromorpha) dates from about 390 Ma (Devonian). The estimated node ages of the orders and families predate the fossil data considerably (Van Damme and Kotov 2016), with the nodes of the stem and crown group of the Cladocera falling in the Paleozoic (Fig. 3). The MRCA of extant Cladoceromorpha is estimated around 362 Ma (Late Devonian) and that of the Cladocera at about 325 Ma (Early Carboniferous near the end of the Mississippian). All cladoceran orders and families included in our analysis diverged over a period of about 120 Ma around the Paleozoic-Mesozoic boundary, between the Carboniferous and the Triassic (210–330 Ma).
Cladoceromorpha) dates from about 390 Ma (Devonian). The estimated node ages of the orders and families predate the fossil data considerably (Van Damme and Kotov 2016), with the nodes of the stem and crown group of the Cladocera falling in the Paleozoic (Fig. 3). The MRCA of extant Cladoceromorpha is estimated around 362 Ma (Late Devonian) and that of the Cladocera at about 325 Ma (Early Carboniferous near the end of the Mississippian). All cladoceran orders and families included in our analysis diverged over a period of about 120 Ma around the Paleozoic-Mesozoic boundary, between the Carboniferous and the Triassic (210–330 Ma).
Diversification of the cladoceran orders is estimated towards the end of the Paleozoic between 260 Ma and 330 Ma (Carboniferous—Permian), with several events that led to main lineages occurring within a relatively short time frame. For example, the divergence between the Haplopoda and Ctenopoda, the MRCA of the two Onychopoda families in our study, and the MRCA of the Anomopoda are similarly situated around 260–280 Ma, diverging over ca. 20 Myr, in the Permian (Table S5 of the Supplementary material available on Dryad). Divergence estimates of the anomopod families fall in the early Mesozoic about 215–250 Ma (Triassic). Among the taxa considered, the Ilyocryptidae and Bosminidae appear as the first anomopod families at the Perm/Triassic (P/T) boundary (ca. 250 Ma) (Fig. 3). The two subfamilies in the Chydoridae (Aloninae-Chydorinae) are estimated to have separated around 223 Ma (Late Triassic), shortly after the MRCA of the Eurycercoidea (Eurycercidae—Chydoridae; ca. 230 Ma); the latter appears around the same time as the MRCA of Daphniidae, Moinidae, and Macrothricidae (233 Ma) and just before the MRCA of Moinidae and Macrothricidae (217 Ma) in the Late Triassic. The MRCA of the five Daphniidae genera is estimated at 162 Ma (Late Jurassic) at least.
The obtained divergence rates are sensitive to the choice of codon position. When we used the highly saturated 4-fold degenerate sites (4fds) as input in BEAST, the Cladocera MRCA appeared to occur around 410–440 Ma (Fig. S8 of the Supplementary material available on Dryad), about 100 Myr earlier than the output shown here (Fig. 3). The origins of the cladoceran orders and families therefore seem considerably older in the 4fds tree, with differences in topology in the positions of Moina, Bosmina, and Scapholeberis as well (Fig. S8 of the Supplementary material available on Dryad). The origins of the Daphniidae genera also skew older using the nodes in the 4fds data set (Fig. S8 of the Supplementary material available on Dryad; Cornetti et al. 2019) than in the 1st–2nd codon positions (Fig. 3). Time estimates should, thus, be interpreted with care, taking codon positions into account, as different molecular clock approaches may produce different time estimates (Van Damme and Kotov 2016; Cornetti et al. 2019). In our findings, saturation clearly affected the topology of the tree and timing of the nodes; thus, we consider our analysis using the combined 1st and 2nd codon positions the most accurate and conservative approach to the molecular clock, producing relatively younger minimal estimates for the data set at hand (Fig. 3). Saturation made the 4fds less reliable for a time calibration analysis over this time span (as the 3d codon positions; Fig. S2 of the Supplementary material available on Dryad).
Discussion
Our study sought to resolve the evolutionary history of a morphologically and ecologically diversified group of arthropods, the Cladocera, whose deepest branching events go back over 300 Ma. Although limited and often biased information make such deep phylogenetic events notoriously difficult to study, we overcame this problem by using whole genome sequencing and an analysis based on more than 800 conserved genes. We found that a combined use of approaches, including fossil-calibrated divergence time estimates, provided a useful way to discover the evolutionary history of ancient clades and assess the potential presence of convergent evolution. In recent studies, a critical comparison of different methods helped identify some of the more difficult relationships among organisms and avoided potential artifacts linked to concatenation approaches (Arcila et al. 2017; Abadi et al. 2019). Assessing gene tree-species tree discordance is vital to such multifaceted phylogenomic approaches (Pease et al. 2018; Morales-Briones et al. 2021). We applied concatenated and coalescent phylogenomic approaches and compared species versus gene tree discordance of nucleotide and amino acid tree inferences based on single-copy orthologs, combining genome- with transcriptome data.
In comparing the confidence in coalescent versus concatenated trees, we noticed the presence of two recalcitrant nodes, yet the low confidence in the coalescent trees did not contradict our main hypothesis or the observation of independent evolution (Fig. 1; Table S4 of the Supplementary material available on Dryad). We found strong and consistent support for all deepest nodes, the relationships between the branchiopod orders. Besides a potential bias due to taxon undersampling, large multilocus data sets can be prone to errors and phylogenetic noise that sometimes create conflicts even between trees with well-supported topologies depending on the analysis type. For example, the choice of outgroup has been shown to influence certain types of phylogenetic analysis (Fernández et al. 2018; Gillung et al. 2018). We used all non-cladoceran Branchiopoda as outgroup for this reason, including the sister lineage Cyclestherida; containing 11 cladoceran families, our data set is the most comprehensive to date to understand the deeper levels in the group.
The only incongruence we observed was at the intermediate (intrafamilial) level in the phylogeny. Two out of 22 nodes were discordant while the topology remained consistent. Both nodes showed strong bootstrap support in RAxML-NG for the nucleotide data set yet low support in ASTRAL-III. Only one incongruence of this type remained in the amino acid data set but did not affect the higher grouping of the clade (Macrothrix; Table S4 of the Supplementary material available on Dryad). In our case, this difference in confidence could be related to potential incomplete lineage sorting as a result of abrupt cladogenesis around a Permian-Triassic boundary extinction event, as suggested from our molecular clock estimation. Our results suggest extreme forms of phenotypic homoplasy (e.g., the body plan of the predatory cladocerans) in arthropods and confirm ancient rapid radiation in the group, supporting the increasing evidence that homoplasy and rapid cladogenesis may be more common in nature than previously thought, occurring at all depths of the Tree of Life (Alda et al. 2019; Kusy et al. 2019; Hime et al. 2021; Ramírez et al. 2021). Thus, our work here helps illuminate long-obscured relationships, unveiling independent evolution among arthropod lineages in the branchiopod tree, which spans several hundreds of millions of years.
Independent evolution can only be revealed by incongruence between different data sets, for example by comparing morphological, developmental, and molecular data. Observing these discordances is key to our understanding of evolution and diversity, compelling us to ask new questions and examine the potential mechanisms and constraints of morphological diversity in nature (Shubin et al. 2009; Wake et al. 2011). Phylogenetic data derived from cladoceran development are sparse (Kotov 2017). Previously suggested groupings of several cladoceran lineages due to morphological similarities and functional links to specific niches are not supported by our genomic approach (Figs. S9 and S10 of the Supplementary material available on Dryad). The discrepancy between functionally important traits and the tree topology derived here clarify that, while several features correlate with defined taxa (e.g., number of limb pairs, mode of reproduction), other important characteristics have a polyphyletic origin, such as being planktonic or having a predatory life-style (Fig. S10 of the Supplementary material available on Dryad).
The well-supported phylogenetic evidence presented here raises important questions about the extent and limits of morphological forms in an ancient arthropod group. Homoplasy may be an underestimated influence on diversity and phenotypic evolution in Cladocera. Underlying genetic and developmental mechanisms—for example in the transition from suspension feeders with leaf-like limbs to highly specialized predators with strong reductions and rod-shaped limbs—remain poorly known. Other examples exist in arthropod groups where there is little morphological support for a highly reliable branch based on genomic data (e.g., Copepoda—Eyun 2017; Myriapoda—Fernández et al. 2018). Although conflicting results raise important questions, groupings supported by different independent data sets are more likely to be correct (Meusemann et al. 2010; Lee and Palci 2015). Importantly, data from morphology and development remain crucial to compare against molecular data sets, while incongruences between different data sets help us examine systematics from all perspectives (Pisani et al. 2007; Wake et al. 2011). As the BUSCO gene-set used here consists of conserved near-universal markers that are present in a single copy, it is generally considered a reliable set of phylogenetically informative genes and has proved useful in phylogenies, including arthropods (Fernández et al. 2018; Waterhouse et al. 2018).
Implications for Cladocera Evolution
Using a kaleidoscopic phylogenomic approach, we found new, robust evidence for independent evolution of main feeding modes in the Cladocera and the accompanying body plans. Functional morphology, apparent in limb characters, forms the basis of cladoceran systematics; our findings contradict the classical systematics in the group at the deeper levels.
The pelagic predatory lifestyle arose twice independently, as the Gymnomera, containing the orders Haplopoda and Onychopoda, are polyphyletic. This convergence is an important result of our study; it is expressed in similar phenotypes with large eyes, stenopodous raptorial limbs and a reduced carapace to enable predation in the open water. Also planktonic herbivory appeared several times independently, in the orders Ctenopoda and in the Anomopoda, sometimes grouped together under the polyphyletic Calyptomera, and at least twice at the family level in the Anomopoda (e.g., in the Bosminidae and the Daphniidae). The scraping-gathering lifestyle of the littoral lineages may have arisen at least twice as well, as the suborder Radopoda appeared polyphyletic in our study, due to the unexpected position of the Macrothricidae as a sister lineage to specialized filter-feeders Daphniidae or Moinidae.
Our analysis supported the general hypothesis that Cladocera are monophyletic and confirmed their sister relationship with the monotypic Cyclestherida, hence the monophyly of the Cladoceromorpha. We found no support for the sister relationship between Ctenopoda and Anomopoda, which is accepted by several authors, nor for a sister relationship between the Ctenopoda and the remaining three cladoceran orders (Table S1 of the Supplementary material available on Dryad). We provide robust evidence for a sister relationship between Anomopoda and Onychopoda and a relationship between the Haplopoda and Ctenopoda. Indeed, the ML trees (Fig. 1; Figs. S4 and S5 of the Supplementary material available on Dryad) do not support two widely accepted hypotheses in cladoceran higher systematics: the Gymnomera and Calyptomera concepts (Fig. S1 of the Supplementary material available on Dryad). In particular, the monophyly of the Gymnomera is a generally accepted hypothesis (Olesen 1998; Kotov 2013; alternative in Wesenberg-Lund 1952; Table S1 of the Supplementary material available on Dryad), based mainly on morphological features of a predatory lifestyle in the pelagic and often supported by molecular phylogenies using a few mitochondrial and/or nuclear markers (e.g., Swain and Taylor 2003; Richter et al. 2007).
The new relationships are maximally supported in our protein and nucleotide trees using different approaches (RAxML-NG, ASTRAL-III, PhyloBayes; Fig. 1; Table S4 of the Supplementary material available on Dryad). We suggest, for practical use and future reference, names for the clades supported by our genomic analysis. We suggest a new superorder “Oligopoda” Van Damme et Ebert, derived from the Greek prefix oligo (“few”) and poda (“feet”) to contain the orders Onychopoda and Anomopoda. This name refers to the strong oligomerization of body and limbs, with the sixth limb being strongly reduced or absent; even the fifth limb may be reduced (Fig. S10 of the Supplementary material available on Dryad). The second proposed group would be the superorder “Magipoda” Van Damme et Ebert from the Latin magis (“more”) and poda (“feet”), which would entail the Haplopoda and Ctenopoda, referring to six serial limbs. Our divergence estimates situate the most recent common ancestor of the Magipoda and Oligopoda towards the end of the Early Carboniferous.
Besides compelling evidence for independent evolution, the divergence time estimates in our study add a new dimension to the interpretation of branchiopod evolution. An important implication of our time estimates is that the Cladocera’s basic body plans—the predatory and basic suspension/deposit-feeding models adapted to freshwater pelagic and littoral areas—must already have been present in the late Paleozoic. The pelagic predatory lifestyle, including the remarkable transition from phyllopodous to stenopodous limbs (Olesen et al. 2001) and reduction of the carapace, which we now know happened more than once independently, is likely an innovation dating back to the Paleozoic. For the nonpredatory lifestyles, the ancestral lineages may have combined both deposit- and suspension-feeding in the pelagic and possibly also near the sediment (Kotov 2009, 2013), yet the highly specialized limb arrangements we observe in the Daphniidae and Chydoridae, seem to have a Mesozoic signature, strengthening the theory that diversification of the successful herbivorous anomopod cladocerans is linked with the Lacustrine Mesozoic Revolution (Van Damme and Kotov 2016). Besides a Paleozoic diversification, we found that an important Triassic explosive radiation in freshwater ecosystems gave rise to the ancestors of several extant cladoceran families. In addition, our study shifts the interpretation of evolution in several lineages, as it provides evidence that several lineages diverged hundreds of millions of years earlier (Onychopoda families) or later (Chydoridae subfamilies) than previously thought (Cristescu and Hebert 2002; Sacherová and Hebert 2003; Text ST2 of the Supplementary material available on Dryad
Although the presented phylogeny should be interpreted with the usual care, independent evolution needs to be considered at deeper systematic levels in the Cladocera. Each of the major forms with predatory, suspension-feeding and deposit-feeding lifestyles may have evolved more than once independently in Cladocera. Examining these traits through state-of-the-art phylogenetic reconstruction presents their evolution and inferred niches in a new perspective and allows us to assess the role of homoplasy in diversification within a calibrated temporal context. We show several examples of a strong mismatch between morphological and phylogenetic similarity, underscoring that independent evolution is an important aspect of the evolution of biodiversity. As phylogenomic methods improve, more taxa are added and other important data layers (e.g., development) are explored, future analyses may shed further light on the relationships among cladoceran families, suborders, and orders. The current study is therefore not an end-point but rather a first step to testing hypothetical relationships and timings of divergence in the deep cladoceran lineages using whole-genome data. As our results are unorthodox compared to previous studies exploring Cladocera systematics, we provide a detailed account of the implications of our study for the evolution of body plans, divergence times, and functional diversity in the major clades; a more elaborate discussion is provided (as a Text ST2 of the Supplementary material available on Dryad), including the importance of our fossil-calibrated molecular phylogeny for cladoceran lineages and evolution.
Acknowledgments
We thank Jürgen Hottinger, Urs Stiefel, Anu Vehmaa (A.V. in Table S2 of the Supplementary material available on Dryad), Jaana Koistinen, and Joanna Norkko for support in the field and in the laboratory. We highly appreciate the language editing by Suzanne Zweizig and the revision by the reviewers and by the editors Dr B. Carstens and Dr J. Bond.
Supplementary Material
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.mkkwh711b.
Funding
This work was supported by the Swiss National Science Foundation (SNSF) DE [310030B_166677 to D.E.] and by a Senior Research Fellow Grant [H5771/VanDamme/4700 to K.V.D.] from the Finnish Pro Mare Balticum Walter and Andrée de Nottbeck Foundation, funding the project “Cladocera of the Baltic Sea – Taxonomy and Diversity” at Tvärminne Zoological Station (University of Helsinki), Finland.
References
- Abadi S., Azouri D., Pupko T., Mayrose I.. 2019. Model selection may not be a mandatory step for phylogeny reconstruction. Nat. Commun. 10:934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alda F., Tagliacollo V.A., Bernt M.J., Waltz B.T., Ludt W.B., Faircloth B.C., Alfaro M.E., Albert J.S., Chakrabarty P.. 2019. Resolving deep nodes in an ancient radiation of neotropical fishes in the presence of conflicting signals from incomplete lineage sorting. Syst. Biol. 68:573–593. [DOI] [PubMed] [Google Scholar]
- Arcila D., Ortí G., Vari R., Armbruster J.W., Stiassny M.L.J., Ko K.D., Sabaj M.H., Lundberg J., Revell L.J., Betancur-R R.. 2017. Genome-wide interrogation advances resolution of recalcitrant groups in the tree of life. Nat. Ecol. Evol. 1:0020. [DOI] [PubMed] [Google Scholar]
- Bouckaert R., Heled J., Kühnert D., Vaughan T., Wu C.H., Xie D., Suchard M.A., Rambaut A., Drummond A.J.. 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbourne J.K., Pfrender M.E., Gilbert D., Thomas W.K., Tucker A., Oakley T.H., Tokishita S., Aerts A., Arnold G.J., Basu M.K., Bauer D.J., Caceres C.E., Carmel L., Casola C., Choi J.-H., Detter J.C., Dong Q., Dusheyko S., Eads B.D., Froehlich T., Geiler-Samerotte K.A., Gerlach D., Hatcher P., Jogdeo S., Krijgsveld J., Kriventseva E.V., Kueltz D., Laforsch C., Lindquist E., Lopez J., Manak, J.R., Muller J., Pangilinan J., Patwardhan R.P., Pitluck S., Pritham E.J., Rechtsteiner A., Rho M., Rogozin I.B., Sakarya O., Salamov A., Schaack S., Shapiro H., Shiga Y., Skalitzky C., Smith Z., Souvorov A., Sung W., Tang Z., Tsuchiya D., Tu H., Vos H., Wang M., Wolf Y.I., Yamagata H., Yamada T., Ye Y., Shaw J.R., Andrews J., Crease T.J., Tang H., Lucas S.M., Robertson H.M., Bork P., Koonin E.V., Zdobnov E.M., Grigoriev I.V., Lynch M., Boore J.L.. 2011. The ecoresponsive genome of Daphnia pulex. Science 331:555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornetti L., Fields P.D., Van Damme K., Ebert D.. 2019. A fossil-calibrated phylogenomic analysis of Daphnia and the Daphniidae. Mol. Phylogenet. Evol. 137:250–262. [DOI] [PubMed] [Google Scholar]
- Cristescu M.E.A., Hebert P.D.N.. 2002. Phylogeny and adaptive radiation in the Onychopoda (Crustacea, Cladocera): evidence from multiple gene sequences. J. Evol. Biol. 15:838–849. [Google Scholar]
- Delsuc, F., Brinkmann, H., Philippe, H.. 2005. Phylogenomics and the reconstruction of the tree of life. Nat. Rev. Genet. 6:361–375. [DOI] [PubMed] [Google Scholar]
- de Waard, J.R., Sacherová, V., Cristescu, M.E.A., Remigio, E.A., Crease, T.J., Hebert, P.D.N.. 2006. Probing the relationships of the branchiopod crustaceans. Mol. Phylogenet. Evol. 39:491–502. [DOI] [PubMed] [Google Scholar]
- Duchêne D.A., Bragg J.G., Duchêne S., Neaves L.E., Potter S., Moritz C., Johnson R.N., Ho S.Y.W., Eldridge M.D.B.. 2018. Analysis of phylogenomic tree space resolves relationships among marsupial families. Syst. Biol. 67: 400–412. [DOI] [PubMed] [Google Scholar]
- Dumont H.J., Negrea S.V.. 2002. Introduction to the class Branchiopoda. In: Dumont H.J., editor. Guides to the identification of the microinvertebrates of the continental waters of the world, vol. 19. Leiden, The Netherlands: Backhuys Publishers. 397 pp. ISBN: 9789057821127. [Google Scholar]
- Dumont H.J., Silva-Briano M.. 1998. A reclassification of the anomopod families Macrotrichidae and Chydoridae, with the creation of a new suborder, the Radopoda (Crustacea: Branchiopoda). Hydrobiologia 384:119–149. [Google Scholar]
- Eyun S.I. 2017. Phylogenomic analysis of Copepoda (Arthropoda, Crustacea) reveals unexpected similarities with earlier proposed morphological phylogenies. BMC Evol. Biol. 17:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández R., Edgecombe G.D., Giribet G.. 2018. Phylogenomics illuminates the backbone of the Myriapoda Tree of Life and reconciles morphological and molecular phylogenies. Sci. Rep. 8:83. doi: 10.1038/s41598-017-18562-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forró L., Korovchinsky N.M., Kotov A.A., Petrusek A.. 2008. Global diversity of cladocerans (Cladocera; Crustacea) in freshwater. Hydrobiologia 595:177–184. [Google Scholar]
- Fryer G. 1968. Evolution of adaptive radiation in the Chydoridae (Crustacea: Cladocera): a study in comparative functional morphology and ecology. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 254:221–385. [DOI] [PubMed] [Google Scholar]
- Fryer G. 1987a. A new classification of the branchiopod Crustacea. Zool. J. Linn. Soc. 91:357–381. [Google Scholar]
- Fryer G. 1987b. Morphology and the classification of the so-called Cladocera. Hydrobiologia 145:19–28. [Google Scholar]
- Fryer G. 1991. Functional morphology and the adaptive radiation of the Daphniidae (Branchiopoda Anomopoda). Philos. Trans. R. Soc. Lond., B, Biol. Sci. 331:1–99. [Google Scholar]
- Fryer G. 1995. Phylogeny and adaptive radiation within the Anomopoda: a preliminary exploration. Hydrobiologia 307:57–68. [Google Scholar]
- Fryer G. 1999. A comment on a recent phylogenetic analysis of certain orders of the branchiopod Crustacea. Crustaceana 72:1039–1050. [Google Scholar]
- Gillung J.P., Winterton S.L., Bayless K.M., Khouri Z., Borowiec M.L., Yeates D., Kimsey L.S., Misof B., Shin S., Zhou X., Mayer C., Petersen M., Wiegmann B.M.. 2018. Anchored phylogenomics unravels the evolution of spider flies (Diptera, Acroceridae) and reveals discordance between nucleotides and amino acids. Mol. Phyl. Evol. 128:233–245. [DOI] [PubMed] [Google Scholar]
- Giribet G., Edgecombe G.D.. 2019. The phylogeny and evolutionary history of arthropods. Curr. Biol. 29:R592–R602. [DOI] [PubMed] [Google Scholar]
- Harvey T.H.P., Vélez M.I., Butterfield N.J.. 2012. Exceptionally preserved crustaceans from western Canada reveal a cryptic Cambrian radiation. Proc. Natl. Acad. Sci. USA 109:1589–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havird J.C., Santos S.R.. 2016. Here we are, but where do we go? A systematic review of crustacean transcriptomic studies from 2014-2015. Integr. Comp. Biol. 56:1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hime P.M., Lemmon A.R., Lemmon E.C.M., Prendini E., Brown J.M., Thomson R.C., Kratovil J.D., Noonan B.P., Pyron R.A., Peloso P.L.V., Kortyna M.L., Keogh J.S., Donnellan S.C., Mueller R.L., Raxworthy C.J., Kunte K., Ron S.R., Das S., Gaitonde N., Green D.M., Labisko J., Che J., Weisrock D.W.. 2021. Phylogenomics reveals ancient gene tree discordance in the amphibian tree of life. Syst. Biol. 70:49–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kainer D., Lanfear R.. 2015. The effects of partitioning on phylogenetic inference. Mol. Biol. Evol. 32:1611–1627. [DOI] [PubMed] [Google Scholar]
- Kapli P., Yang Z., Telford M.J.. 2020. Phylogenetic tree building in the genomic age. Nat. Rev. Genet. 21:428-444. [DOI] [PubMed] [Google Scholar]
- Korovchinsky N.M. 2006. The Cladocera (Crustacea: Branchiopoda) as a relict group. Zool. J. Linn. Soc. 147:109–124. [Google Scholar]
- Kotov A.A. 2009. A revision of the extinct Mesozoic family Prochydoridae Smirnov, 1992 (Crustacea: Cladocera) with a discussion of its phylogenetic position. Zool. J. Linn. Soc. 155:253–265. [Google Scholar]
- Kotov A.A. 2013. Morphology and phylogeny of the Anomopoda (Crustacea: Cladocera). Moscow: KMK. 638 pp. (In Russian). [Google Scholar]
- Kotov A.A. 2017. Notes on the physiology of embryogenesis. In: Smirnov N.N., editor. Physiology of the Cladocera 2nd ed. Amsterdam, The Netherlands: Academic Press, Elsevier; Inc. p. 281–297. [Google Scholar]
- Kotov A.A., Forró L., Korovchinsky N.M., Petrusek A.. 2013. World checklist of freshwater Cladocera species. Available from: http://fada.biodiversity.be/group/show/17 (date accessed: February10, 2020). [Google Scholar]
- Kozlov A.M., Darriba D., Flouri T., Morel B.. 2019. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35:4453–4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusy D., Motyka M., Bocek M., Masek M., Bocak L.. 2019. Phylogenomic analysis resolves the relationships among net-winged beetles (Coleoptera: Lycidae) and reveals the parallel evolution of morphological traits. Syst. Biol. 44:911–925. [Google Scholar]
- Lampert W. 2011. Daphnia: development of a model organism in ecology and evolution. Excellence in Ecology, vol. 21. Oldendorf/Luhe: International Ecology Institute Publishers. 250 pp. [Google Scholar]
- Lanfear R., Frandsen P.B., Wright A.M., Senfeld T., Calcott B.. 2017. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34:772–773. [DOI] [PubMed] [Google Scholar]
- Lartillot N., Rodrigue N., Stubbs D., Richer J.. 2013. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 62:611–615. [DOI] [PubMed] [Google Scholar]
- Lee M.S.Y., Palci A.. 2015. Morphological phylogenetics in the genomic age. Curr. Biol. 25:R922–R929. [DOI] [PubMed] [Google Scholar]
- Meusemann K., von Reumont B.M., Simon S., Roeding F., Straus, S., Kück P., Ebersberger I., Walzl M., Pass G., Breuers S., Achter V., von Haeseler A., Burmester T., Hadrys H., Wägele J.W., Misof B.. 2010. A phylogenomic approach to resolve the arthropod tree of life. Mol. Biol. Evol. 27:2451–2464. [DOI] [PubMed] [Google Scholar]
- Miner B.E., De Meester L., Pfrender M.E., Lampert W., Hairston N.G. Jr.. 2012. Linking genes to communities and ecosystems: Daphnia as an ecogenomic model. Proc. Biol. Sci. 279:1873–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy E.K., Warnow T.. 2018. To include or not to include: the impact of gene filtering on species tree estimation methods. Syst. Biol. 67:285–303. [DOI] [PubMed] [Google Scholar]
- Morales-Briones D.F., Kadereit G., Tefarikis D.T., Moore M.J., Smith S.A., Brockington S.F., Timoneda A., Yim W.C., Cushman J.C., Yang Y.. 2021. Disentangling sources of gene tree discordance in phylogenomic data sets: testing ancient hybridizations in Amaranthaceae s.l. Syst. Biol. doi: 70:219–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrea S., Butnariuc N., Dumont H.J.. 1999. On the phylogeny, evolution and classification of the Branchiopoda (Crustacea). Hydrobiologia 412:191–212. [Google Scholar]
- Noah K.E., Hao J., Li L., Sun X., Foley B., Yang Q., Xia X.. 2020. Major revisions in arthropod phylogeny through improved supermatrix, with support for two possible waves of land invasion by chelicerates. Evol. Bioinf. 16:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J. 1998. A phylogenetic analysis of the Conchostraca and Cladocera (Crustacea, Branchiopoda, Diplostraca). Zool. J. Linn. Soc. 122:491–536. [Google Scholar]
- Olesen J. 2009. Phylogeny of Branchiopoda (Crustacea) – character evolution and contribution of uniquely preserved fossils. Arthropod Syst. Phyl. 67:3–39. [Google Scholar]
- Olesen J., Richter S., Scholtz G.. 2001. The evolutionary transformation of phyllopodous to stenopodous limbs in the Branchiopoda (Crustacea) - is there a common mechanism for early limb development in arthropods? Int. J. Dev. Biol. 45:869–876. [PubMed] [Google Scholar]
- Pease J.B., Brown J.W., Walker J.F., Hinchliff C.E., Smith, S.A.. 2018. Quartet sampling distinguishes lack of support from conflicting support in the green plant tree of life. Am. J. Bot. 105:385–403. [DOI] [PubMed] [Google Scholar]
- Pisani D., Benton M.J., Wilkinson M.. 2007. Congruence of morphological and molecular phylogenies. Acta Biotheor. 55:269–281. [DOI] [PubMed] [Google Scholar]
- Ramírez M.J., Magalhae I.L.F., Derkarabetian S., Ledford J., Griswold C.E., Wood H.M., Hedin M.. 2021. Sequence capture phylogenomics of true spiders reveals convergent evolution of respiratory systems. Syst. Biol. 70:14–20. [DOI] [PubMed] [Google Scholar]
- Richter S., Olesen J., Wheeler W.C.. 2007. Phylogeny of Branchiopoda (Crustacea) based on a combined analysis of morphological data and six molecular loci. Cladistics 23:301–336. [DOI] [PubMed] [Google Scholar]
- Robinson D.F., Foulds, L.R.. 1981. Comparison of phylogenetic trees. Math. Biosci. 53:131–147. [Google Scholar]
- Rotllant G., Palero F., Mather P.B., Bracken-Grissom H.D., Begoña Santos M.. 2018. Preface: recent advances in crustacean genomics. Hydrobiologia 825:1–4. [Google Scholar]
- Roycroft E.J., Moussalli A., Rowe K.C.. 2020. Phylogenomics uncovers confidence and conflict in the rapid radiation of Australo-Papuan rodents. Syst. Biol. 69:431-444. [DOI] [PubMed] [Google Scholar]
- Sacherová V., Hebert P.D.N.. 2003. The evolutionary history of the Chydoridae (Crustacea: Cladocera). Biol. J. Linn. Soc. 79:629–643. [Google Scholar]
- Sars G.O. 1865. Norges ferskvandskrebsdyr. Første afsnit. Branchiopoda I. Cladocera Ctenopoda (Fam. Sididae og Holopedidae). – Universitetsprogram for 1ste halvår 1863. 71 pp., pls. 1–4. [Google Scholar]
- Schwentner M., Richter S., Rogers D.C., Giribet G.. 2018. Tetraconatan phylogeny with special focus on Malacostraca and Branchiopoda: highlighting the strength of taxon-specific matrices in phylogenomics. Proc. R. Soc. Lond. B Biol. Sci. 285:20181524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shubin N., Tabin C., Carroll S.. 2009. Deep homology and the origins of evolutionary novelty. Nature 457:818. [DOI] [PubMed] [Google Scholar]
- Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. [DOI] [PubMed] [Google Scholar]
- Smith S.A., Moore M.J., Brown J.W., Yang Y.. 2015. Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 15:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain T.D., Taylor D.J.. 2003. Structural rRNA characters support monophyly of raptorial limbs and paraphyly of limb specialization in water fleas. Proc. R. Soc. Lond. B. Biol. Sci. 270:887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telford M.J., Budd G.E., Philippe H.. 2015. Phylogenomic insights into animal evolution. Curr. Biol. 25:R876–R887. [DOI] [PubMed] [Google Scholar]
- Thomas G.W.C., Dohmen E., Hughes D.S.T., Murali S.W., Poelchau M., Glastad K., Anstead C.A., Ayoub N.A., Batterham P., Bellair M., Binford G.J., Chao H., Chen Y.H., Childers C., Dinh H., Doddapaneni H.V., Duan J.J., Dugan S., Esposito L.A., Friedrich M., Garb J., Gasser R.B., Goodisman M.A.D., Gundersen-Rindal D.E., Han Y., Handler A.M., Hatakeyama M., Hering L., Hunter W.B., Ioannidis P., Jayaseelan J.C., Kalra D., Khila A., Korhonen P.K., Lee C.E., Lee S.L., Li Y., Lindsey A.R.I., Mayer G., McGregor A.P., McKenna D.D., Misof B., Munidasa M., Munoz-Torres M., Muzny D.M., Niehuis O., Osuji-Lacy N., Palli S.R., Panfilio K.A., Pechmann M., Perry T., Peters R.S., Poynton H.C., Prpic N.-M.Qu J., Rotenberg D., Schal C., Schoville S.D., Scully E.D., Skinner E., Sloan D.B., Stouthamer R., Strand M.R., Szucsich N.U., Wijeratne A., Young N.D., Zattara E.E., Benoit J.B., Zdobnov E.M., Pfrender M.E., Hackett K.J., Werren J.H., Worley K.C., Gibbs R.A., Chipman A.D., Waterhouse R.M., Bornberg-Bauer E., Hahn M.W., Richards S.. 2020. Gene content evolution in the arthropods. Genome Biol. 21:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme K., Kotov A.A.. 2016. The fossil record of the Cladocera (Crustacea: Branchiopoda): evidence and hypotheses. Earth-Sci. Rev. 163:162–189. [Google Scholar]
- Wake D.B., Wake M.H., Specht C.D.. 2011. Homoplasy: from detecting pattern to determining process and mechanism of evolution. Science 331:1032–1035. [DOI] [PubMed] [Google Scholar]
- Waterhouse R.M., Seppey M., Simão F.A., Zdobnov E.M.. 2019. Using BUSCO to assess insect genomic resources. In: Brown S., Pfrender M., editors., Insect genomics. Methods in molecular biology, vol. 1858. New York, USA: Humana Press. p. 59–74. [DOI] [PubMed] [Google Scholar]
- Waterhouse R.M., Seppey M., Simão F.A., Manni M., Ioannidis P., Klioutchnikov G., Kriventseva E.V., Zdobnov E.M.. 2018. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
Wesenberg-Lund C.
1952. De Danske Søers Og Dammes Dyriske Plankton. Copenhagen, Denmark: Nordlundes Bogtrykkeri, Ejnar Munksgaard. 182 pp.
 XXVII plates (In Danish). [Google Scholar]
XXVII plates (In Danish). [Google Scholar] - Whitfield J.B., Lockheart P.J.. 2007. Deciphering ancient rapid radiations. Trends Ecol. Evol. 22:5. [DOI] [PubMed] [Google Scholar]
- Widhelm T.J., Huang J.-P., Mercado-Díaz J.A., Goffinet B., Lücking R., Moncada B., Mason-Gamer R., Lumbsch H.T.. 2019. Multiple historical processes obscure phylogenetic relationships in a taxonomically difficult group (Lobariaceae, Ascomycota). Sci. Rep. 9:8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe J.M., Daley A.C., Legg D.A., Edgecombe G.D.. 2016. Fossil calibrations for the arthropod tree of life. Earth-Sci. Rev. 160:43–110. [Google Scholar]
- Xi Z., Liu L., Rest J.S., Davis C.C.. 2014. Coalescent versus concatenation methods and the placement of Amborella as sister to water lilies. Syst. Biol. 63:919–932. [DOI] [PubMed] [Google Scholar]
- Zhang C., Rabiee M., Sayyari E., Mirarab S.. 2018. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 19:15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]