

SUMMARY
Hematopoietic stem cells (HSCs) regenerate blood cells upon hematopoietic injuries. During homeostasis, HSCs are maintained in a low reactive oxygen species (ROS) state to prevent exhaustion. However, the role of nitric oxide (NO) in controlling HSC regeneration is still unclear. Here, we find increased NO during HSC regeneration with an accumulation of protein aggregation. S-nitrosoglutathione reductase (GSNOR)-deleted HSCs exhibit a reduced reconstitution capacity and loss of self-renewal after chemotherapeutic injury, which is resolved by inhibition of NO synthesis. Deletion of GSNOR enhances protein S-nitrosylation, resulting in an accumulation of protein aggregation and activation of unfolded protein response (UPR). Treatment of taurocholic acid (TCA), a chemical chaperone, rescues the regeneration defect of Gsnor−/− HSCs after 5-fluorouracil (5-FU) treatment. Deletion of C/EBP homologous protein (Chop) restores the reconstitution capacity of Gsnor−/− HSCs. These findings establish a link between S-nitrosylation and protein aggregation in HSC in the context of blood regeneration.
In brief
Yi et al. reveal a role of NO-induced protein S-nitrosylation in HSC regeneration. Deficiency of GSNOR, a denitrosylase, causes HSC reconstitution decrease, which is restored by NO inhibition, TCA treatment, or Chop deletion. The findings link S-nitrosylation, protein aggregation, and cellular viability in HSC during blood regeneration.
Graphical Abstract
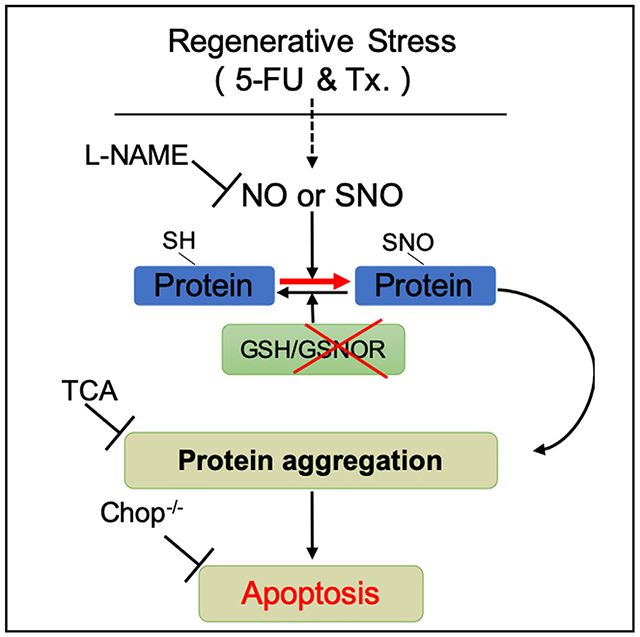
INTRODUCTION
Hematopoietic stem cells (HSCs) have the ability to self-renew and differentiate to generate all types of mature blood cells through the production of a series of committed progenitor cells (Orkin and Zon, 2008). The regenerative capacity of HSC is essential for the replenishment of the hematopoietic system upon various hematopoietic stresses, including bone marrow (BM) transplantation of the lethally irradiated recipients and chemotherapy-induced myeloablation (Mohrin et al., 2010; Yahata et al., 2011). At homeostasis condition, HSCs are maintained in a protective dormant state in order to limit the oxidative stress-induced damage. Increased reactive oxygen species (ROS) impairs HSC self-renewal capacity (Ito et al., 2004). Although the importance of ROS in HSC biology has been extensively studied, the significance of reactive nitrogen species (RNS) in the function and maintenance of HSC remains rather obscure.
Nitric oxide (NO) and its derivatives are the major source of RNS in cells. NO over-production by the increased NO synthase (NOS) leads to cellular nitrosative stress and aberrantly enhanced S-nitrosylation contributing to cellular damage and diseases (Li et al., 2017; Rizza and Filomeni, 2017; Yang et al., 2015). NO exerts its biological activity through S-nitrosylating (SNO) protein cysteine residues (Benhar et al., 2009), which are involved in a wide range of biological processes (Hess and Stamler, 2012; Nakamura et al., 2013; Wang et al., 2009; Zhang et al., 2017). S-nitrosoglutathione reductase (GSNOR), as a major denitrosylase, reverts S-nitrosylation of SNO proteins. Although GSNOR expression is dispensable for the tissue maintenance during steady state, experimental evidence showed that GSNOR-deficient mice exhibited an increased mortality correlated with elevations of SNO under lipopolysaccharide (LPS)-induced endotoxic shock (Liu et al., 2004).
In hematopoietic system, NO is required for HSC emerging during the embryogenesis (North et al., 2009). Experimental induction of NO leads to an activated mobilization of hematopoietic stem and progenitor cells (HSPCs) (Gur-Cohen et al., 2015; Xu et al., 2021), enhanced proliferation of HSC, and an inclination of HSC to myeloid differentiation (Nogueira-Pedro et al., 2014). However, as a downstream response of NO, SNO and its turnover in controlling NO-associated bioactivity in adult stem cells, especially in HSCs, has been largely unknown. Previous study reported GSNOR function as a formaldehyde dehydrogenase in HSCs in the context of DNA damage response (DDR) (Pontel et al., 2015), but the function of GSNOR, as a denitrosylase, in HSCs regeneration remains to be delineated. Here, we showed that hematopoietic stress strongly increased NO level in HSCs. Gsnor−/− HSCs exhibited an increased level of NO and failure to regenerate the blood system after transplantation and 5-fluorouracil (5-FU)-induced myeloablation. These changes were associated with an enhanced protein S-nitrosylation, which promoted cell death via protein-aggregation-mediated apoptosis in Gsnor−/− HSCs. Chemical chaperone taurocholic acid (TCA) treatment or Chop deletion attenuated the Gsnor−/− HSCs protein aggregation and restored their regenerative capacity. NO synthesis inhibitor, L-NG-nitroarginine methyl ester (L-NAME), reduced the protein S-nitrosylation level and restored the repopulating defect of Gsnor−/− HSCs. Taken together, our study discovers a previously unknown mechanism of NO-induced and GSNOR-mediated protein aggregation in regulating HSC regeneration.
RESULTS
Increase of NO level and protein aggregation in regenerating HSCs
To assess the role of NO in the hematopoietic system, we first examined the levels of NO in hematopoietic cells from wild-type (WT) mice (2 months old) using the NO-sensitive fluorescent dye 3-amino, 4-aminomethyl-2’,7’-difluorescein diacetate (DAF-FM DA). We found the NO level was lower in long-term HSCs (LT-HSCs) (CD150+CD48−Lineage−Sca1+cKit+ cells) compared with LSK (Lineage−Sca1+cKit+), Lin− (Lineage−) cells, and Lin+ (Lineage+) cells (Figure 1A), suggesting a lower oxidative stress in HSC at steady state. To further examine the levels of NO in hematopoietic cells under hematopoietic stress, we treated the WT mice with a single-dose of 5-FU (150 mg/kg), and the levels of NO in BM were checked at day 9 post-treatment. In line with the previous finding, we also found that 5-FU strongly induced HSC proliferation (Figure 1B), which led to an increased absolute number of HSCs (Figure S1A). The ratio of LT-HSCs in BM was increased, and a trend of increased ratio of granulocyte-macrophage progenitor (GMP), common myeloid progenitor (CMP), and megakaryocyte-erythrocyte progenitor (MEP) was detected in BM of the mice after 5-FU treatment (Figure S1B). Interestingly, the levels of NO were significantly elevated in HSPCs, with the highest increase in LT-HSCs (Figure 1C). As NO exerts its role by S-nitrosylating protein cysteine residues, we therefore detected the protein S-nitrosylation in Lin− BM cells at day 9 post-5-FU treatment using a biotin switch method (Zhang et al., 2017). As expected, we observed a significant increased level of protein S-nitrosylation in the Lin− cells of WT mice treated with 5-FU compared with Lin− cells from control mice (Figure 1D). Taken together, these data indicate a specific role of NO, which contributes to the increased protein S-nitrosylation in HSCs during regeneration.
Figure 1. Increase of NO level and protein aggregation in regenerating HSCs.
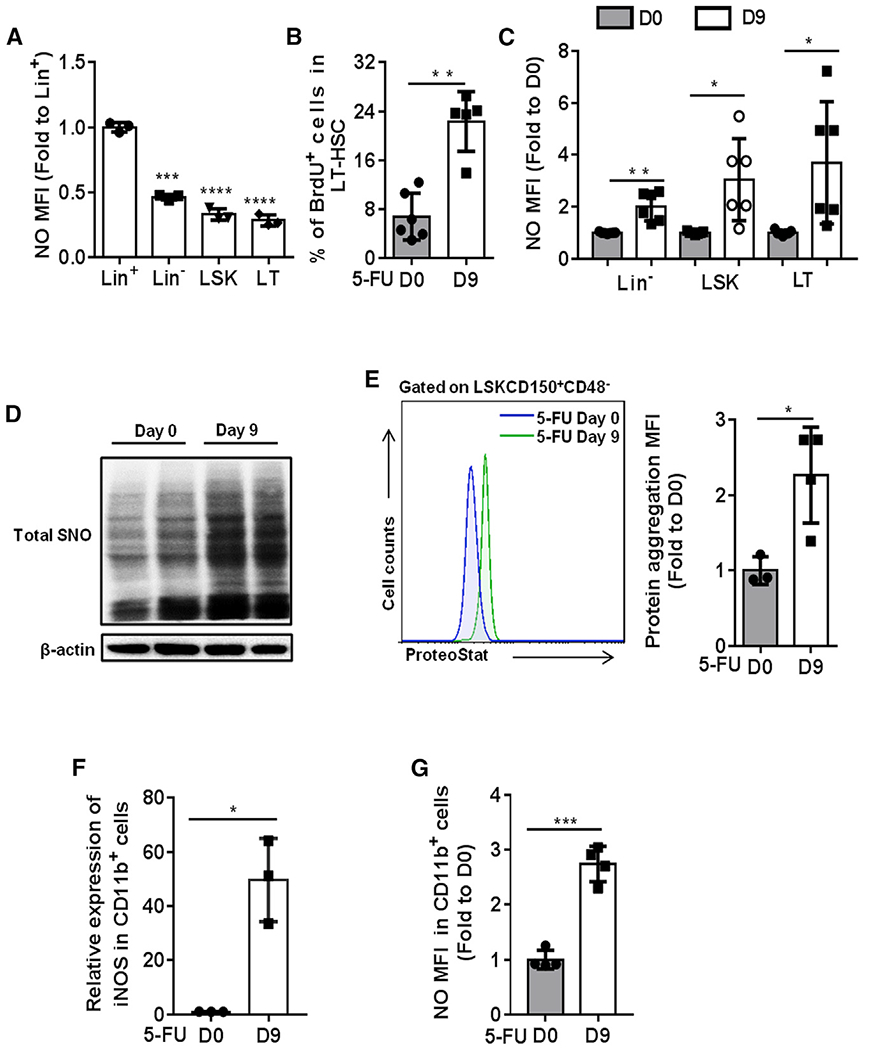
(A) The levels of NO in freshly isolated Lineage+ (Lin+), Lineage− (Lin−), Lineage−cKit+Sca1+ (LSK), and CD48−CD150+LSK (LT-HSCs) cells from 2-month-old WT mice were detected by DAF-FM DA probe. The data show the relative changes of NO levels in the indicated groups (fold change to the NO level in Lin+ cells; n = 3 per group).
(B) WT mice were treated with a single dose of 5-FU (150 mg/kg) and sacrificed at day 9 post-treatment. BrdU (100 mg/kg) was injected 16 h prior to mice sacrifice. The results present the percentage of BrdU-positive cells in LT-HSCs at the indicated time points after 5-FU treatment (n = 5-6 per group).
(C–G) WT mice were treated with a single dose of 5-FU (150 mg/kg) and sacrificed at day 9 post-treatment.
(C) The data show the relative change of NO levels in the indicated populations (fold change to the NO level in the same population at day 0 post-5-FU treatment; n = 6–7 per group from 2 independent experiments).
(D) The S-nitrosylated proteins in Lin− cells were purified by a biotin-switch method, and the total SNO was detected by western blot with antibody against biotin (n = 2 pools; n = 3 mice per pool).
(E) Representative fluorescence-activated cell sorting (FACS) histogram (left) and the quantification of the level of protein aggresome (right) in LT-HSCs. The value of protein aggresome in LT-HSCs was normalized to the value detected at day 0 (n = 3–4 per group).
(F) The mRNA expression of iNOS relative to β-actin was analyzed in CD11b+ BM cells from the indicated groups (n = 3 per group).
(G) The level of NO was examined in CD11b+ BM cells from the indicated groups (n = 4 per group).
(A–G) Data were tested for normal distribution via Shapiro-Wilk normality test. Statistical significance between two groups of normally distributed data was assessed by Student’s t test with Welch’s correction. Statistical significance of the non-normally distributed data was assessed by Wilcoxon/Mann-Whitney test (B). All data are shown as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Recent study showed that the level of aggregated protein was quickly increased in proliferating fetal liver HSCs, indicating an important role of protein homeostasis (proteostasis) in HSC regeneration (Sigurdsson et al., 2016). Considering 5-FU treatment strongly stimulates HSC proliferation, we hypothesize that there would be an accumulation of protein aggregation in HSCs. We then performed protein aggregation assay in LT-HSCs at day 9 post-5-FU treatment using ProteoStat staining, a specific fluorescent dye sensitive for detecting protein aggregation. As compared with LT-HSCs from untreated mice, a significantly higher level of protein aggregation was detected in LT-HSCs from 5-FU-treated mice (Figure 1E). Considering the increased level of NO and protein aggregation in HSCs after 5-FU treatment, we believe that NO and its induced protein S-nitrosylation plays a unique role in HSC regeneration, which might be involved in the regulation of protein homeostasis.
To verify the source of NO in HSCs after 5-FU treatment, we checked the expression of NO synthases (iNOS [inducible NOS], eNOS [endothelial NOS], and nNOS [neuronal NOS]) in HSCs at day 9 post-5-FU treatment. The expression levels of nNOS and iNOS were too low to be detected in LT-HSCs, and the expression level of eNOS was not increased in LT-HSCs after 5-FU treatment (Figure S1C). These results suggest that the increased level of NO in HSCs was not due to cell-autonomous production. Previous studies showed that genotoxic stresses promote NO production in CD11b+ myeloid BM cells via inducing iNOS expression (Gorbunov et al., 2000; Punjabi et al., 1994). Because CD11b+ BM cells are well known as HSC niche components (Winkler et al., 2010), we hypothesis that 5-FU treatment may promote iNOS expression in CD11b+ myeloid cells, leading to NO induction in HSC niches, and subsequently caused the increased level of NO in HSCs due to the permeability of NO. Indeed, the result showed that 5-FU treatment strongly induced iNOS expression in CD11b+ BM cells (Figure 1F), which led to a dramatic induction of NO (Figure 1G). In order to further confirm these conclusions, we also examined the level of NO in cultured HSPCs when the exogenous NO supply is limited. Lin− cells from WT mice were cultured for 24 h, and protein S-nitrosylation was checked afterward. We found that, compared with freshly isolated Lin− cells, cultured Lin− cells did not show an induction of protein S-nitrosylation (Figure S1D). This result further supports the idea that the increased NO level in HSCs after 5-FU treatment was not due to cell-autonomous generation.
Gsnor−/− mice show normal HSPC phenotypes under homeostatic condition
Considering that NO induction led to increased protein S-nitrosylation in HPSCs, we then examined the expression of GSNOR, thioredoxin (TRX1), thioredoxin reductase (TRXR), and thioredoxin interacting protein (TXNIP), which are required for reducing the protein S-nitrosylation. We found that the expressions of GSNOR, TRX1, TRXR, and TXNIP were increased in Lin− cells from WT mice 9 days after 5-FU treatment. Of note, GSNOR was the most upregulated denitrosylase, suggesting GSNOR maybe the main denitrosylase in response to the 5-FU-induced HSC regeneration (Figure 2A). We then took advantage of a genetically manipulated mouse model lacking GSNOR to examine its effect on the maintenance of hematopoiesis. We analyzed the BM cells in Gsnor+/+ and Gsnor−/− mice and found that there was no change in the absolute number of both LSK cells and LT-HSCs (Figures S2A–S2D). After further analysis of the progenitor cells, including CMP, GMP, MEP, and common lymphoid progenitor (CLP), we also did not find any significant differences between Gsnor+/+ and Gsnor−/− mice (Figures S2E–S2H). Meanwhile, there was no significant difference of the NO level between the Gsnor+/+ and Gsnor−/− LSK cells (Figure S2I). These data suggest that GSNOR is dispensable for the homeostatic maintenance of HSPCs at steady state.
Figure 2. GSNOR deficiency impairs HSC regeneration after 5-FU treatment.
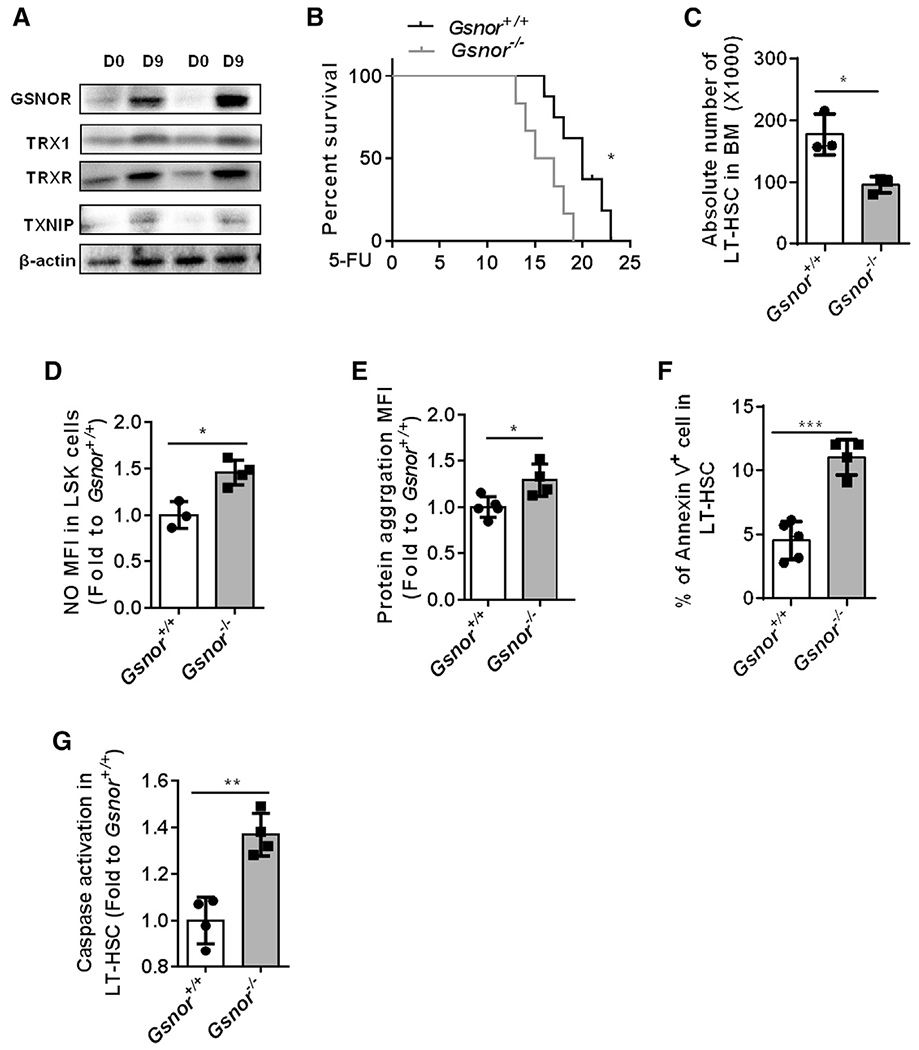
(A) Western blot was performed to detect the expression of GSNOR, TRX1, TRXR, and TXNIP in freshly isolated Lin− cells from WT mice post-5-FU treatment. WT mice received an intraperitoneal (i.p.) injection of 5-FU (150 mg/kg) and were sacrificed at the indicated time points (n = 2 pools; n = 3 mice per pool).
(B) Gsnor+/+ and Gsnor−/− mice (2 to 3 months old) were treated with 5-FU (150 mg/kg) twice with a 1-week interval. The results show the survival rate of Gsnor+/+ and Gsnor−/− mice after 5-FU treatment (n = 6–7 per group).
(C–G) Gsnor+/+ and Gsnor−/− mice (2 to 3 months old) were treated with a single dose of 5-FU (150 mg/kg), and the following assays were performed at day 9 post-5-FU treatment.
(C) The data show the absolute number of LT-HSCs in indicated groups (n = 3 per group).
(D) The data show the levels of NO in indicated groups (the values of NO were normalized to the mean value of NO in Gsnor+/+ LSK cells (n = 3–4 mice per group).
(E) The data show the level of protein aggresomes in the indicated groups (the values of protein aggresomes were normalized to the mean value of protein aggresomes in Gsnor+/+ LT-HSCs; n = 4–5 mice per group).
(F) The data show the percentage of annexin-V-positive cells in LT-HSCs in the indicated groups (n = 4–5 mice per group).
(G) The data show the activation of caspase 3/7 in LT-HSCs in the indicated groups (values were normalized to Gsnor+/+ LT-HSCs; n = 4 mice per group).
(C–G) Data were tested for normal distribution via Shapiro-Wilk normality test. Statistical significance of the two groups of normally distributed data was assessed by Student’s t test with Welch’s correction.
Data are shown as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001.
GSNOR deficiency impairs HSC regeneration after 5-FU treatment
To investigate the role of GSNOR in HSCs regeneration, we treated the Gsnor+/+ and Gsnor−/− mice with 5-FU and recorded the survival status afterward. Notably, Gsnor−/− mice died significantly earlier than Gsnor+/+ mice after two times of 5-FU treatment, suggesting that GSNOR-deficient mice are more sensitive to 5-FU treatment (Figure 2B). To further verify the effect of 5-FU treatment on HSC regeneration, we treated Gsnor+/+ and Gsnor−/− mice with a single dose of 5-FU and assessed the number of HSCs. We found that the absolute number of LT-HSCs and side population (SP)-HSCs were less in Gsnor−/− mice compared to Gsnor+/+ mice at day 3 following 5-FU treatment (Figure S3A). At day 9 post-5-FU treatment, a significant reduction in the absolute number of LT-HSCs was detected in Gsnor−/− mice compared with Gsnor+/+ mice, which was associated with a higher level of NO in Gsnor−/− LSK cells compared with Gsnor+/+ LSK cells (Figures 2C and 2D). Importantly, the level of protein aggregation was significantly higher in Gsnor−/− LT-HSCs than Gsnor+/+ LT-HSCs at day 9 after 5-FU treatment (Figure 2E). Taken together, the above data showed that GSNOR deficiency results in loss of HSC regeneration capacity under a highly regenerative stress associated with NO and protein aggregation induction.
To understand the cause of decreased HSC number in the Gsnor−/− mice post-5-FU treatment, we examined the proliferation and apoptosis rate in HSCs. Ki67 staining and 5-bromo-2-deoxyuridine (BrdU) labeling analyses showed no significant difference between Gsnor+/+ and Gsnor−/− LT-HSCs at day 9 after 5-FU treatment (Figures S3B and S3C). Interestingly, annexin V staining analysis indicated a significant increase of apoptosis rate in Gsnor−/− LT-HSC compared to Gsnor+/+ LT-HSC (Figure 2F), which was further confirmed by the enhanced caspase-3/7 activity detected in the Gsnor−/− LT-HSC compared with Gsnor+/+ LT-HSC (Figure 2G). GSNOR also functions as a formaldehyde dehydrogenase, which has been reported to protect DNA damage caused by formaldehyde in FANCD2 (Fanconi anemia complementation group D2) -deficient mice (Pontel et al., 2015). We therefore speculate that the increased apoptosis in Gsnor−/− HSCs might attribute to the DDR in regenerating HSCs after 5-FU treatment. However, the gamma-H2AX staining analysis revealed a comparable level of DDR in LT-HSCs between Gsnor+/+ and Gsnor−/− mice (Figure S3D), indicating that GSNOR deficiency was unlikely through its function as a formaldehyde dehydrogenase to impair HSCs regeneration after 5-FU treatment. Taken together, these data indicate that increase in apoptosis accounts for the impaired HSC recovery in Gsnor−/− mice after 5-FU treatment. Lastly, to test whether other denitrosylases may also play a role in HSC regeneration after 5-FU treatment when GSNOR is absent, we examined the expression of TRX1, TRXR, and TXNIP in Lin− cells from Gsnor+/+ and Gsnor−/− mice at day 9 post-5-FU treatment. The result showed that only a mild increase in the expression of TRXR with no difference of TRX1 and TXNIP was observed in Gsnor−/− Lin− cells compared with Gsnor+/+ Lin− cells at day 9 post-5-FU treatment (Figure S3E). These data further confirm that GSNOR plays a key role in the context of 5-FU-induced HSC regeneration.
Deletion of GSNOR impairs the repopulating ability of HSCs
To examine the impact of GSNOR deficiency on the long-term regenerative capacity of HSC in vivo, we performed serial competitive transplantation experiments, in which the transplanted HSCs experience extensive long-term regenerative stress (Figure 3A). In contrast to the Gsnor+/+ HSCs that made robust peripheral blood (PB) contribution after the first round of transplantation, the Gsnor−/− HSCs showed a progressive decline in the percentage of donor-derived PB cells (Figure 3B). Further analysis revealed a multi-lineage defect in the reconstitution ability of Gsnor−/− donor HSCs (Figures S4A–S4C). In secondary transplantation, the chimerism of donor-derived cells showed a further decrease in recipient mice that were transplanted with HSCs from Gsnor−/− mice compared with the recipients transplanted with HSCs from Gsnor+/+ mice (Figures 3C and S4D). To examine the self-renewal and differentiation ability of HSCs post-transplantation, we analyzed the chimerisms of donor cells in BM from the primary and secondary recipients. We found a significant reduction in the percentage of donor-derived CD34−LSK, LSK, LK, Lin−, and BM cells in Gsnor−/− HSCs-transplanted recipients as compared with the Gsnor+/+ HSCs-transplanted controls in both first and second round of transplantation (Figures 3D and 3E). Taken together, these data demonstrate that GSNOR deficiency impairs the long-term repopulating capacity of HSC.
Figure 3. Deletion of GSNOR impairs the repopulating ability of HSCs.
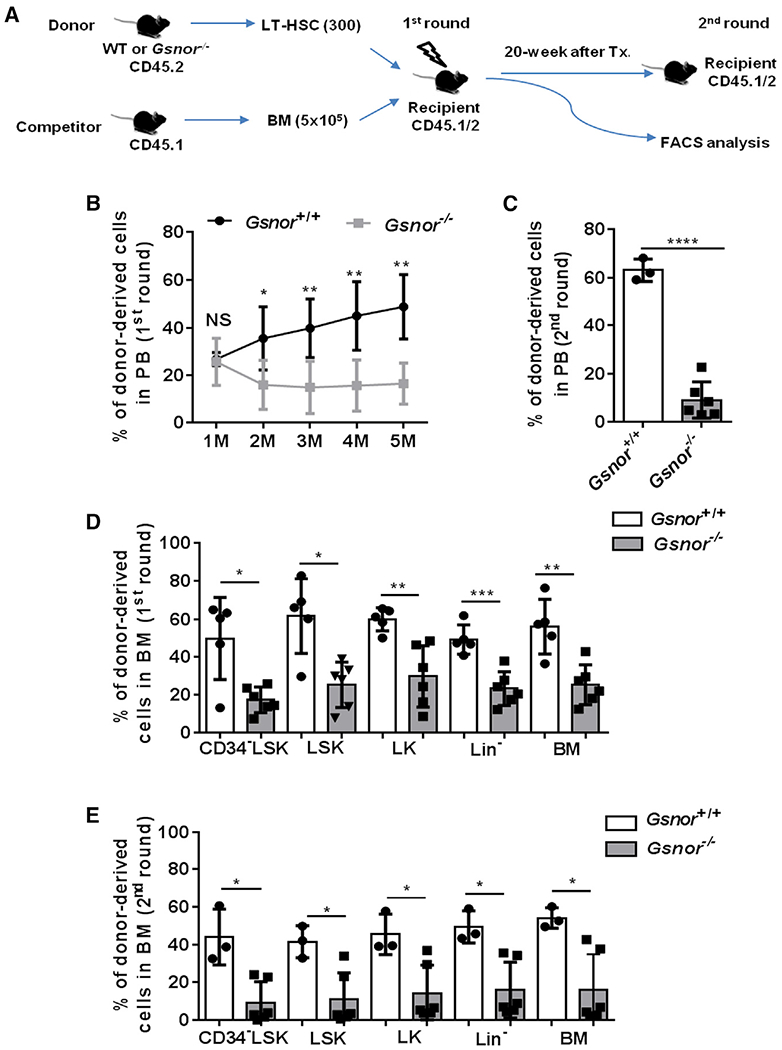
(A) Schematic diagram showing the serial competitive transplantation assay. In brief, 300 LT-HSCs from Gsnor+/+ mice or Gsnor−/− mice (2 to 3 months old; CD45.2) were transplanted into lethally irradiated recipients (CD45.1/2) along with 5 × 105 competitor BM cells (CD45.1). 5 months post-transplantation, 1 × 106 BM cells from the primary recipients were transplanted into the second round of recipient mice.
(B and C) The chimerism of donor-derived cells in PB was examined at the indicated time points after the first and second round of transplantation (n = 3–6 recipients per group).
(D) The chimerisms of donor-derived cells in the indicated populations were analyzed at 5 months after the primary transplantation (n = 5–6 per group).
(E) The chimerisms of donor-derived cells in the indicated populations were analyzed at 8 months after the 2nd transplantation (n = 3–6 per group).
(B–E) Data were tested for normal distribution via Shapiro-Wilk normality test. Statistical significance of the two groups of normally distributed data was assessed by Student’s t test with Welch’s correction (C and D). Logit transformation was performed with the non-normally distributed data, and the Shapiro-Wilk normality test was used for testing the normal distribution. Student’s t test with Welch’s correction was used to compare the statistical significance of the logit-transformed normally distributed data (B). Statistical significance of the non-normally distributed data was assessed by Wilcoxon/Mann-Whitney test (E). Data are shown as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; NS, not significant.
Inhibiting NO synthesis restores regenerative capacity of Gsnor−/− HSCs
Given that GSNOR-deficient HSCs exhibited increased level of NO and impaired regeneration upon regeneration stress (Figures 2 and 3), we speculated that excessive NO might be responsible for the reconstitution defect of Gsnor−/− HSCs. To establish the direct link between NO-mediated protein S-nitrosylation and HSC regeneration, we treated the serial transplanted recipient mice with NOS inhibitor L-NAME, which has been widely used to inhibit NO synthesis (Gomes et al., 2013), and examined the donor contribution in PB and BM (Figure S5A). The chimerism of Gsnor−/− donor-derived cells in PB was significantly increased by L-NAME treatment compared with PBS treatment in both the first and second rounds of transplantation (Figures 4A and S5B). Similarly, the multi-lineage reconstitution capacity of Gsnor−/− donor HSCs was increased by L-NAME treatment (Figures S5C–S5E). Consistently, the percentage of Gsnor−/− donor-derived CD34−LSK, LSK, LK, Lin−, and total BM were significantly increased after L-NAME treatment compared with PBS treatment (Figure 4B). In contrast, the L-NAME versus PBS treatment had no significant effect on the chimerism of mice that were transplanted with Gsnor+/+ HSCs (Figures 4A, 4B, and S5B–S5E). These data indicate that induction of NO contributes to the impairment of the regenerative capacity of Gsnor−/− HSCs.
Figure 4. Inhibiting NO synthesis restores regenerative capacity of Gsnor−/− HSCs.
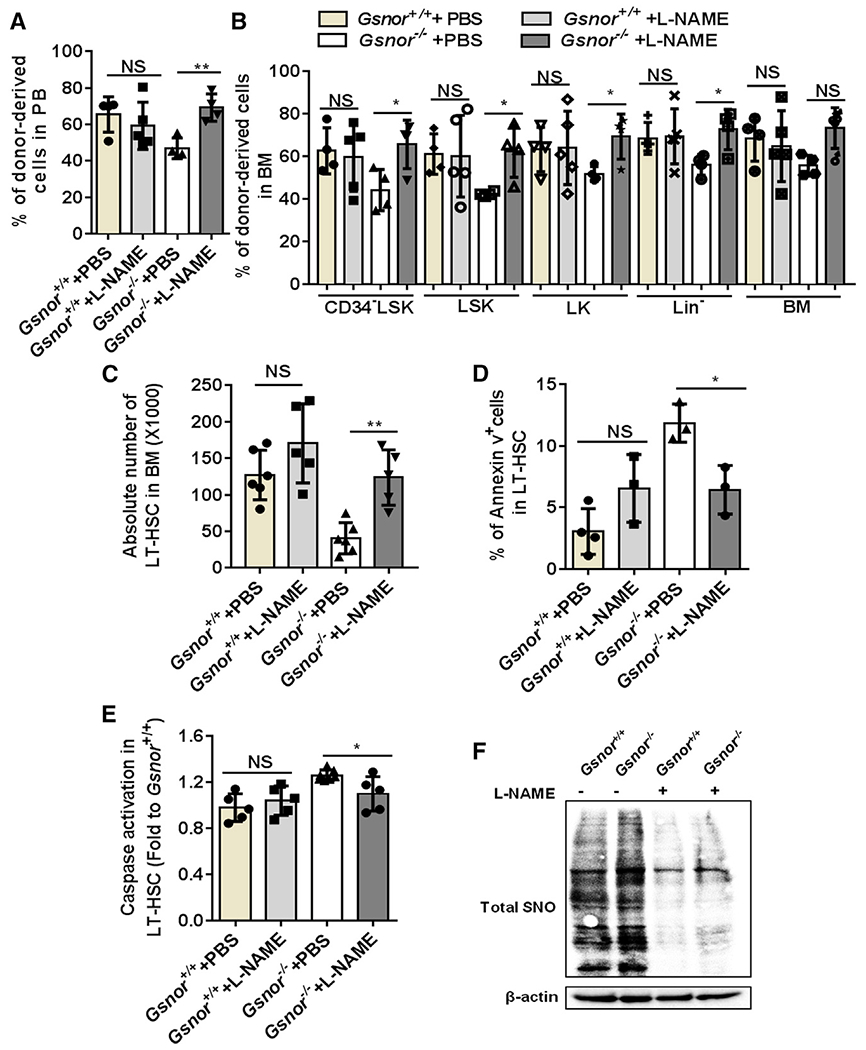
(A and B) Three hundred LT-HSCs from Gsnor+/+ and Gsnor−/− mice were transplanted into lethally irradiated recipients along with 5 × 105 competitor cells (LT-HSCs were pooled from 3 to 4 donor mice). 3 weeks after transplantation, mice were treated with L-NAME (10 mg/kg) or PBS every other day. The chimerisms of donor-derived cells in PB and BM were analyzed at 16 weeks after transplantation (n = 4–5 per group).
(A) Percentage of donor-derived cells in PB.
(B) Percentage of donor-derived cells in the indicated populations.
(C–F) Gsnor+/+ and Gsnor−/− mice received an i.p. injection of 5-FU (150 mg/kg). L-NAME (10 mg/kg) or PBS injection were performed with the mice every other day after 5-FU treatment. Mice were sacrificed for further analysis at day 9 post-5-FU treatment.
(C) Data show the absolute number of LT-HSCs in indicated groups (n = 5–6 per group from two independent experiment).
(D) Data show the percentage of annexin-V-positive cells in LT-HSCs in indicated groups (n = 3–4 per group).
(E) Data show the activation of cleaved-caspase-3/7 in LT-HSCs in indicated groups (values were normalized to WT LT-HSCs; n = 5–7 per group).
(F) Total S-nitrosylated proteins (SNO) were purified from Lin− cells by a biotin-switch method. Total SNO was detected by western blot with an antibody against biotin (n = 3 mice per pool; n = 2 independent experiments; one of the two is shown, the other shows a similar result).
(A–E) Data were tested for normal distribution via Shapiro-Wilk normality test. (A and B) Statistical significance of the non-normally distributed data was assessed by Wilcoxon/Mann-Whitney test.
(C–E) To compare the significance between two treatments that passed normality test, two-way ANOVA followed by Tukey’s multiple comparison test was used. Data are shown as mean ± SD. *p < 0.05; **p < 0.01.
Next, we examined the effect of L-NAME treatment on hematopoietic recovery in the Gsnor−/− and Gsnor+/+ mice following 5-FU treatment. L-NAME-treated Gsnor+/+ mice showed a reduction of NO level in LT-HSCs, suggesting a successful inhibition of NO by L-NAME (Figure S5F). Further analysis on the absolute number of hematopoietic stem/progenitor cells (LT-HSCs, GMP, CMP, and MEPs) and mature cells (B cells, myeloid cells, and T cells) showed no significant difference between L-NAME-treated mice and PBS-treated mice (Figures S5G and S5H), indicating L-NAME treatment does not have detrimental effect on hematopoietic system. Gsnor−/− mice showed a decreased number of LT-HSCs in response to 5-FU treatment (Figure 4C). However, the reduction of LT-HSCs in Gsnor−/− mice was restored by L-NAME treatment (Figure 4C). Consistently, L-NAME treatment ameliorated the GSNOR-deficiency-induced apoptosis, as indicated by both the reduction of annexin-V-positive cells and activation of caspase-3/7 (Figures 4D and 4E). To further determine the effect of L-NAME on NO inhibition, we detected the total protein S-nitrosylation in Lin− cells from Gsnor−/− and Gsnor+/+ mice treated with L-NAME and PBS at day 9 post-5-FU treatment. The results showed that, as compared with PBS-treated group, the total protein S-nitrosylation level was decreased after L-NAME treatment in both Gsnor−/− and Gsnor+/+ group (Figure 4F), indicating an inhibition of protein S-nitrosylation via L-NAME treatment. In conclusion, these data suggest that NOS inhibitor L-NAME treatment improves the impaired recovery of Gsnor−/− HSCs after 5-FU treatment.
Chemical chaperone rescues the regeneration defect of Gsnor−/− HSCs
As shown above (Figure 2), a defect of HSC recovery and accumulation of protein aggregation were detected in Gsnor−/− mice in response to 5-FU treatment. Therefore, we reasoned that the defect of HSC recovery in Gsnor−/− mice after 5-FU treatment might be due to protein aggregation. To verify this hypothesis, after 5-FU treatment, we treated Gsnor+/+ and Gsnor−/− mice with TCA, which has been used to reduce the level of protein aggregation in fetal liver HSCs (Sigurdsson et al., 2016; Figure 5A). We found that TCA treatment significantly inhibited the increased protein aggregation in Gsnor−/− HSCs (Figure 5B). We then analyzed the absolute number of HSCs in Gsnor+/+ and Gsnor−/− mice after 5-FU followed by TCA treatment. The number of LT-HSCs in Gsnor−/− mice treated with TCA was significantly increased compared with Gsnor−/− mice treated with PBS, suggesting that TCA treatment enhanced HSC recovery in Gsnor−/− mice in response to 5-FU treatment (Figure 5C). Of note, TCA treatment strongly diminished the elevated apoptosis in Gsnor−/− HSCs upon 5-FU treatment, as indicated by the decreased annexin-V-positive cells (Figure 5D) and alleviated activation of caspase-3/7 (Figure 5E). In contrast, TCA versus PBS treatment had no significant effect on the protein aggregation, LT-HSCs number, and apoptosis rate in Gsnor+/+ mice (Figures 5B–5E). Collectively, these data indicate that the accumulation of protein aggregation and consequently enhanced apoptosis are responsible for the impaired regenerative capacity of Gsnor−/− HSCs.
Figure 5. Chemical chaperone rescues the regeneration defect of Gsnor−/− HSCs.
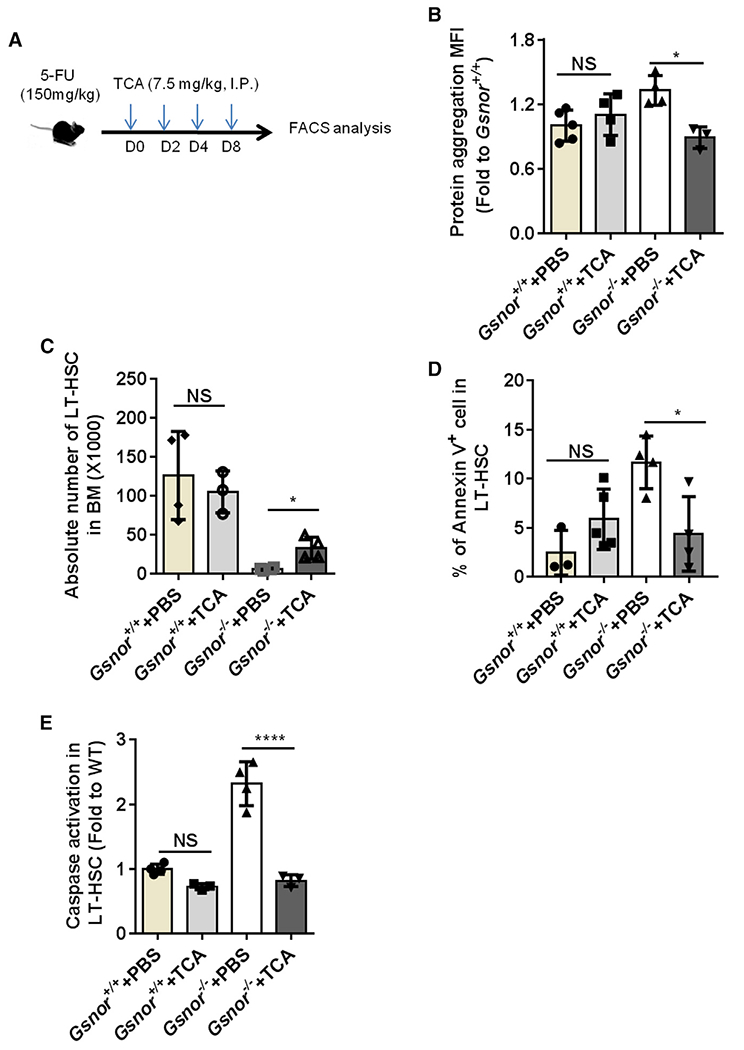
(A) Schematic diagram of chemical chaperone taurocholic acid (TCA) treatment post-5-FU treatment. Gsnor+/+ and Gsnor−/− mice were treated with a single-dose of 5-FU (150 mg/kg). The mice were then treated with TCA (7.5 mg/kg) or PBS every other day after 5-FU treatment. Mice were sacrificed at day 9 post-5-FU treatment for further analyses.
(B) Data show the quantification of protein aggregation in LT-HSCs of mice in the indicated groups (values were normalized to the mean value in Gsnor+/+ PBS group; n = 3–5 mice per group from two independent experiments).
(C) Data show the absolute number of LT-HSCs in the indicated groups (n = 3–4 per group from three independent experiments).
(D) Data show the percentage of annexin-V-positive cells in LT-HSCs in the indicated groups (n = 3–5 mice per group from two independent experiments).
(E) Data show the activation of caspase 3/7 in LT-HSCs in the indicated groups (the values were normalized to the mean value of LT-HSCs in PBS-treated Gsnor+/+ mice; n = 3–4 per group).
(B–E) Data were tested for normal distribution via Shapiro-Wilk normality test. To compare the significance between two treatments that passed normality test, two-way ANOVA followed by Tukey’s multiple comparison test was used. Data are shown as mean ± SD. *p < 0.05; ****p < 0.0001.
Chop deletion rescues the impaired function of Gsnor−/− HSCs
It has been shown that accumulation of protein aggregation activates endoplasmic reticulum (ER)-stress-induced apoptosis in fetal liver HSCs (Sigurdsson et al., 2016). We also found decreased absolute number of LT-HSCs in Gsnor−/− fetal liver compared with Gsnor+/+ fetal liver at embryonic day 14.5 (E14.5) (Figure S6A). Similar like 5-FU-treatment-induced HSC proliferation (Figure 1B), HSCs at fetal liver stage are highly proliferative (Pietras et al., 2011). This may lead to NO production and cause protein S-nitrosylation, which cannot be efficiently removed in Gsnor−/− fetal liver and results in HSC number reduction via ER-stress-mediated apoptosis. Therefore, we reasoned that ER stress response could mediate the apoptosis of Gsnor−/− HSCs. To test this hypothesis, Gsnor+/+ and Gsnor−/− mice were treated with a single dose of 5-FU, and LT-HSCs were purified at day 9 post-5-FU treatment for unfolded protein response (UPR)-related gene expression analysis. As expected, the expression of many UPR-related genes was markedly upregulated in LT-HSCs from Gsnor−/− mice compared with LT-HSCs from Gsnor+/+ mice (Figure 6A). In order to examine the induction of UPR activation at protein level, we treated the Gsnor+/+ and Gsnor−/− mouse embryonic fibroblast (MEF) cells with GSNO and examined the expression of UPR-related proteins, such as Bip, ATF4, Chop, and the level of protein S-nitrosylation 24 h after GSNO treatment. Compared with PBS treatment, an increase of the total protein S-nitrosylation in both Gsnor−/− and Gsnor+/+ MEF cells was detected after GSNO treatment (Figure 6B). Meanwhile, increased expression of Bip, ATF4, and Chop were detected in GSNO-treated Gsnor−/− MEF cells compared with GSNO-treated Gsnor+/+ MEF cells (Figure 6B). Among the upregulated proteins, Chop has been reported as an important factor regulating UPR-induced apoptosis (van Galen et al., 2014). To test whether the ER-stress-induced apoptosis is responsible for the regeneration defect of Gsnor−/− HSCs, we generated Gsnor and Chop double-knockout mice (Gsnor−/−Chop−/−) and performed serial competitive transplantation to examine the regenerative capacity of Gsnor−/−Chop−/− HSCs. We found that the chimerism of donor-derived cells in PB was significantly increased in Gsnor−/−Chop−/− recipient mice compared with Gsnor−/−Chop+/+ recipient mice in both the first and second rounds of transplantation (Figures 6C and 6D). These data suggest that Chop deletion rescues the reconstitution defect of Gsnor−/−Chop+/+ HSCs. To further determine the HSC recovery in Gsnor−/−Chop−/− mice, we treated the Gsnor−/−Chop+/+ and Gsnor−/−Chop−/− mice with a single dose of 5-FU and calculated the number of LT-HSCs at day 9 post-5-FU treatment. We found that the number of LT-HSCs was strongly increased in Gsnor−/−Chop−/− mice compared with Gsnor−/−Chop+/+ mice (Figure 6E). Furthermore, the activity of caspase-3/7 in LT-HSCs was markedly reduced in Gsnor−/−Chop−/− mice compared with LT-HSCs in Gsnor−/−Chop+/+ mice (Figure 6F). In line with these results, either L-NAME or TCA treatment reduced the expression of Chop in LT-HSCs of Gsnor−/− mice (Figure S6B). Taken together, these data indicate that GSNOR regulates HSC regeneration via Chop-mediated apoptosis.
Figure 6. Chop deletion rescues the impaired function of Gsnor−/− HSCs.
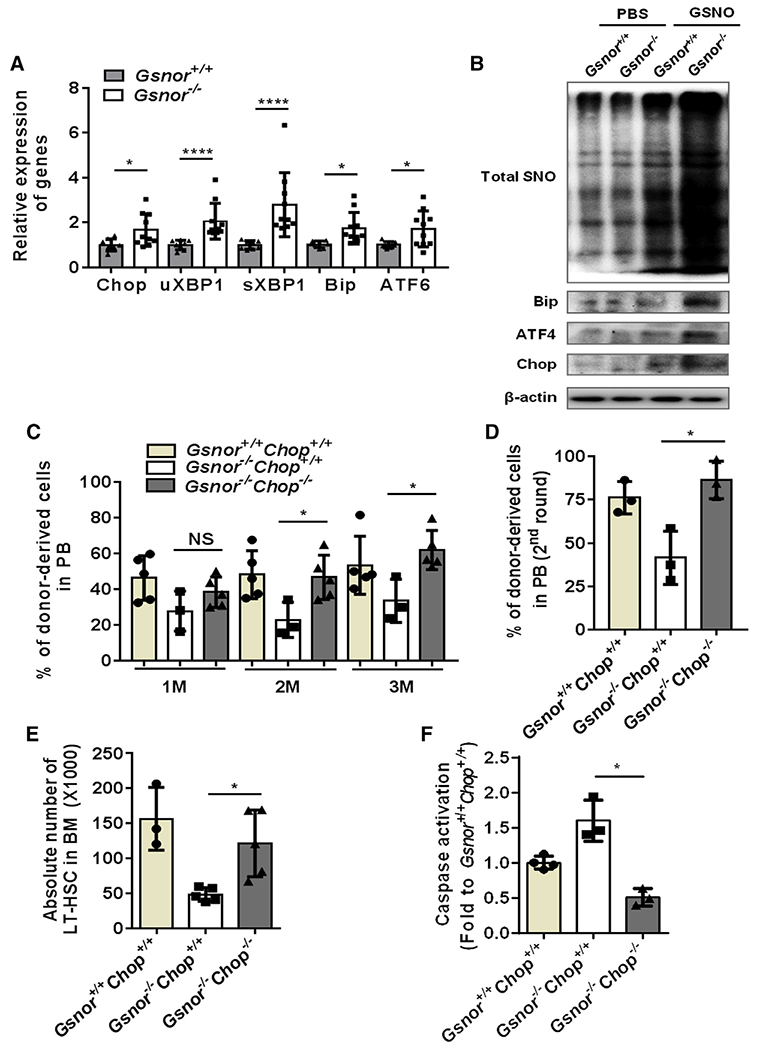
(A) Gsnor+/+ mice and Gsnor−/− mice received an i.p. injection of 5-FU (150 mg/kg). The relative expression of the indicated genes to β-actin was measured by qPCR in freshly isolated LT-HSCs from the mice 9 days after 5-FU treatment (n = 8–10 per group).
(B) Gsnor+/+ and Gsnor−/− MEF cells were treated with or without chemical NO donor S-nitrosoglutathione (GSNO), and the total SNO level was detected. Total SNO was detected by western blot with an antibody against biotin. Western blot was performed to detect the expression level of Bip, ATF4, and Chop in Gsnor+/+ and Gsnor−/− MEF cells in the presence or absence of GSNO (n = 2 independent experiments; one of the two is shown, the other shows a similar result).
(C and D) Three hundred LT-HSCs from Gsnor+/+Chop+/+, Gsnor−/−Chop+/+, and Gsnor−/−Chop−/− mice were transplanted into lethally irradiated recipient mice along with 5 × 105 competitor BM cells. 12 weeks later, 1 × 106 BM cells from the primary recipients were transplanted into the secondary recipients.
(C) The results show the percentage of donor-derived cells in PB at the indicated time point after first round of transplantation (n = 3–5 per group).
(D) The data show the chimerism of donor-derived cells in PB 8 weeks after the second round of transplantation (n = 3 per group).
(E and F) Gsnor+/+Chop+/+ mice, Gsnor−/−Chop+/+ mice, and Gsnor−/−Chop−/− mice were treated with a single dose of 5-FU (150 mg/kg) and sacrificed at day 9 post-5-FU treatment for further analyses.
(E) The data show the absolute number of LT-HSCs in the indicated groups (n = 3–5 per group).
(F) The data show the activation of cleaved-caspase-3/7 in LT-HSCs in the indicated groups (the values were normalized to the mean value in LT-HSCs from Gsnor+/+Chop+/+ mice; n = 3–4 group).
(A–F) Data were tested for normal distribution via Shapiro-Wilk normality test. Statistical significance of the two groups of normally distributed data was assessed by Student’s t test with Welch’s correction. Statistical significance of the non-normally distributed data was assessed by Wilcoxon/Mann-Whitney test (A). Data are shown as mean ± SD. *p < 0.05; ****p < 0.0001.
DISCUSSION
In this study, we found that increased NO level induced in LT-HSCs after 5-FU treatment contributed to the increased protein S-nitrosylation as well as the protein aggregation (Figures 1G and 1H). In GSNOR-deficient mice, the loss of protein denitrosylation ability further exacerbated the accumulation of protein S-nitrosylation, which eventually triggered apoptosis in HSCs. The rescue of apoptosis by chemical chaperone in Gsnor−/− HSCs suggested that proteostatic collapse is responsible for the protein-S-nitrosylation-induced apoptosis. Furthermore, the genetic ablation of Chop rescued the regeneration defect of Gsnor−/− HSCs, indicating that proteostatic collapse impairs HSC regeneration via ER-stress-induced apoptosis.
The previous studies revealed that NO stimulates HSC proliferation, mobilization, and myeloid differentiation (Aicher et al., 2003; Nogueira-Pedro et al., 2014). We detected an increased level of NO (Figure 1C), enhanced HSC proliferation (Figure 1B), and increased HSC absolute number (Figure S1A) in 5-FU-treated mice, which was consistent with previous studies. In our study, GSNOR deficiency does not affect the homeostasis of HSC although results in a dramatic cell death in HSCs under regenerative stresses. A reasonable possibility is, at steady state, most of HSCs remain in quiescent stage with limited protein synthesis, and once experiencing proliferative stress, sharply increased protein synthesis in HSCs requires a boost of the protein folding machinery, whereas the enhanced NO accompanied by HSC proliferation leads to increased S-nitrosylation-compromised protein folding and results in an accumulation of protein aggregation and eventually leads to UPRER-mediated apoptosis. These data demonstrate a unique role of GSNOR in maintaining the balance between HSC proliferation and survival through the regulation of proteostasis upon regenerative stresses.
GSNOR, functioning as a denitrosylase, plays an important role in many physiological processes by denitrosylating target proteins. GSNOR deficiency impairs mitochondrial dynamics and mitophagy in aging cells by promoting the S-nitrosylation of Drp1 and Parkin (Rizza et al., 2018). In lymphatic system, GSNOR deletion enhances the S-nitrosylation of GAPDH (Glyceraldehyde-3-phosphate dehydrogenase), which results in thymocytes apoptosis and impairs lymphocyte development (Yang et al., 2010). GSNOR deletion increases HIF-alpha S-nitrosylation, which results in improvement in angiogenesis after myocardial injury (Lima et al., 2009). In this study, we show that GSNOR, by exerting its catalytic efficiency and specificity for GSNO reduction, regulates the protein S-nitrosylation during HSC regeneration. Consistently, MSCs from Gsnor−/− mice showed decreased capacity during vasculogenesis due to downregulation of PDGFRα (platelet derived growth factor receptor alpha) related to NO/GSNOR imbalance (Gomes et al., 2013). Together, these studies suggest that GSNOR regulates different stem cell functions according to the distinct denitrosylated target proteins in a tissue- and physiological-condition-dependent manner.
Protein quality and proteostasis are essential for the maintenance of cell function (García-Prat et al., 2017; Higuchi-Sanabria et al., 2018). A research showed an accumulation of protein aggregation in proliferative HSCs (Liu et al., 2019). In this study, GSNOR deficiency resulted in a significant increase of protein aggregation and defected reconstitution capacity in response to HSC regenerative stress (Figures 2E and 5B). UPR-related genes were highly expressed in Gsnor−/− HSCs after 5-FU treatment (Figure 6A), which may be due to the accumulation of protein aggregation. A recent study showed that chemical chaperone TCA can significantly diminish the increased level of protein aggregation in fetal liver HSCs (Sigurdsson et al., 2016). Consistent with this finding, we found that TCA treatment attenuated the accumulation of protein aggregation and rescued the defect of HSC recovery in Gsnor−/− mice (Figure 5). These data demonstrate a clear and mostly linear relationship between the accumulation of protein aggregation and the defected reconstitution capacity of GSNOR-deficient HSCs.
ER utilizes its protein folding status as a signal to orchestrate downstream adaptive or apoptotic response. Previous studies showed that, compared to the progenitor compartment, HSCs are susceptible to misfolded protein induced by ER stress and have a strong activation of apoptotic response (van Galen et al., 2014). Consistently, our data showed an enhanced ER-stress response and apoptosis in Gsnor−/− HSCs after 5-FU treatment (Figure 6A), suggesting a potential role of UPR in GSNOR-deficient HSCs. Among the upregulated UPR genes, Chop is most functionally relevant to the reduced viability of Gsnor−/− HSCs, as it is the key factor mediating UPRER-induced apoptosis. Deletion of Chop significantly rescued the defect of Gsnor−/− HSCs, demonstrating that the UPR-induced apoptosis is essential in suppressing HSC regeneration.
As a posttranslational modification, S-nitrosylation is widely considered as an important mediator of signal transduction pathways. However, aberrant S-nitrosylation often affects protein misfolding and causes ER stress, which results in an impairment of cell function (Ryan et al., 2013). It has been reported that excessive S-nitrosylation of IRE1a induced by obesity-associated inflammation results in compromising UPR function and alters ER homeostasis (Yang et al., 2015). S-nitrosylation of PDI (protein-disulphide isomerase) and parkin plays an essential role in protein homeostasis maintenance, trigger protein misfolding, and ER stress under neurodegeneration conditions (Uehara et al., 2006; Chung et al., 2004; Yao et al., 2004). In line with the above findings, our study showed an increased level of protein S-nitrosylation and protein aggregation in HSPCs under regeneration stress, which reinforces the relationship between the increased protein S-nitrosylation and impaired protein folding. In our study, several S-nitrosylated proteins were observed in Gsnor−/− HSPCs during regeneration process (Figure S7A; Table S1). Interestingly, a few heat shock proteins, HSP60, HSP70, and HSP90 were highly S-nitrosylated in Gsnor−/− Lin− BM cells(Table S1). One of them, the S-nitrosylation of HSP90 and its contribution in protein aggregation, was further measured. Previous study showed S-nitrosylation on the catalytic domain (Cys 596/597) and the C-terminal domain of HSP90 inhibits its ATPase activity (Martínez-Ruiz et al., 2005; Retzlaff et al., 2009). Here, we reported new S-nitrosylation residues of HSP90 located at its M domain (Cys521/590/591) in GSNOR deficiency cells, which affected the protein folding function of HSP90 and caused an accumulation of protein aggregation (Figures S7B–S7D). Reversal of the S-nitrosylation by mutation of the nitrosylation residues of HSP90 ameliorated the protein aggregation induced by GSNOR deficiency (Figures S7E–S7G). Taken together, we found that S-nitrosylation of HSP90 compromises its protein-folding activity, contributing to the accumulation of protein aggregation in Gsnor−/− cells upon stress. These data suggest that the increased level of S-nitrosylated UPR-related proteins might lead to an impairment of protein folding and contribute to the induction of protein aggregation. Nevertheless, given that GSNOR deficiency induces protein aggregation via increased S-nitrosylation of many proteins, identifying other essential targets of GSNOR that regulate the UPR will be of great interest.
In summary, our data established a mechanistic link between the GSNOR-mediated protein S-nitrosylation and the Chop-mediated UPR in regulating HSC viability during regeneration process. This finding further emphasized the importance of proper regulation of proteostasis in HSCs under proliferative stress, which is also physiologically important for maintaining HSC stress responses in vivo. Interfering with S-nitrosylation modification may help to achieve HSC functional expansion without losing its viability under proliferative stresses.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Zhenyu Ju (zhenyuju@163.com).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate datasets or codes.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
MICE Gsnor−/− mice were provided by Dr. Limin Liu (University of California, San Francisco)(Liu et al., 2004). Chop deficient mice were purchased from Nanjing institute of biomedical research, Nanjing University. 8-week-old CD45.1 mice or CD45.1/2 were used as recipient or competitor mice in HSC transplantation assay. Both female and male mice were used. All mice were maintained in a specific pathogen-free facility. The Animal Care and Ethics Committee at Hangzhou Normal University approved all animal experiments in our study.
METHOD DETAILS
Flow cytometry and cell sorting
BM cells were incubated in a lineage cocktail containing antibodies against CD4 (RAM4-5), CD8 (53-6.7), Ter119 (TER119), CD11b (M1/70), Gr-1 (RB6-8C5), and B220 (RA3-6B2) for 30 min. After washing, the cells were incubated with CD48 (HM48-1), CD150 (TC15-12F12.2), CD45.2 (104), CD45.1 (A20), Sca1 (E13-161.7), Flt3 (A2F10), IL-7R (A7R34), c-Kit (ACK2), CD16/32 (93), CD34 (RAM34), and streptavidin. Side population cell analysis was performed using Hoechst 33342 (5ug/ml, Sigma-Aldrich). All monoclonal antibodies were purchased from BD Bioscience or eBioscience. Before cell sorting, BM cells were first enriched with anti-antigen-presenting cell microbeads (MiltenyiBiotec) and then stained with surface markers. FACS analysis was performed with BD Influx cell sorter and BD LSRFortessa. Data were analyzed with FlowJo software.
Transplantation Analysis
Competitive transplantation was performed by transplanting 300 LT-HSCs sorted from 2-3-month old Gsnor+/+ and Gsnor−/− mice (donor, CD45.2) along with 5 × 105 BM cells from young (2-3-month old) competitor mice (CD45.1 or CD45.1/CD45.2). Recipient mice (CD45.1/CD45.2 or CD45.1) were lethally (8Gy) irradiated before transplantation and treated with antibiotic water for 2 weeks after transplantation. The chimerisms of donor-derived cells in PB and BM were examined at indicated time points after transplantation.
Apoptosis and cell cycle assays
Apoptosis assays were performed via Annexin V/DAPI (Annexin V Apoptosis Detection Kit, eBioscience) and caspase- Glo 3/7 (Promega) staining. Cell cycle analyses was performed via BrdU (BD Cytofix/Cytoperm kit) and Ki67 staining (BD Cytofix/Cytoperm kit). For BrdU assay, BrdU (100mg/kg, BD Bioscience) was injected via intraperitoneal (i.p.) 16 hours before mice sacrifice.
Lentivirus production
Lentivirus were produced in 293T cells after transfection of 7 μg HSP90WTor HSP90M plasmid, 5 μg pspAX2 helper plasmid, and 3 μg Pmd2g.23. The virus was collected 48 hours after transfection and concentrated by centrifugation at 25,000 rpm for 2.5 hours at 4°C, and the virus pellet was suspended in PBS. LSK cells from WT or Gsnor−/− mice were infected with HSP90WT and/or HSP90M lentivirus and then transplanted into lethally irradiated mice 48 hours after infection. The recipient mice were treated with 5-FU two months later, 1000 of the transduced LT-HSC (GFP+) cells were sorted, and caspase-3/7 assay were performed.
NO level detection assay
A total of 5 × 106 BM cells were stained with surface markers and washed with cold PBS, cells were then stained with NO-sensitive fluorescent dye DAF-FM DA (Sigma-Aldrich, 1 μM) at 37°C for 30 min. DAF-FM DA signal was analyzed by FACS.
5-FU treatment
For survival assay, the recipient mice were i.p. injected with two rounds of 5-FU (Sigma Aldrich, 150 mg/kg). For HSC regeneration assay, mice were i.p. injected with a single-dose of 5-FU (Sigma Aldrich, 150 mg/kg).
L-NAME and TCA treatment
The eNOS inhibitor L-NAME (Sigma Aldrich) was dissolved in PBS with 50mg/ml as a stock solution. Intraperitoneal injected with 10 mg/kg per mouse. For the HSC engraftment assay, mice were injected with L-NAME every 3 times per week at 3 weeks post transplantation;For the 5-FU and L-NAME treatment assay, mice were injected with L-NAME every other days after 5-FU treatment. TCA (Sigma-Aldrich) was dissolved in PBS with 5mg/ml as a stock solution. The stock solution was diluted to 500 μg/ml, and 300ul of the diluents was intraperitoneal injected into mice. Mice were euthanized and BM cells were collected for subsequent analysis.
Protein Aggregation (ProteoStat Staining)
Cells were fixed and permeabilized using BD Fixation/Permeabilization Solution kit (BD, 554714). The levels of protein aggregation in indicated populations were analyzed by FACS using ProteoStat Dye (Enzo Life Sciences).
RNA isolation and Real-time PCR analysis
Total RNA was isolated from HSCs using RNeasy Micro Kit (QIAGEN) and reversed using PrimeScriptcDNA synthesis Kit (Takara). Real-time-PCR was performed with Eva-Green probe on a 7300 Real-Time PCR system for 40 cycles. A mouse internal β-actin was included in every reaction for normalization. Each experiment was performed in at least three biological replicates.
Immunofluorescence assay
For immunostaining, MEF cells were cultured in 24 wells plate with coverslips. After cell seeding for 24 hours, the cells were further treated with GSNO donor (Sigma Aldrich, 10uM) for 24 hours. Cells were fixed with 4% paraformaldehyde (Sigma Aldrich), and blocked with a blocking solution (normal goat serum) accompanied with 0.1% saponin for permeabilization. After blocking, the cells were incubated in 0.1% saponin with primary antibody: anti-FLAG (Medical & Biological Laboratories), secondary antibody and fluorescence-probe: Alex Flour 633 goat anti-mouse, ProteoStat Dye (1:1000), DAPI (1ug/ml). Images were taken in multi-tracking mode on a laser scanning confocal microscope (LSM710, Carl Zeiss) with a 63 plan apochromat 1.4 NA objective.
Western Blot assay
The cells were lysed in RIPA buffer and subjected to SDS-PAGE. Proteins were transferred to nitrocellulose filter membrane (Millipore). Subsequently, the membrane was blocked in 5% skimmed milk and followed by incubation with anti-GSNOR (ABclonal, WA-20754D), anti-TRX1 (Proteintech, 14999-1-AP), anti-TRXR (Proteintech, 67728-1-Ig), anti-TXNIP (Proteintech, 18243-1-AP), β-actin (Sungene Biotech, KM9001) antibodies overnight. On the next day, the membranes were incubated with secondary antibody. Finally, the protein bands were visualized with an ECL Western Blot detection kit (Pierce).
Quantitative S-nitrosylation proteomics and data analysis
The iodo TMT labeling was conducted by using iodoacetyl tandemmass tag (iodoTMT) reagents (90103, Thermo Scientific, USA) as described previously(Qu et al., 2014). Briefly, isolated WT and Gsnor−/− Lin− cells were lysed in HENS buffer, free cysteine was blocked with methyl methanethiosulfonate (MMTS), and S-nitrosylated cysteine were labeled with different iodo TMT reagent and enriched with anti-TMT antibody. The multiplexed quantitative mass spectrometry data were collected by using Q Exactive mass spectrometer equipped with an easy n-LC 1000HPLC system (Thermo Scientific) on data-dependent acquisition mode. The raw data from Q Exactive were analyzed with Proteome Discovery version 1.4 using Sequest HT search engine for protein identification and Percolator for false discovery rate analysis. The Uniprot miceprotein database was used for searching data from the mice sample. Protein quantification was also performed on Proteome Discovery version 1.4 using the ratio of the intensity of reporter ions from the MS/MS spectra. Only unique peptides of proteins or protein groups were selected for protein relative quantification.
The biotin switch assay
The IBP for detecting the S-nitrosylation was performed as previously described. cells were lysed in HEN buffer (250mM HEPES pH 7.7, 0.1mM EDTA, 10mM neocuproine) with 1% NP40 (Nonidet P-40), protease inhibitor cocktail and 20mM MMTS for 20min at 4°C, then centrifuged at 12000 × g for 10 minutes at 4°C. The supernatant were incubated with 2.5% SDS at 50°C for 30 minutes with frequent vortex (every 5 minutes), and excess MMTS was removed by ice-cold acetone precipitation followed by centrifugation at 2000 × g for 10 minutes. This precipitation was repeated three times. The precipitate was recovered in HEN buffer containing 2.5% SDS with 0.4mM biotin- maleimide and 10mM ascorbate and incubated at 37°C for 1 hour or at room temperature for 2 hours. The excess biotin-maleimide was removed by ice-cold acetone precipitation as previously described. The pellet was resuspended in HEN buffer with 200mM DTT and incubated for 15 minutes at 100°C, followed by addition neutralization buffer (250mM HEPES pH 7.7, 100mM NaCl, 0.1mM EDTA, 10mM neocuproine) and streptavidin-agarose (50-100 μl/sample) to purify the biotinylated proteins. This material was incubated at room temperature for 2 hours. The agarose was washed 3 times(800 × g for 1 minute)with neutralization buffer with 0.05% SDS and the proteins were eluted by HEN buffer containing 2.5% SDS at 100°C for 15 minutes. The eluted mixture was analyzed by SDS-PAGE, followed by immunoblotting with anti-HSP90 (CST, 5087), anti-Bip (CST, 3177), anti-ATF4 (CST, 11815), anti-Chop (CST, 2895) antibodies.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as the mean ± SD. All data were tested for the normal distribution using the Shapiro-Wilk normality test. For data that did not pass normality test, we performed log (for cell number or ratios) or logit transformation (for proportions), and tested for the normal distribution using the Shapiro-Wilk normality test with the transformed data before statistical analysis. We used parametric tests for the data that passed normality test. Statistical significance between two groups was assessed by Student’s t test with Welch’s correction. To assess the statistical difference between two conditions or treatments, we performed two-way ANOVA followed by Tukey’s multiple comparison test. The non-normally distributed data were assessed Wilcoxon/Mann-Whitney test. All statistical analyses were performed with GraphPad Prism 7 software. The significance level was set at 0.05. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| B220-APC | eBioscience | RA3-6B2; RRID: AB_469394 |
| B220-PE-cy7 | Biolegend | RA3-6B2; RRID: AB_313004 |
| B220-biotin | Biolegend | RA3-6B2; RRID: AB_312989 |
| Flt3-PE | eBioscience | A2F10; RRID: AB_465860 |
| IL-7R-Percp-Cy5.5 | Biolegend | A7R34; RRID: AB_1937273 |
| CD16/32- Percp-Cy5.5 | eBioscience | 93; RRID: AB_996661 |
| CD16/32- Alexafluo700 | eBioscience | 93; RRID: AB_493994 |
| cKit-APC | eBioscience | ACK2; RRID: AB_469433 |
| cKit-PE-cy7 | Biolegend | 2B8; RRID: AB_893228 |
| Streptavidin-APC-cy7 | Biolegend | 405208 |
| CD34-FITC | eBioscience | RAM34; RRID: AB_465021 |
| CD34- Alexafluo700 | eBioscience | RAM34: RRID: AB_2815232 |
| Sca1-FITC | Biolegend | D7: RRID: AB_756190 |
| Sca1-PE-cy7 | Biolegend | D7; RRID: AB_493597 |
| CD150-PE | Biolegend | TC15-12F12.2: RRID: AB_313683 |
| CD150-BV605 | Biolegend | TC15-12F12.2; RRID: AB_11204248 |
| Ki67- Alexafluo488 | Biolegend | 11F6; RRID: AB_2566800 |
| CD48-FITC | Biolegend | HM48-1; RRID: AB_313019 |
| Annexin V-FITC | BD Bioscience | 556419 |
| CD11b-biotin | Biolegend | M1/70; RRID: AB_312787 |
| CD4-biotin | Biolegend | RM4-5; RRID: AB_312711 |
| CD8-biotin | Biolegend | 53-6.7; RRID: AB_312743 |
| Gr1-biotin | Biolegend | RB6-8C5; RRID: AB_313369 |
| Ter119-biotin | Biolegend | TER119; RRID: AB_313705 |
| CD11b-APC-cy7 | Biolegend | M1/70; RRID: AB_830642 |
| CD4-FITC | Biolegend | RM4-5; RRID: AB_312713 |
| CD8-FITC | Biolegend | 53-6.7: RRID: AB_312745 |
| CD45.1-PE | eBioscience | A20; RRID: AB_465675 |
| CD45.2-Percp-Cy5.5 | BD Bioscience | 104 |
| BrdU-FITC | BD Bioscience | Bu20a |
| Chemicals, peptides, and recombinant proteins | ||
| Anti-APC magnetic microbeads | Miltenyi Biotec | 130-090-855 |
| 5-Fluorouracil | Sigma-Aldrich | F6627 |
| verapamil | Sigma-Aldrich | V105 |
| Hoechst 33342 | Sigma-Aldrich | B2261 |
| L-NAME | Sigma-Aldrich | 483125 |
| TCA | Sigma-Aldrich | T4009 |
| GSNO | Sigma-Aldrich | N4148 |
| Critical commercial assays | ||
| Caspase Glo3/7 | Promega | G8090 |
| BD Cytofix/Cytoperm Kit | BD Biosciences | 554714 |
| ROS detection Reagents | Invitrogen | C10446 |
| DAF-FM-DA-NO detection Kit | Beyotime | S0019 |
| ProteoStat Aggregation Detection Kit | Enzo | ENZ-51035-K100 |
| Annexin V Apoptosis Detection Kit I | eBioscience | 00-0055-56 |
| RNeasy Micro Kit | QIAGEN | 74004 |
| Plasmid Maxi Kit | Omega | D6926-03 |
| PrimeScript cDNA synthesis Kit | Takara | RR047A |
| Software | ||
| BD FACSDiva | BD | N/A |
| Flowjo v.10 | Treestar | N/A |
| GraphPad Prism v.7 | Graphpad Software | N/A |
Highlights.
S-nitrosylation and protein aggregation are increased during HSC regeneration
GSNOR deficiency augments protein S-nitrosylation, leading to abnormal HSC regeneration
Reduction of S-nitrosylation via NO inhibition enhances the function of Gsnor−/− HSC
TCA treatment or Chop deletion rescues regeneration defect of GSNOR-deficient HSC
ACKNOWLEDGMENTS
We thank Dr. Limin Liu (University of California, San Francisco) for kindly providing the Gsnor−/− mice and Jifeng Wang (Institute of Biophysics Core Facility) for assistance with the Q-Exactive mass spectrometer analysis. This work was supported by the National Key R&D Program (2016YFA0100602, 2017YFA0103302, 2017YFA0504000, and 2020YFA0112404); the National Natural Science Foundation of China (91749203, 82030039, 92049304, 31570857, 31701203, 81901403, 91849203, and 92049112); Innovative Team Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory (2018GZR110103002); Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2017ZT07S347); the Zhejiang Province College Students’ Science and Technology Innovation Activities (2018R413067); and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB39000000).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108922.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, and Dimmeler S (2003). Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat. Med 9, 1370–1376. [DOI] [PubMed] [Google Scholar]
- Benhar M, Forrester MT, and Stamler JS (2009). Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol 10, 721–732. [DOI] [PubMed] [Google Scholar]
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, and Dawson TM (2004). S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science 304, 1328–1331. [DOI] [PubMed] [Google Scholar]
- García-Prat L, Sousa-Victor P, and Muñoz-Cánoves P (2017). Proteostatic and metabolic control of stemness. Cell Stem Cell 20, 593–608. [DOI] [PubMed] [Google Scholar]
- Gomes SA, Rangel EB, Premer C, Dulce RA, Cao Y, Florea V, Balkan W, Rodrigues CO, Schally AV, and Hare JM (2013). S-nitrosoglutathione reductase (GSNOR) enhances vasculogenesis by mesenchymal stem cells. Proc. Natl. Acad. Sci. USA 110, 2834–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbunov NV, Pogue-Geile KL, Epperly MW, Bigbee WL, Draviam R, Day BW, Wald N, Watkins SC, and Greenberger JS (2000). Activation of the nitric oxide synthase 2 pathway in the response of bone marrow stromal cells to high doses of ionizing radiation. Radiat. Res 154, 73–86. [DOI] [PubMed] [Google Scholar]
- Gur-Cohen S, Itkin T, Chakrabarty S, Graf C, Kollet O, Ludin A, Golan K, Kalinkovich A, Ledergor G, Wong E, et al. (2015). PAR1 signaling regulates the retention and recruitment of EPCR-expressing bone marrow hematopoietic stem cells. Nat. Med 21, 1307–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess DT, and Stamler JS (2012). Regulation by S-nitrosylation of protein post-translational modification. J. Biol. Chem 287, 4411–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi-Sanabria R, Frankino PA, Paul JW 3rd, Tronnes SU, and Dillin A (2018). A futile battle? Protein quality control and the stress of aging. Dev. Cell 44, 139–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, et al. (2004). Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431, 997–1002. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang Y, Wang L, Wang P, Xue Y, Li X, Qiao X, Zhang X, Xu T, Liu G, et al. (2017). Autophagy impairment mediated by S-nitrosation of ATG4B leads to neurotoxicity in response to hyperglycemia. Autophagy 13, 1145–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, Messina E, Bowles D, Kontos CD, Hare JM, et al. (2009). Endogenous S-nitrosothiols protect against myocardial injury. Proc. Natl. Acad. Sci. USA 106, 6297–6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, and Stamler JS (2004). Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116, 617–628. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhao M, Jin X, Ney G, Yang KB, Peng F, Cao J, Iwawaki T, Del Valle J, Chen X, et al. (2019). Adaptive endoplasmic reticulum stress signalling via IRE1α-XBP1 preserves self-renewal of haematopoietic and pre-leukaemic stem cells. Nat. Cell Biol 21, 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Ruiz A, Villanueva L, Gonzálezde Orduña C, López-Ferrer D, Higueras MA, Tarín C, Rodríguez-Crespo I, Vázquez J, and Lamas S (2005). S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc. Natl. Acad. Sci. USA 102, 8525–8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M, Bourke E, Alexander D, Warr MR, Barry-Holson K, Le Beau MM, Morrison CG, and Passeguá E (2010). Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell 7, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, and Lipton SA (2013). Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 78, 596–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira-Pedro A, Dias CC, Regina H, Segreto C, Addios PC, Lungato L, D’Almeida V, Barros CC, Higa EM, Buri MV, et al. (2014). Nitric oxide-induced murine hematopoietic stem cell fate involves multiple signaling proteins, gene expression, and redox modulation. Stem Cells 32, 2949–2960. [DOI] [PubMed] [Google Scholar]
- North TE, Goessling W, Peeters M, Li P, Ceol C, Lord AM, Weber GJ, Harris J, Cutting CC, Huang P, et al. (2009). Hematopoietic stem cell development is dependent on blood flow. Cell 137, 736–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH, and Zon LI (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Warr MR, and Passeguá E (2011). Cell cycle regulation in hematopoietic stem cells. J. Cell Biol 195, 709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontel LB, Rosado IV, Burgos-Barragan G, Garaycoechea JI, Yu R, Arends MJ, Chandrasekaran G, Broecker V, Wei W, Liu L, et al. (2015). Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Mol. Cell 60, 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjabi CJ, Laskin JD, Hwang SM, MacEachern L, and Laskin DL (1994). Enhanced production of nitric oxide by bone marrow cells and increased sensitivity to macrophage colony-stimulating factor (CSF) and granulocyte-macrophage CSF after benzene treatment of mice. Blood 83, 3255–3263. [PubMed] [Google Scholar]
- Qu Z, Meng F, Bomgarden RD, Viner RI, Li J, Rogers JC, Cheng J, Greenlief CM, Cui J, Lubahn DB, et al. (2014). Proteomic quantification and site-mapping of S-nitrosylated proteins using isobaric iodo TMT reagents. J. Proteome Res 13, 3200–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retzlaff M, Stahl M, Eberl HC, Lagleder S, Beck J, Kessler H, and Buchner J (2009). Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Rep. 10, 1147–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizza S, and Filomeni G (2017). Chronicles of a reductase: biochemistry, genetics and physio-pathological role of GSNOR. Free Radic. Biol. Med 110, 19–30. [DOI] [PubMed] [Google Scholar]
- Rizza S, Cardaci S, Montagna C, Di Giacomo G, De Zio D, Bordi M, Maiani E, Campello S, Borreca A, Puca AA, et al. (2018). S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 115, E3388–E3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, Soldner F, Sunico CR, Nagar S, Talantova M, et al. (2013). Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 155, 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson V, Takei H, Soboleva S, Radulovic V, Galeev R, Siva K, Leeb-Lundberg LM, Iida T, Nittono H, and Miharada K (2016). Bile acids protect expanding hematopoietic stem cells from unfolded protein stress in fetal liver. Cell Stem Cell 18, 522–532. [DOI] [PubMed] [Google Scholar]
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, and Lipton SA (2006). S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441, 513–517. [DOI] [PubMed] [Google Scholar]
- van Galen P, Kreso A, Mbong N, Kent DG, Fitzmaurice T, Chambers JE, Xie S, Laurenti E, Hermans K, Eppert K, et al. (2014). The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 510, 268–272. [DOI] [PubMed] [Google Scholar]
- Wang P, Liu G-H, Wu K, Qu J, Huang B, Zhang X, Zhou X, Gerace L, and Chen C (2009). Repression of classical nuclear export by S-nitrosylation of CRM1. J. Cell Sci 122, 3772–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, Poulton IJ, van Rooijen N, Alexander KA, Raggatt LJ, and Lévesque JP (2010). Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 116, 4815–4828. [DOI] [PubMed] [Google Scholar]
- Xu D, Yang M, Capitano M, Guo B, Liu S, Wan J, Broxmeyer HE, and Huang X (2021). Pharmacological activation of nitric oxide signaling promotes human hematopoietic stem cell homing and engraftment. Leukemia 35, 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahata T, Takanashi T, Muguruma Y, Ibrahim AA, Matsuzawa H, Uno T, Sheng Y, Onizuka M, Ito M, Kato S, and Ando K (2011). Accumulation of oxidative DNA damage restricts the self-renewal capacity of human hematopoietic stem cells. Blood 118, 2941–2950. [DOI] [PubMed] [Google Scholar]
- Yang Z, Wang ZE, Doulias PT, Wei W, Ischiropoulos H, Locksley RM, and Liu L (2010). Lymphocyte development requires S-nitrosoglutathione reductase. J. Immunol 185, 6664–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Calay ES, Fan J, Arduini A, Kunz RC, Gygi SP, Yalcin A, Fu S, and Hotamisligil GS (2015). METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 349, 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao D, Gu Z, Nakamura T, Shi ZQ, Ma Y, Gaston B, Palmer LA, Rockenstein EM, Zhang Z, Masliah E, et al. (2004). Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 101, 10810–10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wu K, Su W, Zhang DF, Wang P, Qiao X, Yao Q, Yuan Z, Yao YG, Liu G, et al. (2017). Increased GSNOR expression during aging impairs cognitive function and decreases S-nitrosation of CaMKIIα. J. Neurosci 37, 9741–9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets or codes.