

Abstract
Spinal cord injury (SCI) can have profound effects on the autonomic and cardiovascular systems, notably with injuries above high-thoracic levels that result in the development of autonomic dysreflexia (AD) characterized by volatile hypertension in response to exaggerated sympathetic reflexes triggered by afferent stimulation below the injury level. Pathophysiological changes associated with the development of AD include sprouting of both nociceptive afferents and ascending propriospinal ‘relay’ neurons below the injury, as well as dynamic changes in synaptic inputs onto sympathetic preganglionic neurons. However, it remains uncertain whether synapse formation between sprouted c-fibers and propriospinal neurons contributes to the development of exaggerated sympathetic reflexes produced during AD. We previously reported that once daily treatment with the anti-epileptic and neuropathic pain medication, gabapentin (GBP), at low dosage (50 mg/kg) mitigates experimentally induced AD soon after injections, likely by impeding glutamatergic signaling. Since much higher doses of GBP are reported to block the formation of excitatory synapses, we hypothesized that continuous, high dosage GBP treatment after SCI might prevent the formation of aforementioned aberrant synapses and, accordingly, reduce the incidence and severity of AD. Adult female rats implanted with aortic telemetry probes for hemodynamic monitoring underwent T4-transection SCI and immediately received 100 mg/kg (i.p.) of GBP and then every six hours (400 mg/kg/day) for 4-weeks after injury. We assessed daily body weight, mean arterial pressure, heart rate, frequency of spontaneous AD, and hemodynamic changes during colorectal distension (CRD) to establish whether high dose GBP treatment prophylactically mitigates both AD and associated aberrant synaptic plasticity. This regimen significantly reduced both the absolute blood pressure reached during experimentally induced AD and the time required to return to baseline afterwards. Conversely, GBP prevented return to pre-injury body weights and paradoxically increased the frequency of spontaneously occurring AD. While there were significant decreases in the densities of excitatory and inhibitory pre-synaptic markers in the lumbosacral dorsal horn following injury alone, they were unaltered by continuous GBP treatment. This indicates distinct mechanisms of action for acute GBP to mitigate induced AD whereas chronic GBP increases non-induced AD frequencies. While high dose prophylactic GBP is not recommended to treat AD, acute low dose GBP may hold therapeutic value to mitigate evoked AD, notably during iatrogenic procedures under controlled clinical conditions.
Keywords: Neurontin, synapse, autonomic, plasticity, sympathetic
Introduction
Along with sensory and motor impairments, traumatic spinal cord injury (SCI) results in a constellation of cardiovascular and autonomic dysfunctions (Hou and Rabchevsky, 2014; Krassioukov et al., 2003; Weaver et al., 2012), notably with injuries above the sixth thoracic (T6) segment that frequently result in the development of a condition termed autonomic dysreflexia (AD). This syndrome is characterized by episodes of volatile and potentially lethal hypertension in response to exaggerated sympathetic reflexes triggered by unperceived afferent stimuli below the injury level. Affected individuals may experience cardiac arrhythmia, pounding headache, anxiety, flushing of the skin and profuse sweating above the lesion during an episode of AD. This episodic disorder can occur frequently throughout the day due to regular filling of the bowel and bladder creating noxious stimuli, or by irregular and less predictable triggers such as pressure sores or ingrown toe nails (Karlsson, 1999). Because of the unpleasant and potentially dangerous manifestations, prevention or effective treatment of AD is one of the highest priorities in the SCI community for enhancing quality of life (Anderson, 2004).
Mechanisms known to contribute to the development of AD include the loss of descending vasomotor modulatory pathways, hyperreactivity of peripheral vasculature to adrenergic stimulation, and a number of maladaptive changes within spinal circuitry influencing sympathetic outflow below the lesion (Krassioukov et al., 1999; Schramm, 2006). Intraspinal changes include sprouting of both unmyelinated afferent c-fibers and ascending propriospinal ‘relay’ projections towards sympathetically-correlated interneurons in the thoracic spinal cord, as well as dynamic alterations of synaptic inputs to decentralized sympathetic preganglionic neurons (SPN) in the thoracolumbar spinal cord. Synapses derived from descending supraspinal projections onto SPN are eliminated within one week after complete spinal transection at the fourth thoracic (T4) segment, but by two weeks after injury there is restoration of synaptic terminals onto SPN derived from spinal interneurons and/or primary afferents below the injury (Llewellyn-Smith and Weaver, 2001). Because the development of AD occurs over weeks after SCI in rodents, it has been suggested that injury induced synaptogenesis onto decentralized SPN contributes to AD pathophysiology (Weaver et al., 1997). Based on seminal electrophysiological studies (Krassioukov et al., 2002), a model of AD development has emerged in which the loss of supraspinal control, coupled with convergence from both primary afferents and propriospinal neurons onto sympathetically correlated interneurons, enhances the transmission of noxious stimuli below the injury to SPN, triggering unrestricted sympathetic reflexes during AD (Rabchevsky, 2006); however, this has yet to be thoroughly examined.
We have reported that weeks following complete T4 spinal transection in rats, the administration of the anticonvulsant neuropathic pain medication gabapentin (GBP) significantly reduces the magnitude of colorectal distension (CRD) induced AD and tail spasticity shortly after treatment, lasting several hours (Rabchevsky et al., 2011; Rabchevsky et al., 2012). The mechanism of this clinically valuable effect, however, remains uncertain. GBP was first developed for the treatment of epilepsy and gained subsequent traction as a treatment for neuropathic pain (reviewed in Sirven, 2010). Despite being a structural analog of y-aminobutyric-acid (GABA) with enhanced blood-brain barrier penetration (Crawford et al., 1987), most studies suggest that its actions are independent of GABAergic modulation. GBP’s high-affinity binding site is the L-type calcium channel α2δ subunits on presynaptic terminals (Gee et al., 1996), blocking it decreases intraspinal glutamatergic neurotransmission (Coderre et al., 2007), and large daily doses of GBP are reported to prevent the formation of excitatory glutamatergic synapses in the developing CNS by binding to the α2δ1 calcium channel subunit (Eroglu et al., 2009). However, we have found that injured rats treated with GBP an hour prior to CRD weeks later showed significantly reduced AD, whether or not they were treated once daily with saline versus low-dose (50 mg/kg, i.p.) GBP after SCI (Rabchevsky et al., 2012). Because this appears to favor the notion that acute GBP treatment transiently blocks intraspinal glutamatergic neurotransmission which underlies AD (Maiorov et al., 1997), but not synaptogenesis in the injured spinal cord, per se, we sought to determine whether chronic high-dose GBP treatment beginning immediately after SCI alters the development of AD by modulating synaptogenesis of aforementioned intraspinal pathways.
Methods and Materials
Telemetry probe implantation:
All animal housing conditions, surgical procedures, and postoperative care were conducted according to the University of Kentucky Institutional Animal Care and Use Committee and the National Institutes of Health animal care guidelines. Animals were housed in a temperature and humidity-controlled room with a 12/12-hour light/dark cycle. Efforts were made to minimize unnecessary foot traffic and other potential environmental disturbances in the room. One week prior to SCI, a total of 24 naïve 3 to 3.5 month old female Wistar rats (250-275 grams) were anesthetized (2% isoflurane) and implanted with telemetric blood pressure transmitters (model HD-S10, Data Sciences International, Inc., St. Paul, MN) into the descending abdominal aorta according to the manufacturer’s surgical guidelines as previously reported (Eldahan et al., 2018; Rabchevsky et al., 2012). Telemetry probes were secured intra-abdominally by suturing to the ventral abdominal wall using non-absorbable silk sutures before closing the skin with 3-0 vicryl sutures. Animals were treated post-operatively as described below.
Spinal cord injury:
All rats recovered from telemetry probe implantation for one week. A total of n=24 rats were anesthetized (ketamine, 80 mg/kg; xylazine 7 mg/kg, i.p.) and underwent a T3 laminectomy prior to complete transection of the T4 spinal cord (T4Tx) using a scalpel blade, as previously described (Cameron et al., 2006; Rabchevsky et al., 2012). Two independent observers confirmed complete spinal cord transection of each rat based on full separation of the rostral and caudal stumps. Immediately afterwards, gelfoam was placed into the transection site to achieve hemostasis before the overlying muscles were sutured with 3-0 vicryl and the skin stapled with wound clips (Stoelting, Wood Dale, IL).
Cardiophysiological monitoring:
Hemodynamic data was recorded 24/7 (500Hz sampling rate) using implanted telemetric probes as described above using Dataquest A.R.T. acquisition system (Data Sciences International, Inc., St. Paul, MN). Rats were single-housed, with each cage placed directly on top of its corresponding data receiver plate (model RPC-1, Data Sciences International, Inc., St. Paul, MN). Raw telemetry files were stored in a single folder containing data for the entire study period to analyze daily blood pressure, heart rate, colorectal distension, and spontaneous AD (sAD).
Daily blood pressure and heart rate values were calculated with Dataquest A.R.T. analysis system (Data Sciences International, Inc., St. Paul, MN) by averaging the corresponding 24-hour period. CRD data was calculated using the 30-second period immediately prior to balloon catheter inflation as a baseline for comparison with the 60-second average values during the duration of balloon inflation. The effect of experimental handling on resting blood pressure was evaluated by comparing the averaged mean arterial pressure during the 30-minute period prior to entering the animal housing room with the 30-minute period following handling and catheter insertion when animals were returned to their home cage for catheter acclimation. Un-induced sAD was analyzed with a modified version of our previously reported algorithm (Eldahan et al., 2018; Rabchevsky et al., 2012) implemented in Matlab software (The MathWorks, Inc., Natwick, MA). Briefly, this algorithm simultaneously processes 24-hour mean arterial pressure (MAP) and heart rate (HR) traces for instances where a rise in MAP of 20 mmHg or more above baseline is accompanied by a decrease in HR of at least 20 beats per minute (bpm). While this algorithm does not require such events to be sustained above baseline for a specific duration, it does require the rise in MAP over baseline to occur within a 35-second window. MAP rises accompanied by tachycardia (an increase in HR) were not included in these sAD detections. Notably, our modified algorithm screens for instances where multiple sAD detections occur within a 120-second window such that events beginning within two minutes of each other are counted as a single unified event. This modification is similar to recent sAD algorithms described by (Mironets et al., 2018; West et al., 2015). Following automated computer detected sAD events, a single human observer manually screened all detections to remove false-positive events stemming from technical artifacts, such as non-physiological rises in MAP associated with sudden animal movements. For each of the four daily injections (12:00 a.m., 6:00 a.m., 12:00 p.m. and 6:00 p.m.), recordings were stopped simultaneously for all animals and an “event marker” was placed for each animal immediately after injection to allow for accurate tracking of all injection time points. Routine animal care, including twice-daily bladder expression, was performed at 6 am and 6 pm along with corresponding treatment injections. Cardiophysiological recordings were paused during routine animal care and handling to minimize data artifacts.
Drug administration and colorectal distension (CRD):
Gabapentin (GBP; 100 mg/kg, i.p, 4x daily, Spectrum Chemicals, New Brunswick, NJ) was injected after injury and then every 6 hours for 4 weeks. Daily body weights were recorded immediately prior to drug administration to ensure accurate dosages. On days 14, 21 and 28 post-injury, rats underwent 2 trials of CRD one hour after treatment. Rats were gently restrained using surgical towels leaving the lower body exposed for observation of hindlimb spasticity during CRD. After removing fecal pellets in the distal rectum, silicone balloon catheters (Coloplast Cysto-Care Folysil, 2-Way Pediatric Silicone Foley Catheter, Fr10 3 cc) were inserted 2.5cm into the rectum and secured to the tail with Transpore™ tape. Bladder expression was performed immediately prior to catheter insertion. Following restraint and catheter insertion, rats acclimated for 20-30 minutes. All efforts were made to minimize distractions and potentially stressful stimuli. A hood fan was left running to provide white noise and buffer noise occurring outside of the experimental room. All CRD testing was performed in the same room that the animals were housed in regularly. For each CRD trial, the catheter balloon was inflated with 2.2 mL of air over the course of 5 seconds. Each distention trial lasted 60 seconds, separated by 30 minutes of rest. At 28 DPI, following the second CRD trial, a prolonged 90-minute session of intermittent CRD (30 second inflation periods separated by 60 seconds of rest) was performed to induce intraspinal c-Fos expression prior to euthanasia (Eldahan et al., 2018; Hou et al., 2008).
Animal Care:
All rats which underwent ketamine/xylazine anesthesia prior to cord transection received i.p. injections of the xylazine reversal agent antisedan (0.5 mg/kg Zoetis Inc., Kalamazoo, MI). Immediately after all surgical procedures, rats received 10 mL of subcutaneous lactated Ringer’s solution. Post-surgical pain management was achieved with twice-daily buprenorphine injections (0.03mg/kg, s.c.; Reckitt-Benckiser Pharmaceuticals Inc., Richmond, VA) for three days. All rats received two daily injections of the antibiotic cefazolin (33.3 mg/kg, s.c.; WG Critical Care, LLC, Paramus, NJ) for 5 days post-surgery and twice daily manual bladder expression for 2-3 weeks or until they regained spontaneous bladder voiding reflexes without signs of urinary tract infection. Rats were housed one per cage with food (Envigo Tekland, 18% Protein Rodent Diet) and water ad libitum. A heating pad and surgical towel was placed between each cage and its corresponding telemetric receiver plate for the duration of the study period. Following telemetry implantation, body weight was recorded every morning during routine animal care procedures.
Tissue collection and immunohistochemistry:
Terminal body weights were recorded, and rats received a fatal overdose of sodium pentobarbital within 15 minutes following the end of the 90-minute CRD session. After the loss of pinch-reflex in the forelimbs, the spleen was rapidly dissected after applying Bulldog clamps to the vascular bundle serving the spleen. Rats were then transcardially perfused with 0.1M PBS, the telemetry probes were rapidly explanted according to manufacturer’s guidelines, which was followed by perfusion with 4% paraformaldehyde (PFA) in 0.1M PBS. Excess fat and connective tissue were removed from the spleens and raw spleen weights were recorded prior to soaking them in 4% PFA overnight before flash freezing. The spinal cord caudal to the T4-transection site was carefully dissected and post-fixed for 3-4 hours in 4% PFA before being placed in 0.2M PB solution overnight at 4°C. Cords were then cryopreserved in 20% sucrose (in 0.1M PBS) with 0.02% sodium azide at 4°C until all cords sank, after which they were embedded in gum tragacanth mixed with 20% sucrose and stored at −80°C until cryosectioning (Eldahan et al., 2018). Every fifth serial coronal cryosection (20uM) was collected and placed on a series of Superfrost Plus™ slides (Thermo Fisher Scientific, Waltham, MA). This cutting scheme results in 10 adjacent sets of slides with each slide containing 10 consecutive spinal sections separated by 1mm. Slides were stored at −20°C until further use.
For immunohistochemistry, slides were removed from the freezer and warmed on a 37°C slide warmer for 10 minutes then 3 x 30-min before a Pap-pen (Ted Pella, Redding, CA) border was applied to all tissue slides before washing in 0.1M PBS with gentle agitation. Slides were then blocked for 1-hour at room temperature in 5% normal serum and 0.3% Triton X-100 in 0.1M PBS. Primary antibodies against presynaptic excitatory and inhibitory neurotransmitters were incubated for either 24-hours (synaptophysin and VGLUT2) or 72-hours (VGAT) then 24-hours (synaptophysin) in buffer containing 1 ug/ml mouse anti-synaptophysin (SYN) (Clone SY3; Synaptic Systems Cat #101-011) and 2 ug/ml rabbit anti-VGLUT2 (Synaptic Systems Cat #135-403) or 2 ug/ml rabbit anti-VGAT (Synaptic Systems Cat #131-002). Excess primary antibodies were washed 3x10 minutes in 0.1 M PBS before incubation with secondary antibody (goat anti-rabbit-Alexa488 (1:500) or biotinylated goat anti-mouse (1:200) for 24-hours at 4°C. Following removal of excess secondary antibody with 3x10 minute 0.1 M PBS washes, slides were incubated in tertiary solution containing 1:200 streptavidin conjugated Texas Red in 0.1 M PBS. Two series of tissue staining were performed, one for SYN and VGLUT2 and the second for SYN and VGAT, and all treatment groups were stained simultaneously using identical buffers and antibody lots; different SYN antibody lots were used in each staining set due to availability.
Image acquisition and analysis
High-resolution (2048 x 2048 pixels) images were captured with a 40x oil-immersion lens using a Nikon Ti Eclipse inverted confocal microscope paired with a C2+ controller system and Nikon NIS Elements Advanced Research software (Nikon Inc., Melville, NY, USA). Laser settings for each fluorescent marker were optimized on naïve spinal cord tissues and maintained constant for all sections assessed. The pinhole size was maintained at 40 μM and pixel dwell time set to 4.4 seconds/pixel. A histogram was used to monitor pixel intensity and ensure that the total fraction of saturated pixels remained below 2% for all images.
From the central plane of the tissue section, two additional optical sections 2 μM above and below (4 μM total thickness) were captured. Each optical section represented a thickness of 0.86 μM and 0.16 μM individual pixel size. After image acquisition, the optical plane with the highest mean fluorescence intensity was selected for analysis. Selection of the optical plane with the brightest staining helps to correct for potential issues with antibody penetration (Ferguson et al., 2008). Synaptic quantification was performed using densitometric analysis. For each subject, 3 spinal sections centered on the L6/S1 spinal segment and separated by 1 mm were analyzed (see Figure 1). Cross-sections of the right dorsal horn were used for all animals with the rare exception of folded tissue sections, in which case the left dorsal horn was used. A local contrast was applied using a radius of 1.0 μM to more clearly delineate the border of individual SYN+, VGLUT2+ and VGAT+ puncta. Thereafter, each fluorescent channel was thresholded to exclude non-specific background signal. All contrast and threshold settings were optimized on naïve spinal cord tissues and remained constant for all sections (see Figure 2). After applying a threshold for each fluorescent channel, a binary layer mask was created for each marker (SYN and VGLUT2 or VGAT) indicating the location of pixels above threshold. These two layers were then overlaid to create a third layer representing the area of overlap between markers. Using these three binary layers, either the area occupied by SYN, VGLUT2 or VGAT or the area occupied by the overlap of VGLUT2/SYN or VGAT/SYN puncta were measured using a standardized region of interest (ROI) measuring 175 x 175 μM. One ROI, designated “R1”, was placed the upper dorsal horn (laminae I-II) and the other, designated “R2”, was placed in the dorsal gray commissure (laminae X just above the central canal (see Figure 1). To control for potential variation in antibody labeling efficiency between sets of synaptic staining, data was normalized to naïve control values within a respective set of staining. These methods were adapted from McKillop et al. (2016) who used a similar threshold-based method to identify and quantify areas occupied by presynaptic (SYN) terminals in the mouse spinal ventral horns after SCI as either glutamatergic (VGLUT1) or GABAergic (VGAT).
Figure 1. Diagrammatic representation of synaptic quantification scheme.

(A,B) Tissue from the L6/S1 spinal segment was sampled for histological analysis of synaptic density. High-resolution confocal images were acquired from laminae I-II of the upper right dorsal horn (B, R1) and the laminae X region containing the dorsal gray commissure (B, R2). (C) Volumetric reconstruction shows co-localization of VGLUT2 and Synaptophysin in 3D, indicating the co-occurrence of these presynaptic markers. Scale bars in C = 4 uM or 1 uM (inset).
Figure 2. Representative threshold and overlap identification of excitatory and inhibitory presynaptic terminals.

Adjacent tissue sections were double-labeled with VGLUT2/Synaptophysin (A-F) or VGAT/Synaptophysin (G-L). Contrasted images (top rows A-C; G-I) were thresholded (bottom rows D-F; J-L) to segregate positive staining from background, and a mask was applied representing supra-threshold pixels in VGLUT2 (D), VGAT (J) and Synaptophysin (E,K) channels. Co-localization of VGLUT2/Synaptophysin or VGAT/Synaptophysin was determined based on overlapping of signal (F,L). Images represent cropped and enlarged areas taken from the R1 field as shown in Figure 1B. Scale bar in F = 5 uM and applies to all photos.
To validate the densitometric analyses, quantitative co-localization analyses were also performed using the JACoP plugin (https://imagej.nih.gov/ij/plugins/track/jacop.html) implemented in the ImageJ/FIJI platform. This plugin, which has previously been described in depth (Bolte and Cordelieres, 2006), contains a collection of commonly used tools to measure intensity correlation and co-occurrence of dual-color molecular probes. The same ROIs used for density analysis were cropped for JACoP analysis. To reduce confounding influences of non-specific background pixels, a threshold was applied to restrict analysis to pixels containing positive staining. The Costes’ automated thresholding algorithm was determined to yield inappropriately low thresholds, likely due to the high density of labeling and variability of pixel intensity reported to be problematic for appropriate automated thresholding and image segmentation (Dunn et al., 2011). Therefore, threshold values were determined based on the background pixel intensity in a set of negative primary antibody control slides processed and imaged using identical settings. These threshold values were visually validated on a set of naive group images and subsequently applied equally to all images/treatment groups. The Manders’ overlap coefficient (MOC) and fractional overlap coefficients (tM1 and tM2) were calculated using supra-threshold pixels with positive staining to avoid inflated results due to low-level background signal (Bolte and Cordelieres, 2006). The Costes’ randomization test was applied to a subset of images representing each experimental group (i.e. 3-4 images selected from 3 animals per group), which resulted in P values of 100%, along with R-values of 0.0 +/− 0.001, indicating that the positive co-localization signal measured in our images was not attributed to statistical chance or haphazard signal overlap.
Statistical Analyses
For daily body-weight, MAP, HR, spontaneous AD and spasticity measurements, a repeated measures two-way analysis of variance (2-way RM ANOVA) was performed with Sidak’s multiple comparisons post-hoc test when appropriate. The Geisser-Greenhouse correction was used for repeated measures analyses to provide additional rigor and minimize type-I errors. Unpaired t-tests were performed on CRD and spleen weight data. One-way ANOVA tests were used for synaptic histology, with Sidak’s post-hoc analysis when appropriate. All statistical analyses were conducted with GraphPad Prism 7 software using alpha = 0.05.
Results
We had a 13% attrition rate with deaths occurring during recovery in the first week after injury and with similar frequency in the saline and GBP groups (n=1-2 per group).
Effects on body weight recovery after SCI
We compared the effects of time and continuous high-dose GBP treatment on the change in body weight after complete SCI and observed a significant effect of time (p<0.0001) but no significant treatment effect (p=0.4197) (Figure 3). There was, however, a significant interaction between time and treatment (p<0.0001). Within one-week after SCI, both groups lost a significant amount of body weight. Compared to pre-injury (−1 DPI) values, saline and GBP treated rats had significantly lower body weights by 5 DPI and 7 DPI, respectively (p<0.05). Whereas vehicle treated rats returned to pre-injury weight by 20 DPI, continuous GBP treatment prevented return to baseline body weights by the end of the study period.
Figure 3. Daily change in body weight after SCI.
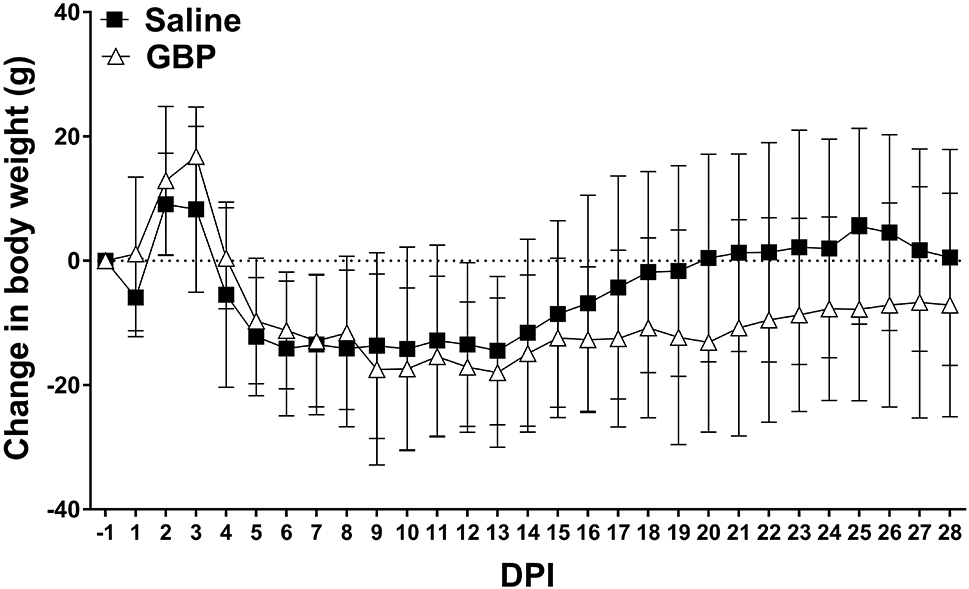
All rats experienced an initial gain in weight in the first 2-3 days post-injury (DPI) before losing body weight for the first 2 weeks. Saline treated rats started to regain body weight by 14 days post injury and returned to pre-injury weights (dotted line) by 20 DPI. In contrast, injured GBP treated rats maintained reduced body weight below pre-injury levels. N=11 Saline, N=10 GBP. Symbols are mean ± SD.
Effects on daily hemodynamic measures after SCI
We examined whether continuous high-dose GBP had temporal effects on daily hemodynamics post-injury. There was an overall significant effect of time post-injury (p<0.0001) on daily MAP (Figure 4A), with no treatment effect (p=0.65). Compared to pre-injury values, the daily MAP was significantly lower for the first two days post-injury (DPI) in both groups (p<0.05) before stabilizing to slightly below naïve levels after the first two weeks post-injury. There was a significant effect of time post-injury on daily HR (p<0.0001), with no treatment effect (p=0.65). While the mean daily HR of the GBP treated group was maintained approximately 20 bpm higher than the saline group at all DPI, this observation was not statistically significant (p>0.05).
Figure 4. Effects of SCI and GBP treatment on daily hemodynamics.
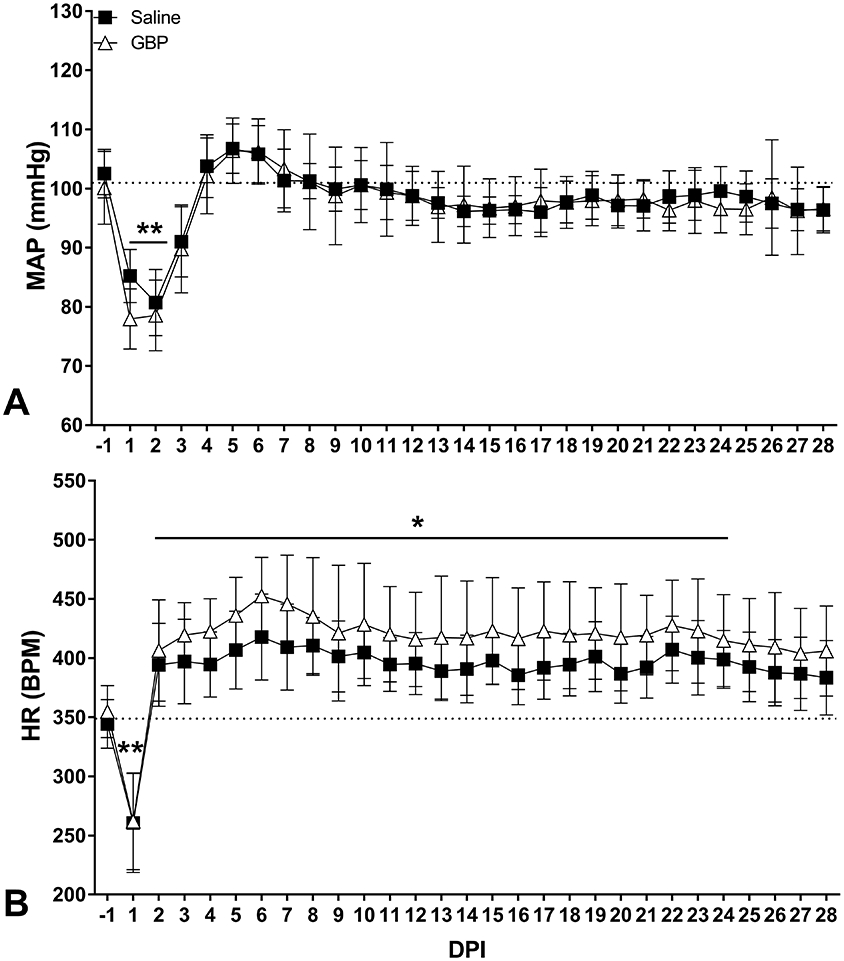
Daily (24-hour) mean arterial pressure (MAP, A) and heart rate (HR, B) measured across days post-injury (DPI). Daily MAP dropped significantly in the first two days post-injury before returning to normal values after one week. The daily HR sharply decreased the first day after injury before rising significantly above pre-injury levels beginning at 2 DPI. Horizontal dotted lines indicate pre-injury control values. N=11 Saline, N=10 GBP. Symbols are mean ± SD. *p<0.05, **p<0.01 vs pre-injury values.
Effects on incidences of spontaneous autonomic dysreflexia (sAD)
Daily MAP and HR traces were analyzed with a computer algorithm to identify sAD events 24/7 (Figure 5A). Threshold settings employed to detect relatively moderate events (MAP increase ≥ 20 mmHg with HR drop ≥ 20 BPM) (Figure 5B). We observed an overall significant effect of time (p<0.0001) and treatment (p<0.005; Figure 5C) with GBP treated rats having significantly more sAD events compared to saline treated injured rats.
Figure 5. Effects of GBP on the frequency of non-induced AD after injury.

(A) Representative 24-hour mean arterial pressure (MAP) traces show more overall sAD events with GBP treatment compared to saline controls; vertical bars indicate detected events. (B) A typical event with a MAP rise >20 mmHg and heart rate (HR) drop >20 beats per minute (bpm). (C) The frequency of daily sAD detections across days post-injury (DPI) was significantly higher with GBP compared to saline treatment. N=10 GBP, N=11 Saline. Symbols represent mean ± SD.
Effects on colorectal distension induced AD
When we examined the severity of AD as assessed by the magnitude of hypertension during experimentally-induced AD, there was an overall significant treatment effect on the MAP reached during noxious colorectal distension (CRD) stimulation (p<0.0001; Figure 6A). GBP treated rats had significantly lower MAP during CRD stimulation at 21 (p<0.01) and 28 DPI (p<0.001) compared to saline treated; in the latter group the absolute MAP reached during CRD increased over time with a significantly higher value at 28 DPI compared to 14 DPI (p<0.01). In contrast, the CRD-induced MAP did not increase over time in the GBP treated group (p>0.05). In parallel, the baseline MAP measured immediately prior to CRD revealed an overall treatment effect (p<0.0005; Figure 6B), with GBP treated rats having significantly lower baseline MAP at 21 (p<0.01) and 28 DPI (p<0.01). Whereas the CRD baseline MAP in saline treated rats did not change over time (p>0.05), it was significantly lower at 28 vs 14 DPI (p<0.05) in the GBP group. There was a significant effect of time (p<0.0001) but not treatment (p>0.05) on the CRD-induced changes in MAP relative to baselines (Figure 6C). The magnitude of change increased over time in both groups, with significantly larger changes in MAP at 28 DPI compared to 14 DPI in saline (p<0.001) and GBP (p<0.001) treated rats. Unlike the significant blood pressure changes, there was no effect of time (p>0.05) or treatment (p>0.05) on the highly variable changes in HR during CRD (Figure 6D).
Figure 6. Effects of GBP on hypertension induced by noxious colorectal distension.
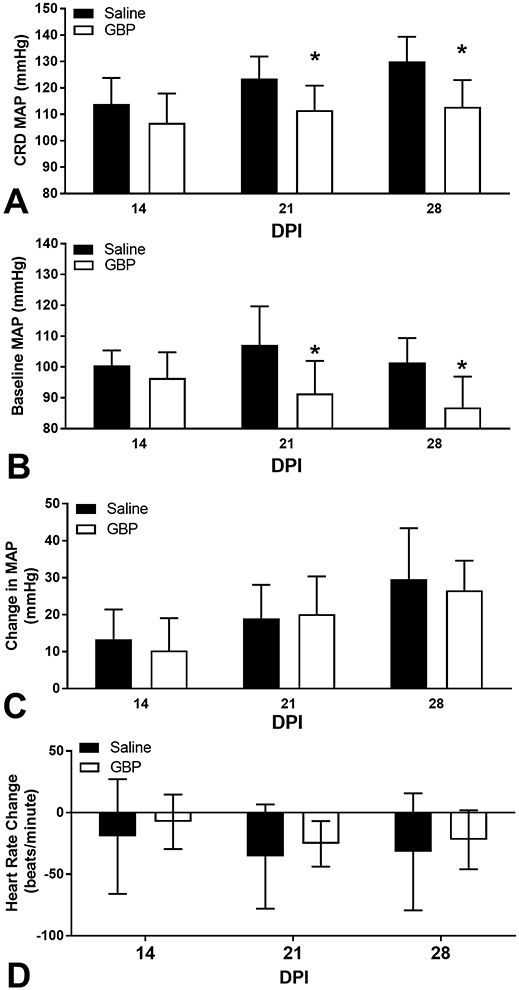
Autonomic dysreflexia was experimentally induced on 14, 21- and 28-days post-injury (DPI) through noxious colorectal distension (CRD). GBP significantly reduced both the (A) absolute mean arterial pressure (MAP) reached during CRD across DPI. (B) The baseline MAP calculated from the 30 seconds immediately prior to each CRD trial was significantly lower in GBP treated rats. (C) The CRD-induced change in MAP relative to baseline was not different between groups, however this parameter increased significantly over time. (D) CRD-induced changes in heart rate were not significantly altered by GBP over time. N=11 Saline, N=10 GBP. Bars are mean ± SD. *p<0.05 vs saline.
Effects on resting blood pressure during experimental handling and restraint
We compared the resting MAP before the start of weekly CRD testing sessions to the MAP after placing rats under gentle restraint for the CRD procedures. There was an overall effect of treatment (p<0.05), but not time (p=0.94) on pressor responses to experimental handling (Figure 7). Notably, during handling procedures the GBP treated rats had significantly lower MAP elevations at 21 (p<0.005) and 28 (p<0.05) DPI.
Figure 7. Effect of experimental handling on blood pressure prior to colorectal distension.
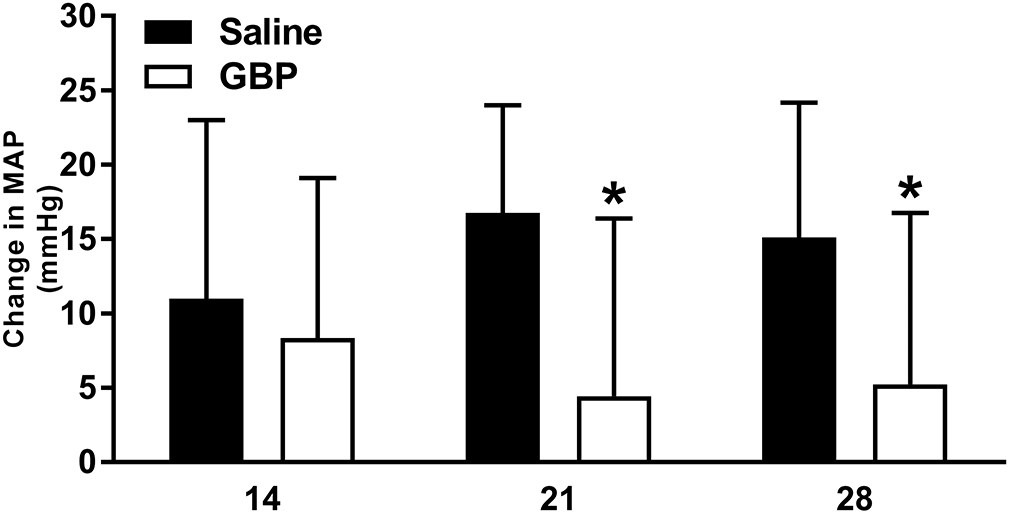
The MAP measured in the mornings before the start of weekly CRD testing sessions compared to that following experimental handling during CRD test preparation showed that it induced an increase in basal MAP of 10-20 mmHg in vehicle-treated injured rats whereas GBP treatment reduced the hypertension significantly at 21 and 28 DPI. N=11 Saline, N=10 GBP. *p<0.05 vs saline. Bars are mean ± SD.
Effects of SCI and GBP on spleen weight
Based on reports of immunosuppression and splenic atrophy linked with AD in mice (Zhang et al., 2013), we measured spleen size as a surrogate marker of immunosuppression in rats but did not observe an effect of injury on terminal spleen mass (Figure 8; p>0.05). However, GBP treated SCI rats had significantly (p<0.05) heavier spleens compared to both saline treated SCI rats and uninjured naïve controls.
Figure 8. Effects of injury and GBP on terminal spleen weights.
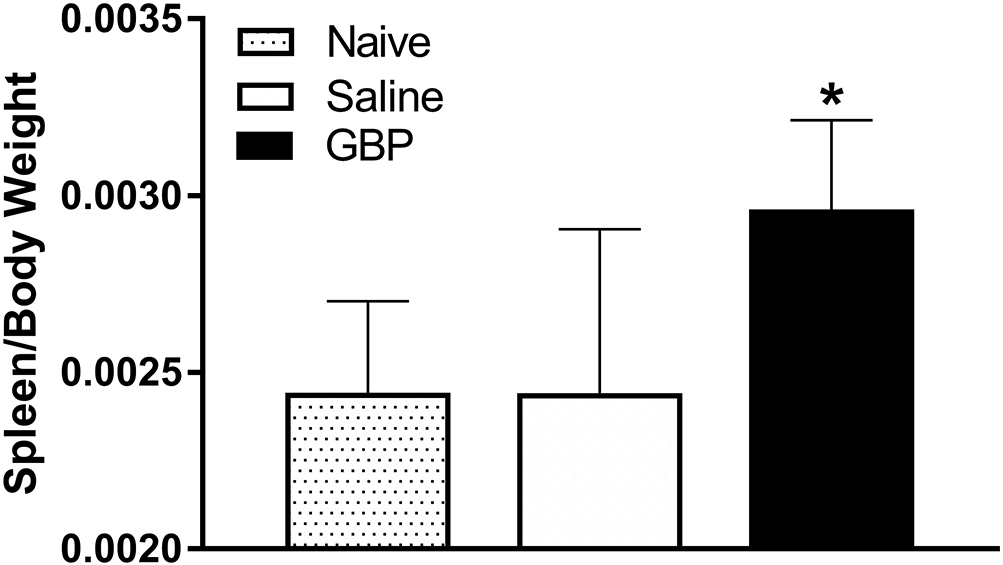
Spleen wet weights were recorded and normalized to terminal body weights at 4 weeks post-injury. While there was no injury effect, spleens from the GBP treated injured rats were significantly heavier in comparison to both saline treated injured and age/body-weight matched naïve controls. N=8 Naïve, N=11 Saline, N=10 GBP. *p<0.05 vs naive control and saline treated SCI rats. Bars are mean ± SD.
Effects of SCI and GBP treatment on neuronal activity and pre-synaptic densities
While c-Fos immunoreactivity was seen throughout the lumbosacral spinal cord following terminal CRD procedures in all injured spinal cords, there were no apparent effects of GBP on c-Fos expression (unpublished observations; (Eldahan et al., 2018)). We measured the immunoreactive densities of presynaptic synaptophysin (SYN)+, VGLUT2+ (glutamatergic) and VGAT+ (GABAergic) puncta in the dorsal horn (lamina I-II; R1) and dorsal gray commissure (lamina X; R2) of the L6/S1 spinal cord (see Figures 1 and 9). These regions contain the putative central terminals of c-fibers conveying colorectal afferent stimuli into the dorsal horn (R1) and propriospinal neurons (R2) believed to relay afferent stimuli towards the thoracolumbar SPN. Our analysis of R1 was based on location of putative spinal sympathetic interneurons in these upper laminae (Chau et al., 2000; Tang et al., 2003). Representative images taken from R1 demonstrate conspicuous SCI-induced decreases in immunoreactivities for SYN, VGLUT2 and VGAT compared to naïve cords (Figure 9A-H), but GBP did not alter the reduced synaptic densities (not shown). The normalized percent area covered by SYN+, VGLUT2+ and VGAT+ in both R1 and R2 was significantly (p<0.01) reduced by SCI (Figure 9I,L). In the VGAT staining set, SYN+ coverage was significantly lower in both injury groups compared to naïve controls (p<0.001; Figure 9L); however, this reduction was not significant in the VGLUT2 set (p=0.09; Figure 9I). GBP treatment did not alter (p>0.05) the reduced SYN+, VGLUT2+ or VGAT+ (Figure 9J,M) densities observed in either staining set. Similarly, the area occupied by the overlay puncta of SYN and VGLUT2 or VGAT immunoreactivities was significantly reduced in both regions of interest after injury (p<0.05; Figure 9K,N), indicating an overall decrease in excitatory and inhibitory synapses that was not altered by GBP treatment (p>0.5).
Figure 9. Density of pre-synaptic puncta following injury.

Representative photomicrographs (A-H) in the lumbosacral dorsal horn (inset, top left) demonstrate punctate Synaptophysin (A,B and E,F), VGLUT2 (C,D) and VGAT (G,H) immunoreactivities in naïve dorsal gray matter (R1) that were reduced 4 weeks after injury (SCI + Vehicle). Quantitative analyses showed that injury significantly decreased the densities of Synaptophysin (I,L) VGLUT2 (J), VGAT (M), and the overlap of VGLUT/Synaptophysin (K) and VGAT/Synaptophysin (N) in both R1 and R2 of dorsal gray matter. Immunoreactive densities were unaltered by continuous gabapentin (GBP) treatment. N=8 naïve, N=11 Saline, N=10 GBP. *p<0.05 vs naive. Scale bars = 10 uM Bars are mean ± SD.
Effects of SCI and GBP treatment on pre-synaptic co-localization
To corroborate density measures, the Manders’ coefficients, tM1 and tM2, and Manders’ overlap coefficient (MOC) were calculated for presynaptic immunoreactivities. The fraction of total VGLUT2 signal overlapping with SYN (tM1) was not significantly altered by SCI or treatment in either region of interest (p>0.05), whereas the fraction of total VGAT signal overlapping with SYN was significantly decreased by SCI in the R2 region (p<0.05; Figure 10A). No effect of GBP treatment was observed on tM1 in the VGLUT2 or VGAT staining sets (p>0.1). The converse relationship, defined as the fraction of total SYN signal overlapping with VGLUT2 or VGAT (tM2; Figure 10B) showed that tM2 was significantly decreased after injury in both regions of interest in both the VGLUT2/SYN and VGAT/SYN staining sets (p<0.01). Like tM1, no significant effect of GBP treatment was observed on the tM2 overlap coefficient (p>0.05). The MOC for each pair of markers (VGLUT2/SYN and VGAT/SYN) indicated a high degree of signal overlap (0.9) in R1 and R2 regions of interest (Figure 10C). Injury significantly increased the MOC of VGLUT2/SYN (p<0.001) and VGAT/SYN (p<0.01) relative to naïve controls in R1, but MOC was unaffected by GBP treatment in either staining set (p>0.05).
Figure 10. Quantitative co-localization analyses of synaptic immunostaining.
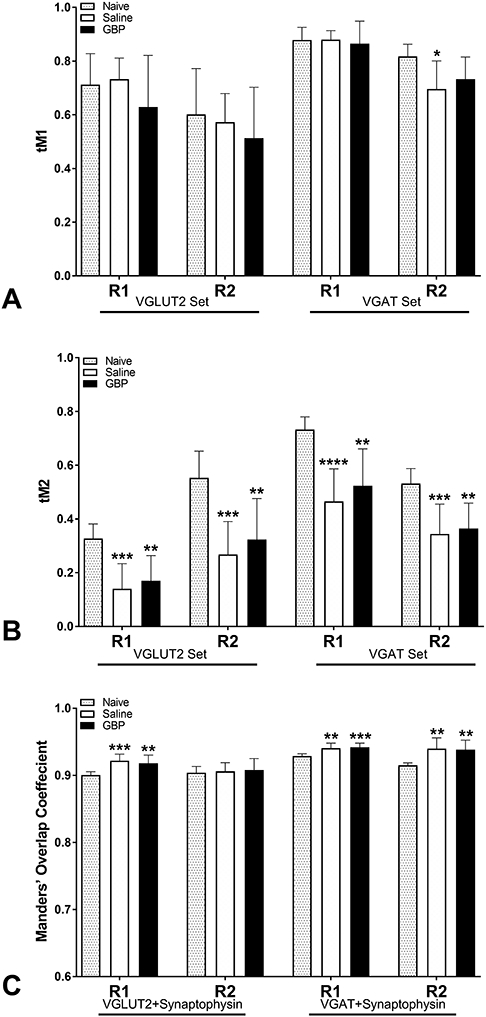
(A,B) The Manders’ fractional overlap coefficients, tM1 and tM2, and Manders’ overall overlap coefficient (MOC; C) were calculated to assess the co-localization of VGLUT2/Synaptophysin and VGAT/Synaptophysin staining in the same regions of interest (R1 and R2) used for density measurements. (A) The fraction of total VGLUT2 or VGAT signal overlapping with Synaptophysin (tM1) was largely unaffected by injury or GBP treatment. (B) Conversely, injury significantly decreased the fraction of Synaptophysin signal overlapping with VGLUT2 or VGAT (tM2). (C) MOC was greater than 0.9 in all groups and significantly increased after injury, with no effect of GBP treatment. N=8 naïve, N=11 Saline, N=10 GBP. *p<0.05; **p<0.01; ***p<0.001; ****p<0.001 vs naive control. Bars are mean ± SD.
Discussion
Based on our documentation that acute GBP administration reduces the severity of experimentally induced AD (Rabchevsky et al., 2011; Rabchevsky et al., 2012), and that daily treatment with high dosage GBP can block the formation of excitatory synapses in the brain and spinal cord (Eroglu et al., 2009; Lau et al., 2017; Yu et al., 2018), we sought to determine whether continuous delivery of high-dose GBP can mitigate the development of AD in association with altered synaptic densities in putative dorsal horn relay pathways. In the weeks following complete high thoracic SCI, we found that high-dose GBP treatment significantly reduced both the absolute magnitude of hypertension reached during weekly CRD-induced AD. However, this regimen also mitigated the reestablishment of pre-injury body weight and, paradoxically, was associated with a higher frequency of daily sAD events. While we found injury induced reductions in the densities of excitatory and inhibitory presynaptic markers in the lumbosacral dorsal horn, it is uncertain whether such alterations contributed to the incidence or severity of AD; notably continuous high-dose GBP did not change the reduced synaptic densities.
Herein, we increased both the treatment frequency and drug dosage (4x daily, 100 mg/kg) based on reports that 100-400 mg/kg of GBP daily prevents excitatory synaptogenesis in CNS of developing mice (Eroglu et al., 2009; Takahashi et al., 2018). We also delivered daily dosages across 4 injections to maintain relatively stable GBP concentrations due to the 2-3 hour half-life in rats (Vollmer et al., 1986). In clinical studies, efficacy to treat neuropathic pain has been demonstrated over a range of GBP doses from 1800-3600 mg/day with comparable effects across the dose range (Wiffen et al., 2017). The significant reductions in CRD-induced AD that we observed is consistent with our previous reports (Rabchevsky et al., 2011; Rabchevsky et al., 2012). However, when we assessed the MAP changes relative to baseline prior to CRD, we did not observe treatment effects, and we determined that the decrease in the baseline MAP is due to the anxiolytic effects of GBP (Houghton et al., 2017). Notably, GBP treated rats had significantly smaller increases in blood pressure from experimental handling and restraint just prior to CRD. Because our manipulations can present considerable stressors for rodents, pre-treatment with GBP likely attenuated anxiety levels and consequent cardiovascular responses. As with all drugs that interact directly with the central nervous system, GBP has potential side effects that can include somnolence, dizziness and ataxia.
Saline treated rats started to reach pre-injury body weight levels by three weeks, whereas GBP treated rats never returned to pre-injury body weight by termination at 4 weeks post-injury. The gradual return to pre-injury weight beginning approximately 2-weeks after SCI is parallel to the development of fulminant muscle spasticity in rats after SCI (Bennett et al., 2004). Therefore, given the continuous 4 x daily GBP administration in our current study, it is feasible that the failure of GBP treated rats to return to pre-injury weight is due to a lack of spasticity-induced muscle preservation. GBP (Neurontin®; Pfizer) is approved for the treatment of epilepsy and is widely used for the treatment of neuropathic pain (Levendoglu et al., 2004; Morello et al., 1999). GBP has been shown to inhibit presynaptic glutamate release by modulating calcium channels (Tran-Van-Minh and Dolphin, 2010), and it demonstrates the potential to help decrease the manifestation of spasticity in individuals with SCI (Gruenthal et al., 1997). However, there has been no systematic examination of the efficacy of GBP as a single treatment for the management of SCI-induced spasticity (Rabchevsky and Kitzman, 2011). It is important to note that continuous GBP dosage regimens could abrogate the potentially beneficial effects of muscle spasticity, as preservation of muscle tone helps prevent musculoskeletal issues (Pingel et al., 2017).
Synaptic plasticity
Consistent with previous reports (Beauparlant et al., 2013; McKillop et al., 2016), we observed significantly reduced densities of putative excitatory (VGLUT2+) and inhibitory (VGAT+) pre-synaptic puncta in dorsal horn regions of the lumbosacral spinal cord thought to be involved in the viscero-sympathetic relay pathways after T4 SCI (Krassioukov et al., 2002; Weaver et al., 2002). However, continuous high dose GBP did not alter synaptic re-organization, suggesting that it acts to reduce induced hypertensive crises by blocking intraspinal glutamatergic neurotransmission underlying AD (Maiorov et al., 1997). We assessed VGLUT2+ synaptic puncta based on previous studies showing that this is the predominant VGLUT expressed in the upper dorsal horn and lamina X regions of interest in our model (Alvarez et al., 2004; Llewellyn-Smith et al., 2007). Since VGLUT2 in the dorsal cord is expressed by excitatory interneurons (Maxwell et al., 2007), much of the reduced VGLUT2 density that we observed is likely attributed to propriospinal loss. Notably, decreases in the number of dorsal inhibitory neurons after SCI is proposed to contribute to the development of injury-induced neuropathic pain (Meisner et al., 2010). We cannot rule out that our chosen regions of interest limited to the superficial laminae in the dorsal horn and lamina X may not have sampled other putative synaptic relays that might be altered by both injury and GBP. Nevertheless, it remains uncertain how such dynamic alterations in the lumbosacral spinal cord, in parallel to plasticity of SPN or sympathetically correlated interneurons, contributed to both reduced magnitude of induced AD in parallel to increased frequency of sAD events with chronic high-dose GBP treatment.
Cardiovascular consequences
Abnormal fluctuations in blood pressure associated with SCI may contribute to cerebrovascular dysfunction (Phillips et al., 2016), indicating that the increased sAD events detected with continuous GBP treatment may be clinically relevant outcome measures. Therefore, our finding that chronic administration of high dose GBP increases the frequency of moderate hemodynamic fluctuations after SCI warrants consideration of potentially negative effects on cardiovascular outcome measures. Classical definitions of hypertensive AD have included bradycardia as a cardinal sign for diagnosis, however bradycardia is not a consistent indicator. Our results showed highly variable HR responses during CRD-induced AD with many instances of accompanying tachycardia, notably at earliest time point evaluated. As posited by Karlsson (1999), it is conceivable that tachycardia during AD may result from the propagation of reflex arcs towards the SPN responsible for cardiac sympathetic drive in the upper thoracic spinal cord. In our model of T4-transection SCI, it is possible that spared and decentralized cardiac SPN in the T4-T6 spinal segments (Coote and Chauhan, 2016) retain the ability to provide reflex-driven accelerated heart rate during CRD-induced AD. Accordingly, contemporary diagnostic guidelines for AD include both bradycardia and tachycardia during hypertensive crises (Murray et al., 2019).
The α2δ1 calcium channel subunit that binds GBP is found ubiquitously in many tissues and is highly abundant in the myocardium, vascular smooth muscle, skeletal muscle, and many brain regions, and they are essential for vasoregulation by calcium channels (Bannister et al., 2009; Taylor and Garrido, 2008). Fuller-Bicer et al. (2009) used knockout mice with targeted disruption of α2δ1 to show both decreased calcium currents in cardiomyocytes, as well as reduced basal contractility and relaxation parameters in ex-vivo heart preparations compared to wild-type. This suggests important roles of α2δ1 in cardiac function, indicating that perhaps continuous high-dose GBP influenced daily HR directly in our model, though increased tachycardic trends were not significant.
Based on our previous reports with much lower dose, we hypothesized that continuous high-dose GBP treatment would reduce the frequency of sAD. However, the current treatment paradigm, which delivers eight times the amount of daily GBP, had the unexpected effect of increasing daily sAD frequency. The exact mechanisms of sAD generation are uncertain, aside from bladder filling and/or fecal impaction. AD is thought to be caused, in part, by increased excitatory input to SPNs from afferents (via propriospinal relays), and GBP decreases intraspinal release of glutamate from primary afferent terminals (Coderre et al., 2007). Accordingly, these observations present the possibility that continuous GBP treatment decreases afferent stimulation of SPN or sympathetically correlated interneurons to increase their intrinsic excitability. For example, large scale reductions in neuronal network activity induce homeostatic responses, whereby individual neurons increase their membrane excitability in attempt to maintain “normal” activity (Braegelmann et al., 2017). We did not test in our model, but prolonged deprivation of the SPN from afferent excitation during continuous high-dose GBP treatment may have increased their excitability and spontaneous firing, manifesting as increased sAD events.
Among the many variables involved in cardiovascular regulation, psychological and emotional stressors can introduce acute alterations in MAP and HR. Controlling for potential sources of stress during monitoring of the cardiovascular or autonomic systems is important, and GBP treated rats had significantly reduced pressor responses to experimental manipulations prior to CRD. GBP is well known to have anxiolytic effects in humans and rodents (Greenblatt and Greenblatt, 2018; SM et al., 2011), and evidence suggests that >95% of individuals with SCI above T6 experience episodes of AD during penile vibrostimulation procedures for sperm retrieval (Ekland et al., 2008). While immediate-release nifedipine (Procardia, Adalat, Afeditab), also a calcium channel blocker, is the most commonly prescribed agent for management of ongoing AD episodes, it lowers MAP for up to 5 h that can result in dizziness, fatigue, and weakness (Krassioukov et al., 2009). An alternative to nifedipine is prazosin (Minipress), a selective adrenergic blocker with a slower and less abrupt suppressive effect on MAP, and clinical studies suggest that prazosin is a more suitable option than nifedipine for treatment of AD secondary to sexual stimulation in this population (Phillips et al., 2015). However, because prazosin requires a “first-dose response” the day prior iatrogenic to procedures, it may not be suitable for alleviating fulminant AD since it acts peripherally to dilate blood vessels. Like GBP, its side effects of are largely limited to dizziness, potential for fainting, as well as increased inotropy, and possibly altered bladder function. Therefore, since GBP is widely prescribed to the SCI population with high tolerance even long-term at high dosages with a half-life of 2-3 hours, then it is conceivable that acute low dose GBP treatment in the clinical setting may provide similar benefits as nifedipine or prazosin to mitigate AD that is evoked by intentional stimulation to the genitals and/or pelvic visceral organs in the clinical setting.
Immunological consequences
SCI is linked to immunosuppression in humans, with chronic SCI individuals being at a higher risk of potentially lethal infections (Brommer et al., 2016). Spinal cord injury-induced immune depression syndrome (SCI-IDS) is dependent upon the level of injury, with injuries above upper-thoracic levels that decentralize the major sympathetic outflow having higher risks of developing SCI-IDS (Brommer et al., 2016). This phenomenon has been observed in experimental models of SCI in mice, which similarly undergo diminished immune function as determined by reduced lymphocytic markers and spleen size associated temporally with the development of AD (Lucin et al., 2007; Ueno et al., 2016; Zhang et al., 2013). Accordingly, using changes in spleen size as a metric of immunomodulation, we observed no injury effects, suggesting that SCI/AD does not contribute to splenic atrophy in rats, per se. On the contrary, continuous high dose GBP treatment after SCI caused splenomegaly (enlarged spleen). Although there are no readily apparent connections between GBP and spleen size, and the α2δ1 subunit to which GBP binds is sparsely expressed in the spleen (Taylor and Garrido, 2008), there are clinical reports of splenomegaly associated with GBP administration; daily 200 mg/kg GBP can elicit GBP hypersensitivity syndrome (Ragucci and Cohen, 2001), which includes splenomegaly and elevated hepatic enzymes that return to normal rapidly after discontinuation of GBP. Irrespective of GBP effects, future experiments are required to establish the role of splenic atrophy/hypertrophy in the development of AD and its relationship to chronic immunosuppression in SCI rats.
Conclusions
Continuous high-dose GBP treatment after complete high-thoracic SCI was found to offer significant benefit in reducing the magnitude of AD evoked by noxious stimulation of the pelvic viscera weeks after injury, which was reflected in anxiolytic effects on hemodynamics during the handling procedures. Both low- and high-dose acute GBP treatments appear to mitigate experimentally induced AD, which may be relevant in controlled clinical settings such as iatrogenic procedures. However, the potential deleterious effects of chronic high-dose GBP to impair weight gain, reduce preservation of muscle tone, promote splenomegaly, and increase sAD events must be considered cautiously for use in the SCI population which is prescribed this medication for a myriad of indications. The absence of high-dose GBP effects on the reduced synaptic densities in the lumbosacral cord after SCI suggests that it acts through distinct mechanisms to alter induced AD and sAD events, perhaps affecting peripheral vasculature. Experiments to assess GBP influences on cardiac contractility, stroke volume or aortic stiffness in this AD model are required, along with more refined approaches to study synaptic connectivity to provide a better characterization of potential remodeling in specific neuronal pathways thought to contribute to the pathophysiology of AD.
Acknowledgments
Supported by: NIH 5T32 NS077889 (KCE); KSCHIRT #10-10 (AGR); SCoBIRC Chair Endowment (AGR); NIH/NINDS 2P30NS051220.
References
- Alvarez FJ, Villalba RM, Zerda R, Schneider SP, 2004. Vesicular glutamate transporters in the spinal cord, with special reference to sensory primary afferent synapses. J Comp Neurol 472, 257–280. [DOI] [PubMed] [Google Scholar]
- Anderson KD, 2004. Targeting recovery: priorities of the spinal cord-injured population. Journal of neurotrauma 21, 1371–1383. [DOI] [PubMed] [Google Scholar]
- Bannister JP, Adebiyi A, Zhao G, Narayanan D, Thomas CM, Feng JY, Jaggar JH, 2009. Smooth muscle cell alpha2delta-1 subunits are essential for vasoregulation by CaV1.2 channels. Circ Res 105, 948–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauparlant J, van den Brand R, Barraud Q, Friedli L, Musienko P, Dietz V, Courtine G, 2013. Undirected compensatory plasticity contributes to neuronal dysfunction after severe spinal cord injury. Brain 136, 3347–3361. [DOI] [PubMed] [Google Scholar]
- Bennett DJ, Sanelli L, Cooke CL, Harvey PJ, Gorassini MA, 2004. Spastic long-lasting reflexes in the awake rat after sacral spinal cord injury. J Neurophysiol 91, 2247–2258. [DOI] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP, 2006. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224, 213–232. [DOI] [PubMed] [Google Scholar]
- Braegelmann KM, Streeter KA, Fields DP, Baker TL, 2017. Plasticity in respiratory motor neurons in response to reduced synaptic inputs: A form of homeostatic plasticity in respiratory control? Exp Neurol 287, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brommer B, Engel O, Kopp MA, Watzlawick R, Muller S, Pruss H, Chen Y, DeVivo MJ, Finkenstaedt FW, Dirnagl U, Liebscher T, Meisel A, Schwab JM, 2016. Spinal cord injury-induced immune deficiency syndrome enhances infection susceptibility dependent on lesion level. Brain 139, 692–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron AA, Smith GM, Randall DC, Brown DR, Rabchevsky AG, 2006. Genetic manipulation of intraspinal plasticity after spinal cord injury alters the severity of autonomic dysreflexia. J Neurosci 26, 2923–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau D, Johns DG, Schramm LP, 2000. Ongoing and stimulus-evoked activity of sympathetically correlated neurons in the intermediate zone and dorsal horn of acutely spinalized rats. J Neurophysiol 83, 2699–2707. [DOI] [PubMed] [Google Scholar]
- Coderre TJ, Kumar N, Lefebvre CD, Yu JS, 2007. A comparison of the glutamate release inhibition and anti-allodynic effects of gabapentin, lamotrigine, and riluzole in a model of neuropathic pain. J Neurochem 100, 1289–1299. [DOI] [PubMed] [Google Scholar]
- Coote JH, Chauhan RA, 2016. The sympathetic innervation of the heart: Important new insights. Auton Neurosci 199, 17–23. [DOI] [PubMed] [Google Scholar]
- Crawford P, Ghadiali E, Lane R, Blumhardt L, Chadwick D, 1987. Gabapentin as an antiepileptic drug in man. J Neurol Neurosurg Psychiatry 50, 682–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KW, Kamocka MM, McDonald JH, 2011. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300, C723–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekland MB, Krassioukov AV, McBride KE, Elliott SL, 2008. Incidence of autonomic dysreflexia and silent autonomic dysreflexia in men with spinal cord injury undergoing sperm retrieval: implications for clinical practice. J Spinal Cord Med 31, 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldahan KC, Cox DH, Gollihue JL, Patel SP, Rabchevsky AG, 2018. Rapamycin Exacerbates Cardiovascular Dysfunction after Complete High-Thoracic Spinal Cord Injury. J Neurotrauma 35, 842–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA, 2009. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, Bresnahan JC, Beattie MS, 2008. Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neurosci 28, 11391–11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller-Bicer GA, Varadi G, Koch SE, Ishii M, Bodi I, Kadeer N, Muth JN, Mikala G, Petrashevskaya NN, Jordan MA, Zhang SP, Qin N, Flores CM, Isaacsohn I, Varadi M, Mori Y, Jones WK, Schwartz A, 2009. Targeted disruption of the voltage-dependent calcium channel alpha2/delta-1-subunit. Am J Physiol Heart Circ Physiol 297, H117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN, 1996. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J Biol Chem 271, 5768–5776. [DOI] [PubMed] [Google Scholar]
- Greenblatt HK, Greenblatt DJ, 2018. Gabapentin and Pregabalin for the Treatment of Anxiety Disorders. Clin Pharmacol Drug Dev 7, 228–232. [DOI] [PubMed] [Google Scholar]
- Gruenthal M, Mueller M, Olson WL, Priebe MM, Sherwood AM, Olson WH, 1997. Gabapentin for the treatment of spasticity in patients with spinal cord injury. Spinal Cord 35, 686–689. [DOI] [PubMed] [Google Scholar]
- Hou S, Duale H, Cameron AA, Abshire SM, Lyttle TS, Rabchevsky AG, 2008. Plasticity of lumbosacral propriospinal neurons is associated with the development of autonomic dysreflexia after thoracic spinal cord transection. J Comp Neurol 509, 382–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S, Rabchevsky AG, 2014. Autonomic consequences of spinal cord injury. Compr Physiol 4, 1419–1453. [DOI] [PubMed] [Google Scholar]
- Houghton KT, Forrest A, Awad A, Atkinson LZ, Stockton S, Harrison PJ, Geddes JR, Cipriani A, 2017. Biological rationale and potential clinical use of gabapentin and pregabalin in bipolar disorder, insomnia and anxiety: protocol for a systematic review and meta-analysis. BMJ Open 7, e013433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson AK, 1999. Autonomic dysreflexia. Spinal Cord 37, 383. [DOI] [PubMed] [Google Scholar]
- Krassioukov A, Warburton DE, Teasell R, Eng JJ, Spinal Cord Injury Rehabilitation Evidence Research, T., 2009. A systematic review of the management of autonomic dysreflexia after spinal cord injury. Arch Phys Med Rehabil 90, 682–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krassioukov AV, Bunge RP, Pucket WR, Bygrave MA, 1999. The changes in human spinal sympathetic preganglionic neurons after spinal cord injury. Spinal Cord 37, 6–13. [DOI] [PubMed] [Google Scholar]
- Krassioukov AV, Furlan JC, Fehlings MG, 2003. Autonomic dysreflexia in acute spinal cord injury: an under-recognized clinical entity. J Neurotrauma 20, 707–716. [DOI] [PubMed] [Google Scholar]
- Krassioukov AV, Johns DG, Schramm LP, 2002. Sensitivity of sympathetically correlated spinal interneurons, renal sympathetic nerve activity, and arterial pressure to somatic and visceral stimuli after chronic spinal injury. J Neurotrauma 19, 1521–1529. [DOI] [PubMed] [Google Scholar]
- Lau LA, Noubary F, Wang D, Dulla CG, 2017. alpha2delta-1 Signaling Drives Cell Death, Synaptogenesis, Circuit Reorganization, and Gabapentin-Mediated Neuroprotection in a Model of Insult-Induced Cortical Malformation. eNeuro 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levendoglu F, Ogun CO, Ozerbil O, Ogun TC, Ugurlu H, 2004. Gabapentin is a first line drug for the treatment of neuropathic pain in spinal cord injury. Spine (Phila Pa 1976) 29, 743–751. [DOI] [PubMed] [Google Scholar]
- Llewellyn-Smith IJ, Martin CL, Fenwick NM, Dicarlo SE, Lujan HL, Schreihofer AM, 2007. VGLUT1 and VGLUT2 innervation in autonomic regions of intact and transected rat spinal cord. J Comp Neurol 503, 741–767. [DOI] [PubMed] [Google Scholar]
- Llewellyn-Smith IJ, Weaver LC, 2001. Changes in synaptic inputs to sympathetic preganglionic neurons after spinal cord injury. J Comp Neurol 435, 226–240. [DOI] [PubMed] [Google Scholar]
- Lucin KM, Sanders VM, Jones TB, Malarkey WB, Popovich PG, 2007. Impaired antibody synthesis after spinal cord injury is level dependent and is due to sympathetic nervous system dysregulation. Exp Neurol 207, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiorov DN, Krenz NR, Krassioukov AV, Weaver LC, 1997. Role of spinal NMDA and AMPA receptors in episodic hypertension in conscious spinal rats. Am J Physiol 273, H1266–1274. [DOI] [PubMed] [Google Scholar]
- Maxwell DJ, Belle MD, Cheunsuang O, Stewart A, Morris R, 2007. Morphology of inhibitory and excitatory interneurons in superficial laminae of the rat dorsal horn. J Physiol 584, 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKillop WM, York EM, Rubinger L, Liu T, Ossowski NM, Xu K, Hryciw T, Brown A, 2016. Conditional Sox9 ablation improves locomotor recovery after spinal cord injury by increasing reactive sprouting. Exp Neurol 283, 1–15. [DOI] [PubMed] [Google Scholar]
- Meisner JG, Marsh AD, Marsh DR, 2010. Loss of GABAergic interneurons in laminae I-III of the spinal cord dorsal horn contributes to reduced GABAergic tone and neuropathic pain after spinal cord injury. J Neurotrauma 27, 729–737. [DOI] [PubMed] [Google Scholar]
- Mironets E, Osei-Owusu P, Bracchi-Ricard V, Fischer R, Owens EA, Ricard J, Wu D, Saltos T, Collyer E, Hou S, Bethea JR, Tom VJ, 2018. Soluble TNFalpha Signaling within the Spinal Cord Contributes to the Development of Autonomic Dysreflexia and Ensuing Vascular and Immune Dysfunction after Spinal Cord Injury. J Neurosci 38, 4146–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello CM, Leckband SG, Stoner CP, Moorhouse DF, Sahagian GA, 1999. Randomized double-blind study comparing the efficacy of gabapentin with amitriptyline on diabetic peripheral neuropathy pain. Arch Intern Med 159, 1931–1937. [DOI] [PubMed] [Google Scholar]
- Murray TE, Krassioukov AV, Pang EHT, Zwirewich CV, Chang SD, 2019. Autonomic Dysreflexia in Patients With Spinal Cord Injury: What the Radiologist Needs to Know. AJR Am J Roentgenol, 1–5. [DOI] [PubMed] [Google Scholar]
- Phillips AA, Elliott SL, Zheng MM, Krassioukov AV, 2015. Selective alpha adrenergic antagonist reduces severity of transient hypertension during sexual stimulation after spinal cord injury. J Neurotrauma 32, 392–396. [DOI] [PubMed] [Google Scholar]
- Phillips AA, Matin N, Frias B, Zheng MM, Jia M, West C, Dorrance AM, Laher I, Krassioukov AV, 2016. Rigid and remodelled: cerebrovascular structure and function after experimental high-thoracic spinal cord transection. J Physiol 594, 1677–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingel J, Bartels EM, Nielsen JB, 2017. New perspectives on the development of muscle contractures following central motor lesions. J Physiol 595, 1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabchevsky AG, 2006. Segmental organization of spinal reflexes mediating autonomic dysreflexia after spinal cord injury. 152, 265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabchevsky AG, Kitzman PH, 2011. Latest approaches for the treatment of spasticity and autonomic dysreflexia in chronic spinal cord injury. Neurotherapeutics 8, 274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabchevsky AG, Patel SP, Duale H, Lyttle TS, O'Dell CR, Kitzman PH, 2011. Gabapentin for spasticity and autonomic dysreflexia after severe spinal cord injury. Spinal Cord 49, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabchevsky AG, Patel SP, Lyttle TS, Eldahan KC, O'Dell CR, Zhang Y, Popovich PG, Kitzman PH, Donohue KD, 2012. Effects of gabapentin on muscle spasticity and both induced as well as spontaneous autonomic dysreflexia after complete spinal cord injury. Front Physiol 3, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragucci MV, Cohen JM, 2001. Gabapentin-induced hypersensitivity syndrome. Clin Neuropharmacol 24, 103–105. [DOI] [PubMed] [Google Scholar]
- Schramm LP, 2006. Spinal sympathetic interneurons: their identification and roles after spinal cord injury. Prog Brain Res 152, 27–37. [DOI] [PubMed] [Google Scholar]
- Sirven JI, 2010. New uses for older drugs: the tales of aspirin, thalidomide, and gabapentin. Mayo Clin Proc 85, 508–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SM OM, Coelho AM, Fitzgerald P, Lee K, Winchester W, Dinan TG, Cryan JF, 2011. The effects of gabapentin in two animal models of co-morbid anxiety and visceral hypersensitivity. Eur J Pharmacol 667, 169–174. [DOI] [PubMed] [Google Scholar]
- Takahashi DK, Jin S, Prince DA, 2018. Gabapentin Prevents Progressive Increases in Excitatory Connectivity and Epileptogenesis Following Neocortical Trauma. Cereb Cortex 28, 2725–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Neckel ND, Schramm LP, 2003. Locations and morphologies of sympathetically correlated neurons in the T(10) spinal segment of the rat. Brain Res 976, 185–193. [DOI] [PubMed] [Google Scholar]
- Taylor CP, Garrido R, 2008. Immunostaining of rat brain, spinal cord, sensory neurons and skeletal muscle for calcium channel alpha2-delta (alpha2-delta) type 1 protein. Neuroscience 155, 510–521. [DOI] [PubMed] [Google Scholar]
- Tran-Van-Minh A, Dolphin AC, 2010. The alpha2delta ligand gabapentin inhibits the Rab11-dependent recycling of the calcium channel subunit alpha2delta-2. J Neurosci 30, 12856–12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Ueno-Nakamura Y, Niehaus J, Popovich PG, Yoshida Y, 2016. Silencing spinal interneurons inhibits immune suppressive autonomic reflexes caused by spinal cord injury. Nat Neurosci 19, 784–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer KO, von Hodenberg A, Kolle EU, 1986. Pharmacokinetics and metabolism of gabapentin in rat, dog and man. Arzneimittelforschung 36, 830–839. [PubMed] [Google Scholar]
- Weaver LC, Cassam AK, Krassioukov AV, Llewellyn-Smith IJ, 1997. Changes in immunoreactivity for growth associated protein-43 suggest reorganization of synapses on spinal sympathetic neurons after cord transection. Neuroscience 81, 535–551. [DOI] [PubMed] [Google Scholar]
- Weaver LC, Fleming JC, Mathias CJ, Krassioukov AV, 2012. Disordered cardiovascular control after spinal cord injury. Handb Clin Neurol 109, 213–233. [DOI] [PubMed] [Google Scholar]
- Weaver LC, Marsh DR, Gris D, Meakin SO, Dekaban GA, 2002. Central mechanisms for autonomic dysreflexia after spinal cord injury. Prog Brain Res 137, 83–95. [DOI] [PubMed] [Google Scholar]
- West CR, Popok D, Crawford MA, Krassioukov AV, 2015. Characterizing the temporal development of cardiovascular dysfunction in response to spinal cord injury. J Neurotrauma 32, 922–930. [DOI] [PubMed] [Google Scholar]
- Wiffen PJ, Derry S, Bell RF, Rice AS, Tolle TR, Phillips T, Moore RA, 2017. Gabapentin for chronic neuropathic pain in adults. Cochrane Database Syst Rev 6, CD007938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YP, Gong N, Kweon TD, Vo B, Luo ZD, 2018. Gabapentin prevents synaptogenesis between sensory and spinal cord neurons induced by thrombospondin-4 acting on pre-synaptic Cav alpha2 delta1 subunits and involving T-type Ca(2+) channels. Br J Pharmacol 175, 2348–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Guan Z, Reader B, Shawler T, Mandrekar-Colucci S, Huang K, Weil Z, Bratasz A, Wells J, Powell ND, Sheridan JF, Whitacre CC, Rabchevsky AG, Nash MS, Popovich PG, 2013. Autonomic dysreflexia causes chronic immune suppression after spinal cord injury. J Neurosci 33, 12970–12981. [DOI] [PMC free article] [PubMed] [Google Scholar]