

Abstract

Radical S-adenosyl-l-methionine (SAM) enzymes catalyze a diverse group of complex transformations in all aspects of cellular physiology. These metalloenzymes bind SAM to a 4Fe–4S cluster and reductively cleave SAM to generate a 5′-deoxyadenosyl radical, which generally initiates the catalytic cycle by catalyzing a H atom to activate the substrate for subsequent chemistry. This perspective will focus on our discovery of several members of this superfamily of enzymes, with a particular emphasis on the current state of the field, challenges, and outlook.
Keywords: radical SAM superfamily, radical mediated reactions, enzymes
I observed my 50th birthday last year amidst the pandemic. I say observed and not celebrated because it felt less like a milestone and more like a bank holiday! When Squire invited me to write this perspective, I started reflecting on the 20 years since the Sofia paper1 and the birth of the radical SAM (RS) superfamily. It was remarkable to think back to my days as a young graduate student at the Enzyme Institute (Figure 1) at the University of Wisconsin and realize how much of the early work in the field I had observed in real-time. It has been thrilling to see some of the beautiful work from many investigators on this topic that are being highlighted in this special issue. In contrast to my midlife birthday, this 20-year celebration of the birth of the radical SAM superfamily is truly momentous.
Figure 1.

Institute for Enzyme Research at the University of Wisconsin—Madison. The Frey and Reed laboratories were located on the 5th and 4th floors. The photo from Cory Coyle/CC-BY-SA-3.0 was used without modification.
My own journey with radical SAM enzymes was accidental. In 1995, Squire Booker had arrived at the Enzyme Institute to carry out a postdoctoral stint with Perry Frey. Since the anaerobic chambers were all located on the fourth floor of the Institute, near the Reed lab, I had daily interactions with Squire and other members of the Frey lab to discuss their most recent experimental results. I did not realize that I had a front-row seat to what would become the sprawling radical SAM field. However, when I left UW Madison, I had seen and heard enough to know that radical SAM enzymes were difficult to work with and as my graduate advisor George Reed used to say, “there are easier ways to earn a living.”
I briefly collaborated with Perry’s lab on studies involving substrate radicals that formed with alternate substrates. In the Reed lab, I had been working on identifying and characterizing radical intermediates in the B12-dependent ethanolamine ammonia lyase. Perry and George had been collaborating on EPR spectroscopic studies of lysine 2,3-aminomutase, which at the time was known as LAM but has since been renamed KAM.2−4 They tasked me with simulating and assigning the spectra of the radical intermediates observed with two alternate substrates of LAM, 4-thialysine and allyllysine.5 These studies have influenced my thinking about enzymatic reaction mechanisms a great deal in the years since and were the inspiration for the cyclopropylglycine radical clock experiment described later.
Before I left to begin my graduate studies at UW Madison, my undergraduate research advisor at California State University in Los Angeles, Scott Grover, had told me to be on the lookout for “nuggets” that I could file as possible research ideas. The direction of my research program was set unwittingly sometime in 1995, when I was introduced, in a journal club, to a paper that suggested a B12-dependent enzyme was involved in the biosynthesis of the hypermodified RNA base, queuosine. This was one such nugget. I remember discussing this with Squire after that seminar, and we both considered discovering this mysterious B12-dependent enzyme in our independent careers. Later, when I described the idea of studying this enzyme to my postdoctoral advisor at the University of Michigan, Rowena Matthews, her encouragement was a further sign that it would be a good starting project.
That single nugget pulled me and my research program in the area of radical SAM enzymes. In this perspective, I will outline my lab’s nearly 20-year journey, some of the key findings from my laboratory that have contributed to the field, my perspective on these enzymes, and the future of the field.
Discovery of CDG Synthase
I started my independent program at the University of Arizona focused on discovering the enzymes involved in the biosynthesis of the pyrrolopyrimidine natural products.6 The first known 7-deazapurine-containing natural product, toyocamycin, was discovered from soil samples in 1956.7 Active research programs by Pfizer and others led to the discovery of many more related compounds. Starting in the late 1960s and into the 1970s, studies on modified RNA bases had led to the identification of queuosine, another 7-deazapurine, which is a widely distributed base8 found in the wobble position of tRNA Asp, Asn, His, and Tyr.9,10 A related nucleoside, archaeosine, was also uncovered in Archaea.11
While arising from seemingly disparate biological niches, independent studies in the laboratories of Suhadolnik and Nishimura had indicated that all 7-deazapurines arise from a purine in a process that entails the loss of C-8 of the nucleoside.12−14 Using bioinformatic approaches, de Crécy-Lagard had identified four genes in B. subtilis, which, when knocked out, led to the loss of queuosine from RNA.15 At the time the knockout experiments were published, my lab was focused on uncovering the biosynthetic pathway for the related 7-deazapurines toyocamycin and sangivamycin from S. rimosus. Suhadolnik had shown that the organism makes both, and that crude extracts exhibited a toyocamycin nitrile hydratase activity.16 Using protein sequence information from the purified hydratase, my student Reid McCarty was able to find a putative deazapurine biosynthetic cluster.17 To our surprise, of the cluster of genes we had identified, three appeared to be homologous to those identified bioinformatically in B. subtilis as being involved in the biosynthesis of queuosine.15 Through a series of in vitro experiments, Reid succeeded in reconstructing the pathway from GTP to 7-carboxy-7-deazaguanine (CDG). These experiments led to the discovery of CDG synthase (QueE), a RS enzyme that catalyzes the conversion of 6-carboxy-5,6,7,8-tetrahydropterin (CPH4) to CDG (Figure 2). Zach Miles in my lab would discover the mysterious B12-dependent epoxyqueuosine (oQ) reductase enzyme that is responsible for catalyzing the final step of the biogenesis of the modified base queuosine,18−20 whose identity had initially set us on this journey. oQ reductase, while also an iron/sulfur-containing enzyme, is not a member of the RS superfamily.
Figure 2.
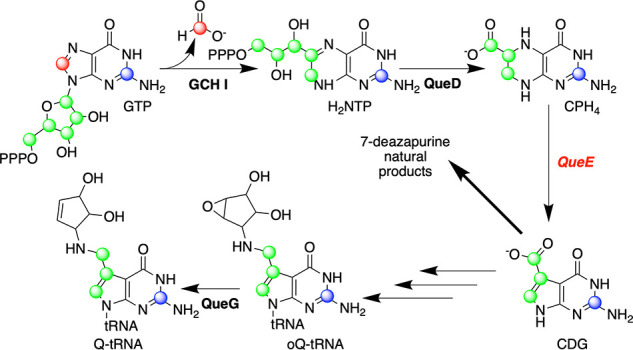
Biosynthesis of 7-deazapurines. GTP cyclohydrolase I (GCH I), CPH4 synthase (QueD), and 7-carboxy-7-deazaguanine (CDG) synthase catalyze the conversion of GTP to CDG, which is a common precursor to 7-deazapurines. QueG catalyzes the B12-dependent reduction of oQ-tRNA to the Q-tRNA.
Our first foray into the RS space occurred during the reconstruction of this biosynthetic pathway to deazapurines.21,22 CDG synthase catalyzes a complex radical-mediated ring contraction to convert CPH4 to CDG (Figure 3).23 Biochemical studies with isotopologues of the substrate have delineated key aspects of the mechanism. The enzyme initiates the catalytic cycle by H atom transfer from the C-6 of the substrate to the 5′-deoxyadenosyl radical (dAdo•), activating it for ring contraction. While the mechanism of the rearrangement mechanism is not known, we have proposed that the radical intermediate is converted to a gem-aminocarboxylate intermediate via a product radical, which is quenched by H atom transfer from dAdo to regenerate the SAM. Deamination and aromatization follow, forming CDG. These later steps appear to involve stereoselective removal of the proR hydrogen of at C-7 of the starting substrate, as the proS hydrogen remains associated with the product. In contrast to many other radical SAM enzymes, SAM is used catalytically by CDG synthase, as evidenced by ability of an equivalent of SAM to support formation of multiple equivalents of CDG, and by the appearance of multiply deuterated dAdo in the presence of the C-6-deuterated substrate. The details of the rearrangement remain an active area of research in the lab.
Figure 3.
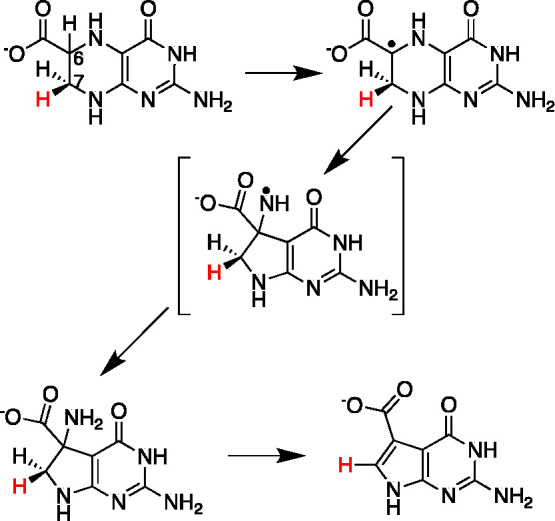
CDG synthase catalyzes a radical-mediated ring contraction reaction.
CDG synthase has been extensively characterized by X-ray crystallography in collaboration with Cathy Drennan’s lab at MIT.24−26 The structural work on CDG synthase was carried out by Daniel Dowling and continued by Tsehai Grell. This collaboration has led to X-ray crystal structures of CDG synthase homologues from E. coli, B. multivorans, and B. subtilis in various ligand-occupied states. The structures all show that the protein is composed of a dimer of TIM barrel-like proteins (the structure of the B. subtilis protein is shown in Figure 4A as an example). While the details of the barrel structure are variable, the active sites of all the homologues are remarkably similar. A CxxxCxxC motif binds a 4Fe–4S cluster, with the α-amino and α-carboxylate of SAM occupying the remaining open coordination site. The structures of the enzyme bound to CPH4/SAM (Figure 4B), or CDG/SAM (Figure 4C) reveal that the substrate is bound through interactions with an essential Mg2+ divalent cation, and that the exocyclic amino group and the pyrimidine nitrogen form H-bonding interactions with the C-terminal carboxylate. The side chain of a conserved arginine residue interacts with the carboxylate of the substrate/product.
Figure 4.

Structure of Burkholderia multivorans CDG synthase bound to substrate (CPH4) or product (CDG). (A) CDG synthase is a dimer of identical subunits. The yellow/orange spheres in the structure denote the iron–sulfur cluster, and the green sphere is the required magnesium divalent cation. The substrate (B) and product (C) bind similarly and make identical interactions with a small number of residues. The separation between C-5′ of SAM and C-6 hydrogen of the substrate is shown by the dashed line. The H was modeled with the PyMol Molecular Graphics System (version 2.0 Schrödinger, LLC).
The most intriguing structural finding with CDG synthase is that the C-6 hydrogen of CPH4 is within 3.1 Å of the C-5′ of the SAM (see dashed line in Figure 4B), suggesting that very little movement of the dAdo• is needed to promote H atom transfer. Moreover, the structure of the enzyme with CDG bound shows that the substrate undergoes minimal displacement during the conversion to the product, and that the interactions with the divalent cation and the C-terminal carboxylate are maintained (compare Figure 4B,C). Moreover, very few residues within the enzyme are maintained as close to the substrate/product as the SAM. Two glutamate side chains (E15 and E116 in B. multivorans numbering) are near the substrate and may be involved in the stereoselective deprotonation that leads to the product. This paucity of interactions between the substrate and the enzyme and the use of a non-redox-active metal ion to template/bind the substrate are noteworthy. In effect, Nature evolved an environment in which the radical-mediated rearrangement can occur with limited enzymatic intervention to minimize off-pathway chemistry.
One experiment that highlights the significance of the seemingly minimal but precise arrangement of the interactions in the active site is the reaction of CDG synthase with the substrate analogue 6-carboxypterin (6-CP). 6-CP is a fully oxidized analogue of CPH4.25 When 6-CP is incubated with the protein, its fate is dependent on whether reductant is present or not. In the absence of reductant, the 6-carboxypterin-5′-deoxyadenosyl ester forms (Figure 5). This is presumably because of the juxtaposition of the carboxylate of the analogue and the C-5′ of SAM, which is activated by attachment to the sulfonium. This adduct was, in fact, also observed in the X-ray crystal structure of the protein solved with 6-CP.25 However, in the presence of a reductant, the analogue instead undergoes radical addition with dAdo•, which, following oxidative decarboxylation, forms 6-deoxyadenosylpterin. EPR spectroscopic analysis of the reaction carried out in collaboration with David Britt and Jarret Wilcoxen in his lab at UC Davis revealed the initial deoxyadenosyl adduct of 6-CP under reducing conditions.27 However, a detailed analysis of the structure proved difficult, as the initial radical formed is delocalized into the aromatic π system. Nevertheless, the divergent fates of 6-CP under nonreducing and reducing conditions highlight the need for radical SAM enzymes to maintain active sites that are sparse—with only those residues that are needed for chemistry to be near the reacting species. Without such care, even small misalignments could lead to alternate off-pathway chemistry. In this case, in the absence of reductant, the proximity of the carboxylate to the activated dAdo promotes group transfer. However, in the presence of reductant, 6-CP undergoes radical addition with dAdo•.
Figure 5.
Divergent fates of 6-CP incubated with CDG synthase. In the absence of reductant, the analogue is adenosylated by SAM to form a C–O bond. In the presence of reductant, radical addition followed by oxidative decarboxylation leads to a C–C linked dAdo adduct.
As with other radical SAM enzymes, the mechanism by which CDG synthase is reductively activated to cleave SAM remains unknown. Activation of many radical SAM enzymes can be accomplished by chemical means (dithionite, titanium citrate) or enzymatically using flavodoxin. The E. coli flavodoxin has been used successfully in many systems (see chapters in ref (28)). However, the structural data on the three homologues of CDG synthase show that the proteins have very distinct surfaces near the iron–sulfur cluster (see Figure 9 in ref (26)). It is important to note that E. coli and many other organisms encode two homologues of flavodoxin. In E. coli, deletion of fldA is lethal, whereas fldB is not essential. The two flavodoxin homologues likely perform non-overlapping functions, as overexpression of fldB does not rescue the lethality associated with the loss of fldA.29 To date, only the product of the fldA gene has been used extensively in studies of radical SAM enzymes. The B. subtilis genome also encodes two flavodoxin homologues (ykuN and ykuP). My postdoctoral fellow Nate Bruender carried out biochemical studies which show that while both can activate CDG synthase, one (YkuN) is more effective than the other (YkuP).30 Studies exploring the structure–activity relationships in the activation pathway and, more generally, those aimed at identifying the physiological electron donors for radical SAM enzymes remain an underexplored area and an important future direction of the field.
Discovery of TYW1
Wybutosine (yW) and its derivatives are modified bases found in the anticodon loop of tRNAPhe in higher organisms.31,32 Prior to our work, the modified base was shown to be introduced by a series of five enzymes in Saccharomyces cerevisiae(31,33) (Figure 6). However, the key enzyme that introduces the imidazoline moiety, TYW1, had eluded in vitro studies because the source of the two carbons that constitute the imidazoline moiety was unknown. In 2011, a new graduate student, Anthony Young, joined the group, and one of his projects was to identify the substrate for TYW1. Our initial hypothesis for the two-carbon donor focused mainly on biologically common intermediates, such as acetyl CoA and acetyl phosphate. When we were unable to see any products after multiple attempts, the search expanded to PEP but with no effect. At this point, Anthony suggested to me that pyruvate was the source of the two carbons incorporated into yW. If memory serves me correctly, I told him pyruvate was probably not the two-carbon source and that it was likely PEP, which is more activated, but that he should try it since pyruvate is inexpensive. Of course, as with any story that starts with the advisor telling a student something was not going to work, the outcome was the opposite. PEP did not work, but pyruvate did.34 In subsequent studies, studies with various isotopically enriched forms of pyruvate and N1-methylguanosine (m1G) have led to a detailed picture of the source of carbon and hydrogen atoms that are found in the newly formed ring35 (summarized in Figure 6).
Figure 6.
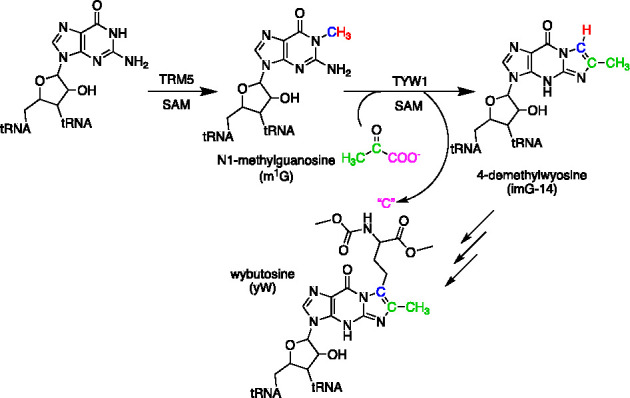
Biosynthesis of wybutosine. The biosynthetic pathway to yW consists of five SAM-dependent steps. TYW1 catalyzes the second step, which entails condensation of pyruvate with m1G to form imG-14. The reaction product retains C-2 and C-3 of pyruvate. The fate of C-1 of pyruvate remains to be established.
At the time, we initiated work on this protein, and two X-ray crystal structures of TYW1 homologues from thermophilic organisms had been published.36,37 Unlike CDG synthase, which has a single 4Fe–4S cluster, TYW1 had been predicted to have multiple Fe–S clusters. However, neither of the two published structures showed either cluster or provided any clues about the mechanism of this enzyme.
We initiated a collaboration with the Drennan lab to solve the crystal structure of TYW1. Tsehai Grell solved multiple X-ray crystal structures of the protein, which clearly showed electron density for two clusters.38 Unfortunately, in all of the structures that she was able to solve, the SAM-binding cluster appeared to be partially occupied, presumably because of the sensitivity of the cluster toward degradation during the extended periods of time that are required to obtain crystals and diffraction data. However, she noted that as was predicted based on conserved Cys residues and Fe/S occupancy, a second 4Fe–4S cluster was present on the opposite end of the active site from the RS cluster.
While the density for the second cluster was clear in the data, it was also clear that the cluster, which had three Cys thiolates ligated to three iron ions, appeared to interact with an unusual ligand. The fourth iron ion of this cluster appeared to be bound to an unknown entity, with density that was contiguous with the side chain of Lys41 (Methanococcus jannaschii numbering). This was an intriguing observation, as in vivo experiments with the M. jannaschii homologue had shown that this Lys residue is essential for activity.36 In our initial paper reporting the discovery of pyruvate as a substrate for TYW1, we had hypothesized that this conserved Lys could be binding the pyruvate as an imine, facilitating the transformation through covalent catalysis.34 Intriguingly, the additional positive electron density in the model could be readily interpreted to arise from modification of the Lys side chain by pyruvate to form an imine, but because of the resolution of the structure, the oxidation state of the adduct could not be determined unambiguously.
The X-ray crystallographic observation of the Lys41 adduct was further confirmed by LC-MS/MS studies after proteolytic digestion of the protein.38 Intriguingly, we observed a reduced imine in the as-purified protein. This suggests that the adduct observed in the structural studies was likely in the reduced form, as well. The mechanism by which this reduction occurs is not known. However, when TYW1 was incubated with various isotopologues of pyruvate, followed by reduction to stabilize the imine, we clearly observed adducts that were formed by chemistry between TYW1 and the proffered pyruvate.
The structural and biochemical studies support a catalytic mechanism (Figure 7), whereby pyruvate forms a Schiff base with the conserved Lys residue. The adduct interacts with the auxiliary cluster via the imino nitrogen and carboxylate moieties. The catalytic cycle is initiated by the reductive cleavage of SAM generates a dAdo•, which abstracts a hydrogen atom from the methyl group of m1G. The ensuing radical addition to the pyruvate leads to a tRNA–pyruvate adduct, which can be resolved by cluster-assisted formation of a HCOOH or CO2 from C-1 of pyruvate. ImG-14 forms following transimination, which also regenerates the active site Lys residue.
Figure 7.

Mechanism of TYW1. The enzyme binds pyruvate as a Schiff base to a conserved Lys residue in the active site. The substrate is activated for radical addition by H atom transfer from m1G to dAdo•. Radical addition is followed by C–C bond cleavage to release C-1 of the pyruvate as a yet unknown product. Transimination and aromatization complete the catalytic cycle.
In retrospect, one of the reasons I had been intrigued with TYW1 in the first place was because there were indications that the protein, at least in eukaryotic organisms, is present as fusion between a flavodoxin domain and the radical SAM domain. Perhaps it was fortuitous that all the initial work could be done with the archaea homologues of TYW1, which are by far easier to express and purify. However, the role of the flavodoxin domain in TYW1 had remained a mystery that we were tackling in parallel, though until recently without success.
Many of our initial attempts to heterologously express the eukaryotic TYW1 either yielded either insoluble protein or no protein at all. The breakthrough was when we introduced a second plasmid encoding the suf operon in the expression strains. This led to the purification of the Saccharomyces cerevisiae TYW1 (ScTYW1), which is now the first characterized radical SAM flavoenzyme.39 After initially establishing that the protein carries out the same reaction as the archaeal homologue, we focused on the role of the flavodoxin domain. Remarkably, the formation of imG-14 with ScTYW1 requires only the presence of NAD(P)H, pyruvate, and m1G-containing substrate. The flavodoxin domain is presumably able to catalyze the reduction of the enzyme, from electrons derived from the pyridine nucleotide to support turnover. To our knowledge, this is the first instance where activation of a radical SAM enzyme does not require a strong reductant, such as dithionite or an additional protein. Future studies on this enzyme will delineate the role of the flavodoxin domain in catalysis, as this protein affords a unique window into the reductive activation mechanism, which is taking place in trans.
Studies on Peptide Maturases and Discovery of SbtM
My interest in RS peptide maturases was piqued in by a paper by Dan Haft, in which using computational methods he discovered a series of RS enzymes that appeared to colocalize with short protein-coding orfs.40 I had the good fortune to attend the 2012 GRC on Protein Cofactors, Radicals and Quinones, where I heard him speak. If memory serves me correctly, Judith Klinman was the one who introduced me to Dan at the conference, and we had several phone conversations on these enzymes after the meeting. I was intrigued by the biosynthetic clusters that were highlighted in the Haft paper and tasked a new postdoctoral fellow, Nathan Bruender, to begin characterizing them. Our work on RS RiPP maturases intensified after my move from Arizona to the Department of Chemistry at the University of Utah. In my lab, Nate worked on the biogenesis of several thioether-containing natural products and mycofactocin, a natural product whose presence was first suggested by Haft. As the details of these systems are the subject of other contributions in this issue, I will highlight only the most recent discovery of a selenium-containing RiPP.
Of the systems highlighted by Haft in his study,40 one stood out to us because it was proposed that it likely involved a selenium-containing precursor peptide. In the original paper, Haft showed that the radical SAM homologue, sbtM, appeared to colocalize with a peptide that had all the hallmarks of a selenium-containing protein. The gene encoded a UAG stop codon and a selenocysteine insertion sequence immediately downstream of the amber codon. More recent annotations suggest that the peptide substrate is, in fact, significantly longer than initially proposed and the natural product likely has at least two SeCys residues. The maturase/peptide pair appears to be limited to strains of Geobacter and Desulfbacteria. The SeCys is generally embedded in a conserved GGUG motif.
One of my graduate students, Julia Lewis, expressed an interest in characterizing this protein. To simplify the initial analysis of the peptide, we synthesized a minimal peptide by solid-phase peptide synthesis with a Cys instead of SeCys. Incubation of the peptide with the maturase led to a loss of 4 amu from the peptide. Remarkably, the modification is accompanied by a peak at 304 nm for the peptide. The same modification occurs with SeCys, and the UV–visible feature of the peptide red shifts by 9 nm. MS/MS sequencing localized the modification to within the conserved −Gly–Cys(SeCys)– motif of the peptide. The structure of the modified peptide was explored by a series of isotopically labeled analogues. Incubation of SbtM with a peptide containing deuterated Gly revealed that the Gly retains the deuteriums. By contrast, when Cys that is deuterated at the β-carbon was used, we observed a loss of 6 amu. Analysis of the resulting dAdo from the reaction mixtures showed incorporation of deuterium from the β-carbon of Cys, supporting the intermediacy of H atom transfer from the peptide to dAdo•.
Additional structural insights were obtained from NMR. 1H HSQC analysis of the modified peptide with 15N-Cys and 15N-Gly confirmed that, by contrast to the amide hydrogen of the Gly, that of the Cys is not retained in the product. Additionally, the NMR of the 13C-containing Cys revealed three peaks in the aromatic region (165, 120, and 118 ppm), suggesting that the modification entails the formation of an aromatic structure. These resonances are similar to those that have been observed with peptidyl oxazoles previously.41,4213C HSQC revealed a correlation between the protons on the Gly and the 165 ppm resonance, which is derived from the peptide carboxyl carbon of the Gly. As with the unmodified peptide, the modified peptide retains the ability to react with iodoacetamide. These observations collectively suggest that SbtM catalyzes the radical-mediated formation of a thio(seleno)oxazole in the peptide. The current working model for this transformation involves undergoing two rounds of oxidation (Figure 8). In the first, the Cys is oxidized, allowing for the formation of a thio(seleno)oxazoline. In the second, the thio(seleno)oxazoline is converted to the observed product. Many aspects of this reaction remain to be established, but at least on the surface, SbtM appears to catalyze a transformation that is similar to peptide anaerobic sulfatase maturing enzyme (AnSME, oxidation of a thiol), but that formally, this reactivity supports formation of an oxazole in the peptide. To our knowledge, this is the first SeCys-containing RiPP natural product installed by a RS enzyme.
Figure 8.

Reaction catalyzed by SbtM. The reaction catalyzed by SbtM is hypothesized to occur via oxidation of the Cys/SeCys side chain, followed by ring formation and an additional oxidation step to form the product. Each of the oxidation steps are thought to utilize SAM and involve H atom transfer from the β-carbon of the side chain to dAdo•. A thio/selenoaldehyde-like intermediate is proposed on the pathway to the final product.
SbtM contains several conserved Cys residues, and the purified protein has enough Fe/S to make up three 4Fe–4S clusters. With this enzyme, as with many of the others RS enzymes, it appears that there are likely multiple Fe/S clusters. Three of the Cys residues are within the conserved CxxxCxxC motif, which binds and activates SAM. The other two are likely in the SPASM domain, which has been shown biochemically and structurally to be responsible for binding the auxiliary clusters in other proteins.43,44 In SbtM, we favor a model where one of these auxiliary clusters binds the Cys residue and activates it for oxidation. This is like what is observed with other RS enzymes, such as the thioether-forming RiPPs, wherein this cluster has also been proposed to be involved in oxidation (see, for example, refs (45) and (46)). The other is likely involved in shuttling reducing electrons to or from the active site. The discovery of the function of SbtM highlights the many opportunities that remain in the RS field, where there are likely dozens of other systems to be discovered and characterized. In the case of most of the RS-encoding RiPP maturase producing clusters, the biological function(s), or for that matter, the fully formed product(s) are not known and highlight an additional area of opportunity in future studies.
Negative Catalysis and Radical-Mediated Transformations
In the 20 years since the recognition that radical SAM enzymes represent a large superfamily, it has become increasingly clear that these groups of enzymes catalyze complex transformations that very often have very few (if any) counterparts in polar chemistry. In general, these enzymes appear to have evolved to catalyze transformations that occur at unactivated carbon atoms. In nearly all cases, the reaction is initiated by reductive cleavage of SAM, which is a high-energy process. Solution electrochemical measurements suggest that the peak potential for the one-electron cleavage of SAM to produce dAdo• is approximately −1.4 V (versus SHE).47 The dAdo•, in turn, activates the substrate to generate an additional high-energy intermediate on the substrate.
Given the energetically unfavorable nature of these unstable species, one would have expected significant “off-pathway” reactions to take place. To be sure, the cleavage of SAM leading to dAdo in what has been termed “abortive cleavage” is one such off-pathway event which results from H atom abstraction from an alternate site when the cleavage of SAM occurs in the absence of substrate or if the substrate is somehow not in a productive conformation. Recent reports suggest that one can “trap” these unstable intermediates by various means, such as through reaction with spin traps or dithionite.48 Biochemical studies of radical SAM enzymes with alternate substrates are rare. However, in cases where they have been accessible, the observed products follow from the known reactivity of the intermediate. For example, when my graduate student Will Kincannon incubated SkfB, which forms a thioether between a Cys residue and the α-carbon of a methionine, with a substrate where cyclopropylglycine replaces the methionine. In this reaction, instead of forming a thioether, the cyclopropane ring opening results.49 This outcome is precisely what one would expect from the physical organic chemistry literature, where cyclopropane moieties have been used extensively to study radical reactions.50 Additionally, the study with 6-CP with CDG synthase highlighted above is another example of “unleashing” alternative reactivity when substrate structure is altered.25,27 Both studies were inspired by work from the Frey and Reed on lysine 2,3-aminomutase, whereby substrate analogues were used to pinpoint key aspects of mechanism.3,5
Perhaps the remarkable selection against alternative or off-pathway products is a result of the exquisite control that these enzymes exercise on the position of the substrate relative to the dAdo•. Where available, 3D structures of radical SAM enzymes show that the 5′-carbon of the dAdo moiety in SAM is within van der Waals distance of the hydrogen atom to be abstracted. Upon activation, the reactions likely require little by way of enzymatic intervention or movement to form product. The binding energy of the substrate and cofactor is leveraged toward preorganizing the active site for the reaction and minimizing off pathway intermediates. This is reminiscent of the negative catalysis concept that Rétey introduced to explain the exquisite specificity and absence of significant off pathway reactions by B12-dependent enzymes.51
Lessons Learned
In the two decades since the founding of the RS superfamily by Sofia1 and the insightful studies by Moss and Frey,52,53 prior to that leading to the identification of common mechanistic features of the RS superfamily (see ref (54) for review), we have learned a great deal about many aspects of catalysis by RS enzymes. However, I would venture to guess that none of the contributors to this issue would contest the statement that these enzymes are temperamental, slow, and generally a pain in the ass to work with! This is because while we are a great deal savvier about how to treat these enzymes, there are many aspects of the function of RS enzymes that we have yet to understand.
The first roadblock to studying a RS enzyme is knowing the substrate. We were fortunate that with CDG synthase, we had a good guess of the substrate based on a knowledge of the functions of the three preceding enzymes in the pathway. However, the discovery of pyruvate as the substrate for TYW1 was good fortune. The studies on the RiPP maturase enzymes are facilitated by the presence of the orfs in the biosynthetic clusters that encode the RS enzyme, which are suggestive of at least the precursor to the natural product. However, I would predict, in many cases that are yet to be discovered, that it is not the peptide substrate that will be a challenge but identifying the second or third substrate that is required in the biosynthetic pathway. Since in most of the cases the structure of the mature natural product is not known, one has very little to guide the search to the required substrates. However, recent advances in bioinformatics tools that permit visualization of genome neighborhoods should be very helpful in at least providing possible candidates (see, for example, radicalsam.org). This approach was recently applied to the discovery of the function of viperin, for example.55
The second hurdle is obtaining an enzyme preparation that is replete with the required cofactors. We and others are constantly hampered by the challenges of obtaining cofactor-replete proteins, particularly in cases where multiple clusters are present. Many organisms have overlapping systems for incorporating Fe and S into metalloenzymes, and it is often not clear which one would be best for heterologous overproduction. An additional complication is that, while many of the auxiliary clusters discovered to date are 4Fe–4S clusters, those in biotin synthase,56 MiaB,57 and probably many others are not. In some, the auxiliary clusters undergo degradation as part of the catalytic cycle, necessitating a rebuild on each turnover. There have been recent advances in some of these cases, such as the role of NfuA in lipoic acid synthase,58,59 in understanding the rebuilding process. However, we are a long way from having robust catalytic turnover in many cases. I recently edited an entire volume of Methods in Enzymology devoted to practical aspects of working with RS enzymes.28 Perhaps the most striking conclusion to emerge from the contributions is the diversity in preparation and reconstitution methods that are employed in each case, underscoring the challenge of working on these systems.
Even if one can obtain sufficient quantities of cofactor-replete protein, it is often challenging to see any activity in these proteins because the details of the reductive activation step are not known. In most cases, for example, we do not know the identity of the in vivo reducing system that activates the radical SAM enzyme. Considering that many use SAM stoichiometrically and require activation on each turnover, this is a significant challenge. Strong reductants, such as dithionite, come with their own problems, which include reactivity of the reductant with the unstable intermediates that are being produced or, more worrisome, the possibility for over-reduction of the protein rendering it catalytically inactive. This is particularly concerning with enzymes that, in addition to the radical SAM cluster, also have one or more auxiliary clusters. There is, a priori, no reason to expect that the reduced form of all these clusters is required during turnover.
The E. coli flavodoxin/flavodoxin reductase pair is often used to achieve reductive activation with electrons derived from NADPH. This is based on work by Knappe and co-workers that established this system as useful for activation of ribonucleotide reductase activase.60 However, consider that there are two flavodoxin homologues in E. coli and other organisms. In our studies, we found differences between the two flavodoxins from B. subtilis in their ability to support the activity of the B. subtilis CDG synthase.30 The reductive activation requires binding of the flavodoxin to the RS enzyme, in such a way as to promote reduction of the cluster by the bound flavin. It is unreasonable to assume that the surfaces of RS proteins from every organism will be compatible with FldA, which is the flavodoxin that is most commonly employed in studies published on RS enzymes. Indeed, experimental evidence with three CDG synthase homologues suggests significant differences in surface profiles.26
In summary, while great strides have been made by my lab and others in characterizing these proteins, many challenges, in vivo cofactor reconstitution, reductive activation, and the identification of the substrates, hamper more rapid progress.
Outlook
“At nyght was come into that hostelrye
Wel nyne and twenty in a compaignye
Of sondry folk, by aventure yfalle
In felaweship, and pilgrimes were they alle,
That toward Caunterbury wolden ryde.”
Geoffrey Chaucer, Prologue,”Canterbury Tales”
Each of us have arrived at the study of RS enzymes by a unique route. Whether as Chaucer would have said by fate (aventure) or by chance (cas), we are pilgrims on a road toward a more complete understanding of the complex and fascinating reactions that are catalyzed by the RS enzymes. In the last 20 years, I have learned that the best way to approach these systems is with patience. To borrow a sentiment from Martin Kamen, “a cupful of luck and a pinch of sagacity”61 are the most useful ingredients on this pilgrimage. As I have indicated above, there are many unknowns in this field, which will provide opportunities for future discovery. While in the last 20 years, we have really been in “discovery” mode, it is perhaps an understatement that we are on the first stop of the pilgrimage toward a complete understanding of structure, mechanism, and function. As we gather at this first stop, around a fire to tell our stories, it is exciting to imagine the many new and unanticipated stops that remain and the tales that are yet to be written by us and others who join us on this pilgrimage.
Acknowledgments
V.B. gratefully acknowledges lab members for proofreading the many versions of this manuscript. This manuscript is dedicated to my late student Reid McCarty, whose early contributions were critical to this journey.
Glossary
Abbreviations
- CDG
7-carboyx-7-deazaguanine
- 6-CP
6-carboxypterin
- CPH4
6-carboxy-5,6,7,8-tetrahydropterin
- dAdo
5′-deoxyadenosine
- dAdo•
5′-deoxyadenosyl radical
- orf
open reading frame
- SAM
S-adenosyl-l-methionine
- Q
queuosine
- oQ
epoxyqueuosine
- RiPP
ribosomally synthesized and posttranslationally modified polypeptide
- yW
wybutosine
National Institute of General Medical Sciences of the National Institutes of Health (Grant R35 GM126956 to V.B.).
The author declares no competing financial interest.
References
- Sofia H. J.; Chen G.; Hetzler B. G.; Reyes-Spindola J. F.; Miller N. E. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001, 29, 1097–1106. 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinger M. D.; Reed G. H.; Frey P. A. An organic radical in the lysine 2,3-aminomutase reaction. Biochemistry 1992, 31, 949–953. 10.1021/bi00119a001. [DOI] [PubMed] [Google Scholar]
- Wu W.; Lieder K. W.; Reed G. H.; Frey P. A. Observation of a second substrate radical intermediate in the reaction of lysine 2,3-aminomutase: a radical centered on the beta-carbon of the alternative substrate, 4-thia-L-lysine. Biochemistry 1995, 34, 10532–10537. 10.1021/bi00033a027. [DOI] [PubMed] [Google Scholar]
- Ballinger M. D.; Frey P. A.; Reed G. H. Structure of a substrate radical intermediate in the reaction of lysine 2,3-aminomutase. Biochemistry 1992, 31, 10782–10789. 10.1021/bi00159a020. [DOI] [PubMed] [Google Scholar]
- Miller J.; Bandarian V.; Reed G.; Frey P. Inhibition of lysine 2,3-aminomutase by the alternative substrate 4-thialysine and characterization of the 4-thialysyl radical intermediate. Arch. Biochem. Biophys. 2001, 387, 281–288. 10.1006/abbi.2001.2261. [DOI] [PubMed] [Google Scholar]
- McCarty R. M.; Bandarian V. Biosynthesis of pyrrolopyrimidines. Bioorg Chem. 2012, 43, 15–25. 10.1016/j.bioorg.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura H.; Katagiri K.; Sato K.; Mayama M.; Shimoka N. Toyocamycin, a new anti-candida antibiotics. J. Antibiot. 1956, 9, 60–62. [PubMed] [Google Scholar]
- Kasai H.; Kuchino Y.; Nihei K.; Nishimura S. Distribution of the modified nucleoside Q and its derivatives in animal and plant transfer RNA’s. Nucleic Acids Res. 1975, 2, 1931–1940. 10.1093/nar/2.10.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RajBhandary U. L., Chang H. J., Gross F., Harada F., Kimura S., Nishimura S.. E. coli tyrosine transfer RNA-primary sequence and direct evidence for base pairing between terminal sequences. Fed Proc. Fed Amer Soc. Exp Biol. 1969, 28. [Google Scholar]
- Harada F.; Nishimura S. Possible anticodon sequences of tRNA His, tRNA Asn, and tRNA Asp from Escherichia coli B. Universal presence of nucleoside Q in the first poistion of the anticondons of these transfer ribonucleic acids. Biochemistry 1972, 11, 301–308. 10.1021/bi00752a024. [DOI] [PubMed] [Google Scholar]
- Gregson J.; Crain P.; Edmonds C.; Gupta R.; Hashizume T.; Phillipson D.; McCloskey J. Structure of the archaeal transfer RNA nucleoside G*-15 (2-amino-4,7-dihydro- 4-oxo-7-beta-D-ribofuranosyl-1H-pyrrolo[2,3-d]pyrimidine-5-carboximidamide (archaeosine)). J. Biol. Chem. 1993, 268, 10076–10086. 10.1016/S0021-9258(18)82174-3. [DOI] [PubMed] [Google Scholar]
- Suhadolnik R. J.; Uematsu T. Biosynthesis of the pyrrolopyrimidine nucleoside antibiotic, toyocamycin. VII. Origin of the pyrrole carbons and the cyano carbon. J. Biol. Chem. 1970, 245, 4365–4371. 10.1016/S0021-9258(19)63804-4. [DOI] [PubMed] [Google Scholar]
- Uematsu T.; Suhadolnik R. J. Nucleoside antibiotics. VI. Biosynthesis of the pyrrolopyrimidine nucleoside antibiotic toyocamycin by Streptomyces rimosus. Biochemistry 1970, 9, 1260–1266. 10.1021/bi00807a030. [DOI] [PubMed] [Google Scholar]
- Kuchino Y.; Kasai H.; Nihei K.; Nishimura S. Biosynthesis of the modified nucleoside Q in transfer RNA. Nucleic Acids Res. 1976, 3, 393–398. 10.1093/nar/3.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reader J. S.; Metzgar D.; Schimmel P.; de Crécy-Lagard V. Identification of four genes necessary for biosynthesis of the modified nucleoside queuosine. J. Biol. Chem. 2004, 279, 6280–6285. 10.1074/jbc.M310858200. [DOI] [PubMed] [Google Scholar]
- Uematsu T.; Suhadolnik R. Toyocamycin nitrile hydrolase. Meth Enzymol 1975, 43, 759–762. 10.1016/0076-6879(75)43143-3. [DOI] [PubMed] [Google Scholar]
- McCarty R. M.; Bandarian V. Deciphering deazapurine biosynthesis: pathway for pyrrolopyrimidine nucleosides toyocamycin and sangivamycin. Chem. Biol. 2008, 15, 790–798. 10.1016/j.chembiol.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles Z. D.; Myers W. K.; Kincannon W. M.; Britt R.; Bandarian V. Biochemical and Spectroscopic Studies of Epoxyqueuosine Reductase: A Novel Iron-Sulfur Cluster- and Cobalamin-Containing Protein Involved in the Biosynthesis of Queuosine. Biochemistry 2015, 54, 4927–4935. 10.1021/acs.biochem.5b00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles Z. D.; McCarty R. M.; Molnar G.; Bandarian V. Discovery of epoxyqueuosine (oQ) reductase reveals parallels between halorespiration and tRNA modification. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 7368–7372. 10.1073/pnas.1018636108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling D. P.; Miles Z. D.; Köhrer C.; Maiocco S. J.; Elliott S. J.; Bandarian V.; Drennan C. L. Molecular basis of cobalamin-dependent RNA modification. Nucleic Acids Res. 2016, 44, 9965–9976. 10.1093/nar/gkw806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty R. M.; Somogyi A.; Bandarian V. Escherichia coli QueD is a 6-carboxy-5,6,7,8-tetrahydropterin synthase. Biochemistry 2009, 48, 2301–2303. 10.1021/bi9001437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty R. M.; Somogyi A.; Lin G.; Jacobsen N. E.; Bandarian V. The deazapurine biosynthetic pathway revealed: in vitro enzymatic synthesis of PreQ(0) from guanosine 5′-triphosphate in four steps. Biochemistry 2009, 48, 3847–3852. 10.1021/bi900400e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty R. M.; Krebs C.; Bandarian V. Spectroscopic, steady-state kinetic, and mechanistic characterization of the radical SAM enzyme QueE, which catalyzes a complex cyclization reaction in the biosynthesis of 7-deazapurines. Biochemistry 2013, 52, 188–198. 10.1021/bi301156w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling D. P.; Bruender N. A.; Young A. P.; McCarty R. M.; Bandarian V.; Drennan C. L. Radical SAM enzyme QueE defines a new minimal core fold and metal-dependent mechanism. Nat. Struct. Biol. 2014, 10, 106–112. 10.1038/nchembio.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruender N. A.; Grell T. A.; Dowling D. P.; McCarty R. M.; Drennan C. L.; Bandarian V. 7-Carboxy-7-deazaguanine Synthase: A Radical S-Adenosyl-L-methionine Enzyme with Polar Tendencies. J. Am. Chem. Soc. 2017, 139, 1912–1920. 10.1021/jacs.6b11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grell T. A. J. A.; Bell B. N.; Nguyen C.; Dowling D. P.; Bruender N. A.; Bandarian V.; Drennan C. L. Crystal structure of AdoMet radical enzyme 7-carboxy-7-deazaguanine synthase from Escherichia coli suggests how modifications near [4Fe-4S] cluster engender flavodoxin specificity. Protein Sci. 2019, 28, 202–215. 10.1002/pro.3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcoxen J.; Bruender N. A.; Bandarian V.; Britt D. R. A Radical Intermediate in Bacillus subtilis QueE during Turnover with the Substrate Analogue 6-Carboxypterin. J. Am. Chem. Soc. 2018, 140, 1753–1759. 10.1021/jacs.7b10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandarian V., Ed. Radical SAM enzymes; Elsevier, 2018. [Google Scholar]
- Gaudu P.; Weiss B. Flavodoxin Mutants of Escherichia coli K-12. J. Bacteriol. 2000, 182, 1788–1793. 10.1128/JB.182.7.1788-1793.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruender N. A.; Young A. P.; Bandarian V. Chemical and Biological Reduction of the Radical SAM Enzyme 7-Carboxy-7-deazaguanine [corrected] Synthase. Biochemistry 2015, 54, 2903–2910. 10.1021/acs.biochem.5b00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A.; Kirino Y.; Ikeuchi Y.; Suzuki T. Biosynthesis of wybutosine, a hyper-modified nucleoside in eukaryotic phenylalanine tRNA. EMBO J. 2006, 25, 2142–2154. 10.1038/sj.emboj.7601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Crécy-Lagard V.; Brochier-Armanet C.; Urbonavičius J.; Fernandez B.; Phillips G.; Lyons B.; Noma A.; Alvarez S.; Droogmans L.; Armengaud J.; Grosjean H. Biosynthesis of Wyosine Derivatives in tRNA: An Ancient and Highly Diverse Pathway in Archaea. Mol. Biol. Evol. 2010, 27, 2062–2077. 10.1093/molbev/msq096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waas W. F.; de Crécy-Lagard V.; Schimmel P. Discovery of a gene family critical to wyosine base formation in a subset of phenylalanine-specific transfer RNAs. J. Biol. Chem. 2005, 280, 37616–37622. 10.1074/jbc.M506939200. [DOI] [PubMed] [Google Scholar]
- Young A. P.; Bandarian V. Pyruvate is the source of the two carbons that are required for formation of the imidazoline ring of 4-demethylwyosine. Biochemistry 2011, 50, 10573–10575. 10.1021/bi2015053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A. P.; Bandarian V. Mechanistic Studies of the Radical S-Adenosyl-L-methionine Enzyme 4-Demethylwyosine Synthase Reveal the Site of Hydrogen Atom Abstraction. Biochemistry 2015, 54, 3569–3572. 10.1021/acs.biochem.5b00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y.; Noma A.; Suzuki T.; Senda M.; Senda T.; Ishitani R.; Nureki O. Crystal structure of the radical SAM enzyme catalyzing tricyclic modified base formation in tRNA. J. Mol. Biol. 2007, 372, 1204–1214. 10.1016/j.jmb.2007.07.024. [DOI] [PubMed] [Google Scholar]
- Goto-Ito S.; Ishii R.; Ito T.; Shibata R.; Fusatomi E.; Sekine S. I.; Bessho Y.; Yokoyama S. Structure of an archaeal TYW1, the enzyme catalyzing the second step of wye-base biosynthesis. Acta crystallographica. Section D, Biological crystallography 2007, 63, 1059–1068. 10.1107/S0907444907040668. [DOI] [PubMed] [Google Scholar]
- Grell T. A.; Young A. P.; Drennan C. L.; Bandarian V. Biochemical and Structural Characterization of a Schiff Base in the Radical-Mediated Biosynthesis of 4-Demethylwyosine by TYW1. J. Am. Chem. Soc. 2018, 140, 6842–6852. 10.1021/jacs.8b01493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A. P.; Bandarian V. Eukaryotic TYW1 Is a Radical SAM Flavoenzyme. Biochemistry 2021, 60, 2179–2185. 10.1021/acs.biochem.1c00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haft D. H.; Basu M. Biological systems discovery in silico: radical S-adenosylmethionine protein families and their target peptides for posttranslational modification. J. Bacteriol. 2011, 193, 2745–2755. 10.1128/JB.00040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila A.; Andsten R.-M.; Jumppanen M.; Assante M.; Jokela J.; Wahlsten M.; Mikula K. M.; Sigindere C.; Kwak D. H.; Gugger M.; Koskela H.; Sivonen K.; Liu X.; Yli-Kauhaluoma J.; Iwaï H.; Fewer D. P. Biosynthesis of the Bis-Prenylated Alkaloids Muscoride A and B. ACS Chem. Biol. 2019, 14, 2683–2690. 10.1021/acschembio.9b00620. [DOI] [PubMed] [Google Scholar]
- Brachmann A. O.; Probst S. I.; Rüthi J.; Dudko D.; Bode H. B.; Piel J. A Desaturase-Like Enzyme Catalyzes Oxazole Formation in Pseudomonas Indolyloxazole Alkaloids. Angewandte Chemie Int. Ed 2021, 60, 8781–8785. 10.1002/anie.202014491. [DOI] [PubMed] [Google Scholar]
- Dowling D. P.; Vey J. L.; Croft A. K.; Drennan C. L. Structural diversity in the AdoMet radical enzyme superfamily. Biochim. Biophys. Acta 2012, 1824, 1178–1195. 10.1016/j.bbapap.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grell T. A.; Goldman P. J.; Drennan C. L. SPASM and Twitch Domains in S-adenosylmethionine (SAM) Radical Enzymes. J. Biol. Chem. 2015, 290, 3964–3971. 10.1074/jbc.R114.581249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluhe L.; Knappe T. A.; Gattner M. J.; Schafer A.; Burghaus O.; Linne U.; Marahiel M. A. The radical SAM enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat. Chem. Biol. 2012, 8, 350–357. 10.1038/nchembio.798. [DOI] [PubMed] [Google Scholar]
- Flühe L.; Burghaus O.; Wieckowski B. M.; Giessen T. W.; Linne U.; Marahiel M. A. Two [4Fe-4S] clusters containing radical SAM enzyme SkfB catalyze thioether bond formation during the maturation of the sporulation killing factor. J. Am. Chem. Soc. 2013, 135, 959–962. 10.1021/ja310542g. [DOI] [PubMed] [Google Scholar]
- Miller S. A.; Bandarian V. Analysis of Electrochemical Properties of S-Adenosyl-l-methionine and Implications for Its Role in Radical SAM Enzymes. J. Am. Chem. Soc. 2019, 141, 11019–11026. 10.1021/jacs.9b00933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S.; Fedoseyenko D.; Sharma V.; Nesbit M. A.; Britt R. D.; Begley T. P. Menaquinone Biosynthesis: New Strategies to Trap Radical Intermediates in the MqnE-Catalyzed Reaction. Biochemistry 2021, 60, 1642–1646. 10.1021/acs.biochem.1c00181. [DOI] [PubMed] [Google Scholar]
- Kincannon W. M.; Bruender N. A.; Bandarian V. A Radical Clock Probe Uncouples H Atom Abstraction from Thioether Crosslink Formation by the Radical S-Adenosyl-L-methionine Enzyme SkfB. Biochemistry 2018, 57, 4816–4823. 10.1021/acs.biochem.8b00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowry V. W.; Lusztyk J.; Ingold K. U. Calibration of a new horologery of fast radical clocks. Ring-opening rates for ring- and α-alkyl-substituted cyclopropylcarbinyl radicals and for the bicyclo[2.1.0]pent-2-yl radical. J. Am. Chem. Soc. 1991, 113, 5687–5698. 10.1021/ja00015a024. [DOI] [Google Scholar]
- Rétey J. Enzymatic Reaction Selectivity by Negative Catalysis or How Do Enzymes Deal with Highly Reactive Intermediates?. Angew. Chem., Int. Ed. Engl. 1990, 29, 355–361. 10.1002/anie.199003551. [DOI] [Google Scholar]
- Frey P. A.; Moss M. L. S-adenosylmethionine and the Mechanism of Hydrogen Transfer in the Lysine 2,3-Aminomutase Reaction. Cold Spring Harb Symp. Quant Biol. 1987, 52, 571–577. 10.1101/SQB.1987.052.01.065. [DOI] [PubMed] [Google Scholar]
- Moss M.; Frey P. A. The role of S-adenosylmethionine in the lysine 2,3-aminomutase reaction. J. Biol. Chem. 1987, 262, 14859–14862. 10.1016/S0021-9258(18)48103-3. [DOI] [PubMed] [Google Scholar]
- Frey P. A. Travels with carbon-centered radicals. 5′-deoxyadenosine and 5′-deoxyadenosine-5′-yl in radical enzymology. Acc. Chem. Res. 2014, 47, 540–549. 10.1021/ar400194k. [DOI] [PubMed] [Google Scholar]
- Gizzi A. S.; Grove T. L.; Arnold J. J.; Jose J.; Jangra R. K.; Garforth S. J.; Du Q.; Cahill S. M.; Dulyaninova N. G.; Love J. D.; Chandran K.; Bresnick A. R.; Cameron C. E.; Almo S. C. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 2018, 558, 610–614. 10.1038/s41586-018-0238-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovitch F.; Nicolet Y.; Wan J. T.; Jarrett J. T.; Drennan C. L. Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science 2004, 303, 76–79. 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esakova O. A.; Grove T. L.; Yennawar N. H.; Arcinas A. J.; Wang B.; Krebs C.; Almo S. C.; Booker S. J. Structural basis for tRNA methylthiolation by the radical SAM enzyme MiaB. Nature 2021, 597, 566–570. 10.1038/s41586-021-03904-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy E. L.; Booker S. J. Destruction and reformation of an iron-sulfur cluster during catalysis by lipoyl synthase. Science 2017, 358, 373–377. 10.1126/science.aan4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy E. L.; Booker S. J. Biochemical Approaches for Understanding Iron-Sulfur Cluster Regeneration in Escherichia coli Lipoyl Synthase During Catalysis. Methods Enzymol 2018, 606, 217–239. 10.1016/bs.mie.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschkowski H. P.; Knappe J.; Ludwig-Festl M.; Neuer G. Routes of Flavodoxin and Ferredoxin Reduction in Escherichia coli. Eur. J. Biochem. 1982, 123, 563–569. 10.1111/j.1432-1033.1982.tb06569.x. [DOI] [PubMed] [Google Scholar]
- Kamen M. D. A Cupful of Luck, A Pinch of Sagacity. Annu. Rev. Biochem. 1986, 55, 1–35. 10.1146/annurev.bi.55.070186.000245. [DOI] [PubMed] [Google Scholar]