

Abstract

Increased interest in the trifluoromethoxy group in organic synthesis and medicinal chemistry has induced a demand for new, selective, general, and faster methods applicable to natural products and highly functionalized compounds at a later stage of hit-to-lead campaigns. Applying pyrylium tetrafluoroborate, we have developed a mechanochemical protocol to selectively substitute the aromatic amino group with the OCF3 functionality. The scope of our method includes 31 examples of ring-substituted anilines, including amides and sulfonamides. Expected SNAr products were obtained in excellent yields. The presented concise method opens a pathway to new chemical spaces for the pharmaceutical industry.
1. Introduction
Introducing fluorine and fluorine-containing substituents into the structures of biologically active molecules is a standard strategy in drug design to modify properties such as acidity or basicity (which influences binding affinity, pharmacokinetics, and bioavailability), lipophilicity, steric properties, conformational constraint and metabolic stability.1−4 According to recent estimates, 20% of prescribed or clinically administered pharmaceuticals contain at least one fluorine atom. Moreover, in general, 30–50% of the most profitable drugs (depending on the sales period) contain fluorine.5,6
Among all the fluorine-containing substituents, the trifluoromethoxy group (OCF3) is the least-investigated and least-understood moiety. However, this “exotic” entity has attracted more and more attention,7 partially due to its specific features. Apart from the high electronegativity8 and excellent lipophilicity,9 in aryl trifluoromethyl ethers, the OCF3 moiety adopts an orthogonal orientation relative to the aromatic ring.10,11 In contrast to CH3, this group is not conjugated to the aromatic ring because the oxygen p-electrons are delocalized in the σ*-orbitals of the C–F bonds.12
In addition to biologically active molecules in medicine and agrochemicals, the OCF3 group can be found in compounds with applications as electro-optical materials.13,14 Moreover, in recent years, significant progress has been made in C–F bond activation strategies.15−20 Selective and efficient monodefluorination in (hetero)aryl di- and trifluoromethyl ethers is possible with frustrated Lewis pair (FLP) chemistry.21,22 This methodology was also shown to be valid for Ar-OCF3 ethers, opening new synthetic possibilities.23 Particularly, the monosubstitution of fluorine in OCF3 by a variety of nucleophiles can provide diversely substituted derivatives (including handles for further reactions) or chain elongation protocols. A transformation sequence that provides an efficient OCF3 introduction method in combination with C–F activation can give access to unexplored chemical space based on the −OCF2– connection. Additionally, such protocols could be potentially realized in a one-pot fashion if only they were independent of each other’s reactants and byproducts. Due to the large interest in the introduction of the difluoromethylenoxy moiety, -OCF2- intermediates are already finding applications as reagents, for example, in oxidative C–H aryloxydifluoromethylation with α,α-difluorophenoxyacetic acids.24
According to known methods, the synthesis of the aryl trifluoromethyl ethers can proceed in several ways, assuming the formation of a C–F, C–OCF3, or O–CF3 bond or a combination thereof. Chronologically, an aryl–OCF3 ether was obtained for the first time by Yagupolskii in 1955.25 In the first step of this method, the substituted anisole derivatives are converted to trichloromethyl intermediates, which are then submitted to a halogen exchange step (Scheme 1a). Similarly, the whole sequence can be realized in one-pot by heating phenols as starting materials in a CCl4/anhydrous HF mixture in a pressure vessel with BF3 as a catalyst.26 The trichloromethyl intermediate can also be obtained from chlorothionoformates,27 however, the high toxicities of those reagents limit their use substantially. The CF3 group can also be “constructed” on the phenol oxygen via nucleophilic fluorination using aryl fluoroformates28,29 or dithiocarbonates as intermediates (Scheme 1b).30−33 Unfortunately, these methods have limited scopes, require harsh conditions, and often suffer from low yields, precluding their use in any late-stage modifications of drug candidates. More versatile and useful methods for the direct formation of the ArO–CF3 bond rely on reagents that deliver the complete CF3 synthon as an electrophile, such as Umemoto’s oxonium reagents34 and Togni’s benziodoxolon reagent II35 (Scheme 1c and d, respectively), or proceed via silver-mediated oxidative trifluoromethylation with the Ruppert-Prakash reagent36 (Scheme 1e), which is formally a source of the nucleophilic CF3 synthon. Additionally, a two-step procedure catalyzed by silver salts was devised by combining O-carboxydifluoromethylation with subsequent decarboxylative fluorination with SelectFluor II (Scheme 1f).37,38
Scheme 1. Known-to-Date Ar-OCF3 Ether Formation Methods.

In the case of OCF3 transfer agents, the radical trifluoromethoxylation of arenes with trifluoromethyl-hypofluorite39 and the nucleophilic reaction of various trifluoromethoxylate salts with arynes40 offer low selectivities and limited scopes. The two-step OCF3 migration method is limited to N-aryl-N-hydroxyl amines that react with Togni’s reagent II to give O-trifluoromethylated adducts, which subsequently undergo thermal-induced migration.41 The direct silver-mediated trifluoromethoxylation of aryl precursors (boronic acids and stannanes) accepts a variety of starting materials and offers good yields (Scheme 1g).42 Additionally, the C–H trifluoromethoxylation of arenes using a transition-metal redox-active catalyst and a •OCF3 radical-generating photoactivated reagent (R1R2N–OCF3) was described (Scheme 1h).43,44 Later, in 2022, Qing and co-workers developed the C–H trifluoromethoxylation of arenes by combining trifluoromethyl 2-pyridyl sulfone with oxygen as a convenient trifluoromethyl source utilizing a unique electrochemical protocol with a graphite anode and a platinum cathode.45 A very interesting approach was proposed that used (hetero)aryldiazonium tetrafluoroborate salts as starting materials (Scheme 1i). Those methods use various trifluoromethyl alkyl- and arylsulfonates (R–SO2–OCF3) to generate the CF3OM salt in situ.46−49 Furthermore, very recently, two other papers worth mentioning were published. Togni and co-workers communicated the straightforward trifluoromethoxylation of aromatic substrates using the bench-stable pyridinium-based trifluoromethoxylation reagent via the non-directed functionalization of C–H bonds utilizing Ru(II)- and Ru(III)-mediated photoredox catalysis.50 This process involves the formation of OCF3 radicals. The work by Hu describes an original concept for the nucleophilic trifluoromethoxylation of alkyl (pseudo)halides and cross-coupling with aryl stannanes, where trifluoromethyl benzoate is used as an efficient and readily available trifluoromethoxylation reagent.51
In contrast to the diazonium salts, which are temperature-unstable, shock-sensitive, explosive, and require the use of strong acids and oxidants for generation, the pyridinium salts can be generated via condensation with pyrylium salts (Pyry-BF4) under relatively mild conditions in ethanol and used in situ in the nucleophilic aromatic substitution.52,53 The pyrylium tetrafluoroborate reagent can be prepared in large quantities and safely stored for long periods. Moreover, Pyry-BF4 selectively activates amino groups in synthetic and natural aminoheterocycles and therefore can also be used in the late-stage modification of drugs and drug candidates.
2. Results and Discussion
Inspired by the recent successful development of a deaminative chlorination protocol for aminoheterocycles that used Pyry-BF4,54 we envisioned that a similar method could be used to introduce the trifluoromethoxy group. In the current paper, we present a new methodology that enables the efficient installation of the OCF3 functionality onto aromatic substrates through the conversion of the NH2 group using a readily available and commercialized pyrylium tetrafluoroborate reagent (Pyry-BF4).
The activation of the C(sp2)–NH2 bond is complicated due to its low nucleophilicity. However, condensation with the pyrylium reagent (Pyry-BF4) gives pyridinium salts as intermediates in good yields for use in aromatic substitution reactions (SNAr).52 The reaction of those salts with a variety of nucleophiles results in C–O, C–N, C–S, and C–SO2R bond formation. Those two steps can be realized in one-pot without isolation of the pyridinium salt. The simplicity and generality of this strategy allow for the selective functionalization of aromatic NH2 groups. Moreover, the Pyry-BF4 reagent is easy to prepare, stable and nowadays commercially available. We envisioned a similar substitution of pyridinium salts with ⊖OCF3.
Our first attempts to realize the title transformation were unsuccessful. The utilization of an isolated pyridinium salt, 1-(4-(methylsulfonyl)phenyl)pyridin-1-ium tetrafluoroborate, as well as one-pot approach starting from (methylsulfonyl)aniline 1e in different solvents (DMF, DMA, dichloromethane, MeOH and acetonitrile) under a range of temperatures (also under reflux) did not result in the formation of the desired substitution product (for more details, see Table S1, entries 13–31 in the SI). Of note, only 1,4-dioxane showed promising results (Table S1, entries 18, 19), as the model compound was isolated in 8% and 32% yields, respectively.
On the basis of our previous experiences and the literature on mechanochemical realizations of SNAr reactions,55−59 and as part of our general policy to search for green methodologies, we conducted a routine experiment under mechanochemical conditions (Table S1, entry 1) in a one-pot fashion, starting from the same substrate 1e. To our content, the evident presence of the target product in the reaction mixture was demonstrated by TLC and confirmed after separation (12% yield). To the best of our knowledge, there are no other successful examples or attempts to realize such SNAr pyridinium salt substitution with the ⊖OCF3 donor in the literature, neither in solution nor in the solid state. Encouraged, we conducted an optimization of the mechanochemical reaction conditions by adjusting the reagent equivalents and the ⊖OCF3 source (see Table S1, entries 1–12). Since some of the starting materials are liquids at r.t. and a molar equivalent of water is produced in the first step of the process (Scheme 4), we applied a grinding auxiliary material to both improve mixing and energy transfer and prevent the reaction mass from forming a gum or paste. Among the following oxides, ZrO2 gave the best yield (19%, 27%, 28%, and 34%, respectively; Table S1, entries 2–5): CeO2, TiO2, Yb2O3 and ZrO2.
Scheme 4. Proposed Mechanism of the Second Step.

The applied ⊖OCF3 sources included tetramethylammonium trifluoromethanolate (3a), 1-methyl-quinuclidin-1-ium trifluoromethanolate (3b), 1-methyl-1,4-diaza-bicyclo[2.2.2]octan-1-ium trifluoromethanolate (3c), 1,1-dimethyl-pyrrolidin-1-ium trifluoromethanolate (3d), tris(dimethylamino)sulfonium trifluoro-methanolate (3e), potassium trifluoromethanolate (3f), rubidium trifluoromethanolate (3g), and cesium trifluoromethanolate (3h) (Scheme 2). The best conditions, comprising 1e (1.0 equiv), Pyry-BF4 (1.1 equiv), ZrO2 (1.0 equiv), and 3c (1.5 equiv) ground at 30 Hz for 90 min, gave a 78% yield (Table S1, entry 7). Of note, the superiority of ZrO2 within this protocol might be explained by the stabilizing effect it has on the parental OCF3 anion, most likely as a result of the interaction of Zr with both the oxygen and fluorine atoms. Four-coordinate Zr has good affinity to aliphatic C–F bonds. It is also known that differences in the structures of the inorganic materials result in different friction energies.
Scheme 2. Optimization of the Reaction Conditions.

With the optimized conditions in hand, we embarked on an assessment of the method’s scope. Several substituted anilines were subjected to the protocol, yielding OCF3-substituted products 4a–4q in 58–92% isolated yields (Scheme 3). The two-step one-pot mechanochemical protocol makes impossible any inquires on the efficiency of consecutive steps without the isolation of every single intermediate pyridinium salt. However, conclusions from the optimization and the literature on pyridinium salt generation and stability52 suggest that the first formal step (the generation of the pyridinium salt) is not the limiting one. On the other hand, one must remember that solid-state reaction kinetics is governed by entirely different rules than in-solution processes.60−63 Evidently, lower yields (Scheme 3) were observed for ortho-substituted starting materials 4l (58%), 4n (65%), 4x (65%), 4g (73%), 4h (78%), 4i (77%), etc. This could be explained by the bulkiness of the adjacent ortho-substituent and the hydrogen bond donor or acceptor properties of the COOH, OH, and C(O)NHR groups, which could interfere with all steps that lead to the desired product. The developed methodology is also feasible on the gram scale; thus, compounds 4e (73%), 4i (72%), 4q (74%), 4s (80%), and 4ae (73%) were prepared in acceptable yields.
Scheme 3. Reaction Scope.
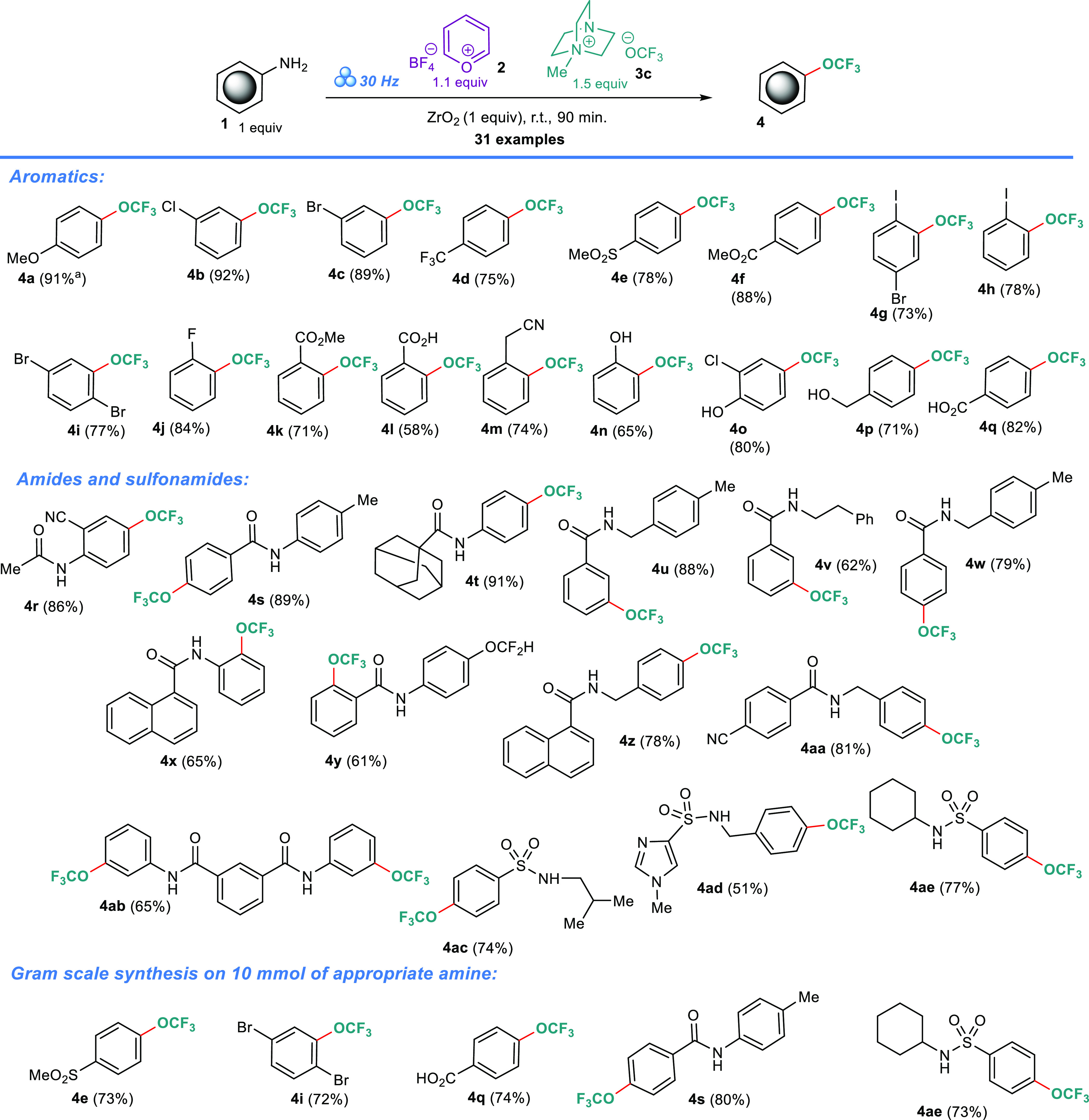
In fact, the higher yield for the ortho-COOMe derivative 4k (71%) compared to that of 4l could suggest that intramolecular hydrogen bond formation stabilizes the starting material, interfering with the first step (Scheme 4, pyridinium salt formation). In contrast, the bulkiness makes the nucleophile approach difficult in the irreversible second step, leading to a quaternary carbon product (Scheme 4b). The nucleophilic attack of ⊖OCF3 on the 2- and 4-positions of the pyridinium ring is reversible, thus those equilibria are not responsible for diminishing the overall process efficacy significantly. However, upon analysis of the TLC and NMR profiles of the crude mixtures, a byproduct of the same type was observed as a trace and identified as the F substitution product. To explain the occurrence of this product, we performed a simple test reaction under mechanochemical conditions starting from [1,1′-biphenyl]-4-amine, without adding the trifluoromethoxyl anion source 3c (Scheme 5). The TLC control revealed the presence of the byproduct after 1 h, which was accompanied by almost complete conversion of the starting material (to the pyridinium salt). After the reaction proceeded for a prolonged time (6 h), a 4-fluoro-1,1′-biphenyl 5 was isolated in a 70% yield, as evidenced by the NMR spectra of the purified sample (SI, spectral data for compound 5). This concurrent reaction can be explained by the slow process of including ⊖BF4 as the fluoride nucleophile source (Scheme 4c), which possibly enters some equilibria or undergoes dimerization in the presence of water64−66
Scheme 5. Test Reaction Excluding the ⊖OCF3 Source.

Even taking the above explained interference into account, our mechanochemical protocol gives good results for anilines with simple, small substituents, as well as for starting materials with amide (4r–4ab) and sulfonamide (4ac–4ae) connections. In the case of 4ab, a 74% yield is quite high considering that four reaction events are required to produce this double-substitution product. Notably, we also tried several aminoheterocycles, such as 2-aminopyridine, benzo[d]thiazol-2-amine, etc. To our great disappointment, the title reaction experienced a failure, and we did not observe the formation of the desired OCF3 products.
To elucidate the mechanism of the synthesis, we performed computational modeling (Figure 1). Since the first step of the reaction mechanism is clearly established in the literature, we focused on the second step, namely the substitution and dissociation of the pyridium salts into respective products. We studied two possible mechanistic routes: (i) single electron transfer (SET) followed by radical formation and (ii) the direct nucleophilic attack of the ⊖OCF3 anion.
Figure 1.

Energy profile diagram. All energies (kcal/mol) are from isolated reactants at a 0 kcal/mol reference. The barriers were calculated for 298 K.
The adiabatic (including molecular relaxation) electron affinity of unsubstituted pyridinium salt was calculated as 4.86 eV at the CCSD(T)/def2-TZVPP level of theory in the gas phase. From the species in the reaction mixture, the ⊖OCF3 anion has a sufficiently low ionization potential of 4.21 eV in the gas phase, allowing SET. This situation is reversed by effects of the environment. Toluene as nonpolar solvent stabilizes the small ⊖OCF3 anion to the extent that its ionization potential becomes larger than the electron affinity of the larger pyridinium salt, minimizing the possibility of SET in the ground state of the system. Several authors studied the behavior of pyridinium salts following SET. Lorance et al. found that N-methoxypyridinium salts dissociate with minimal activation energy upon SET, giving rise to pyridine and a methoxy radical.67,68 We tested our methodology on these systems and consistently found either none or a minimal barrier for methoxy radical formation upon SET, in agreement with the mentioned experiment. When we approached systems studied here with the identical methodology, we observed a barrier of 35 kcal/mol that hindered the formation of the phenyl radical as a possible reactive intermediate. Moreover, the dissociation of the N-phenylpyridium radical into pyridine and the phenyl radical is also thermodynamically disfavored (ΔGr = 18.1 kcal/mol). This is in line with results of Sevov et al., who used structurally comparable pyridinium salts as anolytes to sustain many cycles of charging and discharging.69
Next, we turned our attention to the possible nucleophilic attack of ⊖OCF3 on the formed pyridinium salt. All possible substitution positions were considered. From a thermochemical viewpoint, nucleophilic attacks on carbons that already had hydrogens led to metastable intermediates relatively high in energy in case of the ortho- and para-positions (ΔG ≈ 5 kcal/mol) on pyridine and the para-position on phenyl (ΔG ≈ 14 kcal/mol). Such intermediates are protected from disintegration by a very small barrier less than 2 kcal/mol and require another particle, which would leave with hydrogen or proton to stabilize into the products. For other positions, it was impossible to stabilize nucleophilic substitution intermediate, with one notable exception that led to the observed products.
The nucleophilic attack on a carbon bonded to the pyridine nitrogen provides a channel to the stable products without any intermediate, and pyridine and the corresponding OCF3 derivative are generated directly. The reaction ΔG favors such a splitting by −10 kcal/mol compared to the reactants. Not only the ⊖OCF3 anion is capable of achieving such a dissociative substitution. Calculations using the fluorine anion corroborate the experimental finding that fluorinated products can be reached in the absence of an OCF3 source.
To gain more insight, we tried to replicate the non-reactive behavior of pyridinium salt (leading to 4e) toward the ⊖OCF3 anion, which was observed for a range of different solvents and temperatures. In polar solvents (ACN and MeOH), separate ionic reactants tend to be over-stabilized compared to the transition state, and the resulting activation ΔG‡ reaches 37.6 kcal/mol, according to our calculations. Nonpolar solvents such as 1,4-dioxane seem to be the rational choice to overcome this issue. However, in dioxane, the hydrogen-bonded F3CO⊖···H–pyridinium complex stabilizes reactants, and the activation ΔG‡ reaches a value of 29.1 kcal/mol, which is still too high for a straightforward reaction. This was proven by the corresponding experiments in solution (Table S1, entries 18 and 19). The mechanochemical setup allows this reaction to proceed in a high yield in a short time. It is difficult to establish the exact reasons behind this. As an effect of the higher concentration, more frequent collisions70 of reactants can increase the prefactor in the Eyring equation, leading to effective acceleration.
It has been shown that concentration effects alone may not be sufficient to explain the observed changes in kinetics.71 The effects of unoriented mechanical forces experienced by the system between the walls of the grinding balls can modify the transition-state position and lower the activation energy. Here we note that the imaginary vibration in the transition state has a value of 242 cm–1, while the intermolecular vibrations of reactants mostly have values under 100 cm–1, meaning that relatively low force constants and thus external forces can significantly divert the system from the equilibrium position.
3. Conclusion
In summary, we have developed a mechanochemical one-pot procedure for the selective and highly efficient substitution of an aromatic amine group with an OCF3 substituent via the pyridinium salt intermediate. 1-Methyl-1,4-diazabicyclo[2.2.2]octan-1-ium trifluoromethoxide salt was selected as the best ⊖OCF3 nucleophile source. Our method accepts a variety of functionalities that can act as entry points for further transformations (including Br, I, and COOH groups) as well as moieties such as amide and sulfonamide. The lower yields for ortho-substituted starting materials point to steric hindrance as a limiting factor. Surprisingly, the developed procedure works only in the solid state; further studies must be conducted to explain this behavior and possibly harness its benefits in other synthetic applications. Nevertheless, the mechanochemical conditions ensure a mild temperature, a reduced workup time and solvent economy, corresponding to the principles of green chemistry. The generality of our protocol will be confirmed by further studies; however, the selectivity, efficiency and robustness of the presented method will indeed have an impact on medicinal chemistry in the near future considering the significance of the OCF3 substituent and the -OCF2- linkage as pharmacophores.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c02611.
General information; Cartesian coordinates; HRMS and MG MS data; spectral data; and copies of 1H, 19F{1H} and 13C{1H} NMR spectra (PDF)
This research is supported by a grant from National Science Centre (NSC) Poland within the framework of the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Grant 665778 (POLONEZ 2 Grant, no. 2016/21/P/ST5/00630). The computational part of this work was supported by the Slovak Research and Development Agency and the Scientific Grant Agency through VEGA 1/0562/20 and APVV-20-0098, respectively.
The authors declare no competing financial interest.
Supplementary Material
References
- Wang J.; Sánchez-Roselló M.; Aceña J. L.; Del Pozo C.; Sorochinsky A. E.; Fustero S.; Soloshonok V. A.; Liu H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Jeffries B.; Wang Z.; Felstead H. R.; Le Questel J. Y.; Scott J. S.; Chiarparin E.; Graton J.; Linclau B. Systematic Investigation of Lipophilicity Modulation by Aliphatic Fluorination Motifs. J. Med. Chem. 2020, 63, 1002–1031. 10.1021/acs.jmedchem.9b01172. [DOI] [PubMed] [Google Scholar]
- Mei H.; Remete A. M.; Zou Y.; Moriwaki H.; Fustero S.; Kiss L.; Soloshonok V. A.; Han J. Fluorine-containing drugs approved by the FDA in 2019. Chin. Chem. Lett. 2020, 31, 2401–2413. 10.1016/j.cclet.2020.03.050. [DOI] [Google Scholar]
- O’Hagan D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 2010, 131, 1071–1081. 10.1016/j.jfluchem.2010.03.003. [DOI] [Google Scholar]
- Jeschke P.; Baston E.; Leroux F. α-Fluorinated Ethers as “Exotic” Entity in Medicinal Chemistry. Mini-Reviews Med. Chem. 2007, 7, 1027–1034. 10.2174/138955707782110150. [DOI] [PubMed] [Google Scholar]
- McClinton M. A.; McClinton D. A. Trifluoromethylations and related reactions in organic chemistry. Tetrahedron 1992, 48, 6555–6666. 10.1016/S0040-4020(01)80011-9. [DOI] [Google Scholar]
- Flynn G. L. Substituent Constants for Correlation Analysis in Chemistry and Biology. J. Pharm. Sci. 1980, 69, 1109. 10.1002/jps.2600690938. [DOI] [Google Scholar]
- Manteau B.; Genix P.; Brelot L.; Vors J. P.; Pazenok S.; Giornal F.; Leuenberger C.; Leroux F. R. A General Approach to (Trifluoromethoxy)pyridines: First X-ray Structure Determinations and Quantum Chemistry Studies. Eur. J. Org. Chem. 2010, 2010, 6043–6066. 10.1002/ejoc.201000958. [DOI] [Google Scholar]
- Federsel D.; Herrmann A.; Christen D.; Sander S.; Willner H.; Oberhammer H. Structure and conformation of α,α,α-trifluoroanisol, C6H5OCF3. J. Mol. Struct. 2001, 567–568, 127–136. 10.1016/S0022-2860(01)00541-5. [DOI] [Google Scholar]
- Marrec O.; Billard T.; Vors J. P.; Pazenok S.; Langlois B. R. A deeper insight into direct trifluoromethoxylation with trifluoromethyl triflate. J. Fluor. Chem. 2010, 131, 200–207. 10.1016/j.jfluchem.2009.11.006. [DOI] [Google Scholar]
- Kirsch P.; Bremer M. Nematic Liquid Crystals for Active Matrix Displays: Molecular Design and Synthesis. Angew. Chemie - Int. Ed. 2000, 39, 4216–4235. . [DOI] [PubMed] [Google Scholar]
- Mamada M.; Shima H.; Yoneda Y.; Shimano T.; Yamada N.; Kakita K.; Machida T.; Tanaka Y.; Aotsuka S.; Kumaki D.; Tokito S. A Unique Solution-Processable n-Type Semiconductor Material Design for High-Performance Organic Field-Effect Transistors. Chem. Mater. 2015, 27, 141–147. 10.1021/cm503579m. [DOI] [Google Scholar]
- Yoshida S.; Shimomori K.; Kim Y.; Hosoya T. Single C–F Bond Cleavage of Trifluoromethylarenes with an ortho-Silyl Group. Angew. Chemie - Int. Ed. 2016, 55, 10406–10409. 10.1002/anie.201604776. [DOI] [PubMed] [Google Scholar]
- Dang H.; Whittaker A. M.; Lalic G. Catalytic activation of a single C–F bond in trifluoromethyl arenes. Chem. Sci. 2016, 7, 505–509. 10.1039/C5SC03415A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallov I.; Ruddy A. J.; Zhu H.; Grimme S.; Stephan D. W. Activation of Alkyl C–F Bonds by B(C6F5)3: Stoichiometric and Catalytic Transformations. Chem. - A Eur. J. 2017, 23, 17692–17696. 10.1002/chem.201705276. [DOI] [PubMed] [Google Scholar]
- Chen K.; Berg N.; Gschwind R.; König B. Selective Single C(sp3)–F Bond Cleavage in Trifluoromethylarenes: Merging Visible-Light Catalysis with Lewis Acid Activation. J. Am. Chem. Soc. 2017, 139, 18444–18447. 10.1021/jacs.7b10755. [DOI] [PubMed] [Google Scholar]
- Vogt D. B.; Seath C. P.; Wang H.; Jui N. T. Selective C–F Functionalization of Unactivated Trifluoromethylarenes. J. Am. Chem. Soc. 2019, 141, 13203–13211. 10.1021/jacs.9b06004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C.; Bandar J. S. Selective Defluoroallylation of Trifluoromethylarenes. J. Am. Chem. Soc. 2019, 141, 14120–14125. 10.1021/jacs.9b07766. [DOI] [PubMed] [Google Scholar]
- Mandal D.; Gupta R.; Young R. D. Selective Monodefluorination and Wittig Functionalization of gem-Difluoromethyl Groups to Generate Monofluoroalkenes. J. Am. Chem. Soc. 2018, 140, 10682–10686. 10.1021/jacs.8b06770. [DOI] [PubMed] [Google Scholar]
- Mandal D.; Gupta R.; Jaiswal A. K.; Young R. D. Frustrated Lewis-Pair-Meditated Selective Single Fluoride Substitution in Trifluoromethyl Groups. J. Am. Chem. Soc. 2020, 142, 2572–2578. 10.1021/jacs.9b12167. [DOI] [PubMed] [Google Scholar]
- Gupta R.; Jaiswal A. K.; Mandal D.; Young R. D. A Frustrated Lewis Pair Solution to a Frustrating Problem: Mono-Selective Functionalization of C–F Bonds in Di- and Trifluoromethyl Groups. Synlett. 2020, 31, 933–937. 10.1055/s-0039-1690811. [DOI] [Google Scholar]
- Zhu X. L.; Huang Y.; Xu X. H.; Qing F. L. Silver-Catalyzed C–H Aryloxydifluoromethylation and Arylthiodifluoromethylation of Heteroarenes. Org. Lett. 2020, 22, 5451–5455. 10.1021/acs.orglett.0c01826. [DOI] [PubMed] [Google Scholar]
- Yagupolskii L. M. Sintez proizvodnykh feniltriftormetilovogo efira. Dokl. Akad. Nauk. SSSR 1955, 105, 100–102. [Google Scholar]
- Feiring A. E. Chemistry in hydrogen fluoride. 7. A novel synthesis of aryl trifluoromethyl ethers. J. Org. Chem. 1979, 44, 2907–2910. 10.1021/jo01330a017. [DOI] [Google Scholar]
- Yarovenko N. N.; Vasileva A. S. A new method of introduction of trihalomethyl group into organic compounds. Zh. Obs. Khim. 1958, 28, 2502–2504. [Google Scholar]
- Sheppard W. A. α-Fluorinated Ethers. I. Aryl Fluoroalkyl Ethers. J. Org. Chem. 1964, 29, 1–11. 10.1021/jo01024a001. [DOI] [Google Scholar]
- Aldrich P. E.; Sheppard W. A. α-Fluorinated Ethers. II. Alkyl Fluoroalkyl Ethers. J. Org. Chem. 1964, 29, 11–15. 10.1021/jo01024a002. [DOI] [Google Scholar]
- Kanie K.; Tanaka Y.; Suzuki K.; Kuroboshi M.; Hiyama T. A Convenient Synthesis of Trifluoromethyl Ethers by Oxidative Desulfurization-Fluorination of Dithiocarbonates. Bull. Chem. Soc. Jpn. 2000, 73, 471–484. 10.1246/bcsj.73.471. [DOI] [Google Scholar]
- Kuroboshi M.; Kanie K.; Hiyama T. Oxidative Desulfurization-Fluorination: A Facile Entry to a Wide Variety of Organofluorine Compounds Leading to Novel Liquid-Crystalline Materials. Adv. Synth. Catal. 2001, 343, 235–250. . [DOI] [Google Scholar]
- Kuroboshi M.; Suzuki K.; Hiyama T. Oxidative desulfurization-fluorination of xanthates. A convenient synthesis of trifluoromethyl ethers and difluoro(methylthio)methyl ethers. Tetrahedron Lett. 1992, 33, 4173–4176. 10.1016/S0040-4039(00)74681-8. [DOI] [Google Scholar]
- Shimizu M.; Hiyama T. Modern Synthetic Methods for Fluorine-Substituted Target Molecules. Angew. Chemie - Int. Ed. 2005, 44, 214–231. 10.1002/anie.200460441. [DOI] [PubMed] [Google Scholar]
- Umemoto T.; Adachi K.; Ishihara S. CF3 Oxonium Salts, O-(Trifluoromethyl)dibenzofuranium Salts: In Situ Synthesis, Properties, and Application as a Real CF3+ Species Reagent. J. Org. Chem. 2007, 72, 6905–6917. 10.1021/jo070896r. [DOI] [PubMed] [Google Scholar]
- Stanek K.; Koller R.; Togni A. Reactivity of a 10-I-3 Hypervalent Iodine Trifluoromethylation Reagent With Phenols. J. Org. Chem. 2008, 73, 7678–7685. 10.1021/jo8014825. [DOI] [PubMed] [Google Scholar]
- Liu J. B.; Chen C.; Chu L.; Chen Z. H.; Xu X. H.; Qing F. L. Silver-Mediated Oxidative Trifluoromethylation of Phenols: Direct Synthesis of Aryl Trifluoromethyl Ethers. Angew. Chemie - Int. Ed. 2015, 54, 11839–11842. 10.1002/anie.201506329. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Ni C.; He Z.; Hu J. O-Trifluoromethylation of Phenols: Access to Aryl Trifluoromethyl Ethers by O-Carboxydifluoromethylation and Decarboxylative Fluorination. Org. Lett. 2016, 18, 3754–3757. 10.1021/acs.orglett.6b01779. [DOI] [PubMed] [Google Scholar]
- Zhang Q. W.; Brusoe A. T.; Mascitti V.; Hesp K. D.; Blakemore D. C.; Kohrt J. T.; Hartwig J. F. Fluorodecarboxylation for the Synthesis of Trifluoromethyl Aryl Ethers. Angew. Chemie - Int. Ed. 2016, 55, 9758–9762. 10.1002/anie.201604793. [DOI] [PubMed] [Google Scholar]
- Venturini F.; Navarrini W.; Famulari A.; Sansotera M.; Dardani P.; Tortelli V. Direct trifluoro-methoxylation of aromatics with perfluoro-methyl-hypofluorite. J. Fluor. Chem. 2012, 140, 43–48. 10.1016/j.jfluchem.2012.04.008. [DOI] [Google Scholar]
- Kolomeitsev A. A.; Vorobyev M.; Gillandt H. Versatile application of trifluoromethyl triflate. Tetrahedron Lett. 2008, 49, 449–454. 10.1016/j.tetlet.2007.11.105. [DOI] [Google Scholar]
- Lee K. N.; Lee J. W.; Ngai M. Y. Synthesis of Trifluoromethoxylated (Hetero)Arenes via OCF3 Migration. Synlett 2016, 27 (3), 313–319. 10.1055/s-0035-1560516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Liang T.; Harada S.; Lee E.; Ritter T. Silver-Mediated Trifluoromethoxylation of Aryl Stannanes and Arylboronic Acids. J. Am. Chem. Soc. 2011, 133, 13308–13310. 10.1021/ja204861a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Morales-Rivera C. A.; Lee J. W.; Liu P.; Ngai M. Y. Catalytic C-H Trifluoromethoxylation of Arenes and Heteroarenes. Angew. Chemie - Int. Ed. 2018, 57, 9645–9649. 10.1002/anie.201800598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Lee J. W.; Morales-Rivera C. A.; Liu P.; Ngai M.-Y. Redox-Active Reagents for Photocatalytic Generation of the OCF3 Radical and (Hetero)Aryl C–H Trifluoromethoxylation. Angew. Chemie - Int. Ed. 2018, 57, 13795–13799. 10.1002/anie.201808495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Y.; Xu X.-H.; Qing F.-L. Electrochemical Trifluoromethoxylation of (Hetero)aromatics with a Trifluoromethyl Source and Oxygen. Angew. Chemie - Int. Ed. 2022, 61 (3), e202114048. 10.1002/anie.202114048. [DOI] [PubMed] [Google Scholar]
- Yang Y. M.; Yao J. F.; Yan W.; Luo Z.; Tang Z.-Y. Silver-Mediated Trifluoromethoxylation of (Hetero)aryldiazonium Tetrafluoroborates. Org. Lett. 2019, 21, 8003–8007. 10.1021/acs.orglett.9b03000. [DOI] [PubMed] [Google Scholar]
- Yang S.; Chen M.; Tang P. Visible-Light Photoredox-Catalyzed and Copper-Promoted Trifluoromethoxylation of Arenediazonium Tetrafluoroborates. Angew. Chemie - Int. Ed. 2019, 58, 7840–7844. 10.1002/anie.201901447. [DOI] [PubMed] [Google Scholar]
- Mo F.; Qiu D.; Zhang L.; Wang J. Recent Development of Aryl Diazonium Chemistry for the Derivatization of Aromatic Compounds. Chem. Rev. 2021, 121 (10), 5741–5829. 10.1021/acs.chemrev.0c01030. [DOI] [PubMed] [Google Scholar]
- Deng Z.; Zhao M.; Wang F.; Tang P. Selective C-H trifluoromethoxylation of (hetero)arenes as limiting reagent. Nat. Commun. 2020, 11, 2569. 10.1038/s41467-020-16451-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelier B. J.; Tripet P. F.; Pietrasiak E.; Franzoni I.; Jeschke G.; Togni A. Angew. Chemie - Int. Ed. 2018, 57, 13784–13789. 10.1002/anie.201806296. [DOI] [PubMed] [Google Scholar]
- Zhou M.; Ni C.; Zeng Y.; Hu J. Trifluoromethyl Benzoate: A Versatile Trifluoromethoxylation Reagent. J. Am. Chem. Soc. 2018, 140, 6801–6805. 10.1021/jacs.8b04000. [DOI] [PubMed] [Google Scholar]
- Moser D.; Duan Y.; Wang F.; Ma Y.; O’Neill M. J.; Cornella J. Selective Functionalization of Aminoheterocycles by a Pyrylium Salt. Angew. Chemie - Int. Ed. 2018, 57, 11035–11039. 10.1002/anie.201806271. [DOI] [PubMed] [Google Scholar]
- Sowmiah S.; Esperança J. M. S. S.; Rebelo L. P. N.; Afonso C. A. M. Pyridinium salts: from synthesis to reactivity and applications. Org. Chem. Front. 2018, 5, 453–493. 10.1039/C7QO00836H. [DOI] [Google Scholar]
- Ghiazza C.; Faber T.; Gómez-Palomino A.; Cornella J. Deaminative chlorination of aminoheterocycles. Nat. Chem. 2022, 14, 78–84. 10.1038/s41557-021-00812-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. W. Mechanochemical organic synthesis. Chem. Soc. Rev. 2013, 42, 7668–7700. 10.1039/c3cs35526h. [DOI] [PubMed] [Google Scholar]
- Porcheddu A.; Colacino E.; De Luca L.; Delogu F. Metal-Mediated and Metal-Catalyzed Reactions Under Mechanochemical Conditions. ACS Catal. 2020, 10, 8344–8394. 10.1021/acscatal.0c00142. [DOI] [Google Scholar]
- Lee S.-C.; Hsu C.-M.; Kao H.; Li L.-Y.; Tsai Z.-N.; Tsao Y.-T.; Hsu Y.-L.; Miñoza S.; Chiu C.-C.; Liao H.-H.. Studies on the Synthesis of Perfluoroaryl Sulfides and their Application in Desulfurative Nickel-Catalyzed Reductive Cross-Coupling. ChemRxiv, August 11, 2021, ver. 1. 10.26434/chemrxiv-2021-fdd2c (accessed 2022-05-12). [DOI]
- Achar T. K.; Bose A.; Mal P. Mechanochemical synthesis of small organic molecules. Beilstein J. Org. Chem. 2017, 13, 1907–1931. 10.3762/bjoc.13.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J. M.; Starbuck H. F. Rate and Yield Enhancements in Nucleophilic Aromatic Substitution Reactions via Mechanochemistry. J. Org. Chem. 2021, 86 (20), 13983–13989. 10.1021/acs.joc.0c02996. [DOI] [PubMed] [Google Scholar]
- Todres Z. V.Organic Mechanochemistry and Its Practical Applications; CRC Press: Boca Raton, FL, 2006; p 170. [Google Scholar]
- Boldyreva E. Mechanochemistry of inorganic and organic systems: what is similar, what is different?. Chem. Soc. Rev. 2013, 42, 7719–7738. 10.1039/c3cs60052a. [DOI] [PubMed] [Google Scholar]
- Do J. L.; Friščić T. Mechanochemistry: A Force of Synthesis. ACS Cent. Sci. 2017, 3, 13–19. 10.1021/acscentsci.6b00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta M.; Colacino E.; Delogu F.; Porcheddu A. Kinetics of mechanochemical transformations. Phys. Chem. Chem. Phys. 2020, 22, 14489–14502. 10.1039/D0CP01658F. [DOI] [PubMed] [Google Scholar]
- Mesmer R. E.; Rutenberg A. C. Nuclear magnetic resonance study on the interconversion of the complexes [Ni(N3P)X]B(C6H5)4 and the crystal structure of the complex [Ni(N3P)Br]B(C6H5)4. Inorg. Chem. 1973, 12, 699–702. 10.1021/ic50121a047. [DOI] [Google Scholar]
- Bassett R. L. The ionization of boric acid in NaCl, NaCaCl and NaMgCl solutions at 25°C. Geochim. Cosmochim. Acta 1980, 44, 1151–1160. 10.1016/0016-7037(80)90069-1. [DOI] [Google Scholar]
- Kuhlmann K.; Grant D. M. Spin-Spin Coupling in the Tetrafluoroborate Ion. J. Phys. Chem. 1964, 68, 3208–3213. 10.1021/j100793a021. [DOI] [Google Scholar]
- Lorance E. D.; Kramer W. H.; Gould I. R. Barrierless Electron Transfer Bond Fragmentation Reactions. J. Am. Chem. Soc. 2004, 126 (43), 14071–14078. 10.1021/ja030438+. [DOI] [PubMed] [Google Scholar]
- Lorance E. D.; Hendrickson K.; Gould I. R. Nonracemic Betti Base as a New Chiral Auxiliary: Application to Total Syntheses of Enantiopure (2S,6R)-Dihydropinidine and (2S,6R)-Isosolenopsins. J. Org. Chem. 2005, 70 (6), 2014–2020. 10.1021/jo040259q. [DOI] [PubMed] [Google Scholar]
- Sevov C. S.; Hickey D. P.; Cook M. E.; Robinson S. G.; Barnett S.; Minteer S. D.; Sigman M. S.; Sanford M. S. Physical Organic Approach to Persistent, Cyclable, Low-Potential Electrolytes for Flow Battery Applications. J. Am. Chem. Soc. 2017, 139 (8), 2924–2927. 10.1021/jacs.7b00147. [DOI] [PubMed] [Google Scholar]
- Andersen J. M.; Mack J. Decoupling the Arrhenius equation via mechanochemistry. Chem. Sci. 2017, 8, 5447–5453. 10.1039/C7SC00538E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J. M.; Starbuck H. F. Rate and Yield Enhancements in Nucleophilic Aromatic Substitution Reactions via Mechanochemistry. J. Org. Chem. 2021, 86 (20), 13983–13989. 10.1021/acs.joc.0c02996. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Lee S.-C.; Hsu C.-M.; Kao H.; Li L.-Y.; Tsai Z.-N.; Tsao Y.-T.; Hsu Y.-L.; Miñoza S.; Chiu C.-C.; Liao H.-H.. Studies on the Synthesis of Perfluoroaryl Sulfides and their Application in Desulfurative Nickel-Catalyzed Reductive Cross-Coupling. ChemRxiv, August 11, 2021, ver. 1. 10.26434/chemrxiv-2021-fdd2c (accessed 2022-05-12). [DOI]