

Abstract
Quinolone antibiotics disrupt bacterial DNA synthesis by interacting with DNA gyrase and topoisomerase IV. However, in addition, they have been shown to act as inhibitors of pentameric ligand-gated ion channels such as GABAA receptors and the α7 nicotinic acetylcholine receptor (nAChR). In the present study, we have examined the effects of quinolone antibiotics on the human α4β2 nAChR, an important subtype that is widely expressed in the central nervous system. A key feature of α4β2 nAChRs is their ability to coassemble into two distinct stoichiometries, (α4)2(β2)3 and (α4)3(β2)2, which results in differing affinities for acetylcholine. The effects of nine quinolone antibiotics were examined on both stoichiometries of the α4β2 receptor by two-electrode voltage-clamp recording. All compounds exhibited significant inhibition of α4β2 nAChRs. However, all of the fluoroquinolone antibiotics examined (ciprofloxacin, enoxacin, enrofloxacin, difloxacin, norfloxacin, pefloxacin, and sparfloxacin) were significantly more potent inhibitors of (α4)2(β2)3 nAChRs than of (α4)3(β2)2 nAChRs. This stoichiometry-selective effect was most pronounced with pefloxacin, which inhibited (α4)2(β2)3 nAChRs with an IC50 of 26.4 ± 3.4 μM but displayed no significant inhibition of (α4)3(β2)2 nAChRs. In contrast, two nonfluorinated quinolone antibiotics (cinoxacin and oxolinic acid) exhibited no selectivity in their inhibition of the two stoichiometries of α4β2. Computational docking studies suggest that pefloxacin interacts selectively with an allosteric transmembrane site at the β2(+)/β2(−) subunit interface, which is consistent with its selective inhibition of (α4)2(β2)3. These findings concerning the antagonist effects of fluoroquinolones provide further evidence that differences in the subunit stoichiometry of heteromeric nAChRs can result in substantial differences in pharmacological properties.
Keywords: Nicotinic acetylcholine receptor, subunit stoichiometry, antagonist, quinolone, antibiotic, pefloxacin
Introduction
Nicotinic acetylcholine receptors (nAChRs) form part of the superfamily of pentameric ligand-gated ion channels, which includes receptors for 5-hydroxytryptamine (5-HT), γ-aminobutyric acid (GABA), and glycine.1 Seventeen nAChR subunits have been identified in vertebrates (α1−α10, β1−β4, γ, δ, and ε) that can coassemble in a variety of combinations to generate a diverse family of pharmacologically distinct nAChR subtypes, including both heteromeric subunit combinations (such as α4β2) and homomeric complexes (such as α7).2 Further complexity can arise as a consequence of nAChR subunits coassembling with different stoichiometries. For example, the α4 and β2 subunits can coassemble into pentameric complexes containing either two α4 and three β2 subunits ((α4)2(β2)3) or three α4 and two β2 subunits ((α4)2(β2)3).3 As has been reported previously, the two stoichiometries of α4β2 nAChR differ in their sensitivity to acetylcholine (ACh) and, as a consequence, are often referred to as “high-sensitivity” and “low-sensitivity” subtypes, respectively.4 Receptors containing α4 and β2 subunits mediate the effects of nicotine associated with tobacco smoking and are the site of action of drugs used to assist with smoking cessation.5 In addition, α4β2 nAChRs are targets for drug discovery in areas such as cognition, attention, and pain.6−8 In recent years, considerable attention has focused on studies of allosteric modulators of nAChRs that are thought to bind within the receptor’s transmembrane domain.9,10
Quinolone antibiotics interact with two distinct targets within bacterial cells, DNA gyrase (DNAG) and topoisomerase IV, both of which are involved in bacterial DNA synthesis.11 Quinolones inhibit DNA synthesis by stabilizing complexes of DNA and topoisomerase IV or DNAG which blocks the progression of the replication fork.11 However, previous studies have indicated that quinolone antibiotics can also modulate pentameric neurotransmitter-gated ion channels. For example, they have been reported to inhibit ionotropic receptors for GABA (GABAA receptors)12−15 and also human α7 nAChRs.16 In the case of α7 nAChRs, pefloxacin was identified as a potential allosteric modulator (interacting with the α7 nAChR transmembrane domain) on the basis of virtual screening,16 performed with a revised homology model of the α7 nAChR,17 and was subsequently shown to act as a noncompetitive antagonist on α7 nAChRs.16 Here, we have examined the effects of a series of nine quinolone antibiotics (Figure 1), including pefloxacin, on the two stoichiometries of the human α4β2 nAChR by two-electrode voltage-clamp recording of cloned receptor subunits expressed in Xenopus oocytes.
Figure 1.

Chemical structures of quinolone antibiotics. The effects of seven fluoroquinolone antibiotics (ciprofloxacin, difloxacin, enoxacin, enrofloxacin, norfloxacin, pefloxacin, and sparfloxacin) and two nonfluorinated quinolone antibiotics (cinoxacin and oxolinic acid) were examined in the present study.
Materials and Methods
Plasmids and Reagents
Ciprofloxacin, enrofloxacin, difloxacin, and sparfloxacin were purchased from Sigma-Aldrich (Gillingham, U.K.). Pefloxacin, cinoxacin, and oxolinic acid were purchased from Santa-Cruz Biotechnology (Dallas, TX, USA). Enoxacin was purchased from TOKU-E (Washington, USA), and norfloxacin was purchased from Merck Life Science UK Ltd. (Southampton, U.K.). Stock solutions of antibiotics (100 mM) were prepared in DMSO, with the exception of enoxacin which was prepared in 1 M NaOH. Stock solutions were stored at −20 °C before use.
Plasmids and Site-Directed Mutagenesis
Human nAChR subunit cDNAs in plasmid expression vector pSP64GL (pSP64GL-α4 and pSP64GL-β2) have been described previously.18 Site-directed mutagenesis (to generate plasmids pSP64GL-α4L283A, pSP64GL-α4S284A, and pSP64GL-β2V278A) was performed using the QuikChange mutagenesis kit (Agilent Technologies) and verified by nucleotide sequencing (Source Bioscience). Note that the numbering of these amino acids in the human nAChR α4 and β2 subunits is based on the intact protein sequence (including the signal sequence), as indicated in the EMBL/GenBank database entries NP_000735.1 and NP_000739.1, respectively.
RNA Synthesis and Oocyte Expression
Plasmid expression vectors were linearized by restriction enzyme digestion at sites downstream from the inserted cDNA. Linearized plasmids were purified with QIAQuik PCR purification kit (Qiagen), and transcription of cRNA was carried out using mMESSAGE mMACHINE SP6 kit (Ambion, Life Technologies). To achieve heterologous expression of human α4β2 nAChRs in two distinct subunit stoichiometries [(α4)2(β2)3 and (α4)3(β2)2], a well-established protocol was employed in which Xenopus laevis oocytes were injected with α4 and β2 cRNA in ratios of 1:10 and 10:1, respectively.19,20 A similar approach was employed to generate two stoichiometries of nAChRs containing mutated subunits (α4L283A, α4S284A, or β2V278A). Oocytes were injected, using a Drummond variable volume microinjector, with 32.2 nL of cRNA containing either a mixture of 30 ng/μL human α4 and 300 ng/μL human β2 or 300 ng/μL human α4 and 30 ng/μL human β2 cRNAs.
Oocyte Electrophysiology
Adult female Xenopus laevis frogs were obtained from the European Xenopus Resource Centre at the University of Portsmouth. Animals were sacrificed using Schedule 1 procedures approved by the Animals (Scientific Procedures) Act 1986 and by the UCL Research Ethics Committee. Xenopus were anesthetized by immersion in 0.2% MS222 for 15 min (or until complete anesthesia was confirmed by absence of leg-withdrawal and righting reflex), followed by cranial concussion, decapitation, and pithing. Xenopus oocytes were isolated, maintained, and injected with cRNA, as described previously.21 Two-electrode voltage-clamp recordings were performed using a Warner Instruments OC-725C amplifier (Harvard Apparatus) with the oocyte membrane potential held at −60 mV, as described previously.22 Oocytes were continuously perfused with a modified Ringer’s solution (115 mM NaCl, 2.5 mM KCl, 1.8 mM BaCl2, and 10 mM HEPES, pH 7.3). Application of compounds was controlled by LabChart software (AD Instruments) using a BPS-8 solenoid valve solution exchange system (ALA Scientific Inc.). Typically, agonists were applied for 5 s or until a plateau in the response was observed. Antagonists were preapplied for 30 s and then coapplied with agonist for 5 s or until a plateau in the response was observed. Where data has been normalized to a maximum ACh response, the maximum response was determined from a minimum of three independent ACh dose–response curves.
Statistical Analysis
For individual pairwise comparisons, statistical significance was determined using unpaired Student’s t tests or ANOVA for multiple comparisons. Dose–response curves were fitted by GraphPad Prism, using the following equation (where I is the current, Imax is the maximum current, the EC50 is the concentration of agonist that elicits a half-maximal response, and nH is the Hill coefficient):
Small Molecule Docking
To identify potential binding sites for quinolone antibiotics in the human (α4)2(β2)3 and (α4)3(β2)2 nAChRs, computational docking was performed with protein structures that have been determined previously by cryoelectron microscopy (Protein Data Bank codes 6CNJ and 6CNK, respectively).23 Small molecule computer docking was performed using AutoDock Vina (Molecular Graphics Lab at Scripps Research Institute, La Jolla, CA) and PLANTS (Protein–Ligand ANT System; Universität Tübingen, Germany). Docking was performed within a search area of 18 Å radius centered on the γ-carbon of T286 (α4) or T277 (β2) of the subunit corresponding to the principal (+) side of the subunit interface. This covered the inter- and intrasubunit cavities of the β2/α4 and α4/α4 interfaces of (α4)3(β2)2 (PDB code 6CNK) and the β2/α4 and β2/β2 interfaces of (α4)2(β2)3 (PDB code 6CNJ). With both docking programs, ligands were allowed to be fully flexible and the maximum search efficiency was used. One-thousand protein–ligand conformations were produced by each docking program for each interface query and analyzed with a previously described consensus docking protocol.17 This in-house script allows for a consensus binding mode or cluster to be identified from the protein–ligand conformations produced from the two independent docking programs. The rationale for this approach is to identify predicted binding sites for which there is a consensus between two docking programs that employ different scoring functions. The most highly populated consensus cluster of solutions (determined by RMSD with a cutoff of 2 Å between the two docking programmes) and highest ranked (by either PLANTS or AutoDock Vina scoring function) was taken to represent the active conformation of the ligand in each receptor stoichiometry.
Results
Antagonist Effects of Quinolone Antibiotics on (α4)2(β2)3 and (α4)3(β2)2 nAChRs
The effects of nine quinolone antibiotics (Figure 1) were examined by two-electrode voltage-clamp recording on heteromeric α4β2 nAChRs expressed in Xenopus oocytes. As has been reported previously, α4β2 nAChRs assemble into two subunit stoichiometries ((α4)2(β2)3 and (α4)3(β2)2) and these two distinct receptor populations can be generated in Xenopus oocytes by injection of differing ratios of α4 and β2 subunit cRNAs. In agreement with previous studies,3,4,20 oocytes expressing (α4)2(β2)3 nAChRs (injected with α4 and β2 cRNAs in a ratio of 1:10) were activated by ACh with an EC50 value of 1.6 ± 0.1 μM (n = 5), whereas oocytes expressing (α4)3(β2)2 nAChRs (injected with α4 and β2 cRNAs in a ratio of 10:1) were activated by ACh with an EC50 value of 37.1 ± 6.8 μM (n = 3).
When applied alone to heterologously expressed α4β2 nAChRs, none of the quinolone antibiotics had any significant effect. However, when preapplied and coapplied with ACh, all displayed significant antagonist effects on at least one stoichiometry of α4β2 nAChRs (Figure 2 and Table 1). In initial studies, 100 μM of each antibiotic was coapplied with an EC50 concentration of ACh (1 μM ACh for (α4)2(β2)3 and 40 μM ACh for (α4)2(β2)3). Of the nine quinolone antibiotics examined, seven were fluoroquinolones (ciprofloxacin, difloxacin, enoxacin, enrofloxacin, norfloxacin, pefloxacin, and sparfloxacin) and all of these fluoroquinolone compounds displayed significantly greater antagonism on (α4)2(β2)3 nAChRs than on (α4)3(β2)2 nAChRs (Figure 2). This stoichiometry-selective antagonism was most apparent for pefloxacin, which inhibited responses to ACh on (α4)2(β2)3 nAChRs by 73.3 ± 1.9% (n = 4), whereas responses to ACh on (α4)3(β2)2 nAChRs were not significantly different in the presence or absence of pefloxacin (Figure 2H and Table 1). In addition, two nonfluorinated quinolone antibiotics were examined (cinoxacin and oxolinic acid). Once again, significant antagonist effects were observed, but in contrast to the fluoroquinolone antibiotics, there was no significant difference in the level of antagonism observed with (α4)2(β2)3 and (α4)3(β2)2 nAChRs (Figure 2A,G). In summary, stoichiometry-selective antagonism was displayed by all seven fluoroquinolone antibiotics examined, whereas nonselective antagonism was observed with both of the nonfluorinated quinolone antibiotics.
Figure 2.

Inhibitory effects of quinolone antibiotics on α4β2 nAChRs: bar charts illustrating the effects of quinolone antibiotics on (α4)2(β2)3 nAChRs (white bars) and (α4)3(β2)2 nAChRs (black bars) expressed in Xenopus oocytes. Antibiotics (100 μM) were preapplied for 30 s and then coapplied with agonist (an EC50 concentration of ACh) for 5 s or until a plateau in the response. Responses are normalized to responses to ACh in the absence of antibiotic. Data are the mean ± SEM from at least three individual experiments (as indicated). Significant differences are indicated (∗∗ = P < 0.01, ∗∗∗ = P < 0.001, ns = not significant).
Table 1. Inhibitory Effects of Quinolone Antibiotics on (α4)2(β2)3 and (α4)3(β2)2 nAChRsa.
| antibiotic | (α4)2(β2)3 nAChR (% control response to ACh) | (α4)3(β2)2 nAChR (% control response ACh) |
|---|---|---|
| cinoxacin | 58.2 ± 1.20 (n = 8)*** | 60.4 ± 0.61 (n = 4)*** |
| ciprofloxacin | 28.6 ± 0.99 (n = 4)*** | 78.2 ± 0.53 (n = 9)*** |
| enoxacin | 23.5 ± 1.65 (n = 4)*** | 74.6 ± 1.88 (n = 3)*** |
| enrofloxacin | 13.2 ± 0.46 (n = 4)*** | 64.8 ± 1.01 (n = 5)*** |
| difloxacin | 39.9 ± 3.02 (n = 4)*** | 57.2 ± 0.97 (n = 3)*** |
| norfloxacin | 13.0 ± 1.0 (n = 8)*** | 54.1 ± 3.40 (n = 6)*** |
| oxolinic acid | 59.5 ± 0.99 (n = 6)*** | 60.4 ± 0.72 (n = 4)*** |
| pefloxacin | 26.7 ± 1.9 (n = 4)*** | 104.1 ± 1.6 (n = 8)NS |
| sparfloxacin | 17.4 ± 1.12 (n = 4)*** | 58.4 ± 1.72 (n = 8)*** |
In all cases, inhibition was examined with 100 μM antibiotic coapplied with an EC50 concentration of ACh (1 μM for (α4)2(β2)3 and 40 μM for (α4)3(β2)2). Data are the mean ± SEM. Significant differences from control responses in the absence of antibiotic are indicated (***P < 0.001; NS = not significant).
Following our initial studies with a range of quinolone antibiotics, two compounds were selected for more detailed studies. These were pefloxacin, which displayed selective antagonism of (α4)2(β2)3 nAChRs, and cinoxacin, which displayed nonselective antagonism on (α4)2(β2)3 and (α4)3(β2)2 nAChRs.
Antagonism of α4β2 nAChRs by Pefloxacin
Oocytes expressing either (α4)2(β2)3 or (α4)2(β2)3 nAChRs were examined by coapplying a range of concentrations of pefloxacin with an EC50 concentration of ACh. With (α4)3(β2)2 nAChRs, pefloxacin showed no significant effect on responses to EC50 concentrations of ACh (Figure 3A). In contrast, with (α4)2(β2)3 nAChRs, pefloxacin inhibited responses with an IC50 value of 26.4 ± 3.4 μM, n = 4 (Figure 3A). When a fixed concentration of pefloxacin (100 μM) was coapplied with a range of ACh concentrations to (α4)2(β2)3 nAChRs, it caused a rightward shift of the ACh dose–response curve, together with a reduced maximal response to ACh (Figure 3B). Pefloxacin (100 μM) caused a significant shift in the ACh EC50 from 1.6 ± 0.1 μM (n = 5) to 6.4 ± 0.7 μM (n = 4) (P < 0.001) and reduced the maximal normalized ACh response to 91.0 ± 1.3% (n = 4; P < 0.001). Pefloxacin also caused a significant change (P < 0.0001) in the Hill coefficient from 0.83 ± 0.2 (n = 5) to 1.3 ± 0.1 (n = 4) (Figure 3B). In contrast, with (α4)3(β2)2 nAChRs, pefloxacin had no significant effect on responses to ACh, causing no changes in maximal response, EC50, or Hill coefficient (Figure 3B). This is consistent with pefloxacin acting as a selective noncompetitive antagonist of (α4)2(β2)3 nAChRs. Representative traces of ACh responses in the absence and presence of pefloxacin are shown (Figure 3D,E).
Figure 3.

Effects of pefloxacin on (α4)2(β2)3 and (α4)3(β2)2 nAChRs expressed in Xenopus oocytes. (A) Effects of a range of concentrations of pefloxacin, preapplied and coapplied with an EC50 concentration of ACh on (α4)2(β2)3 (open circles) and (α4)3(β2)2 (filled circles). Data are the mean ± SEM of a least three experiments. (B) ACh dose–response curve with (α4)2(β2)3 nAChRs in the absence (open circles) and presence (filled circles) of pefloxacin (100 μM). Data are the mean ± SEM of a least three experiments. (C) ACh dose–response curve with (α4)3(β2)2 nAChRs in the absence (open circles) and presence (filled circles) of pefloxacin (100 μM). Data are the mean ± SEM of a least three independent experiments. (D) Representative traces from (α4)2(β2)3 nAChRs showing responses to an EC50 concentration of ACh in the absence (left) and presence (right) of pefloxacin (100 μM). Scale bars: 500 nA (vertical) and 5 s (horizontal). (E) Representative traces from (α4)3(β2)2 nAChRs showing responses to an EC50 concentration of ACh in the absence (left) and presence (right) of pefloxacin (100 μM). Scale bars: 500 nA (vertical) and 5 s (horizontal).
Antagonism of α4β2 nAChRs by Cinoxacin
A similar series of experiments were performed with cinoxacin. Oocytes expressing either (α4)2(β2)3 or (α4)3(β2)2 nAChRs were examined by coapplying a range of concentrations of the cinoxacin with an EC50 concentration of ACh. The level of antagonism observed with cinoxacin was similar on the two receptor populations. Cinoxacin (1 mM) inhibited (α4)2(β2)3 by 50.5 ± 4.5% (n = 4) and (α4)2(β2)3 by 50.0 ± 2.5% (n = 4) (Figure 4A). When a fixed concentration of cinoxacin (100 μM) was coapplied with a range of ACh concentrations, it resulted in an insurmountable antagonism of ACh responses with both (α4)2(β2)3 and (α4)3(β2)2 nAChRs (Figure 4B,C). Cinoxacin (100 μM) caused a significant shift in the ACh EC50 on (α4)2(β2)3 nAChRs from 1.6 ± 0.1 μM (n = 5) to 2.1 ± 0.1 μM (n = 4) (P = 0.01) and reduced the maximal normalized ACh response to 76.0 ± 1.5% (n = 4; P < 0.001). In addition, cinoxacin caused a significant change in the Hill coefficient from 0.83 ± 0.07 (n = 5) to 1.0 ± 0.1 (n = 4; P = 0.01) (Figure 4B). Cinoxacin (100 μM) also caused a significant shift in the ACh EC50 on (α4)3(β2)2 nAChRs from 37.1 ± 6.8 μM (n = 3) to 94.5 ± 3.1 μM (n = 4; P = 0.01) and reduced the maximal normalized ACh response to 80.0 ± 0.9% (n = 4; P < 0.001). There was also a significant change in the Hill coefficient from 0.74 ± 0. (n = 3) to 0.62 ± 0.2 (n = 4; P < 0.001) (Figure 4B). These findings are consistent with cinoxacin acting as a nonselective, noncompetitive antagonist of both (α4)2(β2)3 and (α4)3(β2)2 nAChRs. Representative traces of ACh responses in the absence and presence of cinoxacin are shown (Figure 4D,E).
Figure 4.

Effects of cinoxacin on human (α4)2(β2)3 and (α4)3(β2)2 nAChRs expressed in Xenopus oocytes. (A) Effects of a range of concentrations of cinoxacin, preapplied and coapplied with an EC50 concentration of ACh on (α4)2(β2)3 (open circles) and (α4)3(β2)2 (filled circles). Data are the mean ± SEM of a least three experiments. (B) ACh dose–response curve with (α4)2(β2)3 nAChRs in the absence (open circles) and presence (filled circles) of cinoxacin (100 μM). Data are the mean ± SEM of a least three experiments. (C) ACh dose–response curve with (α4)3(β2)2 nAChRs in the absence (open circles) and presence (filled circles) of cinoxacin (100 μM). Data are the mean ± SEM of a least three independent experiments. (D) Representative traces from (α4)2(β2)3 nAChRs showing responses to an EC50 concentration of ACh in the absence (left) and presence (right) of cinoxacin (100 μM). Scale bars: 500 nA (vertical) and 5 s (horizontal). (E) Representative traces from (α4)3(β2)2 nAChRs showing responses to an EC50 concentration of ACh in the absence (left) and presence (right) of cinoxacin (100 μM). Scale bars: 500 nA (vertical) and 5 s (horizontal).
Docking of Quinolone Antibiotics into α4β2 nAChR Structures
Computational docking studies were performed with three-dimensional atomic models of the (α4)2(β2)3 and (α4)3(β2)2 nAChRs that had been determined previously by cryoelectron microscopy (PDB codes 6CNJ and 6CNK, respectively).23 A consensus docking approach17 was employed, involving two independent docking methods (AutoDock Vina and PLANTS). Since previous studies had identified the intersubunit transmembrane region as being the most plausible binding site for allosteric modulators such as pefloxacin in the α7 nAChR,16,17 docking studies were performed within a search area of 18 Å radius centered in this region (see Materials and Methods). When results were compared from the two computational docking studies, no consensus binding site for pefloxacin was identified in the (α4)3(β2)2 nAChR subtype, whereas a single plausible consensus binding site was identified in (α4)2(β2)3 at the β2/β2 interface (Figure 5) at a location similar to that identified previously for allosteric modulators of nAChRs.16,17 These findings are consistent with evidence that pefloxacin is a selective antagonist of the (α4)2(β2)3 nAChR subtype. In contrast, docking studies with cinoxacin identified plausible binding sites in both receptor structures. Again, this is consistent with the finding that these compounds display no selectivity in their antagonist effects on (α4)2(β2)3 and (α4)3(β2)2 nAChRs. Three binding sites were identified within (α4)2(β2)3 (one within the β2/β2 interface and two within the β2/α4 interface) (Figure 5), and two binding sites were identified in the (α4)3(β2)2 nAChR (both within the β2/α4 interface) (Figure 5).
Figure 5.
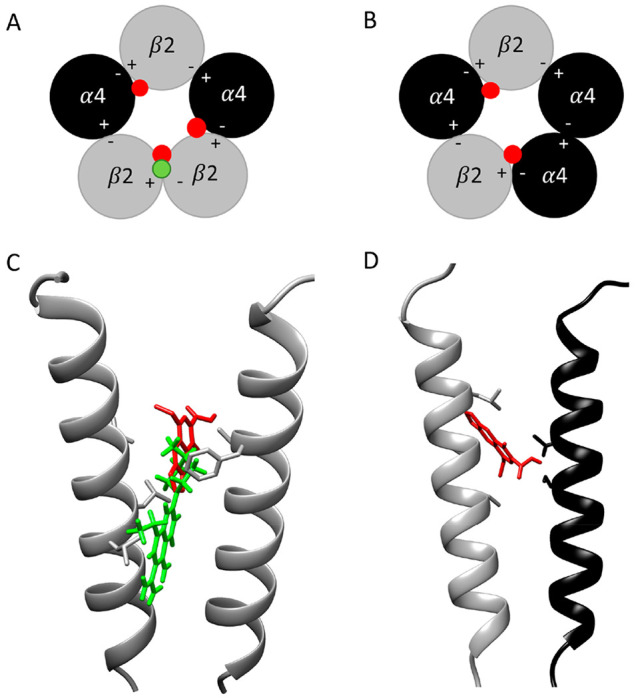
Computational docking of pefloxacin and cinoxacin into human (α4)2(β2)3 (PDB code 6CNJ) and (α4)3(β2)2 nAChRs structures (PDB code 6CNK). (A) Schematic representation of the docking sites of cinoxacin identified in the (α4)2(β2)3 nAChRs structures (red dots) and one site for pefloxacin (green dot). (B) Schematic representation of the docking sites of cinoxacin identified in the (α4)3(β2)2 nAChRs structures (red dots). (C, D) Docked positions of cinoxacin (red) and pefloxacin (green) in the transmembrane regions of (α4)2(β2)3 and (α4)3(β2)2, respectively. The images show the pore-lining TM2 transmembrane region of the β2(+)/β2(−) interface of (α4)2(β2)3 (C) and the β2(+)/α4(−) interface of (α4)3(β2)2 (D).
Further docking studies were performed with the other seven quinolone antibiotics that had been examined on nAChRs expressed in Xenopus oocytes (ciprofloxacin, difloxacin, enoxacin, enrofloxacin, norfloxacin, oxolinic acid, and sparfloxacin). These are compounds that, like cinoxacin, displayed antagonist effects on both α4β2 stoichiometries. As was observed with docking studies with cinoxacin (but in contrast to pefloxacin), plausible binding sites were identified for all seven of these compounds in both α4β2 stoichiometries and in positions that closely resembled those that had been identified with cinoxacin.
Effects of Pefloxacin and Cinoxacin on Mutant α4β2 nAChRs
A possible explanation for the nonselective antagonism by compounds such as cinoxacin and for the selective antagonism by pefloxacin might be that cinoxacin is able to bind to subunit interfaces containing the α4 subunit, whereas pefloxacin binds selectively at the interface of two β2 subunits. Such an explanation would also be consistent with the computer docking studies. With the aim of testing this hypothesis, the influence of α4 subunit mutations was examined on the antagonist effects of pefloxacin and cinoxacin. Two amino acids within the transmembrane domain of the α4 subunit were selected for site-directed mutagenesis (L283 and S284) due to their close proximity to the predicted binding sites of cinoxacin and the lack of proximity to the predicted binding site for pefloxacin. A further reason for selecting these two amino acids was that mutagenesis of the analogous amino acids in α7 nAChRs has been shown to alter allosteric modulation by compounds such as pefloxacin.16 Both amino acids were mutated individually to alanine to create α4L283A and α4S284A. In addition, an amino acid within the transmembrane domain of the β2 subunit (V278) was selected for site-directed mutagenesis due to its proximity to the predicted binding site of both cinoxacin and pefloxacin and was mutated to alanine to create β2V278A.
Receptors containing transmembrane mutations were generated by injecting cRNA encoding α4L283A, α4S284A, or β2V278A along with wild-type subunit cRNA in the ratio 1:10 or 10:1, and dose–response curves to ACh were generated (Figure 6). The α4S284A mutation had no significant effect on the EC50 value for ACh compared with that of wild-type α4β2 (Figure 6C,D), but the α4L283A and β2V278A mutations caused a significant leftward shift in the ACh concentration–response curve for both stoichiometries (Figure 6). Receptors containing α4L283A with the assumed stoichiometry of (α4L283A)2(β2)3 had an ACh EC50 of 422.3 ± 42.9 nM (n = 3), which is significantly different (P = 0.0009) from that of the wild-type (α4)2(β2)3 nAChR. In addition, those with the assumed stoichiometry of (α4L283A)3(β2)2 had an ACh EC50 of 11.42 ± 3.5 μM (n = 3), which is significantly different (P = 0.029) from that of the corresponding wild-type nAChR. Similarly, receptors containing β2V278A with an assumed stoichiometry of (α4)2(β2V278A)3 had an ACh EC50 of 24.9 ± 4.1 nM (n = 3), which is significantly different (P < 0.001) from that of the wild-type (α4)2(β2)3 nAChR. In addition, those with an assumed stoichiometry of (α4)3(β2V278A)2 had an ACh EC50 value of 294.8 ± 34.3 nM (n = 3), which is significantly different (P < 0.001) from that of the corresponding wild-type nAChR.
Figure 6.

Agonist (ACh) sensitivity of α4β2 nAChRs containing α4L283A, α4S284A, or β2V278A mutations. ACh concentration–response curve for (A) (α4)2(β2)3 (open circles) and (α4L283A)2(β2)3 (filled circles), (B) (α4)3(β2)2 (open circles) and (α4L283A)3(β2)2 (filled circles), (C) (α4)2(β2)3 (open circles) and (α4S284A)2(β2)3 (closed circles), (D) (α4)3(β2)2 (open circles) and (α4S284A)3(β2)2 (filled circles), (E) (α4)2(β2)3 (open circles) and (α4)2(β2V278A)3 (filled circles), and (F) (α4)3(β2)2 (open circles) and (α4)3(β2V278A)2 (filled circles). All data are normalized to the maximum ACh response and are the mean ± SEM of at least three independent experiments.
As was found with wild-type α4β2 nAChRs, cinoxacin had no effect when applied alone to α4β2 nAChRs containing mutated α4L283A, α4S284A, or β2V278A subunits. Similarly, pefloxacin had no effect when applied alone to α4β2 nAChRs containing mutated α4L283A or α4S284A subunits. In contrast, when pefloxacin was applied alone to α4β2 nAChRs containing the β2V278A subunit, weak agonist effects were observed (Figure 7). When applied alone to (α4)2(β2V278A)3, pefloxacin generated maximal normalized responses of 10.1 ± 1.1% (n = 4) with an EC50 of 15.9 ± 1.2 μM (n = 4) (Figure 7A,C). Similarly, on (α4)3(β2V278A)2 pefloxacin generated maximal normalized responses of 48.9 ± 1.8% (n = 4) with an EC50 of 15.9 ± 1.2 μM (n = 4) (Figure 7B,D). These findings indicate that, in contrast to the two transmembrane mutations examined on the α4 subunit, the β2V278A transmembrane mutation converts pefloxacin (but not cinoxacin) into a partial agonist.
Figure 7.

Agonist effects of pefloxacin on α4β2 nAChRs containing the β2V278A mutation. (A) Agonist dose–response curves for ACh (open circles) and pefloxacin (filled circles) on (α4)2(β2V278A)3. Data are normalized to maximal ACh responses (100 μM) and are the mean ± SEM of at least three experiments. (B) Representative traces of maximal ACh response (100 μM) and maximal pefloxacin response (1 mM) of (α4)2(β2V278A)3. Scale bars: 500 nA (vertical) and 5 s (horizontal). (C) Agonist dose–response curves for ACh (open circles) and pefloxacin (filled circles) on (α4)2(β2V278A)3. Data are normalized to maximal ACh responses (1 mM) and are the mean ± SEM of at least three experiments. (D) Representative traces of maximal ACh response (1 mM) and maximal pefloxacin response (1 mM) of (α4)3(β2V278A)2. Scale bars: 500 nA (vertical) and 5 s (horizontal).
The effects of cinoxacin (100 μM) on responses to an EC50 concentration of ACh were examined in α4β2 nAChRs containing mutated α4L283A, α4S284A, or β2V278A subunits in both stoichiometries. Each of the three transmembrane mutations abolished the antagonist effect of cinoxacin in both stoichiometries of α4β2 nAChRs (Figure 8A,B). In contrast, when pefloxacin was coapplied with ACh, neither of the α4 subunit mutations had a significant effect on the antagonist effect of pefloxacin (Figure 8C,D). For receptors containing a mutated α4 subunit in the stoichiometry (α4)2(β2)3, pefloxacin exhibited antagonist effects that were not significantly different from those observed with wild-type (α4)2(β2)3 nAChRs (Figure 7C). For receptors containing a mutated α4 subunit in the stoichiometry (α4)3(β2)2, pefloxacin exerted no significant antagonist effect (Figure 8D). Examining the inhibitory effect of pefloxacin on receptors containing the β2V278A mutation may be harder to interpret due to this mutation converting pefloxacin into a partial agonist (as was described earlier). Nevertheless, the effects of pefloxacin (100 μM) on responses to an EC50 concentration of ACh were examined on α4β2 nAChRs containing the β2V278A and were broadly similar to those observed with the α4 mutations (Figure 8C,D). As was the case with both wild-type (α4)3(β2)2 nAChRs and (α4)3(β2)2 nAChRs containing an α4 mutation, pefloxacin caused no significant inhibition of responses to ACh on (α4)3(β2V278A)2 (Figure 8D). Pefloxacin acted as an inhibitor of ACh responses on (α4)2(β2V278A)3 nAChRs, as it did with wild-type (α4)2(β2)3 nAChRs and (α4)2(β2)3 nAChRs containing an α4 mutation, but caused a significantly lower level of inhibition (P < 0.001) (Figure 8C). Therefore, all three transmembrane mutations produce effects that are consistent with the hypothesis that pefloxacin and cinoxacin modulate α4β2 nAChRs though different binding sites or mechanisms.
Figure 8.

Influence of cinoxacin and pefloxacin of α4β2 nAChRs containing α4L283A, α4S284A, or β2V278A mutations. Bar graphs illustrate the effects of 100 μM cinoxacin (A, B) and pefloxacin (C, D) on responses to an EC50 concentration of ACh. Data are presented for (α4)2(β2)3 (A, C; white bars), (α4L283A)2(β2)3 (A, C; black bars), (α4S284A)2(β2)3 (A, C; red bars), (α4)2(β2V278A)3 (A, C; blue bars), (α4)3(β2)2 (B, D; white bars), (α4L283A)3(β2)2 (B, D; black bars), (α4S284A)3(β2)2 (B, D; red bars), and (α4)3(β2V278A)2 (B, D; blue bars). All data are normalized to responses to an EC50 concentration of ACh and are the mean ± SEM of at least three independent experiments. Significant differences are indicated (∗∗∗ = P < 0.001, ns = not significant).
Discussion
A notable aspect of the present study is that fluoroquinolone antibiotics exhibit stoichiometry-selective antagonism of α4β2 nAChRs. The effect was most pronounced for pefloxacin, which exhibits complete selectivity for α4β2 nAChRs in the (α4)2(β2)3 stoichiometry. The primary difference between the two α4β2 nAChR stoichiometries is the presence of a β2/β2 interface in (α4)2(β2)3 and an α4/α4 interface in the (α4)3(β2)2. It is of interest, therefore, that computational docking studies are consistent with the possibility that pefloxacin binds preferentially to a site at the β2/β2 interface in (α4)2(β2)3 nAChRs, whereas less selective and nonselective quinolone antibiotics were predicted to interact with sites at both the β2/β2 and β2/α4 subunit interfaces. Although plausible binding sites were identified in (α4)2(β2)3 for all nine quinolone antibiotics examined, the predicted binding site for pefloxacin is qualitatively distinct from that of the other compounds, extending deeper into the intersubunit cavity within the β2/β2 subunit interface. In addition, while cinoxacin and the other quinolone antibiotics are predicted to interact with TM2 of both subunits at the β2/β2 interface, pefloxacin is predicted to also interact with the TM1 and TM3 helices of the complementary and primary subunits, respectively. This supports the possibility that pefloxacin may make important interactions with the β2/β2 interface that are distinct from that of the other antibiotics examined.
A prediction, based on our docking results, was that mutations in the α4 subunit and close to the predicted intersubunit transmembrane binding site of quinolone antibiotics might have a more profound effect on nonselective antibiotics such as cinoxacin (that were predicted to bind at both the β2/α4 and β2/β2 subunit interfaces) than pefloxacin (that was predicted to bind exclusively at the β2/β2 subunit interface), having found that two such mutations (α4L283A and α4S284A) abolish the antagonist effects of cinoxacin but have no significant effect on pefloxacin supports the predictions. These particular amino acids were selected for mutagenesis studies because they are at positions in the α4 subunit that are analogous to two amino acids in the α7 nAChR (α4S248 and α7L247) that have been shown previously to modulate the effects of compounds predicted to bind in the intersubunit transmembrane cavity.16 In addition, a mutation was made within the β2 subunit (V278A) at a site that is in close proximity to the predicted binding site of both cinoxacin and pefloxacin. As was seen with nAChRs containing α4 transmembrane mutations, the inhibitory effects of cinoxacin were abolished by this mutation in both stoichiometries. Interestingly, the β2V278A mutation converted pefloxacin but not cinoxacin into a partial agonist. There are previous examples of transmembrane mutations converting antagonists into agonist, a finding that is probably a consequence of the mutations causing conformational changes that alter the energy barrier for transitions between open and closed states following ligand binding or by allowing bound ligands to more easily stabilize the open conformation. One of the best characterized examples is a transmembrane mutation in the nAChR α7 subunit (L247T) that causes increase spontaneous openings, reduces receptor desensitization, alters temperature sensitivity, and converts antagonists into agonists.24−27 Similarly, this α7 nAChR mutation can convert both positive allosteric modulators and silent allosteric modulators into allosteric agonists.28−30 In addition, several other nAChR transmembrane mutations have been reported that convert positive allosteric modulators into either agonists or antagonists.17,31
It is of interest that whereas some degree of selectivity for (α4)3(β2)2 nAChRs was observed with all fluoroquinolone antibiotics, no selectivity between (α4)2(β2)3 and (α4)3(β2)2 was seen with the two nonfluorinated quinolone antibiotics (cinoxacin and oxolinic acid). However, in contrast to the situation with pefloxacin, our docking studies do not provide a comprehensive explanation for this difference. Indeed, all of these antibiotics (with the exception of pefloxacin) were predicted to bind in broadly similar locations. However, it may be worth noting that cinoxacin and oxolinic acid were predicted to bind at positions in the transmembrane intersubunit cavity, while larger fluoroquinolones bound at position closer to the central ion channel and extended into the pore. Our findings extend previous evidence demonstrating that a variety of nicotinic ligands can show selectivity for the different stoichiometries of α4β2 nAChRs. This includes evidence for the stoichiometry-selective modulation of α4β2 nAChRs by agonists,3,4,19,20,32−34 competitive antagonists,3,19 divalent cations,32,35 and positive allosteric modulators.36−42
It has been estimated that between 1% and 4% of individuals treated with quinolone antibiotics display adverse side effects, including headaches, insomnia, and in some cases convulsions that become more prevalent when quinolone antibiotics are coadministered with nonsteroidal anti-inflammatory drugs.43−45 It has been suggested that these side effects are mediated via interactions with GABAA receptors, since inhibitors of these receptors are proconvulsant, whereas potentiators are anxiolytic and sedative.46 Radioligand binding experiments have demonstrated that quinolone antibiotics can inhibit the binding of [3H]GABA or [3H]muscimol to GABAA receptors in preparations of rat or mouse brain synaptic membranes. Furthermore, this inhibition was shown to be more potent when the antibiotics were coadministered with biphenylacetic acid, a nonsteroidal anti-inflammatory drug.12,13 Subsequently, whole-cell voltage-clamp recordings of rat dorsal root ganglion neurons and hippocampal neurons have demonstrated inhibition of GABA-evoked responses of GABAA receptors by quinolone antibiotics, an effect that was also increased by the presence of biphenylacetic acid.13,15,47 In contrast, radioligand binding experiments have shown no effects of quinolone antibiotics on agonist binding to excitatory glutamate receptors, muscarinic acetylcholine receptors, and GABAB receptors.48,49 It is unclear whether the antagonist effects of quinolone antibiotics observed on nAChRs have any relevance to the side effects that are sometimes reported, but it is of interest that they can exert significant effects on both inhibitory GABAA receptors and excitatory nAChRs, both members of the superfamily of pentameric ligand-gated ion channels.
In previous studies, pefloxacin has been shown to be a noncompetitive antagonist of α7 nAChR and was originally identified on the basis of virtual screening for compounds predicted to interact with an allosteric transmembrane site on the α7 nAChR.16 Here we have obtained evidence of insurmountable antagonism with both pefloxacin and cinoxacin on α4β2 nAChRs that is consistent with them acting as noncompetitive antagonists of α4β2 nAChRs. It is well-known that the pharmacological properties of nAChRs are influenced by subunit composition, but the present study provides further evidence that such properties can also be influenced by the same subunits being arranged in different stoichiometries.
Acknowledgments
Work was supported by a Ph.D. studentship awarded to V.R.S. from the Biotechnology and Biological Sciences Research Council (BBSRC) that is associated with the London Interdisciplinary Doctoral Program (Grant BB/M009513/1). Support was also provided by a grant to M.T. from the Wellcome Trust (Grant 209250/Z/17/Z).
Author Present Address
∥ Centre for Structural Systems Biology, Leibniz-Institut für Experimentelle Virologie (LIV), Hamburg, Germany
Author Present Address
# Centre for Structural Systems Biology, Leibniz-Institut für Experimentelle Virologie (LIV) and Universitaẗsklinikum Hamburg-Eppendorf (UKE), 20246 Hamburg, Germany
Author Contributions
V.R.S. conducted the molecular biological and electrophysiological experiments. V.R.S. and A.S. conducted the computer docking experiments. V.R.S., A.S., M.T., and N.S.M. designed the study, interpreted the findings, performed data analysis, and wrote the manuscript.
The authors declare no competing financial interest.
References
- Changeux J.-P. The nicotinic acetylcholine receptor: the founding father of the pentameric ligand-gated ion channel superfamily. J. Biol. Chem. 2012, 287, 40207–40215. 10.1074/jbc.R112.407668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar N. S.; Gotti C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacol 2009, 56, 237–246. 10.1016/j.neuropharm.2008.07.041. [DOI] [PubMed] [Google Scholar]
- Zwart R.; Vijverberg H. P. M. Four pharmacologically distinct subtypes of α4β2 nicotinic acetylcholine receptor expressed in Xenopus laevis oocytes. Mol. Pharmacol. 1998, 54, 1124–1131. 10.1124/mol.54.6.1124. [DOI] [PubMed] [Google Scholar]
- Nelson M. E.; Kuryatov A.; Choi C. H.; Zhou Y.; Lindstrom J. Alternate stoichiometries of α4β2 nicotinic acetylcholine receptors. Mol. Pharmacol. 2003, 63, 332–341. 10.1124/mol.63.2.332. [DOI] [PubMed] [Google Scholar]
- Rollema H.; Coe J. W.; Chambers L. K.; Hurst R. S.; Stahl S. M.; Williams K. E. Rationale, pharmacology and clinical efficacy of partial agonists of α4β2 nACh receptors for smoking cessation. Trends Phramacol. Sci. 2007, 28, 316–325. 10.1016/j.tips.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Arneric S. P.; Holladay M.; Williams M. Neuronal nicotinic receptors: a perspective on two decades of drug discovery research. Biochem. Pharmacol. 2007, 74, 1092–1101. 10.1016/j.bcp.2007.06.033. [DOI] [PubMed] [Google Scholar]
- D’hoedt D.; Bertrand D. Nicotinic acetylcholine receptors: an overview on drug discovery. Expert Opin. Ther. Targets 2009, 13, 395–411. 10.1517/14728220902841045. [DOI] [PubMed] [Google Scholar]
- Haydar S. N.; Dunlop J. Neuronal nicotinic acetylcholine receptors - targets for the development of drugs to treat cognitive impairment associated with schizophrenia and Alzheimer’s disease. Curr. Top. Med. Chem. 2010, 10, 144–152. 10.2174/156802610790410983. [DOI] [PubMed] [Google Scholar]
- Williams D. K.; Wang J.; Papke R. L. Positive allosteric modulators as an approach to nicotinic acetylcholine receptor-targeted therapeutics: advantages and limitations. Biochem. Pharmacol. 2011, 82, 915–930. 10.1016/j.bcp.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzidaki A.; Millar N. S. Allosteric modulation of nicotinic acetylcholine receptors. Biochem. Pharmacol. 2015, 97, 408–417. 10.1016/j.bcp.2015.07.028. [DOI] [PubMed] [Google Scholar]
- Hooper D. C. Mechanisms of action of antimicrobials: focus on fluoroquinolones. Clin. Infect. Dis 2001, 32 (Suppl. 1), S9–S15. 10.1086/319370. [DOI] [PubMed] [Google Scholar]
- Hori S.; Kizu J.; Kawamura M. Effects of anti-inflammatory drugs on convulsant activity of quinolones: a comparative study of drug interaction between quinolones and anti-inflammatory drugs. J. Infect. Chemother 2003, 9, 314–320. 10.1007/s10156-003-0275-1. [DOI] [PubMed] [Google Scholar]
- Akahane K.; Sekiguchi M.; Une T.; Osada Y. Structure-epileptogenicity relationship of quinolones with special reference to their interaction with gamma-aminobutyric acid receptor sites. Antimicrob. Agents Chemother. 1989, 33, 1704–1708. 10.1128/AAC.33.10.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike N.; Shirasaki T.; Yakushiji T. Quinolones and fenbufen interact with GABAA receptor in dissociated hippocampal cells of rat. J. Neurophysiol. 1991, 66, 497–504. 10.1152/jn.1991.66.2.497. [DOI] [PubMed] [Google Scholar]
- Halliwell R. F.; Davey P. G.; Lambert J. J. The effects of quinolones and NSAIDs upon GABA-evoked currents recorded from rat dorsal root ganglion neurones. J. Antimicrob. Chemother. 1991, 27, 209–218. 10.1093/jac/27.2.209. [DOI] [PubMed] [Google Scholar]
- Smelt C. L. C.; Sanders V. R.; Newcombe J.; Burt R. P.; Sheppard T. D.; Topf M.; Millar N. S. Identification by virtual screening and functional characterisation of novel positive and negative allosteric modulators of the α7 nicotinic acetylcholine receptor. Neuropharmacol 2018, 139, 194–204. 10.1016/j.neuropharm.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcombe J.; Chatzidaki A.; Sheppard T. D.; Topf M.; Millar N. S. Diversity of nicotinic acetylcholine receptor positive allosteric modulators revealed by mutagenesis and a revised structural model. Mol. Pharmacol. 2018, 93, 128–140. 10.1124/mol.117.110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbent S.; Groot-Kormelink P. J.; Krashia P. A.; Harkness P. C.; Millar N. S.; Beato M.; Sivilotti L. G. Incorporation of the β3 subunit has a dominant-negative effect on the function of recombinant central-type neuronal nicotinic receptors. Mol. Pharmacol. 2006, 70, 1350–1356. 10.1124/mol.106.026682. [DOI] [PubMed] [Google Scholar]
- Moroni M.; Zwart R.; Sher E.; Cassels B. K.; Bermudez I. α4β2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol. Pharmacol. 2006, 70, 755–768. 10.1124/mol.106.023044. [DOI] [PubMed] [Google Scholar]
- Zwart R.; Carbone A. L.; Moroni M.; Bermudez I.; Mogg A. J.; Folly E. A.; Broad L. M.; Williams A. C.; Zhang D.; Ding C.; Heinz B. A.; Sher S.E. Sazetidine-A is a potent and selective agonist at native and recombinant α4β2 nicotinic acetylcholine receptors. Mol. Pharmacol. 2008, 73, 1838–1843. 10.1124/mol.108.045104. [DOI] [PubMed] [Google Scholar]
- Young G. T.; Broad L. M.; Zwart R.; Astles P. C.; Bodkin M.; Sher E.; Millar N. S. Species selectivity of a nicotinic acetylcholine receptor agonist is conferred by two adjacent extracellular β4 amino acids that are implicated in the coupling of binding to channel gating. Mol. Pharmacol. 2007, 71, 389–397. 10.1124/mol.106.030809. [DOI] [PubMed] [Google Scholar]
- Gill J. K.; Dhankher P.; Sheppard T. D.; Sher E.; Millar N. S. A series of α7 nicotinic acetylcholine receptor allosteric modulators with close chemical similarity but diverse pharmacological properties. Mol. Pharmacol. 2012, 81, 710–718. 10.1124/mol.111.076026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh R. M.; Roh S.-H.; Gharpure A.; Morales-Perez C. L.; Teng J.; Hibbs R. E. Structural principles of distinct assemblies of the human α4β2 nicotinic receptor. Nature 2018, 557, 261–165. 10.1038/s41586-018-0081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revah F.; Bertrand D.; Galzi J. L.; Devillers-Thiery A.; Mulle C.; Hussy N.; Bertrand S.; Ballivet M.; Changeux J.-P. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature 1991, 353 (6347), 846–849. 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- Bertrand D.; Devillers-Thiery A.; Revah F.; Galzi J. L.; Hussy N.; Mulle C.; Bertrand S.; Ballivet M.; Changeux J. P. Unconventional pharmacology of a neuronal nicotinic receptor mutated in the channel domain. Proc. Natl. Acad. Sci. U.S.A. 1992, 89 (4), 1261–5. 10.1073/pnas.89.4.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand S.; Devillers-Thiéry A.; Palma E.; Buisson B.; Edelstein S. J.; Corringer P.-J.; Changeux J.-P.; Bertrand D. Paradoxical allosteric effects of competitive inhibitors on neuronal α7 nicotinic receptor mutants. NeuroReport 1997, 8, 3591–3596. 10.1097/00001756-199711100-00034. [DOI] [PubMed] [Google Scholar]
- Jindrichova M.; Lansdell S. J.; Millar N. S. Changes in temperature have opposing effects on current amplitude in α7 and α4β2 nicotinic acetylcholine receptors. PLoS One 2012, 7, e32073. 10.1371/journal.pone.0032073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill J. K.; Savolainen M.; Young G. T.; Zwart R.; Sher E.; Millar N. S. Agonist activation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 5867–5872. 10.1073/pnas.1017975108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzidaki A.; Fouillet A.; Li J.; Dage J.; Millar N. S.; Sher E.; Ursu D. Pharmacological characterisation of nicotinic acetylcholine receptors expressed in human iPSC-derived neurons. PLoS One 2015, 10 (4), e0125116 10.1371/journal.pone.0125116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill-Thind J. K.; Dhankher P.; D’Oyley J. M.; Sheppard T. D.; Millar N. S. Structurally similar allosteric modulators of α7 nicotinic acetylcholine receptors exhibit five distinct pharmacological effects. J. Biol. Chem. 2015, 290, 3552–3562. 10.1074/jbc.M114.619221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins T.; Millar N. S. Nicotinic acetylcholine receptor transmembrane mutations convert ivermectin from a positive to a negative allosteric modulator. Mol. Pharmacol. 2010, 78, 198–204. 10.1124/mol.110.064295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzaferro S.; Benallegue N.; Carbone A.; Gasparri F.; Vijayan R.; Biggin P. C.; Moroni M.; Bermudez I. Additional acetylcholine (ACh) binding site at alpha4/alpha4 interface of (α4β2)2α4 nicotinic receptor influences agonist sensitivity. J. Biol. Chem. 2011, 286, 31043–31054. 10.1074/jbc.M111.262014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzaferro S.; Gasparri F.; New K.; Alcaino C.; Faundez M.; Vasquez P. I.; Vijayan R.; Biggin P. C.; Bermudez I. Non-equivalent ligand selectivity of agonist sites in (α4β2)2α4 nicotinic acetylcholine receptors: a key determinant of agonist efficacy. J. Biol. Chem. 2014, 289, 21795–21806. 10.1074/jbc.M114.555136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indurthi D. C.; Lewis T. M.; Ahring P. K.; Balle T.; Chebib M.; Absalom N. L. Ligand Binding at the α4-α4 Agonist-Binding Site of the α4β2 nAChR Triggers Receptor Activation through a Pre-Activated Conformational State. PLoS One 2016, 11, e0161154. 10.1371/journal.pone.0161154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzaferro S.; Strikwerda J. R.; Sine S. M. Stoichiometry-selective modulation of α4β2 nicotinic ACh receptors by divalent cations. Br. J. Pharmacol. 2022, 179, 1353–1370. 10.1111/bph.15723. [DOI] [PubMed] [Google Scholar]
- Moroni M.; Vijayan R.; Carbone A.-L.; Zwart R.; Biggin P. C.; Bermudez I. Non-agonist-binding subunit interfaces confer distinct functional signatures to the alternate stoichiometries of the α4β2 nicotinic receptor: an α4-α4 interface is required for Zn2+ potentiation. J. Neurosci. 2008, 28, 6884–6894. 10.1523/JNEUROSCI.1228-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. A.; Kastrup J. S.; Peters D.; Gajhede M.; Balle T.; Ahring P. K. Two distinct allosteric binding sites at α4β2 nicotinic acetylcholine receptors revealed by NS206 and NS9283 give unique insights to binding activity-associated linkage at Cys-loop receptors. J. Biol. Chem. 2013, 288, 35997–36006. 10.1074/jbc.M113.498618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. A.; Ahring P. K.; Kastrup J. S.; Gajhede M.; Balle T. Structural and functional studies of the modulator NS9283 reveal agonist-like mechanism of action at α4β2 nicotinic acetylcholine receptors. J. Biol. Chem. 2014, 289, 24911–24921. 10.1074/jbc.M114.568097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermann D. B.; Sandager-Nielsen K.; Dyhring T.; Smith M.; Jacobsen A.-M.; Nielsen E. Ø.; Grunnet M.; Christensen J. K.; Peters D.; Kohlhaas K.; Olsen G. M.; Ahring P. K. Augmentation of cognitive function by NS9283, a stoichiometry-dependent positive allosteric modulator of α2- and α4-containing nicotinic acetylcholine receptors. Br. J. Pharmacol. 2012, 167, 164–182. 10.1111/j.1476-5381.2012.01989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.-J.; Deba F.; Mohamed T. S.; Chiara D. C.; Ramos K.; Hamouda A. K. Unraveling amino acid residues critical for allosteric potentiation of (α4)3(β2)2-type nicotinic acetylcholine receptor responses. J. Biol. Chem. 2017, 292, 9988–10001. 10.1074/jbc.M116.771246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzaferro S.; Bermudez I.; Sine S. M. Potentiation of a neuronal nicotinic receptor via pseudo-agonist site. Cell. Mol. Life Sci. 2019, 76, 1151–1167. 10.1007/s00018-018-2993-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzaferro S.; Bermudez I.; Sine S. M. α4β2 Nicotinic Acetylcholine Receptors: Relationships between subunit stoichiometry and function at the single channel level. J. Biol. Chem. 2017, 292, 2729–2740. 10.1074/jbc.M116.764183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball P. Ciprofloxacin: an overview of adverse experiences. J. Antimicrob. Chemother. 1986, 18, 187–193. 10.1093/jac/18.SD.187. [DOI] [PubMed] [Google Scholar]
- Christ W. Central nervous system toxicity of quinolones: human and animal findings. J. Antimicrob. Chemother. 1990, 26, 219–225. 10.1093/jac/26.suppl_B.219. [DOI] [PubMed] [Google Scholar]
- von Rosenstiel N.; Adam D. Quinolone antibacterials. An update of their pharmacology and therapeutic use. Drugs 1994, 47, 872–901. 10.2165/00003495-199447060-00003. [DOI] [PubMed] [Google Scholar]
- Simmonds M. A.; Turner J. P. Potentiators of responses to activation of gamma-aminobutyric acid (GABAA) receptors. Neuropharmacol 1987, 26, 923–930. 10.1016/0028-3908(87)90071-2. [DOI] [PubMed] [Google Scholar]
- Halliwell R. F.; Davey P. G.; Lambert J. J. Antagonism of GABAA receptors by 4-quinolones. J. Antimicrob. Chemother. 1993, 31, 457–462. 10.1093/jac/31.4.457. [DOI] [PubMed] [Google Scholar]
- Dodd P. R.; Davies L. P.; Watson W. E.; Nielsen B.; Dyer J. A.; Wong L. S.; Johnston G. A. Neurochemical studies on quinolone antibiotics: effects on glutamate, GABA and adenosine systems in mammalian CNS. Pharmacol. Toxicol. 1989, 64, 404–411. 10.1111/j.1600-0773.1989.tb00676.x. [DOI] [PubMed] [Google Scholar]
- Segev S.; Rehavi M.; Rubinstein E. Quinolones, theophylline, and diclofenac interactions with the gamma-aminobutyric acid receptor. Antimicrob. Agents Chemother. 1988, 32, 1624–1626. 10.1128/AAC.32.11.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]