

Abstract
Blood vessels in the developing retina are formed in concert with neural growth, resulting in functional neurovascular network. Disruption of the neurovascular coordination contributes to the pathogenesis of retinopathy of prematurity (ROP), a potentially blinding retinal neovascular disease in preterm infants that currently lacks an approved drug therapy in the USA. Despite vasculopathy as predominant clinical manifestations, an increasing number of studies revealed complex neurovascular interplays among neurons, glial cells and blood vessels during ROP. Coordinated expression of glia-derived vascular endothelial growth factor (VEGF) in spatio-temporal gradients is pivotal to the formation of well-organized vascular plexuses in the healthy retina, whereas uncoordinated VEGF expression triggers pathological angiogenesis with disorganized vascular tufts in ROP. In contrast with VEGF driving both pathological and physiological angiogenesis, neuron-derived angiogenic factor secretogranin III (Scg3) stringently regulates ROP but not healthy retinal vessels in animal models. Anti-VEGF and anti-Scg3 therapies confer similar high efficacies to alleviate ROP in preclinical studies but are distinct in their disease selectivity and safety. This review discusses neurovascular communication among retinal blood vessels, neurons and glial cells during retinal development and ROP pathogenesis and summarizes the current and emerging therapies to address unmet clinical needs for the disease.
Keywords: Retinopathy of prematurity, oxygen-induced retinopathy, neurovascular interaction, vascular endothelial growth factor, secretogranin III, anti-angiogenic therapy
Introduction
Retinopathy of prematurity (ROP), the leading cause of blindness in children, was first described in 1942 as “retrolental fibroplasia” and characterized by pathological retinal neovascularization (RNV) [1]. The disease primarily affects premature infants weighing approximately 1250 grams or less born before 31 weeks of gestation. In 2010, an estimated 184,700 out of 14.9 million preterm babies globally developed ROP of any stages, including 20,000 of these babies suffering from severe visual impairment or blindness and another 12,300 developing mild or moderate visual impairment [2]. The pathophysiology of ROP can be separated into two phases. In Phase Ⅰ, hyperoxia-induced vaso-obliteration occurs immediately following premature birth and is further exacerbated by supplemental oxygen used to improve survival of preterm infants suffering from respiratory distress syndrome. In Phase II, withdrawal of supplemental oxygen coupled with vaso-obliteration and downregulated expression of angiogenic factors in the developing retina with increased retinal metabolic demands creates relative hypoxia that induces angiogenic factors and pathological RNV.
The major clinical hallmark of ROP is abnormal retinal vasculature, which is mainly manifested as pathological RNV and intravitreal neovascularization (IVNV) (Fig. 1A and B), retinal folding, arterial tortuosity, vein dilation (Fig. 1C), and retinal or vitreous haemorrhage. Despite vascular manifestations, accumulating evidence indicates a crucial role of the neural retina in ROP progression. The standard treatment for ROP is laser ablation of the peripheral avascular retina, which scarifies the peripheral visual fields to save the central vision. Vascular endothelial growth factor (VEGF) inhibitors are applied for the clinical treatment of ROP, but their safety remains a major concern [3–7]. In spite of treatment, children with low birth weight suffer from ongoing visual impairments, suggesting disruption of normal retinal neuronal development to some extent [8]. Animal models of oxygen-induced retinopathy (OIR), which mimic the key pathological vascular and neuronal features of human ROP, exhibit significant retinal dysfunction and structural abnormalities in the inner retinal thickness, persistent ectopic synapses, prolonged cellular apoptosis and retinal gliosis even after full peripheral revascularization [9]. Hence, therapeutic strategies for ROP need to not only address vasculopathy but also restore retinal structure and functions. The aim of this review is to discuss intricate roles of neurons and glial cells during normal retinal development and pathological angiogenesis in ROP, elaborate neurovascular crosstalk, summarize the current and emerging therapies and propose strategies to promote the recovery and development of the immature injured retina.
Fig. 1.
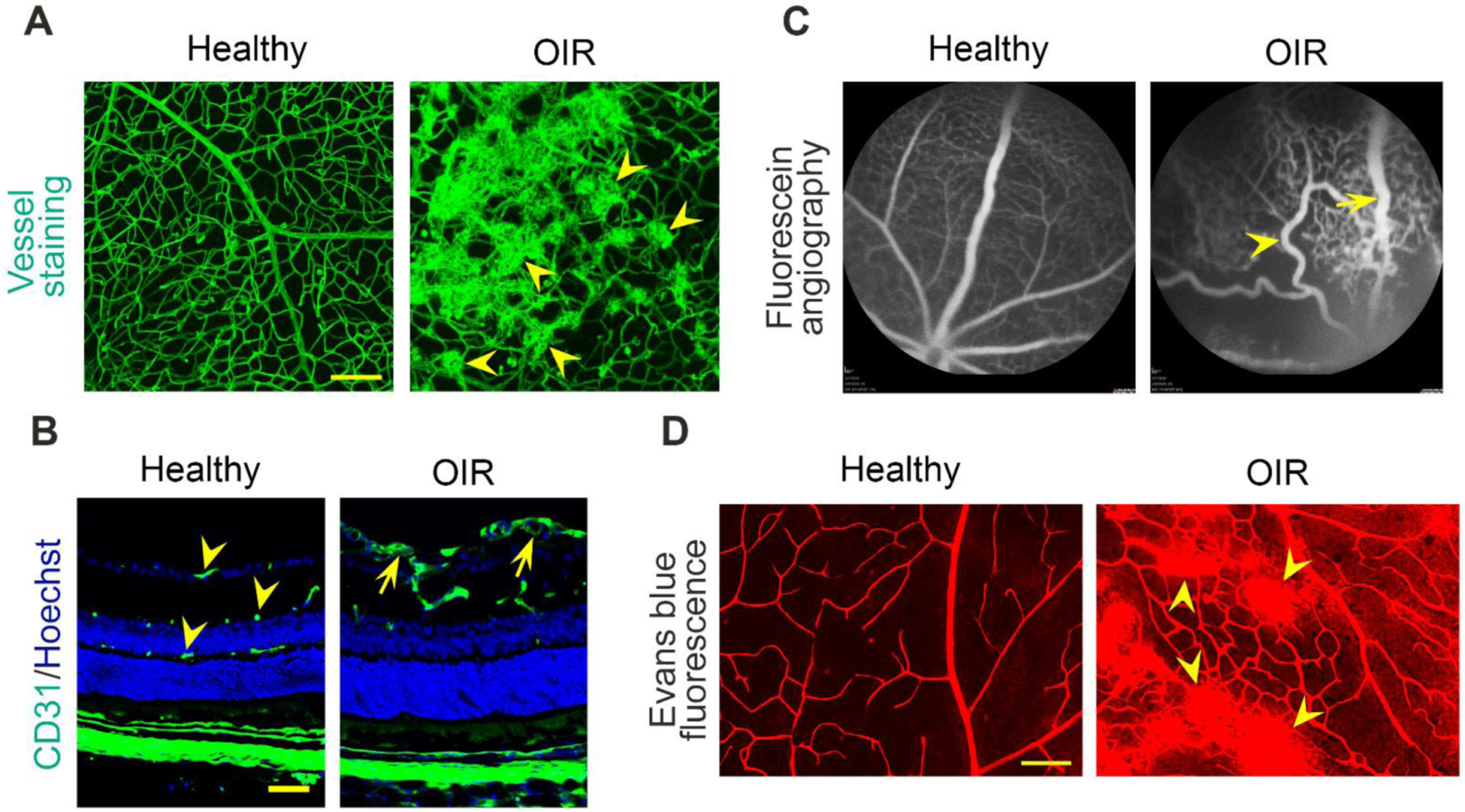
Vascular characteristics of ROP. A Flat-mount retinas of healthy and OIR mice stained with Alexa Flour 488-conjugated isolectin B4. Scale bar= 100 μm. Arrowheads indicate pathological retinal neovascularization. B Retinal sections of healthy vs. OIR mice stained with anti-CD31 monoclonal antibody and Hoechst. Scale bar= 50 μm. Arrowheads indicate normal superficial, intermediate and deep retinal plexuses. Arrows indicate pathological neovascularization in the superficial layer. C Fluorescein angiography images of healthy vs. OIR mice. The arrowhead indicates a tortuous artery. The arrow indicates a dilated vein. D Images showing the leakage of fluorescent Evans blue dye from retinal vessels in healthy and OIR mice. Arrowheads indicate the leaking sites in the OIR retina. Scale bar= 100 μm.
Neurovascular development of the retina
Retinal structure
Cells in the vertebrate retina are organized and structured into different layers that work in parallel and combination to produce complex visual outputs and maintain homeostasis (Fig. 2). Photoreceptors are located above the retinal pigment epithelium (RPE), which is the outermost layer of the retina, and are beneath the inner retinal layers of bipolar cells, amacrine cells and horizontal cells. Rods and cones are the two types of photoreceptors to sense light and transmit visual signals to bipolar cells, and this process is modulated by horizontal cells [10]. Retinal ganglion cells (RGCs) in the innermost layer of the retina receive signals from bipolar and amacrine cells and project axons into the visual centres in the brain through the optical nerve [10]. Müller cells are the dominant glial cells in the vertebrate retina, spanning the entire thickness of the retina and contacting all retinal neuronal somata and processes [11]. The processes of astrocytes and microglia constitute the perivascular glia limitans in the mature retina [12].
Fig. 2.
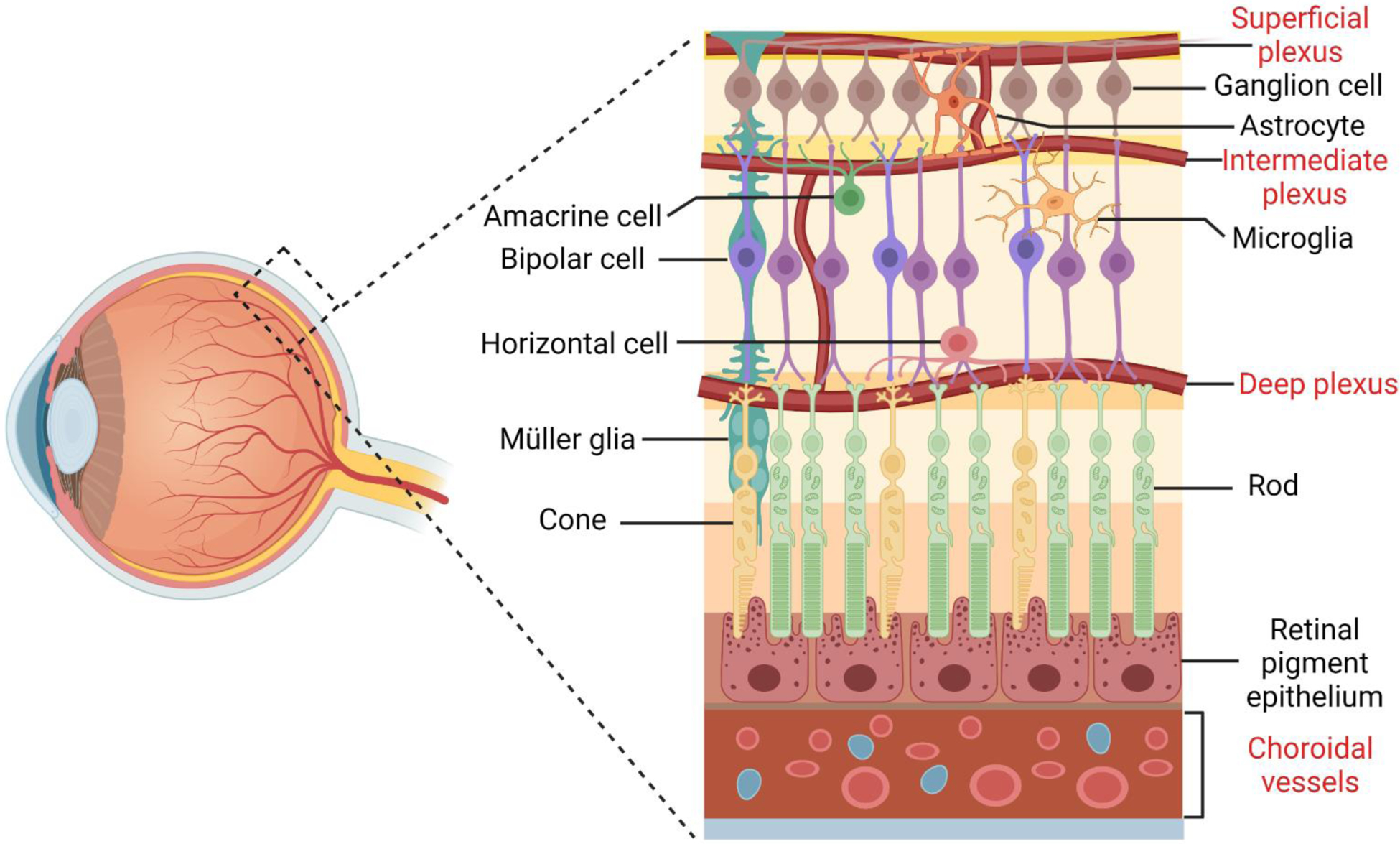
Cellular organization of the neuronal and vascular retina. There are six major types of neurons in the retina: rod and cone photoreceptors, bipolar cells, retinal ganglion cells, horizontal cells and amacrine cells. These neurons form complex neural circuities to detect light, generate electric impulse and transmit visual signals. Three basic types of glial cells are present in the human retina: Müller cells, astrocytes and microglia. The processes of astrocytes and microglia contribute to the blood-retina barrier together with pericytes and endothelial cells of the superficial, intermediate and deep vascular plexuses. Retinal pigmental epithelial cells are at the outer most layer of the retina, beneath which are choroidal vessels.
The retina has dual blood supplies: i) the central retinal artery enters the eye from the optical nerve head, expands radially toward the periphery on the retinal surface, penetrates into the inner retina and forms three layers of vascular plexuses and ii) the choriocapillaris supports the RPE and photoreceptors in the outer retina with oxygen and nutrients through diffusion (Fig. 2). As a result, the inner retina is vascularized, whereas the outer retina is avascular. In humans, primary retinal vessels form at the nerve fibre layer/RGC layer interface. Sprouts from the primary plexus veins penetrate into the retina and establish two deep laminar networks on either side of the inner nuclear layer (INL) [13]. In mice, the retinal vessels develop in a similar way as in humans, but the process begins postnatally, which makes it plausible to elicit OIR after birth [14]. While retinal vascularization is not complete until 38–40 weeks of gestation in humans [13], the choroidal vasculature is fully developed by 22 weeks with mature and fenestrated endothelial cells (ECs) [15], implying that retinal oxygenation in preterm infants mainly depends on the choroidal circulation. However, the absence of autoregulation of choroidal blood flow and the immature retina devoid of well-developed antioxidant systems in preterm babies favour retinal hyperoxygenation and peroxidation. This results in vasoconstriction and hyperoxia-induced vaso-obliteration during the supplemental oxygen in Phase I of ROP and could further develop into vaso-proliferative retinopathy in hypoxic Phase II [16]. Several clinical studies reported that the severity of ROP and the level of visual impairment are inversely correlated with choroidal thickness [17–19], whereas such findings were not confirmed by others [20, 21]. Therefore, a causal relationship among choroidal thickness, ROP severity and visual impairment needs to be further investigated in clinic.
Neurovascular interaction
The development of the retinal vasculature is a complex process during which ECs, neurons and glial cells, including Müller cells, astrocytes and microglia, interact and functionally depend on each other. The neuroretina, but not astrocytes, acts as a primary oxygen sensor that ultimately controls retinal vascular development by regulating an angiogenic astrocyte template [22]. RGC axons guide the formation of an astrocytic network, which subsequently directs retinal vasculature development [23]. RGCs are crucial to normal retinal vascularization as the loss of ganglion cells in Math5−/− mice blocks the development of the primary retinal vessels [24]. RGCs can sense changes in neuronal metabolic demands through G protein-coupled receptor-91 prior to hypoxia-inducible factor (HIF)-1α stabilization and induce the expression of pro-angiogenic factors, so that the vascular supply meets retinal demands [13]. On the other hand, RGCs also produce the anti-angiogenic protein semaphorin 3E to prevent the misdirection of the developing vessels and ensure coordinated retinal vascular sprouting and network formation [25].
Astrocytes play an important role in physiological RNV and appear in the immature and avascular retina before vascular development [26]. Hypoxic astrocytes secrete angiogenic factors that drive the radial migration of ECs into the hypoxic area toward the peripheral retina [14, 27], and in turn RNV and oxygen supply promote astrocytic differentiation in the developing retina [28]. Selective ablation of platelet-derived growth factor receptor alpha (PDGFRα) in astrocytes severely impaired astrocyte patterning and subsequent retinal angiogenesis [29]. Although EC migration, proliferation and maintenance are widely considered to be regulated by VEGF released from glial fibrillary acidic protein (GFAP)-immunopositive astrocytes during retinal development [12–14], data from genetically engineered mice suggest a controversial role of astrocyte-derived VEGF in physiological RNV. Two initial studies reported that mice with the conditional deletion of the VEGF gene in astrocytes have minimal or no impairment on retinal vasculature development [30, 31]. However, a recent study showed that selective elimination of VEGF in neonatal retinal astrocytes has no effect on proliferation or radial migration of astrocytes but completely blocks radial migration of ECs and retinal vessel development [32]. Another study reported that selective deletion of HIF-2α, a transcription factor to promote VEGF expression, in astrocytes significantly interferes with the development of astrocyte networks and reduces retinal vascular development [33]. Because of these conflicting results, the role of astrocyte-derived VEGF in physiological RNV remains elusive.
Although VEGF is expressed by RGCs, astrocytes, retinal pigment epithelial cells and photoreceptor cells, Müller cells are the primary cells that produce VEGF in the retina [34]. Interestingly, Müller cell-derived VEGF does not seem to be critical to ocular vasculogenesis, as mice with the conditional VEGF knockout in the cells have no apparent defects in retinal and choroidal vasculature [35]. Müller cells share the ability of astrocytes to play a major role in the formation of barrier properties in retinal vessels [36].
Proper formation of the retinal vasculature also requires an adequate resident microglial population. Resident microglial cells are located in two horizontal bands and often closely associated with blood vessels [37]. Depletion of resident retinal microglia reduces developmental vessel growth and density, which can be restored by intravitreal microglial injection [38].
Neurovascular abnormalities in ROP
ROP is a two-phase disease: Phase I (vaso-obliteration) is initiated at birth and ends at the withdrawal of supplemental oxygen treatment during approximately 32–34 weeks of gestation and Phase II (vaso-proliferation) immediately follows Phase I and is primarily characterized by pathological RNV and IVNV (Fig. 1A and B) [39]. To mimic the vascular changes in ROP, Smith et al. developed the first mouse model of OIR in 1994 [40], which has become one of the most commonly used animal models for studying the pathology and treatment of ROP. In this model, neonatal mice and their nursing mothers are exposed to 75% oxygen from postnatal day 7 (P7) to P12 and returned to room air at P12. Flat-mounted retinas of OIR mice at P17 exhibit a central zone of vaso-obliteration and pathological RNV at the border between the vascular and avascular zones. To establish rat animal model of ROP, Penn et al. exposed neonatal rats to fluctuating oxygen cycles between 50% and 10% every 24 h for 14 days at birth [41]. The rat model of OIR displays delayed physiological RNV in the peripheral retina and pathological RNV in vascularized areas.
Although the hallmark of ROP is abnormal retinal vasculature, increasing evidence supports a critical role for the neural retina in the disease pathogenesis. Retinas with OIR show changes in Müller cells, astrocytes and microglial cells preceding vascular changes [42, 43] and persistent ectopic synapses between rods and rod-bipolar cells [9]. The formation of ROP-like blood vessels is completely prevented in the retina following N-methyl-D-aspartic acid (NMDA)-induced neuronal cell loss [44]. Therefore, investigating the dysregulated crosstalk among retinal neurons, neuroglia and vessels may provide novel strategies for promoting physiological revascularization, preventing pathological RNV and restoring visual function of retinas in ROP infants.
RGCs
RGCs are particularly susceptible to transient, mild and systemic hypoxia [45] and act as sensors of ischaemic stress. Hypoxia significantly hindered neurite outgrowth and reduced cell viability of RGC-5 in vitro [45]. The hypoxia-evoked RGC loss is partially mediated by increased amounts of tumour necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) derived from activated microglial cells (Fig. 3) [46]. Additionally, hypoxia-mediated induction of neuronal nitric oxide synthase [47] and lysophosphatidic acid receptors under oxygen stress [45] also promote RGC loss. Neurovascular crosstalk between RGCs and ECs governs the progression of retinopathy through different pathways. In hypoxic retinas, accumulated succinate stimulates G protein-coupled receptor-91 (GPR91) primarily expressed in RGCs, modulates the production of various angiogenic factors, including VEGF, angiopoietin-1 (Ang-1) and angiopoietin-2 (Ang-2), and contributes to vessel growth in the settings of both normal retinal development and OIR (Fig. 3) [48]. However, a marked increase in the expression of the suppressor of cytokine signalling 3 (SOCS3) in the RGC layer and INL of OIR retinas inhibits the STAT3-mediated VEGF secretion from neurons and glial cells and limits pathological RNV [49].
Fig. 3.
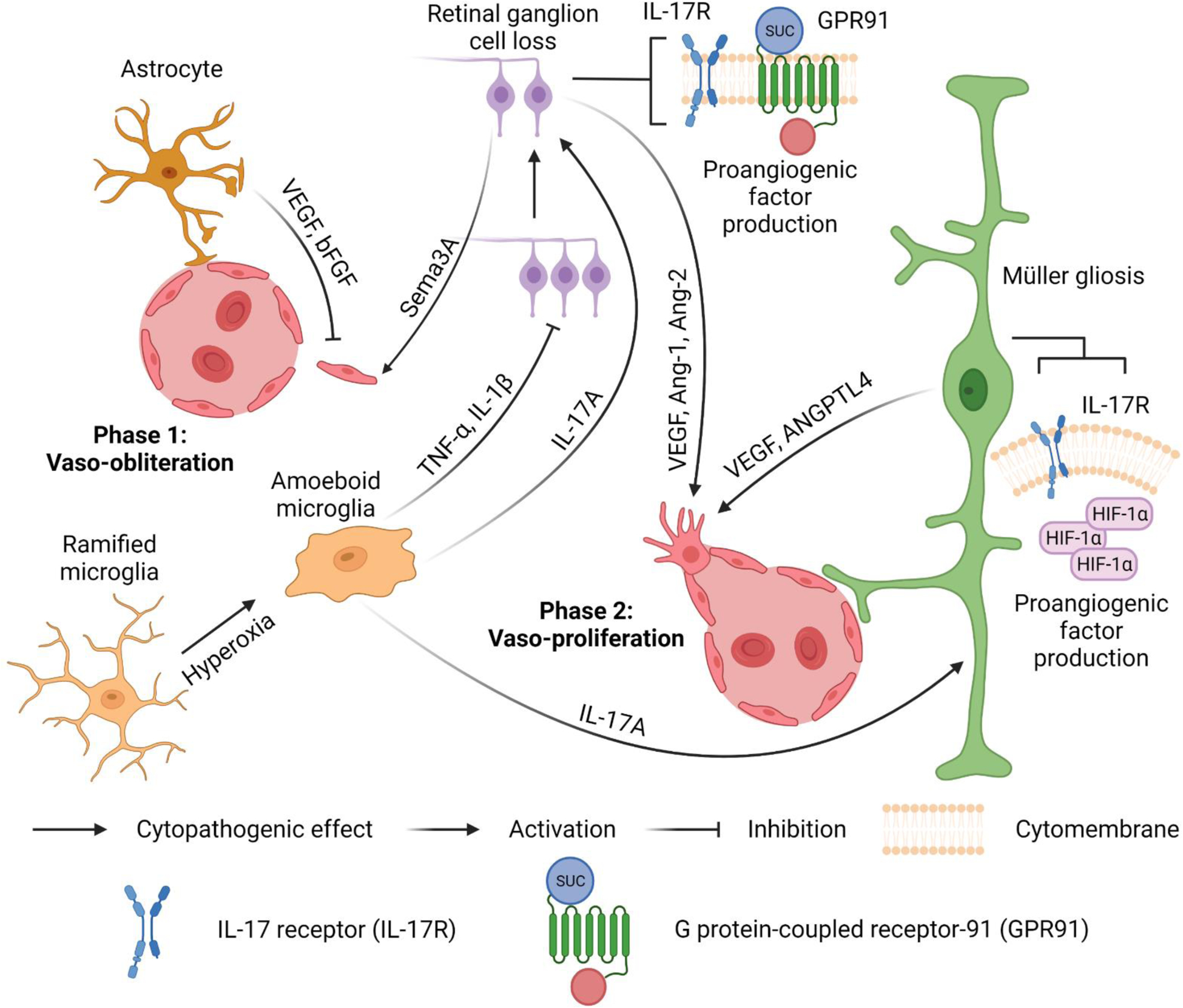
Neurovascular interactions in ROP. Phase 1: Vaso-obliteration. Activated microglia transform from ramified to amoeboid shapes. The tumour necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) secreted from activated amoeboid microglia induce pronounced retinal ganglion cell (RGC) loss and further stimulate the secretion of semaphorin 3A (Sema3A) from adjacent neurons to mediate microvascular injury. Vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) from astrocytes minimize vascular damage. Phase 2: Vaso-proliferation. Accumulated succinate stimulates G protein-coupled receptor-91 (GPR91) on RGCs to facilitate the production of proangiogenic factors, such as VEGF, angiopoietin-1 (Ang-1) and angiopoietin-2 (Ang-2). Stabilized HIF-1α in hypoxic Müller cells promotes the expression of VEGF and angiopoietin-like 4 (ANGPTL4) to induce neovascularization and vascular leakage. Furthermore, interleukin-17A (IL-17A) from amoeboid microglial cells targets interleukin-17 receptors (IL-17Rs) in Müller cells and RGCs to enhance the production of VEGF.
Astrocytes
During the hyperoxic phase in the mouse model of OIR, the cell density of astrocytes decreases to a half of the normal value (400 astrocytes/mm2) due to apoptosis in the central avascular zone and increases only after P17 when OIR regresses [50]. The lowest astrocyte densities are found within the angles between the large vessels of the central avascular area between P14 and P17 [50]. In contrast, a higher density of astrocytes was observed in the peripheral vascularized zone, but astrocyte density is minimally altered at vascular tufts [50]. Although the processes of astrocytes contact and intertwine with each other to form a network-like structure during ROP, the astrocyte network is sparse and features long processes and a reduced association with blood vessels via the end-feet [51].
In the rat OIR model, VEGF mRNA expression is the highest in Müller cells, scattered astrocytes and amacrine cells [52]. VEGF produced by astrocytes is responsible for stabilizing retinal vessels and exerts protective effects in high-oxygen environments. Deletion of astrocyte-derived VEGF dramatically increases hyperoxia-induced obliteration and causes the collapse of normally stable main arteries and veins [30], whereas another study showed that such deletion has no effect on hyperoxia-induced vaso-obliteration but suppresses pathological RNV in OIR mice [31]. The apparently inconsistent results of the vaso-obliteration between these two similar mouse models are yet to be resolved. Interestingly, intravitreal administration of astrocytes or astrocyte-conditioned media rescues endogenous astrocytes from degeneration within the vaso-obliterated zone, accelerates revascularization of normal retinal plexuses and reduces pathological RNV/IVNV, which may be partially attributed to VEGF and basic fibroblast growth factor (bFGF) in the astrocyte-conditioned media (Fig. 3) [42].
Müller glial cells
Müller cell gliosis, which is characterized by upregulation of GFAP expression, indicates that Müller cells have become ‘activated’ or ‘reactive’ in response to pathological alteration of the retina and is associated with virtually every disease of the retina [11]. The number of gliotic Müller cells increases across the entire retina of OIR mice, and these cells are generally localized in the ischaemic central retina rather than the perfused peripheral retina [53]. Hypoxic Müller cells develop a proangiogenic phenotype via activation of HIF-1α and HIF-2α signalling pathways. The expression of HIF-1α/HIF-2α is increased in hypoxic Müller cells [34], and disruption of HIF-1α/HIF-2α expression in the cells attenuates the overproduction of VEGF (Fig. 3) [54, 55]. Müller cell-derived VEGF plays a critical role in ischaemia-induced RNV and vascular leakage (Fig. 1D). In OIR mice, conditional deletion of the VEGF gene in Müller cell significantly inhibits proliferative RNV and attenuates the breakdown of the blood-retina barrier [35]. The expression of angiopoietin-like 4 (ANGPTL4), a potent proangiogenic factor independent of VEGF, is also upregulated by HIF-1α in hypoxic Müller cells (Fig. 3) [53, 56]. Inhibition of ANGPTL4 reduces the angiogenic potential of hypoxic Müller cells, which is additive with inhibition of VEGF expression [56].
Microglia
Microglia are resident macrophages in the retina and play an important role in the maintenance of the neuroretinal homeostasis. Increased number of activated microglial cells switching from ramified to amoeboid cell morphology were observed within the superficial retinal layer of the central avascular zone in OIR mice after the initiation of the hyperoxic phase from P8 to P12 and subsequently in hypoxic phase from P16 to P18 (Fig. 3) [37]. In the peripheral RNV area, activated ameboid microglia are attached to OIR-related vascular tufts, implicating that activated microglial cells may be involved in neovascular tuft formation [57].
Activated microglial cells produce IL-1β, which sustains the activation of microglia and induces microvascular injury through the release of the proapoptotic/repulsive factor semaphorin 3A (Sema3A) from adjacent neurons in the early hyperoxic stages of OIR (Fig. 3) [58]. Hypoxia-exposed microglia in vitro and activated microglia in OIR retinas express interleukin-17A (IL-17A), which interacts with interleukin-17 receptors (IL-17Rs) on Müller cells and RGCs and promotes the secretion of VEGF, thereby inducing vasculopathy (Fig. 3) [59]. On the other hand, exosomes from microglial cells exert protective effects against OIR by attenuating neovascularization and photoreceptor injury, which may result from inhibition of hypoxia-induced expression of inositol-requiring enzyme 1α (IRE1α) in photoreceptors by extremely high levels of microRNA-24–3p [60].
Photoreceptors and the inner retina
Photoreceptors, including rods and cones, can detect and convert photons into electric impulses that are transmitted by neurons to form visual perception in the brain. In the vertebrate retina, rods mediate low-light vision, while cones mediate daylight and colour vision [61]. Photoreceptors are also involved in retinal physiological angiogenesis, as photoreceptor-specific knockdown of platelet-derived growth factor B (PDGFB) inhibits retinal vascular network formation in wild-type mice [62]. ROP, even in mild cases, primarily and persistently affects the structure and function of photoreceptors [63] and has more adverse effects on rods than cone cells [64]. The impairment of rod photoreceptor function varies significantly with the severity during the acute phase of ROP [65]. Lower immature rod sensitivity is associated with higher retinal VEGF expression [66] and can predict development of retinal vascular abnormalities in rat ROP models, implying that aberrant rods may instigate blood vessel abnormalities [67].
Postreceptor cells, including the cells of the inner nuclear and RGC layers that are supplied by the retinal vasculature, are also affected by ROP. In a rat ROP model, postreceptor sensitivity of the retinoelectrography is low at a young age and recovers simultaneously with the resolution of retinal vascular abnormalities [66, 68]. Low postreceptors sensitivity is significantly correlated with high VEGF and Sema3A expression [66].
Current therapies for ROP
Cryotherapy and laser therapy
Cryotherapy and laser photocoagulation ablate the peripheral avascular retina, convert it to nonfunctional scar tissue and inhibit the growth of abnormal blood vessels. The standard treatment for ROP shifted from cryotherapy to laser photocoagulation in Western countries in the 1990s since clinical studies proved that laser therapy is safer and at least as effective as cryotherapy [69, 70]. For threshold ROP, laser-treated eyes exhibit better structural and functional outcomes and less myopia than those treated with cryotherapy [71, 72]. Portability, ease of use, precise delivery and minimal postprocedural adnexal inflammation [73] are further advantages of laser therapy, whereas the common complications of laser therapy include corneal and iris burns, anterior segment haemorrhage and acute pressure rise [74].
Anti-VEGF therapy
VEGF was initially discovered as a vascular permeability factor and subsequently characterized as a potent endothelial mitogenic factor that stimulates proliferation, migration and tube formation of ECs, thereby driving angiogenesis [39]. It was subsequently characterized as a neurotrophic and neuroprotective factor with an important role in neurogenesis [75]. At least five retinal cell types, including RPE, astrocytes, Müller cells, vascular endothelial cells and ganglion cells, have the capacity to produce and secrete VEGF [76]. In vitro studies have demonstrated that Müller cells and astrocytes are major contributors to VEGF secretion under hypoxic conditions [76].
VEGF levels are closely correlated with the progression of ROP, as patients with vascularly active stage 4 ROP demonstrate increased VEGF levels in the vitreous [77]. In OIR mice, VEGF expression is downregulated in the retina after 6 h of exposure to 75% oxygen in Phase I [78], upregulated in response to relative hypoxia in Phase II and remains elevated during the development of neovascularization [79]. During proliferative vasculopathy in ROP, VEGF is mainly expressed by Müller glia and neurons of the ganglion cell layer after degeneration of astrocytes [35, 80]. It was reported that Müller cell-derived VEGF is a significant contributor to pathological RNV in OIR mice [35], whereas the role of astrocyte-derived VEGF in the vascular pathology of OIR remains elusive [30, 31].
To date, six VEGF inhibitors have been approved for clinical therapy of neovascular diseases in adults, including ranibizumab, pegaptanib, brolucizumab, conbercept, aflibercept and bevacizumab. The former five are approved for ocular diseases, such as wet age-related macular degeneration and/or diabetic retinopathy, whereas the latter two are approved for cancer therapy. These agents are often used for off-label treatment of ROP [39]. Compared with traditional laser therapy, the administration of anti-VEGF agents is relatively convenient, requires minimal anaesthesia, has the potential to improve peripheral vision and produces less refractive error [81]. After a randomized, multi-centre Phase 3 clinical trial, ranibizumab was recently approved for ROP therapy in Europe [82].
However, VEGF is of pivotal importance to both physiological and pathological angiogenesis. Compared to neovascular diseases in adults with quiescent and mature healthy vessels, ROP in preterm infants with the developing retina is a unique disease because of concurrent physiological and pathological RNV [39]. Indeed, VEGF binds to OIR and healthy retinal vessels in neonatal mice equally well but minimally to healthy adult vessels [83, 84]. As a result, intravitreal aflibercept in high doses not only delays vascular growth but also alters retinal structure and function in neonatal mice and dogs [83, 85–87]. Clinical trials of ROP also indicated that intravitreal bevacizumab is associated with significant abnormalities in the fovea/macula [3–5]. The adverse effects of anti-VEGF on the retina could result from direct inhibition of retinal neurons due to the blockade of VEGF neurotrophic activity or indirect actions of anti-angiogenesis that limit blood supply to the developing retina.
Additional safety concern is the leakage of intravitreally injected VEGF inhibitors from the eye into the circulation owing to the immature blood-retinal barrier in ROP infants. A recent study reported that serum levels of VEGF in ROP patients decreased for 2 months after intravitreal injections of bevacizumab [6]. The effects of lowering systemic VEGF levels on developing organs of premature infants have raised safety concerns. Clinical studies found that ROP infants treated with bevacizumab have a higher risk of severe neurodevelopmental disabilities than those treated with laser therapy [7]. A recent study also reported that aflibercept via single intravitreal injection can affect body weight gain, the fellow eye and renal vessels in the mouse OIR model [88]. In conclusion, although anti-VEGF agents are effective for ROP therapy, the dosage should be selected individually based on eye and infant size, and further studies are needed to evaluate the long-term safety of these treatments on ocular structure and function and their delayed systemic adverse effects. As a result, anti-VEGF drug has not been approved for ROP therapy in the USA.
HIF regulation and related therapies
VEGF expression is regulated by HIF transcription factor family, of which HIF-1 and 2 are the most characterized [89, 90]. In the presence of oxygen, HIF-1α is hydroxylated on prolyl residues via prolyl hydroxylase domain enzymes (PHDs) and sequestered for ubiquitination and proteasomal degradation by binding to the von Hippel-Lindau E3 ligase complex [90, 91]. During hypoxia, unhydroxylated HIF-1α accumulates in the cytosol and translocates into the nucleus, where it dimerizes with constitutively expressed HIF-1β subunit to form a complex, binds to hypoxia-responsive elements (HREs) of target genes and activates their transcription. HIF-2α is regulated in a similar manner [90].
During retinogenesis, HIF-1α and HIF-2α in retinal progenitor cells respond to hypoxia in avascular retina, upregulate the expression of pro-angiogenic mediators, including VEGF and play essential roles in the capillary development of the inner plexiform layer (IPL) and outer plexiform layer (OPL), respectively [32, 92, 93]. In Phase I of ROP, hyperoxia downregulates HIF-1, thereby suppressing the expression of VEGF and other pro-angiogenic factors with subsequent vaso-obliteration. In Phase II, the withdrawal of supplemental oxygen and the poorly vascularized retina with increasing metabolic demands create a perfect hypoxic environment for HIF-1 upregulation and excessive VEGF production that drives pathological RNV [57]. While HIF-1α demonstrated a rapid but transient accumulation in the inner retina of OIR mice, expression of HIF-2α is relatively delayed but sustains in the retina until the end of the ischaemic stage at P17 [94]. It was reported that HIF-2α promotes RNV in OIR mice by upregulating the expression of VEGF and erythropoietin (EPO) [94, 95].
An effective strategy to mitigate ROP is to lower oxygen saturation targets in premature infants in Phase I for reducing the risk of vasculopathy in Phase II, but this manipulation simultaneously increased the mortality rate [96]. An alternative approach is to mitigate the effects of hyperoxia in Phase I using small-molecule HIF stabilizers without lowering supplemental oxygen, thereby preventing or minimizing the progression to the proliferative stage of the disease [97–100]. Sears et al. first demonstrated that stabilizing hepatic HIF activity specifically with dimethyloxalylglycine (DMOG), a PHD inhibitor, induced the expression of VEGF and EPO during the hyperoxic phase and prevented OIR in mice in the hypoxic phase [97]. PHD inhibitors were subsequently confirmed to prevent OIR, preserve retinal function and structure in multiple animal models with undetectable adverse effects [98–100]. The findings support a low-dose, intermittent approach to prevent OIR with PHD inhibitors. Despite the promising results, this pharmaceutical approach for ROP prevention is yet to be investigated in clinical trials.
Other therapies investigated in clinical trials
A number of other pharmacologic interventions have been investigated in clinical trials for ROP, including EPO [101], the combination of insulin-like growth factor 1 and insulin-like growth factor-binding protein 3 [102], vitamin E [103], vitamin A [104], omega-3 polyunsaturated fatty acids [105] and propranolol [106]. Whereas most of these therapies conferred limited efficacy in clinical trials [101, 103–105, 107], oral propranolol is effective to reduce ROP progression at high doses but is associated with potential safety concerns [108, 109]. Propranolol 0.2% eye micro-drops for topical administration was reported recently to be well-tolerated and appeared to effectively reduce the ROP progression in a Phase IIB trial [106]. Further randomized, controlled multicentre trials are required to confirm the safety and efficacy of propranolol for ROP therapy.
Secretogranin III (Scg3) antagonist as an emerging therapy
Compared to neovascular diseases in adults, such as wet age-related macular degeneration, proliferative diabetic retinopathy and cancer, developing drug therapies for ROP encounters two unique challenges: i) concurrent physiological and pathological angiogenesis [39] and ii) susceptibility of the developing retina to adverse side effects of pharmacological reagents. Anti-Scg3 therapy was recently developed to tackle the challenges.
Scg3 predominantly expressed in neurons, endocrine and neuroendocrine cells is a member of the granin family that regulate the biogenesis of secretory granules [110]. In contrast with the VEGF family, Scg3 shares minimal sequence homology to other proteins, including other granins. Scg3−/− mice exhibit a normal gross phenotype, including viability, fertility, locomotor behaviour or obesity, implicating that Scg3 is not essential for the secretion of neurotransmitters and vital hormones [111]. By applying a novel technology of comparative ligandomics, we recently discovered that Scg3 is a disease-restricted pro-angiogenic factor that selectively binds to diabetic versus healthy retinal vessels [112]. Scg3 preferentially induced angiogenesis and retinal vascular leakage of diabetic but not healthy mice, suggesting that the putative Scg3 receptor is markedly upregulated in diabetic vessels while minimally expressed on normal vessels [112, 113]. Recently, we developed a unique in vivo endothelial ligand binding assay and quantitatively confirmed that the binding of Scg3, but not VEGF, to retinal vessels of OIR vs. healthy mice markedly increases by sevenfold and is fully blocked by Scg3-neutralizing antibody [83]. Functional immunohistochemistry visualized that vessel-bound Scg3 predominantly colocalizes with neovascular tufts and disorganized neovessels in OIR retinas with minimal Scg3 signals detected on well-organized healthy vessels [83]. In contrast, VEGF bound equally to both healthy and OIR neovessels in the developing retina [83].
The differential disease binding selectivity of Scg3 and VEGF has profound implications for disease-targeted therapy. Scg3 selectively binds to upregulated Scg3 receptor(s) stationarily expressed on OIR ECs without cross-reaction with healthy ECs that have low or absent receptor expression, and therefore, Scg3 antagonists should target OIR but not healthy vessels with minimal adverse side effects on healthy vessels. By contrast, VEGF lacks such disease selectivity, and anti-VEGF drugs adversely affect healthy vasculature with collateral side effects as elaborated in the section above.
To investigate the difference in disease-targeted therapy, we developed Scg3-neutralizing monoclonal antibodies (mAbs) and validated their high efficacy to alleviate OIR in mice [86, 112]. Preclinical studies confirmed that anti-Scg3 mAbs have a superior safety profile over aflibercept [86]. To facilitate the translation, one of the optimal anti-Scg3 mAbs was converted into humanized antibody Fab fragment (hFab). Both anti-Scg3 hFab and aflibercept ameliorate pathological RNV, neovascular tufts, retinal arterial tortuosity and vein dilation in OIR mice with similar high efficacies. However, anti-Scg3 hFab selectively blocks pathological but not physiological angiogenesis in OIR mice, whereas aflibercept indiscriminately inhibits both types of angiogenesis [83]. Dose-response curves of efficacy and toxicity revealed that the therapeutic window of anti-Scg3 hFab is at least 10X wider than that of aflibercept. No abnormality of the retinal structure and function in OIR mice treated with anti-Scg3 hFab was detected. Furthermore, the normal phenotype of vascular and retinal development of Scg3-knockout mice attests to the optimal vascular and neuronal safety of anti-Scg3 hFab [83]. These findings suggest that anti-Scg3 hFab has the potential to be developed as the next-generation disease-targeted anti-angiogenic therapy to stringently inhibit pathological but not physiological angiogenesis in ROP. However, the safety and efficacy of this emerging therapy are yet to be fully investigated in clinical trials.
Conclusions
During retinal vascular development, growth factors derived from neurons and glia stimulate endothelial migration, proliferation and development into three layers of retinal plexuses. Although ROP is mainly manifested by vascular symptoms, including pathological IVNV/RNV and tortuosity and dilation of retinal vessels, these abnormalities are increasingly recognized as a result of neurovascular dysregulation with profound therapeutic implications. Hypoxia-induced overexpression of angiogenic factors, such as VEGF, from neurons and glia drives pathological RNV, and many of these factors play important roles in both physiological and pathological RNV in ROP. Recently discovered neuron-derived Scg3 is a disease-selective angiogenic factor that stringently regulates diseased but not healthy retinal vessels. Therefore, anti-Scg3 selectively inhibits pathological but not physiological angiogenesis with minimal adverse effects on healthy vessels, as opposed to anti-VEGF drugs that block both types of angiogenesis with safety concerns. These examples highlight that investigation of neurovascular communication is of therapeutic importance to ROP.
Funding.
This work was partially supported by NIH R01EY027749 (WL), R24EY028764 (WL), R43EY031238 (WL), R43EY032827 (WL), R41EY027665 (WL), NIH P30EY002520, Knights Templar Eye Foundation Endowment in Ophthalmology (WL) and unrestricted institutional grants from Research to Prevent Blindness (RPB) to the Department of Ophthalmology, Baylor College of Medicine.
Abbreviations
- Ang-1
Angiopoietin-1
- Ang-2
Angiopoietin-2
- ANGPTL4
Angiopoietin-like 4
- bFGF
Basic fibroblast growth factor
- ECs
Endothelial cells
- EPO
Erythropoietin
- GFAP
Glial fibrillary acidic protein
- GPR91
G protein-coupled receptor-91
- hFab
Humanized antibody Fab fragment
- HIF
Hypoxia-inducible factor
- IL-17A
Interleukin-17A
- IL-17R
Interleukin-17 receptor
- IL-1β
Interleukin-1β
- INL
Inner nuclear layer
- IVNV
Intravitreal neovascularization
- mAb
Monoclonal antibody
- OIR
Oxygen-induced retinopathy
- PHD
Prolyl hydroxylase domain enzyme
- RGCs
Retinal ganglion cells
- RNV
Retinal neovascularization
- ROP
Retinopathy of prematurity
- RPE
Retinal pigment epithelium
- Scg3
Secretogranin III
- Sema3A
Semaphorin 3A
- TNF-α
Tumour necrosis factor-α
- VEGF
Vascular endothelial growth factor
Footnotes
Conflict of interest. WL is the shareholder of Everglades Biopharma, LLC and LigandomicsRx, LLC. WL is an inventor of issued and pending patents. The remaining authors declare no conflict of interest.
References
- 1.Hartnett ME (2015) Pathophysiology and Mechanisms of Severe Retinopathy of Prematurity. Ophthalmology 122:200–210. 10.1016/j.ophtha.2014.07.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blencowe H, Lawn JE, Vazquez T, et al. (2013) Preterm-associated visual impairment and estimates of retinopathy of prematurity at regional and global levels for 2010. Pediatr Res 74 (Suppl 1):35–49. 10.1038/pr.2013.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lepore D, Quinn GE, Molle F, et al. (2014) Intravitreal Bevacizumab versus Laser Treatment in Type 1 Retinopathy of Prematurity. Ophthalmology 121:2212–2219. 10.1016/j.ophtha.2014.05.015 [DOI] [PubMed] [Google Scholar]
- 4.Vogel RN, Strampe M, Fagbemi OE, et al. (2018) Foveal Development in Infants Treated with Bevacizumab or Laser Photocoagulation for Retinopathy of Prematurity. Ophthalmology 125:444–452. 10.1016/j.ophtha.2017.09.020 [DOI] [PubMed] [Google Scholar]
- 5.Clark A, Wright T, Isaac M, et al. (2017) Macular morphology following unilateral bevacizumab injection for retinopathy of prematurity: an OCT study. J AAPOS 21:499–501.e1. 10.1016/j.jaapos.2017.06.024 [DOI] [PubMed] [Google Scholar]
- 6.Wu W-C, Shih C-P, Lien R, et al. (2017) SERUM VASCULAR ENDOTHELIAL GROWTH FACTOR AFTER BEVACIZUMAB OR RANIBIZUMAB TREATMENT FOR RETINOPATHY OF PREMATURITY. Retina 37:694–701. 10.1097/IAE.0000000000001209 [DOI] [PubMed] [Google Scholar]
- 7.Morin J, Luu TM, Superstein R, et al. (2016) Neurodevelopmental Outcomes Following Bevacizumab Injections for Retinopathy of Prematurity. Pediatrics 137:e20153218. 10.1542/peds.2015-3218 [DOI] [PubMed] [Google Scholar]
- 8.O’Connor AR, Stephenson T, Johnson A, et al. (2002) Long-term ophthalmic outcome of low birth weight children with and without retinopathy of prematurity. Pediatrics 109:12–18. 10.1542/peds.109.1.12 [DOI] [PubMed] [Google Scholar]
- 9.Mezu-Ndubuisi OJ, Macke EL, Kalavacherla R, et al. (2020) Long-term evaluation of retinal morphology and function in a mouse model of oxygen-induced retinopathy. Mol Vis 26:257–276 [PMC free article] [PubMed] [Google Scholar]
- 10.Hoon M, Okawa H, Della Santina L, Wong ROL (2014) Functional architecture of the retina: development and disease. Prog Retin Eye Res 42:44–84. 10.1016/j.preteyeres.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bringmann A, Pannicke T, Grosche J, et al. (2006) Müller cells in the healthy and diseased retina. Prog Retin Eye Res 25:397–424. 10.1016/j.preteyeres.2006.05.003 [DOI] [PubMed] [Google Scholar]
- 12.Provis JM (2001) Development of the primate retinal vasculature. Prog Retin Eye Res 20:799–821. 10.1016/s1350-9462(01)00012-x [DOI] [PubMed] [Google Scholar]
- 13.Selvam S, Kumar T, Fruttiger M (2018) Retinal vasculature development in health and disease. Prog Retin Eye Res 63:1–19. 10.1016/j.preteyeres.2017.11.001 [DOI] [PubMed] [Google Scholar]
- 14.Sapieha P (2012) Eyeing central neurons in vascular growth and reparative angiogenesis. Blood 120:2182–2194. 10.1182/blood-2012-04-396846 [DOI] [PubMed] [Google Scholar]
- 15.Lutty GA, McLeod DS (2018) Development of the hyaloid, choroidal and retinal vasculatures in the fetal human eye. Prog Retin Eye Res 62:58–76. 10.1016/j.preteyeres.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hardy P (2000) Oxidants, nitric oxide and prostanoids in the developing ocular vasculature: a basis for ischemic retinopathy. Cardiovasc Res 47:489–509. 10.1016/S0008-6363(00)00084-5 [DOI] [PubMed] [Google Scholar]
- 17.Wu W-C, Shih C-P, Wang N-K, et al. (2013) Choroidal Thickness in Patients With a History of Retinopathy of Prematurity. JAMA Ophthalmol 131:1451–1458. 10.1001/jamaophthalmol.2013.5052 [DOI] [PubMed] [Google Scholar]
- 18.Erol MK, Coban DT, Ozdemir O, et al. (2016) CHOROIDAL THICKNESS IN INFANTS WITH RETINOPATHY OF PREMATURITY. Retina 36:1191–1198. 10.1097/IAE.0000000000000866 [DOI] [PubMed] [Google Scholar]
- 19.Anderson MF, Ramasamy B, Lythgoe DT, Clark D (2014) Choroidal thickness in regressed retinopathy of prematurity. Eye (Lond) 28:1461–1468. 10.1038/eye.2014.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fieß A, Christian L, Kölb-Keerl R, et al. (2016) Peripapillary Choroidal Thickness in Former Preterm and Full-Term Infants Aged From 4 to 10 Years. Invest Ophthalmol Vis Sci 57:6548–6553. 10.1167/iovs.16-20128 [DOI] [PubMed] [Google Scholar]
- 21.Muslubas IS, Karacorlu M, Hocaoglu M, et al. (2017) Retinal and Choroidal Thickness in Children with a History of Retinopathy of Prematurity and Transscleral Diode Laser Treatment. Eur J of Ophthalmol 27:190–195. 10.5301/ejo.5000843 [DOI] [PubMed] [Google Scholar]
- 22.Nakamura-Ishizu A, Kurihara T, Okuno Y, et al. (2012) The formation of an angiogenic astrocyte template is regulated by the neuroretina in a HIF-1-dependent manner. Dev Biol 363:106–114. 10.1016/j.ydbio.2011.12.027 [DOI] [PubMed] [Google Scholar]
- 23.O’Sullivan ML, Puñal VM, Kerstein PC, et al. (2017) Astrocytes follow ganglion cell axons to establish an angiogenic template during retinal development. Glia 65:1697–1716. 10.1002/glia.23189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edwards MM, McLeod DS, Li R, et al. (2012) The deletion of Math5 disrupts retinal blood vessel and glial development in mice. Exp Eye Res 96:147–156. 10.1016/j.exer.2011.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim J, Oh W-J, Gaiano N, et al. (2011) Semaphorin 3E-Plexin-D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev 25:1399–1411. 10.1101/gad.2042011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorrell MI, Aguilar E, Friedlander M (2002) Retinal vascular development is mediated by endothelial filopodia, a preexisting astrocytic template and specific R-cadherin adhesion. Invest Ophthalmol Vis Sci 43:3500–3510 [PubMed] [Google Scholar]
- 27.Stone J, Itin A, Alon T, et al. (1995) Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci 15:4738–4747. 10.1523/JNEUROSCI.15-07-04738.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duan L-J, Pan SJ, Sato TN, Fong G-H (2017) Retinal Angiogenesis Regulates Astrocytic Differentiation in Neonatal Mouse Retinas by Oxygen Dependent Mechanisms. Sci Rep 7:17608. 10.1038/s41598-017-17962-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tao C, Zhang X (2016) Retinal Proteoglycans Act as Cellular Receptors for Basement Membrane Assembly to Control Astrocyte Migration and Angiogenesis. Cell Rep 17:1832–1844. 10.1016/j.celrep.2016.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott A, Powner MB, Gandhi P, et al. (2010) Astrocyte-derived vascular endothelial growth factor stabilizes vessels in the developing retinal vasculature. PLoS One 5:e11863. 10.1371/journal.pone.0011863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weidemann A, Krohne TU, Aguilar E, et al. (2010) Astrocyte hypoxic response is essential for pathological but not developmental angiogenesis of the retina. Glia 58:1177–1185. 10.1002/glia.20997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rattner A, Williams J, Nathans J (2019) Roles of HIFs and VEGF in angiogenesis in the retina and brain. J Clin Invest 129:3807–3820. 10.1172/JCI126655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duan L-J, Takeda K, Fong G-H (2014) Hypoxia inducible factor-2α regulates the development of retinal astrocytic network by maintaining adequate supply of astrocyte progenitors. PLoS One 9:e84736. 10.1371/journal.pone.0084736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Liu J, Hoh J, Liu J (2019) Müller cells in pathological retinal angiogenesis. Transl Res 207:96–106. 10.1016/j.trsl.2018.12.006 [DOI] [PubMed] [Google Scholar]
- 35.Bai Y, Ma J, Guo J, et al. (2009) Müller cell-derived VEGF is a significant contributor to retinal neovascularization. J Pathol 219:446–454. 10.1002/path.2611 [DOI] [PubMed] [Google Scholar]
- 36.Tout S, Chan-Ling T, Holländer H, Stone J (1993) The role of Müller cells in the formation of the blood-retinal barrier. Neuroscience 55:291–301. 10.1016/0306-4522(93)90473-s [DOI] [PubMed] [Google Scholar]
- 37.Fischer F, Martin G, Agostini HT (2011) Activation of retinal microglia rather than microglial cell density correlates with retinal neovascularization in the mouse model of oxygen-induced retinopathy. J Neuroinflammation 8:120. 10.1186/1742-2094-8-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Checchin D, Sennlaub F, Levavasseur E, et al. (2006) Potential role of microglia in retinal blood vessel formation. Invest Ophthalmol Vis Sci 47:3595–3602. 10.1167/iovs.05-1522 [DOI] [PubMed] [Google Scholar]
- 39.Dai C, Webster KA, Bhatt A, et al. (2021) Concurrent Physiological and Pathological Angiogenesis in Retinopathy of Prematurity and Emerging Therapies. Int J Mol Sci 22:4809. 10.3390/ijms22094809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith LE, Wesolowski E, McLellan A, et al. (1994) Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci 35:101–111 [PubMed] [Google Scholar]
- 41.Penn JS, Henry MM, Tolman BL (1994) Exposure to alternating hypoxia and hyperoxia causes severe proliferative retinopathy in the newborn rat. Pediatr Res 36:724–731. 10.1203/00006450-199412000-00007 [DOI] [PubMed] [Google Scholar]
- 42.Dorrell MI, Aguilar E, Jacobson R, et al. (2010) Maintaining retinal astrocytes normalizes revascularization and prevents vascular pathology associated with oxygen-induced retinopathy. Glia 58:43–54. 10.1002/glia.20900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fletcher EL, Downie LE, Hatzopoulos K, et al. (2010) The significance of neuronal and glial cell changes in the rat retina during oxygen-induced retinopathy. Doc Ophthalmol 120:67–86. 10.1007/s10633-009-9193-6 [DOI] [PubMed] [Google Scholar]
- 44.Nakano A, Asano D, Kondo R, et al. (2018) Retinal neuronal cell loss prevents abnormal retinal vascular growth in a rat model of retinopathy of prematurity. Exp Eye Res 168:115–127. 10.1016/j.exer.2017.12.007 [DOI] [PubMed] [Google Scholar]
- 45.Yang C, Lafleur J, Mwaikambo BR, et al. (2009) The role of lysophosphatidic acid receptor (LPA1) in the oxygen-induced retinal ganglion cell degeneration. Invest Ophthalmol Vis Sci 50:1290–1298. 10.1167/iovs.08-1920 [DOI] [PubMed] [Google Scholar]
- 46.Sivakumar V, Foulds WS, Luu CD, et al. (2011) Retinal ganglion cell death is induced by microglia derived pro-inflammatory cytokines in the hypoxic neonatal retina. J Pathol 224:245–260. 10.1002/path.2858 [DOI] [PubMed] [Google Scholar]
- 47.Rathnasamy G, Sivakumar V, Rangarajan P, et al. (2014) NF-κB-mediated nitric oxide production and activation of caspase-3 cause retinal ganglion cell death in the hypoxic neonatal retina. Invest Ophthalmol Vis Sci 55:5878–5889. 10.1167/iovs.13-13718 [DOI] [PubMed] [Google Scholar]
- 48.Sapieha P, Sirinyan M, Hamel D, et al. (2008) The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat Med 14:1067–1076. 10.1038/nm.1873 [DOI] [PubMed] [Google Scholar]
- 49.Sun Y, Ju M, Lin Z, et al. (2015) SOCS3 in retinal neurons and glial cells suppresses VEGF signaling to prevent pathological neovascular growth. Sci Signal 8:ra94. 10.1126/scisignal.aaa8695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bucher F, Stahl A, Agostini HT, Martin G (2013) Hyperoxia causes reduced density of retinal astrocytes in the central avascular zone in the mouse model of oxygen-induced retinopathy. Mol Cell Neurosci 56:225–233. 10.1016/j.mcn.2013.06.001 [DOI] [PubMed] [Google Scholar]
- 51.Nakano A, Kondo R, Kaneko Y, et al. (2020) Changes in components of the neurovascular unit in the retina in a rat model of retinopathy of prematurity. Cell Tissue Res 379:473–486. 10.1007/s00441-019-03112-9 [DOI] [PubMed] [Google Scholar]
- 52.Dorey CK, Aouididi S, Reynaud X, et al. (1996) Correlation of vascular permeability factor/vascular endothelial growth factor with extraretinal neovascularization in the rat. Arch Ophthalmol 114:1210–1217. 10.1001/archopht.1996.01100140410008 [DOI] [PubMed] [Google Scholar]
- 53.Xin X, Rodrigues M, Umapathi M, et al. (2013) Hypoxic retinal Muller cells promote vascular permeability by HIF-1-dependent up-regulation of angiopoietin-like 4. Proc Natl Acad Sci U S A 110:E3425–3434. 10.1073/pnas.1217091110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin M, Chen Y, Jin J, et al. (2011) Ischaemia-induced retinal neovascularisation and diabetic retinopathy in mice with conditional knockout of hypoxia-inducible factor-1 in retinal Müller cells. Diabetologia 54:1554–1566. 10.1007/s00125-011-2081-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rattner A, Wang Y, Zhou Y, et al. (2014) The role of the hypoxia response in shaping retinal vascular development in the absence of Norrin/Frizzled4 signaling. Invest Ophthalmol Vis Sci 55:8614–8625. 10.1167/iovs.14-15693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Babapoor-Farrokhran S, Jee K, Puchner B, et al. (2015) Angiopoietin-like 4 is a potent angiogenic factor and a novel therapeutic target for patients with proliferative diabetic retinopathy. Proc Natl Acad Sci U S A 112:E3030–3039. 10.1073/pnas.1423765112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu Y, Lu X, Hu Y, et al. (2018) Melatonin attenuated retinal neovascularization and neuroglial dysfunction by inhibition of HIF-1α-VEGF pathway in oxygen-induced retinopathy mice. J Pineal Res 64:e12473. 10.1111/jpi.12473 [DOI] [PubMed] [Google Scholar]
- 58.Rivera JC, Sitaras N, Noueihed B, et al. (2013) Microglia and interleukin-1β in ischemic retinopathy elicit microvascular degeneration through neuronal semaphorin-3A. Arterioscler Thromb Vasc Biol 33:1881–1891. 10.1161/ATVBAHA.113.301331 [DOI] [PubMed] [Google Scholar]
- 59.Talia DM, Deliyanti D, Agrotis A, Wilkinson-Berka JL (2016) Inhibition of the Nuclear Receptor RORγ and Interleukin-17A Suppresses Neovascular Retinopathy: Involvement of Immunocompetent Microglia. Arterioscler Thromb Vasc Biol 36:1186–1196. 10.1161/ATVBAHA.115.307080 [DOI] [PubMed] [Google Scholar]
- 60.Xu W, Wu Y, Hu Z, et al. (2019) Exosomes from Microglia Attenuate Photoreceptor Injury and Neovascularization in an Animal Model of Retinopathy of Prematurity. Mol Ther Nucleic Acids 16:778–790. 10.1016/j.omtn.2019.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu Y (1995) Phototransduction in Rods and Cones. In: Kolb H, Fernandez E, Nelson R (eds) Webvision: The Organization of the Retina and Visual System. University of Utah Health Sciences Center, Salt Lake City (UT) [PubMed] [Google Scholar]
- 62.Fu Z, Löfqvist CA, Liegl R, et al. (2018) Photoreceptor glucose metabolism determines normal retinal vascular growth. EMBO Mol Med 10:76–90. 10.15252/emmm.201707966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fulton AB, Hansen RM, Moskowitz A, Akula JD (2009) The neurovascular retina in retinopathy of prematurity. Prog Retin Eye Res 28:452–482. 10.1016/j.preteyeres.2009.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fulton AB, Hansen RM, Moskowitz A (2008) The cone electroretinogram in retinopathy of prematurity. Invest Ophthalmol Vis Sci 49:814–819. 10.1167/iovs.07-1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fulton AB, Hansen RM, Petersen RA, Vanderveen DK (2001) The rod photoreceptors in retinopathy of prematurity: an electroretinographic study. Arch Ophthalmol 119:499–505. 10.1001/archopht.119.4.499 [DOI] [PubMed] [Google Scholar]
- 66.Akula JD, Mocko JA, Benador IY, et al. (2008) The neurovascular relation in oxygen-induced retinopathy. Mol Vis 14:2499–2508 [PMC free article] [PubMed] [Google Scholar]
- 67.Akula JD, Hansen RM, Martinez-Perez ME, Fulton AB (2007) Rod photoreceptor function predicts blood vessel abnormality in retinopathy of prematurity. Invest Ophthalmol Vis Sci 48:4351–4359. 10.1167/iovs.07-0204 [DOI] [PubMed] [Google Scholar]
- 68.Liu K, Akula JD, Falk C, et al. (2006) The retinal vasculature and function of the neural retina in a rat model of retinopathy of prematurity. Invest Ophthalmol Vis Sci 47:2639–2647. 10.1167/iovs.06-0016 [DOI] [PubMed] [Google Scholar]
- 69.Hunter DG, Repka MX (1993) Diode laser photocoagulation for threshold retinopathy of prematurity. A randomized study. Ophthalmology 100:238–244. 10.1016/s0161-6420(93)31664-7 [DOI] [PubMed] [Google Scholar]
- 70.McNamara JA, Tasman W, Brown GC, Federman JL (1991) Laser photocoagulation for stage 3+ retinopathy of prematurity. Ophthalmology 98:576–580. 10.1016/s0161-6420(91)32247-4 [DOI] [PubMed] [Google Scholar]
- 71.Ng EYJ, Connolly BP, McNamara JA, et al. (2002) A comparison of laser photocoagulation with cryotherapy for threshold retinopathy of prematurity at 10 years: part 1. Visual function and structural outcome. Ophthalmology 109:928–934; discussion 935. 10.1016/s0161-6420(01)01017-x [DOI] [PubMed] [Google Scholar]
- 72.Connolly BP, Ng EYJ, McNamara JA, et al. (2002) A comparison of laser photocoagulation with cryotherapy for threshold retinopathy of prematurity at 10 years: part 2. Refractive outcome. Ophthalmology 109:936–941. 10.1016/s0161-6420(01)01015-6 [DOI] [PubMed] [Google Scholar]
- 73.Capone A, Diaz-Rohena R, Sternberg P, et al. (1993) Diode-laser photocoagulation for zone 1 threshold retinopathy of prematurity. Am J Ophthalmol 116:444–450. 10.1016/s0002-9394(14)71402-3 [DOI] [PubMed] [Google Scholar]
- 74.Chan-Ling T, Gole GA, Quinn GE, et al. (2018) Pathophysiology, screening and treatment of ROP: A multi-disciplinary perspective. Prog Retin Eye Res 62:77–119. 10.1016/j.preteyeres.2017.09.002 [DOI] [PubMed] [Google Scholar]
- 75.Jin K, Zhu Y, Sun Y, et al. (2002) Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci U S A 99:11946–11950. 10.1073/pnas.182296499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Penn JS, Madan A, Caldwell RB, et al. (2008) Vascular endothelial growth factor in eye disease. Prog Retin Eye Res 27:331–371. 10.1016/j.preteyeres.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sonmez K, Drenser KA, Capone A, Trese MT (2008) Vitreous levels of stromal cell-derived factor 1 and vascular endothelial growth factor in patients with retinopathy of prematurity. Ophthalmology 115:1065–1070.e1. 10.1016/j.ophtha.2007.08.050 [DOI] [PubMed] [Google Scholar]
- 78.Pierce EA, Foley ED, Smith LE (1996) Regulation of vascular endothelial growth factor by oxygen in a model of retinopathy of prematurity. Arch Ophthalmol 114:1219–1228. 10.1001/archopht.1996.01100140419009 [DOI] [PubMed] [Google Scholar]
- 79.Pierce EA, Avery RL, Foley ED, et al. (1995) Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci U S A 92:905–909. 10.1073/pnas.92.3.905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stone J, Chan-Ling T, Pe’er J, et al. (1996) Roles of vascular endothelial growth factor and astrocyte degeneration in the genesis of retinopathy of prematurity. Invest Ophthalmol Vis Sci 37:290–299 [PubMed] [Google Scholar]
- 81.Tan H, Blasco P, Lewis T, et al. (2021) Neurodevelopmental outcomes in preterm infants with retinopathy of prematurity. Surv Ophthalmol 66:877–891. 10.1016/j.survophthal.2021.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stahl A, Lepore D, Fielder A, et al. (2019) Ranibizumab versus laser therapy for the treatment of very low birthweight infants with retinopathy of prematurity (RAINBOW): an open-label randomised controlled trial. Lancet 394:1551–1559. 10.1016/S0140-6736(19)31344-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dai C, Waduge P, Ji L, et al. (2022) Secretogranin III stringently regulates pathological but not physiological angiogenesis in oxygen-induced retinopathy. Cell Mol Life Sci 79:63. 10.1007/s00018-021-04111-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ji L, Waduge P, Hao L, et al. (2022) Selectively targeting disease-restricted secretogranin III to alleviate choroidal neovascularization. FASEB J 36:e22106. 10.1096/fj.202101085RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tokunaga CC, Mitton KP, Dailey W, et al. (2014) Effects of anti-VEGF treatment on the recovery of the developing retina following oxygen-induced retinopathy. Invest Ophthalmol Vis Sci 55:1884–1892. 10.1167/iovs.13-13397 [DOI] [PubMed] [Google Scholar]
- 86.Tang F, LeBlanc ME, Wang W, et al. (2019) Anti-secretogranin III therapy of oxygen-induced retinopathy with optimal safety. Angiogenesis 22:369–382. 10.1007/s10456-019-09662-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lutty GA, McLeod DS, Bhutto I, Wiegand SJ (2011) Effect of VEGF Trap on Normal Retinal Vascular Development and Oxygen-Induced Retinopathy in the Dog. Invest Ophthalmol Vis Sci 52:4039–4047. 10.1167/iovs.10-6798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ichiyama Y, Obata S, Saishin Y, et al. (2021) The systemic antiangiogenic effect of intravitreal aflibercept injection in a mouse model of retinopathy of prematurity. FASEB J 35:e21390. 10.1096/fj.202002414R [DOI] [PubMed] [Google Scholar]
- 89.Zimna A, Kurpisz M (2015) Hypoxia-Inducible Factor-1 in Physiological and Pathophysiological Angiogenesis: Applications and Therapies. BioMed Res Int 2015:549412. 10.1155/2015/549412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Befani C, Liakos P (2018) The role of hypoxia-inducible factor-2 alpha in angiogenesis. J Cell Physiol 233:9087–9098. 10.1002/jcp.26805 [DOI] [PubMed] [Google Scholar]
- 91.Koyasu S, Kobayashi M, Goto Y, et al. (2018) Regulatory mechanisms of hypoxia-inducible factor 1 activity: Two decades of knowledge. Cancer Sci 109:560–571. 10.1111/cas.13483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Caprara C, Thiersch M, Lange C, et al. (2011) HIF1A Is Essential for the Development of the Intermediate Plexus of the Retinal Vasculature. Invest Ophthalmol Vis Sci 52:2109–2117. 10.1167/iovs.10-6222 [DOI] [PubMed] [Google Scholar]
- 93.Cristante E, Liyanage SE, Sampson RD, et al. (2018) Late neuroprogenitors contribute to normal retinal vascular development in a Hif2a-dependent manner. Development 145:dev157511. 10.1242/dev.157511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang J, Qin Y, Martinez M, et al. (2021) HIF-1α and HIF-2α redundantly promote retinal neovascularization in patients with ischemic retinal disease. J Clin Invest 131:e139202. 10.1172/JCI139202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Morita M, Ohneda O, Yamashita T, et al. (2003) HLF/HIF-2α is a key factor in retinopathy of prematurity in association with erythropoietin. EMBO J 22:1134–1146. 10.1093/emboj/cdg117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fang JL, Sorita A, Carey WA, et al. (2016) Interventions To Prevent Retinopathy of Prematurity: A Meta-analysis. Pediatrics 137:e20153387. 10.1542/peds.2015-3387 [DOI] [PubMed] [Google Scholar]
- 97.Sears JE, Hoppe G, Ebrahem Q, Anand-Apte B (2008) Prolyl hydroxylase inhibition during hyperoxia prevents oxygen-induced retinopathy. Proc Natl Acad Sci U S A 105:19898–19903. 10.1073/pnas.0805817105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Trichonas G, Lee TJ, Hoppe G, et al. (2013) Prolyl Hydroxylase Inhibition During Hyperoxia Prevents Oxygen-Induced Retinopathy in the Rat 50/10 Model. Invest Ophthalmol Vis Sci 54:4919–4926. 10.1167/iovs.13-12171 [DOI] [PubMed] [Google Scholar]
- 99.Hoppe G, Lee TJ, Yoon S, et al. (2014) Inducing a Visceral Organ to Protect a Peripheral Capillary Bed. Am J Pathol 184:1890–1899. 10.1016/j.ajpath.2014.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hoppe G, Yoon S, Gopalan B, et al. (2016) Comparative systems pharmacology of HIF stabilization in the prevention of retinopathy of prematurity. Proc Natl Acad Sci U S A 113:E2516–E2525. 10.1073/pnas.1523005113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Juul SE, Comstock BA, Wadhawan R, et al. (2020) A Randomized Trial of Erythropoietin for Neuroprotection in Preterm Infants. N Engl J Med 382:233–243. 10.1056/NEJMoa1907423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Horsch S, Parodi A, Hallberg B, et al. (2020) Randomized Control Trial of Postnatal rhIGF-1/rhIGFBP-3 Replacement in Preterm Infants: Post-hoc Analysis of Its Effect on Brain Injury. Front Pediatr 8:517207. 10.3389/fped.2020.517207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schaffer DB, Johnson L, Quinn GE, et al. (1985) Vitamin E and Retinopathy of Prematurity. Ophthalmology 92:1005–1011. 10.1016/S0161-6420(85)33913-1 [DOI] [PubMed] [Google Scholar]
- 104.Sun H, Cheng R, Wang Z (2020) EARLY VITAMIN A SUPPLEMENTATION IMPROVES THE OUTCOME OF RETINOPATHY OF PREMATURITY IN EXTREMELY PRETERM INFANTS. Retina 40:1176–1184. 10.1097/IAE.0000000000002543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Firouzabadi FD, Shab-Bidar S, Jayedi A (2022) The effects of omega-3 polyunsaturated fatty acids supplementation in pregnancy, lactation, and infancy: An umbrella review of meta-analyses of randomized trials. Pharmacol Res 177:106100. 10.1016/j.phrs.2022.106100 [DOI] [PubMed] [Google Scholar]
- 106.Filippi L, Cavallaro G, Berti E, et al. (2019) Propranolol 0.2% Eye Micro-Drops for Retinopathy of Prematurity: A Prospective Phase IIB Study. Front Pediatr 7:180. 10.3389/fped.2019.00180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ley D, Hallberg B, Hansen-Pupp I, et al. (2019) rhIGF-1/rhIGFBP-3 in Preterm Infants: A Phase 2 Randomized Controlled Trial. J Pediatr 206:56–65.e8. 10.1016/j.jpeds.2018.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kong HB, Zheng GY, He BM, et al. (2021) Clinical Efficacy and Safety of Propranolol in the Prevention and Treatment of Retinopathy of Prematurity: A Meta-Analysis of Randomized Controlled Trials. Front Pediatr 9:631673. 10.3389/fped.2021.631673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Filippi L, Cavallaro G, Bagnoli P, et al. (2013) Oral Propranolol for Retinopathy of Prematurity: Risks, Safety Concerns, and Perspectives. J Pediatr 163:1570–1577.e6. 10.1016/j.jpeds.2013.07.049 [DOI] [PubMed] [Google Scholar]
- 110.Li W, Webster KA, LeBlanc ME, Tian H (2018) Secretogranin III: a diabetic retinopathy-selective angiogenic factor. Cell Mol Life Sci 75:635–647. 10.1007/s00018-017-2635-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kingsley DM, Rinchik EM, Russell LB, et al. (1990) Genetic ablation of a mouse gene expressed specifically in brain. EMBO J 9:395–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.LeBlanc ME, Wang W, Chen X, et al. (2017) Secretogranin III as a disease-associated ligand for antiangiogenic therapy of diabetic retinopathy. J Exp Med 214:1029–1047. 10.1084/jem.20161802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rong X, Tian H, Yang L, Li W (2019) Function-first ligandomics for ocular vascular research and drug target discovery. Exp Eye Res 182:57–64. 10.1016/j.exer.2019.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]