

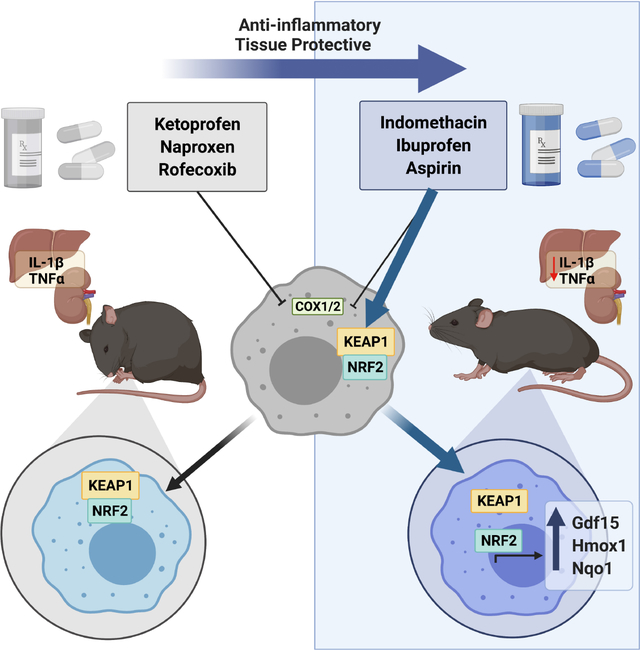
Summary
Nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit cyclooxygenase (COX) enzymes and are ubiquitously used for their anti-inflammatory properties. However, COX inhibition alone fails to explain numerous clinical outcomes of NSAID usage. Screening commonly used NSAIDs in primary human and murine myeloid cells demonstrated that NSAIDs could be differentiated by their ability to induce Growth/differentiation factor 15 (GDF15), independent of COX specificity. Using genetic and pharmacologic approaches, NSAID-mediated GDF15 induction was dependent on activation of Nuclear factor erythroid 2-related factor 2 (NRF2) in myeloid cells. Sensing by Cysteine 151 of the NRF2 chaperone, Kelch-like ECH-associated protein 1 (KEAP1) was required for NSAID activation of NRF2 and subsequent anti-inflammatory effects both in vitro and in vivo. Myeloid-specific deletion of NRF2 abolished NSAID-mediated tissue protection in murine models of gout and endotoxemia. This highlights a noncanonical NRF2-dependent mechanism of action for the anti-inflammatory activity of a subset of commonly used NSAIDs.
Keywords: Nonsteroidal anti-inflammatory drugs, Nuclear factor erythroid 2-related factor 2, Growth/differentiation factor 15
eTOC blurb
The use of nonsteroidal anti-inflammatory drugs (NSAIDs) is ubiquitous. Herein, Eisenstein et al identify a noncanonical mechanism of action for the anti-inflammatory activity of NSAIDs through activation of the transcription factor, Nuclear factor erythroid 2-related factor 2 (NRF2). In endotoxemia and gout models, indomethacin improved inflammation in an NRF2-dependent manner.
Graphical Abstract
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are among the oldest and most commonly used medications in the world. The oldest NSAID, aspirin, is derived from the active agent responsible for the analgesic and antipyretic properties of the willow bark, which has been used as far back as 1900 BC to treat various maladies (Desborough and Keeling, 2017; MacLagan, 1876; Schmidt, 1934; Stone, 1763). Shortly after aspirin’s discovery and commercialization, indomethacin was discovered as a potent NSAID using the first rodent animal model of local inflammation (carrageenan-induced paw inflammation) (T. Y. Shen, 1963; Winter et al., 1962). The subsequent discovery of the ability of NSAIDs to inhibit cyclooxygenase (COX) to prevent inflammatory prostaglandin formation from arachidonic acid then led to the identification of the isozymes COX1 and COX2 and the proliferation of NSAIDs and COX-specific NSAIDs for the treatment of a wide spectrum of inflammatory diseases (Fu et al., 1990; Vane, 1971).
Given the widespread use of NSAIDs across multiple disease states over the last hundred years, many clinical phenomena have arisen that have been difficult to reconcile within the paradigm of COX-inhibition. Multiple studies have demonstrated that equipotent dosing of NSAIDs with the same COX-specificity produce variable outcomes, a phenomenon that had previously been primarily attributed to a combination of pharmacogenetic, pharmacokinetic and pharmacodynamic differences (Bannuru et al., 2015; Li et al., 2020; Rollason et al., 2014). For example, in a large metanalyses of arthritis treatment, diclofenac is the most effective NSAID amongst several other agents with nearly identical COX2 selectivity (da Costa et al., 2017), while amongst post-operative management, the efficacy of similar NSAIDs are quite variable (Ong et al., 2007). Moreover, NSAID use is associated with both beneficial and detrimental outcomes across multiple diseases, and it has been difficult to ascribe these effects to COX inhibition alone. For example, NSAIDs both prevent and cause heart disease in different contexts (Coxib et al., 2013; McGettigan and Henry, 2011). In meta-analyses, ibuprofen and diclofenac increase risk of myocardial infarction, while naproxen–which has similar COX inhibition profiles–does not (Kearney et al., 2006; Trelle et al., 2011). These paradoxes are also observed in cancer, and there has been great interest in understanding the COX-independent anti-tumor mechanisms of NSAIDs (Chubak et al., 2016; Gurpinar et al., 2014). NSAIDs are also implicated in exacerbating allergic disease (Blanca-Lopez et al., 2019; Dona et al., 2012), and NSAIDs with similar COX-profiles have markedly different effects on asthma exacerbation (Lo et al., 2016). Indeed, even NSAID-induced gastrointestinal tract damage, one of the most well-known side effects of NSAIDs, cannot be explained solely by inhibition of COX enzymes (Langenbach et al., 1995). Thus, despite the wealth of data on the potential benefits and harms of NSAIDs, the exact mechanisms underlying these observations is not fully known.
Multiple studies in multiple species have identified non-canonical mechanisms of action of NSAIDs (Gurpinar et al., 2013). The most compelling evidence for this comes from studies utilizing Caenorhabditis elegans, which do not express COX isoforms, where NSAIDs extend longevity (Ching et al., 2011; He et al., 2014). Mechanistically, several COX-independent effects of NSAIDs have been proposed, including suppression of NF-κB (Kopp and Ghosh, 1994) and the inflammasome (Smith et al., 2017), as well as a myriad of other potential off-target pathways (Gao et al., 2018). Given the ubiquitous use of NSAIDs, and the paradoxical but large effect sizes of NSAIDs across multiple diseases, understanding mechanisms of action of NSAIDs is of great biomedical interest.
In a previous report, we showed that Growth/differentiation factor 15 (GDF15) is required for surviving acute systemic inflammation through hepatic metabolic reprograming (Luan et al., 2019). Historically, Gdf15 was identified as an NSAID-inducible gene in studies using colorectal cancer cell lines--many of which do not express COX enzymes (Hanif et al., 1996)--in an attempt to understand the correlation between NSAID use and the decreased incidence of colorectal cancer (Chubak et al., 2016). Fittingly, it was originally named NSAID-activated gene 1 (Nag-1) (Baek et al., 2001). Based on this, we sought to determine if NSAIDs may have different therapeutic efficacy because they differentially induce GDF15. Here we report a noncanonical, COX-independent, mechanism of action of NSAIDs that is dependent on activation of NRF2 in myeloid cells. This has broad implications for the therapeutic use of NSAIDs in a wide range of conditions.
Results
Indomethacin induces GDF15 independent of cyclooxygenases
Given the ability of NSAIDs to induce GDF15 in colorectal cancer cell lines, we sought to determine whether GDF15 could be induced by NSAIDs in primary mouse cells. We challenged bone marrow-derived macrophages (BMDM) with the classical NSAID, indomethacin, and observed robust transcriptional induction (Fig. 1A). Indomethacin is a non-selective COX inhibitor with preference for COX1 (Warner et al., 1999). To test whether the ability to induce GDF15 was a function of COX selectivity, we challenged BMDM with a wide spectrum of NSAIDs spanning COX specificities and across multiple doses, and observed that only a subset of NSAIDs induced GDF15 transcription and protein in a dose-dependent fashion (Fig. 1B and C). We verified that the GDF15-inducing doses of indomethacin and ibuprofen did not cause cell death or induce reactive oxygen species (ROS) (Fig. S1A and S1B). For NSAIDs that did not robustly induce GDF15 in this screen, we could only achieve minimal Gdf15 induction at supratherapeutic doses. Therapeutic doses of ketoprofen, which has a similar COX-specificity profile as indomethacin (Herndon et al., 2008) were unable to induce Gdf15 at any timepoint (Fig. S1C), and was only supratherapeutic, millimolar doses were able to induce GDF15 (Fig. S1D). Similar induction of GDF15 by indomethacin, but not by ketoprofen, was seen in human peripheral blood monocyte-derived macrophages (MDM) (Fig. 1D).
Figure 1. Indomethacin induces GDF15 independent of cyclooxygenases.
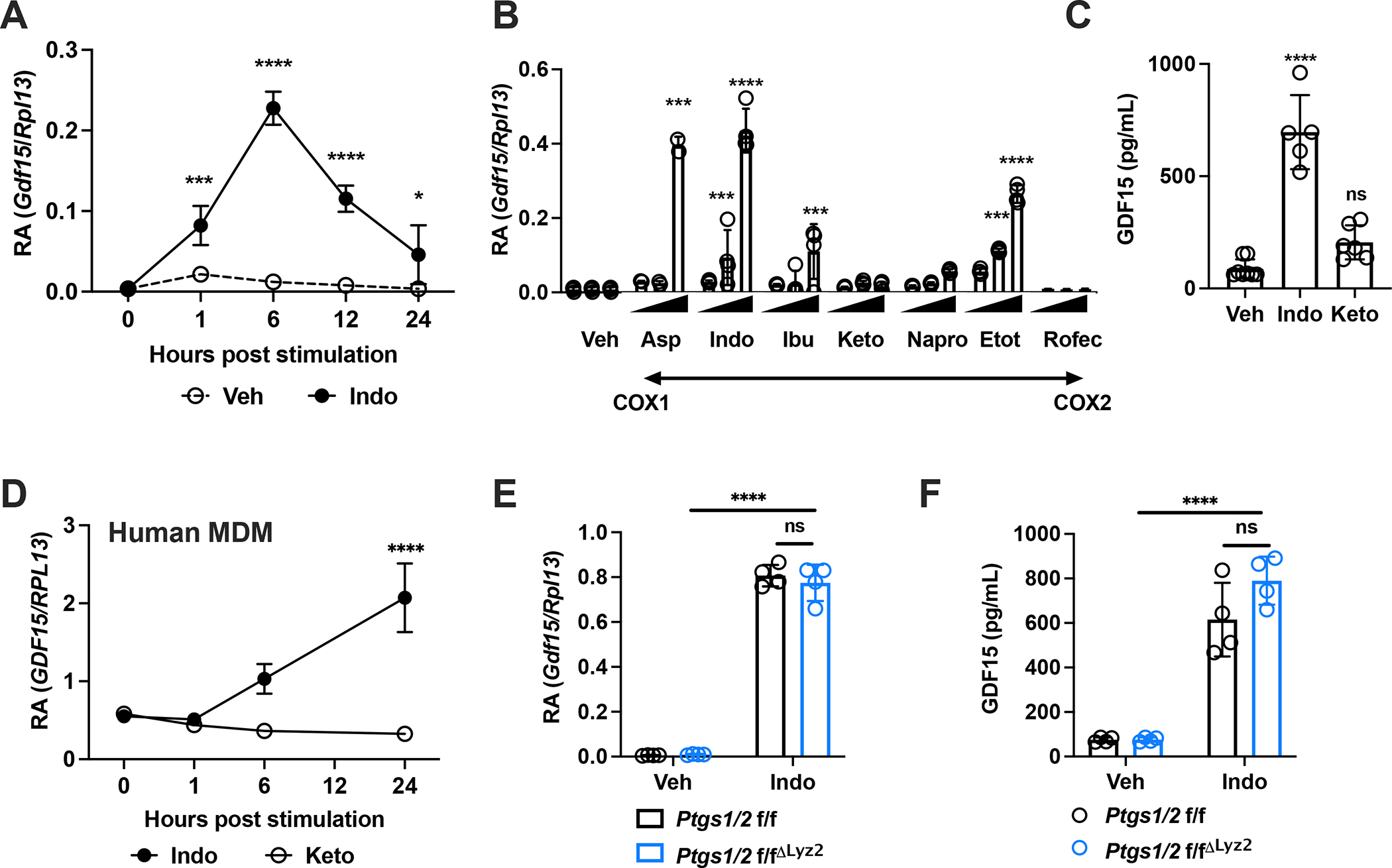
(A) Gdf15 mRNA expression in BMDMs following stimulation with 0.5 mM indomethacin (n=3 per group) and (B) at 6 hours following stimulation with increasing doses of aspirin (0.25mM, 0.5 mM, 1 mM), indomethacin (0.125mM, 0.25mM, 0.5mM), ibuprofen (0.125mM, 0.25mM, 0.5mM), ketoprofen (0.25 mM, 0.5mM, 1mM), naproxen (0.25 mM, 0.5mM, 1mM), etodolac (0.25 mM, 0.5mM, 1mM) and rofecoxib (0.25 mM, 0.5mM, 1mM), in order of increasing specificity of COX2 as compared to COX1 (n=4 per group). (C) Supernatant GDF15 protein levels following 24 hours stimulation with vehicle, 0.5 mM indomethacin and 0.5 mM ketoprofen (n=5–8 per group). (D) GDF15 mRNA expression in human MDM following stimulation by 0.5 mM indomethacin and 0.5 mM ketoprofen (n=3 per group). € Gdf15 mRNA expression of BMDMs stimulated for 6 hours with vehicle or 0.5 mM indomethacin (n=4 per group). (F) Supernatant GDF15 protein levels from BMDMs stimulated for 24 hours with vehicle or 0.5 mM indomethacin (n=4 per group). All experiments are representative and repeated at least once. Statistics by ANOVA. Data are represented as mean ± standard deviation. *P<.05, ***P<.001, ****P<.0001, ns P>.05
Since the ability to induce GDF15 did not depend on COX-specificity, we hypothesized that GDF15 induction may be COX-independent. To test this, we generated animals where Ptgs1 and Ptgs2 (the genes encoding COX1 and 2, respectively) were deleted in Lysozyme 2 (Lyz2)-expressing cells, which include many cells of the myeloid lineage (Abram et al., 2014; Shi et al., 2018), and verified that Ptgs1 and Ptgs2 expression was efficiently, but not completely, decreased (Fig. S1E and S1F). We were unable to detect COX2 in resting BMDM by qPCR or Western Blot, consistent with previous observations that Ptgs2 is expressed at low levels until induced by various inflammatory stimuli (Angel et al., 1994; Arias-Negrete et al., 1995; Inoue et al., 1995). We found that indomethacin itself induced Ptgs2, and that this induction was efficiently, but not completely, diminished (Fig. S1F). Since others have previously demonstrated that residual COX activity and prostaglandin synthesis retain significant functional signaling properties, which can be both pro- and anti-inflammatory, to estimate the functional deletion of prostaglandin signaling, we characterized known prostaglandin-responsive genes after stimulating BMDMs with lipopolysaccharide (LPS), and found that our transgenic system impacted the expression of these targets (Fig. S1G), consistent with our Western blot and qPCR approaches to validating deletion efficiency (Cilenti et al., 2021; Funk, 2001; Zelenay et al., 2015). Despite the significant loss of COX1 and COX2, there was no impact on the ability of indomethacin to induce Gdf15 transcription or on GDF15 secretion into culture supernatant (Fig. 1E and F). These data demonstrated that NSAIDs could be divided into those that could induce Gdf15 and those that could not, and that the induction of Gdf15 was largely independent of COX isozymes, although we cannot definitively exclude their contribution given the incomplete deletion related to limitations of our Cre-loxp approach.
A subset of NSAIDs activate NRF2 and downstream GDF15
Thus, to identify the mechanism of indomethacin-induced Gdf15 upregulation, we generated BMDMs from animals lacking transcription factors and signaling molecules (Patel et al., 2019; Ratnam et al., 2017; Suzuki et al., 2017) previously implicated in GDF15 regulation (Fig. 2A). Gdf15 induction did not require the essential signaling adaptors for almost all toll-like receptors (TLR), MyD88. We included NRF2 deficient BMDMs because most NSAIDs are carboxylic acid-containing electrophiles and a study also implicated NRF2 and GDF15 in the mechanism of action of colchicine in hepatocytes (Weng et al., 2021; Zhou et al., 2005). Kelch-like ECH-associated protein 1 (KEAP1), the chaperone protein which sequesters NRF2 in the cytosol detects excess cytosolic electrophiles through its cysteine moieties and subsequently releases NRF2 into the nucleus where it activates transcriptional programs important in drug detoxification and maintenance of redox balance (Cuadrado et al., 2019; Yamamoto et al., 2018).
Figure 2. A subset of NSAIDs activate NRF2 and downstream GDF15.

(A) Gdf15 mRNA expression from BMDMs derived from B6, Myd88−/−, Chop−/−, Nrf2−/−and Eif2a−/− mice treated for 6h with vehicle or 0.5 mM indomethacin (n=4 per group). (B) Supernatant GDF15 protein levels from BMDMs treated for 24h with vehicle, 0.5 mM indomethacin or 0.5 mM ketoprofen (n=4 per group). (C) Gdf15 mRNA expression from BMDMs treated for 6h with vehicle, sulforaphane or dimethylfumarate (DMF) (n=5–6 per group). (D) Supernatant GDF15 protein levels from BMDMs treated for 24h with vehicle or DMF (n=4 per group). Messenger RNA expression of Cat, Gar, Gclm, Hmox1, Nqo1, Taldo1 from BMDMs derived from (E) B6 and Nrf2−/− mice (n=4 per group) or (F) Ptgs1/2 f/f mice following stimulation with vehicle or 0.5 mM indomethacin for 6h (n=4 per group). (G) Immunofluorescence staining of BMDMs for NRF2 (green) and DAPI (blue) following treatment with vehicle or indomethacin. (H) Chromatin immunoprecipitation of Gdf15 and Nqo1. ChIP was performed on BMDM stimulated for four hours with vehicle or indomethacin using anti-NRF2 (top, red), anti-H3K27Ac (bottom, black) or control (IgG) antibody and qPCR was performed with primers spanning 1 kb upstream and downstream of the identified ARE consensus sequence (Fig. S3) in the regulatory elements of Gdf15 (left) and Nqo1 (right). Representative of 2 independent experiments. All experiments are representative and repeated at least once. Statistics by ANOVA. Data are represented as mean ± standard deviation. *P<.05, **P<0.01, ***P<.001, ****P<.0001, ns P>.05
We found that Gdf15 induction in response to indomethacin required NRF2 (Fig. 2A). Using BMDMs from animals in which NRF2 was deleted only in Lyz2-expressing cells (Fig. S2A), we found that indomethacin-induced GDF15 largely required NRF2 (Fig. 2B). Decreased Gdf15 mRNA and protein were also detected at steady state in these BMDMs (Fig. S2B and S2C), suggesting that while NRF2 contributed significantly, but not solely, to basal Gdf15 expression, it was required for inducibility by NSAIDs. To test if NRF2 activation was sufficient to induce Gdf15, we tested two canonical NRF2 activators, sulforaphane and dimethylfumarate (DMF) (Dinkova-Kostova et al., 2002; Foresti et al., 2013; Linker et al., 2011; Scannevin et al., 2012; Zhang et al., 1992), and found that NRF2 activation was sufficient to induce Gdf15 in both mouse BMDMs (Fig. 2C) and in human MDMs (Fig. S2D). However, the ability of DMF to induce GDF15 depended largely, but not completely, on NRF2, (Fig. 2D and S2E), consistent with other known mechanisms of action of DMF, and consistent with other reported modalities of GDF15 induction. Finally, since NRF2 is responsive to both excess cytosolic electrophiles and also ROS, we tested if ROS could induce Gdf15, and observed that sodium arsenite, 2,3-dimethoxy-1,4-naphthalenedione (DMNQ), and phenazine methosulfate (PMS) could induce Gdf15 in a dose-dependent manner and that this was significantly but incompletely dependent on NRF2 (Fig S2F, S2G). We thus identify NRF2 as a critical transcription factor for the induction of Gdf15 generally and by a subset of NSAIDs specifically in murine BMDMs.
Since NRF2 was required for GDF15 induction, we asked whether GDF15-inducing NSAIDs, like indomethacin and ibuprofen, also activated canonical NRF2 signaling (Itoh et al., 1997; Itoh et al., 2004; Venugopal and Jaiswal, 1996). Indeed, indomethacin and ibuprofen, but not ketoprofen, induced the expression of canonical NRF2 target genes, Cat, Gar, Gclm, Hmox1, Nqo1 and Taldo1 in BMDMs in an NRF2-dependent fashion; the kinetics of other NRF2 target genes activated after indomethacin treatment also mirrored that of Gdf15 (Fig. 2E, S2H–J). As with Gdf15, ketoprofen was unable to induce Hmox1 at any dose (Fig. S2K), suggesting that it does not activate NRF2 signaling in contrast to indomethacin and ibuprofen. Consistent with the observation that a significant deletion of COX expression did not impact Gdf15 induction, the induction of NRF2 target genes by indomethacin and ibuprofen also was not impacted by significant loss of COX1 and COX2 (Fig. 2F and S2L).
Since NRF2 target genes can be induced by other transcription factors such as hypoxia-inducible factor 1a (Toth and Warfel, 2017), we used immunofluorescence and chromatin immunoprecipitation approaches to confirm that indomethacin caused NRF2 nuclear localization and binding to antioxidant response elements (ARE) in the regulatory regions of Gdf15 and the canonical NRF2 target Nqo1. As predicted, indomethacin treatment of BMDMs indeed caused translocation of NRF2 into the nucleus (Fig. 2G). To test if NRF2 bound to regulatory elements in Gdf15, we identified a canonical ARE consensus sequence that overlapped with enhancer signatures after scanning for ARE consensus sequences 10kb upstream and downstream from the transcriptional start site (Fig. S3). We found that four hours after indomethacin stimulation, NRF2 bound to an enhancer containing two ARE consensus sequences of Gdf15; moreover, indomethacin stimulation also resulted in activating histone modifications (H3K27Ac) at this identified control region, indicating that this enhancer is involved in GDF15 transcriptional activation in response to indomethacin (Fig. 2H, left). Since we found that indomethacin activated the NRF2 transcriptional program, we tested if indomethacin would also cause NRF2 binding to the ARE consensus sequence in the promoter of Nqo1, and found that stimulation with indomethacin indeed led to NRF2 binding as well as activating histone modifications (H3K27Ac) at this position (Fig. 2H, right). These data collectively demonstrated that a subset of NSAIDs are NRF2 activators and established Nag-1 (Gdf15) as an NRF2-target gene.
Indomethacin activates NRF2 target genes, including Gdf15, in Lyz2-expressing cells in an NRF2-dependent fashion in vivo
Having established some NSAIDs as NRF2 activators and Gdf15 as an NRF2 target gene in primary mouse and human monocytes/macrophages, we then asked if our in vitro observations could be seen in vivo. As in BMDMs, we observed that, Nrf2−/− mice have less plasma GDF15 levels as compared to B6 mice; furthermore, lack of Nrf2 in only Lyz2 expressing cells also had decreased total serum GDF15 levels (Fig. 3A). As we had observed in BMDMs, indomethacin, but not ketoprofen, robustly induced plasma GDF15, with a peak concentration at 6 hours after intraperitoneal injection (Fig. 3B). Likewise, a single injection of the NRF2 activator sulforaphane also induced GDF15 in vivo in an NRF2-dependent fashion (Fig. S4A). Moreover, bardoxolone, an NRF2 activator in late-stage clinical trials for use in chronic kidney disease, also induced GDF15 in vivo both when given in a single bolus or when administered chronically (Fig. S4B and S4C).
Figure 3. Indomethacin activates NRF2 target genes, including GDF15, in NRF2-depdendent, and COX1/2-independent manner, in Lyz2-expressing cells in vivo.

(A) Baseline GDF15 serum levels (n=3–10 per group). (B) Serum GDF15 protein levels in B6 mice following intraperitoneal injection with vehicle, 15 mg/kg indomethacin or 15 mg/kg ketoprofen (n=3–5 per group). Serum GDF15 protein levels in (C) Nrf2−/− (D) Nrf2 f/f and (E) Ptgs1/2 f/f mice following intraperitoneal injection of 15 mg/kg indomethacin (n=3–6 per group). (F) Serum GDF15 protein levels following injection of 25 mg/kg sulforaphane (n=3–7 per group). (G) Hmox1 RNA expression of bulk liver in animal treated with vehicle or indomethacin at the indicated time points (n=3–4 per group). Kidney Gdf15, Hmox1, and Nqo1 mRNA expression 6h following intraperitoneal injection of 15 mg/kg indomethacin or 50 mg/kg sulforaphane in (H) Nrf f/f mice (n=3–6 per group) or (I) Ptgs1/2 f/f mice (n=5–6 per group). All experiments are representative and repeated at least once. Statistics by ANOVA. Data are represented as mean ± standard deviation. *P<.05, **P<0.01, ***P<.001, ****P<.0001, ns P>.05
Consistent with our in vitro findings, we found that indomethacin-induced GDF15 was dependent on NRF2 (Fig. 3C). Given that Lyz2-expressing cells appeared to be important for basal GDF15 levels, we sought to determine whether lack of NRF2 expression in only Lyz2-expressing cells altered indomethacin-induced GDF15 levels. Indeed, Lyz2-expressing cells produced the majority of plasma GDF15 induced by indomethacin in an NRF2-dependent fashion (Fig. 3D). Furthermore, decreased levels COX1 and 2 enzymes in Lyz2-expressing cells did not have a significant effect on indomethacin-induced GDF15 levels (Fig. 3E). Since inducible GDF15 was completely attenuated in whole body NRF2 deficient mice, and roughly 70% of NSAID-induced GDF15 levels depend on Lyz2-expressing myeloid cells, other NRF2-expressing cells contribute to NSAID-induced GDF15 in vivo. Likewise, the majority of circulating GDF15 induced by sulforaphane, a canonical NRF2 activator, also depended on NRF2 expression specifically in Lyz2-expressing cells (Fig. 3F), suggesting a more general role for this cell lineage in GDF15 regulation and production downstream of NRF2 activation. As we consistently observed transient induction of GDF15 following a single dose of indomethacin, we sought to determine whether this was also true for other NRF2 target genes in vivo. Hmox1 expression in the liver peaked at 6 hours and then decreased, similarly to serum GDF15 (Fig. 3G).
Having established that indomethacin and the canonical NRF2 activator sulforaphane can induce GDF15 in vivo, we explored whether elevated plasma GDF15 levels were indicative of activation of NRF2 programs in response to indomethacin. To test for NRF2 activation in vivo, we first screened organs from B6 mice for transcriptional induction after sulforaphane injection to identify potential organs most responsive to systemic NRF2 activation, and identified robust induction of Gdf15 and Nqo1 primarily in the liver and kidney (Fig. S4D,E). We next sought to determine whether this effect was dependent on NRF2. Consistent with the requirement of NRF2 for NSAID-induced GDF15, both indomethacin and sulforaphane induced NRF2 target genes Gdf15, Hmox1, and Nqo1 in the liver and kidney in a manner dependent on NRF2 in Lyz2 expressing cells (Fig. 3H, S4F). Indomethacin-induced upregulation of Gdf15, Hmox1, and Nqo1 in liver and kidney was independent of COX1 and 2 expression in Lyz2 expressing cells (Fig. 3I, Fig. S4G). Collectively, these data demonstrated that while steady-state GDF15 levels partially require NRF2, NSAID-induced GDF15 required NRF2 primarily in cells of the myeloid lineage, and NSAIDs induced NRF2 target genes primarily in myeloid cells expressing Lyz2 in the liver and kidney in vivo.
The Cysteine 151 (C151) of KEAP1 is necessary for activation of NRF2 target genes by indomethacin
Electrophilic xenobiotic activation of NRF2 requires binding to cysteine moieties in the chaperone protein KEAP1. Of these cysteine residues, C151 has previously been shown to be essential for activation of NRF2 by DMF, sulforaphane, and bardoxolone (Dayalan Naidu et al., 2018; Takaya et al., 2012), all of which we discovered activated GDF15 in vivo and in vitro. Thus, to test the requirement of C151 in indomethacin-induced NRF2 pathway activation, we obtained an animal where a single point mutation was made at that position from cysteine to serine (C151S) (Saito et al., 2016). Using this transgenic animal, we made BMDMs and found that the ability of indomethacin to induce Gdf15 required C151 (Fig.4A). Likewise, the NRF2-mediated transcriptional program also required C151 (Fig. 4B), consistent with results obtained using NRF2-deficient cells (Fig. 2, 3). To test the requirement of C151 in vivo, we challenged C151S mice with indomethacin. We found that basal GDF15 was also partially dependent on C151S (Fig. 4C), suggesting that an endogenous factor may be required to maintain steady state levels of basal GDF15. Consistent with our observation that indomethacin-induced GDF15 was completely dependent on NRF2, we also found that the induction of serum GDF15 by indomethacin was completely dependent on the C151 residue in KEAP1 (Fig. 4D). Finally, we sacrificed C151S animals after indomethacin challenge and assessed the ability of indomethacin to induce NRF2 target genes in the liver, and observed complete abrogation of expression in the setting of C151 disruption (Fig. 4E). Altogether, we demonstrated that a subset of NSAIDs activate GDF15 through NRF2 in a mechanism that is dependent on the C151 residue in KEAP1.
Figure 4. Cysteine 151 of KEAP1 is required for NRF2 activation by indomethacin.

(A) Gdf15 mRNA levels in BMDM generated from C151S KEAP1 transgenic animals and littermate controls 6 hours after stimulation with indomethacin or ketoprofen (n=4 per group). (B) NRF2 target gene expression in BMDM generated from C151S KEAP1 transgenic animals and littermate controls 6 hours after stimulation with indomethacin (n=4 per group). (C) Baseline GDF15 serum levels in C151S KEAP1 mice and controls (n=5–7 per group). (D) Plasma GDF15 levels after an intraperitoneal injection of 15 mg/kg indomethacin at the indicated time points in C151S and wildtype animals (n=5–7 per group). (E) mRNA expression of Gdf15, Hmox1, and Nqo1 at 6 hours from bulk liver of C151S and wildtype animals after treatment with 15 mg/kg indomethacin (n=4 per group). All experiments are representative and repeated at least once. Statistics by ANOVA. Data are represented as mean ± standard deviation. **P<0.01, ***P<.001, ****P<.0001
Indomethacin reduces inflammation in endotoxemia and gout via an NRF2-dependent, KEAP1 C151S-dependent, GDF15-independent mechanism
We next wanted to test the contribution of the NRF2-activating properties of NSAID to its anti-inflammatory effects. NRF2 activation reduces the inflammatory output of myeloid cells via a variety of mechanisms (Kobayashi et al., 2016; Mills et al., 2018). We thus hypothesized that NRF2-activating NSAIDs would be more effective in reducing inflammation in vivo than non-NRF2-activating NSAIDs, and that indomethacin may be less effective in reducing inflammation in the absence of NRF2 in Lyz2-expressing cells.
First, we tested if the same weight-based dose of ketoprofen would be as effective as indomethacin in reducing inflammation using the endotoxemia model of sterile systemic inflammation. We found that indomethacin was more effective than ketoprofen when using loss of core body temperature (CBT), a readout of inflammation and disease severity (Fig. 5A). The therapeutic efficacy of indomethacin was not lost upon the significant reduction of Ptgs1 and Ptgs2 expression in Lyz2-expressing cells (Fig. S5A) but rather was attenuated when Nrf2 was deleted in Lyz2-expressing cells (Fig. 5B), which phenocopied constitutive Nrf2 deletion (Fig. S5B). Likewise, C151 of KEAP1 was also required for the protective effects of indomethacin (Fig. 4C). Having shown that indomethacin activates NRF2 and that Gdf15 is an NRF2 target gene, we next sought to determine whether GDF15 was mediating the protective effects of indomethacin in the endotoxemia model. As such, we treated WT and Nrf2−/− mice with recombinant GDF15 in the endotoxemia model. Recombinant GDF15 did not recapitulate the effects of indomethacin on temperature in the endotoxemia model in WT or Nrf2−/− mice (Fig. 5D).To test if GDF15 was required for the effects of indomethacin, we acutely inhibited GDF15 with anti-GDF15 antibody (Luan et al., 2019), which did not attenuate the benefits of indomethacin treatment on CBT in the endotoxemia model, a result we confirmed using animals with constitutive genetic deletion of Gdf15 (Liu et al., 2020) (Figure S4C, D). These data suggest that while GDF15 may be mediating other physiological processes not assayed for in these experiments, GDF15 is not necessary or sufficient to mediate the beneficial effects of indomethacin in endotoxemia that depend on NRF2 and C151 of KEAP1.
Figure 5. Indomethacin reduces inflammation in endotoxemia and gout via an NRF2-dependent, GDF15-independent and COX1/2-independent mechanism.

(A) Temperature curve for B6 mice following intraperitoneal injection with LPS and vehicle, indomethacin or ketoprofen (AUC p <0.0001, (n=4 per group). Temperature curve for (B) Nrf2 f/f mice (AUC p<0.0085, n=5 per group), (C) C151S mice (AUC<0.001, n=5 per group), and (D) in B6 or Nrf2−/− mice treated with vehicle or indomethacin with or without recombinant GDF15 or protein control (BSA) (AUC p<0.01, n=4–5 per group) (E) Serum IL-1ß levels in B6 and Nrf2−/− mice 24 hours following intraperitoneal injection of LPS and vehicle or indomethacin (n=4–5 per group). (F) Log2 fold change in mRNA expression of inflammatory markers in the liver of B6 and Nrf2−/− mice injected intraperitoneally with LPS and vehicle or indomethacin (n=3–5 per group). (G) Serum alanine transferase (ALT) activity 20 hours following intraperitoneal injection with LPS and vehicle or indomethacin (n=4–5 per group). (H) Liver Nqo1 and Cat mRNA expression following intraperitoneal injection of vehicle, indomethacin or ketoprofen (n=3–5 per group). Footpads of (I) Ptgs1/2 f/f mice (n=3–4 per group) and (J) Nrf2 f/f mice (n=4–8 per group) injected intradermally with PBS or MSU and then treated with intraperitoneal injections of vehicle or indomethacin. Foot diameter was normalized to baseline foot diameter. All experiments are representative and repeated at least once. Statistics by ANOVA. Data are represented as mean ± standard deviation. *P<.05, **P<0.01, ***P<.001, ****P<.0001, ns P>.05
Consistent with improved thermoregulation and protection against loss of CBT, indomethacin also induced inhibition of pro-inflammatory cytokines (Fig. S4E), including circulating IL-1β, in response to endotoxemia. Reduction of IL-1β in response to indomethacin during endotoxemia was dependent on NRF2 (Fig 5E). Since the liver and kidney were identified as the main sites of NRF2 activation after indomethacin injection, we looked at the effect of indomethacin on inflammatory transcripts in the liver and kidney in LPS challenged wildtype and Nrf2-deficient animals. We found that indomethacin decreased a number of inflammatory transcripts in the liver (Fig. 5F). This correlated with reduced liver damage as evidenced by reduced plasma aminotransferase levels (Fig. 5G). Consistent with our findings of decreased systemic inflammation, the reduction in liver inflammation and injury also occurred in an NRF-dependent fashion. We did not detect differences in the number of cellular infiltrates in the liver (Fig. S4F). Similar to our previous findings, indomethacin but not ketoprofen induced NRF2 target genes in the liver of endotoxemic animals (Fig. 5H). We did not detect large differences in the kidney, either with regards to damage or inflammation (Fig. S5G, S5H), potentially because of the negative COX-dependent effects on renal perfusion, particularly in inflammatory settings (Brater et al., 2001). Conversely, we did detect differences in damage and inflammation in the heart. We found that indomethacin, but not ketoprofen, mediated increased cardiac dysfunction and inflammation in an NRF2-dependent fashion (Fig. S5I, S5J). These findings are reminiscent of similar observations seen in clinical trials using the NRF2 activator, bardoxolone, where bardoxolone improved kidney function at the expense of cardiovascular toxicity which ultimately led to trial termination (de Zeeuw et al., 2013).
Finally, we utilized a model of local inflammation, acute gout, similar to the carrageenen-induced paw inflammatory model initially used to discover indomethacin (T. Y. Shen, 1963; Winter et al., 1962). We chose this model because indomethacin remains a first-line agent in the management of acute gout (Schumacher, 2004). In this model, uric acid crystals are injected into the footpad, leading to measurable gross swelling of the foot similar to acute gouty arthritis, which most commonly occurs in the foot of patients with gout (Mallinson et al., 2014). Similar to our findings in the endotoxemia model, mice treated with indomethacin had reduced inflammation in a fashion that was dependent on NRF2 expression and not impacted by significant reduction in Ptgs1 and Ptgs2 expression in Lyz2 expressing cells (Fig. 5I, J). Thus, we demonstrate that NRF2 activation in myeloid cells contributes significantly to the anti-inflammatory properties of certain NSAIDs and that certain NSAIDs may be more potent anti-inflammatory agents than others due to their differential ability to activate NRF2 through KEAP1 C151.
Discussion
NSAIDs are amongst the most ubiquitous medications used in the world. While COX1 and 2 inhibition is clearly a major component of the anti-inflammatory action of NSAIDs, these data suggest an additional COX-independent mechanism of action of some NSAIDs that accounts for at least some of the anti-inflammatory properties of NSAIDs. The differences in the ability of different NSAIDs to activate NRF2, which we demonstrate is dependent on the Cysteine 151 in KEAP1, may in part bely the differential efficacies of NSAIDs in the treatment of inflammatory diseases as well as the unexpected properties of certain NSAIDs when used in a variety of settings, since NRF2 activation has been implicated in cancer (Rojo de la Vega et al., 2018), allergic disease (Al-Harbi et al., 2019; Nagashima et al., 2019; Ogawa et al., 2020; Rangasamy et al., 2005; Wu et al., 2019), and cardiovascular disease (Chen and Maltagliati, 2018).
The biological role of GDF15 in the context of NSAIDs administered during inflammation remains unclear. In our studies, we did not observe large effects of manipulating GDF15 on the response to endotoxemia, but we also did not perform more granular analyses dissecting this specific aspect, since the central hypothesis we were testing centered around the dependence of an NRF2-activating NSAID on NRF2 activation of which GDF15 induction is one part. GDF15 biology is an area of research across disciplines, and, like other endocrine cytokines appears to produce different physiologic effects as a function of the host context (Coll et al., 2020; Day et al., 2019; Luan et al., 2019; Santos et al., 2020; Weng et al., 2021), and its “core” biology remains to be elucidated.
Our observation that NSAIDs activate NRF2 begins to reconcile previous observations that NSAID treatment enhanced lifespan in Caenorhabditis elegans (Ching et al., 2011; He et al., 2014), whose genomes do not encode cyclooxygenase isoforms, but do encode Nrf2. NRF2 activation has been implicated in aging, longevity and reduction of cellular stress associated with detoxification of free radicals (Castiglione et al., 2020; Lewis et al., 2015; Lombard et al., 2020). Indeed, it is possible that one reason that only certain NSAIDs increase longevity in studies that span multiple species, and especially in those that do not encode cyclooxygenase isozymes in their genomes, is because they engage NRF2 pathways, which are simultaneously cytoprotective, through the detoxification of harmful free radicals, and also anti-inflammatory. Since cellular stress and inflammation are highly intertwined, it is possible that our observations in improved organ function and inflammation in the models of acute inflammation we used are simultaneously a result of both the anti-inflammatory and the cytoprotective effects of the NRF2-activating NSAIDs.
Our findings potentially explain long-observed seemingly paradoxical properties of NSAIDs that cannot be explained by COX1 and 2 expression. They may also inform limitations and potential of NRF2 activators in the treatment of inflammatory diseases, which are in clinical trials for diseases ranging from multiple sclerosis, Alzheimer’s Disease, chronic kidney disease, diabetes mellitus type 2, asthma, rheumatoid arthritis, psoriasis and various cancers (Cuadrado et al., 2019; Robledinos-Anton et al., 2019). Likewise, NRF2-activating NSAIDs could be considered for use in diseases that respond to NRF2-activators. Thus, our findings, if they bear out in human studies, may significantly impact prescribing and utilization behaviors of NSAIDs.
Like with COX inhibition, there is likely an optimal amount of NRF2 activation where the antioxidant and anti-inflammatory actions of NRF2 are not negatively counterbalanced by the costs related to excessive activation. This has been observed in the context of oncogenesis, where, initially, low levels of NRF2 activation and subsequent antioxidant and anti-inflammatory properties are beneficial in reducing the development of oncogenic mutations and tumor promoting environment, but with the development of a tumor, mutations that lead to unregulated NRF2 activation likely drive cellular survival and proliferation (Sporn and Liby, 2012). Similarly, NSAIDs have anti-inflammatory properties through COX inhibition, but at the cost of gastrointestinal damage, decreased renal perfusion, increased risk of bleeding and cardiovascular disease (Schjerning et al., 2020).
Finally, we show that GDF15 is a soluble marker of NRF2 activation and implicate other inducers of GDF15 as potential NRF2 activators. Metformin also induces GDF15, and the antidiabetic effects of metformin were partially shown to be dependent on GDF15 (Coll et al., 2020; Day et al., 2019). The transcriptional control of this observation remains unclear. Given our identification of NRF2, a xenobiotic sensor, as the key transcription factor in inducing GDF15 in the context of its original discovery as an NSAID-activated gene, it remains to be seen how many other manmade xenobiotics also exert some of their beneficial or harmful actions by engaging NRF2 pathways. Canagliflozin, an antidiabetic and cardioprotective drug which is thought to primarily exert its mechanism of action by inhibition of the Sodium/Glucose Transport Protein 2, activates NRF2 to mediate antioxidant and anti-inflammatory effects (Behnammanesh et al., 2020; Hasan et al., 2020). Furthermore, colchicine activates GDF15 in hepatocytes and that both NRF2 and GDF15 are required for the anti-inflammatory effects of colchicine (Weng et al., 2021). In this study, GDF15 is proposed to be a hepatokine with the action of GDF15 directly on myeloid cells, implicating another receptor for GDF15 that has yet to be characterized. Although different methods for cell-specific deletion of Nrf2 were employed in this study (siRNA) compared to ours (Cre-loxp), both studies taken together suggest GDF15 is likely produced by both myeloid cells and hepatocytes, and likely other parenchymal cells in the kidney (Mulderrig et al., 2021), with all compartments contributing to the circulating levels. The significance of these respective responding cells in contributing to the absolute level of circulating GDF15, which act distally on neurons in the area postrema (Hsu et al., 2017; Mullican et al., 2017) and possibly on myeloid and other cells in a yet undefined mechanism of action, and whether NRF2 is required in all of these cellular compartments is yet to be understood.
For therapeutics designed to activate NRF2, such as DMF and bardoxolone, and other therapeutics that we and others are now identifying as “off-target” NRF2 activators, it remains to be understood to what extent their therapeutic efficacy are “on-target”, NRF2-dependent, and/or GDF15-dependent. Given the pleiotropic role of GDF15, it is possible that GDF15 may play different roles in different contexts (Borner et al., 2020; Gil et al., 2019; Kim et al., 2018; Luan et al., 2019; Mullican et al., 2017; Patel et al., 2019), and much work remains in elucidating the biology of GDF15 and its therapeutic potential in different disease states. Our identification of a subset of NSAIDs as NRF2 activators and GDF15 as an NRF2 target gene may help reconcile the many paradoxical effects of NSAIDs observed across many disease states and have significant clinical and biological implications for other allopathic xenobiotics.
Limitations of Study
An important limitation in the interpretation of our studies is in the inability to completely delete the Ptgs1 and Ptgs2 isozymes. Thus, while our observed effects definitively require NRF2 and KEAP1 C151, it remains possible that the minimal remaining COX activity is also required as others have shown that even residual COX activity and prostaglandin synthesis have large biological effects (Zelenay et al., 2015). Secondly, while we attempted to identify NSAID adducts to KEAP1 using various pull-down and mass spectroscopic approaches, we were unable to definitively identify novel adducts, and the assumption that C151 forms an adduct with NSAIDs or their modifications once metabolized by cells, is likely but speculative. Thus, the precise chemical basis for our observation that bely why some NSAIDs engage NRF2 and activate GDF15 while others do not, remains to be elucidated. Finally, the translatability of this study to humans beyond the in vitro experiments performed in this study is yet to be determined.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to, and will be fulfilled by the lead contact Andrew Wang (andrew.wang@yale.edu).
Materials Availability
Mouse lines generated in this study are available upon request from lead contact.
Data and Code Availability
This study did not generate or analyze datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experiments were performed in accordance with institutional regulations after protocol review and approval by Yale University’s Institutional Animal Care and Use Committee. C57BL/6J (B6), B6.129X1-Nfe2l2tm1Ywk/J (Nrf2−/−), B6.129S(Cg)-Ddit3tm2.1Dron/J (Chop−/−), B6.129P2-Lyz2tm1(cre)Ifo/J (Lyz2 Cre), C57BL/6-Nfe2l2tm1.1Sred/SbisJ (Nrf2flox), B6;129S4-Ptgs1tm1.1Hahe/J (Ptgs1flox), and B6;129S4-Ptgs2tm1Hahe/J (Ptgs2flox) were obtained from Jackson Laboratory. Dr. Anton Komar provided bone marrow from Eif2a deficient animals. Dr. Masayuki Yamamoto provided the Keap1 C151S transgenic animals. Dr. Andy MacMahon provided the Gdf15 deficient mice. Animals were used between the ages of 8 and 10 weeks. All mice were maintained in specific pathogen-free facility and all animal experiments were performed according to institutional regulations after protocol review and approved by Yale University’s Institutional Animal Care and Use Committee.
Cell culture
Bone marrow derived macrophages (BMDMs) were prepared as previously described (Colegio et al., 2014). For assaying Gdf15 expression in response to various stimuli, BMDMs were plated in either 48 well or 24 well plates at a density of 2.5×105 or 5×105 per well, respectively.
Human monocyte-derived macrophages (MDMs) were derived from human peripheral blood mononuclear cells (PBMCs). Initially, monocytes were isolated from cryopreserved PBMCs as per protocol from a human monocyte isolation kit (130-096-537, Miltenyi Biotec). Monocytes were then cultured at 2.5E6/ml in X-VIVO 15 serum-free hematopoietic cell medium (Lonza) supplemented with 50 ng/ml of human M-CSF (AF-300-25, Peprotech) for five days. Cells were then re-plated prior to cell treatments.
METHOD DETAILS
Cell Stimulation
BMDMs were treated with with 0.125 – 0.5 mM indomethacin (Sigma-Aldrich), 0.25 – 1 mM (S)-ibuprofen (Cayman Chemical), 0.125 – 0.5 mM ketoprofen (Sigma-Aldrich), 0.25 – 1 mM Etodolac (Cayman Chemical), or 0.25 – 1 mM naproxen (Cayman Chemical), 0.25–1 mM rofecoxib, 5 mM sulforaphane (Cayman Chemical), 100 μM dimethylfumarate. NSAIDs were diluted in 1% DMSO. Cells were pretreated with NSAID for 1 hour prior to stimulation with 10 ng/mL LPS (L2880, Sigma-Aldrich). LDH assay performed according to manufacturer’s instructions (601170, Cayman Chemical). Reactive oxygen species (ROS) measured using H2DCFDA (D399, ThermoFisher) as described previously (Kavian et al., 2018). Sodium arsenite (Sigma-Aldrich), naphthalenedione (DMNQ, Cayman Chemical) and phenazine methosulfate (PMS, Cayman Chemical) at the indicated doses were used to induce ROS in BMDM.
MDMs were treated with indicated concentrations of indomethacin (Sigma-Aldrich), ketoprofen (Cayman Chemical), or sulforaphane (Cayman Chemical) for indicated durations.
RNA Extraction and Quantification
Cells were lysed in RLT lysis buffer according to manufacturer’s instructions (QIAGEN). For tissue RNA extraction, tissues were excised into RNA Bee RNA isolation reagent (Tel-Test, Inc.) and disrupted by bead homogenization (Omni, Inc.). RNA was extracted using the RNeasy Kit according to manufacturer’s instructions (QIAGEN). cDNA synthesis was performed using SMARTScribe Reverse Transcriptase (Takara) with oligo(dT) primers. qRT-PCR reactions were performed on either aCFX96 Real-Time System or CFX384 Real-Time System (Bio-Rad) using PerfeCTa SYBR Green SuperMix (Quanta Biosciences) or TaqMan assays, where indicated. Transcript levels were normalized to Rpl13a or RPL13A.
Cytokine and Tissue Injury Marker Analysis
Whole blood was harvested from mice by retro-orbital bleeding and plasma was isolated using lithium heparin coated plasma separator tubes (365985, BD Pharmingen). Plasma GDF15 was quantified using a solid phase sandwich ELISA according to manufacturer’s instructions (MGD150, R&D systems). Plasma IL-1ß was quantified by sandwich ELISA utilizing anti-mouse IL-1ß capture antibody (14–7012-85, Invitrogen), biotinylated rat anti-mouse IL-1ß detection antibody (13-7112-85, eBioscience), HRP-conjugated streptavidin (554066, BD Pharmingen). Plasma Tropinin-I concentration and Alanine Aminotransferase (ALT) activity were assayed using kits according to manufacturers’ protocols (Life Diagnostics and Cayman Chemical, respectively). Plasma creatinine and BUN were assayed using HPLC by The George M. O’Brien Kidney Center at Yale. Mouse multiplex cytokine/chemokine array 31-plex of serum cytokines from B6 and Nrf2−/− mice following intraperitoneal injection of LPS and vehicle or indomethacin was measured by Eve Technologies.
Immunofluorescence
BMDMs were treated for 4 hours with 0.5 mM Indomethacin (Sigma-Aldrich) then fixed with 4% PFA (Electron Microscopy Sciences) for 10 minutes at room temperature. Cells were then washed 3 times in ice-cold PBS, permeabilized with 0.25% Triton-X in PBS for 10 minutes at room temperature. After an additional 3 washes, cells were incubated in blocking buffer (1% BSA, 22.52 mg/mL glycine in PBST) for 30 minutes at room temperature. Cells were then incubated with primary antibody overnight at 4°C in 1% BSA in PBST. Cells were washed 3 times with PBS then incubated with secondary antibody in 1% BSA in PBST for 1 hour at room temperature. Cultures were washed again in PBS before counter staining with 0.1 μg/mL DAPI. After a final wash cells were imaged.
ChIP Quantitative PCR
BMDMs were plated in 15 cm plates at a concentration of 1.5×107 cells per plate. Cells were stimulated with either 0.5 mM Indomethacin or vehicle control (1% DMSO) for 4 hours then fixed in 1% formaldehyde (Electron Microscopy Sciences) for 5 minutes at room temperature with gentle agitation. Post fixation, cells were quenched in 0.125M glycine for 5 minutes at room temperature under gentle agitation. Cells were then washed twice with ice cold PBS, scraped into pellets. Pellets were lysed with nuclear lysis buffer (50 mM Tris HCL, 10 mM EDTA, 0.8% SDS) with Halt™ Protease Inhibitor Cocktail (Sigma-Aldrich) for 15 minutes on ice. Samples were then spun at 11,000 RCF for 20 minutes to precipitate SDS. Protein levels were quantified using a Micro BCA Protein Assay kit (23235, ThermoFisher) and diluted with ChIP dilution buffer (16.7 mM TrisHCl (pH 8.0), 167 mM NaCl, 1.2 mM EDTA, 0.01% SDS, 1.1% Triton X-100) to 100 ug for anti-NRF2 (abcam31163, Abcam) antibody and 25 ug for anti-H3K27Ac antibody (ab4729, Abcam). Precipitation antibody were added to each sample then rotated overnight at 4°C. Dynabeads™ Protein G (Thermofisher) were added to each sample, rotated at 4°C for 2 hours then washed twice in Low Salt Immune Complex Wash Buffer (0.1% SDS, 1% Triton X-100, mM EDTA, 20 mM Tris-HCl, pH 8.1, 140 mM NaCl), once in LiCl Wash Buffer (0.25M LiCl, 1% IGEPAL CA630, 1% deoxycholic acid (sodium salt), 1mM EDTA, 10mM Tris, pH 8.1), and once in TE buffer. After final wash, beads were suspended in 250 uL Nuclear lysis buffer with 20 μg/ml proteinase K (Sigma-Aldrich). All samples were incubated at 55°C for 2 hours and then 65°C overnight. DNA was purified with MinElute PCR Purification Kit (1026476, QIAGEN).
To identify potential NRF2 binding sites in GDF15 regulatory elements (Fig. S3), the ARE consensus sequence was downloaded from JASPAR (Fornes et al., 2020) and queried 10kB upstream and downstream of the GDF15 TSS. Due to the palindromic nature of consensus sequences, the reverse compliment, compliment, and reverse sequence of the JASPAR consensus sequence were included in our search algorithm. Identified potential ARE binding sites were searched against enhancer signatures (H3K4me1 or H3K27Ac) based on ENCODE data from BMDMs. The top twenty putative binding sites were screened using quantitative PCR reactions, which were run on CFX384 Real-Time System (Bio-Rad), and percent input was defined as two to the power of the difference in Cq of input and experimental samples. NRF2 binding was then confirmed by designing primers spanning 1 kb upstream and downstream of the identified binding site to detect a peak of enrichment.
Western blot
BMDMs were lysed in Triton-X Lysis buffer (150 mM NaCl, 0.1% Trition-X, 50 mM Tris HCl) with Halt™ Protease Inhibitor Cocktail (Sigma-Aldrich) then centrifuged at 12,000 rpm for 20 minutes at 4°C. Protein concentration in cell lysates were quantified by Pierce™ BCA Protein Assay Kit (ThermoFisher). Sample volume equivalent to 20 μg were mixed with 4x Leammli buffer (Bio Rad) boiled at 95°C for 5 minutes then run on a 4–20% Mini-PROTEAN® TGX™ Precast Protein Gels (BioRad) in running buffer (25 mM Tris, 190 glycine, 0.1% SDS) using a Mini-PROTEAN Tetra Vertical Electrophoresis Cell (BioRad). Protein was then transferred to blotting membrane using a transfer cassette in transfer buffer (25 mM Tris, 190mM glycine, 20% methanol) for 1.5 hours at 100V. Blot was then blocked for 1 hour at room temperature with 5% milk in PBST, then incubated overnight in primary antibody diluted in 5% milk at 4°C. Blots were then washed 5 times in TBST, incubated in HRP conjugated secondary antibody for 1 hour at room temperature. Blots were imaged with a ChemiDoc Imaging System (17001401, BioRad).
Flow Cytometry
For flow cytometry of CD45+ cells from the liver, mice were treated LPS and indomethacin or vehicle and then sacrificed 20 hours later. Livers were harvested, finely diced using a razor blade, and incubated in 2 mg/ml Collagenase IV (Worthington Biochemical Corporation) at 37°C under constant agitation for 30 minutes. The resultant suspension was then mechanically forced through a 70-micron cell strainer to generate a single cell suspension and washed prior to staining. Functional grade mouse anti-CD16/32 (93) antibody was used for Fc-block. At least 1 × 105 cells were acquired on CD45+ cells within the singlet live gate, as defined by size, granularity and pulse-width. Samples were acquired on an LSRII flow cytometer (BD). Fcs files were analyzed using FlowJo (Tree Star Technologies).
In vivo Inflammatory Mouse Models
For acute response to bardoxolone, C57BL/6 mice were gavaged with vehicle (sesame oil) or bardoxolone methyl (Sigma-Aldrich SMB00376) at the indicated concentrations. For long-term bardoxolone feeding experiments, bardoxolone (Sigma-Aldrich SMB00376) was mixed into moist regular chow at 0.01% wt/wt and dried. This food was then fed to C57BL/6 mice for the indicated number of days. Indomethacin was dosed at 5–15 mg/kg, consistent with the long-standing dosing used in the rodent literature (Crestani et al., 1991; Liang et al., 2015; Winter et al., 1963), which is within linear range of dosing in humans (50 mg TID in a 50 kg adolescent would be 3 mg/kg/d).
For LPS endotoxemia, mice were injected intraperitoneally with sub-lethal doses of LPS derived from Escherichia coli 055:B5 (L2880, Sigma-Aldrich) diluted in PBS. Dosing varies dramatically from lot to lot. New lots are tested for LD50. In these studies, sub-lethal doses are 2.5 mg/kg. Mice were injected intraperitoneally with 5 mg/kg indomethacin or 5 mg/kg ketoprofen diluted in bicarbonate buffer, every 4 hours for the duration of the experiment, starting 15 minutes prior to LPS administration. Temperature was measured at regular intervals using a rectal probe. Mice were euthanized and organs harvested 20 hours later. Validation and dosing of GDF15 neutralizing antibody and recombinant GDF15 are previously described (Luan et al., 2019).
For the gout model, the hind foot pad was injected intradermally with saline (right hind foot) or MSU (left hind foot) (1 mg in 40 uL PBS, Invivogen). Mice were injected intraperitoneally with vehicle or 2.5 mg/kg indomethacin diluted in bicarbonate buffer twice daily. Calipers were used to measure diameter of the foot at baseline and 24 hours after injection.
QUANTIFICATION AND STATISTICAL ANALYSIS
Results were statistically analyzed using Student’s t test or an analysis of variance (ANOVA) test with multiple comparisons where appropriate using Prism 6.0 (GraphPad Software, Inc). p value of < 0.05 was considered to be statistically significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-COX1 | Cell signaling technologies | Cat#4841S |
| Rabbit GAPDH | Cell signaling technologies | Cat#2118S |
| Anti-Nrf2 | Abcam | Cat#ab31163 |
| IL1β capture | Invitrogen | Cat#14-7012-85 |
| IL1β detection | eBioscience | Cat#13-7112-85 |
| HRP-conjugated streptavidin | BD Pharmingen | Cat#554066 |
| CD45-PE | eBioscience | Cat#12-0451-83 |
| CD45-APC | eBioscience | Cat#47-0451-82 |
| CD11b-Pacific Blue | Biolegend | Cat#101224 |
| CD3-Alexa 700 | eBioscience | Cat#56-0032-82 |
| Ly6c-Brilliant Violet 605 | Biolegend | Cat#128035 |
| Ly6g-GR1 FITC | eBioscience | Cat#11-5931-85 |
| Ly6g-PECy7 | BD Pharmingen | Cat#560601 |
| F4/80-APC | eBioscience | Cat#17-4801-82 |
| Anti-CD16/32(93) antibody | eBioscience | Cat#14-0161-86 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| indomethacin | Sigma-Aldrich | Cat#I7378 |
| (S)-ibuprofen | Cayman Chemicals | Cat#16793 |
| ketoprofen | Sigma-Aldrich | Cat#10006661 |
| Etodolac | Cayman Chemicals | Cat#20833 |
| naproxen | Cayman Chemicals | Cat#70290 |
| rofecoxib | Cayman Chemicals | Cat#10010260 |
| sulforaphane | Cayman Chemical | Cat#10496 |
| dimethylfumarate | Cayman Chemical | Cat#14714 |
| LPS | Sigma-Aldrich | Cat#L2880 |
| human M-CSF | Peprotech | Cat#AF-300-25 |
| RNA Bee RNA isolation reagent | Tel-Test, Inc. | Cat#CS-501B |
| SMARTScribe Reverse Transcriptase | Takara | Cat#639538 |
| PerfeCTa SYBR Green SuperMix | Quanta Biosciences | Cat#95054 |
| Halt™ Protease Inhibitor Cocktail | ThermoFisher Scientific | Cat#78442 |
| Collagenase IV | Worthington Biochemical Corporation | Cat#LS004188 |
| Monosodium urate (MSU) | Invivogen | Cat#tlrl-msu |
| Critical Commercial Assays | ||
| Pan Monocyte Isolation Kit, human | Miltenyi Biotec | Cat#130-096-537 |
| H2DCFDA (ROS indicator) | ThermoFisher | Cat#D399 |
| LDH Cytotoxicity Assay Kit | Cayman Chemical | Cat#601170 |
| RNeasy Mini Kit | Qiagen | Cat#74106 |
| Mouse/Rat GDF15 Quantikine ELISA Kit | R&D systems | Cat#MGD150 |
| Mouse Cardiac Troponin I ELISA | Life Diagnostics | Cat#CTNI-1-HSP |
| Alanine Transaminase Colorimetric Activity Assay Kit | Cayman Chemical | Cat#700260 |
| Micro BCA Protein Assay kit | ThermoFisher | Cat#23235 |
| Pierce™ BCA Protein Assay Kit | ThermoFisher | Cat#23225 |
| MinElute PCR Purification Kit | Qiagen | Cat#1026476 |
| Experimental Models: Cell Lines | ||
| Human: Monocyte derived macrophages from human peripheral blood mononuclear cells | N/A | |
| Mouse: Bone Marrow Derived Macrophages from various strains of mouse | Wang Lab | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J (B6) | The Jackson Laboratory and bred in house | JAX: 000664 |
| Mouse: B6.129X1-Nfe2l2tm1Ywk/J (NRF2 KO) | The Jackson Laboratory | JAX: 017009 |
| Mouse: B6.129S(Cg)-Ddit3tm2.1Dron/J (CHOP KO) | The Jackson Laboratory | JAX: 005530 |
| Mouse: B6.129P2-Lyz2tm1(cre)Ifo/J (Lyz2 Cre) | The Jackson Laboratory | JAX: 004781 |
| Mouse: C57BL/6-Nfe2l2tm1.1Sred/SbisJ (Nrf2flox) | The Jackson Laboratory | JAX: 025433 |
| Mouse: B6;129S4-Ptgs1tm1.1Hahe/J (Cox-1flox) | The Jackson Laboratory | JAX: 030884 |
| Mouse: B6;129S4-Ptgs2tm1Hahe/J (Cox-2flox) | The Jackson Laboratory | JAX: 030785 |
| Mouse: B6.129P2(SJL)-Myd88tml.1Defr/J (Myd88−/−) | The Jackson Laboratory | JAX: 009088 |
| Mouse: Eif2a−/− | Dr. Anton Komar | N/A |
| Mouse: Nrf2 f/f Lyz2 | This paper | N/A |
| Mouse: Cox1/2 f/fLyz2 | This paper | N/A |
| Mouse: B6.BD-Keap1<em1(C151 S)Mym> | RIKEN | RBRC09596 |
| Oligonucleotides | ||
| Gdf15 | 5’-CCGAGAGGACTCGAACTCAG-3’ | 5’-GGTTGACGCGGAGTAGCAG-3’ |
| Tnf | 5’-TCTGTCTACTGAACTTCGGGGTG-3’ | 5’-ACTTGGTGGTTTGCTACGACG-3’ |
| Nrf2 | 5’-CTGAACTCCTGGACGGGACTA-3’ | 5’-CGGTGGGTCTCCGTAAATGG-3’ |
| Ptgs1 | 5’-GTCCTGCTCGCAGATCCT-3’ | 5’-GTAGTTGTCGAGGCCAAAGC-3’ |
| Ptgs2 | 5-GACCTGGGTTCACCCGAGGACTG-3’ | 5’-CCAAAGACTTCCTGCCCCACAGC-3’ |
| Il1b | 5’-CAGTTGTCTAATGGGAACGTCA-3’ | 5’-GCACCTTCTTTTCCTTCATCTTT-3’ |
| Nqo1 | 5’-TCCAGACGTTTCTTCCATCC-3’ | 5’-GGTAGCGGCTCCATGTACTC-3’ |
| Hmox1 | 5’-CATCCAAGCCGAGAATGCTG-3’ | 5’-CCTCAGGGAAGTAGAGTGGGG-3’ |
| Cat | 5’-TCCATCCAGCGTTGATTACA-3’ | 5’-ATCCAGGCTCTTCTGGACAA-3’ |
| Gsr | 5’-ACCACGAGGAAGACGAAATG-3’ | 5’-CCTGCAGCATCTCATCACAG-3’ |
| Gclm | 5’-TGGAGCAGCTGTATCAGTGG-3’ | 5’-TTGTTTAGCAAAGGCAGTCAAA-3’ |
| Taldo1 | 5’-GCAAGGACAGAATTCTCATCAA-3’ | 5’-CAGTGTCATGTTGCAGTGGA-3’ |
| Cxcl10 | 5’-AATGAAAGCGTTTAGCCAAAAA-3’ | 5’-GAGGCTCTCTGCTGTCCATC-3’ |
| IL12a | 5’-CTAGACAAGGGCATGCTGGT-3’ | 5’-TCTCCCACAGGAGGTTTCTG-3’ |
| IL12b | 5’-GGTGTAACCAGAAAGGTGCG-3’ | 5’-AAGGTGTCATGATGAACTTAG-3’ |
| Il17a | 5’-AAAGCTCAGCGTGTCCAAAC-3’ | 5’-AGCTTCCCAGATCACAGAGG-3’ |
| Cxcl2 | 5’-AGTTTGCCTTGACCCTGAAGC-3’ | 5’-AGGCTCCTCCTTTCCAGG-3’ |
| Ifng | 5’-CGGCACAGTCATTGAAAGCCTA-3’ | 5’-GTTGCTGATGGCCTGATTGTC-3’ |
| Gpr109a | 5’-ATGGCGAGGCATATCTGTGTAGCA-3’ | 5’-TCCTGCCTGAGCAGAACAAGATGA-3’ |
| Il6 | 5’-GACTTCCATCCAGTTGCCTTCTTGG-3’ | 5’-CCAGTTTGGTAGCATCCATCATTTCT-3’ |
| Ifnb | 5’-CCTGCAACCACCACTCATTC-3’ | 5’-GTCCTCAACTGCTCTCCACT-3’ |
| GDF15 | ThermoFisher Scientific | Hs00171132_m1 |
| RPL13A | ThermoFisher Scientific | Hs04194366_g1 |
Highlights.
A subset of nonsteroidal anti-inflammatory drugs (NSAIDs) activate NRF2.
C151 of the chaperone protein, KEAP1, is necessary for NRF2 activation by indomethacin.
Indomethacin reduces inflammation in endotoxemia and gout models dependent on NRF2.
Acknowledgments
We thank the Wang Lab for valuable discussions. We thank Dr. Anton Komar (Cleveland State University) for providing bone marrow from Eif2a deficient mice. We thank Dr. Masayuki Yamamoto (RIKEN Institute) for the KEAP1 C151S transgenic animals. We thank Dr. Andy MacMahon for the GDF15 deficient animals (University of Southern California). We thank Drs. Ruslan Medzhitov, Michael Brown, Deep Dixit, and Ruth Franklin and Krista Wang for critical review of the manuscript.
Funding:
The Wang lab is supported in part by grants from the NIH (K08 AI128745 and R01 AI162645), the Charles H. Hood Foundation, the Food Allergy Science Initiative, and by the Pew Charitable trusts. AE received support from the following sources: T32 AR007016-43 from NIH/NIAMS, Dermatologist Investigator Research Fellowship from the Dermatology Foundation, Yale Center for Clinical Investigation/Yale Physician Scientist Development Award, and the Robert E Leet and Clara Guthrie Patterson Trust.
Footnotes
Declaration of Interests: PT, HT, and HHL were employees of NGM Biopharmaceuticals at the time this work was done and may hold stock or stock options in this company. All data is available in the main text or the supplementary materials.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abram CL, Roberge GL, Hu Y, and Lowell CA (2014). Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods 408, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Harbi NO, Nadeem A, Ahmad SF, AlThagfan SS, Alqinyah M, Alqahtani F, Ibrahim KE, and Al-Harbi MM (2019). Sulforaphane treatment reverses corticosteroid resistance in a mixed granulocytic mouse model of asthma by upregulation of antioxidants and attenuation of Th17 immune responses in the airways. Eur J Pharmacol 855, 276–284. [DOI] [PubMed] [Google Scholar]
- Angel J, Berenbaum F, Le Denmat C, Nevalainen T, Masliah J, and Fournier C (1994). Interleukin-1-induced prostaglandin E2 biosynthesis in human synovial cells involves the activation of cytosolic phospholipase A2 and cyclooxygenase-2. Eur J Biochem 226, 125–131. [DOI] [PubMed] [Google Scholar]
- Arias-Negrete S, Keller K, and Chadee K (1995). Proinflammatory cytokines regulate cyclooxygenase-2 mRNA expression in human macrophages. Biochem Biophys Res Commun 208, 582–589. [DOI] [PubMed] [Google Scholar]
- Baek SJ, Kim KS, Nixon JB, Wilson LC, and Eling TE (2001). Cyclooxygenase inhibitors regulate the expression of a TGF-beta superfamily member that has proapoptotic and antitumorigenic activities. Mol Pharmacol 59, 901–908. [PubMed] [Google Scholar]
- Bannuru RR, Schmid CH, Kent DM, Vaysbrot EE, Wong JB, and McAlindon TE (2015). Comparative effectiveness of pharmacologic interventions for knee osteoarthritis: a systematic review and network meta-analysis. Ann Intern Med 162, 46–54. [DOI] [PubMed] [Google Scholar]
- Behnammanesh G, Durante GL, Khanna YP, Peyton KJ, and Durante W (2020). Canagliflozin inhibits vascular smooth muscle cell proliferation and migration: Role of heme oxygenase-1. Redox Biol 32, 101527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanca-Lopez N, Soriano V, Garcia-Martin E, Canto G, and Blanca M (2019). NSAID-induced reactions: classification, prevalence, impact, and management strategies. J Asthma Allergy 12, 217–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borner T, Shaulson ED, Ghidewon MY, Barnett AB, Horn CC, Doyle RP, Grill HJ, Hayes MR, and De Jonghe BC (2020). GDF15 Induces Anorexia through Nausea and Emesis. Cell Metab 31, 351–362 e355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brater DC, Harris C, Redfern JS, and Gertz BJ (2001). Renal effects of COX-2-selective inhibitors. Am J Nephrol 21, 1–15. [DOI] [PubMed] [Google Scholar]
- Castiglione GM, Xu Z, Zhou L, and Duh EJ (2020). Adaptation of the master antioxidant response connects metabolism, lifespan and feather development pathways in birds. Nat Commun 11, 2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QM, and Maltagliati AJ (2018). Nrf2 at the heart of oxidative stress and cardiac protection. Physiol Genomics 50, 77–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching TT, Chiang WC, Chen CS, and Hsu AL (2011). Celecoxib extends C. elegans lifespan via inhibition of insulin-like signaling but not cyclooxygenase-2 activity. Aging Cell 10, 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubak J, Whitlock EP, Williams SB, Kamineni A, Burda BU, Buist DS, and Anderson ML (2016). Aspirin for the Prevention of Cancer Incidence and Mortality: Systematic Evidence Reviews for the U.S. Preventive Services Task Force. Ann Intern Med 164, 814–825. [DOI] [PubMed] [Google Scholar]
- Cilenti F, Barbiera G, Caronni N, Iodice D, Montaldo E, Barresi S, Lusito E, Cuzzola V, Vittoria FM, Mezzanzanica L, et al. (2021). A PGE2-MEF2A axis enables context-dependent control of inflammatory gene expression. Immunity 54, 1665–1682 e1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll AP, Chen M, Taskar P, Rimmington D, Patel S, Tadross JA, Cimino I, Yang M, Welsh P, Virtue S, et al. (2020). GDF15 mediates the effects of metformin on body weight and energy balance. Nature 578, 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coxib, traditional, N.T.C., Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, Bombardier C, Cannon C, et al. (2013). Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet 382, 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestani F, Seguy F, and Dantzer R (1991). Behavioural effects of peripherally injected interleukin-1: role of prostaglandins. Brain Res 542, 330–335. [DOI] [PubMed] [Google Scholar]
- Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, Attucks OC, Franklin S, Levonen AL, Kensler TW, et al. (2019). Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov 18, 295–317. [DOI] [PubMed] [Google Scholar]
- da Costa BR, Reichenbach S, Keller N, Nartey L, Wandel S, Juni P, and Trelle S (2017). Effectiveness of non-steroidal anti-inflammatory drugs for the treatment of pain in knee and hip osteoarthritis: a network meta-analysis. Lancet 390, e21–e33. [DOI] [PubMed] [Google Scholar]
- Day EA, Ford RJ, Smith BK, Mohammadi-Shemirani P, Morrow MR, Gutgesell RM, Lu R, Raphenya AR, Kabiri M, McArthur AG, et al. (2019). Metformin-induced increases in GDF15 are important for suppressing appetite and promoting weight loss. Nat Metab 1, 1202–1208. [DOI] [PubMed] [Google Scholar]
- Dayalan Naidu S, Muramatsu A, Saito R, Asami S, Honda T, Hosoya T, Itoh K, Yamamoto M, Suzuki T, and Dinkova-Kostova AT (2018). C151 in KEAP1 is the main cysteine sensor for the cyanoenone class of NRF2 activators, irrespective of molecular size or shape. Sci Rep 8, 8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M, Lambers Heerspink HJ, et al. (2013). Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 369, 2492–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desborough MJR, and Keeling DM (2017). The aspirin story - from willow to wonder drug. Br J Haematol 177, 674–683. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, and Talalay P (2002). Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A 99, 11908–11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dona I, Blanca-Lopez N, Torres MJ, Garcia-Campos J, Garcia-Nunez I, Gomez F, Salas M, Rondon C, Canto MG, and Blanca M (2012). Drug hypersensitivity reactions: response patterns, drug involved, and temporal variations in a large series of patients. J Investig Allergol Clin Immunol 22, 363–371. [PubMed] [Google Scholar]
- Foresti R, Bains SK, Pitchumony TS, de Castro Bras LE, Drago F, Dubois-Rande JL, Bucolo C, and Motterlini R (2013). Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol Res 76, 132–148. [DOI] [PubMed] [Google Scholar]
- Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranasic D, et al. (2020). JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48, D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu JY, Masferrer JL, Seibert K, Raz A, and Needleman P (1990). The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J Biol Chem 265, 16737–16740. [PubMed] [Google Scholar]
- Funk CD (2001). Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294, 1871–1875. [DOI] [PubMed] [Google Scholar]
- Gao J, Mfuh A, Amako Y, and Woo CM (2018). Small Molecule Interactome Mapping by Photoaffinity Labeling Reveals Binding Site Hotspots for the NSAIDs. J Am Chem Soc 140, 4259–4268. [DOI] [PubMed] [Google Scholar]
- Gil CI, Ost M, Kasch J, Schumann S, Heider S, and Klaus S (2019). Role of GDF15 in active lifestyle induced metabolic adaptations and acute exercise response in mice. Sci Rep 9, 20120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurpinar E, Grizzle WE, and Piazza GA (2013). COX-Independent Mechanisms of Cancer Chemoprevention by Anti-Inflammatory Drugs. Front Oncol 3, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurpinar E, Grizzle WE, and Piazza GA (2014). NSAIDs inhibit tumorigenesis, but how? Clin Cancer Res 20, 1104–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanif R, Pittas A, Feng Y, Koutsos MI, Qiao L, Staiano-Coico L, Shiff SI, and Rigas B (1996). Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem Pharmacol 52, 237–245. [DOI] [PubMed] [Google Scholar]
- Hasan R, Lasker S, Hasan A, Zerin F, Zamila M, Chowdhury FI, Nayan SI, Rahman MM, Khan F, Subhan N, et al. (2020). Canagliflozin attenuates isoprenaline-induced cardiac oxidative stress by stimulating multiple antioxidant and anti-inflammatory signaling pathways. Sci Rep 10, 14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Tsuchiyama SK, Nguyen QT, Plyusnina EN, Terrill SR, Sahibzada S, Patel B, Faulkner AR, Shaposhnikov MV, Tian R, et al. (2014). Enhanced longevity by ibuprofen, conserved in multiple species, occurs in yeast through inhibition of tryptophan import. PLoS Genet 10, e1004860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndon CM, Hutchison RW, Berdine HJ, Stacy ZA, Chen JT, Farnsworth DD, Dang D, Fermo JD, Ambulatory Care C, Pain, et al. (2008). Management of chronic nonmalignant pain with nonsteroidal antiinflammatory drugs. Joint opinion statement of the Ambulatory Care, Cardiology, and Pain and Palliative Care Practice and Research Networks of the American College of Clinical Pharmacy. Pharmacotherapy 28, 788–805. [DOI] [PubMed] [Google Scholar]
- Hsu JY, Crawley S, Chen M, Ayupova DA, Lindhout DA, Higbee J, Kutach A, Joo W, Gao Z, Fu D, et al. (2017). Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 550, 255–259. [DOI] [PubMed] [Google Scholar]
- Inoue H, Yokoyama C, Hara S, Tone Y, and Tanabe T (1995). Transcriptional regulation of human prostaglandin-endoperoxide synthase-2 gene by lipopolysaccharide and phorbol ester in vascular endothelial cells. Involvement of both nuclear factor for interleukin-6 expression site and cAMP response element. J Biol Chem 270, 24965–24971. [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et al. (1997). An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236, 313–322. [DOI] [PubMed] [Google Scholar]
- Itoh K, Tong KI, and Yamamoto M (2004). Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic Biol Med 36, 1208–1213. [DOI] [PubMed] [Google Scholar]
- Kavian N, Mehlal S, Jeljeli M, Saidu NEB, Nicco C, Cerles O, Chouzenoux S, Cauvet A, Camus C, Ait-Djoudi M, et al. (2018). The Nrf2-Antioxidant Response Element Signaling Pathway Controls Fibrosis and Autoimmunity in Scleroderma. Front Immunol 9, 1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, and Patrono C (2006). Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ 332, 1302–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Kim SH, Han DH, Jo YS, Lee YH, and Lee MS (2018). Growth differentiation factor 15 ameliorates nonalcoholic steatohepatitis and related metabolic disorders in mice. Sci Rep 8, 6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, et al. (2016). Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun 7, 11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp E, and Ghosh S (1994). Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 265, 956–959. [DOI] [PubMed] [Google Scholar]
- Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, et al. (1995). Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell 83, 483–492. [DOI] [PubMed] [Google Scholar]
- Lewis KN, Wason E, Edrey YH, Kristan DM, Nevo E, and Buffenstein R (2015). Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proc Natl Acad Sci U S A 112, 3722–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Yu C, and Zeng X (2020). Comparative efficacy of traditional non-selective NSAIDs and selective cyclo-oxygenase-2 inhibitors in patients with acute gout: a systematic review and meta-analysis. BMJ Open 10, e036748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Bittinger K, Li X, Abernethy DR, Bushman FD, and FitzGerald GA (2015). Bidirectional interactions between indomethacin and the murine intestinal microbiota. Elife 4, e08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S, et al. (2011). Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 134, 678–692. [DOI] [PubMed] [Google Scholar]
- Liu J, Kumar S, Heinzel A, Gao M, Guo J, Alvarado GF, Reindl-Schwaighofer R, Krautzberger AM, Cippa PE, McMahon J, et al. (2020). Renoprotective and Immunomodulatory Effects of GDF15 following AKI Invoked by Ischemia-Reperfusion Injury. J Am Soc Nephrol 31, 701–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo PC, Tsai YT, Lin SK, and Lai JN (2016). Risk of asthma exacerbation associated with nonsteroidal anti-inflammatory drugs in childhood asthma: A nationwide population-based cohort study in Taiwan. Medicine (Baltimore) 95, e5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Kohler WJ, Guo AH, Gendron C, Han M, Ding W, Lyu Y, Ching TT, Wang FY, Chakraborty TS, et al. (2020). High-throughput small molecule screening reveals Nrf2-dependent and -independent pathways of cellular stress resistance. Sci Adv 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM, Herzog EL, Kang I, Pisani MA, Yu S, et al. (2019). GDF15 Is an Inflammation-Induced Central Mediator of Tissue Tolerance. Cell 178, 1231–1244 e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLagan T (1876). The treatment of acute rheumatism by salicin. Lancet I, 585. [Google Scholar]
- Mallinson PI, Reagan AC, Coupal T, Munk PL, Ouellette H, and Nicolaou S (2014). The distribution of urate deposition within the extremities in gout: a review of 148 dual-energy CT cases. Skeletal Radiol 43, 277–281. [DOI] [PubMed] [Google Scholar]
- McGettigan P, and Henry D (2011). Cardiovascular risk with non-steroidal anti-inflammatory drugs: systematic review of population-based controlled observational studies. PLoS Med 8, e1001098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, et al. (2018). Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulderrig L, Garaycoechea JI, Tuong ZK, Millington CL, Dingler FA, Ferdinand JR, Gaul L, Tadross JA, Arends MJ, O’Rahilly S, et al. (2021). Aldehyde-driven transcriptional stress triggers an anorexic DNA damage response. Nature 600, 158–163. [DOI] [PubMed] [Google Scholar]
- Mullican SE, Lin-Schmidt X, Chin CN, Chavez JA, Furman JL, Armstrong AA, Beck SC, South VJ, Dinh TQ, Cash-Mason TD, et al. (2017). GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat Med 23, 1150–1157. [DOI] [PubMed] [Google Scholar]
- Nagashima R, Kosai H, Masuo M, Izumiyama K, Noshikawaji T, Morimoto M, Kumaki S, Miyazaki Y, Motohashi H, Yamamoto M, et al. (2019). Nrf2 Suppresses Allergic Lung Inflammation by Attenuating the Type 2 Innate Lymphoid Cell Response. J Immunol 202, 1331–1339. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Ishitsuka Y, Nakamura Y, Kubota N, Saito A, Fujisawa Y, Watanabe R, Okiyama N, Suga Y, Roop DR, et al. (2020). NRF2 Augments Epidermal Antioxidant Defenses and Promotes Atopy. J Immunol 205, 907–914. [DOI] [PubMed] [Google Scholar]
- Ong CK, Lirk P, Tan CH, and Seymour RA (2007). An evidence-based update on nonsteroidal anti-inflammatory drugs. Clin Med Res 5, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Alvarez-Guaita A, Melvin A, Rimmington D, Dattilo A, Miedzybrodzka EL, Cimino I, Maurin AC, Roberts GP, Meek CL, et al. (2019). GDF15 Provides an Endocrine Signal of Nutritional Stress in Mice and Humans. Cell Metab 29, 707–718 e708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, et al. (2005). Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med 202, 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnam NM, Peterson JM, Talbert EE, Ladner KJ, Rajasekera PV, Schmidt CR, Dillhoff ME, Swanson BJ, Haverick E, Kladney RD, et al. (2017). NF-kappaB regulates GDF-15 to suppress macrophage surveillance during early tumor development. J Clin Invest 127, 3796–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robledinos-Anton N, Fernandez-Gines R, Manda G, and Cuadrado A (2019). Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid Med Cell Longev 2019, 9372182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo de la Vega M, Chapman E, and Zhang DD (2018). NRF2 and the Hallmarks of Cancer. Cancer Cell 34, 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollason V, Samer CF, Daali Y, and Desmeules JA (2014). Prediction by pharmacogenetics of safety and efficacy of non-steroidal anti- inflammatory drugs: a review. Curr Drug Metab 15, 326–343. [DOI] [PubMed] [Google Scholar]
- Saito R, Suzuki T, Hiramoto K, Asami S, Naganuma E, Suda H, Iso T, Yamamoto H, Morita M, Baird L, et al. (2016). Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol Cell Biol 36, 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos I, Colaco HG, Neves-Costa A, Seixas E, Velho TR, Pedroso D, Barros A, Martins R, Carvalho N, Payen D, et al. (2020). CXCL5-mediated recruitment of neutrophils into the peritoneal cavity of Gdf15-deficient mice protects against abdominal sepsis. Proc Natl Acad Sci U S A 117, 12281–12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, Zeng W, Ryan S, Yamamoto M, Lukashev M, et al. (2012). Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther 341, 274–284. [DOI] [PubMed] [Google Scholar]
- Schjerning AM, McGettigan P, and Gislason G (2020). Cardiovascular effects and safety of (non-aspirin) NSAIDs. Nat Rev Cardiol 17, 574–584. [DOI] [PubMed] [Google Scholar]
- Schmidt A (1934). Die Industrielle Chemie in ihrer Bedeutung im Weltbild und Erinnerungen an ihren Aufbau. (Berlin: De Greuter; ). [Google Scholar]
- Schumacher HR Jr. (2004). Management strategies for osteoarthritis, ankylosing spondylitis, and gouty arthritis. J Clin Rheumatol 10, S18–25. [DOI] [PubMed] [Google Scholar]
- Shi J, Hua L, Harmer D, Li P, and Ren G (2018). Cre Driver Mice Targeting Macrophages. Methods Mol Biol 1784, 263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE, Soti S, Jones TA, Nakagawa A, Xue D, and Yin H (2017). Non-steroidal Anti-inflammatory Drugs Are Caspase Inhibitors. Cell Chem Biol 24, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sporn MB, and Liby KT (2012). NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer 12, 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone E (1763). An account of the succefs of the bark of the willow in the cure of agues. Philosophical Transactions 53, 195–200. [Google Scholar]
- Suzuki T, Gao J, Ishigaki Y, Kondo K, Sawada S, Izumi T, Uno K, Kaneko K, Tsukita S, Takahashi K, et al. (2017). ER Stress Protein CHOP Mediates Insulin Resistance by Modulating Adipose Tissue Macrophage Polarity. Cell Rep 18, 2045–2057. [DOI] [PubMed] [Google Scholar]
- Shen TY, T.B.W., Rosegay A, Witzel BE, Wilson AN, Willett JD, Holtz WJ, Ellis RL, Matzuk AR, Lucas S, Stammer CH, Holly FW, Sarett LH, Risley EA, Nuss GW, and Winter CA (1963). Non-Steroid Anti-Inflammatory Agents. J Am Chem Soc 85, 488–489. [Google Scholar]
- Takaya K, Suzuki T, Motohashi H, Onodera K, Satomi S, Kensler TW, and Yamamoto M (2012). Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic Biol Med 53, 817–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth RK, and Warfel NA (2017). Strange Bedfellows: Nuclear Factor, Erythroid 2-Like 2 (Nrf2) and Hypoxia-Inducible Factor 1 (HIF-1) in Tumor Hypoxia. Antioxidants (Basel) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trelle S, Reichenbach S, Wandel S, Hildebrand P, Tschannen B, Villiger PM, Egger M, and Juni P (2011). Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ 342, c7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR (1971). Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231, 232–235. [DOI] [PubMed] [Google Scholar]
- Venugopal R, and Jaiswal AK (1996). Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A 93, 14960–14965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, and Vane JR (1999). Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci U S A 96, 7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng JH, Koch PD, Luan HH, Tu HC, Shimada K, Ngan I, Ventura R, Jiang R, and Mitchison TJ (2021). Colchicine acts selectively in the liver to induce hepatokines that inhibit myeloid cell activation. Nat Metab 3, 513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter CA, Risley EA, and Nuss GW (1962). Carrageenin-induced edema in hind paw of the rat as an assay for antiiflammatory drugs. Proc Soc Exp Biol Med 111, 544–547. [DOI] [PubMed] [Google Scholar]
- Winter CA, Risley EA, and Nuss GW (1963). Anti-Inflammatory and Antipyretic Activities of Indomethacin, 1-(P-Chlorobenzoyl)-5-Methoxy-2-Methylindole-3-Acetic Acid. J Pharmacol Exp Ther 141, 369–376. [PubMed] [Google Scholar]
- Wu W, Peng G, Yang F, Zhang Y, Mu Z, and Han X (2019). Sulforaphane has a therapeutic effect in an atopic dermatitis murine model and activates the Nrf2/HO1 axis. Mol Med Rep 20, 1761–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kensler TW, and Motohashi H (2018). The KEAP1-NRF2 System: a Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol Rev 98, 1169–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, et al. (2015). Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 162, 1257–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Talalay P, Cho CG, and Posner GH (1992). A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci U S A 89, 2399–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Chan E, Duan W, Huang M, and Chen YZ (2005). Drug bioactivation, covalent binding to target proteins and toxicity relevance. Drug Metab Rev 37, 41–213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze datasets or code.