

Abstract
The Angiotensin Converting Enzyme 2 (ACE2) assists the regulation of blood pressure and is the main target of the coronaviruses responsible for SARS and COVID19. The catalytic function of ACE2 relies on the opening and closing motion of its peptidase domain (PD). In this study we investigated the possibility to the allosteric control of the ACE2 PD functional dynamics. After confirming that ACE2 PD binding site opening-closing motion is dominant in characterizing its conformational landscape, we observed that few mutations in the viral receptor binding domain fragments were able to impart different effects on the binding site opening of ACE2 PD. This showed that binding to the solvent exposed area of ACE2 PD can effectively alter the conformational profile of the protein, and thus likely its catalytic function. Using a targeted machine learning model and relative entropy-based statistical analysis, we proposed the mechanism for the allosteric perturbation that regulates the ACE2 PD binding site dynamics at atomistic level. The key residues and the source of the allosteric regulation of ACE PD dynamics are also presented.
1. Introduction
Angiotensin-converting enzyme 2 (ACE2) is a key player in a variety of crucial biological systems.1 Recently, ACE2 has gained considerable attention as the human receptor for a number of coronaviruses, including the ones responsible for the Severe Acute Respiratory Syndrome (SARS) and the current Coronavirus Disease 2019 (COVID19).1–3 During coronaviruses infection, the viral spike protein fragment S13–5 binds to ACE2 at the host cell surface first. Then the viral capsid fuses with the membrane and injects the viral load into the cell.6
The severity in pathology for these respiratory coronaviruses, and in particular for COVID19, was correlated with comorbidities that are related to the Renin-Angiotensin-Aldosterone System (RAAS). RAAS is the hormonal system responsible for the regulation of blood pressure as well as numerous other essential aspects of human physiology.7 The connection between the RAAS anomalies and coronaviruses severity lies in ACE2. In RAAS, ACE2 catalyzes the hormonal peptide angiotensin-II, a vasoconstrictor, to angiotensin (1–7), a vasodilator, to lower blood pressure.1,5 Due to this dual role of ACE2, ACE2 inhibitors were proposed as potential treatment against coronaviruses infection.8
ACE2 is located at the extracellular side of cell membranes and interacts with the transmembrane sodium-dependent neutral amino acid transporter B(0)AT1, which ACE2 helps as chaperone.1,9,10 ACE2 is composed of an extracellular N-terminal peptidase domain (PD, residues 19–615) and a C-terminal collectrin-like domain (CLD residues 616 to 768). The CLD domain is further divided into the neck domain (residues 616 to 726) and a transmembrane helix (residues 726 to 768). 3 The CLD domain anchors ACE2 to the cell membrane and aids B(0)AT1 in transporting amino acids.3,9,10
PD domain is responsible for ACE2 catalytic activity and is the target for coronavirus S1 receptor binding domain (RBD) binding.3,11 Alteration of the this structural region has the potential to negate the catalytic ability of ACE2 by restricting the access to the binding site, which can ultimately impact ACE2 related pathologies. This could occur via protein allostery, a biological mechanism of protein dynamical and conformational space change via binding to a secondary allosteric site.
In this study, we present a computational mechanistic investigation to probe allosteric differences in ACE2 PD via targeted machine learning, structural, and statistical approaches. Four systems are considered: the ACE2 PD in the apo state11, the ACE2 PD in presence of the inhibitor MLN-4760,11 and ACE2 PD complexed with the S1 fragments of SARS-CoV-112 or SARS-CoV-23. Our mechanistic investigations are based on the ACE2 simulation data provided by D. E. Shaw Research13 as part of the global initiative that sparked the scientific community to effectively study coronaviruses, ACE2, and their interaction.14–17
Molecular dynamics simulations serve as a powerful tool to explore the relations between molecular structures, their motions, and protein function by providing the evolution of a system over time at an atomistic level.18–20 Machine learning approaches have been applied to the study of biomolecular system and have helped to reveal the underlying biological information by tackling the high complexity of molecular dynamics simulations data.21–23 Among the multitude of machine learning approaches, Convolutional Neural Networks (CNN) have gained special interests due to their ability to extract local patterns in the data structure.24,25 Herein, we developed a Collective Variable-guided CNN (CV-CNN) model as a novel scheme to capture the functional and structural differences of ACE2 PD. Principal component analysis was used to visualize the high-dimensional protein conformational space. Markov state model was used to characterize the kinetics of ACE2 PD dynamics within its conformational space. Lastly, the relative entropy-based dynamical allosteric network model was employed to obtain the pathway information of residue-residue interactions that characterize ACE2 PD functional dynamics.
This ensemble of computational tools for analyzing molecular dynamics simulations enabled us to validate and integrate ACE2 PD functional dynamics with atomistic-level details, which are otherwise inaccessible from experimental investigations. Our results detail the possibility of the allosteric control of the functional dynamics of ACE2: where key interactions in the solvent exposed region of the ACE2 PD can force the closing of the catalytic domain.
2. Materials and Methods
2.1. Data Acquisition
The molecular dynamics trajectories were obtained from D.E. Shaw research.13 Four systems were obtained based on the following initial structure: ACE2 ectodomain (peptidase domain) in an apo open state (PDB ID: 1R42)11, ACE2 ectodomain in an inhibitor-bound closed state (PDB ID: 1R4L)11, human ACE2 in complex with the Receptor Binding Domain of spike protein from SARS-CoV-1 (PDB ID: 2AJF)12, and ACE2 in complex with the Receptor Binding Domain of spike protein from SARS-CoV-2 (PDB ID: 6M17)3. The glycosylation states of the systems are shown in Fig. S1. The simulations details are provided in the reference 17 and are briefly listed as the following. The simulations used the Amber ff99SB-ILDN force field26 for proteins, the TIP3P model 27 for water, and the generalized Amber force field28 for glycosylated asparagine. The carboxylate and amino peptide termini, including those exposed due to missing loops in the crystal structures, are capped with amide and acetyl groups, respectively. The system was neutralized and salted with NaCl at a final concentration of 0.15 M. Each system was simulated for 10 μs. The interval between frames is 1.2 ns. The simulations were conducted at 310 K in the isothermal-isobaric (NPT) ensemble. In the present study, the naming of the secondary structure follows the UniProt29 classification and can be found in the Supporting Table S1.
2.2. CV-CNN: Collective Variable-Guided Multi-Task Convolutional Learning
A novel deep learning protocol was developed and implemented in the present study to obtain critical biologically relevant information regarding the ACE2 dynamics.
2.2.1. Data Featurization
The first step to achieve model interpretability is to provide the learner with reasonable training data representation. In this study, the following data featurization scheme is adopted. Each frame of the simulations is represented by a N×N contact matrix for N number of residues. This contact matrix initially contains residue-to-residue distances, expressed in angstrom (Å), between the closest heavy-atoms. Subsequently, the interaction distances exceeding 4 Å were set to zero. This cutoff was chosen as it represents the upper limit for significant inter-molecular interactions, such as salt bridges and hydrogen bonding.30,31 This featurization strategy enforces the learning of local bonding patterns correlated with large protein motions. The distances were computed using the python package MDTraj 1.9.3.32
2.2.2. Model Architecture
Motivated by the unique feature of convolutional filters to extract detailed local patterns in the data structure while retaining the high-level semantics24,25, a multi-task Convolutional Neural Network (CNN) model was implemented in the current study. Convolution is a linear operation used for feature extraction. In a convolutional layer, a set of matrices called filters (or kernels) are applied across the input matrix. For each overlap between a filter and a portion of the input matrix, an element-wise product is calculated to obtain the output value in the corresponding position of the output matrix, called the feature map.33 The latter is then delivered as the input to the following layers. Because the convolutional filters extract local feature patterns, we expect that the model to learn specific short-range bonding patterns buried implicitly in the data. Accordingly, we defined two goals for the learning model: The first is a goal of classification that the CNN model is expected to correctly identify the state of each molecular dynamics snapshot, which are the two metastable states of ACE2 in the presence of SARS-CoV-1 and SARS-CoV-2 RBD fragments. The second is a goal of regression where the collective variable, which describes the opening of the ACE2 binding site, should be correctly predicted by the learning model. Accordingly, a multi-task CNN architecture was developed to achieve these two goals simultaneously (Fig. 1). These two outputs are connected to the convolutional section of the model via a fully connected (dense) layer that functions as the latent space. The latter is expected to select for and optimize the feature patterns that help both distinguishing protein functional states and correlation with the collective variable. For these reasons the machine learning model in our study is referred as CV-CNN.
Fig. 1.
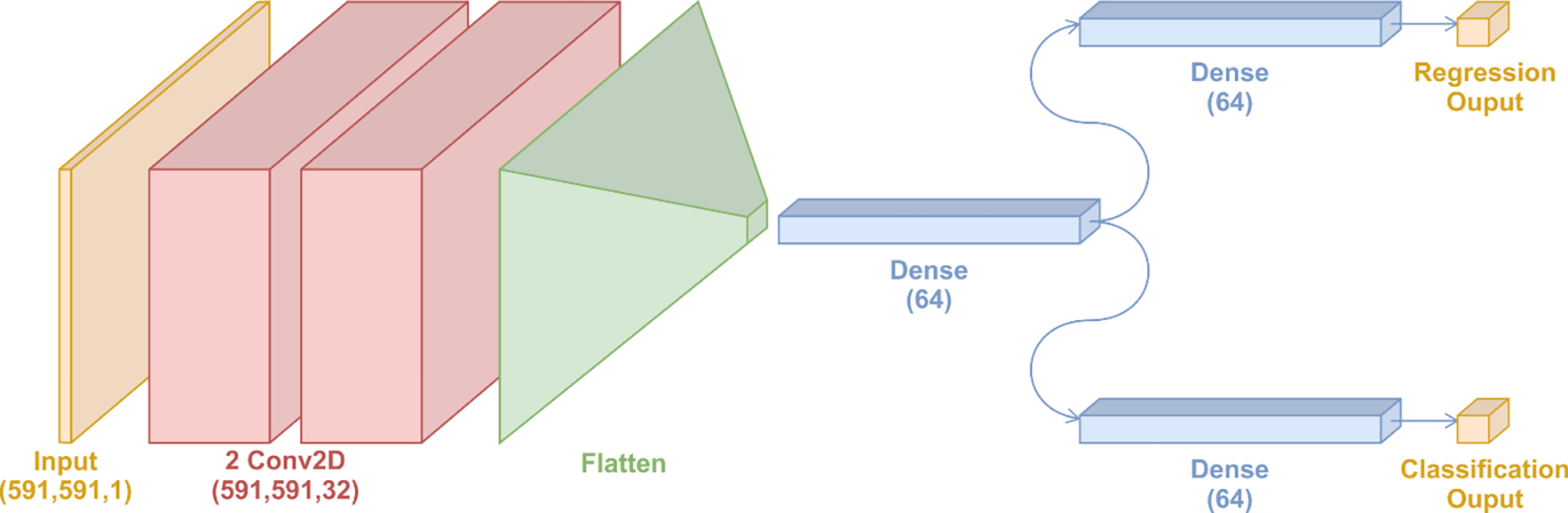
The architecture of the collective variable-guided multi-task convolutional neural network (CV-CNN).
The outline of the architecture is illustrated in Fig. 1. We used 32 filters, each of size 4×4, for all convolutional layers. The dense layers were composed of 64 neurons each. A rectified linear unit (ReLU) activation function is used for both convolutional and latent dense layer. The normalized exponential function (softmax) and the linear activation function were used for the classification and regression layers, respectively. In Table S2, the details for each layer are shown. The dropout and L1-regularization strategies were implemented to avoid overfitting and to promote generalizability of the model.34–36 The details on these techniques can be found in the supporting information. For the training process, 80% of the data was used for training and validation. The test set, composed of the remaining 20%, was used for further analysis. The machine learning analysis here described was performed using the GPU-accelerated version of TensorFlow 2.0.37
2.2.3. Explainable Artificial Intelligence (XAI)
A crucial component of our machine learning implementation is to attribute the contributions of each input dimension to the predictive outcome of our model, thus revealing the chemical interactions that are distinct between states upon binding of different S1 domains and are correlated with the pocket opening of the ACE2. Extracting learned information from machine learning, especially the deep learning models, is a major ongoing research direction known as the Explainable Artificial Intelligence (XAI).38 XAI unravels the black box nature of deep learning and plays a fundamental role in both practical and ethical AI by serving as a tool for rational improvement of deep learning models and for granting transparency in the interpretation of the results.38–41 In the present study, the XAI technique Gradient-weighted Class Activation Mapping (Grad-CAM) was implemented and applied to our ACE2-learning CV-CNN model.25 Grad-CAM is a well-known XAI method in image processing and has recently shown potential for the analysis of protein structures.42 Formally, for a datapoint k, Grad-CAM computes its gradient score yc for a certain class c with respect to the feature map activation of the last convolutional layer Ak, which yields the neuron importance weights .
| (Eq. 1) |
In Eq. 1, i and j are indices for the row and columns in the activation matrix, 1/Z is the normalization factor. The importance weights are then calculated for each layer by the dot product between the weight matrices and the backpropagated gradients with respect to the activation function. The outcome of the Grad-CAM importance attribution is a heatmap of regions in the input space that have positive influence in the recognition of a certain class.25 For each class, the importance heatmap is a square matrix of size N×N (with N the number of residues), where the contribution for each pair of residues is the normalized average importance over all frames. Grad-CAM is a post hoc XAI method with the advantages of generally applicability to any CNN architecture and not needing to re-train the model. In this study, Grad-CAM activation is applied on the test sets of the classes investigated. The implementation of this method using Keras/TensorFlow was employed in this study.43
2.3. Relative Entropy-Based Dynamical Allosteric Network (REDAN)
The REDAN model developed by Zhou et al.44 was used to build the quantitative model to accurately describe the ACE2 PD differences in conformational dynamics upon different S1 domain binding. Relative entropy, or Kullback–Leibler divergence, is defined as a measure of similarity between the distributions p for system P and q for system Q.45
| (Eq. 2) |
In the context of molecular dynamics simulations, protein conformations can be represented as collections of pair-wise residue-residue distances. For each pair of residues, the distribution of the distances can be compared between different states. High relative entropy values indicate that the interaction between a residue pair is significantly different between two states.
In the REDAN implementation, the Dijkstra algorithm46 is used to identify the shortest path of interactions that connects two residues. The Dijkstra algorithm computes the path with the lowest cost between two nodes in a graph by iteratively looping all possible paths connecting these two nodes and calculating their costs.47 In our application, the inverse of the relative entropy DKL between a pair of nodes i and j based on their distance distributions in states P and Q is calculated as the cost for the connection between them:
| (Eq. 3) |
If a distance distribution is strongly perturbed between the two biological states, the interaction considered is likely important in the propagation of the structural changes in the protein. This will be reflected by high relative entropy values, which in turn will cause the cost to be low, making this particular interaction to be favored in the finding of the allosteric path with the lowest-cost.
Furthermore, the relative entropy for a pair of nodes i and j can be weighted by their associated machine learning importance, , obtained from Grad-CAM attributions for each class. As mentioned above, describes the structural regions crucial for the functional dynamics of a certain class. Therefore, this strategy would reveal the ability of a certain class to use the allosteric path identified and allow the comparison of allosteric communication for between different classes. The states of ACE2 in presence of the SARS-CoV-1 or SARS-CoV-2 S1 domains are considered as different classes. This weighted REDAN model allows to compare the cost associated with the allosteric perturbation when different coronavirus S1 domains are present. The machine learning weighted cost for a class c is calculated as
| (Eq. 4) |
and is used to build machine learning weighted REDAN model.
2.4. Principal Component Analysis (PCA)
PCA reduces the dimensionality of the data by projecting each data point onto a few principal components as a lower-dimensional representation of the original data while preserving its distribution variation.48 The principal components are linear combinations of input variables and are orthogonal to each other. Given two variables, x and y, their covariances measure how these two variables vary in relation to each other using n data points:
| (Eq. 5) |
where xi and yi are the values in ith data point, and and are the averaged values for variables x and y, respectively.
In PCA, the covariance matrix C is subsequently constructed. In this symmetric matrix, each element is a covariance between two variables. For the N variables in the given dataset with each variable represented as ki, the covariance matrix has the following form:
| (Eq. 6) |
The eigenvectors of C are the components of PCA. The eigenvalues of C measure the contribution of each component in the dataset. The larger the magnitude of eigenvalue, the higher the contribution of its corresponding component, i.e., eigenvector. Generally, the eigenvectors with the largest eigenvalues are designated as principal components to form two-dimensional (2D) or three-dimensional (3D) space for data projection. The PCA was performed using Scikit-learn implemented in python.49
2.5. Markov State Model (MSM)
MSM50–53 has become increasingly useful network models to describe the transitions among functional states during allosteric events.53–56 The MSM provides the transition probabilities among macro-states.52,57 The collection of the transition probabilities among n macrostates is represented as the transition matrix T. The element of T is calculated as , where cik is the count of the number of times the trajectories transition from state i to state j within a certain time interval (referred as lag time τ).
In this study, the first two components of each PCA model were used as collective variables to construct the MSMs. The discretization of the conformational space into 200 microstates was performed using k-means clustering implemented in scikit-learn.58 The PyEmma python package was used to build the MSM.59 The default hyper-parameters provided were used for the analysis. The ergodic cutoff was turned on and the Maximum Likelihood method was used to achieve the reversibility of the transition matrix. A lag time of 200 ns was chosen. The MSM was used as a mean to obtain a kinetical clustering of the conformational space. The metastable states, also referred to as macrostates, were created using the Perron-cluster cluster analysis (PCCA) implemented in the PyEmma package.59
2.6. Root Mean Square Deviation (RMSD)
For a system represented in Cartesian coordinates, RMSD is calculated to measure the deviation from a reference structure by taking the square root of the averaged difference between the atomic coordinates vectors of a reference structure, and of the structure in the ith frame among total of N atoms, ri,
| (Eq. 7) |
Ui is the rotation and translation matrix to superimpose the structure in the ith frame against the reference structure.
2.7. Root Mean Square Fluctuation (RMSF)
The RMSF of atom i is calculated as its averaged fluctuation among T frames.
| (Eq. 8) |
2.8. Radius of Gyration
The protein compactness is measured using the radius of gyration, .
| (Eq. 9) |
mi is the mass of the ith atoms, M is the mass of the atoms in the protein, Rc are the coordinates of the center of mass of the system, and ri are the coordinates of the ith atom.
3. Results
ACE2 plays a role in regulation of blood pressure in the RAAS system by catalyzing peptide hydrolysis of signaling peptide angiotensin II to angiotensin (1–7).1,11,60,61 The role of ACE2 in RAAS system has been studied as a target for cardiovascular diseases as well as the role of ACE2 in coronavirus pathology as an antiviral target.60,61 To explore these aspects of ACE2 biology, most ACE2 PD structural studies focus on screening of inhibitors62–67 and on the energetics of complexing with viral spike proteins.68–73 As the conformational ensemble of proteins are crucial for their biological functions, we investigated how orthosteric and allosteric binding can affect the functional ACE2 PD conformational dynamics using the inhibitor MLN-4760 and S1 RBD fragments from SARS-CoV-1 and SARS-CoV-2.
3.1. Orthosteric and allosteric effects on the conformational space explored by ACE2 PD
The conformational ensemble that proteins explore are crucial to biological function. The conformational space explored by ACE2 during extensive molecular dynamics simulations was examined. Four different systems were considered: ACE2 in its apo state (ACE2 apo), ACE2 bound to the inhibitor MLN-4760 (ACE2: inhibitor), ACE2 in complex with the S1 RBD fragment from SARS-CoV-1 (ACE2:COV1-S1), and ACE2 in complex with the S1 RBD fragment from SARS-CoV-2 (ACE2:COV2-S1). Only the PD was considered as both enzymatic and viral entry mechanisms occur through domain.3 In all systems, the simulations are stable and reached equilibrium (Fig. S2).
The RMSF analysis in Fig. 2a displays the dynamic fingerprint of the ACE2 PD. α1, α2, β5, β6, α20, the α17–α18 loop, and the α20–β21 loop are the structural regions responsible for the majority of the protein motions as shown by the highest fluctuation during the simulations (Fig. 2b). A common characteristic for these structural components is their location in proximity to the ACE2 PD entrance (Fig. 3a). This agrees with previous studies of ACE2, where the catalytic functionality of ACE2 was proposed to rely on the opening and closing motion of the ACE2 PD binding site. 11 Thus, the binding site opening motions of ACE2 PD in the different systems are analyzed and compared. The front and the back of the ACE2 PD catalytic binding site entrance (Fig. 3a) were considered separately as they demonstrate distinct dynamical profiles in the RMSF analysis (Fig. 2a, 2b). The distance distribution analysis of the front and back opening/closing motions of the entrance show a non-synchronized movement (Fig. 3b, 3c). ACE2 PD is free to explore its open conformation in the apo state (Fig. 3b). On the other hand, in the ACE2:inhibitor complexes the MLN-4760 ortho-sterically closes the binding site, as expected from crystal studies.11
Fig. 2.
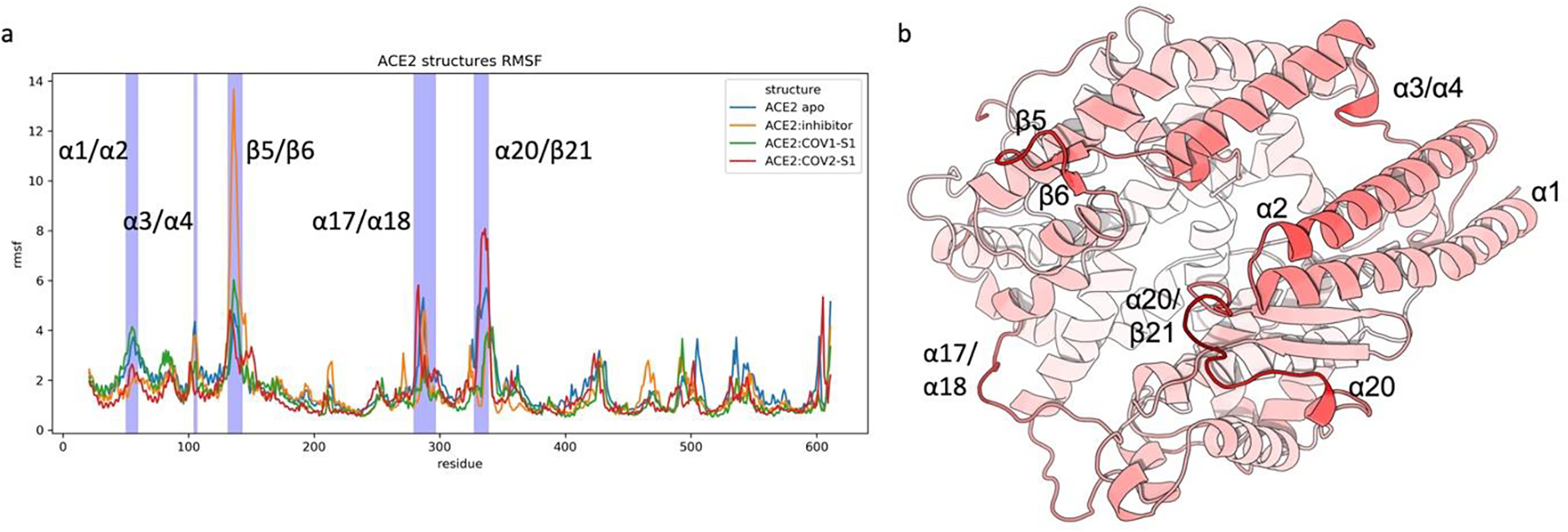
Root Mean Squared Fluctuation (RMSF) analysis of ACE2 PD in different systems. a) RMSF plot vs ACE2 PD residue for ACE2:apo, ACE2:inhibitor, ACE2:COV1-S1, and ACE2:COV2:S1. b) ACE2 PD fluctuation trends in its apo form. The color gradient from white to red represents the degree of flexibility from low to high, respectively. Protein rendered with 3D Protein Imaging.74
Fig. 3.
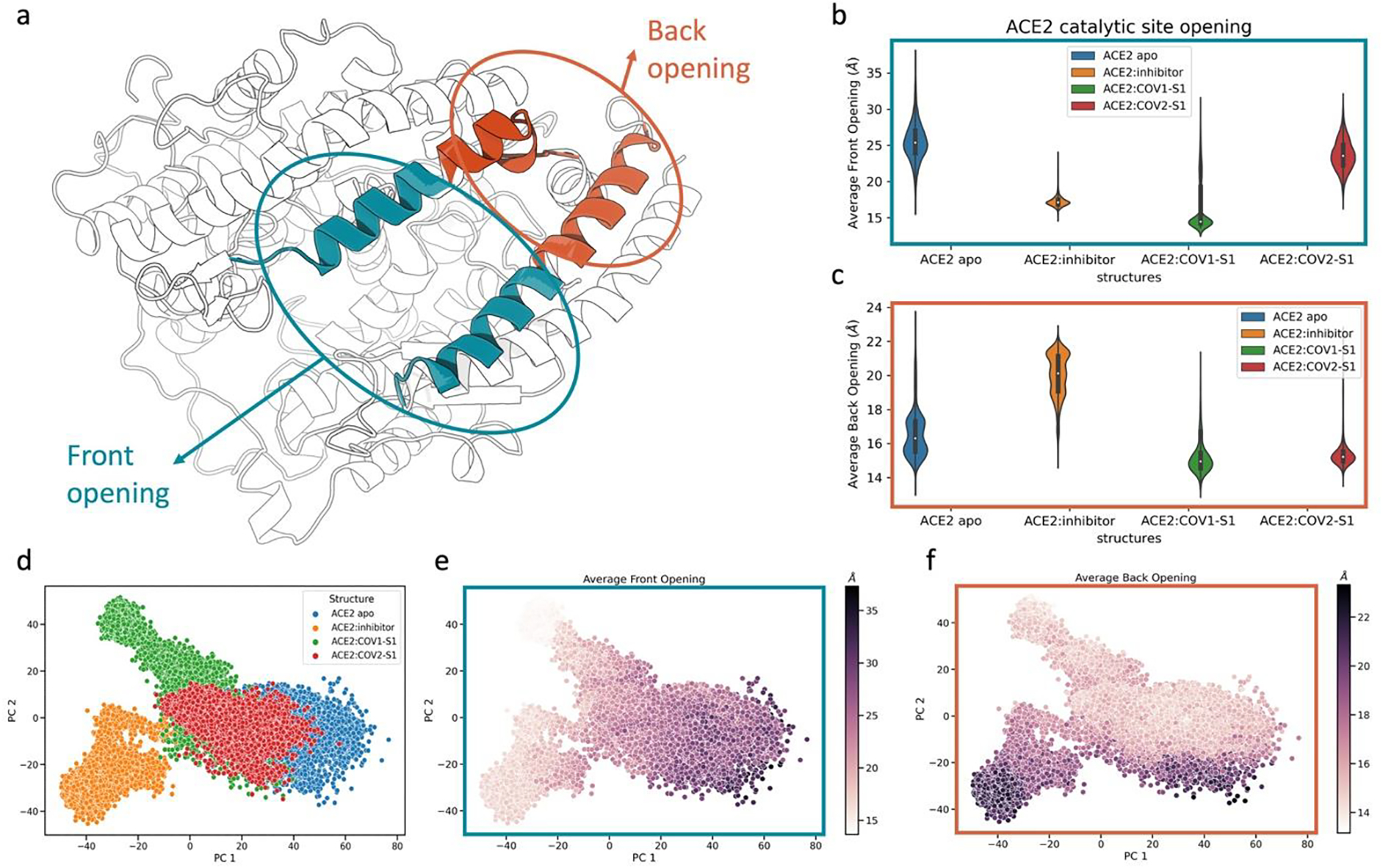
Conformational space of ACE2 related to the front and back openings of its binding site in different states. a) Structure of ACE2 peptidase domain (PD). The helices involving the front and back opening are colored in blue and orange, respectively. The helices involved are the α2 and α4 (Table S1). b) Distance distributions of the front opening in the different ACE2 states. The distances reported are the average distance between the residues the residues 69 to 84 of the α2 and the residues 101 to 116 of α4. c) Distance distributions of the back opening in the different ACE2 structures. The distances reported are the average distance between the residues 53 to 68 of the α2 and the residues 117 to 132 of α4. d) Projection of each state onto the conformational space represented by the first two components of principal component analysis. Each data point represents a frame (conformation) of ACE2 PD. Colored scheme: blue for ACE2 apo, orange for ACE2:inhibitor, green for ACE2:COV1-S1, red for ACE2:COV2-S1. e) Projection of conformational space explored using the first two components of principal component analysis. The data points are colored based on the value of average distance between the carbons of the helices at the entrance of ACE2 binding site (front opening). f) Projection of conformational space explored using the first two components of principal component analysis. The data points are colored based on the value of average distance between the carbons of the helices at the back of ACE2 binding site (back opening). Points are colored from light to dark for small to large distances, respectively. Protein rendered with 3D Protein Imaging.74
Interestingly, different RBD fragments allosterically affect the conformational landscape of ACE2 PD in distinct ways. The presence of SARS-CoV-2 S1 RBD fragment (ACE2:COV2-S1) causes a wide distance distribution of the binding site opening similar to the apo state (Fig. 3b). This contrasts with the presence of the SARS-CoV-1 S1 RBD fragment (ACE2:COV1-S1), where the front of the binding site was observed to adopt a closed conformation, similarly to the ACE2:inhibitor complexes. This indicates that the binding to the ACE2 PD surface can allosterically cause a population shift on the free energy landscape, ultimately causing the closing of the protein catalytic site. This trend also affects the protein compactness, as shown by the radius of gyration (Fig. S3).
The interatomic distances between the backbone Cα in each conformation from the molecular dynamic simulations were employed for PCA. The first two principal components, PC1 and PC2, were selected to represent the conformational space of the protein dynamics. The conformational space explored by ACE2 apo overlaps substantially with the conformational space of ACE2:COV2-S1, partially with the conformational space of ACE2:COV1-S1, and very little with the conformational space of ACE2:inhibitor (Fig. 3d). The inhibitor and SARS-CoV-1 S1 RBD fragment drive ACE2 PD to explore different conformational spaces, exclusive to each system. The most dominant component in the ACE2 PD conformational space, PC1, correlates with the opening motion of the front of the binding site. The second component, PC2, is correlated with the motion of the binding site back opening. These correlations are illustrated as the gradual change of the front and back openings along PC1 and PC2, respectively (Fig. 3e, 3f).
In summation, the opening and closing motions of the binding site dominate the conformational landscape of ACE2. The two RBD fragments have significantly different effects on the ACE2 functional conformational space. SARS-CoV-2 S1 RBD fragment does not strongly perturb the ACE2 apo conformational profile, whereas SARS-CoV-1 S1 RBD fragment allosterically induces the closing of the catalytic domain and increase the overall PD rigidity. The ACE2 inhibitor, MLN-4760, induces an ortho-steric closing of the ACE2 catalytic domain.
3.2. Functional Discretization of ACE2 conformational space
The conformation analysis presented above demonstrates striking differences in the effects of SARS-CoV-1/2 S1 RBD fragments binding on the ACE2 PD functional dynamics, despite their high structural and sequence similarities. Thus, specific interactions established with the ACE2 PD surface are expected to be responsible for allosteric control of binding site motions. In the remainder of the study, we pursue a mechanistic explanation of this allosteric phenomenon at atomistic level. Towards this goal, a target machine learning model, coined CV-CNN, was developed and used to extract specific interactions correlated with the S1 RBD-related difference in ACE2 binding site dynamics.
For a machine learning model to learn biologically meaningful information, an appropriate data labeling strategy is needed. A biologically sensible approach is to classify each conformation of the molecular dynamics simulations by the functional metastable state to which they belong. The free energy landscape on the projected conformational surface presented in Fig. 4 suggests that ACE2 PD explores four distinct minima during the simulations. The free energy landscape is further correlated with a kinetical clustering Perron Cluster Cluster analysis (PCCA), which is based on MSMs. This clustering analysis is established on the assumption that structures belonging to the same functional metastable state interconvert more frequently than structure separated by high free energy barriers. From the analysis of the protein relaxation timescales of molecular motions at different lag-times, four distinct macrostates, States 1 through 4, are identified, confirming the qualitative identification based on population density. The details on the micro clustering to build the MSM and the Chapman-Kolmogorov test are presented in Fig. S4.
Fig. 4.
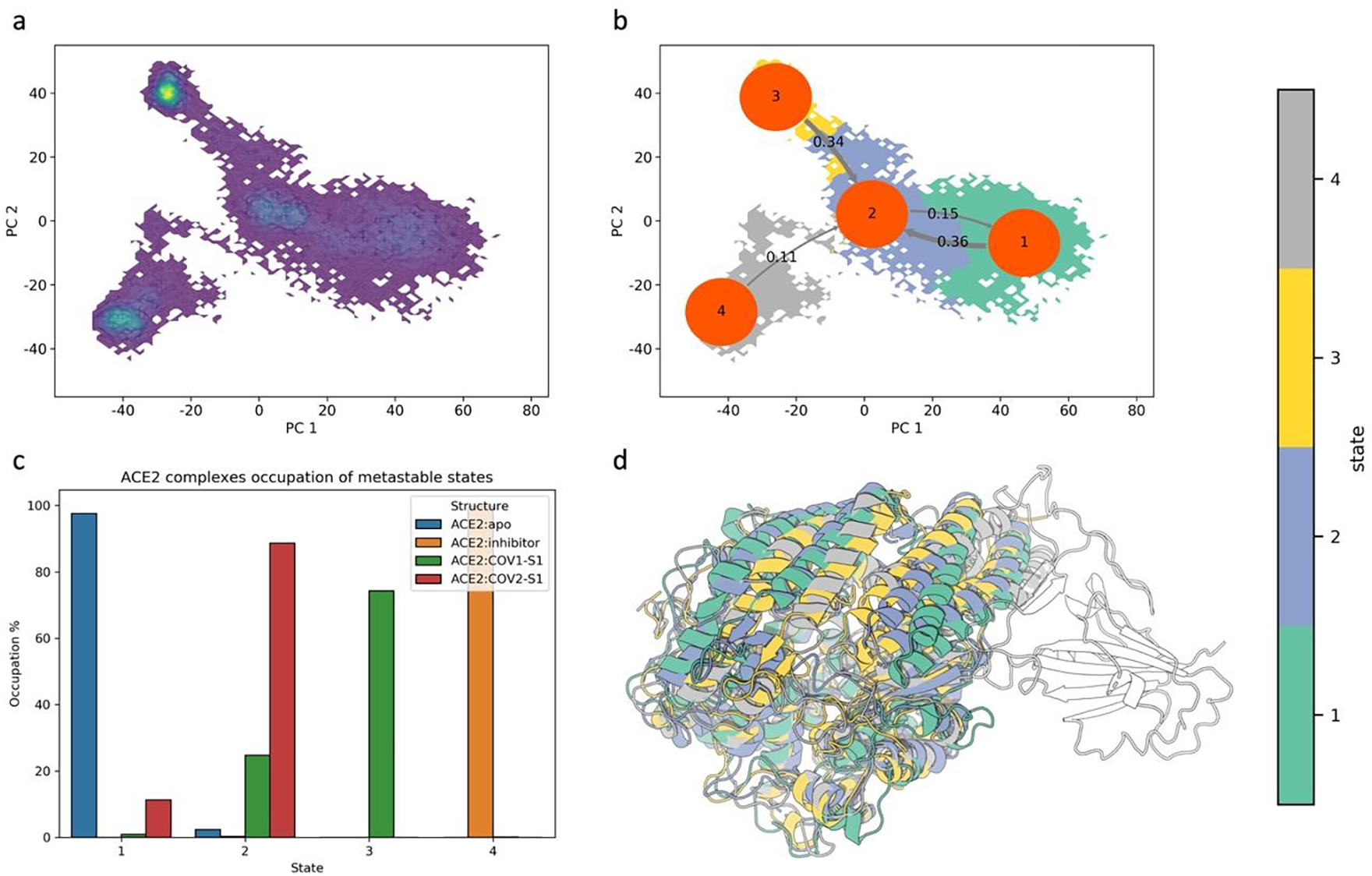
Identification and analysis of ACE2 PD macrostates using kinetical clustering. The clustering method used is PCCA on the assumption that conformations belonging to the same macrostate interconvert rapidly compared to transitioning to another macrostate. a) Visualization of high-density areas on the ACE2 conformational space, which correspond to energy minima. b) Visualization of PCCA macroclusters on the projected conformational space. The flux between the states is included as inverse of mean time passage. The choice of using four clusters was confirmed from the Chapman-Kolmogorov test of our Markov state model (Fig S3). c) ACE2 occupation of the different metastable states. d) Overlap of the ACE2 PB domain of the representative structures for the four macrostates identified. Macrostates representative structures are colored based on PCCA clustering. Protein rendered with 3D Protein Imaging.74
The effect of inhibitor or S1 fragment domain complex on the peptide binding site of ACE2 is preserved by the clustering analysis (Fig. 4c). We observe that with exception for State 2, all the other states are uniquely populated. State 2, which is mostly populated by ACE2:COV2-S1, is populated by the ACE2 apo and ACE2:COV1-S1 as well.
3.3. CV-CNN and REDAN structural characterization of ACE2 PD allostery
After assigning ACE2 PD conformations to functional metastable states on the protein conformational landscape, the CV-CNN model was implemented to guide the learning of the structural features that influence the ACE2 PD allostery when different S1 RBD fragments are bound. ACE2 PD conformations corresponding to State 2 (mainly populated by ACE2:COV2-S1) and State 3 (mainly populated by ACE2:COV1-S1) were used to train our novel CV-CNN model. The selected conformations were featurized to express short range interactions as described in the method section. The CV-CNN learning process has a dual goal. It optimizes the learning of pairwise residues interaction patterns that distinguish ACE2:COV1-S1 from ACE2:COV2-S1 and are also correlated with the functional ACE2 PD binding site opening. CV-CNN achieved a 99% accuracy in its classification task and 0.4 Å error in predicting the degree of opening of the binding site (Fig. S5).
Grad-CAM, an explainable artificial intelligence method, was adopted to extract the structural information found by our CV-CNN model to be crucial in distinguishing ACE2:COV1-S1 and ACE2:COV2-S1 functional dynamics. To extract which areas of the protein are found to be more important in distinguishing the dynamics between these two states, the Grad-CAM results were averaged and pooled over the test set for each class. The ACE2 PD structural regions identified by Grad-CAM validate that our CV-CNN model learned structurally insightful information. These structural regions with high importance are located either in the proximity of the binding site entrance (Fig. 5a, 5b) or are directly involved in the binding site opening motion (Fig. 5c). Further, CV-CNN recognized that ACE2 PD spike binding locus (Fig. 5a) distinguishes ACE2:COV1-S1 and ACE2:COV2-S1 dynamics. Interestingly, the residues in this region bind to several spike mutations, suggesting that this region likely constitutes the origin of the allosteric phenomenon observed in ACE2 PD dynamics.
Fig. 5.
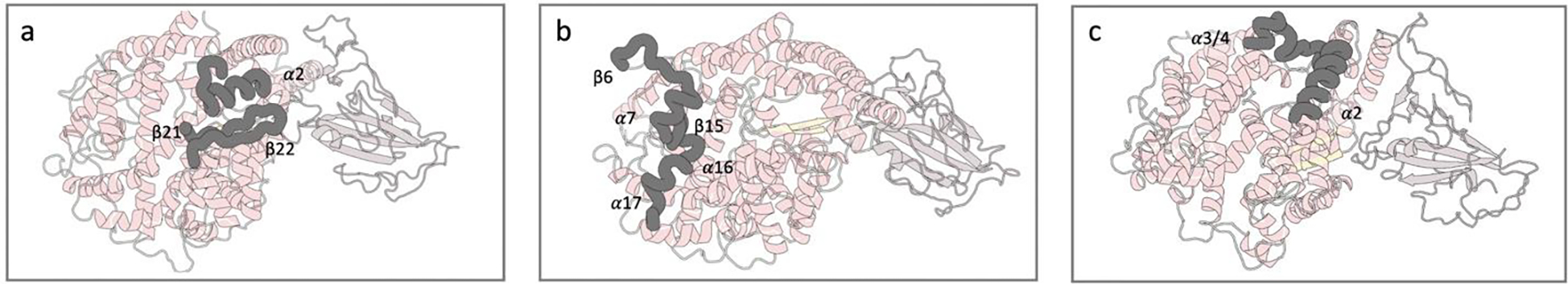
Structural regions found important by the CV-CNN model using Grad-CAM. The Grad-CAM feature importance for State 3 was obtained by averaging the importance heatmap for each conformation in the test set for this state. The regions were obtained by pooling the averaged heatmap into groups of 19 residues each. a) Most important region includes α1 (residues 40 to 58) and β21 and β22 (residues 344 to 362). These groups are adjacent to one of the S1 domain binding regions. b) Second most important region includes residues 268 to 286 and residues 136 to 153, these groups are located at the entrance of the binding site region. c) Third most important region includes residues 59 to 77 and 97 to 115. The residue groups involved are located on the opposite side of the binding site, making this distance representative for the opening and closing of the binding site.
To complement the CV-CNN structural insights and to obtain a complete picture of ACE2 PD allostery, a mechanistic investigation of this allosteric perturbation is performed using the REDAN model of Zhou et al.44 REDAN compares the distributions of residue-residue distances between two different states, in our case the states with SARS-CoV-1/2 RBD fragments, to identify the allosteric pathways contributing to their dynamical differences. This comparison is carried via relative entropy to identify which residues pairs are most affected by interaction differences between the S1 RBD fragments and ACE2 PD surface. The affected residue pairs will likely be part of the network of residue interactions which regulate overall ACE2 PD conformational dynamics.
To explore the connection among these residues, REDAN utilizes the shortest path Dijkstra algorithm. In the characterization of the allosteric path, the starting and ending points need to be determined. The starting point is selected from the α1, β21 and β22 highlighted in our CV-CNN model. These secondary structures are part of the spike-binding locus of ACE2 PD, and their residues interact with the mutations that differentiate the two S1 RBD fragments. All the residues in these regions were considered as starting point for the allosteric perturbation, the residue that provided the lower cost associated with the pathway was selected for further analysis (Table S3). The residues selected is Asp350. The ending point is Tyr127, which lies at the entrance of ACE2 PD binding site on the opposite side of the spike binding region.
The allosteric pathway connecting Asp350 and Tyr127 is illustrated in Fig 6. In this pathway composed of 13 residues in addition to Asp350 and Tyr127 (Fig. 6a, 6b), three groups of interactions are observed:
Fig. 6.
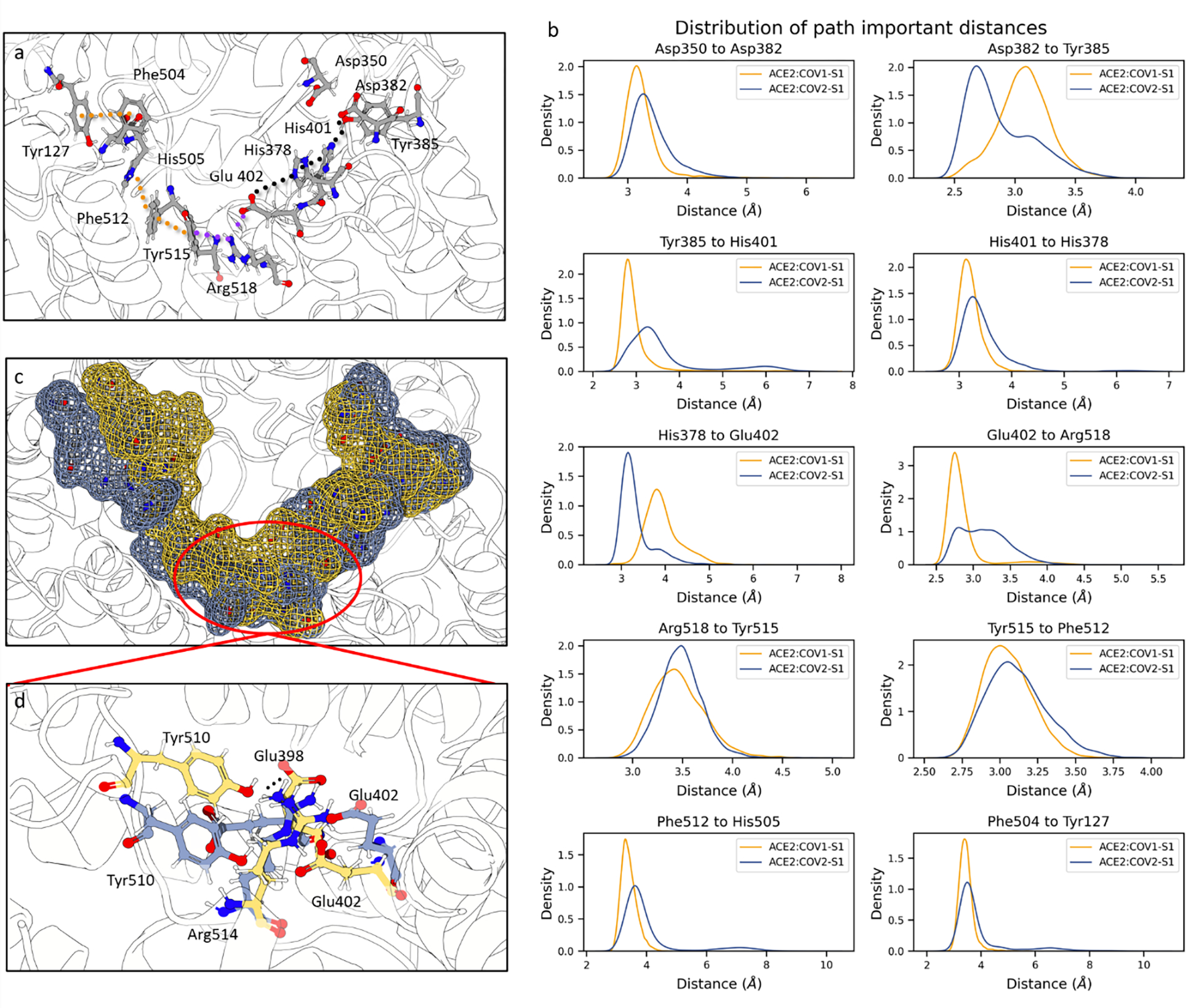
Proposed allosteric path used by ACE2 to dictate the closing of the binding site obtained from the REDAN analysis and its structural details. a) Residues involved in the allosteric path and their interaction. The path from REDAN was refined to ensure that chemically meaningful interactions are captured. The raw path can be found in Table S3. The dotted lines represent interactions. Black dots include the interaction of residues of the S1 domain-binding side of the ACE2 binding site. The purple indicates interactions that bridge the two sides of ACE2 binding site. The orange dots show the π-stacking interactions that reach the opposite side of the binding site. b) Distance density distributions of key interactions. c) Surface mesh representation of the residues involved in the path to show the movement responsible for the binding site closing. The yellow represents State 3, occupied by ACE2:COV1-S1, whilst blue represents State 2, occupied mostly by ACE2:COV2-S1. d) Details of key interactions in the ACE2 PD bridging area between the two binding site sides that stabilize the closed state in presence of SARS-CoV-1. Residues in blue and yellow belong to State 2 and 3, respectively. Protein rendered with 3D Protein Imaging.74
Interactions starting from the spike protein complexing locus to the ACE2 core (black dotted lines). These interactions involve residues Asp350, Asp382, Tyr385, His401, and His378;
Bridging interactions, bridging interaction that connect the two sides of the binding site and cause the pulling of the opposite side of the ACE2 binding domain (purple dotted lines). These interactions involve residues Glu402, Arg518, and Tyr515;
π-stacking network interaction that propagates the perturbation to the opening of the binding site (yellow dotted lines). These interactions involve residues Tyr515, Phe512, His505, Phe504, and Tyr127.
The overall effect of these interactions is a pull of the binding site region opposite to the spike complex locus as shown by the mesh surface of the residues involved in the allosteric path (Fig. 6c). Glu402 is identified as the pivotal residue in the opening closing mechanism of the binding site (Fig 6b, 6d). This residue interacts with both sides of the catalytic domain. Its interaction with His378, located in the catalytic domain, decreases in presence of SARS-CoV-1 S1 RBD fragment. Consequently, the Glu402 side chain is free to rotate to form a salt bridge with Arg518 and partially a ternary π-cation-anion75 interaction with Tyr515 (Fig. 6b), both located on the opposite side of the catalytic domain (Fig 6a). We propose that the pulling exerted by the bridging residues is stabilized by neighboring residues. Specifically, upon the movement of Glu402, sidechain rotation of Glu398 occurs. This motion allows Tyr510 to establish a hydrogen bond with Glu398, uniquely observed in the presence of SARS-CoV-1 S1 fragment (Fig. S6).
A comparative analysis of the allostery prowess between the ACE2:COV1-S1 and ACE2:COV2-S1 states was performed by weighting the relative entropy in this REDAN model with the machine learning feature importance for each state. The allosteric pathway identified above remains as the dominant pathway in ACE2:COV1-S1 but has lower cost (Table S4). This indicates that the residues involved in the path are the key residues needed to define the dynamic behavior of ACE2:COV1-S1 compared to ACE2:COV2-S1, suggesting a stronger allosteric communication along the proposed path for ACE2:COV1-S1.
3.4. Bridging S1 domain binding with ACE2 binding site motion
From our CV-CNN machine learning analysis, the ACE2 region adjacent to the RBD binding locus was found to differentiate the conformational dynamics between ACE2:COV1-S1 and ACE2:COV2-S1 and therefore considered the potential focus of allosteric control of ACE2 PD functional dynamics. Our targeted machine learning approach CV-CNN combined with the graph-based statistical analysis REDAN elucidated an allosteric path that regulates ACE2 PD closing. Aiming to detect the main interactions at the root of these findings, we performed a contact analysis between mutated residues in the spike S1 domain fragment and ACE2 PD the α1, β21, and β22.
From comparing the top contacts between ACE2 and the S1 RBD fragments (Fig. 7a, 7b), SARS-CoV-2 S1 RBD fragment establishes more energetically favorable interactions compared to SARS-CoV-1. The interactions among the ACE2 PD Lys353 and Asp38 and the SARS-CoV-2 Gln498 and Asn501 could exert a pulling force on α1, β21, and β22. On the other hand, the SARS-CoV-1 S1 RBD fragment lacks these interactions. Instead, Thr487, equivalent to Asn501 in SARS-CoV-2 S1, might sterically push β21 and β22 towards the binding site. The superposition between these two ACE2 states (Fig 7c) reveals the concerted movement of β21 and β22. In the representative structure for ACE2:COV2-S1 (blue structure in Fig 7c), α1 and the neighboring β21 and β22 are away from the ACE2 core compared to the ACE2:COV1-S1 (yellow structure in Fig 7c). This shift of β21 and β22 allows for mechanistic description of allosteric propagation. The translation of these secondary structures shown in Fig 7d is accompanied by movement of Phe356. Phe356 is observed to sterically cause a shift in Asp382, which loses its hydrogen bond with Tyr385. The latter, in turn, switches its hydrogen bond towards His401, explaining the start of the allosteric path described above.
Fig. 7.
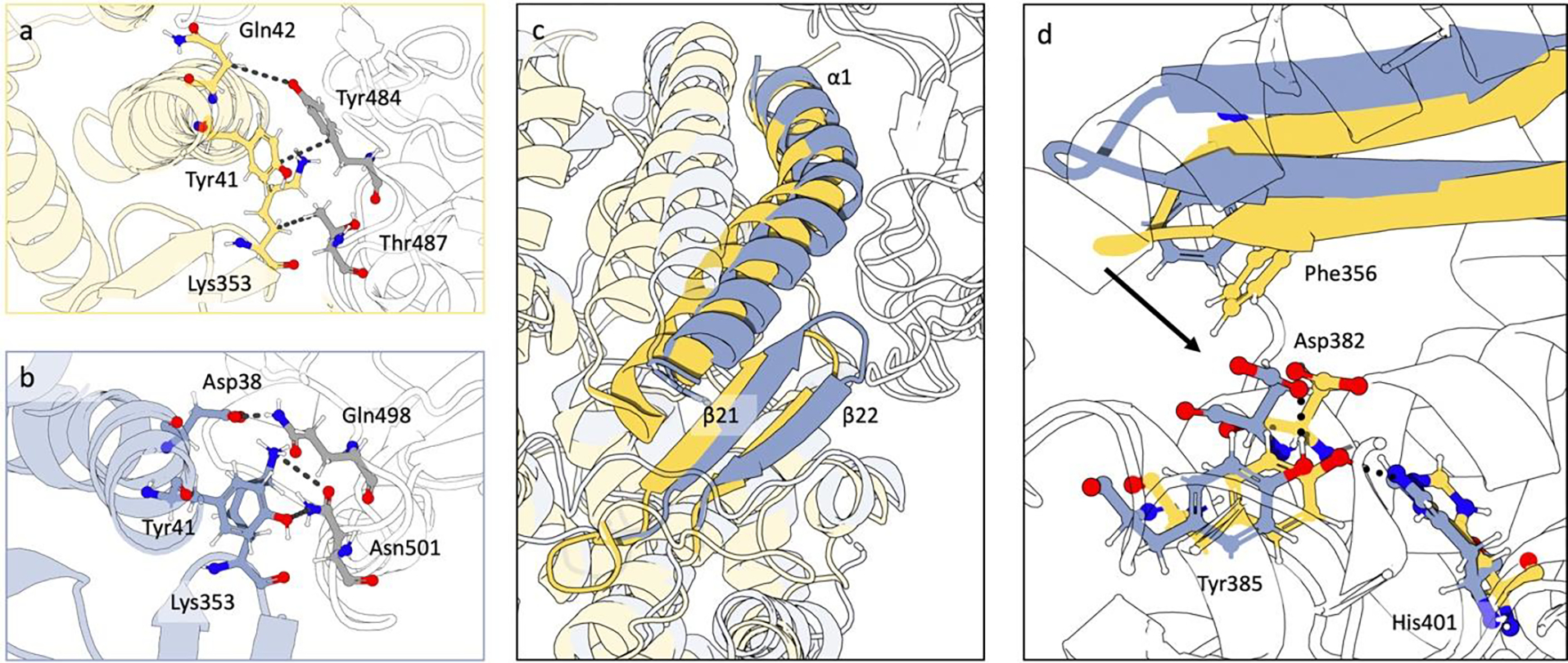
Interaction between coronaviruses S1 domains and ACE2 PD via contact analysis and SASA. a) Contact analysis between the residues in α1, β21, and β22 of ACE2 PD and the SARS-CoV-1 spike S1 residues Tyr484 and Thr487. Residues in yellow belong to ACE2, residues in grey belong to viral spike S1 fragment. Contacts less than 4 Å for more than 50% of the frames were shown. From the contacts shown only a π-stacking interaction was found as energetically favored interaction. b) Contact analysis between the residues in α1, β21, and β22 of ACE2 PD and the SARS-CoV-2 spike S1 residues Gln498 and Asn501. Residues in blue belong to ACE2, residues in grey belong to viral spike fragment. Contacts less than 4 Å for more than 50% of the frames were shown. In this case, a strong network of energetically favorable interaction was identified. The spike S1 residues considered differ between the two coronaviruses spikes. c) Details of the overlap of ACE2:COV1-S1 (in yellow) and ACE2:COV2-S1 (in blue). The secondary structures represented are α1, β21, and β22. d) Illustration of the effects of β21 and β22 shift on residues involved in the allosteric path. Protein rendered with 3D Protein Imaging.74
Ulterior difference between ACE2:COV-S1 and ACE2:COV2-S1 systems investigated are the glycosylation sites, which must be considered when researching the cause of the allosteric perturbation to the binding site dynamics. It is important to stress that the glycols molecules analyzed in this study do not depict the full glycosylation profile, and thus the analysis is to find the cause of the specific ACE2 PD allosteric pathway identified above and not to delineate biological relevant differences between coronaviruses. Glycosylation sites on residues 53, 90, 322, and 546 in ACE2:COV1-S1 are all present in ACE2:COV2-S1, excluding the ability of these glycols to cause the allosteric perturbation observed (Fig. S1).
Additional glycosylation on residue 103, which lies in the back of the binding site, is present in ACE2:COV2-S1 but not in ACE2:COV1-S1. Therefore, the ability of this glycosylation site to cause the conformational differences observed between ACE2:COV2-S1 and ACE2:COV1-S1 is further scrutinized. The conformational profile of the back of the binding site, where the glycosylation is located, is conserved in the two systems (Fig. 3c). Furthermore, the possibility of this glycosylation to prevent the closing of the binding site in ACE2:COV2-S1 can be ruled out because of the presence of this glycosylation in ACE2:inhibitor (Fig. S1), which is able to adopt the closed conformation (Fig. 3b). The present results suggest that the conformational differences in the two systems considered does not derive from their glycosylation differences but from the contact analysis detailed above.
In summary, the different interactions between ACE2 and RBD fragments can explain the structural differences observed in the ACE2 spike complexing regions, specifically in α1, β21, and β22, which are identified in the CV-CNN machine learning model. These regions are crucial in the binding site opening mechanism, identified as the main component of the ACE2 functional conformational landscape. The allosteric propagation pathways from the spike binging region to the opposite side of the binding sited identified using REDAN models complete the picture of allosteric mechanism of ACE2 PD dynamics.
4. Discussion and Conclusion
The peptide hormone angiotensin II is a crucial part of RAAS where it acts as vasoconstrictor. Its mis-regulation can cause hypo- and hyper-tension, potentially leading to heart failure. To ensure proper regulation of this hormone, RAAS employs the protein ACE2. ACE2 plays a crucial role in the regulation of blood pressure via converting the angiotensin II to the vasodilator angiotensin (1–7). Past structural studies of inhibitors binding showed that functional motions of the peptidase domain of ACE2 play a role in its catalytic ability. In addition to ligands’ orthosteric control of binding site opening, simulations of the ACE2 PD in complex with RBD fragments of the S1 domains of SARS-CoV-1 and SARS-CoV-2 can be leveraged to investigate the possibility of allosteric control ACE2 PD functional motions.
In this study, we investigated the dynamic profiles of the ACE2 PD and identified selective allosteric control on the human enzyme using RBD fragments of the S1 domains via advanced computational approaches. We combined standard methods of trajectory analysis with a custom CV-CNN and statistical REDAN analysis to identify key residues in the allosteric network.
The investigations of ACE2 conformational dynamics showed differences in binding site motions upon inhibitor or S1 RBD binding. The projection of the principal components that define the ACE2 conformational space showed that these ACE2 systems fell into distinct functional metastable states. The ACE2 PD binding site opening-closing motion was identified to be dominant in characterizing the conformational landscape of the protein. Strikingly, different S1 domain RBD fragments imparted distinct effects on the opening of ACE2 PD, despite their similarity. Specifically, the SARS-CoV-1 RBD fragment caused the closing of ACE2 PD catalytic domain, similar to its response when an inhibitor is bound within the binding site. As ACE2 catalytic activity is contingent on these binding site dynamics, this evidence for allosteric-induced binding site closing might open the possibility to the allosteric inhibition of ACE2.
Using a novel CV-CNN machine learning model and REDAN statistical analysis, we propose the mechanism for the allosteric perturbation that regulates the ACE2 PD binding site dynamics. The allosteric path for closing the ACE2 binding site is based on the flip of residues Glu402, which interacts with Tyr515 and Arg518 to close the binding. Further, we proposed a series of side-chain motions, namely Tyr510 and Glu398, that assist the stabilization of the closed states. The difference in the interactions between the allosteric binders and the human receptor are proposed to be the cause of the difference in allosteric effect observed. Our CV-CNN machine learning model identified the region in the ACE2 surface where the allosteric activation is most likely to start. This region, composed α1, β21, and β22, is in contact with some mutations in the RBD fragments considered, further validating this region as an allosteric center in ACE2. In our contact analysis between the ACE2 PD α1, β21, and β22 and the RBD fragments, we propose that binding to these surface secondary structures can induce a concerted shift of the β21 and β22 due to interactions between ACE2 PD Tyr41 and RBD Tyr 484 (Fig. 7a).
The similarities between ACE2:inhibitor and ACE2:COV1-S1 dynamic profiles show an additional structural factor linked to the ACE2 PD binding site closing. Tyr515, identified as part of the ACE2 PD allosteric path in our analyses, is a key residue in the interaction with the ACE2 inhibitor MLN-4760.11 As MLN-4760 causes the binding site to close, it supports the involvement of Tyr515 in the closing of ACE2 PD binding site. This suggests that the proposed allosteric pathway is intrinsic of ACE2 functional dynamics. Furthermore, the orthosteric binding of the ligand causes a decrease in flexibility of the disordered region adjacent to β21 and β22, which are located on the ACE2 PD surface. The same decrease in flexibility in ACE2:COV1-S1 is likely caused by the interaction of Glu329 and Arg426 (Fig. S7). This comparison invigorates the allosteric connection proposed between ACE2 PD surface and binding site opening motion and shows the bidirectionality of the allosteric phenomenon.
This study represents an advancement of the understanding of ACE2 conformational dynamics, its implications, and can be leveraged to guide further studies. However, potential limitations of the current work must be considered. The systems analyzed lack a complete description of the microheterogeneity of the glycosylated residues. This leads to an incomplete interaction profile between ACE2 PD and viral S1 RBD fragments.77–80 Furthermore, the consideration ACE2 in membrane-bound dimeric form, full length viral spike, with other partners such as TMPRSS2, can impact ACE2 PD dynamics as well. Due to these limitations, the results presented should not be elaborated in the context of coronaviruses pathology without further testing but should be interpreted as a demonstration of the possibility to allosterically control ACE2 PD conformational dynamics and how the allosteric perturbation can propagate within the domain.
In summary, ACE2 is essential in regulating the RAAS system by converting the vasoconstrictor Angiotensin-II into the vasodilator Angiotensin (1–7). The accessibility of the binding site is a fundamental prerequisite for this protein to exert its catalytic activity, thus the modulation of binding site opening via allosteric binding is of interest. The knowledge of the residues involved in the closing mechanism of ACE2 could help design ACE2 PD variants to induce the closing of the binding site. This, for instance, could be used to design ACE2 variants that are able to complex coronaviruses while not interfering with the RAAS system. A potential therapeutic application for such variants lies in ACE2 administration therapy.76 Furthermore, the identification of common residue Tyr515 used by both the coronaviruses spikes and ACE2 inhibitor to induce closing of the catalytic domain could help the development of ACE2 inhibitors by rationally strengthening interactions that might trigger closing of the binding site.
Supplementary Material
Table S1. Naming convention for ACE2 PD.
Table S2. CV-CNN layers details.
Table S3. Un-refined REDAN paths with respective costs.
Table S4. Cost comparison for best path between REDAN and ML-weighted REDAN.
Machine learning model regularization
Fig. S1. Glycosylation profile of the systems analyzed.
Fig. S2. Root Mean Square Deviation (RMSD) analysis of ACE2 PD in different systems.
Fig. S3. Radius of Gyration analysis for ACE2 PD in multiple systems.
Fig. S4. Kinetical clustering of ACE2 PD conformational space. a) K-means microclustering of ACE2 PD conformational space. b) Relaxation timescales of ACE2 PD at different lag times. c) Log values of relaxation timescales of ACE2 PD at different lag times.
Fig. S5. Validation of CV-CNN model. a) Mean Average Error of binding site opening regression task. b) Classification accuracy; c) Model validation loss.
Fig. S6. Interaction between Glu398 and Ty510. This interaction is measured as percentage of frames where these two residues are found at a distance lower than 4 Å.
Fig. S7. a) Interaction between ACE2 PD Glu329 with SARS-CoV-1 spike Arg426 (yellow) vs SARS-CoV-2 spike Asn439 (blue). b) Percentage of bond occurrence between ACE2 PD Glu329 with SARS-CoV-1 spike Arg426 (ACE2:COV1-S1) vs SARS-CoV-2 spike Asn439 (ACE2:COV2-S1). The percentage is measured based on the number of frames where the pair of residues was at a distance smaller than 4 Å.
Acknowledgments
Computational time was generously provided by Southern Methodist University’s Center for Research Computing.
Funding Sources
Research reported in this paper was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award No. R15GM122013.
Footnotes
Conflicts of Interest
There are no conflicts of interest to declare.
References
- 1.Clarke NE & Turner AJ Angiotensin-Converting Enzyme 2: The First Decade. Int. J. Hypertens 2012, 1–12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donoghue M et al. UltraRapid Communication A Novel Angiotensin-Converting Enzyme – Related to Angiotensin 1–9. Circ Res 87, e1–e9 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Yan R et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 367, 1444–1448 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li W et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 24, 1634–1643 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turner AJ, Hiscox JA & Hooper NM ACE2: From vasopeptidase to SARS virus receptor. Trends Pharmacol. Sci 25, 291–294 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.V’kovski P, Kratzel A, Steiner S, Stalder H & Thiel V Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol 19, 155–170 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel S, Rauf A, Khan H & Abu-Izneid T Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother 94, 317–325 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Pang J, Liu M, Ling W & Jin T Friend or foe? ACE2 inhibitors and GLP-1R agonists in COVID-19 treatment. Obes. Med 22, 100312 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malakauskas SM et al. Aminoaciduria and altered renal expression of luminal amino acid transporters in mice lacking novel gene collectrin. Am. J. Physiol. - Ren. Physiol 292, 533–544 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Camargo SMR et al. Tissue-Specific Amino Acid Transporter Partners ACE2 and Collectrin Differentially Interact With Hartnup Mutations. Gastroenterology 136, 872–882.e3 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Towler P et al. ACE2 X-Ray Structures Reveal a Large Hinge-bending Motion Important for Inhibitor Binding and Catalysis. J. Biol. Chem 279, 17996–18007 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li F, Li W, Farzan M & Harrison SC Structure of SARS Coronavirus Spike Receptor-Binding Domain Complexed with Receptor. Science. 309, 1864–1868 (2005). [DOI] [PubMed] [Google Scholar]
- 13.D. E. Shaw Research. Molecular dynamics simulations related to Sars-Cov-2. Shaw Res. Tech. Data http://www.deshawresearch.com/resources_sarscov2.html (2020).
- 14.Mulholland AJ & Amaro RE COVID19 - Computational Chemists Meet the Moment. J. Chem. Inf. Model 60, 5724–5726 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Turoňová B et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science. 370, 203–208 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casalino L et al. Beyond shielding: The roles of glycans in the SARS-CoV-2 spike protein. ACS Cent. Sci 6, 1722–1734 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zimmerman MI & Bowman G SARS-CoV-2 Simulations go Exascale to Capture Spike Opening and Reveal Cryptic Pockets Across the Proteome. Biophys. J 120, 299a (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansson T, Oostenbrink C & Van Gunsteren WF Molecular dynamics simulations. Current Opinion in Structural Biology (2002) doi: 10.1016/S0959-440X(02)00308-1. [DOI] [PubMed] [Google Scholar]
- 19.Karplus M & Kuriyan J Molecular dynamics and protein function. Proc. Natl. Acad. Sci 102, 6679–6685 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klepeis JL, Lindorff-Larsen K, Dror RO & Shaw DE Long-timescale molecular dynamics simulations of protein structure and function. Current Opinion in Structural Biology vol. 19 120–127 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Tsuchiya Y, Taneishi K & Yonezawa Y Autoencoder-Based Detection of Dynamic Allostery Triggered by Ligand Binding Based on Molecular Dynamics. J. Chem. Inf. Model 59, 4043–4051 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Ramaswamy VK, Musson SC, Willcocks CG & Degiacomi MT Deep Learning Protein Conformational Space with Convolutions and Latent Interpolations. Phys. Rev X 11, 11052 (2021). [Google Scholar]
- 23.Tian H et al. Explore protein conformational space with variational autoencoder. ChemRIX 1–21 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albawi S, Mohammed TA & Al-Zawi S Understanding of a convolutional neural network. Proc. 2017 Int. Conf. Eng. Technol. ICET 2017 2018-Janua, 1–6 (2018). [Google Scholar]
- 25.Selvaraju RR et al. Grad-CAM: Visual Explanations from Deep Networks via Gradient-Based Localization. Int. J. Comput. Vis 128, 336–359 (2020). [Google Scholar]
- 26.Lindorff-Larsen K et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinforma 78, 1950–1958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW & Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926–935 (1983). [Google Scholar]
- 28.Wang J, Wolf RM, Caldwell JW, Kollman PA & Case DA Development and testing of a general amber force field. J. Comput. Chem 25, 1157–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Bateman A et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480–D489 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kajander T et al. Buried charged surface in proteins. Structure 8, 1203–1214 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Kumar S & Nussinov R Close-Range Electrostatic Interactions in Proteins. ChemBioChem 3, 604 (2002). [DOI] [PubMed] [Google Scholar]
- 32.McGibbon RT et al. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J 109, 1528–1532 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamashita R, Nishio M, Do RKG & Togashi K Convolutional neural networks: an overview and application in radiology. Insights Imaging 9, 611–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klein EB, Stone WN, Hicks MW & Pritchard IL Understanding Dropouts. J. Ment. Heal. Couns 25, 89–100 (2003). [Google Scholar]
- 35.Srivastava N, Hinton G, Krizhevsky A, Sutskever I & Salakhutdinov R Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res 15, 1929–1958 (2014). [Google Scholar]
- 36.Ranstam J & Cook JA LASSO regression. Br. J. Surg 105, 1348 (2018). [Google Scholar]
- 37.Abadi M et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems. (2016).
- 38.Goebel R et al. Explainable AI: The New 42? in Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics) vol. 11015 LNCS; 295–303 (2018). [Google Scholar]
- 39.Samek W, Montavon G, Lapuschkin S, Anders CJ & Müller KR Explaining Deep Neural Networks and Beyond: A Review of Methods and Applications. Proc. IEEE 109, 247–278 (2021). [Google Scholar]
- 40.Gunning D et al. XAI—Explainable artificial intelligence. Sci. Robot 4, (2019). [DOI] [PubMed] [Google Scholar]
- 41.Murdoch WJ, Singh C, Kumbier K, Abbasi-Asl R & Yu B Definitions, methods, and applications in interpretable machine learning. Proc. Natl. Acad. Sci 116, 22071–22080 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verma N et al. SSnet: A Deep Learning Approach for Protein-Ligand Interaction Prediction. Int. J. Mol. Sci 22, 1392 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chollet F Deep learning with Python. (Simon and Schuster, 2021). [Google Scholar]
- 44.Zhou H & Tao P REDAN: relative entropy-based dynamical allosteric network model. Mol. Phys 117, 1334–1343 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cover TM & Thomas J Chapter 2 Entropy, Relative Entropy and Mutual Information. Entropy vol. 1 (1991). [Google Scholar]
- 46.Dijkstra EW A note on two problems in connexion with graphs. Numer. Math 1, 269–271 (1959). [Google Scholar]
- 47.Noto M & Sato H Method for the shortest path search by extended Dijkstra algorithm. Proc. IEEE Int. Conf. Syst. Man Cybern 3, 2316–2320 (2000). [Google Scholar]
- 48.Wold S, Esbensen K & Geladi P Principal component analysis. Chemom. Intell. Lab. Syst 2, 37–52 (1987). [Google Scholar]
- 49.Pedregosa F et al. Scikit-learn: Machine Learning in Python. the Journal of machine Learning research vol. 12 (2011). [Google Scholar]
- 50.Prinz JH et al. Markov models of molecular kinetics: Generation and validation. J. Chem. Phys 134, (2011). [DOI] [PubMed] [Google Scholar]
- 51.Shukla S, Shamsi Z, Moffett AS, Selvam B & Shukla D Application of Hidden Markov models in biomolecular simulations. in Methods in Molecular Biology vol. 1552 29–41 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Husic BE & Pande VS Markov State Models: From an Art to a Science. Journal of the American Chemical Society vol. 140 2386–2396 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Pande VS, Beauchamp K & Bowman GR Everything you wanted to know about Markov State Models but were afraid to ask. Methods vol. 52 99–105 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bowman GR, Bolin ER, Hart KM, Maguire BC & Marqusee S Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc. Natl. Acad. Sci. U.S.A 112, 2734–2739 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bowman GR & Noé F Software for Building Markov State Models. in An Introduction to Markov State Models and Their Application to Long Timescale Molecular Simulation 139 (Springer, 2014). [Google Scholar]
- 56.Sengupta U & Strodel B Markov models for the elucidation of allosteric regulation. Philos. Trans. R. Soc. Lond., B, Biol. Sci 373, 20170178 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harrigan MP et al. MSMBuilder: Statistical Models for Biomolecular Dynamics. Biophys. J 112, 10–15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pedregosa F et al. Scikit-learn: Machine Learning in {P}ython. J. Mach. Learn. Res 12, 2825–2830 (2011). [Google Scholar]
- 59.Scherer MK et al. PyEMMA 2: A software package for estimation, validation, and analysis of Markov models. J. Chem. Theory Comput 11, 5525–5542 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Kuba K, Imai Y & Penninger JM Multiple functions of angiotensin-converting enzyme 2 and its relevance in cardiovascular diseases. Circ. J 77, 301–308 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Turner AJ ACE2 Cell Biology, Regulation, and Physiological Functions. Prot. Arm Renin Angiotensin Syst. Funct. Asp. Ther. Implic 185–189 (2015) doi: 10.1016/B978-0-12-801364-9.00025-0. [DOI] [Google Scholar]
- 62.Choudhary S, Malik YS & Tomar S Identification of SARS-CoV-2 Cell Entry Inhibitors by Drug Repurposing Using in silico Structure-Based Virtual Screening Approach. Front. Immunol 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prajapat M et al. Virtual screening and molecular dynamics study of approved drugs as inhibitors of spike protein S1 domain and ACE2 interaction in SARS-CoV-2. J. Mol. Graph. Model 101, 107716 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsegay KB et al. A Repurposed Drug Screen Identifies Compounds That Inhibit the Binding of the COVID-19 Spike Protein to ACE2. Front. Pharmacol 12, 1–7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Teralı K, Baddal B & Gülcan HO Prioritizing potential ACE2 inhibitors in the COVID-19 pandemic: Insights from a molecular mechanics-assisted structure-based virtual screening experiment. J. Mol. Graph. Model 100, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karki N et al. Predicting Potential SARS-COV-2 Drugs—In Depth Drug Database Screening Using Deep Neural Network Framework SSnet, Classical Virtual Screening and Docking. Int. J. Mol. Sci 22, 1573 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang C et al. Human Cathelicidin Inhibits SARS-CoV-2 Infection: Killing Two Birds with One Stone. ACS Infect. Dis 7, 1545–1554 (2021). [DOI] [PubMed] [Google Scholar]
- 68.Shang J et al. Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nguyen HL et al. Does SARS-CoV-2 bind to human ACE2 more strongly than does SARS-CoV? J. Phys. Chem. B 124, 7336–7347 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Loganathan SK et al. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science 367, 1264–1269 (2020). [DOI] [PubMed] [Google Scholar]
- 71.Walls AC et al. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 181, 281–292.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khan A et al. The SARS-CoV-2 B.1.618 variant slightly alters the spike RBD–ACE2 binding affinity and is an antibody escaping variant: a computational structural perspective. RSC Adv. 11, 30132–30147 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khan A et al. Computational modelling of potentially emerging SARS-CoV-2 spike protein RBDs mutations with higher binding affinity towards ACE2: A structural modelling study. Comput. Biol. Med 141, 105163 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tomasello G, Armenia I & Molla G The Protein Imager: a full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics 36, 2909–2911 (2020). [DOI] [PubMed] [Google Scholar]
- 75.Philip V et al. A Survey of Aspartate–Phenylalanine and Glutamate–Phenylalanine Interactions in the Protein Data Bank: Searching for Anion–π Pairs. Biochemistry 50, 2939–2950 (2011). [DOI] [PubMed] [Google Scholar]
- 76.Higuchi Y et al. Engineered ACE2 receptor therapy overcomes mutational escape of SARS-CoV-2. Nat. Commun 12, 3802 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao P et al. Virus-Receptor Interactions of Glycosylated SARS-CoV-2 Spike and Human ACE2 Receptor. Cell Host Microbe 28, 586–601.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barros EP et al. The flexibility of ACE2 in the context of SARS-CoV-2 infection. Biophys. J 120, 1072–1084 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mehdipour AR & Hummer G Dual nature of human ACE2 glycosylation in binding to SARS-CoV-2 spike. Proc. Natl. Acad. Sci 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gong Y, Qin S, Dai L & Tian Z The glycosylation in SARS-CoV-2 and its receptor ACE2. Signal Transduct. Target. Ther 6, 396 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Naming convention for ACE2 PD.
Table S2. CV-CNN layers details.
Table S3. Un-refined REDAN paths with respective costs.
Table S4. Cost comparison for best path between REDAN and ML-weighted REDAN.
Machine learning model regularization
Fig. S1. Glycosylation profile of the systems analyzed.
Fig. S2. Root Mean Square Deviation (RMSD) analysis of ACE2 PD in different systems.
Fig. S3. Radius of Gyration analysis for ACE2 PD in multiple systems.
Fig. S4. Kinetical clustering of ACE2 PD conformational space. a) K-means microclustering of ACE2 PD conformational space. b) Relaxation timescales of ACE2 PD at different lag times. c) Log values of relaxation timescales of ACE2 PD at different lag times.
Fig. S5. Validation of CV-CNN model. a) Mean Average Error of binding site opening regression task. b) Classification accuracy; c) Model validation loss.
Fig. S6. Interaction between Glu398 and Ty510. This interaction is measured as percentage of frames where these two residues are found at a distance lower than 4 Å.
Fig. S7. a) Interaction between ACE2 PD Glu329 with SARS-CoV-1 spike Arg426 (yellow) vs SARS-CoV-2 spike Asn439 (blue). b) Percentage of bond occurrence between ACE2 PD Glu329 with SARS-CoV-1 spike Arg426 (ACE2:COV1-S1) vs SARS-CoV-2 spike Asn439 (ACE2:COV2-S1). The percentage is measured based on the number of frames where the pair of residues was at a distance smaller than 4 Å.