

Abstract
Proteoglycans are heterogeneous macromolecular glycoconjugates that orchestrate many important cellular processes. While much attention has focused on the poly-sulfated glycosaminoglycan chains that decorate proteoglycans, other important elements of their architecture, such as core proteins and membrane localization, have garnered less emphasis. Hence, comprehensive structure-function relationships that consider the replete proteoglycan architecture as glycoconjugates are limited. Here, we present an extensive approach to study proteoglycan structure and biology by fabricating defined semi-synthetic modular proteoglycans that can be tailored for cell surface display. The expression of proteoglycan core proteins with unnatural amino acids permits bioorthogonal click chemistry with functionalized glycosaminoglycans for methodical dissection of the parameters required for optimal binding and function of various proteoglycan-binding proteins. We demonstrate that these sophisticated materials can recapitulate the functions of native proteoglycan ectodomains in mouse embryonic stem cell differentiation and cancer cell spreading while permitting the analysis of the contributing architectural elements toward function.
Graphical Abstract
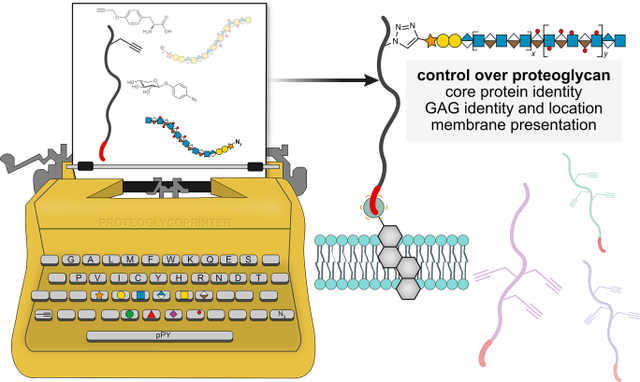
Introduction
Proteoglycans (PGs) are ubiquitous macromolecular glycoconjugates that serve dynamic and important roles in cell biology1. There is incredible detail embedded within the architecture of PG ectodomains, as it is composed of a core protein covalently decorated with poly-sulfated glycosaminoglycans (GAGs). Moreover, the glycoconjugate product can be differentially displayed as soluble or membrane-anchored molecules. Despite the overwhelming presence of PGs at the glycocalyx2, the systematic exploration of the roles of PG architecture as replete glycoconjugates is lacking. Because of their polymorphic nature, current approaches focus on achieving structural resolution of the individual components (either the protein or GAG chains), but in doing so, they sacrifice the total architecture of PGs. The core proteins organize the GAG chains into multivalent copies, and this arrangement can be important for function3. Different core proteins can exhibit their own set of unique functions, dictate the extent of GAG sulfation4, and some protein ligands preferentially bind deglycosylated PGs5. The decorating GAGs, on the other hand, can encode binding motifs for proteins such as fibroblast growth factor-2 (FGF2)6. PG biology is further complicated by the ability of ectodomains to be shed, and soluble ectodomains can function as autocrine effectors, or compete with cell surface counterparts7. Through these various structural elements, PGs can mediate and fine-tune a range of biological processes.
Control over PG architecture is difficult to achieve, and efficient methods to access defined PG glycoconjugates that permit a holistic way to modulate their architectures are lacking. PG biosynthesis begins with the ribosomal synthesis of the core protein, which is composed of an extracellular facing ectodomain and a membrane-anchoring portion (Fig. 1A). The core protein is then post-translationally modified with a tetrasaccharide linker (GlcA-Gal-Gal-Xyl) at serine residues and extensively elongated with disaccharide repeating units of a glucuronic acid (GlcA or IdoA) and an amino sugar (GlcNAc or GalNAc). The disaccharide compositions and glycosidic linkages dictate whether the GAG is classified as a heparan sulfate (HS), chondroitin sulfate (CS), dermatan sulfate (DS), or keratan sulfate (KS), and the GAG chains are further modified with sulfate groups at various positions (e.g., 2-, 3-, and 6-O, or N-sulfation)8. In total, a symphony of >40 enzymes is responsible for modulating PG architecture, and neither conventional genetic, chemical or chemoenzymatic strategies provide direct means to manipulate the core protein, GAGs, nor membrane presentation. Failure to control the sulfation patterns of GAGs can lead to contradictory experimental observations9,10. The over-expression of PG core proteins may result in dysregulated GAG chains, whereas targeting the GAG biosynthetic machinery can alter PG expression patterns or cause lethality11. Moreover, manipulation of sheddases, the enzymes that cleave cell surface-anchored ectodomains to soluble fragments, often results in the off-target removal of many other proteins12.
Figure 1. Production of pPY-functionalized proteoglycan (PG) core protein and azido-GAGs to generate defined ectodomains.
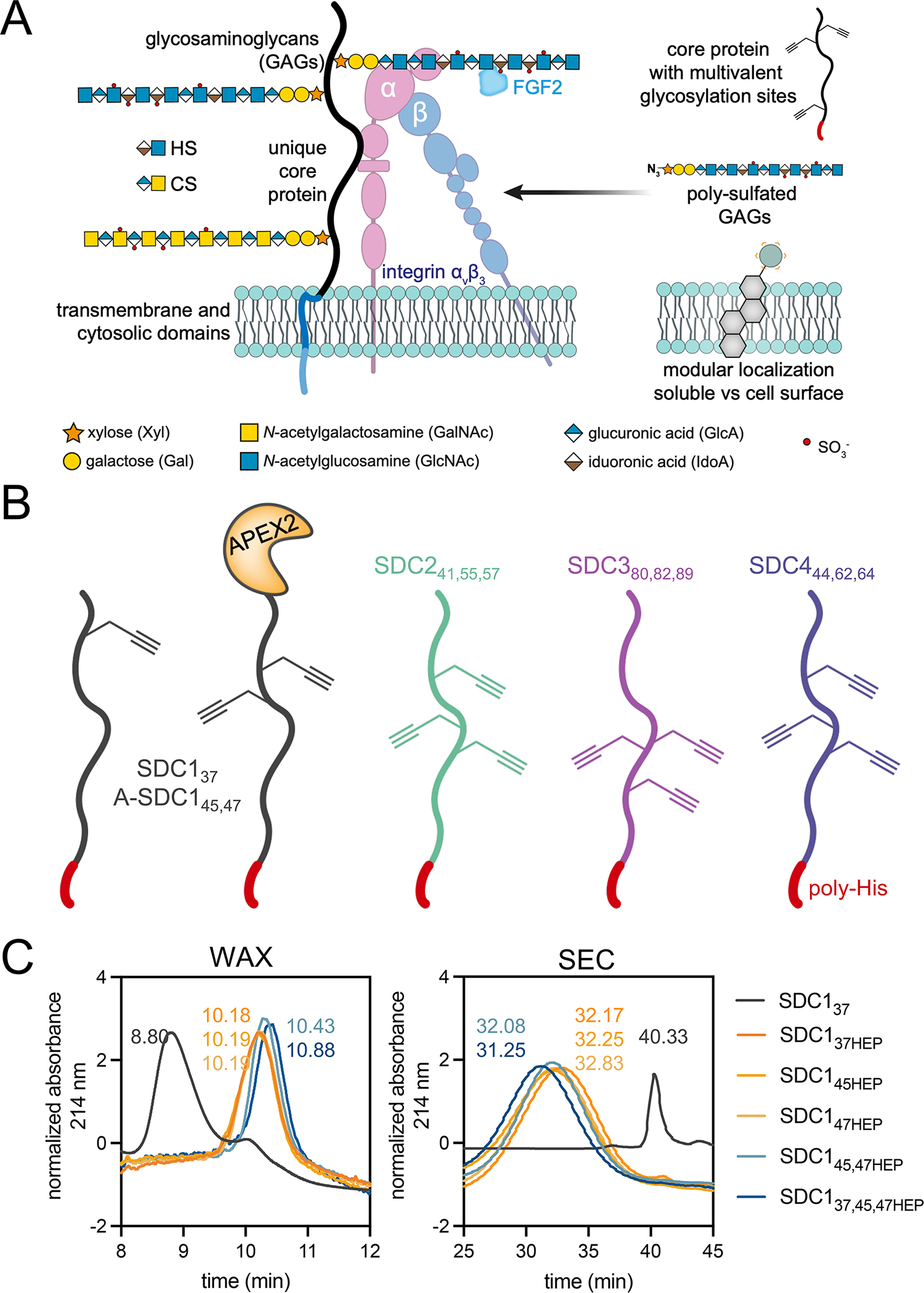
(A) PG architecture is composed of a core protein covalently decorated with glycosaminoglycan (GAG) chains, with a common tetrasaccharide (GlcA-Gal-Gal-Xyl) linker, followed by disaccharide repeating units. The elongated GAG chains are typically heparan (HS, IdoA-GlcNAc) or chondroitin sulfate (CS, GlcA-GalNAc). SDC1 N-terminal GAG attachment sites (S37,45,47) are usually occupied by HS3. PGs can bind a variety of biomolecules (e.g. FGF2, αvβ3 integrin), yet which components are responsible for binding and how GAG chain multivalency influences these interactions, are unclear. The modular platform outlined herein utilizes protein engineering to produce PG ectodomains with p-propargyltyrosine (pPY)-functionalized amino acids for bioorthogonal glycosylation with azido-GAGs. The presentation of native PGs as membrane-anchored or soluble ectodomains is mimicked by remodeling cells with or without a lipid anchor, respectively. (B) The incorporation of pPY into E. coli expressed PG ectodomains at the conserved GAG attachment sites (indicated by subscripts) provides a handle for chemical glycosylation. (C) Weak anion exchange (WAX, left) and size exclusion (SEC, right) chromatography discern deglycosylated (gray), monovalent (orange, SDC137HEP, SDC145HEP, SDC147HEP), divalent (SDC145,47HEP, light blue) and trivalent (SDC137,45,47, dark blue) glycoconjugates. Representative data from duplicate experiments.
Here, we present a straightforward platform to control all the components of the PG ectodomain - the core protein, the appended GAGs, and its membrane localization (Fig. 1A). This platform, rooted in chemical biology, offers unique opportunities to study the biological functions of PGs in high molecular detail, while maintaining a focus on the structures of the individual components. To generate functionalized PG ectodomains, we use unnatural amino acid (UAA) incorporation to install site-specific bioorthogonal reactive handles. To recapitulate the structure of native GAGs, we hijack the cellular biosynthetic machinery using a synthetic azidoxyloside. Finally, to tune the display of the edited PGs onto cell surfaces, we employ a lipid-based cell surface engineering strategy. The engineered PGs permit a renewed understanding of how PG architectures influence binding and functions mediated by heparan sulfate proteoglycan (HSPG)-binding proteins, such as FGF2, FGFR1, vitronectin (VN) and the αvβ3 integrin. We catalogue their corresponding structure-binding relationships and their functional effects towards mouse embryonic stem cell (mESC) differentiation and human mammary carcinoma cell spreading.
Results
Modular synthesis of proteoglycan ectodomains
We designed a modular platform that permits the site-specific installation of GAGs onto sequence-defined ectodomains, produced in E. coli as deglycosylated proteins due to the lack of a GAG glycosylation machinery (Fig. 1A, Supplementary Table 1). We utilized amber codon (TAG) reassignment to install the alkyne functionalized amino acid p-propargyl tyrosine (pPY (1); Supplementary Note 1) at canonical sites of GAG glycosylation in PG ectodomains, with C-terminal poly-histidine (poly-His) tags for nickel-nitriloacetic acid (NTA)-based purification and complexation to cells (Supplementary Table 2)13,14. We prepared pPY-modified versions of all four members of the murine SDC family (SDC1–4), with each protein incorporating one to three pPY amino acids at residues known to incorporate either a HS or CS GAG (Fig. 1B; modified residues depicted by subscripts). The SDCs are a four-membered family of type I transmembrane proteins4, of which SDC1 is the most studied. We focused our efforts towards the three GAG sites of SDC1 at residues 37, 45, 47, which are thought to be most important for function3. We prepared different monovalent versions of SDC1 with a single pPY site (SDC137, SDC145, SDC147; Extended Data Fig. 1A–C), as well as constructs bearing two (SDC145,47) or three (SDC137,45,47) pPY sites. We also prepared SDC1 fused with the engineered ascorbate peroxidase enzyme APEX215, separated by a TEV protease-cleavable tag (A-SDC145,47, A-SDC137,45,47; Fig. 1B, Supplementary Table 2, 3, Extended Data Fig. 1B) for future applications in proximity tagging, and additionally expressed the remaining murine SDCs as trivalent proteins (SDC241,55,57, SDC380,82,89, SDC444,62,64; Extended Data Fig. 1A–C). The pPY-modified SDCs were expressed in moderate to high yields (0.3–4.0 mg/L, Supplementary Table 3) and efficiently purified using Ni/NTA-based chromatography. We confirmed the incorporation of pPY by copper(I)-catalyzed [3+2] cycloaddition (CuAAC)16 with a tetramethylrhodamine (TMR)-azide fluorophore, in addition to Western blot and mass spectrometry (MS) analyses (Extended Data Fig. 1A–C). We also confirmed that SDC137 retained its capacity to be selectively processed by sheddases (Extended Data Fig. 1D).These proteins migrated at higher-than-expected molecular weights by SDS-PAGE due to their high (~65%) intrinsic disorder, which was observed by circular dichroism spectroscopy (Extended Data Fig. 1E–F)17.
We prepared recombinant azide-primed HS (azHS) and CS (azCS) GAGs using a metabolic oligosaccharide engineering (MOE) strategy, where we hijacked GAG biosynthesis (Extended Data Fig. 2A) using synthetic small molecules. Previous work has shown that xylosides derivatized with hydrophobic aglycones can efficiently serve as decoys that compete for native GAG elongation processes18,19. We posited that this strategy could be co-opted towards the production of GAGs with functionalizable handles by simply appending the hydrophobic moieties with azide groups. Starting from a bromo-xyloside intermediate and p-azidophenol, we generated p-azidophenol-xyloside 2 (Extended Data Fig. 2B, Supplementary Note 1). Compound 2 docks into the β4GalT7 galactosyltransferase active site with an observed ΔG = −6.3 kcal/mol, similar to the phenyl derivative lacking the azide moiety (Extended Data Fig. 2C), suggesting that 2 could be used for the production of recombinant GAGs. We incubated 2 (400 μM, 72 hrs) with suspension Chinese hamster ovary (CHO-S) cells, purified the conditioned media (CM; Extended Data Fig. 2D), and observed broad, diffuse smears upon reaction with a functionalized fluorophore, indicative of the extensive polydispersity of native GAGs (Extended Data Fig. 2E–F).
The resulting azide-modified HS (azHS) and CS (azCS) produced from 1 recapitulate native GAGs produced by host cells. Consistent with the Ext1-hemizygous nature of CHO cells, a majority of the material isolated is CS, as confirmed by selective digestion with heparinase (HSase) or chondroitinase (CSase; Extended Data Fig. 2E–F) and GAG disaccharide analysis20. Extended Data Fig. 2G). While only ~27% of total HS is functionalized with azide groups (Extended Data Fig. 2G, ~16.6 kDa), the non-primed GAGs are inert towards CuAAC and thus our azide-primed materials can be used without further purification. No significant changes in HS or CS sulfation were observed upon treatment with 2 (Extended Data Fig. 2H–K). We also prepared azido-heparin (azHEP) as a comparative control by functionalization of commercial heparin with 4-azidomethyl benzhydrazide21 (3, Supplementary Note 1, Supplementary Fig. 1). As expected, azHEP was largely composed of the trisulfated D2S6 disaccharide, compared to our azHS, which largely consisted of the non-sulfated D0A0 (Extended Data Fig. 2H–I). We also analyzed the disaccharide composition of a commercial glycosylated mouse SDC1 ectodomain (mSDC1) as a comparative control (Extended Data Fig. 2I, I–L, Supplementary Note 2).
With these materials in hand, we chemically glycosylated our SDC1 ectodomains through CuAAC with azHEP or azHS and analyzed the resulting purified products by weak anion exchange chromatography (WAX; Fig. 1C, Supplementary Fig. 2) and size-exclusion chromatography (SEC; Fig. 1C, Supplementary Fig. 3). Consistent with their highly disordered nature resulting in larger hydrodynamic radii22, the molecular weights determined using SDS-PAGE and SEC (Supplementary Fig. 2) were larger than their atomic mass (Supplementary Fig. 3, Supplementary Table 2). We consistently observed high conversion yields (~90%) regardless of the GAG, core protein, number of conjugation sites (Extended Data Fig. 3A–E), or the poly-anionic nature of the GAGs and the SDC1 ectodomain (pI = 4.7). Indeed, we observed full occupancy of divalent SDC1 constructs upon conjugation (Supplementary Fig. 4). Further, we observed similar levels of conformational disorder between SDC137 and its heparin-conjugated product, SDC137HEP (Extended Data Fig. 3F–G).
Defined PGs permit dissection of binding relationships
We performed AlphaScreen and ELISA binding assays to dissect structure-binding relationships with HSPG-binding proteins (Fig. 2). We first evaluated binary interactions using an AlphaScreen assay, which consists of streptavidin donor and Ni-NTA acceptor beads individually immobilized with biotinylated FGF2 or His-tagged SDC1, respectively (Extended Data Fig. 4A). The affinity between the two molecules brings the beads in close proximity, allowing for transfer of singlet oxygen and subsequent luminescence upon irradiation. Consistent with the affinity of FGF2 for sulfated GAGs6, we observed minimal binding of FGF2 to the deglycosylated SDC137 core protein (EC50 >100 nM; Fig. 2A). All heparin conjugates bound FGF2 maximally (EC50 ~ 0.3 – 0.4 nM) and significantly tighter than their HS conjugate counterparts (EC50 ~ 4 – 30 nM). We similarly observed a strong requirement for GAG sulfation of SDC1 ectodomains in binding biotinylated FGFR1, as affinity was only apparent in heparin conjugates (EC50 ~ 0.3 – 0.4 nM), with minimal binding to deglycosylated SDC137 or HS glycoconjugates (Fig. 2B)23. There appears to be some positional importance in binding FGF2 among monovalent SDC1 ectodomains, as HS occupancy at site 37 seems to impart greater affinity (SDC137HS vs. SDC147HS: 4.2 vs 27 nM). Although increasing HS chain valency from 1 to 2 (SDC137 or SDC147 vs SDC145,47) imparted some enhancement in binding FGF2, trivalent GAG display decreased affinity (Fig. 2C). A significant role for GAG chain valency in enhancing interactions with FGFR1 was not directly observed.
Figure 2. Binding of SDC1 to HSPG-binding proteins is influenced by GAG identity and multivalency.

AlphaScreen assays performed with (A) His-tagged SDC1 ectodomains and biotinylated FGF2, or (B) His-tagged SDC1 ectodomains and biotinylated FGFR1. (C) Scatter plots of average EC50 values depicting multivalent GAG chain relationships with binding FGF2 or FGFR1. (D) AlphaScreen assay to probe for ternary complex formation among His-tagged SDC1 ectodomains, biotinylated FGFR1, and soluble FGF2. (E) Scatter plots of average EC50 values depicting multivalent GAG chain relationships with ternary complex formation. (F) AlphaScreen assay performed with His-tagged SDC1 ectodomains and biotinylated VN. (G) ELISA performed with immobilized SDC1 and recombinant αvβ3 integrin. (H) Scatter plots of average EC50 values depicting multivalent GAG chain relationships with VN and αvβ3 integrin. All data points were constructed as the average of two experimental duplicates. Graphs were fitted using non-linear regression plotted using GraphPad Prism 9.
To evaluate the formation of ternary FGF2-FGFR1-SDC1 complexes, we added non-tagged soluble FGF2 to the aforementioned system (Fig. 2D). While minimal complexation was observed with deglycosylated SDC1 (EC50 > 20 nM), we observed positive ternary complex formation in all heparin-conjugated SDC1 ectodomains (EC50 < 8 pM). Here, we observe a significant effect from GAG valency, as the divalent heparin conjugate (SDC145,47HEP) displayed tighter interactions than its monovalent counterparts (SDC137HEP, SDC147HEP; Fig. 2E). In HS glycoconjugates, we observed a requirement for divalent chains to detect meaningful ternary complex formation (EC50: 1.5 nM), and a decrease in affinity with the trivalent HS conjugate (EC50: 2.7 nM).
We also evaluated binding of VN to the SDC1 ectodomain variants (Fig. 2F). VN is thought to bind HS chains24, and as expected, the deglycosylated SDC137 ectodomain alone displayed minimal affinity in the AlphaScreen assay. Only moderate binding was observed with the HS conjugates (EC50 ~ 6 – 30 nM), while maximal binding was observed with the heparin conjugates (EC50 ~ 0.5–1.0 nM). It has been previously reported that SDC1 binds the αvβ3 integrin25. While we observed minimal binding between His-tagged SDC1 and biotinylated αvβ3 integrin using an AlphaScreen assay (Supplementary Fig. 5), we were able to observe positive binding in an ELISA using immobilized SDC1 ectodomains, with all deglycosylated SDC ectodomains displaying EC50 ~ 100 nM (Fig. 2G, Extended Data Fig. 4A). We found significant enhancement of αvβ3 binding to glycosylated ectodomains, with the heparin conjugates displaying EC50 ~ 4.5–10 nM, and the HS conjugates displaying EC50 ~ 37 – 60 nM. The importance of GAG chain valency for enhancing binding was observed with both VN and αvβ3 (Fig. 2H). As expected, divalent and trivalent conjugates demonstrated increased affinity over monovalent ones, where affinity of heparin conjugates for αvβ3 reached EC50 ~ 5 nM, and HS conjugates reached EC50 ~ 40 nM (Table 1).
Table 1.
Apparent affinity constants (EC50) shown for each SDC1 construct and heparan sulfate proteoglycan (HSPG)-binding protein.
| AlphaScreen |
ELISA |
||||
|---|---|---|---|---|---|
| FGF2 | FGFR1 | FGFR1 + FGF2 | VN | αvβ3 | |
| proteoglycan | EC50 (nM) | ||||
|
| |||||
| SDC137 | >100 | >20 | >20 | >100 | 100 |
|
| |||||
| SDC137HEP | 0.4 | 0.3 | 0.007 | 1.0 | 10 |
|
| |||||
| SDC137HS | 4 | >20 | >10 | 6.0 | 60 |
|
| |||||
| SDC147HEP | 0.3 | 0.2 | 0.007 | 0.7 | 5 |
|
| |||||
| SDC147HS | 30 | >20 | >10 | 30 | 40 |
|
| |||||
| SDC145,47HEP | 0.3 | 0.4 | 0.004 | 0.8 | 5 |
|
| |||||
| SDC145,47HS | 6 | >20 | 9 | 8.0 | 40 |
|
| |||||
| SDC137,45,47HEP | 0.3 | 0.3 | 0.008 | 0.5 | 5 |
|
| |||||
| SDC137,45,47HS | 20 | >20 | 3 | 7.0 | 40 |
|
| |||||
| mSDC1 | 7.0 | >20 | 4 | >100 | 100 |
mSDCI represents mammalian expressed recombinant mouse SDC1 ectodomain. All data points were constructed as the average of two experimental duplicates. Graphs were fitted using non-linear regression plotted using Prism 9, error bars represent standard error of means.
We observed overall similar extents of αvβ3 binding to other SDC ectodomains, with SDC380,82,89 and SDC444,62,64, displaying similar affinity enhancements upon heparin or HS conjugation (Extended Data Fig. 4B–F). However, deglycosylated SDC380,82,89 and SDC444,62,64 bound αvβ3 with a higher affinity than SDC1 (EC50: 30 and 20 nM, respectively)26. We generally observed higher affinity HSPG binding interactions with trivalent SDC137,45,47, following the order SDC4 < SDC3 < SDC1 (Extended Data Fig. 4G). These data suggest that protein identity, GAG sulfation and valency can significantly affect the affinity of PG-binding proteins to SDC1. Overall, the commercial mSDC1 construct operated much more closely to the HS compared to the heparin conjugates. In some cases, however, we detected minimal binding of mSDC1, e.g. to VN (EC50 >100 nM) and αvβ3 (EC50 ~ 100 nM). We presume that these differences may be caused by the overwhelming presence of CS rather than HS in this sample (Extended Data Fig. 2L, Extended Data Fig. 4H), and the relatively closer binding affinity of HS and CS to FGF2 and FGFR127, compared to the significantly weaker affinity of VN and αvβ3 towards CS than HS or heparin28.
Probing structure-function relationships in stem cell differentiation
The successful exit from pluripotency and subsequent differentiation of mESCs is dependent on HS GAGs29 (Fig. 3A). However, how GAG chain valency and ectodomain presentation influence this process remains unknown, as does the roles of specific HSPGs involved. To control the presentation of our PG ectodomains (i.e. soluble vs. membrane-anchored), we incubated live mESCs with a functionalized cholesterol lipid (cholesterol-PEG2000-NTA; cholPEGNTA) for the external display of NTA headgroups on the cell surface. Upon incubation of pluripotent mESCs with the His-tagged SDC1 ectodomains in the presence of Ni(OAc)2, the ectodomains are complexed to the cell surface due to the affinity of the C-terminal poly-His tag for the tetradentate Ni-NTA chelating ligand30 (Fig. 3B). We chose cholesterol as a lipid anchor due to its high solubility and the ability of derivatives to passively incorporate into any phospholipid membrane and recycle back to the cell surface upon endocytosis31. We observed that the de novo presentation of proteins at the cell surface is dose-dependent (~650,000 NTA headgroups available), and that the NTA headgroups remain accessible for complexation for ~8 hrs following the initial incubation period (Extended Data Fig. 5A–C). Given previous reports of SDC1 localization to lipid rafts32, we also evaluated whether this cell surface complexation strategy could localize the SDC1 ectodomains into membrane compartments. Indeed, we observed distinct overlap of SDC1-positive membrane fractions with the lipid raft marker caveolin-133 (CAV1; Extended Data Fig. 5D–E), suggesting that endogenous SDC1 is present in lipid rafts. Additionally, cholPEGNTA was detected in the same membrane fraction (Extended Data Fig. 5E), suggesting that this cell surface engineering strategy can recapitulate native SDC1 localization.
Figure 3. Membrane remodeling with engineered SDC1 ectodomains provides insight into regulation of mESC differentiation by proteoglycans.

(A) HS-deficient (Ext1−/−) mESCs fail to differentiate due to their reduced ability to bind FGF2 (blue). The formation of ternary complexes among FGF2, FGFR, and HSPGs promotes differentiation. (B) A two-step complexation strategy is used to present engineered SDC1 ectodomain variants onto cell surfaces. Live mESCs are first incubated with cholPEGNTA (10 μM, 1 hr, 37°C), followed by Ni(OAc)2 (100 μM, teal) and the SDC1 ectodomain equipped with a poly-His tag (red tail; 2 μM, 1 hr, 37°C). Excess material is washed off at each step. (C) A six-day protocol for mESC differentiation, illustrating the loss of pluripotency marker Nanog and gain of SOX1 expression, indicative of neuroectoderm differentiation. Ext1−/− cells are incubated with engineered SDC1 constructs, with or without prior cell surface engineering, for 1 hr on D0 or a single treatment with soluble heparin (5 μg/mL) until D2. (D) GAG-conjugated SDC1 ectodomains (2 μM) can rescue differentiation of Ext1−/− mESCs, as evidenced by expression of neural precursor markers Sox1 (green) and Tubb3 (red). Scale bar: 50 μm. Data representative of three biological replicates. (E) RT-qPCR analysis at D6 of differentiation shows decreased Nanog expression of treated (heparin or SDC1) compared to untreated cells. Cells remodeled with cholPEGNTA before addition of SDC1 proteins demonstrated increased SOX1 expression. (F) RT-qPCR analysis at D6 of differentiation demonstrates increased expression of neural differentiation marker SOX1 with addition of glycosylated SDC1. All proteins are displayed on cell surface (+cholPEGNTA, 10 μM). All experiments performed in technical triplicate in two biological replicates. One-way ANOVA with Tukey’s post-hoc, (*) p <0.0332, (**) p <0.0021, (***) p <0.0002, (****) p <0.0001. Bar graphs represent means and error bars represent SEM.
Using HS-deficient Ext1−/− mESCs that remain pluripotent unless supplemented with HS or heparin, we evaluated the effects of SDC1 glycosylation, GAG chain valency, and membrane presentation in a six-day (D6) adherent differentiation protocol34, (Fig. 3C) where cells are treated with the SDC1 variants for only 1 hr at the onset of differentiation (D0). Upon qualitatively analyzing cells by immunofluorescence at D6, we immediately observed a requirement for HS or heparin, to permit cells to differentiate into Nanog-/SOX1+/TubB3+ neural precursor cells, without a strict requirement for membrane presentation (Fig. 3D, Extended Data Fig. 5F). To provide a quantitative description of the role of membrane presentation, we used qRT-PCR to analyze cells at D6. We observed increased SOX1 expression in cells treated with our membrane-anchored, GAG-conjugated SDC1 constructs compared to soluble constructs (Fig. 3E, Extended Data Fig. 5G). While the monovalent HS-conjugated SDC1 (SDC137HS) resulted in enhanced SOX1 expression over its heparin counterpart (Fig. 3F), both di- and tri-valent heparin conjugates (SDC145,47HEP, SDC137,45,47HEP) caused greater SOX1 expression than their corresponding HS conjugates (SDC145,47HS, SDC137,45,47HS; Fig. 3F). All constructs significantly reduced Nanog expression to similar extents compared to untreated cells (Extended Data Fig. 5H).
Remarkably, the extent of differentiation with our ectodomains (1 hr, D0) exceeded that with soluble heparin despite its prolonged treatment (D0-D2, Fig. 3C). Interestingly, treatment with SDC137 core protein alone also resulted in increased SOX1 expression than untreated cells, or those treated with soluble heparin (Fig. 3F). This observation suggests that SDC core proteins also contribute towards neuronal differentiation (Extended Data Fig. 5G–J). We also observed similar increases in differentiation with SDC380,82,89 and SDC444,62,64 glycoconjugates (Extended Data Fig 5J).
Cell spreading requires cell surface, glycosylated SDC1
Using wild-type MDA-MB-231 cells and SDC1-knockdown (SDC1KD) cells, we dissected the roles of core proteins, GAG composition, and membrane-presentation towards VN-mediated spreading26,25 (Fig. 4A). We focused our efforts on heparin-conjugated SDC constructs over HS counterparts, due to the dramatic increase in affinity of VN and αvβ3 observed (Table 1, Extended Data Fig. 4H). First, using wild-type cells, we observed that incubation with deglycosylated soluble ectodomains displayed minimal effects, whereas the heparin conjugates significantly reduced cell spreading (Fig. 4B–C, Extended Data Fig. 6A). However, the addition of soluble heparin did not replicate inhibition seen with heparin-conjugated SDC1, suggesting a cooperative effect between the GAG and the SDC1 core protein (Fig. 4B). While this spreading phenotype is mostly attributed to SDC125, we observe similar effects with other heparin-conjugated SDC proteins, including SDC241,55,57HEP, SDC380,82,89HEP and SDC444,62,64HEP. Interestingly, we also observed a significant reduction in cell spreading with the deglycosylated SDC380,82,89 (Fig. 4C).
Figure 4. Adhesion and spreading of mammary carcinoma cells.

(A) Cartoon depiction of murine SDC1-mediated cell spreading of MDA-MB-231 mammary carcinoma cells occurring through both protein- and GAG-mediated interactions with integrin αvβ3 and GAG-mediated interactions with vitronectin (VN), for which αvβ3 is the canonical receptor. Note: human and mouse SDC1 are highly homologous (78%) in the region responsible for αvβ3 integrin-mediated activation53. (B) Quantification of cell spreading of wild-type MDA-MB-231 cells on vitronectin surfaces. (C) Representative fluorescence microscopy images of wild-type MDA-MB-231 cells plated in media supplemented with indicated SDC family proteins (2 μM) or heparin (0.4 μM). Adhesion and spreading of cells were inhibited by addition of soluble glycoconjugates and SDC380,82,89. Cells were fixed and stained for phalloidin (red) after 2 hr on VN matrix. Scale bar: 50 μm. Data representative of three biological replicates. (D) Quantification of cell spreading in wild-type (WT) and SDC1 knockdown (SDC1KD) cells. Bar graphs represent means and error bars represent SEM. One-way ANOVA with Tukey’s post-hoc, (*) p <0.0332, (**) p <0.0021, (***) p <0.0002, (****) p <0.0001. Error bars represent SEM. For each condition, n >10 images across two biological replicates.
We also used a previously established assay using SDC1 knockdown cells25 to support the aforementioned observations. Upon hSDC1 siRNA knockdown, we remodeled cells with soluble or membrane-anchored SDC ectodomains and observed a strict requirement for membrane-anchored, glycosylated SDC1 for cell spreading on VN-coated plates (Extended Data Fig. 6B–G). Further, we generated CRISPR-generated SDC1KD cells (Extended Data Fig. 6H–J) and observed enhanced cell spreading with membrane-anchored heparin conjugates, regardless of core protein identity, over untreated cells, with SDC137HEP displaying the most significant rescue (Fig. 4D, Extended Data Fig. 6G). We did not observe a direct effect of GAG chain valency in this limited panel.
Engineered proteoglycans enable live cell proximity tagging
We envision using APEX2-tagged PGs to map interactomes in live cells using proximity tagging.35–37 PGs, such as SDC1, possess many binding partners as a consequence of their multi-component nature38 and may exhibit different interactomes a result of differential glycosylation or membrane localization. As a proof of concept, we performed proximity tagging experiments with A-SDC145,47 and its heparin conjugate A-SDC145,47HEP (Extended Data Fig. 7A) to capture interactomes in live Ext1−/− mESCs. In this approach, the APEX2 peroxidase catalyzes the formation of short-lived (<1 ms) and proximal (<20 nm radius) biotinyl radicals that covalently react with interactors for the SDC1 bait protein (Fig. 5A). The biotinylated proteins can then be probed or enriched.
Figure 5. Proximity tagging with A-SDC145,47 to capture interactomes in live cells.
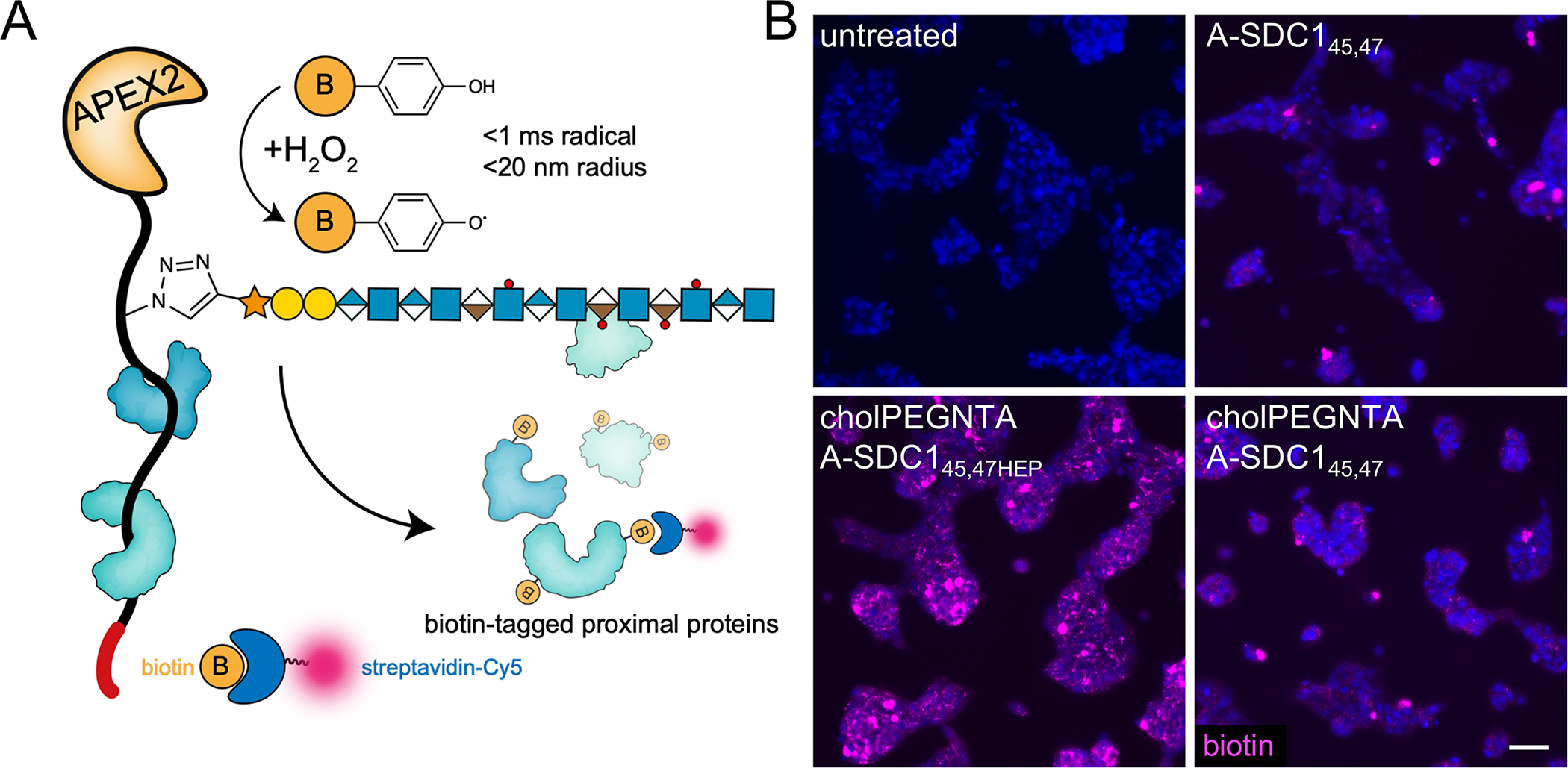
(A) Cartoon depicting proximity tagging with APEX2 fused SDC145,47HEP (A-SDC145,47HEP). After incubation of live cells with A-SDC145,47HEP, excess unbound protein is washed away and cells are incubated with biotin phenol (yellow circle, 0.5 mM, 30 min). The APEX2 peroxidase catalyzes the formation of short lived biotinyl radicals that react with proximal proteins upon addition of H2O2. (B) Ext1−/− mESCs subjected to the proximity tagging protocol qualitatively demonstrate differences in interactomes (biotin, purple) based on membrane anchoring (+ cholPEGNTA) and glycosylation (A-SDC145,47HEP). Data representative of three biological replicates.
We qualitatively compared the differential interactomes of A-SDC145,47 when it is presented as a soluble or a membrane anchored molecule. We observed significant dose-dependent biotinylation of cells following proximity tagging using fluorescence microscopy, probing for the biotinylated interactors (Extended Data Fig. 7B). Fluorescence signals stemmed from the cell surface, consistent with the known localization of SDC1, and we observed varying intensities based on glycosylation and membrane presentation, with highest signals observed from membrane-anchored, glycosylated A-SDC145,47HEP (Fig. 5B). While we have yet to assign the differential interactomes resulting from changes in PG architecture, we demonstrate that fusion proteins of our engineered PGs have tremendous utility and promise to fine-tune our understanding of PG interactions with minimal perturbation.
Discussion
As glycosylation is not template driven, PGs naturally exist as molecular polymorphs of various GAGs attached to a single core protein, which is thought to contribute to their functional diversity. While much heroic work has focused on the generation of GAGs and PG mimetics39–41, such materials fail to recapitulate native structures and overlook the functional roles of the core protein42–44. To systematically dissect the effects of PG molecular heterogeneity, we present a modular platform to chemically edit PGs that considers all aspects of PG architecture, including membrane localization.
Starting with the core proteins of SDC1–4, we incorporated pPY at sites of native GAG glycosylation (Fig. 1B) for bioorthogonal conjugation with azide-modified GAGs, which were produced by MOE with compound 2. We envision the application of this MOE strategy to generate libraries of sulfation-defined GAGs using CRISPR-edited cells45,46. We demonstrate that CuAAC can be used to conjugate highly polyanionic GAGs at up to three pPY sites of a PG core protein (Fig. 1F), and it is compatible with heme-containing proteins, such as the APEX2 peroxidase (Fig. 5, Extended Data Fig. 7). While all multivalent ectodomains produced in this work are modified with the same GAG, we anticipate that other orthogonally functionalized UAAs and new synthetic xylosides will facilitate chemoselective conjugation of different GAGs to construct various proteoglycan hybrids of HS and CS47–49. This production platform is scalable and amenable towards the large-scale production of modified PG ectodomains. Indeed, over the course of these studies we have produced sub-milligram quantities of the most complex material in this series (e.g. SDC137,45,47HS).
In contrast to standard genetic approaches, which can result in off-target effects or aberrant expression of mutants3,50, our platform permits the facile interrogation of the roles of specific GAG structures within the context of native PG proteins51. We observe that the extent of GAG chain sulfation, GAG chain valency and PG identity are important factors in regulating affinity to HSPG binding proteins (Fig. 2, Extended Data Fig. 4). In contrast to isolated PGs, which can be composed of heterogeneous mixtures of HS and CS GAGs, such as mSDC1, our edited PGs offer controlled GAG compositions and enable the facile determination of the impact of sulfation and multivalency on HSPG-binding protein interactions. Coupled with cell surface engineering strategies, our platform facilitates the study of PG interactions and functions at the surface of live cells, especially in those that are not readily amenable to genetic manipulation (e.g. primary cells, stem cells). The ability to perform these structure-activity studies in cellulo is crucial, as many interactions occurring with HS GAGs in vitro have been observed to not interact with native HSPGs52. These differences could be due to additional binding contributions of the core protein itself, the differing fine structure of HS among tissues, or the absence of the multivalent architectural arrangement. To our knowledge, this work represents the most advanced effort toward the generation of full PG glycoconjugates with defined GAG chains.
We have demonstrated the utility of our platform in these two biologically relevant assays, mESC differentiation (Fig. 3) and cancer cell spreading (Fig. 4). Both systems illustrate a dependence for correct GAG compositions, core protein identities, and GAG chain valency, and it is only with VN-mediated cancer cell spreading that we observe an obligate requirement for membrane anchoring (Fig. 4B, C). Curiously, soluble SDC380,82,89 was unique amongst core proteins in its ability to inhibit cell spreading of wild-type mammary carcinoma cells (Fig. 4C). Although interactions with αvβ3 have not been previously explored, SDC3 represents the most similar family member to SDC1 and αvβ3 integrins can still be activated in SDC1−/− mice53, possibly through SDC3. Upon SDC1KD, only glycosylated SDC proteins could rescue cell spreading on VN, suggesting that the relevant biological mechanism requires GAG-conjugated SDCs. We note that the choice of binding assay (i.e. AlphaScreen vs ELISA) to evaluate the edited PGs in vitro requires careful consideration. Given the wide range of affinities in PG-mediated interactions, one assay optimal for a particular PG and binder may not be suitable for another. We could not observe significant interaction between αvβ3 and SDC137 through AlphaScreen (Supplementary Fig. 4), presumably due to relatively weak affinity that is beyond the optimal range for detection (EC50 < 300 nM) or the oriented nature of SDC1 ectodomains on the beads. Using the random orientation of immobilized SDC1 in an ELISA format, we observed binding of the SDC1 core protein, in agreement with previous observations25. The affinity of this interaction is greatly increased by GAG conjugation, with maximal binding occurring with heparin glycoconjugates, which encouragingly concur with contemporary observations of αvβ3 binding to a SDC1-derived glycosylated peptide54. Both our binding and mESC differentiation assays point to divalent GAG attachment to SDC1 for maximal effects, and little to no further enhancements are imparted by the third GAG attachment site. Moreover, we determine that maximal binding occurs with heparin glycoconjugates (Table 1, Extended Data Fig. 4H), suggesting that the extent of sulfation, or presence of 3-O-sulfation, can enhance affinity. Given the stark contrasts in affinity displayed between heparin and HS conjugates, however, it is worth noting heparin may artificially over-emphasize affinity beyond what is observed with native HS conjugates.
Lastly, the ability to tag interactors in live cells by combining cell surface engineering with proximity tagging provides new opportunities to study PG interactions in cellulo. The APEX2 approach is especially apt for the capture of glycan-protein interactions that are heavily influenced by three-dimensional presentation of ligands36,37. Notably, SDC1 alone exhibits over 100 interactors in different contexts38. Considering the observation that deglycosylated SDC core proteins unexpectedly promoted mESC differentiation (Fig. 4E–F, Extended Data Fig. 5G–J), we anticipate the application of this proximity labeling to unearth novel molecular mechanisms and interactors underpinning PG-mediated biology.
In summary, we have generated a modular platform that permits the tailored assembly of PG ectodomains, replete with sequence-specific core proteins, defined GAGs, and the ability to present them onto cell surfaces. The process for producing these GAG-conjugated PGs represents the most defined semi-synthetic materials to date. We demonstrate that systematic investigation of GAG composition and chain multivalency on SDC proteins can yield refined insights into structure-function relationships. While there are 16 classically known PGs, of which we have made progress on four, recent work has identified that other proteins not conventionally designated as PGs, can bear GAG chains, and carry out important biological functions55,56. These discoveries therefore create even further opportunities for the use of glycoengineering platforms, like ours, to aid in our understanding the multifaceted functions of PGs in many biological contexts.
Methods
Materials and reagents.
Unless otherwise noted, chemical reagents were used as purchased and obtained from commercial vendors. Recombinant human FGF2 (NovusBio # NBP2–76301) is composed of 154 amino acids (Accession Number: P09038). Recombinant human αvβ3 (R&D Biosystems # 3050-AV-050) is composed of the αv (Phe31-Val992, Accession # NP_002201) and the β3 (Gly27-Asp718, Accession # AAA52589) subunits. Wild-type mESCs and MDA-MB-231 cells were purchased from ATCC, and Ext1−/− mESCs were a gift from Kamil Godula, Catherine Merry, and Jeffrey Esko.
Expression of syndecan ectodomains with unnatural amino acids.
Syndecan ectodomain sequences containing TAG codons at sites of unnatural amino incorporation were cloned into pET28a vectors and co-transformed into BL21 (DE3) competent cells with pULTRA-CNF, a gift from Peter Schultz. Transformed cells were selected on agar supplemented with kanamycin and streptomycin. Single colonies were grown (18 hr, 37°C) stored as stocks (50% glycerol, −80°C) and used to seed overnight 4 mL cultures which were diluted into 1 L of Luria-Bertani media. After growing to an OD600 of 0.6–0.8, IPTG (1 mM) was added, along with pPY (final conc. of 400 mg/L) and an equivalent volume of 10X PBS. The culture was incubated (225 rpm at 30°C for 18 hr) and cells were harvested (20 min, 3600 x g, 4°C). Cells were resuspended in 20 mL of lysis buffer (3X PBS, 25 mM imidazole, 1 mM phenylmethylsulfonyl fluoride, and 5 μg/mL leupeptin, aprotinin, and pepstatin A). Cells were sonicated (Misonix 3000, 1/4” microtip probe, 2.5 min total sonication time, power level 5.5) and centrifuged (17,000 x g, 45 min, 4°C), and the supernatant was passed through a 0.45 μm filter. The lysate was applied to a Ni-NTA column at 4°C (Histrap FF, Cytiva), washed (20 column volumes (CV) of 30 mM imidazole, 3X PBS), and the His-tagged protein was eluted (250 mM imidazole, 3X PBS). The eluate was concentrated and buffer exchanged into PBS with an Amicon 10 kDa centrifugal filter. Absorbance at 280 nm was converted to concentration in mg/mL using molar extinction coefficients calculated with Expasy ProtParam server [3]. Proteins were stored in PBS with 12.5% glycerol, flash frozen, and stored at −80°C.
TEV cleavage.
The A-SDC145,47 and A-SDC137,45,47 fusion proteins were prepared at a concentration of 1 to 15 mg/mL in buffer (0.5 mM EDTA, 1 mM DTT in PBS, pH 8.0) and TEV protease (5U /mL) was added. The reaction was incubated (RT, 6 hr, then 4°C, 18 hr). To remove the TEV, the mixture was incubated with Halolink resin (RT, 45 mins). The resin was removed by centrifugation and the supernatant was diluted ten-fold (3X PBS, 25 mM imidazole). The His-tagged SDC1 protein was purified by a nickel affinity column. The APEX2 product from the TEV cleavage was collected in the Ni column flow through. Samples were buffer exchanged into PBS with a PD-10 desalting column and concentrated by centrifugal filtration.
Mass spectrometry of intact SDC proteins.
Samples were analyzed through both direct injection and LC/MS. For direct injection, purified recombinant protein (10–50 μg) was exchanged into NH4HCO3 buffer (2.5 mM, pH 6.8) at 4°C with Amicon Ultra 3K MWCO spin filters. The concentrated protein (50–75 μL) was collected and mixed with an equal volume of methanol with 2% formic acid, prior to injection into a LTQ Orbitrap XL (Thermo Fisher). For LC/MS, samples were at 1 to 5 mg/mL in PBS, injected onto an AccuCore 150 C4 column (Thermo, 100 × 2.1 mm) at 60°C, incorporated into an Ultimate 3000 UHPLC system. Flow rate was 0.4 mL/min. Column was equilibrated with 90% buffer A (H2O, 0.1% formic acid), and a 23 min gradient to 60% buffer B (acetonitrile, 0.1% formic acid) was employed. UV was monitored at 214 nm. Data was acquired with Xcalibur 2.1 software.
MMP-9/Sheddase treatment of SDC1.
SDC137TMR (10 μM in 50 mM Tris pH 7.4,4 mM CaCl2, 150 mM NaCl, 0.05% Triton X-100) was treated with MMP-9 (35 μg/mL), ADAM-TS1 (28 μg/mL), or trypsin (85 ug/mL). MMP-9 was first activated by treatment with 1.0 mM 4-aminophenylmercuric acetate (PAPMA, 2 h, 37°C). For samples treated with MMP-9 and trypsin, His-tagged SDC137TMR was captured after incubation with the enzyme with 50 μL of Ni-NTA resin, washed (3X with 250 μL PBS), and imaged. For ADAM-TS1 treated sample, the SDC137TMR was reacted while bound to the Ni-NTA resin, washed (3X with 250 μL PBS), and imaged.
Circular dichroism.
SDC1 proteins were buffer exchanged into CD buffer (55 mM KH2PO4, pH 7.4) to remove glycerol from storage buffer. Circular dichroism (CD) assays were performed on a Jasco J-815 spectropolarimeter. Proteins were diluted in CD buffer to 0.1mg/mL. 200 mL of each protein sample were then loaded into a 0.1 cm quartz cuvette for analysis. Experimental scanning parameters were set to 300–185 nm, with 50 nm/min scanning speed, 0.5 second response, 5 nm bandwidth, and 0.1 nm data pitch. Compartment chamber was set to 20°C. Predicted structures from the spectra were analyzed using the web-based CAPITO analysis tool57.
Production, purification, and analysis of recombinant GAGs from CHO-S cells.
Azidoxyloside 2 (400 μM) was added to Freestyle CHO-S cells in Gibco Freestyle CHO expression media with added GlutaMAX (1×106 cells/ml). Following incubation (72 hr), the conditioned media was collected, filtered (45 μm) and loaded onto a 5 mL Hitrap DEAE Sepharose FF (Cytiva) equilibrated with PBS at a flow rate of 4 mL/min. The column was washed (25 CVs PBS), and GAGs were eluted (6 CVs of 1.5 M NaCl in PBS). The eluate was dialyzed for 24 hr against 10 mM ammonium bicarbonate (3500 MWCO Snakeskin, ThermoFisher, 68035), changing the buffer 3 times. Following dialysis, the sample was lyophilized and reconstituted in PBS. To isolate heparan sulfate (HS) and chondroitin sulfate (CS), the purified GAGs were digested with chondroitinase or heparinase, respectively. To purify HS, GAGs were digested using 1.0 U/mL chondroitinase ABC in PBS with 50 mM sodium acetate. To purify CS GAGs, HS was digested with a mixture of heparinase I, II, and III at 2.5 U/mL each in PBS (+ Ca+2, Mg+2). Degraded GAGs were removed with an SEC column (Hiprep 16/60, Sephacryl S-200 HR) in PBS buffer. Fractions containing full-size GAGs were combined and concentrated with a 10 kDa MWCO centrifugal filter for a yield of 1.5 μg HS and 2.6 μg CS per mL of cell media.
Molecular docking of azidoxyloside 2 with β4GalT7.
Compound 2 was docked into the active site of β4GalT7 in complex with UDP-galactose (PDB ID 4M4K)58 using Autodock Vina59.
CuAAC click reactions.
Alkyne and azido-functionalized reactants were dissolved in PBS with aminoguanidine HCl (alkyne-protein at 25 μM, azido-GAG at 20 eqs per alkyne, 5.0 mM aminoguanidine HCl). For conjugations with azido-HS, 1.1 eqs per alkyne group were used. The copper catalyst was prepared in H2O sparged with N2 and added to the reaction mixture (320 μM CuSO4 1600 μM Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA), 21 mM sodium ascorbate). The reaction was incubated in a sealed Eppendorf tube (300 rpm, 37 °C for 4 hr). Protein-GAG conjugate was purified DEAE column (WAX-10 4×250 mm Thermo Scientific) on an Ultimate 3000 UHPLC system at 1.0 mL/min in 20 mM tris pH 7.5 buffer. To remove excess GAG, DEAE fractions were loaded onto a Ni-NTA column (Histrap FF, Cytiva) and washed with 20 column volumes of 1 M NaCl (with 25 mM imidazole, 20 mM Tris, pH 7.5). The product was eluted with 5 column volumes of elution buffer (400 mM imidazole, 500 mM NaCl, 25 mM Tris at pH 7.5). This was exchanged into PBS with a Sephadex G-25 PD-10 column, and concentrated to approximately 50 μM with a 10 kDa centrifugal filter. Absorbance at 280 nm was measured by Nanodrop and converted to molar concentration. For CuAAC conjugation to TMR azide 5-isomer, the procedure was essentially the same, (10 mM TMR azide 5-isomer in DMSO diluted into the reaction mixture to give 200 μM final concentration). After 1 to 2 hr, an aliquot of the reaction was mixed with SDS-PAGE sample buffer (10% 2-ME) and loaded onto the gel. Azido-GAGs (azGAGs) were labelled with AlexaFluor-488-yne using CuAAC as described above, with azGAGs dissolved in 200 μl PBS and reacted with 200 μM AlexaFluor-488-yne. Excess fluorophore was removed with a PD MiniRrap G-10 column (Cytiva). Copper-free click chemistry labeling of azGAGs with was performed by incubating with DBCO-PEG4-Fluor 545 (200 μM, 20 hr, 37 °C).
Weak anion exchange (WAX) monitoring of CuAAC reactions.
Click reactions between alkyne-displaying proteins and azide-GAGs were analyzed with a Propac WAX-10 column (4 × 250 mm on an Ultimate 3000 UHPLC system (Thermo) at 1.0 mL/min with a gradient from 0–1.75 M NaCl (20 mM Tris pH 7.5) with fluorescent detection (λex=280 nm, λem=350 nm).
Size exclusion chromatography.
Samples were analyzed on a Superose 6 Increase 10/300 GL SEC column (on an Ultimate 3000 UPHLC (0.4 mL/min, 2X PBS). Injections consisted of 1 to 2 μg sample in 20 μL. Eluate was monitored with fluorescence (λex = 280 nm, λem = 350 nm) and UV detection (λ = 214 nm). TMR-labelled GAGs were detected with λex = 545 nm, λem = 567 nm. For size estimation of recombinant azido-GAGs, calibration was performed using dextran standards (5, 12, 25, 50, 80 and 150 kDa).
Preparation of chondroitinase ABC and heparinase cocktail.
A mixture of heparinases was prepared (1 mL PBS per 100 U vial of Heparinase I and III). After mixing, 1 mL of the above solution was added to 100 U of heparinase II. Chondroitinase ABC stock solutions were prepared at 20.0 U/mL (60 mM Tris, 50 mM sodium acetate, pH 7.9). Aliquots of the enzymes were stored at −80°C.
Enzymatic digestion of GAGs for disaccharide analysis.
GAGs (soluble or covalently attached to beads) were resuspended in 400 μL of PBS (+ Ca+2, Mg+2) with 1 U of heparinases I, II, and III or chondroitinase ABC. The samples were rotated (37°C, 18 hr), and centrifuged (2000 rpm, 2 min). The supernatant was then transferred to Amicon 3K MWCO spin filters (14,000 x g, 30 mins) to separate undigested material. The digested samples (eluate) were lyophilized and stored at −80°C.
GAG disaccharide labeling.
Disaccharides were fluorescently labelled with a 1.0 M solution of NaBH3CN and 0.2 M anthranilamide (30% acetic acid in DMSO)20. 35μL of the labelling mixture was added to each dried sample, which was heated (70°C, 2 hr). A column for each sample was equipped with Whatman Grade 3 filter paper pre-equilibrated with water (1 mL), 30% acetic acid (aq) (2.5 mL), and acetonitrile (2 mL). Sample was added to the filter paper and allowed to adsorb for 15–20 minutes, then washed four times (2 mL acetonitrile). Samples were eluted (1 mL H2O) and stored at –20°C.
GAG disaccharide analysis
Samples were injected onto a Propac-PA1 column (4 × 150 mm, Thermo) affixed with a guard column (4 × 50 mm, Thermo) on an Ultimate 3000 UHPLC system (1.0 mL/min, gradient from 0 to 1.0 mM NaCl in 50 mM NaOH) with fluorescence detection (λex = 348 nm, λem = 440 nm)45. Retention times for the disaccharides were confirmed using anthranilamide labelled disaccharide standard mixes (Galen). Absolute quantification of disaccharides was based on calibration curves constructed with known quantities of disaccharide.
Biotinylation of FGF2.
A HEPES buffered (200 mM, pH 8.4, 50 μL) solution of FGF2 (100–150 μg, 35 μL) was prepared with heparin (20 mg/mL in H2O, 10 μL) and sulfo-NHS-biotin (4 mg/mL H2O, 5 μL) and reacted (2 hr, RT). Reaction quenched with glycine solution (20 μL, 10 mg/mL). The solution was then purified and eluted (3 M NaCl, 0.2% BSA, 20 mM HEPES, pH 7.4) using a Heparin Sepharose 6 column and stored at 4°C prior to use.
Biotinylation of vitronectin, FGFR1, and αvβ3 integrin.
Proteins (2 – 5 mg/mL) were prepared in PBS, to which sulfo-NHS-biotin (20 eqs, 2 mg/mL sulfo-NHS-biotin, H2O) was added. This reaction was allowed to proceed (1hr, RT). Excess NHS-biotin was removed with a PD-10 desalting column, followed by concentration with a 10 kDa MWCO filter.
AlphaScreen.
Dilution series of the SDC constructs in buffer (0.5% BSA in PBS) were aliquoted to 1/2 Area OptiPlate-96 well plates, followed by addition of biotinylated proteins. Samples were run in duplicate. For assays where a non-His-tagged, non-biotinylated protein was present, all three components were added to the plate in succession. The plate was covered, briefly centrifuged (250 x g, 10 s), and the mixture was incubated (1hr, RT) on an orbital shaker. AlphaScreen beads consisted of streptavidin donor beads and nickel-chelate acceptor beads, which were diluted into buffer (5 μg/mL) prior to use and employed under dim-light conditions. Acceptor beads were added and incubated (1 hr, RT, with shaking). Subsequently streptavidin beads were added, followed by a third incubation performed as above. Positive control consisted of biotinylated, poly-His tagged peptide used at 5 nM. Plates were read on a Perkin Elmer Envision 2101 multilabel plater reader, under the software Envision Manager Version 1.13.3. Data was normalized and fit to a four-parameter sigmoidal curve (log(agonist) vs normalized response – variable slope) in GraphPad Prism. All curve fits gave an R2 value >0.9.
ELISA
Each SDC construct was immobilized in duplicate onto 96-well Maxisorp high-binding plates (4°C, overnight, rocking, 50 μL, 5 μg/mL in Na2CO3 buffer, pH 9.6). Following blocking (2% BSA/PBST, 100 μL) and washing 3 times with PBST (100 μL), wells were incubated with titrations of αvβ3 (PBST, 4°C, overnight, rocking, 50 μL). Wells were washed, then probed with biotinylated anti-αvβ3 antibody (1:1000, RT, 1hr, rocking, 100 μL) followed by washing and incubation with HRP-streptavidin (1:1000, RT, 1hr, rocking). Plates were washed, then incubated with a TMB substrate (100 μL) until a solid blue color developed, and the reaction was quenched with H2SO4 (2N, 100 μL). Absorbance at 450 nm was evaluated and subtracted from blank wells without immobilized SDC. Data was normalized and fit to a four-parameter sigmoidal curve (log(agonist) vs normalized response – variable slope) in GraphPad Prism. All curve fits gave an R2 value >0.98.
Neuronal differentiation of mouse embryonic stem cells (mESCs).
Cells were seeded overnight (37°C, 5% CO2, ~18 hr) in maintenance media (MM) at a density of 1×104 cells/cm2 in 24-well gelatin-coated 24-well plates42. The following day (D0), cells were washed with 1X DPBS and remodeled with cholPEGNTA (Nanocs #PG2-CSNT-2k; 10 μM, 1 hr, 37°C). After washing twice with DPBS, cells were incubated with SDC constructs in KO-DMEM media (1 hr, 37°C), or heparin (5 μg/ml, 48h, 37°C) Cells were washed twice and grown in N2B27 neural differentiation media for 6 days. Subsequent washes and media replacements were performed at D2, D4, and the cells were analyzed at D6.
Flow cytometry.
Cells were harvested using non-enzymatic cell dissociation buffer (MDA-MB-231) or EDTA-free 0.05% trypsin (remodeled mESCs) into 96-well round bottom plates. After centrifugation (500 xg, 5 mins, 4°C) the supernatant was removed and cells were fixed (4% PFA/PBS, 10 mins, RT). Cells were washed in 1X DPBS twice before incubation with antibodies (3% BSA, 1hr, on ice). Cells were washed twice in 1X DPBS before secondary antibody incubation (1h, on ice, in dark). Cells were washed twice with 1X DPBS before re-suspension (5% FBS/PBS) and analysed on an BD Accuri C6. Data analysis was performed using FlowJo and GraphPad Prism 9.
Lipid raft/caveolae isolation.
mESCs were seeded in a 10cm2 plate at 90% confluency and incubated with 3 μM GFP(His)6 (1hr,37°C), with or without prior incubation with cholPEGNTA (10 μM, 1hr). Cells were processed using protocol provided by manufacturer using Optiprep density gradient medium60. Lysate was centrifuged (200,000xg, 4hr, 4°C) in a Beckman-Coulter Optima MAX-XP centrifuge fitted with an MLA-50 rotor. The resulting fractions were collected into pre-cooled Eppendorfs for dot blot analysis using 5 μL of each fraction on Immobilon-FL membranes activated in methanol. Blots were probed with anti-Caveolin-1 and anti-SDC1 antibodies and fluorescent secondaries (5% BSA/TBST). Quantification was performed using Image Lab 6.1 software (BioRad). GFP(His)6 fluorescence was quantified by fluorescence plate reader using of 20 μL of each fraction.
Immunofluorescence (IF) staining.
After fixation, cells were blocked in IF buffer (3% BSA, 0.1% Triton in PBS) for 1hr at RT with rocking. Cells were then incubated with primary antibodies (See Table S1) in IF buffer for 1hr at RT, or overnight at 4°C, with rocking. Cells were washed twice with 1X DPBS before incubation with secondary antibody (1:1000) in IF buffer (1 hr, RT, with rocking, in the dark). Nuclei were stained using Hoescht 33342 (5 mins, RT, with rocking). Cells were washed twice with 1X PBS before imaging. All cell imaging was performed on a benchtop EVOS M5000 microscope.
RT-qPCR.
RNA was extracted using QIAGEN RNeasy Kit, followed by genomic DNA removal (TURBO DNA-free™ Kit) and quantification by NanoDrop One. RNA was normalized for reverse transcription using High-Capacity cDNA Reverse Transcription Kit. qPCR was performed with 5 ng cDNA, 10 μM forward and reverse primers (Supplementary Table 4) and 1X Fast SYBR Green Master Mix in 384-well plates on an Applied Biosystems 7900HT Fast Real Time PCR System.
Cell spreading assay.
24 hr after DsiRNA transfection, 24-well plates were coated in 1X poly-D-lysine (15 mins, RT), washed twice with 1X PBS before incubation with vitronectin (VN; 10 μg/mL; overnight at 4°C, rocking). The following day, the plate was washed twice with PBS and blocked (1 hr, 37°C, DMEM, 1% BSA). MDA-MB-231 cells harvested using non-enzymatic cell dissociation buffer and remodeled with SDC proteins (2 μΜ, 1 hr), with or without pre-incubation with cholPEGNTA (10 μM, 1 hr). Remodeling was performed in a 96-well round bottom plate, with 1X PBS washing between stages. Cells were resuspended in DMEM and plated onto VN at a density of 1.5×105 cells per well. Cells were allowed to adhere (2 hr, 37°C) before being washed with 1X PBS and fixed (4% PFA/PBS). Cell spreading was visualized by detection of actin filaments using rhodamine-conjugated phalloidin (1:40), followed by Hoechst staining on an EVOS M5000 fluorescence imager (Thermo Fisher Scientific).
Mouse and Human SDC1 sequence comparison:
Mouse (UniProtKB #P18828) and human (#P18827) SDC1 were aligned via protein-protein BLAST, yielding 76% overall sequence identify. Comparing the αvβ3 integrin binding site directly (mouse 92–119 LPAGEKPEEGEPVLHVEAEPGFTARDKE, human 93–120) shows 67% sequence similarity between the two species in this region. These sequences were compared by protein-protein BLAST with a BLOSUM62 scoring matrix.
Knockdown of SDC1 by DsiRNA.
Three Dicer substrate RNAs (DsiRNAs) from TRiFECTa DsiRNA kit designed for hSDC1 (hs.Ri.SDC1.13, Supplementary Table 5) were pooled to concentrations of 100 nM and 200 nM. 150 μL of DsiRNA was mixed with 150 μL of Opti-MEM supplemented with 9 μL Lipofectamine RNAiMAX (Invitrogen) and incubated (5 mins, RT). 250 μL of DsiRNA-lipid complex was added drop-wise to cells seeded in a 24-well plate. A scrambled DsiRNA (10nM) was used as a negative control, alongside a fluorescently labelled TYE563 transfection control (10 nM). Cells were harvested after 48hr.
Generation of SDC1KD cells.
MD-MBA-231 cells were seeded (2.5×105 cells/wel)l in a 6-well plate and allowed to adhere overnight (37°C, 5% CO2, ~18 hr) before spinoculation (2000 rpm, 2 hr, 37°C) with guideRNA targeting SDC1 exon 5 (guideRNA sequence GTTCCGGCGGTCAGGCTCCA) in lentiCRISPR v2 backbone ordered from the GenCRISPR™ Plasmid Collection. Cells were kept in lentiviral-supplemented DMEM overnight before being changed to DMEM/10% FBS (96 hr). Transduced cells were selected with 5 μg/mL puromycin before a clonal population was obtained by limiting dilution.
Quantification of cell spreading.
Microscopy images were analysed using ImageJ software. Individual channel microscopy images were converted to 8-bit greyscale and segmented with the threshold function, with holes filled. The ‘analyze particles’ function was then used (size 150-infinity) to quantify cell spreading. This was performed on both Hoechst and rhodamine-conjugated phalloidin images to quantify number of cells and extent of spreading, respectively. Extent of spreading was quantified by dividing the total rhodamine-positive area by the number of Hoechst-stained nuclei. Normalization was performed relative to the least spread cells (i.e. untreated SDC1 KD cells). Measurements were taken from (>10) images of distinct areas of cells across two biological replicates. Statistical analyses and graphs were generated using GraphPad Prism 9.
Live cell proximity labeling.
Proximity tagging was adapted from previously published procedures15,37. mESCs seeded on gelatin were treated with cholPEGNTA (10 μM, 1 hr, 37°C), washed twice with PBS, and further incubated with A-SDC145,47 constructs (10 μM, 1 hr, 37°C). After further PBS washes, cells were incubated with biotin phenol (500 μM in KO-DMEM, 30 mins, 37°C), followed by H2O2 (1 mM, 1 min, RT). The reaction was stopped with 3X washes of quenching solution (5 mM Trolox, 10 mM sodium ascorbate, and 10 mM sodium azide in PBS). mESCs were then fixed and probed for biotinylated interactors by Cy5-streptavidin and imaged by fluorescence microscopy.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its Supplementary Information files. Please contact the corresponding author (M.H.) for access of raw data, which will be made available upon reasonable request.
Extended Data
Extended Data Fig. 1.
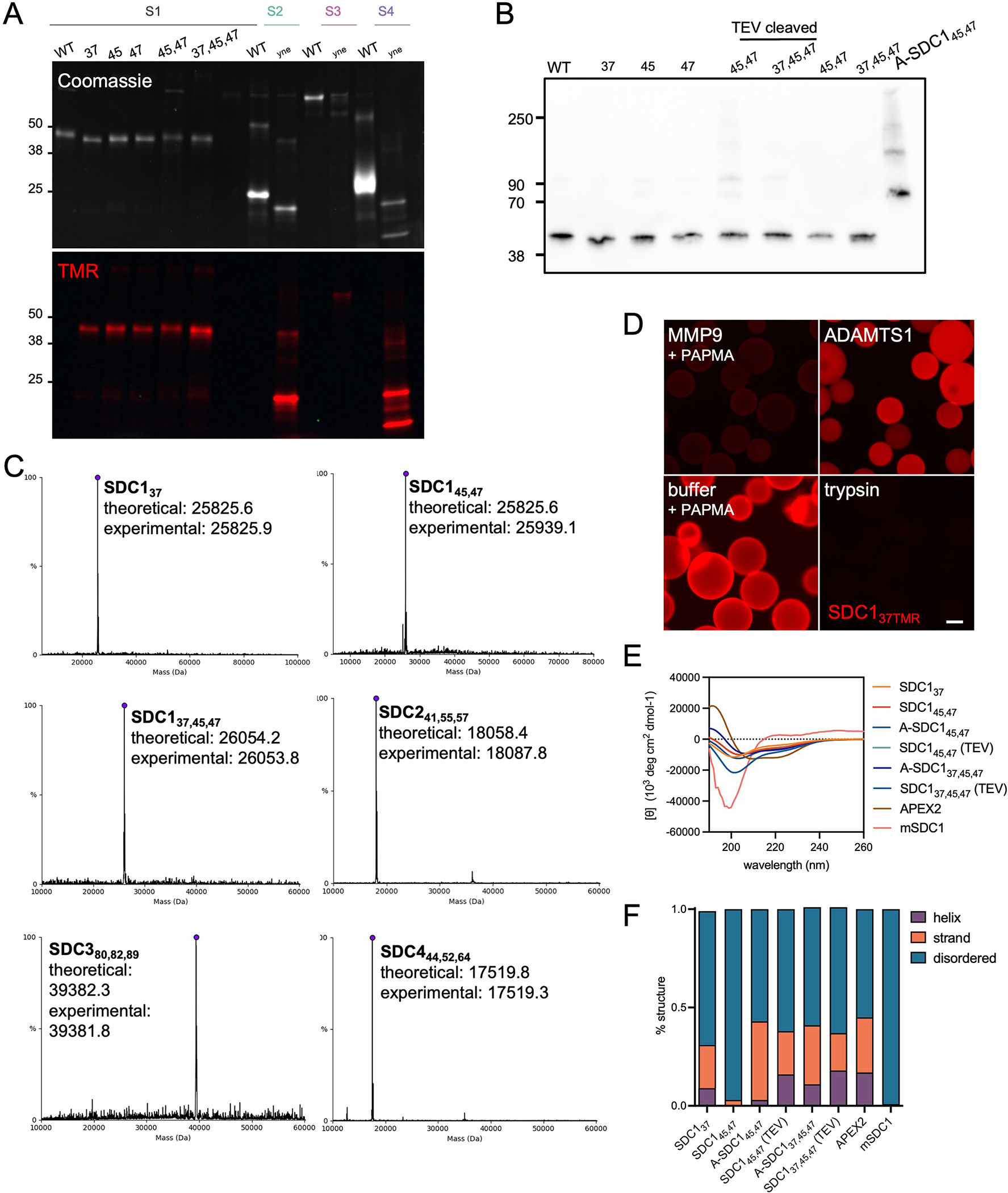
Characterization of engineered proteoglycan proteins (A) SDS-PAGE analysis and subsequent Coomassie (top) and fluorescence (bottom) imaging of SDC1, SDC2, SDC3 and SDC4 ectodomains reacted with TMR-azide confirms successful incorporation of pPY. As highly disordered proteins, SDCs run anomalously by SDS-PAGE. Representative of two technical replicates, molecular weight ladder in kDa. (B) Western blot analyses of SDC1 ectodomains probed with anti-SDC1 clone 281–2. (C) Intact mass spectrometry of SDC ectodomains confirms expected masses and demonstrates fidelity of pPY incorporation. Representative of two technical replicates, molecular weight ladder in kDa. N-terminal MQ residues were missing in some constructs, consistent with production and processing by E. coli. (D) Fluorescence microscopy images of beads following treatment with proteases or buffer with p-aminophenylmercuric acetate (PAPMA, required to activate MMP) shows SDC137TMR is only cleaved by MMP9 and trypsin (positive control). The TMR fluorophore is released from the beads after being washed, as it does not contain a poly-His tag sequence that enables the C-terminal fragment to remain anchored onto the nickel bead. (E) Circular dichroism (CD) spectrum of SDC1 ectodomains show >57% disordered conformations. APEX2 fusion proteins (A-SDC145,47 and A-SDC137,45,47) display enhanced helical structure relative to their non-fused counterparts due to the helical APEX220 domain. Ten readings were taken per sample to create an average CD spectrum. (F) Quantification of predicted secondary structures, generated by CAPITO analysis57.
Extended Data Fig. 2.

Characterization of recombinant and azido-GAG (A) Graphical representation of heparan sulfate (HS) glycosaminoglycan biosynthesis in the endoplasmic reticulum (ER) and Golgi apparatus. The common HS and CS tetrasaccharide linker (Xyl-Gal-Gal-GlcA) is synthesized in the ER before being elongated by Ext1/2 enzyme complexes to create the HS repeating disaccharide motif. (B) Synthetic strategy to produce azidoxyloside 2. (C) Xyloside 2 was docked (AutoDock Vina) into the active site of β4GalT7 in complex with the UDP-Gal donor (teal, PDB ID 4M4K). Top poses for the docked compounds showed positioning of the xyloside C4 oxygen within 3–4 Å of the C1 atom on the galactose ring of UDP-galactose. The binding affinity of the five lowest energy poses for each simulation was averaged. Comparison of the average binding energies showed the addition of azido group into the aromatic ring component did not exact a substantial binding penalty. (D) Suspension CHO (CHO-S) cells are incubated with 2 (400 μM; 72 hr) and conditioned media is harvested for isolation of soluble recombinant, azido-GAGs (azGAGs). (E) SDS-PAGE analysis using AF546-appended rGAGs confirms that azide-primed GAGs are present mostly in the conditioned medium (CM) compared to cell extracts (E). Selective degradation of rGAGs by chondroitinase (Csase) results in a collapsed signal, indicative of our CM consisting primarily of CS. Representative of two biological replicates, molecular weight ladder in kDa. (F) azGAGs were analyzed by SDS-PAGE after copper-catalyzed azide-alkyne cycloaddition (CuAAC) with AF546 fluorophore and digestion with chondroitinase (CSase) or heparinase (HSase). The presence of a collapsed lower molecular-weight band upon CSase digestion suggests large amounts of primed CS. Representative of two biological replicates, molecular weight ladder in kDa. (G) Through dibenzocyclooctyne (DBCO) bead capture and subsequent disaccharide analysis, the proportion of azide-functionalized HS (orange) and CS (green) in the rGAG mixture can be quantified, with 27% being azido-HS (azHS), which mimics endogenous GAG ratios. (H) Standard curve of HS disaccharides. (I) Disaccharide analysis of endogenous HS (grey) from untreated cells, soluble non-primed HS (blue), azHS (orange) show similar sulfation profiles. Similarly, functionalization of free heparin (HEP, green) to azido-primed heparin (azHEP, yellow) did not affect sulfation. As expected, heparin is substantially more sulfated than HS. HS chains for mSDC1 (pink) were also analyzed. (J) Standard curve of CS disaccharides. (K) Disaccharide analysis of CS from endogenous (grey), azCS (green), rCS (blue) and mSDC1 (pink). (L) Proportion of CS and HS GAGs isolated from commercial, mammalian expressed mSDC1, calculated by disaccharide analysis. Analyses and graphs generated with GraphPad Prism 9. Bar graphs represent means and error bars represent SEM representative from two technical replicates.
Extended Data Fig. 3.
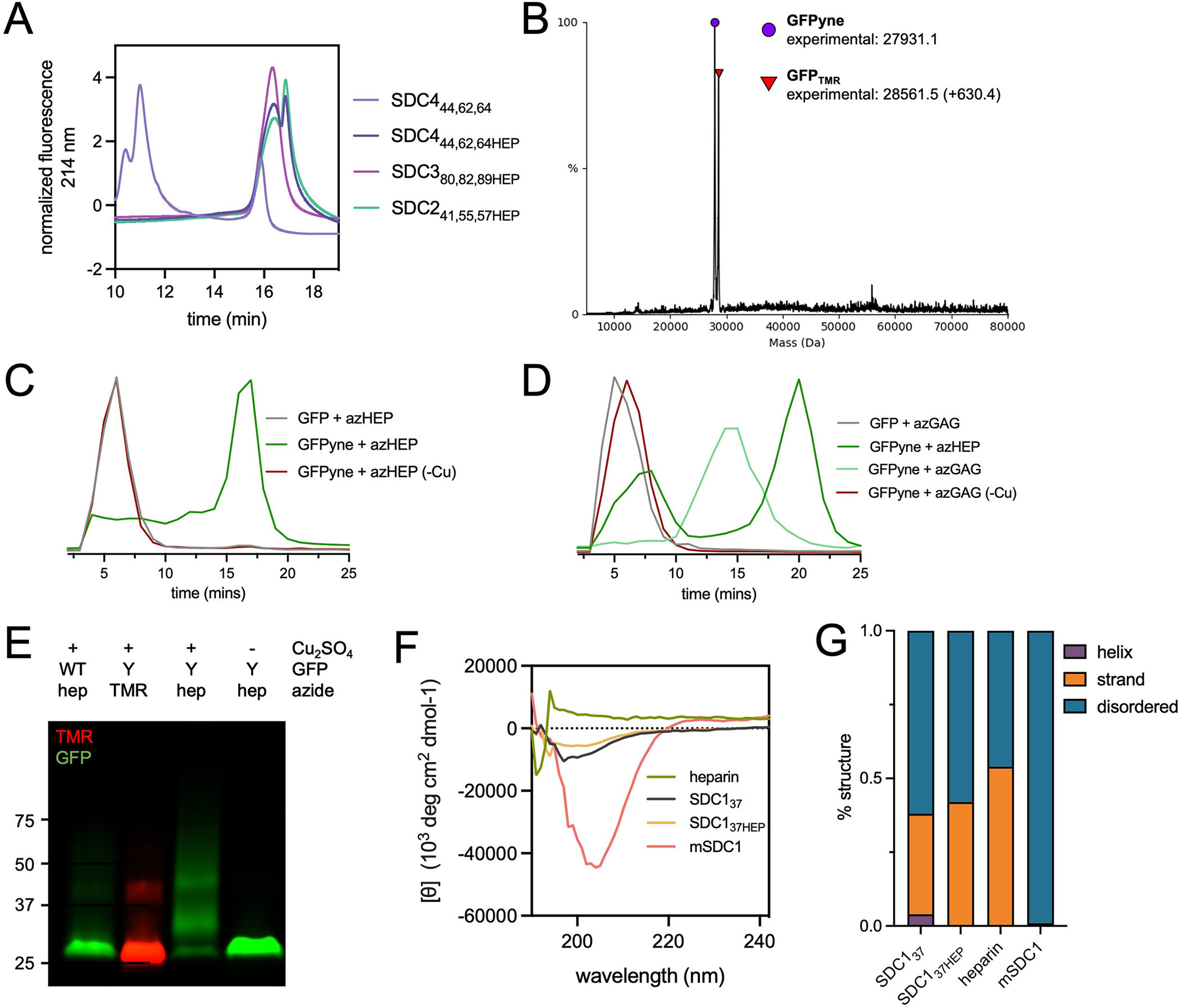
Glycoconjugation of pPY-modified proteins by CuAAC (A) Weak anion exchange (WAX) traces demonstrating successful conjugation of azHEP to form SDC241,55,57HEP (mint), SDC380,82,89HEP (pink) and SDC444,62,64HEP (dark purple). SDC444,62,64 core protein (light purple) included as reference point for SDC core proteins (. (B) Intact mass spectrometry of pPY-modified GFP (GFPyne(His)6), used as a model protein, confirms the appropriate mass shift (+630.4 Da) when reacted with tetramethylrhodamine (TMR)-azide. (C) WAX traces after reaction of GFPyne(His)6 with azHEP demonstrates a shift to the more anionic product (dark green). When the reaction is performed without copper (-Cu, red), the trace overlaps with WT GFP (grey). (D) WAX traces after reaction with azGAG (green) demonstrate a less anionic glycoconjugate than GFPyne + azHEP (dark green). Copper-free controls overlap with WT GFP peak. (E) SDS-PAGE analysis of WT and GFPyne(His)6 (Y) confirms the incorporation of pPY by TMR detection only in Y. azHEP conjugation results in a higher molecular weight smear, only in the presence of copper. Representative of two technical replicates, molecular weight ladder indicates kDa. (F) Circular dichroism spectra of heparin (red), SDC137 (grey), SDC137HEP (orange) and HEK239T expressed mSDC1 (pink). (G) Analysis of the predicted structures from CD spectra indicates that glycosylation of SDC137 does not impart a change, or increase, in protein structures and native, glycosylated SDC1 is highly disordered.
Extended Data Fig. 4.
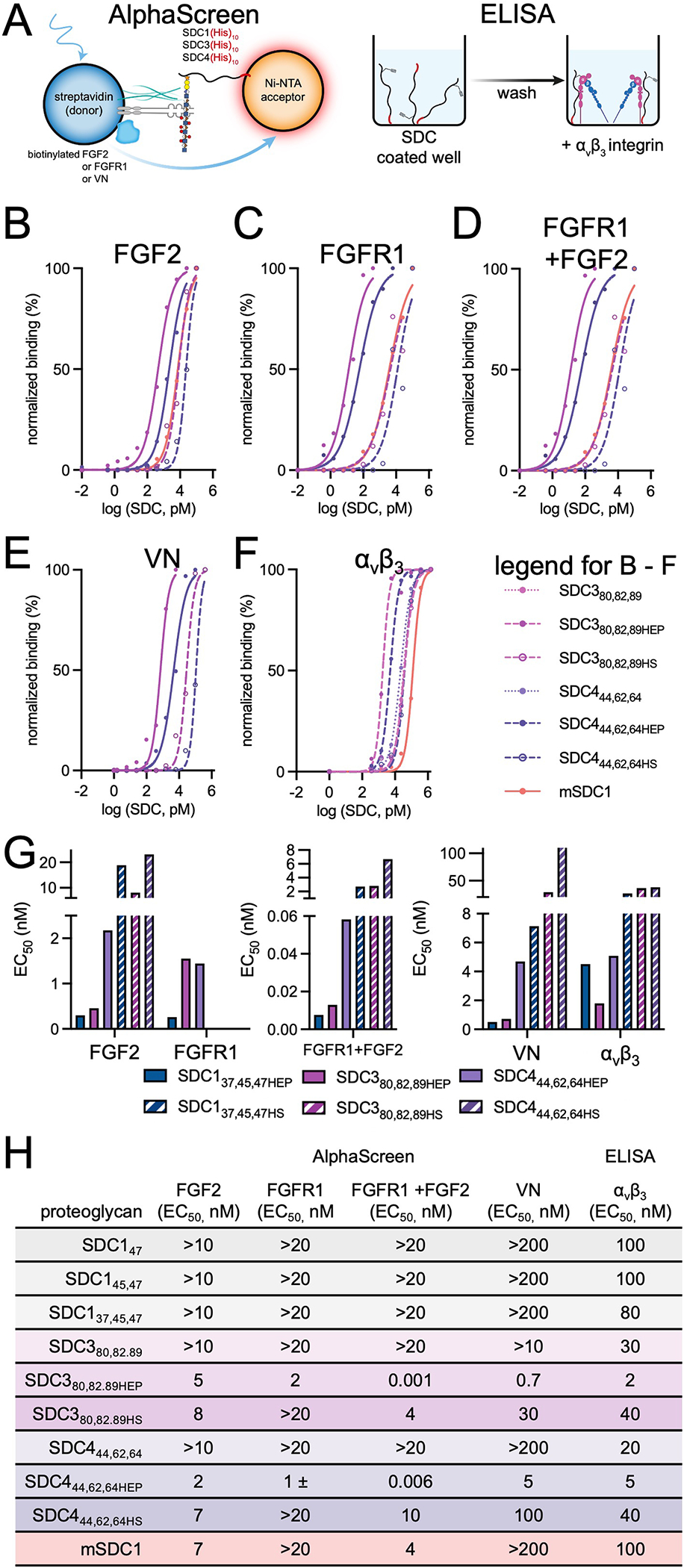
Additional AlphaScreen and ELISA binding data, including SDC380,82,89 and SDC444,62,64 glycoconjugates (A) Left: Graphical depiction of AlphaScreen assay using biotinylated interactors (FGF2, FGFR, vitronectin (VN)) attached to streptavidin donor beads and Ni-NTA acceptor beads which bind His-tagged SDC proteins and present them uniformly. Right: Graphical depiction of ELISA performed with randomly oriented immobilized SDC1 and recombinant integrin αvβ3. (B) AlphaScreens performed with biotinylated FGF2. (C) AlphaScreen performed with biotinylated FGFR1. (D) AlphaScreen performed with biotinylated FGFR1 with the addition of non-tagged FGF2 to stimulate ternary complex formation. (E) AlphaScreen performed with His-tagged SDC1 ectodomains and biotinylated vitronectin (VN). (F) ELISA performed with immobilized SDC1 and recombinant integrin αvβ3. Curves represent core proteins (filled, dotted), heparin conjugates (filled, dashed) and HS conjugates (open, dashed) of SDC380,82,89 (purple) and SDC444,62,64 (pink). (G) Bar graphs of EC50 from trivalent heparin (solid) and HS (dashed) SDC137,45,47, −380,,82,89 and −444,62,64 glycoconjugates. Graphs were fitted using non-linear regression plotted using GraphPad Prism 9. Bar graphs represent means and error bars represent SEM generated from two technical replicates. (H) Tabulated apparent affinity constants (EC50) shown for SDC1 core proteins, SDC3 and SDC4 constructs, and heparan sulfate proteoglycan (HSPG)-binding proteins; FGF2, FGFR vitronectin (VN) and integrin αvβ3. mSDC1 represents HEK239T expressed recombinant mouse SDC1 ectodomain. EC50 tabulated at 1 significant figure.
Extended Data Fig. 5.
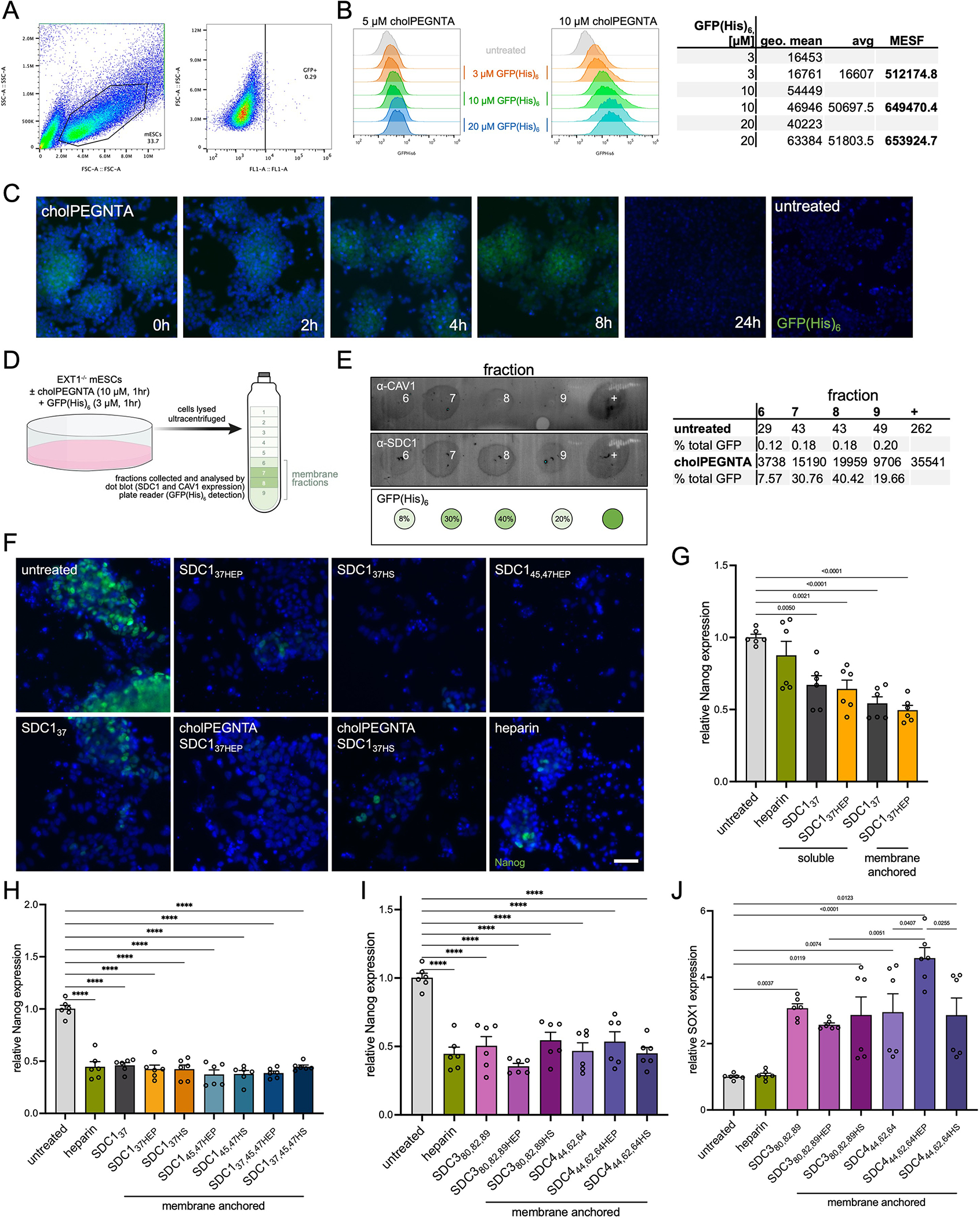
Characterization of mESC remodeling by cholPEGNTA and additional differentiation data (A) Flow cytometry gating of EXT1−/− mESCs incubated with 10 μM cholPEGNTA for single cells (left), and GFP-negative population (right). (B) Treatment of EXT1−/− mESCs with 10 μM cholPEGNTA alone (untreated), or further incubation with varying concentrations of GFP(His)6 (3, 10 and 20 μM) demonstrates dose-dependent fluorescence. Comparisons of geometric mean GFP fluorescence from cells treated with 10 μM cholPEGNTA to GFP calibration beads allows for quantification of molecules of equivalent soluble fluorochrome (MESF) of protein on each cell. Saturation is observed at 10 μM GFP(His)6 at ~650,000 MESF. (C) Representative microscopy image of EXT1−/− cells treated with 10 μM cholPEGNTA for 1 hr and fixed at the indicated time points (hours after end of cholPEGNTA treatment) with 4% PFA/PBS. Cells were incubated with 10 μM GFP(His)6 and 100 μM Ni(OAc)2 for 1 hr in PBS before Hoechst staining. NTA headgroup remains accessible to His-tagged proteins for at least 8 hr. Data representative of two biological replicates. (D) Cartoon representation of EXT1−/− mESC treatment and ultracentrifugation for isolation of lipid rafts/caveolae in membrane fractions (green). (E) Dot blots from cholPEGNTA and GFP(His)6 treated EXT1−/− mESCs demonstrates significant overlap of CAV-1 (top) and SDC1 (middle) in lipid rafts. GFP(His)6 (bottom) was detected by fluorescence plate reader and quantified as a percentage of cholPEGNTA in each fraction (right). (F) Representative fluorescence microscopy images of mESCs on D6 of neuronal differentiation. Untreated EXT1−/− cells retain high Nanog expression, indicative of a pluripotent state, whilst mESCs differentiated with SDC1 constructs or soluble heparin lose Nanog expression (green). (G) RT-qPCR analysis of differentiated cells demonstrates decreased Nanog expression compared to untreated. Cells remodeled with cholPEGNTA for cell surface display of SDC1 proteins had lesser Nanog expression. (H) RT-qPCR analysis demonstrates significantly decreased expression of pluripotency marker Nanog at D6 upon treatment with heparin or SDC1 constructs. (I) Similar results to SDC1 are observed for Nanog expression when cells are treated with SDC3 and SDC4 proteins. (J) RT-qPCR analysis at D6 shows increased SOX1 expression when cells are treated with SDC3 and SDC4 proteins, both deglycosylated and as glycoconjugates. SDC4 shows significant differences between core protein (light purple) and its azHEP conjugate (purple), and between azHEP and azHS conjugates (dark purple). All experiments performed in technical triplicate in two biological replicates. One-sided ANOVA with Tukey’s post-hoc, p values indicated on graph, (****) p <0.0001. Bar graphs represent means and error bars represent SEM.
Extended Data Fig. 6.
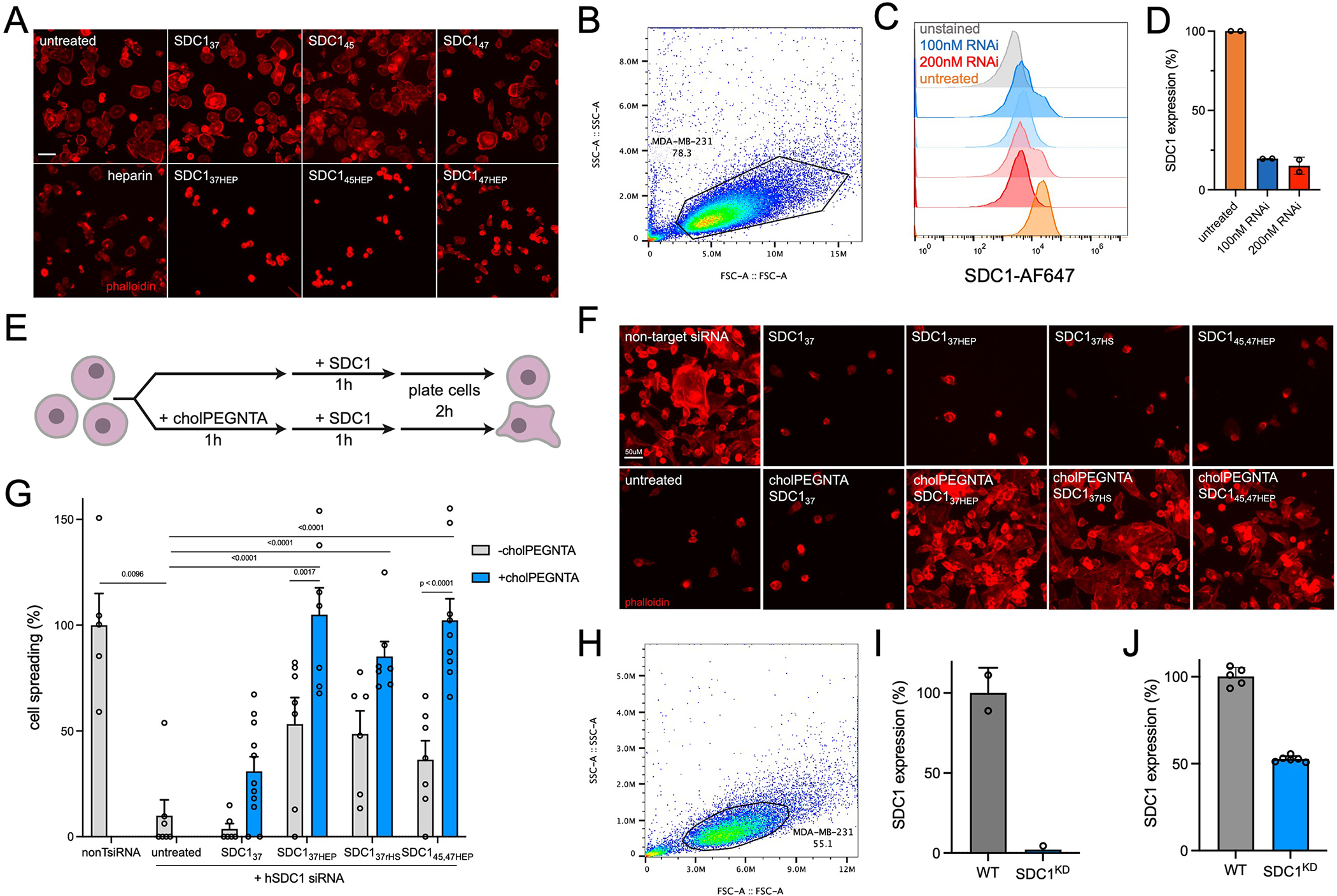
Characterization of SDC1 knockdown by RNAi and CRISPR (SDC1KD) and additional cell spreading experiments (A) Representative microscopy images from soluble addition of monovalent SDC1 to wild-type MDA-MB-231. Data representative of three biological replicates. (B) Flow cytometry gating of wild-type MDA-MB-231 cells. (C) MDA-MB-231 cells treated with 100 nM (blue) or 200 nM (red) pooled SDC1 dsRNAi exhibit reduced SDC1 expression compared to non-targeting DsiRNA control (orange). Unstained cells (gray) are those incubated with secondary antibody only. (D) Quantification of SDC1 expression in MDA-MB-231 cells after knockdown with 100 nM and 200 nM pooled DsiRNA as a percent of non-targeting DsiRNA control. (E) Cartoon depiction of remodeling strategy of MDA-MB-231 cells, performed whilst cells are suspended in 96-well round bottom plates. Remodeled cells are plated on vitronectin-coated surfaces and allowed to spread for 2 hr. (F) Representative microscopy images of MDA-MB-231 cells treated with 200 nM hSDC1 siRNA or non-target. Only cell surface, glycosylated SDC1 proteins can rescue cell spreading. Data representative of three biological replicates. (G) Quantification of cell spreading on vitronectin. (H) Flow cytometry gating of wild-type MDA-MB-231 cells. (I) Quantification of SDC1 protein levels in CRISPR-generated SDC1KD cells by flow cytometry. Data represents the mean fluorescence intensity noramalised to WT MDA-MB-231 cells. (J) qRT-PCR confirms knockdown of SDC1 in SDC1KD cells using two primer sets targeting SDC1. Data presented is fold change of SDC1 mRNA as a percent of WT MDA-MB-231 cells, as calculated by delta delta CT. Bar graphs represent means and error bars represent SEM. One-sided ANOVA with Tukey’s post-hoc test with Šidák correction for multiple comparisons was performed; p values indicated on graph, (****) p <0.0001. For each condition, n >10 images examined across two biological replicates.
Extended Data Fig. 7.
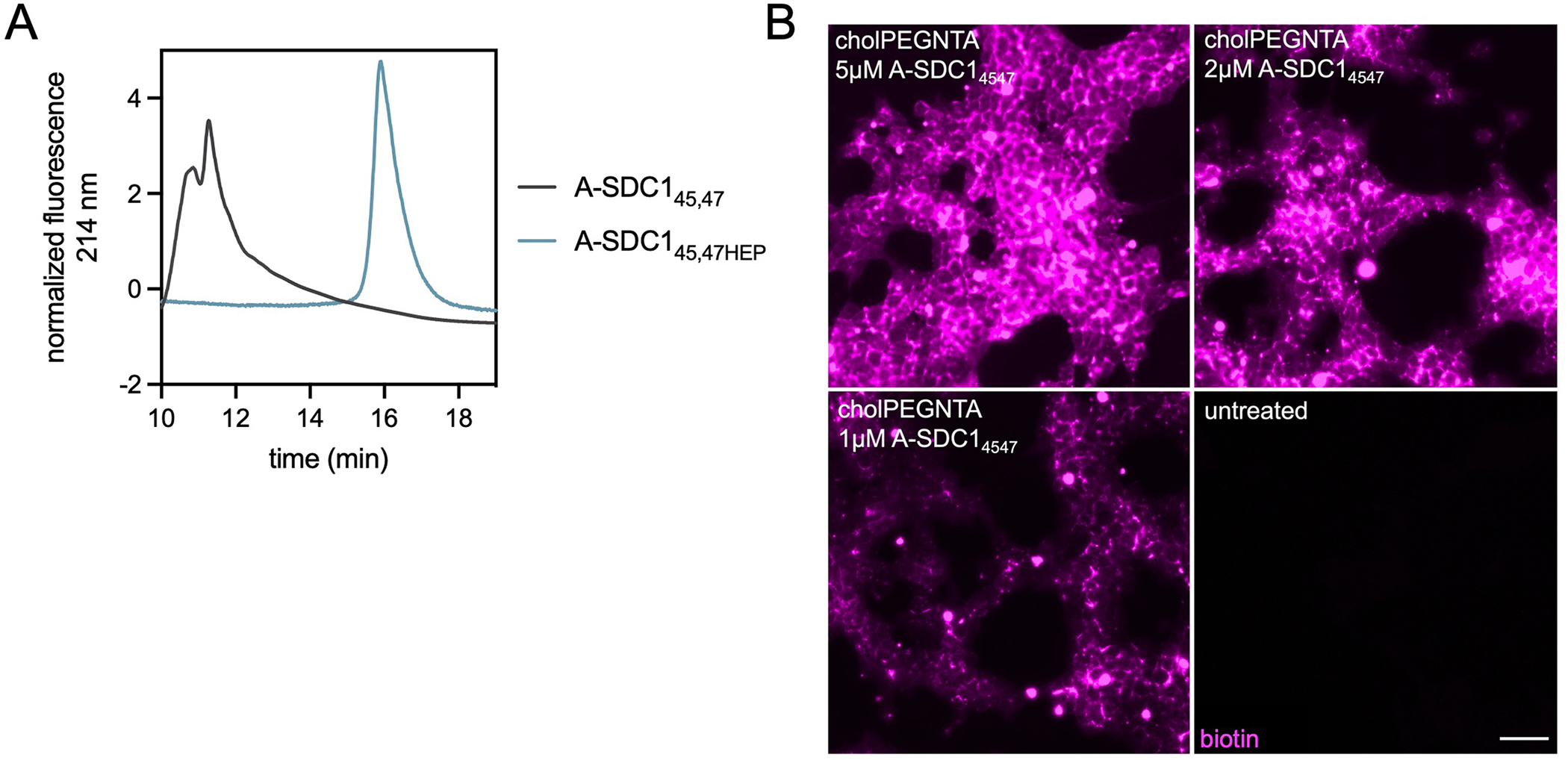
APEX-SDC145,47 glycoconjugation yields and additional proximity labeling data (A) Weak anion exchange (WAX) traces demonstrating successful conjugation of azHEP to A-SDC145,47 (grey) and formation of the more anionic product A-SDC145,47HEP (blue). CuAAC performed at quantitative yields. (B) Dose dependent fluorescence generated after live cell proximity labeling with cell surface (10 μM cholPEGNTA) A-SDC145,47 at 5 μM, 2 μM and 1 μM. Biotinylation is detected by Cy5-streptavidin (pink) with fluorescence mostly localized to cell surfaces. Representative images from three biological replicates.
Supplementary Material
Acknowledgements
We thank Dr Sourav Chatterjee for assistance in preparing compound 2. We thank Farin S. Ahmed and Dr John A. Hammond of the Scripps Research Biophysics and Biochemistry Core and Douglas S. Kojetin for assistance with circular dichroism experiments. This work, T.N.S. (HD090292-S1), M.C., and M.L.H are supported by the NIH K99/R00 Pathway to Independence Award (R00-HD090292). The acquisition of 600MHz NMR spectra was supported by NIH S10 OD021550. M.L.H is grateful for the support and scientific counsel of Profs. Kamil Godula and Jeffrey Esko for this work.
Footnotes
Competing Interests Statement
The authors declare no competing interests.
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41589-022-01023-5.
References:
- 1.Hacker U, Nybakken K & Perrimon N Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol 6, 530–541, doi: 10.1038/nrm1681 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Critcher M, O’Leary T & Huang ML Glycoengineering: scratching the surface. Biochem J 478, 703–719, doi: 10.1042/BCJ20200612 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Langford JK, Stanley MJ, Cao D & Sanderson RD Multiple heparan sulfate chains are required for optimal syndecan-1 function. J Biol Chem 273, 29965–29971, doi: 10.1074/jbc.273.45.29965 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Bernfield M et al. Biology of the Syndecans - a Family of Transmembrane Heparan-Sulfate Proteoglycans. Annu Rev Cell Biol 8, 365–393, doi:DOI 10.1146/annurev.cb.08.110192.002053 (1992). [DOI] [PubMed] [Google Scholar]
- 5.Zhang YH et al. Targeting of Heparanase-modified Syndecan-1 by Prosecretory Mitogen Lacritin Requires Conserved Core GAGAL plus Heparan and Chondroitin Sulfate as a Novel Hybrid Binding Site That Enhances Selectivity. Journal of Biological Chemistry 288, 12090–12101, doi: 10.1074/jbc.M112.422717 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gama CI et al. Sulfation patterns of glycosaminoglycans encode molecular recognition and activity. Nat Chem Biol 2, 467–473, doi: 10.1038/nchembio810 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Manon-Jensen T, Itoh Y & Couchman JR Proteoglycans in health and disease: the multiple roles of syndecan shedding. FEBS J 277, 3876–3889, doi: 10.1111/j.1742-4658.2010.07798.x (2010). [DOI] [PubMed] [Google Scholar]
- 8.Salmivirta M, Lidholt K & Lindahl U Heparan sulfate: a piece of information. FASEB J 10, 1270–1279, doi: 10.1096/fasebj.10.11.8836040 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Nadanaka S, Clement A, Masayama K, Faissner A & Sugahara K Characteristic hexasaccharide sequences in octasaccharides derived from shark cartilage chondroitin sulfate D with a neurite outgrowth promoting activity. Journal of Biological Chemistry 273, 3296–3307, doi:DOI 10.1074/jbc.273.6.3296 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Brittis PA, Canning DR & Silver J Chondroitin Sulfate as a Regulator of Neuronal Patterning in the Retina. Science 255, 733–736, doi:DOI 10.1126/science.1738848 (1992). [DOI] [PubMed] [Google Scholar]
- 11.Forsberg E & Kjellen L Heparan sulfate: lessons from knockout mice. J Clin Invest 108, 175–180, doi: 10.1172/JCI13561 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim JM, Lee K, Kim MY, Shin HI & Jeong D Suppressive effect of syndecan ectodomains and N-desulfated heparins on osteoclastogenesis via direct binding to macrophage-colony stimulating factor. Cell Death Dis 9, doi:ARTN 1119 10.1038/s41419-018-1167-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schultz KC et al. A genetically encoded infrared probe. J Am Chem Soc 128, 13984–13985, doi: 10.1021/ja0636690 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Csizmar CM, Petersburg JR & Wagner CR Programming Cell-Cell Interactions through Non-genetic Membrane Engineering. Cell Chem Biol 25, 931–940, doi: 10.1016/j.chembiol.2018.05.009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lam SS et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12, 51–54, doi: 10.1038/nmeth.3179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bundy BC & Swartz JR Site-Specific Incorporation of p-Propargyloxyphenylalanine in a Cell-Free Environment for Direct Protein-Protein Click Conjugation. Bioconjugate Chem 21, 255–263, doi: 10.1021/bc9002844 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Gondelaud F et al. Extended disorder at the cell surface: The conformational landscape of the ectodomains of syndecans. Matrix Biol Plus 12, 100081, doi: 10.1016/j.mbplus.2021.100081 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fritz TA & Esko JD Xyloside priming of glycosaminoglycan biosynthesis and inhibition of proteoglycan assembly. Methods Mol Biol 171, 317–323, doi: 10.1385/1-59259-209-0:317 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Dahbi S et al. Synthesis of a library of variously modified 4-methylumbelliferyl xylosides and a structure-activity study of human beta 4GalT7. Org Biomol Chem 15, 9653–9669, doi: 10.1039/c7ob02530k (2017). [DOI] [PubMed] [Google Scholar]
- 20.Carnachan SM & Hinkley SFR Heparan Sulfate Identification and Characterisation: Method II. Enzymatic Depolymerisation and SAX-HPLC Analysis to Determine Disaccharide Composition. Bio-Protocol 7, doi:ARTN e2197 10.21769/BioProtoc.2197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trieger GW, Verespy S, Gordts PLSM & Godula K Efficient Synthesis of Heparinoid Bioconjugates for Tailoring FGF2 Activity at the Stem Cell–Matrix Interface. Bioconjugate Chemistry 30, 833–840, doi: 10.1021/acs.bioconjchem.8b00921 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burgess RR A brief practical review of size exclusion chromatography: Rules of thumb, limitations, and troubleshooting. Protein Expr Purif 150, 81–85, doi: 10.1016/j.pep.2018.05.007 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Ibrahimi OA, Zhang F, Hrstka SCL, Mohammadi M & Linhardt RJ Kinetic model for FGF, FGFR, and proteoglycan signal transduction complex assembly. Biochemistry 43, 4724–4730 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Wilkins-Port CE, Sanderson RD, Tominna-Sebald E & McKeown-Longo PJ Vitronectin’s basic domain is a syndecan ligand which functions in trans to regulate vitronectin turnover. Cell Commun Adhes 10, 85–103, doi: 10.1080/cac.10.2.85.103 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Beauvais DM, Burbach BJ & Rapraeger AC The syndecan-1 ectodomain regulates alphavbeta3 integrin activity in human mammary carcinoma cells. J Cell Biol 167, 171–181, doi: 10.1083/jcb.200404171 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beauvais DM & Rapraeger AC Syndecan-1-mediated cell spreading requires signaling by αvβ3 integrins in human breast carcinoma cells. Experimental Cell Research 286, 219–232, doi: 10.1016/s0014-4827(03)00126-5 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Powell AK, Fernig DG & Turnbull JE Fibroblast growth factor receptors 1 and 2 interact differently with heparin/heparan sulfate. Implications for dynamic assembly of a ternary signaling complex. J Biol Chem 277, 28554–28563, doi: 10.1074/jbc.M111754200 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Da Silva MS et al. Heparin Modulates Integrin-Mediated Cellular Adhesion: Specificity of Interactions with α and β Integrin Subunits. Cell Communication & Adhesion 10, 59–67, doi: 10.1080/cac.10.2.59.67 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Johnson CE et al. Essential alterations of heparan sulfate during the differentiation of embryonic stem cells to Sox1-enhanced green fluorescent protein-expressing neural progenitor cells. Stem Cells 25, 1913–1923, doi: 10.1634/stemcells.2006-0445 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Boonyarattanakalin S, Athavankar S, Sun Q & Peterson BR Synthesis of an artificial cell surface receptor that enables oligohistidine affinity tags to function as metal-dependent cell-penetrating peptides. J Am Chem Soc 128, 386–387, doi: 10.1021/ja056126j (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woods EC, Yee NA, Shen J & Bertozzi CR Glycocalyx Engineering with a Recycling Glycopolymer that Increases Cell Survival In Vivo. Angew Chem Int Ed Engl 54, 15782–15788, doi: 10.1002/anie.201508783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hudak A et al. Contribution of syndecans to cellular uptake and fibrillation of alpha-synuclein and tau. Sci Rep 9, 16543, doi: 10.1038/s41598-019-53038-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perez-Verdaguer M et al. Caveolin interaction governs Kv1.3 lipid raft targeting. Sci Rep 6, 22453, doi: 10.1038/srep22453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chatterjee S, Stephenson TN, Michalak AL, Godula K & Huang ML Silencing glycosaminoglycan functions in mouse embryonic stem cells with small molecule antagonists. Method Enzymol 626, 249–270, doi: 10.1016/bs.mie.2019.06.023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hung V et al. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat Protoc 11, 456–475, doi: 10.1038/nprot.2016.018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vilen Z, Joeh E, Critcher M, Parker CG & Huang ML Proximity tagging identifies the glycan-mediated glycoprotein interactors of galectin-1 in muscle stem cells. ACS Chem Biol, doi: 10.1021/acschembio.1c00313 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joeh E et al. Mapping glycan-mediated galectin-3 interactions by live cell proximity labeling. Proc Natl Acad Sci U S A 117, 27329–27338, doi: 10.1073/pnas.2009206117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stepp MA, Pal-Ghosh S, Tadvalkar G & Pajoohesh-Ganji A Syndecan-1 and Its Expanding List of Contacts. Adv Wound Care 4, 235–249, doi: 10.1089/wound.2014.0555 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tully SE et al. A chondroitin sulfate small molecule that stimulates neuronal growth. Journal of the American Chemical Society 126, 7736–7737, doi: 10.1021/ja0484045 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Noti C, de Paz JL, Polito L & Seeberger PH Preparation and use of microarrays containing synthetic heparin oligosaccharides for the rapid analysis of heparin-protein interactions. Chem-Eur J 12, 8664–8686, doi: 10.1002/chem.200601103 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Wang Z et al. Preactivation-Based, One-Pot Combinatorial Synthesis of Heparin-like Hexasaccharides for the Analysis of Heparin-Protein Interactions. Chem-Eur J 16, 8365–8375, doi: 10.1002/chem.201000987 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang ML, Smith RAA, Trieger GW & Godula K Glycocalyx Remodeling with Proteoglycan Mimetics Promotes Neural Specification in Embryonic Stem Cells. Journal of the American Chemical Society 136, 10565–10568, doi: 10.1021/ja505012a (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pulsipher A, Griffin ME, Stone SE, Brown JM & Hsieh-Wilson LC Directing Neuronal Signaling through Cell-Surface Glycan Engineering. Journal of the American Chemical Society 136, 6794–6797, doi: 10.1021/ja5005174 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang WZ et al. Chemical synthesis of human syndecan-4 glycopeptide bearing O-, N-sulfation and multiple aspartic acids for probing impacts of the glycan chain and the core peptide on biological functions. Chemical Science 11, 6393–6404, doi: 10.1039/d0sc01140a (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiu H et al. A mutant-cell library for systematic analysis of heparan sulfate structure-function relationships. Nature Methods 15, 889–+, doi: 10.1038/s41592-018-0189-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen YH et al. The GAGOme: a cell-based library of displayed glycosaminoglycans. Nat Methods 15, 881–888, doi: 10.1038/s41592-018-0086-z (2018). [DOI] [PubMed] [Google Scholar]
- 47.Lang K & Chin JW Cellular Incorporation of Unnatural Amino Acids and Bioorthogonal Labeling of Proteins. Chem Rev 114, 4764–4806, doi: 10.1021/cr400355w (2014). [DOI] [PubMed] [Google Scholar]
- 48.Nguyen TA, Cigler M & Lang K Expanding the Genetic Code to Study Protein-Protein Interactions. Angew Chem Int Edit 57, 14350–14361, doi: 10.1002/anie.201805869 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Chatterjee A, Sun SB, Furman JL, Xiao H & Schultz PG A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry 52, 1828–1837, doi: 10.1021/bi4000244 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eriksson AS & Spillmann D The Mutual Impact of Syndecan-1 and Its Glycosaminoglycan Chains-A Multivariable Puzzle. J Histochem Cytochem 60, 936–942, doi: 10.1369/0022155412460242 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aquino RS, Teng YH & Park PW Glycobiology of syndecan-1 in bacterial infections. Biochem Soc Trans 46, 371–377, doi: 10.1042/BST20170395 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park PW, Pier GB, Hinkes MT & Bernfield M Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature 411, 98–102, doi: 10.1038/35075100 (2001). [DOI] [PubMed] [Google Scholar]
- 53.Beauvais DM, Ell BJ, McWhorter AR & Rapraeger AC Syndecan-1 regulates alphavbeta3 and alphavbeta5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J Exp Med 206, 691–705, doi: 10.1084/jem.20081278 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao J, Xu Y, Liu J & Huang X Convergent chemoenzymatic synthesis and biological evaluation of a heparan sulfate proteoglycan syndecan-1 mimetic. Chem Commun (Camb) 57, 3407–3410, doi: 10.1039/d1cc00796c (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang P et al. Heparan Sulfate Organizes Neuronal Synapses through Neurexin Partnerships. Cell 174, 1450–+, doi: 10.1016/j.cell.2018.07.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Noborn F et al. Identification of Chondroitin Sulfate Linkage Region Glycopeptides Reveals Prohormones as a Novel Class of Proteoglycans. Mol Cell Proteomics 14, 41–49, doi: 10.1074/mcp.M114.043703 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wiedemann C, Bellstedt P & Gorlach M CAPITO--a web server-based analysis and plotting tool for circular dichroism data. Bioinformatics 29, 1750–1757, doi: 10.1093/bioinformatics/btt278 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Siegbahn A et al. Exploration of the active site of β4GalT7: modifications of the aglycon of aromatic xylosides. Organic & Biomolecular Chemistry 13, 3351–3362, doi: 10.1039/C4OB02632B (2015). [DOI] [PubMed] [Google Scholar]
- 59.Trott O & Olson AJ Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J Comput Chem 31, 455–461, doi: 10.1002/jcc.21334 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang J et al. Anti-beta2-microglobulin monoclonal antibodies induce apoptosis in myeloma cells by recruiting MHC class I to and excluding growth and survival cytokine receptors from lipid rafts. Blood 110, 3028–3035, doi: 10.1182/blood-2007-06-094417 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper and its Supplementary Information files. Please contact the corresponding author (M.H.) for access of raw data, which will be made available upon reasonable request.