

Abstract
We disclose here practical strategies toward the synthesis of morpholines and Claisen rearrangement products based on the divergent reactivity of a common halonium intermediate. These reactions employ widely available alkenes in a Lewis acid-catalyzed halo-etherification process that can then transform them into the desired products with exceptional regioselectivity for both activated and unactivated olefins. Our mechanistic probe reveals an interesting regiochemical kinetic resolution process.
Graphical Abstract
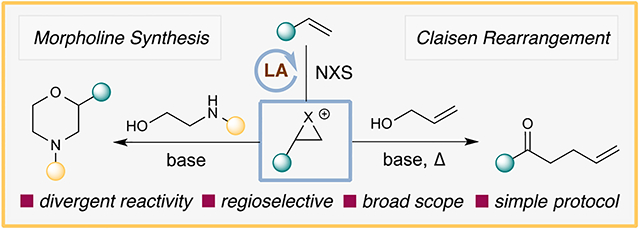
INTRODUCTION
Three-membered cyclic halonium (haliranium) is a classic intermediate that has been widely used in organic synthesis.1 Structural elucidations of these intermediates by Olah, Wynberg, Brown, Nugent, Kochi, and others have significantly benefited our understanding of these unusual structures.2 Variable-temperature 1H NMR studies by Brown and elegant enantiospecific acetolysis experiments by Denmark further unveiled the intricacy of haloniums in olefin-to-olefin transfer processes, illuminating the challenges often encountered in asymmetric halofunctionalizations.3 More recently, haloniums have been used as catalytic templates in olefin difunctionalizations by Muñiz4 and Zhdankin5 and in our recent works.6 To aid our catalytic designs, discovering the practical synthetic utility and novel reactivity of haloniums is highly desirable.
We have long been interested in developing “-onium”-based alkene functionalization reactions that clearly benefit from the use of widely available alkene starting materials.7 Morpholine is a highly valuable motif in bioactive molecules, and numerous synthetic strategies targeting this motif have been developed.8 Several prominent methods, including intramolecular cyclizations,9 the use of the memorable SnAP/SLAP reagents to couple with carbonyls,10 and the adoption of vinyl sulfonium salts with electron-deficient olefins,11 have been developed (Scheme 1a).12 The Claisen rearrangement has long been recognized for its capacity to construct useful synthetic building blocks such as γ,δ-unsaturated carbonyls.13 However, versatile synthetic access to the enol-ether precursor is rather limited.14 An interesting recent example from the Buchwald group involves a cross-coupling strategy with allylic alcohols and vinyl iodides to access the Claisen precursor (Scheme 1b).15 A unified strategy to access both morpholine and Claisen rearrangement products based on an alkene starting material would be an ideal alternative to the existing methods.
Scheme 1.
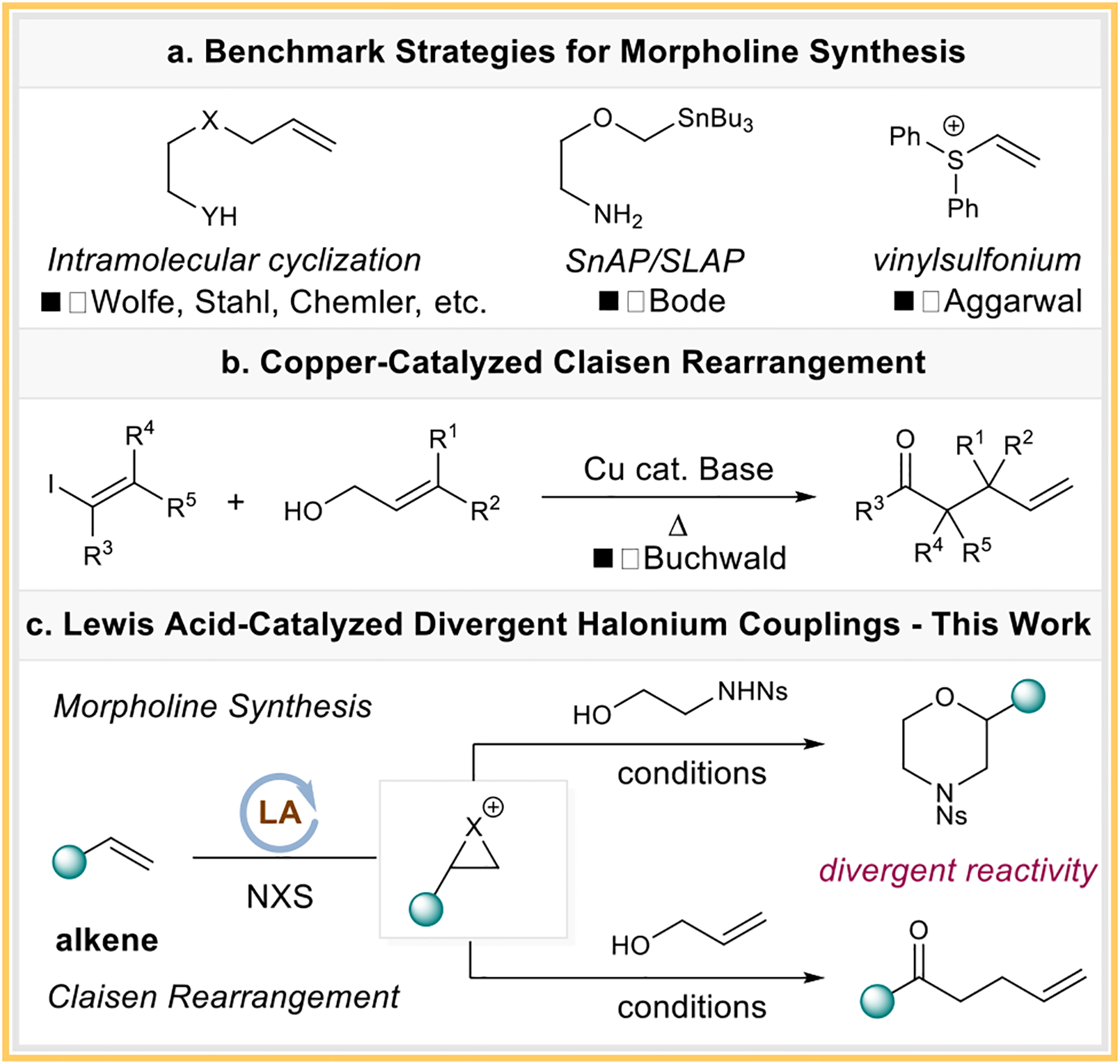
Background on Morpholine Synthesis and Claisen Rearrangement
We envision that complementary reactivity can arise from the halonium by reacting it with either amino alcohols or allylic alcohols to generate the respective haloether intermediate prior to the morpholine synthesis or the Claisen rearrangement (Scheme 1c). We disclose here a halonium-based strategy to achieve the regioselective oxyamination of olefins for morpholine synthesis and the Claisen rearrangement couplings of allylic alcohols and alkenes. Our mechanistic investigation also unveiled an unusual kinetic resolution process that enabled even unactivated olefins to be highly regioselective in the morpholine synthesis.
RESULTS AND DISCUSSION
Our studies began with styrene 1 and 2-nitrobenzene-sulfonyl-protected amino alcohol 2 as the standard substrates. We were delighted to observe the formation of the halofunctionalized intermediate 3 when the NBS reagent was used as the halogen source (Scheme 2, entry 1). However, attempts to optimize the yield of 3 based on NBS alone proved difficult due to a lack of reactivity. To circumvent this issue, we introduced Lewis acids to facilitate the halonium formation.16 Indeed, the introduction of several common Lewis acids significantly increased the production of 3. In(OTf)3 being the optimal Lewis acid, affording a 55% yield of 3 (Scheme 2, entry 2–4). Further tuning of the catalyst loading and the NBS stoichiometry led to the most optimal conditions shown in entry 8 (Scheme 2, entries 4–8). Under these conditions, the direct addition of DBU as a base following the optimal halogenation conditions smoothly produced the desired morpholine product 5 in a 76% isolated yield as a single regioisomer (Scheme 2, entry 9).
Scheme 2. Optimization Studiesa.
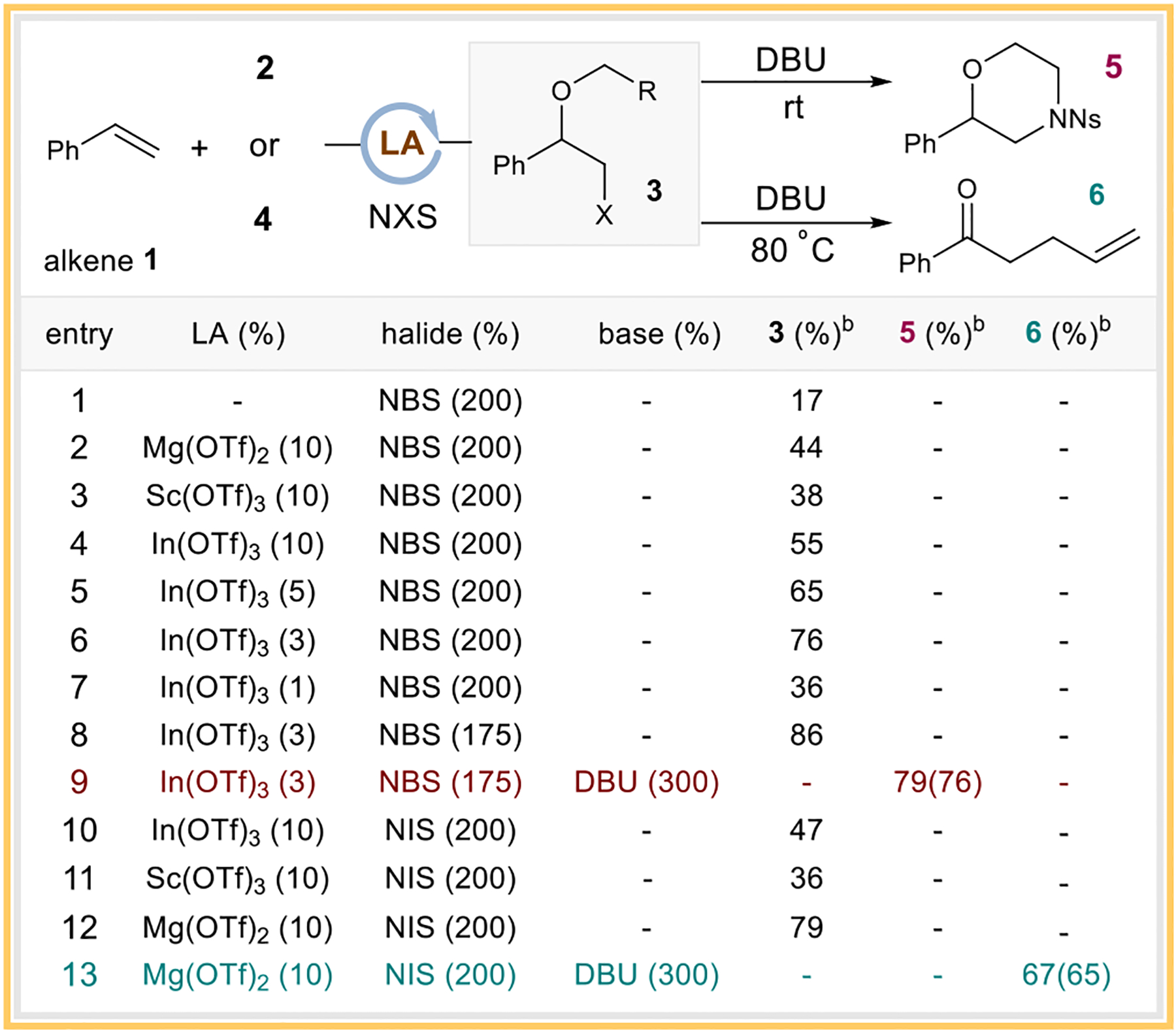
aReaction conditions are as follows: For 5, 1 (0.4375 mmol), 2 (0.25 mmol), In(OTf)3 (3 mol %), NBS (175 mol %), and DCM (1.5 mL), at rt for 1 h, then DBU (0.75 mmol) at rt for 23 h. For 6, 1 (0.5 mmol), 4 (0.25 mmol), Mg(OTf)2 (10 mol %), NIS (200 mol %), and DCM (0.25 mL) at rt for 8 h, then DBU (0.75 mmol) at 80 °C for 36 h. bYields were determined by crude 1H NMR using 1,3-benzodioxole as the internal standard. The yield shown in parentheses was the isolated yield.
During our optimization, we often observed small amounts of the elimination byproduct from 3 when the base was added. While this was problematic for the morpholine synthesis, we speculated that an allylic alcohol such as 2-propen-1-ol 4 could be used instead to obtain the Claisen rearrangement precursor. With that in mind, we switched the halogen source to NIS, which was not only beneficial for the generation of intermediate 3 but also installed a better leaving group for the elimination process. Evaluation of the Lewis acid identity led to Mg(OTf)2 as the optimal catalyst (Scheme 2, entries 10–12). Hence, the addition of DBU as the base at 80 °C effectively promoted both the elimination and the ensuing Claisen rearrangement to afford product 6 in a 65% isolated yield (Scheme 2, entry 13).
With these divergent synthetic applications in hand, we decided to first evaluate the substrate scopes for the morpholine synthesis. We quickly tested several electronically activated alkene substrates. In this regard, a series of cross-coupling-ready functional groups, including p-F, p-Cl, p-Br, and p-OPiv, were all well-tolerated and formed the desired products (Scheme 3, products 7–10, respectively). In addition, α-methylstyrene and 2-vinylnaphthalene afforded the morpholine products 11 and 12, respectively, as single regioisomers (Scheme 3, products 11 and 12). Aliphatic olefins have proven to be a difficult class of substrates in the absence of directing groups for regioselective intermolecular olefin oxyamination reactions.17 Under these conditions using NIS, a number of α-olefins containing alkyl, –OPiv, and –NPhth functional groups smoothly produced the morpholine products in excellent yields, albeit with diminished regioselectivities (Scheme 3, products 13–16, respectively). Interestingly, α-olefins with slightly increased steric hindrance at the α-, β-, or even γ-positions all led to a significant boost in regioselectivity (Scheme 3, products 17–21). Furthermore, several complex olefin substrates derived from estradiol, camphor, and vitamin E all proceeded to the desired products with excellent regioselectivities (Scheme 3, products 22–24, respectively).
Scheme 3. Morpholine Substrate Scopea.
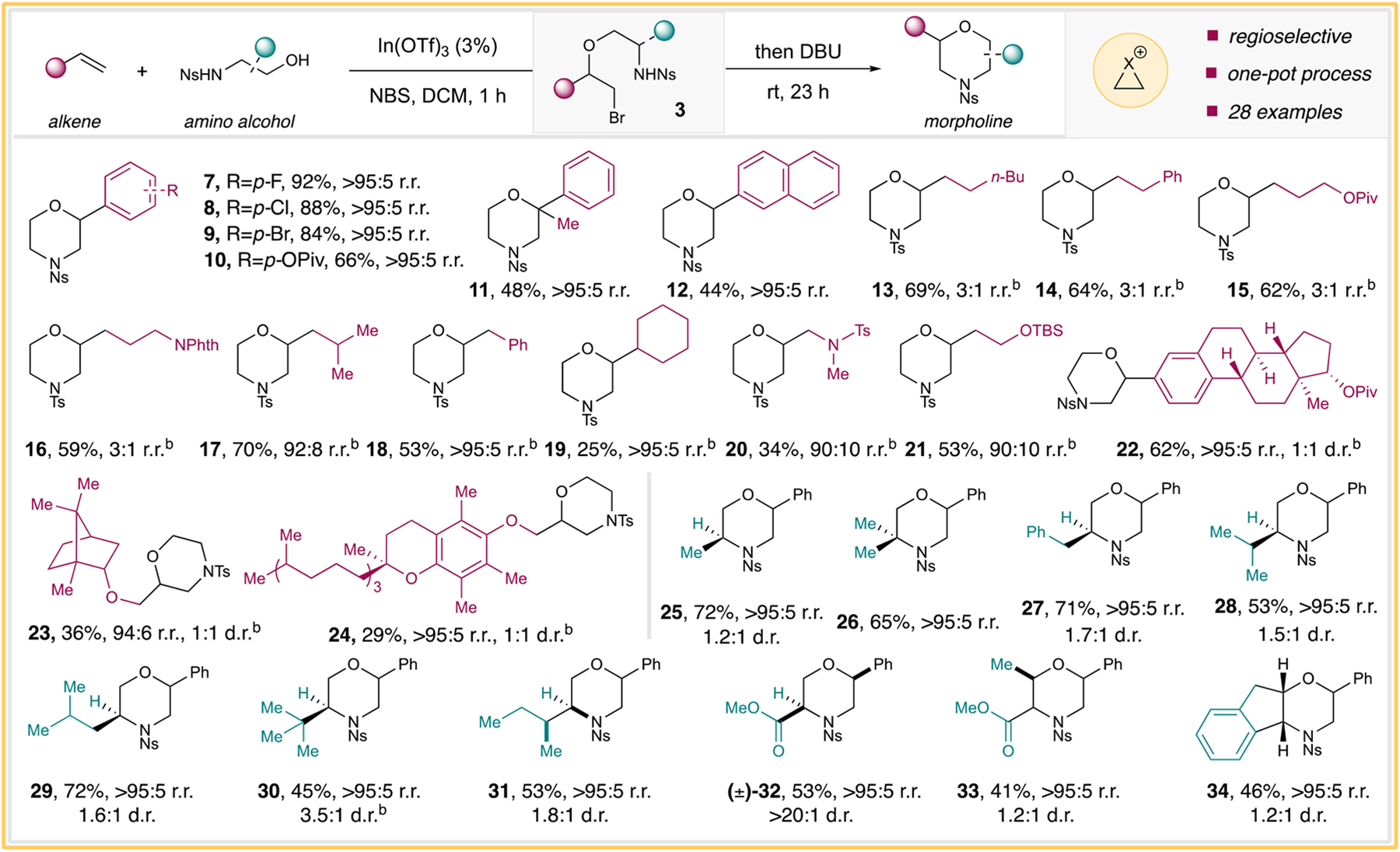
aFor specific reaction conditions, please refer to the Supporting Information. Yields were isolated, and r.r. was assigned by crude 1H NMR using 1,3-benzodioxole as the internal standard. bNIS was used, and isolated yields were for the major regioisomer or stereoisomer.
Encouraged by these findings, we then ventured to evaluate a range of amino alcohols for the morpholine synthesis. In this regard, a number of naturally derived amino alcohols effectively coupled with styrene to generate the desired morpholine products (Scheme 3, products 25–34). For these cases, the stereochemistry was set during the halofunctionalization step, resulting in a mixture of diastereomeric products. For product 32, only the cis-diastereomeric product was observed due to the stereocenter next to the carbonyl group being racemized to avoid steric interactions of the N-nosyl group. Notably, highly sterically hindered amino alcohols could also participate in the reaction with reasonable efficiency (Scheme 3, product 28–31).
With a good substrate scope in hand for the morpholine synthesis, we were intrigued by the regiochemical features observed from the unactivated olefins. (Scheme 3, examples 13–16 versus 17–21). To understand the improved regioselectivities for products 17–21, we carried out the iodoetherification process first and observed the regioselectivities from this process for both olefin substrates 35 (1-octene) and 36 (4-methyl-pent-1-ene) to be around 3:1 r.r. (Scheme 4). The regiochemical outcome here was not surprising given the stereoelectronic contrast of the terminal (sterically favored) versus that of the internal carbon (electronically favored).18 Interestingly when the base was added to both regioisomeric mixtures, the iodoethers 37a and 37b of the less sterically hindered olefin led to the morpholine products in 3:1 r.r., while the iodoethers 38a and 38b of the more sterically hindered olefin led to highly regioselective outcomes. In this case, the cyclization process appeared to be the regio-determining step, suggesting that an apparent kinetic resolution process for the high regioselectivity was taking place.19 Our findings here do raise the prospect that potential dynamic kinetic resolution could be developed for regiochemical purposes. Furthermore, while this may not be a concern for halide-catalyzed reactions that require the halide to be turned over, caution for rationalizing the high regioselectivity in stoichiometric halofunctionalizations must be exercised.
Scheme 4.
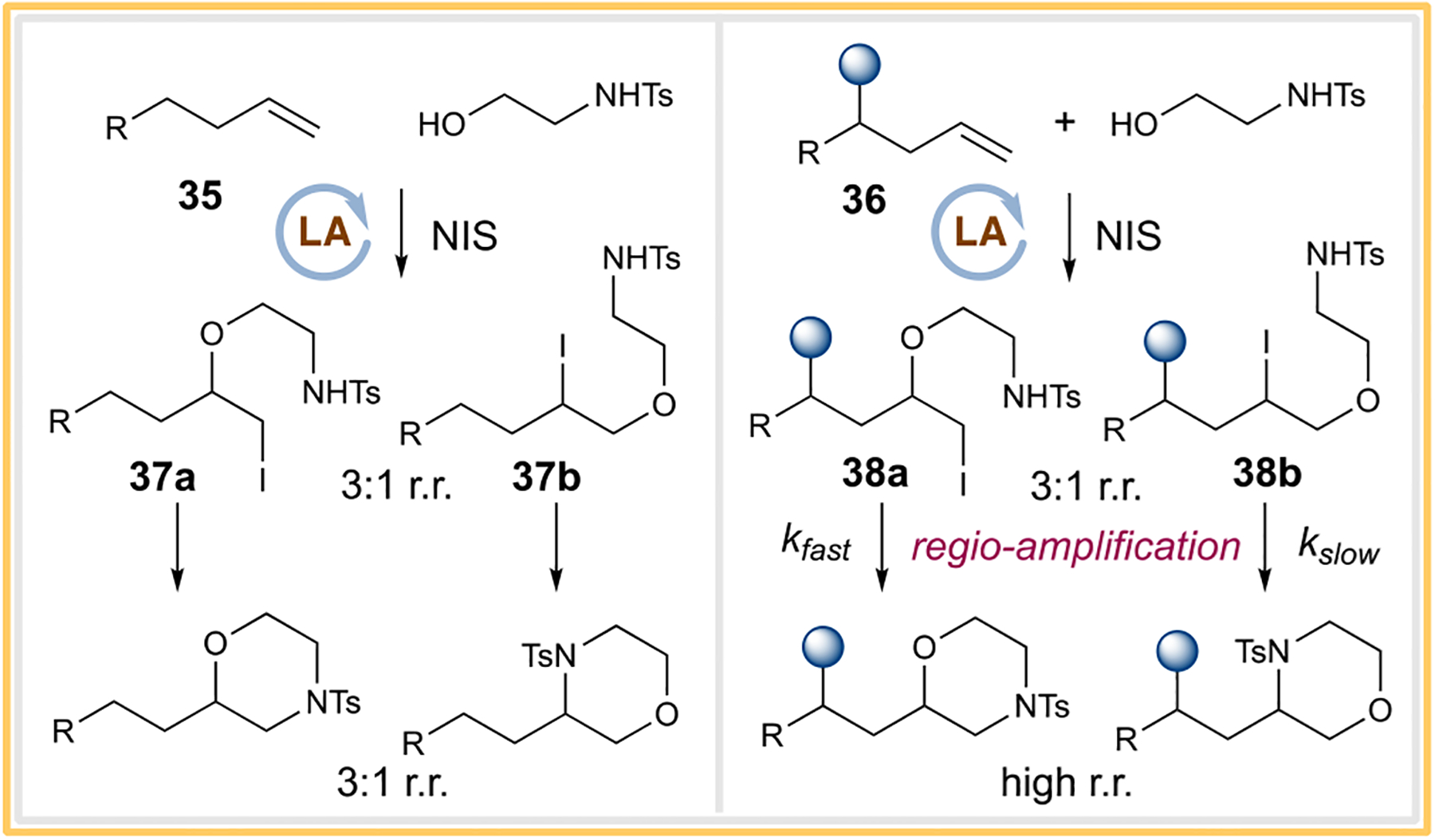
Regiochemical Probe
Satisfied with the morpholine substrate scope, we then turned our attention to the Claisen rearrangement process. Similarly, a range of styrenes bearing para-substitutions with –t-Bu, –OMe, –OPiv, and –F all proceeded smoothly to provide the rearranged products in reasonable yields (Scheme 5, products 39–42, respectively). Reactions with activated olefins containing naphthalene, heteroarene, or β-methylstyrene all resulted in product formation with reasonable yields (Scheme 5, products 43–45, respectively). A number of allylic alcohols also participated in the reaction with similar efficiencies (Scheme 5, products 46–48). Furthermore, estradiol-derived styrene could afford the desired product with an excellent yield (Scheme 5, product 49). Our protocol here demonstrates that simple allylic alcohols can be utilized to couple with alkenes to effect a Claisen rearrangement process to access γ,δ-unsaturated ketones, further highlighting the synthetic utility of halonium intermediates for potential catalytic chemical reaction designs.
Scheme 5. Claisen Rearrangement Scopea.
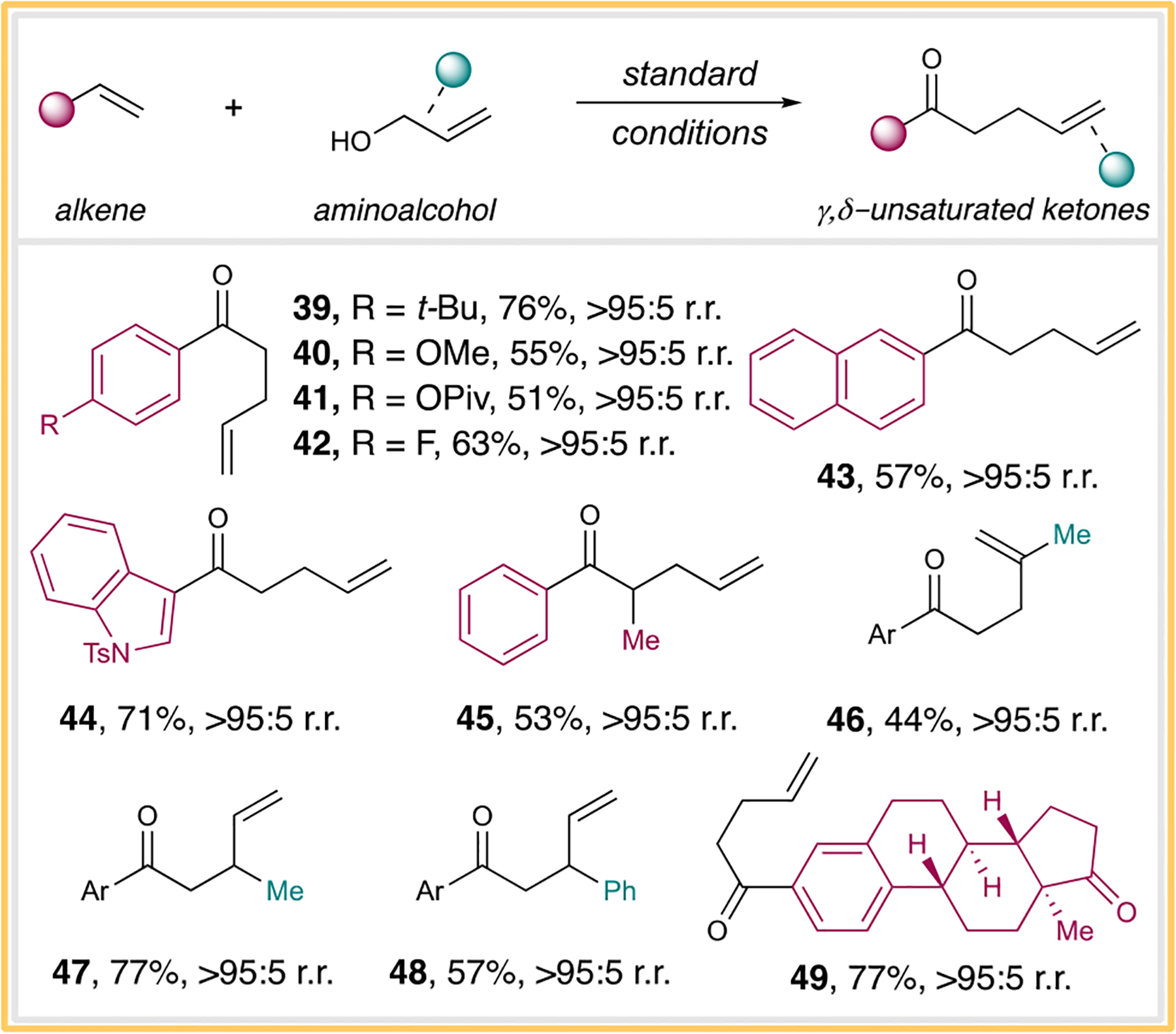
aStandard reaction conditions for the Claisen rearrangement. Ar = para-t-butylphenyl.
In conclusion, we have disclosed here different synthetic utilities based on a common halonium intermediate toward both morpholine synthesis and the Claisen rearrangement. These practical protocols directly furnish useful pharmaceutical motifs and synthetic building blocks while providing interesting mechanistic insights for unexpected regiochemical features. These reaction settings and the mechanistic features will help to guide us in designing future chemical reactions involving halonium catalysis.
EXPERIMENTAL SECTION
General Information.
Commercial reagents and solvents were purchased from Sigma-Aldrich, Oakwood Chemicals, Alfa Aesar, Matrix Scientific, and Acros Organic and were used as received. Alkenes 10,20 15,20 16,21 20,22 21,23 22,6f 23,24 24,6f 39,20 42,6f and 446f were synthesized based on reported literature procedures. All the amino alcohol substrates were tosyl- or nosyl-protected based on a reported literature procedure.25a Amino alcohols 26a, 30a, 31a, 32a, 33a, and 34a were not known previously and are fully characterized here.25b,c Organic solutions were concentrated under reduced pressure on an IKA rotary evaporator using an acetone/dry ice bath. Chromatographic purification of products was accomplished using flash chromatography on 230–400 mesh silica gel. Thin-layer chromatography (TLC) was performed on Analtech 250 mm silica gel HLF UV-250 plates. Visualization of the developed plates was performed using fluorescence quenching and potassium permanganate. 1H and 13C NMR spectra were recorded on a Bruker (600 and 150 MHz) or INOVA 600 (600 and 150 MHz) instrument and were internally referenced to residual protio-solvent signals (for CDCl3, 7.27 and 77.0 ppm, respectively). Data for 1H NMR are reported as follows: chemicals shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, h = heptet, m = multiplet, br = broad), integration, and coupling constant (Hz). 13C spectra were reported as chemical shifts in ppm and multiplicity where appropriate. IR spectra were recorded on a PerkinElmer FT-IR spectrophotometer and reported in terms of the wavenumber of absorption (cm−1). High-resolution mass spectra were obtained on a Waters Synapt time-of-flight (TOF) high-definition mass spectrometer (HDMS) using electrospray ionization at the University of Toledo, OH, and a Bruker MaXis TOF ultra-high-resolution ESI LC/MS at the University of Wisconsin—Madison, WI.
General Procedure for Amino Alcohol Substrate Synthesis.
To a 100 mL round-bottom flask equipped with a stir bar were added the amino alcohol substrate (5.0 mmol) and solvent (DCM, 10 mL). To the mixture was then added Et3N (1 mL, 7.5 mmol) via syringe, followed by 2-nitrobenzenesulfonyl chloride (1.2 g, 5.5 mmol). The reaction mixture was then stirred for 16 h at room temperature. The reaction mixture was diluted with 10 mL of DCM, and the reaction was quenched with 10 mL of 0.1 N HCl. The organic layer was separated, and the aqueous layer was extracted with DCM (2 × 10 mL). The combined organic layer was concentrated under reduced pressure to give the crude product, which was purified by column chromatography on silica gel to afford the pure product.
General Procedure A for Lewis Acid-Catalyzed Morpholine Synthesis.
To an 8 mL vial equipped with a stir bar were added In(OTf)3 (4 mg, 0.0075 mmol), NBS (78 mg, 0.4375 mmol), and the 2-nitrobenzenesulfonyl-protected amino alcohol (0.25 mmol). To the mixture was then added the solvent (DCM, 1.5 mL) via syringe, followed by the alkene (0.4375 mmol). The reaction mixture was then stirred for 1 h at room temperature. To the mixture was added DBU (114 μL, 0.75 mmol) after 1 h, and the mixture continued to stir for another 23 h. The reaction mixture was diluted with 2 mL of DCM, and the reaction was quenched with 2 mL of saturated Na2S2O3 and 0.5 mL of 1 M HCl. The organic layer was separated, and the aqueous layer was extracted with DCM (2 × 2 mL). The combined organic layer was concentrated under reduced pressure to give the crude product, which was purified by column chromatography on silica gel to afford the pure product.
General Procedure B for Lewis Acid-Catalyzed Morpholine Synthesis.
To an 8 mL vial equipped with a stir bar were added In(OTf)3 (4 mg, 0.0075 mmol), NIS (98 mg, 0.4375 mmol), and p-toluenesulfonyl-protected amino alcohol (0.25 mmol). To the mixture was then added the solvent (DCM, 1.5 mL) via syringe, followed by the alkene (0.4375 mmol). The reaction mixture was then stirred for 1 h at room temperature. To the mixture was added DBU (114 μL, 0.75 mmol) after 1 h, and the mixture continued to stir for another 23 h. The reaction mixture was diluted with 2 mL of DCM, and the reaction was quenched with 2 mL of saturated Na2S2O3 and 0.5 mL of 0.5 M HCl. The organic layer was separated, and the aqueous layer was extracted with DCM (2 × 2 mL). Combined organic layer was concentrated under reduced pressure to give the crude product, which was purified by column chromatography on silica gel to afford the pure product.
General Procedure C for the Lewis Acid-Catalyzed Claisen Rearrangement Reaction.
To an 8 mL vial equipped with a stir bar were added Mg(OTf)2 (8 mg, 0.025 mmol) and NIS (113 mg, 0.5 mmol). To the mixture was then added the solvent (DCM, 0.25 mL) via syringe, followed by the alkene (0.5 mmol) and the allylic alcohol (0.25 mmol). The reaction mixture was then stirred for 8 h at room temperature. To the mixture was then added DBU (114 μL, 0.75 mmol) after 8 h, and the mixture continued to stir for another 36 h at 80 °C with heating in a pie-block. The reaction mixture was diluted with 2 mL of EtOAc, and the reaction was quenched with 2 mL of saturated Na2S2O3. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (2 × 2 mL). Combined organic layer was concentrated under reduced pressure to give the crude product, which was purified by column chromatography on silica gel to afford the pure product.
Procedure for Large-Scale Morpholine Synthesis.
To an 10 mL round-bottom flask equipped with a stir bar waere added In(OTf)3 (16 mg, 0.03 mmol), NBS (312 mg, 1.74 mmol), and 2-nitrobenzenesulfonyl-protected 2-amino-1-ethanol (248 mg, 1.0 mmol). To the mixture was then added the solvent (DCM, 6 mL) via syringe, followed by styrene (200 μL, 1.74 mmol). The reaction mixture was then stirred for 1 h at room temperature. To the mixture was added DBU (456 μL, 3.0 mmol) after 1 h, and the mixture continued to stir for another 23 h. The reaction mixture was diluted with 5 mL of DCM, and the reaction was quenched with 5 mL of saturated Na2S2O3 and 2.0 mL of 1 M HCl. The organic layer was separated, and the aqueous layer was extracted with DCM (2 × 5 mL). The combined organic layer was concentrated under reduced pressure to give the crude product, which was purified by column chromatography on SiO2 (10–50% EtOAc/hexanes). The title compound was isolated as a white solid (255 mg, 73% yield).
Procedure for the Large-Scale Claisen Rearrangement Reaction.
To an 8 mL vial equipped with a stir bar were added Mg(OTf)2 (32 mg, 0.1 mmol) and NIS (450 mg, 2.0 mmol). To the mixture was then added the solvent (DCM, 1.0 mL) via syringe, followed by styrene (229 μL, 2.0 mmol) and the allylic alcohol (68 μL, 1.0 mmol). The reaction mixture was then stirred for 8 h at room temperature. To the mixture was added DBU (456 μL, 3.0 mmol) after 8 h, and the mixture continued to stir for another 36 h at 80 °C in metal pie-wedge over a heating plate. The reaction mixture was diluted with 8 mL of EtOAc, and the reaction was quenched with 8 mL of saturated Na2S2O3. The rganic layer was separated, and the aqueous layer was extracted with ethyl acetate (2 × 5 mL). The combined organic layer was concentrated under reduced pressure to give the crude product, which was purified by column chromatography on silica gel (0–2% EtOAc/hexanes). The title compound was isolated as a colorless oil (109 mg, 68% yield).
N-(1-Hydroxy-2-methylpropan-2-yl)-2-nitrobenzenesulfonamide (26a).
This compound was prepared according to the general procedure for starting materials synthesis using 2-amino-2-methylpropan-1-ol (0.5 mL, 5.0 mmol). After purification by column chromatography on SiO2 (70–80% EtOAc/hexanes), the title compound was isolated as a white solid (1.33 g, 97% yield). 1H NMR (600 MHz, CDCl3) δ 8.19 (d, J = 7.7 Hz, 1 H), 7.86 (d, J = 7.3 Hz, 1 H), 7.79−7.69 (m, 2 H), 5.65 (s, 1 H), 3.52 (d, J = 5.5 Hz, 2 H), 2.43 (t, J = 5.9 Hz, 1 H), 1.25 (s, 7 H); 13C{1H} NMR (150 MHz, CDCl3) δ 147.7, 136.5, 133.4, 133.0, 130.4, 125.4, 70.1, 58.9, 24.5; IR (neat) 3595, 3256, 2985, 2910, 1537, 1439, 1366, 1318, 1145, 1125, 1055, 977 cm−1; HRMS (ESI) m/z calcd for C10H15N2O5S [(M + H)+] 275.0702, found 275.0700.
(S)-N-(1-Hydroxy-3,3-dimethylbutan-2-yl)-2-nitrobenzenesulfonamide (30a).
This compound was prepared according the general procedure for starting materials synthesis using (S)-2-amino-3,3-dimethylbutan-1-ol (0.59 g, 5.0 mmol). After purification by column chromatography on SiO2 (70–80% EtOAc/hexanes), the title compound was isolated as a white solid (1.36 g, 90% yield). 1H NMR (600 MHz, CDCl3) δ 8.13 (d, J = 7.3 Hz, 1 H), 7.86 (d, J = 7.3 Hz, 1 H), 7.77−7.68 (m, 2 H), 5.56 (d, J = 8.8 Hz, 1 H), 3.78−3.66 (m, 1 H), 3.58 (dd, J = 7.7, 11.4 Hz, 1 H), 3.34−3.22 (m, 1 H), 1.84 (s, 1 H), 0.91 (s, 9 H); 13C{1H} NMR (150 MHz, CDCl3) δ 147.5, 135.2, 133.2, 132.9, 130.5, 125.2, 65.4, 62.3, 34.0, 26.9; IR (neat) 3559, 1527, 1424, 1363, 1338, 1157, 1087, 1048, 1028 cm−1; HRMS (ESI) m/z calcd for C12H19N2O5S [(M + H)+] 303.1015, found 303.1008.
N-((2S,3R)-1-Hydroxy-3-methylpentan-2-yl)-2-nitrobenzenesulfonamide (31a).
This compound was prepared according to the general procedure for starting materials synthesis using (2S,3R)-2-amino-3-methylpentan-1-ol (0.59 g, 5.0 mmol). After purification by column chromatography on SiO2 (70–80% EtOAc/hexanes), the title compound was isolated as a white solid (1.29 g, 85% yield). 1H NMR (600 MHz, CDCl3) δ 8.20−8.11 (m, 1 H), 7.93−7.85 (m, 1 H), 7.80−7.70 (m, 2 H), 5.53 (d, J = 8.1 Hz, 1 H), 3.69−3.58 (m, 2 H), 3.42−3.35 (m, 1 H), 1.68 (br. s., 1 H), 1.66−1.59 (m, 1 H), 1.54−1.44 (m, 1 H), 1.16−1.05 (m, 1 H), 0.92−0.78 (m, 6 H); 13C{1H} NMR (150 MHz, CDCl3) δ 147.6, 134.7, 133.4, 132.9, 130.6, 125.3, 62.6, 61.0, 36.4, 25.1, 15.2, 11.3; IR (neat) 3337, 3101, 2964, 2934, 1535, 1424, 1361, 1338, 1167, 1120, 1059 cm−1; HRMS (ESI) m/z calcd for C12H19N2O5S [(M + H)+] 303.1015, found 303.1010.
Methyl ((2-Nitrophenyl)sulfonyl)-d-serinate (32a).
This compound was prepared according to the general procedure for starting materials synthesis using methyl d-serinate (0.6 g, 5.0 mmol). After purification by column chromatography on SiO2 (70–100% EtOAc/hexanes), the title compound was isolated as a white solid (1.39 g, 91% yield). 1H NMR (600 MHz, CDCl3) δ 8.18−8.05 (m, 1 H), 8.03−7.91 (m, 1 H), 7.86−7.71 (m, 2 H), 6.51 (d, J = 7.0 Hz, 1 H), 4.36−4.21 (m, 1 H), 4.09−4.01 (m, 1 H), 4.01−3.94 (m, 1 H), 3.61 (s, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 169.7, 147.7, 133.9, 133.8, 133.0, 130.5, 125.7, 63.8, 58.4, 52.9; IR (neat) 3577, 3278, 3091, 2971, 2892, 1738, 1542, 1438, 1353, 1259, 1166, 1123 cm−1; HRMS (ESI) m/z calcd for C10H13N2O7S [(M + H)+] 305.0443, found 305.0439.
Methyl (2R)-3-Hydroxy-2-((4-nitrophenyl)sulfonamido)-butanoate (33a).
This compound was prepared according to the general procedure for starting materials synthesis using methyl (2R)-2-amino-3-hydroxybutanoate (0.67 g, 5.0 mmol). After purification by column chromatography on SiO2 (70–100% EtOAc/hexanes), the title compound was isolated as a white solid (1.48 g, 93% yield). 1H NMR (600 MHz, CDCl3) δ 8.12−8.04 (m, 1 H), 7.99−7.92 (m, 1 H), 7.80−7.70 (m, 2 H), 6.35 (d, J = 9.5 Hz, 1 H), 4.34 (dd, J = 6.4, 2.8 Hz, 1 H), 4.11 (dd, J = 9.4, 2.8 Hz, 1 H), 3.52 (s, 3 H), 1.99 (br. s., 1 H), 1.35 (d, J = 6.2 Hz, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 170.3, 147.5, 134.1, 133.7, 132.9, 130.3, 125.5, 68.1, 61.8, 52.6, 19.9; IR (neat) 3476, 3259, 1726, 1547, 1444, 1359, 1254, 1170, 1090 cm−1; HRMS (ESI) m/z calcd for C11H15N2O7S [(M + H)+] 319.0600, found 319.0606.
N-((1R,2S)-2-Hydroxy-2,3-dihydro-1H-inden-1-yl)-2-nitrobenzenesulfonamide (34a).
This compound was prepared according to the general procedure for starting materials synthesis using (1R,2S)-1-amino-2,3-dihydro-1H-inden-2-ol (0.75 g, 5.0 mmol). After purification by column chromatography on SiO2 (70–80% EtOAc/hexanes), the title compound was isolated as a white solid (1.37 g, 82% yield). 1H NMR (600 MHz, CDCl3) δ 8.28−8.22 (m, 1 H), 7.97−7.91 (m, 1 H), 7.84−7.75 (m, 2 H), 7.29−7.22 (m, 2 H), 7.20 (d, J = 3.3 Hz, 2 H), 6.16 (d, J = 8.1 Hz, 1 H), 4.93 (dd, J = 8.4, 4.8 Hz, 1 H), 4.40 (br. s., 1 H), 3.09 (dd, J = 16.7, 5.0 Hz, 1 H), 2.93 (d, J = 16.5 Hz, 1 H), 2.07 (br. s., 1 H); 13C{1H} NMR (150 MHz, CDCl3) δ 147.8, 139.3, 138.9, 134.5, 133.8, 133.0, 130.7, 128.8, 127.4, 125.6, 125.5, 124.4, 77.2, 76.8, 73.0, 62.3, 39.4; IR (neat) 3527, 3321, 2938, 1535, 1413, 1344, 1161, 1113, 1077 cm−1; HRMS (ESI) m/z calcd for C15H14N2O5SNa [(M + Na)+] 357.0516, found 357.0522.
4-((2-Nitrophenyl)sulfonyl)-2-phenylmorpholine (5).
This compound was prepared according to general procedure A using N-(2-hydroxyethyl)-2-nitrobenzenesulfonamide (62 mg, 0.25 mmol) and styrene (50 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (66 mg, 76% yield). 1H NMR (600 MHz, CDCl3) δ 7.97 (d, J = 7.8 Hz, 1 H), 7.77−7.67 (m, 2 H), 7.64 (d, J = 7.6 Hz, 1 H), 7.40−7.35 (m, 4 H), 7.35−7.30 (m, 1 H), 4.58 (dd, J = 10.4, 2.6 Hz, 1 H), 4.13 (dd, J = 11.7, 2.7 Hz, 1 H), 3.87 (d, J = 12.5 Hz, 1 H), 3.83 (dt, J = 11.8, 2.6 Hz, 1 H), 3.74 (d, J = 12.5 Hz, 1 H), 3.08 (dt, J = 12.1, 3.3 Hz, 1 H), 2.81 (dd, J = 12.3, 10.6, Hz, 1 H); 13C{1H} NMR (150 MHz, CDCl3) δ 148.4, 138.3, 133.9, 131.6, 131.0, 130.9, 128.6, 128.4, 126.0, 124.2, 77.6, 66.5, 51.5, 45.3; IR (neat) 2919, 2857, 1607, 1541, 1511, 1350, 1230, 1163 cm−1; HRMS (ESI) m/z calcd for C16H17N2O5S [(M + H)+] 349.0858, found 349.0861.
2-(4-Fluorophenyl)-4-((2-nitrophenyl)sulfonyl)morpholine (7).
This compound was prepared according to general procedure A using N-(2-hydroxyethyl)-2-nitrobenzenesulfonamide (62 mg, 0.25 mmol) and 4-fluorostyrene (52 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (84 mg, 92% yield). 1H NMR (600 MHz, CDCl3) δ 7.97 (d, J = 7.8 Hz, 1 H), 7.77−7.67 (m, 2 H), 7.64 (d, J = 7.6 Hz, 1 H), 7.34 (dd, J = 8.3, 5.6, Hz, 2 H), 7.05 (t, J = 8.7 Hz, 2 H), 4.55 (dd, J = 10.4, 1.8, Hz, 1 H), 4.11 (dd, J = 11.7, 2.4, Hz, 1 H), 3.84 (d, J = 12.9 Hz, 1 H), 3.81 (dt, J = 12.0, 3.0, Hz, 1 H), 3.72 (d, J = 12.5 Hz, 1 H), 3.06 (dt, J = 12.1, 3.2, Hz, 1 H), 2.78 (t, J = 12.1 Hz, 1 H); 13C{1H} NMR (150 MHz, CDCl3) δ 163.4, 161.7, 148.3, 134.1, 134.1, 134.0, 131.7, 130.9, 130.9, 127.8, 127.8, 124.2, 115.5, 115.4, 66.5, 51.5, 45.2; IR (neat) 2919, 2857, 1607, 1541, 1511, 1350, 1230, 1163 cm−1; HRMS (ESI) m/z calcd for C16H15FN2NaO5S [(M + Na)+] 389.0583, found 389.0570.
2-(4-Chlorophenyl)-4-((2-nitrophenyl)sulfonyl)morpholine (8).
This compound was prepared according to general procedure A using N-(2-hydroxyethyl)-2-nitrobenzenesulfonamide (62 mg, 0.25 mmol) and 4-chlorostyrene (52 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (84 mg, 88% yield). 1H NMR (600 MHz, CDCl3) δ 8.02−7.93 (m, 1 H), 7.79−7.68 (m, 2 H), 7.68−7.61 (m, 1 H), 7.39−7.28 (m, 4 H), 4.55 (dd, J = 10.5, 2.4 Hz, 1 H), 4.11 (dd, J = 11.7, 2.2 Hz, 1 H), 3.88−3.76 (m, 2 H), 3.72 (d, J = 12.5 Hz, 1 H), 3.06 (dt, J = 12.3, 3.3 Hz, 1 H), 2.75 (dd, J = 12.3, 10.5 Hz, 1 H); 13C{1H} NMR (150 MHz, CDCl3) δ 148.4, 136.8, 134.1, 134.0, 131.7, 131.0, 130.9, 128.7, 127.4, 124.2, 76.8, 66.5, 51.4, 45.2; IR (neat) 2914, 2874, 1587, 1547, 1376, 1359, 1165, 1121, cm−1; HRMS (ESI) m/z calcd for C16H15ClN2NaO5S [(M + Na)+] 405.0288, found 405.0287.
2-(4-Bromophenyl)-4-((2-nitrophenyl)sulfonyl)morpholine (9).
This compound was prepared according to general procedure A using N-(2-hydroxyethyl)-2-nitrobenzenesulfonamide (62 mg, 0.25 mmol) and 4-bromostyrene (57 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (90 mg, 84% yield). 1H NMR (600 MHz, CDCl3) δ 7.97 (d, J = 7.8 Hz, 1 H), 7.79−7.68 (m, 2 H), 7.65 (d, J = 7.6 Hz, 1 H), 7.50 (d, J = 8.5 Hz, 2 H), 7.25 (d, J = 8.3 Hz, 2 H), 4.54 (dd, J = 10.5, 2.2 Hz, 1 H), 4.11 (dd, J = 11.8, 2.3 Hz, 1 H), 3.84 (d, J = 12.5 Hz, 1 H), 3.80 (dt, J = 2.7, 11.8 Hz, 1 H), 3.72 (d, J = 12.5 Hz, 1 H), 3.06 (dt, J = 12.1, 3.2 Hz, 1 H), 2.74 (dd, J = 12.2, 10.7 Hz, 1 H); 13C{1H} NMR (150 MHz, CDCl3) δ 148.4, 137.3, 134.0, 131.7, 131.0, 130.9, 127.7, 124.2, 122.3, 76.9, 66.5, 51.4, 45.3; IR (neat) 2998, 2912, 2872, 1740, 1539, 1356, 1265, 1162, 1120, 1069 cm−1; HRMS (ESI) m/z calcd for C16H15BrN2NaO5S [(M + Na)+] 448.9783, found 448.9767.
4-(4-((2-Nitrophenyl)sulfonyl)morpholin-2-yl)phenyl pivalate (10).
This compound was prepared according to general procedure A using N-(2-hydroxyethyl)-2-nitrobenzenesulfonamide (62 mg, 0.25 mmol) and 4-vinylphenyl pivalate (89 mg, 0.4375 mmol). After purification by column chromatography on SiO2 (10–30% EtOAc/hexanes), the title compound was isolated as a white solid (73 mg, 66% yield). 1H NMR (600 MHz, CDCl3) δ 7.95 (d, J = 7.8 Hz, 1 H), 7.75−7.67 (m, 2 H), 7.63 (d, J = 7.8 Hz, 1 H), 7.37 (d, J = 8.5 Hz, 2 H), 7.05 (d, J = 8.5 Hz, 2 H), 4.58 (dd, J = 10.4, 2.1 Hz, 1 H), 4.12 (dd, J = 12.1, 2.1 Hz, 1 H), 3.84 (d, J = 12.5 Hz, 1 H), 3.81 (dt, J = 12.1, 3.1 Hz, 1 H), 3.73 (d, J = 12.5 Hz, 1 H), 3.06 (dt, J = 12.1, 3.1 Hz, 1 H), 2.75 (t, J = 11.4 Hz, 1 H), 1.36 (s, 9 H); 13C{1H} NMR (150 MHz, CDCl3) δ 177.0, 151.0, 148.4, 135.6, 134.0, 131.6, 131.0, 130.7, 127.0, 124.1, 121.6, 66.5, 51.6, 45.3, 39.0, 27.1; IR (neat) 2919, 2973, 2871, 1744, 1543, 1361, 1266, 1164 cm−1; HRMS (ESI) m/z calcd for C21H25N2O7S [(M + H)+] 449.1382, found 449.1376.
2-Methyl-4-((2-nitrophenyl)sulfonyl)-2-phenylmorpholine (11).
This compound was prepared according to general procedure A with a slight modification using N-(2-hydroxyethyl)-2-nitrobenzenesulfonamide (62 mg, 0.25 mmol) and α-methylstyrene (65 μL, 0.4375 mmol). After the first step, to the mixture was added DBU (114 μL, 0.75 mmol) with DMSO (1 mL), and the mixture was stirred for 23 h. After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (43 mg, 48% yield). 1H NMR (600 MHz, CDCl3) δ 7.94 (d, J = 7.8 Hz, 1 H), 7.78−7.68 (m, 2 H), 7.62 (d, J = 7.6 Hz, 1 H), 7.46 (d, J = 7.8 Hz, 2 H), 7.37 (t, J = 7.7 Hz, 2 H), 7.31−7.27 (t, J = 7.8 Hz, 1 H), 3.94 (d, J = 12.5 Hz, 1 H), 3.88−3.81 (m, 1 H), 3.77−3.70 (m, 1 H), 3.40−3.31 (m, 1 H), 3.17 (d, J = 12.5 Hz, 2 H), 1.49 (s, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 148.7, 142.3, 133.9, 131.4, 131.0, 130.1, 128.6, 127.4, 125.7, 124.1, 75.2, 60.7, 52.7, 45.6, 27.6; IR (neat) 2920, 2866, 1749, 1541, 1357, 1163, 1129, 1095, 1071 cm−1; HRMS (ESI) m/z calcd for C17H18N2NaO5S [(M + Na)+] 385.0834, found 385.0823.
2-(Naphthalen-2-yl)-4-((2-nitrophenyl)sulfonyl)morpholine (12).
This compound was prepared according to general procedure A using N-(2-hydroxyethyl)-2-nitrobenzenesulfonamide (62 mg, 0.25 mmol) and 4-vinylnaphthalene (67 mg, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (44 mg, 44% yield). 1H NMR (600 MHz, CDCl3) δ 7.98 (d, J = 8.1 Hz, 1 H), 7.88−7.82 (m, 4 H), 7.74 (t, J = 7.8 Hz, 1 H), 7.69 (t, J = 7.8 Hz, 1 H), 7.65 (d, J = 7.8 Hz, 1 H), 7.53−7.49 (m, 2 H), 7.47 (d, J = 8.5 Hz, 1 H), 4.76 (dd, J = 10.8, 2.4 Hz, 1 H), 4.19 (dd, J = 12.0, 2.7 Hz, 1 H), 3.96 (d, J = 12.5 Hz, 1 H), 3.90 (dt, J = 11.7, 2.7 Hz, 1 H), 3.78 (d, J = 12.5 Hz, 1 H), 3.13 (dt, J = 12.1, 3.3 Hz, 1 H), 2.88 (dd, J = 12.9, 10.5 Hz, 1 H); 13C{1H} NMR (150 MHz, CDCl3) δ 135.7, 133.9, 133.2, 133.1, 131.6, 131.0, 128.4, 128.0, 127.7, 126.3, 126.2, 125.1, 124.2, 123.7, 77.7, 66.6, 51.6, 45.4; IR (neat) 2920, 2855, 1592, 1541, 1439, 1509, 1358, 1271, 1164, 1100 cm−1; HRMS (ESI) m/z calcd for C20H18N2NaO5S [(M + Na)+] 421.0834, found 421.0818.
2-Hexyl-4-tosylmorpholine (13).
This compound was prepared according to general procedure B using N-(2-hydroxyethyl)-4-methylbenzenesulfonamide (54 mg, 0.25 mmol) and 1-octene (69 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (56 mg, 69% yield). Note that the reported spectral data are for the major regioisomer only. 1H NMR (600 MHz, CDCl3) δ 7.63 (d, J = 8.1 Hz, 2 H), 7.34 (d, J = 8.1 Hz, 2 H), 3.89 (dd, J = 11.4, 6.0 Hz, 1 H), 3.66 (dt, J = 11.5, 2.3 Hz, 1 H), 3.58−3.45 (m, 3 H), 2.45 (s, 3 H), 2.37 (dt, J = 11.4, 3.1 Hz, 1 H), 2.02 (t, J = 10.6 Hz, 1 H), 1.48−1.33 (m, 3 H), 1.33−1.21 (m, 8 H), 0.87 (t, J = 6.8 Hz, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 143.8, 132.1, 129.7, 127.8, 75.3, 65.9, 50.4, 45.5, 33.2, 31.6, 29.1, 25.0, 22.5, 21.5, 14.0; IR (neat) 2925, 2857, 1598, 1388, 1340, 1307, 1160, 1089, 1101, 1089 cm−1; HRMS (ESI) m/z calcd for C17H28NO3S [(M + H)+] 326.1790, found 326.1795.
2-Phenethyl-4-tosylmorpholine (14).
This compound was prepared according to general procedure B using N-(2-hydroxyethyl)-4-methylbenzenesulfonamide (54 mg, 0.25 mmol) and 4-phenyl-1-butene (66 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (55 mg, 64% yield). For the major regioisomer: 1H NMR (600 MHz, CDCl3) δ 7.62 (d, J = 8.1 Hz, 2 H), 7.34 (d, J = 8.1 Hz, 2 H), 7.29 (t, J = 7.4 Hz, 2 H), 7.20 (t, J = 7.3 Hz, 1 H), 7.16 (d, J = 7.6 Hz, 2 H), 3.93 (dd, J = 12.0, 3.0 Hz, 1 H), 3.66 (dt, J = 11.5, 2.3 Hz, 1 H), 3.58−3.47 (m, 3 H), 2.81−2.72 (m, 1 H), 2.69−2.60 (m, 1H) 2.45 (s, 3 H), 2.41 (dt, J = 11.5, 3.3 Hz, 1 H), 2.08 (t, J = 10.5 Hz, 1 H), 1.81−1.72 (m, 1 H), 1.72−1.65 (m, 1 H); 13C{1H} NMR (150 MHz, CDCl3) δ 143.8, 141.3, 132.0, 129.7, 128.4, 128.3, 127.8, 126.0, 74.3, 65.8, 50.3, 45.5, 34.7, 31.1, 21.5; IR (neat) 2979, 2916, 2849, 1597, 1497, 1446, 1340, 1160, 1103 cm−1; HRMS (ESI) m/z calcd for C19H24NO3S [(M + H)+] 346.1477, found 346.1485.
For the minor regioisomer: 1H NMR (600 MHz, CDCl3) δ 7.69 (d, J = 8.1 Hz, 2 H), 7.31 (d, J = 8.3 Hz, 3 H), 7.28 (d, J = 7.8 Hz, 2 H), 7.21 (t, J = 6.3 Hz, 1 H), 7.14 (d, J = 7.3 Hz, 2 H), 3.82 (t, J = 6.1 Hz, 1 H), 3.73 (d, J = 9.0 Hz, 1 H), 3.70 (d, J = 11.7 Hz, 1 H), 3.60 (d, J = 11.2 Hz, 1 H), 3.43 (dd, J = 11.6, 2.6 Hz, 1 H), 3.37−3.26 (m, 2 H), 2.65−2.58 (m, 2 H), 2.44 (s, 3 H), 2.05−1.96 (m, 1 H), 1.93−1.83 (m, 1 H); 13C{1H} NMR (150 MHz, CDCl3) δ 143.4, 141.2, 138.0, 129.9, 128.4, 128.3, 127.0, 126.0, 68.3, 66.1, 53.2, 40.8, 32.6, 29.8, 21.5; IR (neat) 2963, 2928, 2861, 2849, 1447, 1345, 1330, 1273, 1156, 1113, 1085 cm−1; HRMS (ESI) m/z calcd for C19H24NO3S [(M + H)+] 346.1477, found 346.1456.
3-(4-Tosylmorpholin-2-yl)propyl pivalate (15).
This compound was prepared according to general procedure B using N-(2-hydroxyethyl)-4-methylbenzenesulfonamide (54 mg, 0.25 mmol) and pent-4-en-1-yl pivalate (74 mg, 0.4375 mmol). After purification by column chromatography on SiO2 (20–30% EtOAc/hexanes), the title compound was isolated as a white solid (59 mg, 62% yield). Note that the reported spectral data are for the major regioisomer only. 1H NMR (600 MHz, CDCl3) δ 7.63 (d, J = 8.1 Hz, 2 H), 7.35 (d, J = 8.1 Hz, 2 H), 4.03 (t, J = 6.1 Hz, 2 H), 3.92−3.89 (dd, J = 12.1, 2.4 Hz, 1 H), 3.65 (dt, J = 12.1, 2.4 Hz, 1 H), 3.57−3.48 (m, 3 H), 2.44 (s, 3 H), 2.37 (dt, J = 11.5, 3.2 Hz, 1 H), 2.03 (t, J = 10.5 Hz, 1 H), 1.82−1.73 (m, 1 H), 1.70−1.60 (m, 1 H), 1.46 (q, J = 7.3 Hz, 2 H), 1.18 (s, 9 H); 13C{1H} NMR (150 MHz, CDCl3) δ 178.5, 143.9, 132.0, 129.7, 127.8, 74.8, 65.9, 63.9, 50.3, 45.5, 38.7, 29.6, 27.1, 24.5, 21.5; IR (neat) 2957, 2865, 1721, 1447, 1479, 1344, 1287, 1162, 1098, 954 cm−1; HRMS (ESI) m/z calcd for C19H29NO5S [(M + Na)+] 406.1664, found 406.1664.
2-(3-(4-Tosylmorpholin-2-yl)propyl)isoindoline-1,3-dione (16).
This compound was prepared according to general procedure B using N-(2-hydroxyethyl)-4-methylbenzenesulfonamide (54 mg, 0.25 mmol) and 2-(pent-4-en-1-yl)isoindoline-1,3-dione (94 mg, 0.4375 mmol). After purification by column chromatography on SiO2 (10–30% EtOAc/hexanes), the title compound was isolated as a white solid (63 mg, 59% yield). Note that the reported spectral data are for the major regioisomer only. 1H NMR (600 MHz, CDCl3) δ 7.86−7.81 (m, 2 H), 7.74−7.70 (m, 2 H), 7.62 (d, J = 8.1 Hz, 2 H), 7.34 (d, J = 8.1 Hz, 2 H), 3.85 (dd, J = 11.7, 2.0 Hz, 1 H), 3.68 (dt, J = 7.0, 2.9 Hz, 2 H), 3.63 (dt, J = 11.5, 2.3 Hz, 1 H), 3.56−3.46 (m, 3 H), 2.45 (s, 3 H), 2.34 (dt, J = 11.5, 3.2 Hz, 1 H), 2.01 (t, J = 11.1 Hz, 1 H), 1.91−1.79 (m, 1 H), 1.77−1.66 (m, 1 H), 1.50−1.37 (m, 2 H); 13C{1H} NMR (150 MHz, CDCl3) δ 168.4, 143.9, 133.9, 132.0, 129.7, 127.8, 123.2, 74.8, 65.8, 50.2, 45.5, 37.7, 30.4, 24.5, 21.5; IR (neat) 2922, 2871, 1771, 1708, 1432, 1465, 1455, 1400, 1331, 1163, 1111 cm−1; HRMS (ESI) m/z calcd for C22H24N2O5S [(M + Na)+] 451.1304, found 451.1302.
2-Isobutyl-4-tosylmorpholine (17).
This compound was prepared according to general procedure B using N-(2-hydroxyethyl)-4-methylbenzenesulfonamide (54 mg, 0.25 mmol) and 4-methyl-1-pentene (56 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10% EtOAc/hexanes), the title compound was isolated as a white solid (52 mg, 70% yield). The spectral data of this compound matched those from previously reported literature.8 1H NMR (600 MHz, CDCl3) δ 7.63 (d, J = 8.4 Hz, 2 H), 7.34 (d, J = 8.1 Hz, 2 H), 3.88 (dd, J = 11.6, 2.0 Hz, 1 H), 3.65 (dt, J = 11.5, 2.4 Hz, 1 H), 3.59−3.54 (m, 1 H), 3.52 (d, J = 11.7 Hz, 2 H), 2.44 (s, 3 H), 2.37 (dt, J = 11.6, 3.3 Hz, 1 H), 2.01 (t, J = 10.6 Hz, 1 H), 1.79−1.70 (m, 1 H), 1.40−1.32 (m, 1 H), 1.16−1.09 (m, 1 H), 0.90 (d, J = 3.3 Hz, 3 H), 0.89 (d, J = 3.3 Hz, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 143.8, 132.0, 129.7, 127.8, 73.5, 65.8, 50.6, 45.5, 42.1, 24.0, 23.1, 22.1, 21.5; IR (neat) 2953, 2865, 1706, 1597, 1454, 1346, 1336, 1160, 1102, 1086 cm−1.
2-Benzyl-4-tosylmorpholine (18).
This compound was prepared according to general procedure B using N-(2-hydroxyethyl)-4-methylbenzenesulfonamide (54 mg, 0.25 mmol) and allylbenzene (59 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10% EtOAc/hexanes), the title compound was isolated as a white solid (44 mg, 53% yield). The spectral data of this compound matched those from previously reported literature.26 1H NMR (600 MHz, CDCl3) δ 7.62 (d, J = 8.1 Hz, 2 H), 7.36 (d, J = 7.8 Hz, 2 H), 7.31 (t, J = 7.4 Hz, 2 H), 7.24 (t, J = 7.2 Hz, 1 H), 7.17 (d, J = 7.3 Hz, 2 H), 3.90 (d, J = 11.5 Hz, 1 H), 3.81−3.73 (m, 1 H), 3.64 (dt, J = 11.5, 2.2 Hz, 1 H), 3.56 (d, J = 11.5 Hz, 1 H), 3.50 (d, J = 11.5 Hz, 1 H), 2.79 (dd, J = 13.9, 7.3 Hz, 1 H), 2.67 (dd, J = 13.9, 5.6 Hz, 1 H), 2.46 (s, 3 H), 2.42 (dt, J = 11.4, 2.8 Hz, 1 H), 2.14 (t, J = 10.6 Hz, 1 H); 13C{1H} NMR (150 MHz, CDCl3) δ 143.9, 136.9, 132.1, 129.7, 129.1, 128.5, 127.8, 126.6, 75.9, 65.9, 49.9, 45.4, 39.6, 21.5; IR (neat) 2967, 2858, 1724, 1596, 1445, 1345, 1164, 1117, 1097, 1068 cm−1.
2-Cyclohexyl-4-tosylmorpholine (19).
This compound was prepared according to general procedure B using N-(2-hydroxyethyl)-4-methylbenzenesulfonamide (54 mg, 0.25 mmol) and vinylcyclohexane (60 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (20 mg, 25% yield). 1H NMR (600 MHz, CDCl3) δ 7.64 (d, J = 8.1 Hz, 2 H), 7.35 (d, J = 7.8 Hz, 2 H), 3.92−3.86 (dd, J = 12.1, 1.8 Hz, 1 H), 3.67−3.57 (m, 2 H), 3.51 (d, J = 11.5 Hz, 1 H), 3.28−3.22 (m, 1 H), 2.45 (s, 3 H), 2.35 (dt, J = 11.4, 3.3 Hz, 1 H), 2.08 (t, J = 10.7 Hz, 1 H), 1.83 (d, J = 12.9 Hz, 1 H), 1.77−1.68 (m, 2 H), 1.67−1.56 (m, 2 H), 1.39−1.29 (m, 1 H), 1.23−1.10 (m, 3 H), 1.07−0.91 (m, 2 H); 13C{1H} NMR (150 MHz, CDCl3) δ 143.8, 132.2, 129.7, 127.8, 79.4, 65.9, 48.3, 45.6, 40.8, 28.7, 28.5, 26.3, 25.9, 25.8, 21.5; IR (neat) 2921, 2846, 1600, 1441, 1338, 1308, 1162, 1101, 1092, 1063 cm−1; HRMS (ESI) m/z calcd for C17H26NO3S [(M + H)+] 324.1633, found 324.1613.
N, 4-Dimethyl-N-((4-tosylmorpholin-2-yl)methyl)-benzenesulfonamide (20).
This compound was prepared according to general procedure B using N-(2-hydroxyethyl)-4-methylbenzenesulfonamide (54 mg, 0.25 mmol) and N-allyl-N,4-dimethylbenzenesulfonamide (99 mg, 0.4375 mmol). After purification by column chromatography on SiO2 (10–30% EtOAc/hexanes), the title compound was isolated as a white solid (37 mg, 34% yield). 1H NMR (600 MHz, CDCl3) δ 7.64 (d, J = 8.1 Hz, 2 H), 7.35 (d, J = 7.8 Hz, 2 H), 3.92−3.86 (dd, J = 12.1, 1.8 Hz, 1 H), 3.67−3.57 (m, 2 H), 3.51 (d, J = 11.5 Hz, 1 H), 3.28−3.22 (m, 1 H), 2.45 (s, 3 H), 2.35 (dt, J = 11.4, 3.3 Hz, 1 H), 2.08 (t, J = 10.7 Hz, 1 H), 1.83 (d, J = 12.9 Hz, 1 H), 1.77−1.68 (m, 2 H), 1.67−1.56 (m, 2 H), 1.39−1.29 (m, 1 H), 1.23−1.10 (m, 3 H), 1.07−0.91 (m, 2 H); 13C{1H} NMR (150 MHz, CDCl3) δ 143.8, 132.2, 129.7, 127.8, 79.4, 65.9, 48.3, 45.6, 40.8, 28.7, 28.5, 26.3, 25.9, 25.8, 21.5; IR (neat) 2920, 2844, 1596, 1453, 1345, 1329, 1153, 1108, 1087 cm−1; HRMS (ESI) m/z calcd for C20H27N2O5S2 [(M + H)+] 439.1361, found 439.1364.
2-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-4-tosylmorpholine (21).
This compound was prepared according to general procedure B using N-(2-hydroxyethyl)-4-methylbenzenesulfonamide (54 mg, 0.25 mmol) and tert-butyldimethylsilyl-protected homoallylic alcohol (82 mg, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (53 mg, 53% yield). 1H NMR (600 MHz, CDCl3) δ 7.64 (d, J = 7.8 Hz, 2 H), 7.35 (d, J = 8.1 Hz, 2 H), 3.88 (d, J = 10.3 Hz, 1 H), 3.73−3.63 (m, 4 H), 3.60 (d, J = 11.2 Hz, 1 H), 3.53 (d, J = 11.2 Hz, 1 H), 2.45 (s, 3 H), 2.42−2.35 (m, 1 H), 2.09 (t, J = 10.7 Hz, 1 H), 1.67−1.53 (m, 2 H), 0.90 (s, 9 H), 0.05 (s, 3 H), 0.04 (s, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 143.8, 132.2, 129.7, 127.8, 72.5, 65.8, 58.7, 50.5, 45.5, 36.2, 25.9, 21.5, 18.3, −5.4; IR (neat) 2953, 2929, 2887, 2857, 1597, 1454, 1349, 1250, 1161, 1110 cm−1; HRMS (ESI) m/z calcd for C19H34NO4SSi [(M + H)+] 400.1978, found 400.1958.
(8R,9S,13S,14S,17S)-13-Methyl-3-(4-((2-nitrophenyl)sulfonyl)-morpholin-2-yl)-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-17-yl Pivalate (22).
This compound was prepared according to general procedure A using N-(2-hydroxyethyl)-2-nitrobenzenesulfonamide (62 mg, 0.25 mmol) and (8R,9S,13S,14S,17S)-13-methyl-3-vinyl-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-17-yl pivalate (160 mg, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20%EtOAc/hexanes), the title compound was isolated as a white solid (94 mg, 62% yield). 1H NMR (600 MHz, CDCl3) δ 7.94 (d, J = 7.8 Hz, 1 H), 7.71 (t, J = 7.6 Hz, 1 H), 7.67 (t, J = 7.6 Hz, 1 H), 7.62 (d, J = 7.8 Hz, 1 H), 7.25 (s, 1 H), 7.10 (d, J = 7.8 Hz, 1 H), 7.06 (br. s., 1 H), 4.65 (t, J = 8.3 Hz, 1 H), 4.49 (d, J = 9.8 Hz, 1 H), 4.09 (d, J = 11.5 Hz, 1 H), 3.85−3.75 (m, 2 H), 3.71 (d, J = 12.5 Hz, 1 H), 3.05 (t, J = 12.0 Hz, 1 H), 2.86 (d, J = 5.1 Hz, 2 H), 2.79 (t, J = 11.5 Hz, 1 H), 2.29−2.17 (m, 3 H), 1.94−1.86 (m, 2 H), 1.80−1.70 (m, 1 H), 1.53−1.33 (m, 8 H), 1.20 (s, 9 H), 0.83 (s, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 178.6, 148.4, 140.5, 137.0, 135.5, 133.9, 131.6, 131.0, 130.9, 130.9, 126.6, 126.6, 125.6, 124.1, 123.4, 123.3, 82.2, 77.6, 77.5, 66.5, 51.5, 51.5, 49.8, 45.3, 44.2, 43.0, 38.9, 38.2, 36.9, 29.5, 29.4, 27.5, 27.2, 27.1, 25.9, 23.3, 12.1; IR (neat) 2970, 1725, 1544, 1479, 1438, 1370, 1289, 1161, 1130, 1100 cm−1; HRMS (ESI) m/z calcd for C33H43N2O7S [(M + H)+] 611.2791, found 611.2795.
4-Tosyl-2-(((1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl)oxy)-methyl)morpholine (23).
This compound was prepared according to general procedure B using N-(2-hydroxyethyl)-4-methylbenzenesulfonamide (54 mg, 0.25 mmol) and 2-(allyloxy)-1,7,7-trimethylbicyclo[2.2.1]heptane (85 mg, 0.4375 mmol). After purification by column chromatography on SiO2 (10–30% EtOAc/hexanes), the title compound was isolated as a white solid (38 mg, 36% yield). 1H NMR (600 MHz, CDCl3) δ 7.73 (d, J = 9.2 Hz, 2 HMin), 7.65 (d, J = 7.8 Hz, 2 HMaj), 7.35 (d, J = 8.1 Hz, 2 HMaj), 7.32 (d, J = 7.2 Hz, 2 HMin) 3.98 (dd, J = 11.7, 4.2 Hz, 1 HMin), 3.91 (d, J = 11.7 Hz, 1 HMaj), 3.89−3.85 (m, 1 HMin), 3.82−3.64 (m, 3 HMaj + 3 HMin), 3.57−3.47 (m, 3 HMaj), 3.21 (t, J = 12.0 Hz, 1 HMin), 2.45 (s, 3 HMaj), 2.44 (s, 3 HMin), 2.43−2.37 (m, 1 HMaj), 2.20 (q, J = 12.5 Hz, 1 HMaj), 2.12−2.0 (m, 1 HMaj + 1 HMin), 1.94−1.84 (m, 1 HMaj + 1 HMin), 1.75−1.65 (m, 1 HMaj + 1 HMin), 1.65−1.57 (m, 2 HMaj), 1.23−1.12 (m, 2 HMaj + 2 HMin), 1.00−0.90 (m, 1 HMaj + 2 HMin), 0.88−0.77 (6 singlets, 9 HMaj + 9 HMin); 13C{1H} NMR (150 MHz, CDCl3) δ 143.9, 143.5, 137.79, 137.76, 132.1, 132.0, 129.8, 129.7, 127.9, 127.0, 85.7, 85.64, 85.57, 85.1, 74.4, 74.2, 70.6, 70.4, 66.3, 66.2, 66.02, 66.04, 65.9, 65.8, 65.6, 65.1, 52.5, 52.1, 49.2, 48.2, 47.89, 47.86, 47.8, 47.7, 45.58, 45.57, 45.0, 44.9, 41.59, 41.56, 36.08, 36.05, 36.0, 28.2, 26.6, 26.51, 26.48, 21.6, 21.5, 19.7, 18.8, 14.03, 14.01, 14.98, 13.9; IR (neat) 2949, 2872, 1598, 1453, 1388, 1349, 1306, 1164, 1095, 1019 cm−1; HRMS (ESI) m/z calcd for C22H33NNaO4S [(M + Na)+] 430.2028, found 430.2012.
2-((((2S)-2,5,7,8-Tetramethyl-2-(4,8,12-trimethyltridecyl)-chroman-6-yl)oxy)methyl)-4-tosylmorpholine (24).
This compound was prepared according to general procedure B using N-(2-hydroxyethyl)-4-methylbenzenesulfonamide (54 mg, 0.25 mmol) and (S)-6-(allyloxy)-2,5,7,8-tetramethyl-2-(4-methylpentyl)chromane (145 mg, 0.4375 mmol). After purification by column chromatography on SiO2 (10–30% EtOAc/hexanes), the title compound was isolated as a white solid (49 mg, 29% yield). 1H NMR (600 MHz, CDCl3) δ 7.65 (d, J = 8.1 Hz, 2 H), 7.36 (d, J = 7.8 Hz, 2 H), 4.01 (d, J = 11.5 Hz, 1 H), 3.97−3.89 (m, 1 H), 3.82−3.73 (m, 2 H), 3.66 (d, J = 3.0 Hz, 2 H), 3.57 (d, J = 11.2 Hz, 1 H), 2.56 (t, J = 6.6 Hz, 2 H), 2.52−2.40 (m, 5 H), 2.13 (s, 3 H), 2.09 (s, 3 H), 2.07 (s, 3 H), 1.86−1.70 (m, 2 H), 1.63−1.50 (m, 3 H), 1.50−1.41 (m, 2 H), 1.41−1.34 (m, 3 H), 1.34−1.24 (m, 8 H), 1.23 (s, 3 H), 1.19−1.01 (m, 7 H), 0.93−0.80 (4ds, 3*4 = 12 H); 13C{1H} NMR (150 MHz, CDCl3) δ 148.0, 147.4, 144.0, 131.9, 129.8, 127.9, 127.5, 125.6, 123.0, 117.6, 74.8, 74.3, 72.8, 66.0, 47.5, 45.5, 40.0, 39.3, 37.5, 37.4, 37.4, 37.4, 37.3, 37.3, 32.8, 32.7, 32.7, 31.2, 31.1, 28.0, 24.8, 24.8, 24.4, 23.8, 22.7, 22.6, 21.5, 21.0, 21.0, 20.6, 19.7, 19.7, 19.6, 19.6, 19.6, 12.6, 11.8; IR (neat) 2923, 2865, 1598, 1455, 1350, 1377, 1350, 1257, 1167, 1087, 1060 cm−1; HRMS (ESI) m/z calcd for C41H65NNaO5S [(M + Na)+] 706.4481, found 706.4471.
(5S)-5-Methyl-4-((2-nitrophenyl)sulfonyl)-2-phenylmorpholine (25).
This compound was prepared according to general procedure A using 2-nitrobenzenesulfonamide-protected (S)-alaninol (65 mg, 0.25 mmol) and styrene (50 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (65 mg, 72% yield). 1H NMR (600 MHz, CDCl3) δ 8.13 (d, J = 7.8 Hz, 1 HMaj), 8.09 (d, 7.8 Hz, 1 HMin), 7.79−7.66 (m, 3 HMaj + 3 HMin), 7.44−7.29 (m, 5 HMaj + 5 HMin), 4.68 (dd, J = 8.5, 2.4 Hz, 1 Hmaj), 4.47 (dd, J = 11.0, 2.7 Hz, 1 HMin), 4.09−4.02 (m, 1 HMin), 3.97 (dd, J = 12.8, 2.8 Hz, 1 HMaj), 3.89 (dd, J = 12.0, 3.2 Hz, 1 HMaj), 3.86 (m, 2 HMin), 3.71 (dd, J = 13.6, 2.6 Hz, 1 HMin), 3.61 (m, 1 HMaj), 3.41 (dd, J = 11.7, 8.8 Hz, 1 HMaj), 3.27 (dd, J = 13.4, 11.0 Hz, 1 HMin), 3.18 (dd, J = 12.9, 8.8 Hz, 1 HMaj), 1.41 (d, J = 6.8 Hz, 3 HMin), 1.11 (d, J = 6.3 Hz, 3 HMaj); 13C{1H} NMR (150 MHz, CDCl3) δ 147.8, 147.7, 138.5, 138.1, 134.6, 133.8, 133.7, 133.5, 132.0, 131.9, 130.9, 130.2, 128.5, 128.5, 128.4, 128.1, 126.3, 126.0, 124.4, 124.3, 78.0, 76.5, 71.2, 70.7, 52.9, 50.2, 48.8, 46.3, 15.1, 15.0; IR (neat) 2979, 2920, 2864, 1540, 1441, 1386, 1349, 1265, 1162, 1125 cm−1; HRMS (ESI) m/z calcd for C17H19N2O5S [(M + H)+] 363.1015, found 363.1016.
5,5-Dimethyl-4-((2-nitrophenyl)sulfonyl)-2-phenylmorpholine (26).
This compound was prepared according to general procedure A using 2-nitrobenzenesulfonamide-protected 2-amino-2-methylpropan-1-ol (67 mg, 0.25 mmol) and styrene (50 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (61 mg, 65% yield). 1H NMR (600 MHz, CDCl3) δ 8.25−8.19 (m, 1 H), 7.77−7.68 (m, 3 H), 7.47 (d, J = 7.3 Hz, 2 H), 7.40 (t, J = 7.6 Hz, 2 H), 7.37−7.32 (m, 1 H), 4.64 (dd, J = 10.9, 2.6 Hz, 1 H), 3.88 (dd, J = 13.3, 2.8 Hz, 1 H), 3.51 (d, J = 11.7 Hz, 1 H), 3.46 (d, J = 11.5 Hz, 1 H), 3.20 (dd, J = 13.2, 11.0 Hz, 1 H), 1.59 (s, 3 H), 1.14 (s, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 147.5, 138.5, 136.3, 133.5, 132.2, 130.4, 128.5, 128.2, 126.1, 124.5, 77.9, 77.3, 57.7, 48.8, 23.2, 21.2; IR (neat) 2922, 2862, 1725, 1536, 1463, 1366, 1333, 1304, 1159, 1088 cm−1; HRMS (ESI) m/z calcd for C18H20N2NaO5S [(M + Na)+] 399.0991, found 399.0967.
(5S)-5-Benzyl-4-((2-nitrophenyl)sulfonyl)-2-phenylmorpholine (27).
This compound was prepared according to general procedure A using 2-nitrobenzenesulfonamide-protected (S)-phenylalaninol (84 mg, 0.25 mmol) and styrene (50 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (78 mg, 71% yield). 1H NMR (600 MHz, CDCl3) δ 8.07 (d, J = 7.3 Hz, 1 HMaj), 7.93 (d, J = 7.8 Hz, 1 HMin), 7.75−7.56 (m, 3 HMaj + 3 HMin), 7.46−7.41 (m, 1 HMaj), 7.39 (t, J = 7.3 Hz, 1 HMaj), 7.36−7.31 (m, 1 HMin), 7.31−7.12 (m, 8 HMaj + 8 HMin), 4.90 (t, J = 3.8 Hz, 1 HMaj), 4.51 (dd, J = 10.8, 2.4 Hz, 1 HMin) 4.00 (dd, J = 13.4, 3.7 Hz, 1 HMaj + 1 HMin), 3.91 (d, J = 11.7 Hz, 1 HMin), 3.88−3.80 (m, 1 HMaj + 1 HMin), 3.76 (dd, J = 13.4, 4.6 Hz, 1 HMaj), 3.73−3.66 (m, 1 HMaj + 1 HMin), 3.46 (dd, J = 12.0, 4.2 Hz, 1 HMaj), 3.38 (dd, J = 13.7, 11.2 Hz, 1 HMin), 3.26 (dd, J = 13.1, 9.9 Hz, 1 HMin), 3.05−2.97 (m, 1 HMaj + 1 HMin), 2.97−2.92 (m, 1 HMaj); 13C{1H} NMR (150 MHz, CDCl3) δ 147.7, 147.5, 138.5, 137.7, 137.4, 137.0, 133.82, 133.78, 133.63, 133.56, 132.0, 131.9, 130.8, 130.7, 129.5, 129.3, 128.7, 128.59, 128.57, 128.43, 128.40, 127.8, 126.83, 126.76, 126.5, 126.0, 124.49, 124.45, 78.1, 74.0, 67.8, 63.4, 56.9, 55.0, 47.1, 45.7, 35.2; IR (neat) 3029, 2927, 1059, 1539, 1496, 1453, 1345, 1163, 1124, 1085 cm−1; HRMS (ESI) m/z calcd for C23H23N2O5S [(M + H)+] 439.1328, found 439.1303.
(5S)-5-Isopropyl-4-((2-nitrophenyl)sulfonyl)-2-phenylmorpholine (28).
This compound was prepared according to general procedure A using 2-nitrobenzenesulfonamide-protected (S)-valinol (72 mg, 0.25 mmol) and styrene (50 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a colorless oil (52 mg, 53% yield). 1H NMR (600 MHz, CDCl3) δ 8.08−8.10 (m, 1 HMin) 7.95 (d, J = 8.1 Hz, 1 HMaj), 7.765−7.71 (m, 2 HMin), 7.70−7.66 (m, 1 HMin), 7.76 (d, J = 7.5 Hz, 1 HMaj), 7.59 (d, J = 8.3 Hz, 2 HMaj), 7.40−7.35 (m, 1 HMaj + 1 HMin), 7.34−7.30 (m, 1 HMin) 7.26−7.21 (m, 2 HMaj), 7.20−7.14 (m, 2 HMaj + 2 HMin), 4.87 (t, J = 3.3 Hz, 1 HMaj), 4.07 (dd, J = 12.0, 3.4 Hz, 1 HMin), 4.18 (d, J = 11.4 Hz, 1HMin) 4.07 (dd, J = 12.1, 3.3 Hz, 1 HMaj), 4.02−4.94 (m, 1 HMaj + 1 HMin), 3.87−3.79 (m, 2 HMaj) 3.74 (dd, J = 6.2, 3.0 Hz, 1 HMin) 3.45−3.39 (m, 1 HMaj + 1 HMin), 3.18 (dd, J = 15.2, 11.1 Hz, 1 HMin) 2.56−2.48 (m, 1 HMaj), 2.47−2.41 (m, 1 HMin), 1.06 (d, J = 6.9 Hz, 3 HMin) 0.99 (d, J = 6.6 Hz, 3 HMaj), 0.89 (d, J = 6.8 Hz, 3 HMaj), 0.80 (d, J = 6.8 Hz, 1 HMaj); 13C{1H} NMR (150 MHz, CDCl3) δ 147.5, 138.5, 137.9, 134.6, 134.0, 133.5, 133.4, 131.8, 131.7, 131.2, 130.7, 128.5, 128.3, 127.3, 126.1, 125.9, 124.4, 124.3, 77.9, 72.7, 67.5, 62.4, 60.5, 60.0, 47.5, 44.2, 29.7, 26.3, 25.4, 20.1, 19.6, 19.6, 19.3; IR (neat) 2595, 1591, 1539, 1496, 1454, 1344, 1269, 1158, 1125, 1091, 1073 cm−1; HRMS (ESI) m/z calcd for C19H23N2O5S [(M + H)+] 391.1328, found 391.1319.
(5S)-5-Isobutyl-4-((2-nitrophenyl)sulfonyl)-2-phenylmorpholine (29).
This compound was prepared according to general procedure A using 2-nitrobenzenesulfonamide-protected (S)-leucenol (76 mg, 0.25 mmol) and styrene (50 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (65 mg, 72% yield). 1H NMR (600 MHz, CDCl3) δ 8.08 (dd, J = 7.4, 1.3 Hz, 1 HMin), 8.02 (d, J = 8.1 Hz, 1 HMaj), 7.73−7.66 (m, 1 HMaj + 3 HMin), 7.64 (t, J = 9.6 Hz, 2 HMaj), 7.34 (d, J = 4.2 Hz, 2 HMaj), 7.32−7.27 (m, 1 HMaj + 3 HMin), 7.27−7.20 (m, 2 HMaj + 2 HMin), 4.79 (t, J = 4.2 Hz, 1 HMaj), 4.39 (dd, J = 11.1, 2.8 Hz, 1 HMin), 3.91 (dd, J = 13.6, 3.5 Hz, 2 HMin), 3.89−3.83 (m, 1 HMin), 3.80−3.74 (m, 2 HMin), 3.66 (dd, J = 13.6, 4.8 Hz, 2 HMaj), 3.52 (dd, J = 12.0, 4.6 Hz, 1 HMaj), 3.22 (dd, J = 14.0, 11.1 Hz, 1 HMin), 1.88−1.84 (m, 1 HMin), 1.77−1.68 (m, 1 HMaj), 1.62−1.43 (m, 2 HMaj + 1 HMin), 1.29−1.22 (m, 2 HMin), 0.88 (2ds, J = 4.5 Hz, 6 HMin), 0.81 (d, J = 6.6 Hz, 6 Hmaj); 13C{1H} NMR (150 MHz, CDCl3) δ 147.8, 147.7, 138.5, 137.8, 134.3, 134.0, 133.7, 133.6, 131.84, 131.77, 131.0, 130.8, 128.6, 128.5, 128.3, 127.8, 126.5, 126.0, 124.5, 124.4, 77.9, 74.3, 68.9, 65.1, 54.3, 51.6, 46.8, 46.3, 37.7, 37.3, 25.1, 24.8, 23.6, 22.9, 22.2, 21.4; IR (neat) 2957, 2928, 2869, 1541, 1453, 1367, 1349, 1298, 1159, 1098 cm−1; HRMS (ESI) m/z calcd for C20H25N2O5S [(M + H)+] 405.1484, found 405.1500.
(5S)-5-Isobutyl-4-((2-nitrophenyl)sulfonyl)-2-phenylmorpholine (30).
This compound was prepared according to general procedure A using (S)-N-(1-hydroxy-3,3-dimethylbutan-2-yl)-4-nitrobenzenesulfonamide (76 mg, 0.25 mmol) and styrene (50 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (46 mg, 45% yield). Note that the reported spectral data are for the major diastereomer only. 1H NMR (600 MHz, CDCl3) δ 7.84 (d, J = 8.1 Hz, 1 H), 7.60 (t, J = 7.7 Hz, 1 H), 7.53 (d, J = 7.8 Hz, 1 H), 7.43 (t, J = 7.7 Hz, 1 H), 7.15−7.07 (m, 3 H), 7.00 (d, J = 6.1 Hz, 2 H), 4.73 (t, J = 4.5 Hz, 1 H), 4.32−4.23 (m, 1 H), 4.08−3.96 (m, 2 H), 3.87−3.77 (m, 2 H), 1.11 (s, 9 H); 13C{1H} NMR (150 MHz, CDCl3) δ 147.8, 139.4, 133.6, 133.3, 131.9, 131.4, 128.2, 127.0, 125.2, 124.3, 72.2, 62.0, 59.5, 45.4, 36.2, 28.3; IR (neat) 2968, 2875, 1541, 1497, 1368, 1347, 1166, 1123, 1095, 1058 cm−1; HRMS (ESI) m/z calcd for C20H25N2O5S [(M + H)+] 405.1484, found 405.1477.
((5S)-5-((S)-sec-Butyl)-4-((2-nitrophenyl)sulfonyl)-2-phenylmorpholine (31).
This compound was prepared according to general procedure A using 2-nitrobenzenesulfonamide-protected (S)-isoleucenol (76 mg, 0.25 mmol) and styrene (50 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–20% EtOAc/hexanes), the title compound was isolated as a white solid (56 mg, 53% yield). 1H NMR (600 MHz, CDCl3) δ 8.11−8.05 (m, 1 HMin) 7.94 (d, J = 7.8 Hz, 1 HMaj), 7.77−7.70 (m, 2 HMin), 7.66−7.62 (m, 1 HMin) 7.61 (d, J = 7.8 Hz, 1 HMaj), 7.56 (d, J = 7.8 Hz, 1 HMaj), 7.54 (t, J = 7.2 Hz, 1 HMaj), 7.36−7.31 (m, 1 HMaj), 7.31−7.26 (m, 1 HMin), 7.24−7.19 (m, 2 HMaj + 2 HMin), 7.19−7.13 (m, 2 HMaj + 2 HMin), 4.80 (t, J = 4.0 Hz, 1 HMaj), 4.35 (dd, J = 10.8, 3.0 Hz, 1 HMin), 4.15 (d, J = 12.0 Hz, 1 HMin), 4.03 (dd, J = 12.0, 3.4 Hz, 1 HMaj), 3.94 (dd, J = 15.0, 3.0 Hz, 1 HMin), 3.81 (d, J = 4.2 Hz, 2 HMaj), 3.78 (dd, J = 12.0, 3.9 Hz, 1 HMaj), 3.69 (dd, J = 12.0, 2.4 Hz, 1 HMin), 3.50−3.45 (m, 1 HMaj), 3.13 (dd, J = 15.0, 11.4 Hz, 1 HMin), 2.24−2.07 (m, 1 HMaj + 1 HMin), 1.53−1.46 (m, 1 HMaj), 0.99 (d, J = 6.6 Hz, 3 HMin), 0.98−0.89 (m, 1 HMaj + 1 HMin) 0.88 (d, J = 6.6 Hz, 3 HMaj), 0.84 (t, J = 7.2 Hz, 3 HMaj), 0.80 (t, J = 6.0 Hz, 3 HMin); 13C{1H} NMR (150 MHz, CDCl3) δ 147.68, 147.66, 138.5, 138.2, 134.6, 134.0, 133.5, 133.4, 131.8, 131.6, 131.1, 130.8, 128.6, 128.4, 128.3, 127.5, 126.1, 125.8, 124.4, 124.3, 77.7, 73.4, 67.5, 63.0, 59.9, 58.9, 47.5, 45.7, 33.2, 31.8, 25.8, 24.9, 16.0, 15.4, 11.8, 11.4; IR (neat) 2972, 2933, 2876, 1544, 1372, 1348, 1061 cm−1; HRMS (ESI) m/z calcd for C20H24N2NaO5S [(M + Na)+] 427.1304, found 427.1316.
(±)-Methyl (3R,6S)-4-((2-Nitrophenyl)sulfonyl)-6-phenylmorpholine-3-carboxylate (32).
This compound was prepared according to general procedure A using 2-nitrobenzenesulfonamide-protected (d)-serine methyl ester (76 mg, 0.25 mmol) and styrene (50 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–30% EtOAc/hexanes), the title compound was isolated as a white solid (54 mg, 53% yield). 1H NMR (600 MHz, CDCl3) δ 8.01 (dd, J = 7.8, 1.5 Hz, 1 H), 7.77−7.67 (m, 2 H), 7.65 (dd, J = 7.6, 1.2 Hz, 1 H), 7.39−7.29 (m, 5 H), 4.68−4.63 (m, 1 H), 4.59 (d, J = 11.7 Hz, 1 H), 4.54 (dd, J = 11.0, 2.9 Hz, 1 H), 4.01 (dd, J = 11.8, 3.5 Hz, 1 H), 3.83 (dd, J = 12.8, 2.3 Hz, 1 H), 3.72 (s, 3 H), 3.53 (dd, J = 12.7, 11.0 Hz, 1 H); 13C{1H} NMR (150 MHz, CDCl3) δ 169.3, 147.7, 137.9, 133.7, 132.7, 131.8, 130.7, 128.6, 128.6, 126.2, 124.2, 78.2, 68.8, 55.4, 52.6, 48.3; IR (neat) 2923, 1745, 1591, 1541, 1497, 1439, 1352, 1292, 1259, 1218, 1165 cm−1; HRMS (ESI) m/z calcd for C18H19N2O7S [(M + H)+] 407.0913, found 407.0924.
Methyl (2R)-2-Methyl-4-((2-nitrophenyl)sulfonyl)-6-phenylmorpholine-3-carboxylate (33).
This compound was prepared according to general procedure A using methyl ((4-nitrophenyl)sulfonyl)-d-allothreoninate (80 mg, 0.25 mmol) and styrene (50 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–30% EtOAc/hexanes), the title compound was isolated as a colorless oil (43 mg, 41% yield). 1H NMR (600 MHz, CDCl3) δ 8.01 (d, J = 7.8 Hz, 1 HMaj), 7.97 (d, J = 8.1 Hz, 1 HMin), 7.76−7.64 (m, 3 HMaj + 2 HMin), 7.62 (d, J = 7.6 Hz, 1 HMin), 7.41−7.29 (m, 5 HMaj + 5 HMin), 4.98 (dd, J = 11.2, 3.4 Hz, 1 HMin), 4.81 (q, J = 6.8 Hz, 1 HMin), 4.64 (dd, J = 10.7, 3.4 Hz, 1 HMaj), 4.49−4.43 (m, 1 HMaj + 1 HMin), 4.09 (m, 1 HMaj), 3.92 (dd, J = 12.8, 2.8 Hz, 1 HMin), 3.84 (t, J = 11.4 Hz, 1 HMaj), 3.78 (dd, J = 12.9, 3.6 Hz, 1 HMin), 3.68 (s, 3 HMaj), 3.67 (s, 3 HMin), 3.49 (t, J = 12.1 Hz, 1 HMin), 1.58 (d, J = 6.8 Hz, 3 HMin), 1.40 (d, J = 6.6 Hz, 3 HMaj); 13C{1H} NMR (150 MHz, CDCl3) δ 169.5, 168.7, 147.8, 147.7, 138.3, 138.2, 133.8, 133.6, 132.7, 132.4, 131.8, 131.7, 130.9, 130.4, 128.6, 128.5, 126.5, 126.4, 124.3, 124.1, 78.6, 74.3, 70.6, 70.5, 69.4, 58.7, 58.1, 52.5, 51.9, 48.1, 47.4, 18.3, 16.4; IR (neat) 2954, 1743, 1541, 1497, 1439, 1355, 1248, 1165, 1100, 1070 cm−1; HRMS (ESI) m/z calcd for C19H21N2O7S [(M + H)+] 421.1069, found 421.1071.
2-Nitro-4-((4aR,9aS)-2-phenyl-2,3,9,9a-tetrahydroindeno[2,1-b]-[1,4]oxazin-4(4aH)-yl)benzenesulfonic acid (34).
This compound was prepared according to general procedure A using N-((1R,2S)-2-hydroxy-2,3-dihydro-1H-inden-1-yl)-2-nitrobenzenesulfonamide (84 mg, 0.25 mmol) and styrene (50 μL, 0.4375 mmol). After purification by column chromatography on SiO2 (10–30% EtOAc/hexanes), the title compound was isolated as a white solid (52 mg, 46% yield). 1H NMR (600 MHz, CDCl3) δ 8.02 (d, J = 7.8 Hz, 1 H), 7.75−7.65 (m, 2 H), 7.62 (t, J = 8.4 Hz, 1 H), 7.31−7.19 (m, 8 H), 7.18 (t, J = 6.8 Hz, 1 H), 5.19 (d, J = 4.6 Hz, 1 H), 4.86−4.76 (m, 2 H), 3.84−3.69 (m, 2 H), 3.17−3.02 (m, 2 H); 13C{1H} NMR (150 MHz, CDCl3) δ 147.5, 141.0, 137.9, 133.8, 133.5, 131.9, 131.3, 128.5, 128.4, 127.6, 126.9, 126.2, 125.3, 125.2, 124.7, 73.7, 71.2, 60.3, 44.5, 36.8; IR (neat) 2988, 2972, 1740, 1539, 1441, 1365, 1351, 1342, 1161, 1062 cm−1; HRMS (ESI) m/z calcd for C23H20N2NaO5S [(M + Na)+] 459.0991, found 459.0982.
1-Phenylpent-4-en-1-one (6).
This compound was prepared according to general procedure C using allylic alcohol (17 μL, 0.25 mmol) and styrene (58 μL, 0.5 mmol). After purification by column chromatography on SiO2 (0–2% EtOAc/hexanes), the title compound was isolated as a colorless oil (26 mg, 65% yield). The spectral data of this compound matched those from previously reported literature.27 1H NMR (600 MHz, CDCl3) δ 7.98 (d, J = 7.3 Hz, 2 H), 7.57 (t, J = 7.2 Hz, 1 H), 7.48 (t, J = 7.7 Hz, 2 H), 5.96−5.88 (m, 1 H), 5.10 (dd, J = 17.1, 1.5 Hz, 1 H), 5.03 (dd, J = 10.2, 0.6 Hz, 1 H) 3.09 (t, J = 7.3 Hz, 2 H), 2.51 (q, J = 7.1 Hz, 2 H); 13C{1H} NMR (150 MHz, CDCl3) δ 199.4, 137.3, 136.9, 133.0, 128.6, 128.0, 115.3, 37.7, 28.1; IR (neat) 2925, 1732, 1683, 1598, 1449, 1362, 1270, 1206, 1108, 984 cm−1.
1-(4-(tert-Butyl)phenyl)pent-4-en-1-one (39).
This compound was prepared according to general procedure C using allylic alcohol (17 μL, 0.25 mmol) and p-(tert-butyl)styrene (91 μL, 0.5 mmol). After purification by column chromatography on SiO2 (0–2% EtOAc/hexanes), the title compound was isolated as a colorless oil (41 mg, 76% yield). The spectral data of this compound matched those from previously reported literature.28 1H NMR (600 MHz, CDCl3) δ 7.92 (d, J = 8.3 Hz, 2 H), 7.49 (d, J = 8.3 Hz, 2 H), 5.96−5.87 (m, 1 H), 5.10 (dd, J = 17.4, 0.9 Hz, 1 H), 5.02 (d, J = 10.3 Hz, 1 H), 3.06 (t, J = 7.4 Hz, 2 H), 2.51 (q, J = 7.0 Hz, 2 H), 1.35 (s, 9 H); 13C{1H} NMR (150 MHz, CDCl3) δ 199.1, 156.7, 137.4, 134.3, 128.0, 125.5, 115.2, 37.6, 35.1, 31.1, 28.2; IR (neat) 3078, 2963, 2869, 1682, 1605, 1463, 1406, 1270, 1190, 1107 cm−1.
1-(4-Methoxyphenyl)pent-4-en-1-one (40).
This compound was prepared according to general procedure C using allylic alcohol (17 μL, 0.25 mmol) and p-methoxy styrene (67 μL, 0.5 mmol). After purification by column chromatography on SiO2 (5–10% EtOAc/hexanes), the title compound was isolated as a white solid (26 mg, 55% yield). The spectral data of this compound matched those from previously reported literature.28 1H NMR (600 MHz, CDCl3) δ 7.96 (d, J = 8.8 Hz, 2 H), 6.94 (d, J = 8.8 Hz, 2 H), 5.96−5.87 (m, 1 H), 5.09 (dd, J = 17.4, 1.5 Hz, 1 H), 5.01 (d, J = 10.3 Hz, 1 H), 3.88 (s, 3 H), 3.03 (t, J = 7.4 Hz, 2 H), 2.49 (q, J = 7.0 Hz, 2 H); 13C{1H} NMR (150 MHz, CDCl3) δ 198.0, 163.4, 137.5, 130.3, 130.0, 115.1, 113.7, 55.4, 37.4, 28.3; IR (neat) 3060, 3013, 2978, 2838, 1667, 1639, 1599, 1576, 1509, 1419, 1250, 1178, 1029 cm−1.
4-(Pent-4-enoyl)phenyl Pivalate (41).
This compound was prepared according to general procedure C using allylic alcohol (17 μL, 0.25 mmol) and 4-vinylphenyl pivalate (102 mg, 0.5 mmol). After purification by column chromatography on SiO2 (2–5% EtOAc/hexanes), the title compound was isolated as a white solid (33 mg, 51% yield). 1H NMR (600 MHz, CDCl3) δ 8.01 (d, J = 8.5 Hz, 2 H), 7.16 (d, J = 8.5 Hz, 2 H), 5.95−5.86 (m, 1 H), 5.01 (dd, J = 17.1, 1.2 Hz, 1 H), 5.02 (d, J = 10.3 Hz, 1 H), 3.07 (t, J = 7.4 Hz, 2 H), 2.50 (q, J = 7.0 Hz, 2 H), 1.37 (s, 9 H); 13C{1H} NMR (150 MHz, CDCl3) δ 198.2, 176.5, 154.8, 137.2, 134.3, 129.6, 121.7, 115.3, 39.2, 37.7, 28.1, 27.0; IR (neat) 3078, 2974, 2933, 1747, 1682, 1643, 1594, 1478, 1411, 1276, 1201, 1164, 1106 cm−1; HRMS (ESI) m/z calcd for C16H21O3 [(M + H)+] 261.1491, found 261.1477.
1-(4-Fluorophenyl)pent-4-en-1-one (42).
This compound was prepared according to general procedure C using allylic alcohol (17 μL, 0.25 mmol) and p-fluorostyrene (60 μL, 0.5 mmol). After purification by column chromatography on SiO2 (0–2% EtOAc/hexanes), the title compound was isolated as a colorless oil (28 mg, 63% yield). The spectral data of this compound matched those from previously reported literature.29 1H NMR (600 MHz, CDCl3) δ 8.04−7.96 (m, 2 H), 7.14 (t, J = 8.5 Hz, 2 H), 5.95−5.85 (m, 1 H), 5.10 (dd, J = 17.4, 1.5 Hz, 1 H), 5.03 (d, J = 10.3 Hz, 1 H), 3.06 (t, J = 7.3 Hz, 2 H), 2.50 (q, J = 6.8 Hz, 2 H); 13C{1H} NMR (150 MHz, CDCl3) δ 197.8, 165.7 (d, JC,F = 252.0 Hz), 137.1, 133.3, 130.6 (d, JC,F = 9.0 Hz), 115.7 (d, JC,F = 21.0 Hz), 115.4, 37.6, 28.1; IR (neat) 2933, 1685, 1599, 1507, 1468, 1410, 1314, 1237, 1156, 1031 cm−1.
1-(Naphthalen-2-yl)pent-4-en-1-one (43).
This compound was prepared according to general procedure C using allylic alcohol (17 μL, 0.25 mmol) and 2-vinylnaphthalene (77 mg, 0.5 mmol). After purification by column chromatography on SiO2 (0–2% EtOAc/hexanes), the title compound was isolated as a colorless oil (30 mg, 57% yield). The spectral data of this compound matched those from previously reported literature.27 1H NMR (600 MHz, CDCl3) δ 8.49 (s, 1 H), 8.05 (d, J = 8.5 Hz, 1 H), 7.98 (d, J = 8.1 Hz, 1 H), 7.89 (d, J = 8.3 Hz, 1 H), 7.91 (d, J = 8.8 Hz, 1 H), 7.66−7.53 (m, 2 H), 6.02−5.92 (m, 1 H), 5.14 (d, J = 17.1 Hz, 1 H), 5.06 (d, J = 10.3 Hz, 1 H), 3.23 (t, J = 7.4 Hz, 2 H), 2.58 (q, J = 7.1 Hz, 2 H); 13C{1H} NMR (150 MHz, CDCl3) δ 199.4, 137.3, 135.5, 134.2, 132.5, 129.6, 129.5, 128.4, 128.4, 127.7, 126.7, 123.8, 115.3, 37.8, 28.3; IR (neat) 3059, 2917, 1677, 1627, 1468, 1359, 1276, 1171, 1123, 985 cm−1.
1-(1-Tosyl-1H-indol-3-yl)pent-4-en-1-one (44).
This compound was prepared according to general procedure C using allylic alcohol (17 μL, 0.25 mmol) and 1-tosyl-3-vinyl-1H-indole (149 mg, 0.5 mmol). After purification by column chromatography on SiO2 (5–10% EtOAc/hexanes), the title compound was isolated as a white solid (55 mg, 71% yield). 1H NMR (600 MHz, CDCl3) δ 8.31 (d, J = 7.6 Hz, 1 H), 8.22 (s, 1 H), 7.90 (d, J = 8.1 Hz, 1 H), 7.81 (d, J = 8.1 Hz, 2 H), 7.38−7.28 (m, 2 H), 7.25 (d, J = 8.1 Hz, 2 H), 5.94−5.84 (m, 1 H), 5.09 (d, J = 17.3 Hz, 1 H), 5.00 (d, J = 10.0 Hz, 1 H), 2.99 (t, J = 7.4 Hz, 2 H), 2.50 (q, J = 6.8 Hz, 2 H), 2.33 (s, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 199.4, 137.3, 135.5, 134.2, 132.5, 129.6, 129.5, 128.4, 128.4, 127.7, 126.7, 123.8, 115.3, 37.8, 28.3; IR (neat) 3134, 3072, 2920, 1663, 1643, 1596, 1537, 1447, 1384, 1368, 1169 cm−1; HRMS (ESI) m/z calcd for C20H20NO3S [(M + H)+] 354.1164, found 354.1142.
2-Methyl-1-phenylpent-4-en-1-one (45).
This compound was prepared according to general procedure C using allylic alcohol (17 μL, 0.25 mmol) and trans-β-methylstyrene (65 μL, 0.5 mmol). After purification by column chromatography on SiO2 (0–2% EtOAc/hexanes), the title compound was isolated as a colorless oil (23 mg, 53% yield). The spectral data of this compound matched those from previously reported literature.30 1H NMR (600 MHz, CDCl3) δ 7.97 (d, J = 7.6 Hz, 2 H), 7.57 (t, J = 7.4 Hz, 1 H), 7.48 (t, J = 7.7 Hz, 2 H), 5.84−5.75 (m, 1 H), 5.07 (dd, J = 18 Hz, 1.8 Hz, 1 H), 5.02 (d, J = 10.0 Hz, 1 H), 3.59−3.51 (m, 1 H), 2.62−2.52 (m, 1 H), 2.25−2.17 (m, 1 H), 1.22 (d, J = 7.1 Hz, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 203.6, 136.4, 135.8, 132.9, 128.6, 128.3, 116.7, 40.4, 37.6, 17.0; IR (neat) 2962, 1747, 1682, 1605, 1546, 1364, 1270, 1171, 1107, 998 cm−1.
1-(4-(tert-Butyl)phenyl)-4-methylpent-4-en-1-one (46).
This compound was prepared according to general procedure C using 2-methylprop-2-en-1-ol (21 μL, 0.25 mmol) and p-(tert-butyl)styrene (91 μL, 0.5 mmol). After purification by column chromatography on SiO2 (0–2% EtOAc/hexanes), the title compound was isolated as a colorless oil (25 mg, 44% yield). 1H NMR (600 MHz, CDCl3) δ 7.97 (d, J = 7.6 Hz, 2 H), 7.57 (t, J = 7.4 Hz, 1 H), 7.48 (t, J = 7.7 Hz, 2 H), 5.84−5.75 (m, 1 H), 5.07 (dd, J = 18 Hz, 1.8 Hz, 1 H), 5.02 (d, J = 10.0 Hz, 1 H), 3.59−3.51 (m, 1 H), 2.62−2.52 (m, 1 H), 2.25−2.17 (m, 1 H), 1.22 (d, J = 7.1 Hz, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 199.4, 156.7, 144.8, 134.3, 128.0, 125.5, 110.1, 36.7, 35.1, 31.9, 31.1, 22.8; IR (neat) 2963, 2870, 1716, 1683, 1606, 1462, 1408, 1368, 1269, 1188, 1109 cm−1; HRMS (ESI) m/z calcd for C16H23O [(M + H)+] 231.1749, found 231.1734.
1-(4-(tert-Butyl)phenyl)-3-methylpent-4-en-1-one (47).
This compound was prepared according to general procedure C using 2-buten-1-ol (21 μL, 0.25 mmol) and p-(tert-butyl)styrene (91 μL, 0.5 mmol). After purification by column chromatography on SiO2 (0–2% EtOAc/hexanes), the title compound was isolated as a colorless oil (44 mg, 77% yield). 1H NMR (600 MHz, CDCl3) δ 7.91 (d, J = 8.3 Hz, 2 H), 7.49 (d, J = 8.5 Hz, 2 H), 5.90−5.83 (m, 1 H), 5.04 (d, J = 17.1 Hz, 1 H), 4.96 (d, J = 10.3 Hz, 1 H), 3.06−2.98 (m, 1 H), 2.96−2.84 (m, 2 H), 1.35 (s, 9 H), 1.10 (d, J = 6.6 Hz, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 199.0, 156.7, 143.2, 134.7, 128.1, 125.5, 112.9, 45.0, 35.1, 33.6, 31.1, 19.7; IR (neat) 2962, 1680, 1641, 1605, 11544, 1461, 1406, 1363, 1270, 1191 cm−1; HRMS (ESI) m/z calcd for C16H23O [(M + H)+] 231.1749, found 231.1737.
1-(4-(tert-Butyl)phenyl)-3-phenylpent-4-en-1-one (48).
This compound was prepared according to general procedure C using (E)-3-phenylprop-2-en-1-ol (34 mg, 0.25 mmol) and p-(tert-butyl)-styrene (91 μL, 0.5 mmol). After purification by column chromatography on SiO2 (0–2% EtOAc/hexanes), the title compound was isolated as a colorless oil (42 mg, 57% yield). 1H NMR (600 MHz, CDCl3) δ 7.90 (d, J = 8.5 Hz, 2 H), 7.48 (m, J = 8.3 Hz, 2 H), 7.35−7.27 (m, 4 H), 7.22 (t, J = 7.2 Hz, 1 H), 6.11−6.03 (m, 1 H), 5.08 (d, J = 10.3 Hz, 1 H), 5.05 (d, J = 17.3 Hz, 1 H), 4.17 (q, J = 6.7 Hz, 1 H), 3.44 (dd, J = 16.5, 7.7 Hz, 1 H), 3.36 (dd, J = 16.6, 6.3 Hz, 1 H), 1.36 (s, 9 H); 13C{1H} NMR (150 MHz, CDCl3) δ 197.8, 156.7, 143.2, 140.7, 134.5, 128.5, 128.0, 127.7, 126.5, 125.5, 114.6, 44.5, 44.4, 43.9, 35.1, 31.1; IR (neat) 2962, 1681, 1637, 1604, 1492, 1452, 1406, 1363, 1266, 1107 cm−1; HRMS (ESI) m/z calcd for C21H25O [(M + H)+] 293.1905, found 293.1903.
(8R, 9S, 13S, 14S)-13-Methyl-3-(pent-4-enoyl)-6,7,8,9,11,12,13,14,15,16-decahydro-17H-cyclopenta[a]-phenanthren-17-one (49).
This compound was prepared according to general procedure C using allylic alcohol (17 μL, 0.25 mmol) and (8R,9S,13S,14S)-13-methyl-3-vinyl-6,7,8,9,11,12,13,14,15,16-decahydro-17H-cyclopenta[a]phenanthren-17-one (140 mg, 0.5 mmol). After purification by column chromatography on SiO2 (2–5% EtOAc/hexanes), the title compound was isolated as a white solid (60 mg, 77% yield). 1H NMR (600 MHz, CDCl3) 7.74 (d, J = 8.1 Hz, 1 H), 7.71 (s, 1 H), 7.38 (d, J = 8.3 Hz, 1 H), 5.95−5.85 (m, 1 H), 5.09 (dd, J = 17.1, 1.5 Hz, 1 H), 5.01 (d, J = 10.3 Hz, 1 H), 3.05 (t, J = 7.3 Hz, 2 H), 3.03−2.91 (m, 2 H), 2.56−2.43 (m, 4 H), 2.34 (dt, J = 3.5, 10.8 Hz, 1 H), 2.21−2.12 (m, 1 H), 2.12−2.03 (m, 2 H), 2.02−1.96 (m, 1 H), 1.70−1.60 (m, 2 H), 1.59−1.43 (m, 4 H), 0.92 (s, 3 H); 13C{1H} NMR (150 MHz, CDCl3) δ 199.3, 145.3, 137.3, 136.9, 134.5, 128.6, 125.5, 125.5, 115.2, 50.4, 47.8, 44.6, 37.8, 37.6, 35.8, 31.5, 29.3, 28.2, 26.2, 25.5, 21.5, 13.7, −18.4; IR (neat) 2925, 2882, 1730, 1666, 1599, 1539, 1453, 1416, 1370, 1257, 1154 cm−1; HRMS (ESI) m/z calcd for C23H29O2 [(M + H)+] 337.2168, found 337.2151.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Institute of General Medical Sciences of the National Institutes of Health under award no. R15GM139156 for supporting this work. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank the University of Toledo for an internal seed grant from the Summer Research Awards and Fellowship Programs for supporting our initial work. L.P.R. thanks the University of Toledo for an undergraduate summer research fellowship. We thank Dr. Yong W. Kim (University of Toledo) for NMR assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c02804.
1H and 13C NMR spectra of the amino alcohol starting material and the morpholine and Claisen rearrangement products (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.joc.1c02804
The authors declare no competing financial interest.
Contributor Information
Jeewani P. Ariyarathna, Department of Chemistry and Biochemistry, School of Green Chemistry and Engineering, The University of Toledo, Toledo, Ohio 43606, United States
Nur-E Alom, Department of Chemistry and Biochemistry, School of Green Chemistry and Engineering, The University of Toledo, Toledo, Ohio 43606, United States.
Leo P. Roberts, Department of Chemistry and Biochemistry, School of Green Chemistry and Engineering, The University of Toledo, Toledo, Ohio 43606, United States
Navdeep Kaur, Department of Chemistry and Biochemistry, School of Green Chemistry and Engineering, The University of Toledo, Toledo, Ohio 43606, United States.
Fan Wu, Department of Chemistry and Biochemistry, School of Green Chemistry and Engineering, The University of Toledo, Toledo, Ohio 43606, United States;.
Wei Li, Department of Chemistry and Biochemistry, School of Green Chemistry and Engineering, The University of Toledo, Toledo, Ohio 43606, United States;.
REFERENCES
- (1).Roberts I; Kimball GE The Halogenation of Ethylenes. J. Am. Chem. Soc 1937, 59, 947–948. [Google Scholar]
- (2).(a) Olah GA; Bollinger JM Stable Carbonium Ions. XLVIII. Halonium Ion Formation via Neighboring Halogen Participation. Tetramethylethylene Halonium Ions. J. Am. Chem. Soc 1967, 89, 4744–4752. [Google Scholar]; (b) Olah GA; Schilling P; Westerman PW; Lin HC Eletrophilic Reactions at Multiple Bonds. II. Observation and Differentiation of Intermediate. Sigma. and Pi. Complexes in Eletrophilic Additions to Ethene, 2,3-Dimethyl-2-butene, and Adamantylideneadamantane. J. Am. Chem. Soc 1974, 96, 3581–3589. [Google Scholar]; (c) Wieringa JH; Strating J; Wynberg H The Reaction of Chlorine with Admantylideneadamantane. Tetrahedron Lett. 1970, 11, 4579–4582. [Google Scholar]; (d) Brown RS; Nagorski RW; Bennet AJ; McClung RED; Aarts GHM; Klobukowski M; McDonald R; Santarsiero BD Stable Bromonium and Iodonium Ions of the Hindered Olefins Adamantylideneadamantane and Bicyclo[3.3.1]-nonylidenebicyclo[3.3.1]nonane. X-Ray Structure, Transfer of Positive Halogens to Acceptor Olefins, and ab Initio Studies. J. Am. Chem. Soc 1994, 116, 2448–2456. [Google Scholar]; (e) Mori T; Rathore R X-Ray Structure of Bridged 2,2′-Bi(adamant-2-ylidene) Chloronium Cation and Comparison of Its Reactivity with a Singly-Bonded Chloroarenium Cation. Chem. Commun 1998, 927–928. [Google Scholar]
- (3).(a) Denmark SE; Burk MT; Hoover AJ On the Absolute Configurational Stability of Bromonium and Chloronium Ions. J. Am. Chem. Soc 2010, 132, 1232–1233. [DOI] [PubMed] [Google Scholar]; (b) Tao Z; Gilbert BB; Denmark SE Catalytic, Enantioselective syn-Diamination of Alkenes. J. Am. Chem. Soc 2019, 141, 19161–19170. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Denmark SE; Kuester WE; Burk MT Catalytic, Asymmetric Halofunctionalization of Alkenes – A Critical Perspective. Angew. Chem., Int. Ed 2012, 51, 10938–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]; For related comprehensive reviews on hypervalent iodine chemistry, see:; (d) Zhdankin VV; Stang PJ Recent Developments in the Chemistry of Polyvalent Iodine Compounds. Chem. Rev 2002, 102, 2523–2584. [DOI] [PubMed] [Google Scholar]; (e) Zhdankin VV; Stang PJ Chemistry of Polyvalent Iodine. Chem. Rev 2008, 108, 5299–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]; For other interesting studies, see:; (f) Ashtekar KD; Vetticatt M; Yousefi R; Jackson JE; Borhan B Nucleophile-Assisted Alkene Activation: Olefins Alone Are Often Incompetent. J. Am. Chem. Soc 2016, 138, 8114–8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) Muñiz K; Martínez C Development of Intramolecular Vicinal Diamination of Alkenes: From Palladium to Bromine Catalysis. J. Org. Chem 2013, 78, 2168–2174. [DOI] [PubMed] [Google Scholar]; (b) Chávez P; Kirsch J; Hövelmann CH; Streuff J; Martinez-Belmonte M; Escudero-Adán EC; Martin E; Muñiz K Metal-Free Diamination of Alkenes Employing Bromide Catalysis. Chem. Sci 2012, 3, 2375–2382. [Google Scholar]; A very interesting early example using bromine catalysis was also demonstrated by Sharpless:; (c) Jeong JU; Tao B; Sagasser I; Henniges H; Sharpless KB Bromine-Catalyzed Aziridination of Olefins. A Rare Example of Atom-Transfer Redox Catalysis by a Main Group Element. J. Am. Chem. Soc 1998, 120, 6844–6845. [Google Scholar]
- (5).(a) Yoshimura A; Middleton KR; Zhu C; Nemykin VN; Zhdankin VV Hypoiodite-Mediated Metal-Free Catalytic Aziridination of Alkenes. Angew. Chem., Int. Ed 2012, 51, 8059–8062. [DOI] [PubMed] [Google Scholar]; (b) Yoshimura A; Jones TN; Yusubov MS; Zhdankin VV Hypoiodite-Mediated Catalytic Cyclopropanation of Alkenes with Malononitrile. Adv. Synth. Catal 2014, 356, 3336–3340. [Google Scholar]
- (6).(a) Gembreska NR; Vogel AK; Ziegelmeyer EC; Cheng E; Wu F; Roberts LP; Vesoulis MM; Li W Halonium Catalysis: An Underutilized and Underexplored Catalytic Concept in Olefin Functionalizations. Synlett 2021, 32, 539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu F; Stewart S; Ariyarathna JP; Li W Aerobic Copper-Catalyzed Alkene Oxyamination for Amino Lactone Synthesis. ACS Catal. 2018, 8, 1921–1925. [Google Scholar]; (c) Ariyarathna JP; Wu F; Colombo SK; Hillary CM; Li W Aerobic Catalytic Features in Photoredox- and Copper-Catalyzed Iodolactonization Reactions. Org. Lett 2018, 20, 6462–6466. [DOI] [PubMed] [Google Scholar]; (d) Wu F; Ariyarathna JP; Alom N-E; Kaur N; Li W Oxyamination of Unactivated Alkenes with Electron-Rich Amines and Acids via a Catalytic Triiodide Intermedidate. Org. Lett 2020, 22, 884–890. [DOI] [PubMed] [Google Scholar]; (e) Wu F; Alom N-E; Ariyarathna JP; Naß J; Li W Regioselective Formal [3 + 2] Cycloadditions of Ureas with Activated and Unactivated Olefins for Intermolecular Olefin Aminooxygenation. Angew. Chem., Int. Ed 2019, 58, 11676–11680. [DOI] [PubMed] [Google Scholar]; (f) Wu F; Kaur N; Alom N-E; Li W Chiral Hypervalent Iodine Catalysis Enables an Unusual Regiodivergent Intermolecular Olefin Aminooxygenation. J. Am. Chem. Soc. Au 2021, 1, 734–741. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a related intermolecular coupling example, see:; (g) Ye Z; Adhikari S; Xia Y; Dai M Expedient Syntheses of N-Heterocycles via Intermolecular Amphoteric Diamination of Allenes. Nat. Commun 2018, 9, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Kaur N; Wu F; Alom N-E; Ariyarathna JP; Saluga SJ; Li W Intermolecular Alkene Difunctionalizations for the Synthesis of Saturated Heterocycles. Org. Biomol. Chem 2019, 17, 1643–1654. [DOI] [PubMed] [Google Scholar]; (b) Alom N-E; Rina YA; Li W Intermolecular Regio- and Stereoselective Sulfenoamination of Alkenes with Thioimidazoles. Org. Lett 2017, 19, 6204–6207. [DOI] [PubMed] [Google Scholar]; (c) Alom N-E; Kaur N; Wu F; Saluga SJ; Li W Catalytic Regio- and Stereoselective Alkene Sulfenoamination for 1,4-Benzothiazine Synthesis. Chem. Eur. J 2019, 25, 6902–6906. [DOI] [PubMed] [Google Scholar]
- (8).(a) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (b) Holm KJ; Spencer CM Reboxetine. CNS Drugs. 1999, 12 (1), 65. [Google Scholar]; (c) Pal’chikov V Morpholines. Synthesis and Biological Activity. Russ. J. Org. Chem 2013, 49, 787–814. [Google Scholar]
- (9).For examples of intramolecular cyclization strategies, see:; (a) Leathen ML; Rosen BR; Wolfe JP A New Strategy for the Synthesis of Substituted Morpholines. J. Org. Chem 2009, 74, 5107–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lu Z; Stahl SS Intramolecular Pd(II)-Catalyzed Aerobic Oxidative Amination of Alkenes: Synthesis of Six-Membered N-Heterocycles. Org. Lett 2012, 14, 1234–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) O’Reilly MC; Lindsley CW A General, Enantioselective Synthesis of Protected Morpholines and Piperazines. Org. Lett 2012, 14, 2910–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Borah M; Borthakur U; Saikia AK Diastereoselective Synthesis of Substituted Morpholines from N-Tethered Alkenols: Total Synthesis of (±)-Chelonin A. J. Org. Chem 2017, 82, 1330–1339. [DOI] [PubMed] [Google Scholar]; (e) Zhou L; Tan CK; Zhou J; Yeung Y-Y Facile, Efficient, and Catalyst-Free Electrophilic Aminoalkoxylation of Olefins: Scope and Application. J. Am. Chem. Soc 2010, 132, 10245–10247. [DOI] [PubMed] [Google Scholar]; (f) McGhee A; Cochran; Stenmark TA; Michael FE Chem. Commun 2013, 49, 6800–6802. [DOI] [PubMed] [Google Scholar]; (g) Ghosh P; Deka MJ; Saikia AK Lewis Acid Mediated Intramolecular C-O Bond Formation of Alkanol-Epoxide Leading to Substituted Morpholine and 1,4-Oxazepane Derivatives: Total Synthesis of ()-Viloxazine. Tetrahedron. 2016, 72, 690–698. [Google Scholar]
- (10).(a) Siau W-Y; Bode JW One-Step Synthesis of Saturated Spirocyclic N-Heterocycles with Stannyl Amine Protocol (SnAP) Reagents and Ketones. J. Am. Chem. Soc 2014, 136, 17726–17729. [DOI] [PubMed] [Google Scholar]; (b) Luescher MU; Geoghegan K; Nichols PL; Bode JW SnAP Reagents for a Cross-Coupling Approach to the One-Step Synthesis of Saturated N-Heterocycles. Aldrichimica Acta 2015, 48, 43–48. [Google Scholar]; (c) Vo C-VT; Bode JW Synthesis of Saturated N-Heterocycles. J. Org. Chem 2014, 79, 2809–2815. [DOI] [PubMed] [Google Scholar]; (d) Luescher MU; Bode JW Catalytic Synthesis of N-Unprotected Piperazines, Morpholines, and Thiomorpholines from Aldehydes and SnAP Reagents. Angew. Chem., Int. Ed 2015, 54, 10884–10888. [DOI] [PubMed] [Google Scholar]; (e) Hsieh S-Y; Bode JW Silicon Amine Reagents for the Photocatalytic Synthesis of Piperazines from Aldehydes and Ketones. Org. Lett 2016, 18, 2098–2101. [DOI] [PubMed] [Google Scholar]; (f) Jindakun C; Hsieh S-Y; Bode JW Iridium-catalyzed Synthesis of Saturated N-Heterocycles from Aldehydes and SnAP Reagents with Continuous Flow Photochemistry. Org. Lett 2018, 20, 2071–2075. [DOI] [PubMed] [Google Scholar]
- (11).(a) Yar M; McGarrigle EM; Aggarwal VK An Annulation Reaction for the Synthesis of Morpholines, Thiomorpholines, and Piperazines from β-Heteroatom Amino Compounds and Vinyl Sulfonium Salts. Angew. Chem., Int. Ed 2008, 47, 3784–3786. [DOI] [PubMed] [Google Scholar]; (b) Matlock JV; Svejstrup TD; Songara P; Overington S; McGarrigle EM; Aggarwal VK Synthesis of 6- and 7-Membered N-Heterocycles using α-Phenylvinylsulfonium Salts. Org. Lett 2015, 17, 5044–5047. [DOI] [PubMed] [Google Scholar]; For a recent example; (c) Julia F; Yan J; Paulus F; Ritter T Vinyl Thianthrenium Tetrafluoroborate: A Practical and Versatile Vinylating Reagent Made from Ethylene. J. Am. Chem. Soc 2021, 143, 12992–12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Aubineau T; Cossy J A One-Pot Reaction toward the Diastereoselective Synthesis of Substituted Morpholines. Org. Lett 2018, 20, 7419–7423. [DOI] [PubMed] [Google Scholar]; (b) Shen H-C; Wu Y-F; Zhang Y; Fan L-F; Han Z-Y; Gong L-Z Palladium-Catalyzed Asymmetric Aminohydroxylation of 1,3-Dienes. Angew. Chem., Int. Ed 2018, 57, 2372–2376. [DOI] [PubMed] [Google Scholar]; (c) Zhang S; Shan C; Zhang S; Yuan L; Wang J; Tung C-H; Xing L-B; Xu Z Breaking Aziridines to Construct Morpholines with a Gold(I)-Catalyzed Tandem Ring-Openning and Cycloisomerization Reaction. Org. Biomol. Chem 2016, 14, 10973–10980. [DOI] [PubMed] [Google Scholar]
- (13).Claisen L Über Umlagerung von Phenol-allyläthern in C-Allylphenole. Chemische Berichte. 1912, 45, 3157–3166. [Google Scholar]
- (14).For selected examples of different strategies to access the enol-ether precursor, see:; (a) Nelson SG; Wang K Asymmetric Claisen Rearrangements Enabled by Catalytic Asymmetric Di(allyl) Ether Synthesis. J. Am. Chem. Soc 2006, 128, 4232–4233. [DOI] [PubMed] [Google Scholar]; (b) Wei X; Lorenz JC; Kapadia S; Saha A; Haddad N; Busacca CA; Senanayake CH Tandem Pd(II)-Catalyzed Vinyl Ether Exchange-Claisen Rearrangement as a Facile Approach to γ,δ–Unsaturated Aldehydes. J. Org. Chem 2007, 72, 4250–4253. [DOI] [PubMed] [Google Scholar]; (c) Quesada E; Taylor RJK Tandem Horner-Wadsworth-Emmons Olefination/Claisen Rearrangement/Hydrolysis Sequence: Remarkable Acceleration in Water with Microwave Irradiation. Synthesis 2005, 3193–3195. [Google Scholar]; (d) Burns JM; Krenske EH; McGeary RP Aromatic Claisen Rearrangements of Benzylic Ketene Acetals: Conversion of Benzylic Alcohols to (ortho-Tolyl)acetates. Eur. J. Org. Chem 2017, 2017, 252–256. [Google Scholar]; (e) Dupper NJ; Kwon O Functionalized α,α-Dibromo Esters through Claisen Rearrangements of Dibromoketene Acetals. Org. Lett 2015, 17, 1054–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Weiss ME; Kreis LM; Lauber A; Carreira EM Cobalt-Catalyzed Coupling of Alkyl Iodides with Alkenes: Deprotonation of Hydridocobalt Enables Turnover. Angew. Chem., Int. Ed 2011, 50, 11125–11128. [DOI] [PubMed] [Google Scholar]
- (15).Nordmann G; Buchwald SL A Domino Copper-Catalyzed C–O Coupling Claisen Rearrangement Process. J. Am. Chem. Soc 2003, 125, 4978–4979. [DOI] [PubMed] [Google Scholar]
- (16).(a) Tan CK; Yeung Y-Y Recent Advances in Stereoselective Bromofunctionalization of Alkenes Using N-Bromoamide Reagents. Chem. Commun 2013, 49, 7985–7996. [DOI] [PubMed] [Google Scholar]; (b) Ortiz GX Jr.; Kang B; Wang Q One-Pot Synthesis of 3-Azido- and 3-Aminopiperidines by Intramolecular Cyclization of Unsaturated Amines. J. Org. Chem 2014, 79, 571–581. [DOI] [PubMed] [Google Scholar]; (c) Corma A; García H Lewis Acids: From Conventional Homogeneous to Green Homogeneous and Heterogeneous Catalysis. Chem. Rev 2003, 103, 4307–4365. [DOI] [PubMed] [Google Scholar]
- (17).For selected examples of the transition-metal-catalyzed difunctionalization of unactivated alkenes based on directing groups, see:; (a) Zeng T; Liu Z; Schmidt MA; Eastgate MD; Engle KM Directed, Palladium(II)-Catalyzed Intermolecular Aminohydroxylation of Alkenes Using a Mild Oxidation System. Org. Lett 2018, 20, 3853–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dhungana RK; Sapkota RR; Wickham LM; Niroula D; Giri R Ni-Catalyzed Regioselective 1,2-Dialkylation of Alkenes Enabled by the Formation of Two C(sp3)-C(sp3) Bonds. J. Am. Chem. Soc 2020, 142, 20930–20936. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sardini SR; Lambright AL; Trammel GL; Omer HM; Liu P; Brown KM Nickel-Catalyzed Arylboration of Unactivated Alkenes: Scope and Mechanistic Studies. J. Am. Chem. Soc 2019, 141, 9391–9400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) Alom N-E; Wu F; Li W One-Pot Strategy for Thiazoline Synthesis from Alkenes and Thioamides. Org. Lett 2017, 19, 930–933. [DOI] [PubMed] [Google Scholar]; (b) Yu WZ; Cheng YA; Wong MW; Yeung Y-Y Atmosphere- and Temperature-Controlled Regioselective Amino-bromination of Olefins. Adv. Synth. Catal 2017, 359, 234–239. [Google Scholar]
- (19).For a similar stereochemical kinetic resolution process, see our recent work: ; Kaur N; Ziegelmeyer EC; Farinde ON; Truong JT; Huynh MM; Li W Visible Light Bromide Catalysis for Oxazoline, Pyrrolidne, and Dihydrooxazine Syntheses via Csp3–H Functionalizations. Chem. Commun 2021, 57, 10387–10390. [DOI] [PubMed] [Google Scholar]
- (20).Lv Y; Pu W; Cui H; He J; Zhang Q Copper-catalyzed efficient dithiocyanation of styrenes: Synthesis of dithiocyanates. Synth. Commun 2016, 46, 1223–1229. [Google Scholar]
- (21).Mazzocchi PH; Wilson P; Khachik F; Klingler L; Minamikawa S Photochemical Additions of Alkenes to Phthalimides. Mechanistic Investigations on the Stereochemistry of Alkene Additions and the Effect of Aryl Substituents on the Regiochemistry of Alkene Additions. J. Org. Chem 1983, 48, 2981–2989. [Google Scholar]
- (22).Dong X; Sang R; Wang Q; Tang X-Y; Shi M Copper-Catalyzed Trifluoromethylation and Cyclization of Aromatic-Sulfonyl-Group-Tethered Alkenes for the Construction of 1,2-Benzothiazinane Dioxide Type Compounds. Chem. Eur. J 2013, 19, 16910–16915. [DOI] [PubMed] [Google Scholar]
- (23).Baars J; Grimm I; Blunk D; Neudörfl J-M; Schmalz H-G Enantioselective Total Synthesis and Structural Revision of Dysiherbol A. Angew. Chem., Int. Ed 2021, 60, 14915–14920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Su C; Williard PG Isomerization of Allyl Ethers Initiated by Lithium Diisopropylamide. Org. Lett 2010, 12, 5378–5381. [DOI] [PubMed] [Google Scholar]
- (25).(a) Venning ARO; Kwiatkowski MR; Roque Pena JE; Lainhart BC; Guruparan AA; Alexanian EJ Palladium-Catalyzed Carbocyclizations of Unactivated Alkyl Bromides with Alkenes Involving Auto- tandem Catalysis. J. Am. Chem. Soc 2017, 139, 11595–11600. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) James T; MacLellan P; Burslem GM; Simpson I; Grant JA; Warriner S; Sridharan V; Nelson A A Modular Lead-Oriented Synthesis of Diverse Piperazine, 1,4-Diazepane and 1,5-Diazocane Scaffolds. Org. Biomol. Chem 2014, 12, 2584–2591. [DOI] [PubMed] [Google Scholar]; (c) Jain P; Raji IO; Chamakuri S; MacKenzie KR; Ebright BT; Santini C; Young DW Synthesis of Enantiomerically Pure 5-Substituted Piperazine-2-Acetic Acid Esters as Intermediates for Library Production. J. Org. Chem 2019, 84, 6040–6064. [DOI] [PubMed] [Google Scholar]
- (26).Ghorai MK; Shukla D; Das K Enantioselective Syntheses of Morpholines and Their Homologues via SN2-Type Ring Opening of Aziridines and Azetidines with Haloalcohols. J. Org. Chem 2009, 74, 7013–7022. [DOI] [PubMed] [Google Scholar]
- (27).Avilov DV; Malusare MG; Arslancan E; Dittmer DC A Telluride-Triggered Nucleophilic Ring Opening of Monoactivated Cyclopropanes. Org. Lett 2004, 6, 2225–2228. [DOI] [PubMed] [Google Scholar]
- (28).Huang F; Zhang S Iminyl Radicals by Reductive Cleavage of N–O Bond in Oxime Ether Promoted by SmI2: A Straightforward Synthesis of Five-Membered Cyclic Imines. Org. Lett 2019, 21, 7430–7434. [DOI] [PubMed] [Google Scholar]
- (29).Gontala A; Jang GS; Woo SK Visible-Light Photoredox-Catalyzed α-Allylation of α-Bromocarbonyl Compounds Using Allyltrimethylsilane. Bull. Korean Chem. Soc 2021, 42, 506–509. [Google Scholar]
- (30).Reißig H-U; Hippeli C Lithiated 5,6-Dihydro-4H-l,2-oxazines: Synthesis, Highly Diastereoselective Reactions with Electro-philes, and Subsequent Transformations. Chem. Ber 1991, 124, 115–127. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.