

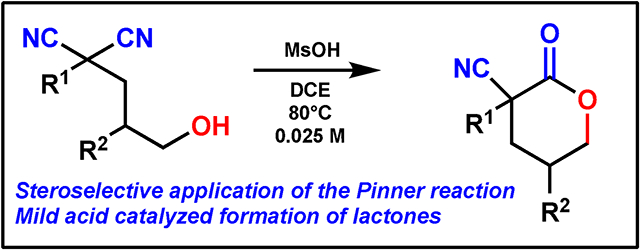
Abstract
Nitriles are important organic functional groups, allowing for installation of nitrogen in organic synthesis. The Pinner reaction transforms nitriles into esters via the imidate group, but in general has previously necessitated harsh acid conditions. This work builds on the utility of the Pinner reaction through a stereoselective desymmetrization of dinitriles to form γ- and δ-lactones in good yields and diastereoselectivites.
Keywords: Pinner Reaction, Desymmetrization, Diastereoselective, Lactones
Graphical Abstract
Introduction
Lactones that contain multiple stereocenters are key frameworks in many bioactive compounds.1 For instance, the lactone derivatives of parthenolide natural products have many anti-cancer properties and are valuable synthetic targets due to this utility (Figure 1A).2-5 To fully unlock the usefulness of these compounds, especially in medicinal or pharmaceutical spaces, synthetic routes giving chiral lactones must be developed. While not usually considered as a substrate, the electrophilic carbon of a nitrile functional group can serve as a template for the formation of chiral lactones.6,7 The Pinner reaction is an underutilized organic transformation of an electrophilic nitrile source (Figure 1B). Here, the nucleophilic attack of an alcohol with an acid forms an imidate salt (Pinner salt).8 Pinner salts can undergo a second nucleophilic attack – such as the hydrolysis of the imidate to form an ester. Due to the harsh conditions of the Pinner reaction, there have been select examples utilizing reaction alternatives (Figure 1B). Watanabe and coworkers used an alternative solvent CMPE which allowed facile isolation of the Pinner salt product by filtration.9 Plaff and coworkers used the Lewis acid TBSOTf to yield the ester, although the conditions require the reaction to be neat in either the nitrile or the alcohol, limiting substrate diversification.10 To our knowledge the only systematic study of the use of an intramolecular lactonization of the Pinner/hydrolysis was reported by Aplander and coworkers using a cationic exchange resin in water.11 A select example of an intramolecular Pinner/hydrolysis includes a lactonization forming Cleistantoxin derivatives using HCl in dioxane.12 Extensive study of mild Pinner/hydrolysis mediators has not been investigated to this point. Mild conditions allow for the exploration of stereoselective desymmetrizations without potential disruption of the nascent stereocenter.
Figure 1:

(A) Lactones are important structural components of several natural products. (B) The Pinner Reaction. (C) This work, a mild acid stereoselective desymmetrization of dinitriles to lactones.
To our understanding, there have been no reports of a nitrile being used as a direct electrophile in the presence of a Brønsted acid stereoselectively. The stereoselective examples are of domino reactions that are Michael/Pinner isomerization reactions to chromene-type compounds.13 While these reactions utilize a Pinner reaction, the Pinner reaction step is not stereoselective. Thus, this work represents the first instance of a stereoseletive Pinner reaction.
Previously, the Petersen lab has reported enantioselective desymmetrizations of diesters to synthesize lactones.14,15 This methodology takes a similar approach but extends these procedures to nitriles which can undergo a diastereoselective intramolecular desymmetrizing cyclization. Here, we present a diastereoselective desymmetrization that generates cyanolactones in good yields and diastereoselectivities.
Results
Our investigation began by establishing a general protocol for the cyclization of hydroxy dinitriles to both γ- and δ-lactones (Scheme 1). δ-Lactones 5 were readily prepared from the corresponding hydroxy dinitrile 4 with para-toluensulfonic acid in good yields. Spontaneous cyclization of TBS protected hydroxy dinitrile 3i-j under deprotection conditions yielded γ-lactones 6 in a single step.
Scheme 1.

A general procedure for synthesis of lactones via the Pinner reaction. δ-Lactones were synthesized from the corresponding alcohol, whereas γ-lactones spontaneously cyclized following TBAF deprotection.
Next, a diastereoselective cyclization of hydroxy dinitriles with a pre-existing stereocenter was tested. Initial efforts focused on the optimization of the stereoselective cyclization of dinitrile (±)-7a to yield lactones (±)-8aa and (±)-8ab (Table 1). Various Brønsted acids were investigated, including several sulfonic acids and trifluoroacetic acid (entry 1-4). These were chosen due to their pKA similarity to chiral acids previously used for stereoselective desymmetrizations, as well as their overall ease of use and accessibility. Based on X-ray analysis, sulfonic acids yielded predominately diastereomer 8aa where the nitrile is cis to the hydrogen (entries 1-3, 5-15). Interestingly, trifluoroacetic acid (entry 4) resulted in formation of the opposite diastereomer 8ab. Methanesulfonic acid was chosen for additional optimization due to the best combination of yield and diastereoselectivity ratio in our initial screen. Reaction time was found to play an important role in the overall diastereoselectivity of the reaction. Longer reaction times would result in lower diastereoselectivity. We hypothesize that this reaction is reversible, allowing for racemization with longer reaction times. The concentration was lowered to avoid undesired intermolecular dimerization which lowered yields. The optimal reaction conditions occurred with methanesulfonic acid in dichloroethane at 80 °C for one hour (entry 10).
Table 1.
Optimization of reaction conditions for formation of lactone. 8aa is the predominant diastereomer. a 2 equivalents acid used. b Unless otherwise noted, isolated yields. cdr measured via NMR. d Yields and dr measured by GC. e Molecular sieves.
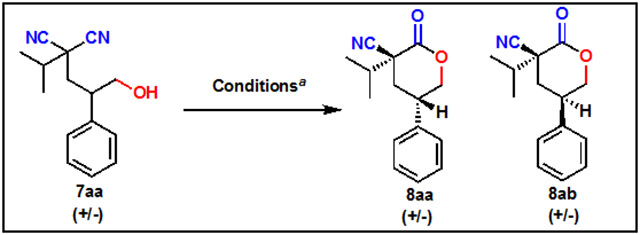
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Acid | Temp. (°C) | Solvent | Conc. (M) | Time (hr) | Yield (%)b | 8aa:8abc |
| 1 | CSA | RT | DCE | 0.1 | 72 | 32 | 3:1 |
| 2 | TsOH | RT | DCE | 0.1 | 72 | 45 | 5:1 |
| 3 | MsOH | RT | DCE | 0.1 | 72 | 51 | 5:1 |
| 4 | TFA | RT | DCE | 0.1 | 72 | 82 | 1:4 |
| 5d | MsOH | RT | DCE | 0.1 | 2 | 20 | 9:1 |
| 6d | MsOH | RT | DCE | 0.1 | 4 | 32 | 7.5:1 |
| 7d | MsOH | RT | DCE | 0.1 | 18 | 57 | 7:1 |
| 8d | MsOH | RT | DCE | 0.1 | 42 | 61 | 6:1 |
| 9 | MsOH | 50 | DCE | 0.025 | 1 | 35 | 5:1 |
| 10 | MsOH | 80 | DCE | 0.025 | 1 | 75 | 7:1 |
| 11 | MsOH | 80 | DCE | 0.025 | 0.5 | 60 | 7:1 |
| 12e | MsOH | 80 | DCE | 0.025 | 1 | 58 | 6:1 |
| 13 | MsOH | 80 | DCE/H2O (20:1) | 0.025 | 1 | Trace | — |
| 14 | MsOH | 80 | DCE | 0.01 | 1 | 54 | 4:1 |
| 15 | MsOH | 80 | Toluene | 0.025 | 1 | 73 | 4:1 |
In order to explore the scope of this diastereoselective cyclization, various substitution patterns were explored (Scheme 2). The cyclization of dinitriles that contained a phenyl group in the R2 position were shown to yield lactone products in good yields and diastereoselectivity (compounds 8aa-ca). The reaction of methanesuflonic acid and dinitriles that contained a methyl group in R2 resulted in excellent yields and good stereoselectivities (compounds 8da-fa).
Scheme 2.

Diastereoselectivity of lactonization.
Next, synthesis of a single enantiomer of a lactone was accomplished by using the commercially available enantioenriched bromide 9. Diastereoselective intramolecular cyclization of hydroxy dinitrile 7f using our optimized reaction conditions yielded enantioenriched lactone 8fa in excellent yields and 99% ee. The diastereomer of this enantioenriched compound, additionally, was able to be separated via silica gel column chromatography.
Conclusions
Here, we have described a stereoselective intramolecular Pinner reaction which forms a lactone in good yields and diastereoselectivites. Additionally, we were able to accomplish this transformation using a mild acid as a catalyst, as opposed to the standard Pinner reaction conditions of HCl. This mild acid catalysis opens the door for further investigation into this method for the synthesis of complex natural product lactones – especially products with beta nitrogen moieties.
Supplementary Material
Scheme 3.

Enantioenriched lactone synthesis.
Acknowledgements
Financial support is gratefully acknowledged from the National Institutes of Health (GM141981-01) and the University of North Carolina at Greensboro. Additionally, we thank Dr. Franklin J. Moy and Dr. Daniel Todd for assistance with NMR and mass spectrometry data analysis. All X-ray crystallography measurements were made in the X-ray Core Laboratory at the University of North Carolina at Chapel Hill and we wish to thank Chun-Hsing Chen for his assistance. Research reported in this publication was supported in part by the National Center for Complementary and Integrative Health of the National Institutes of Health under award number T32AT008938. This research was also supported in part by the National Institutes of Health under award number T34GM113860.
References
- (1).Schulz S; Hötling S Nat. Prod. Rep 2015, 32, 1042–1066. 10.1039/c5np00006h. [DOI] [PubMed] [Google Scholar]
- (2).Ghantous A; Sinjab A; Herceg Z; Darwiche N Drug Discov. Today, 2013, 18, 894–905. 10.1016/j.drudis.2013.05.005. [DOI] [PubMed] [Google Scholar]
- (3).Ding Y; Gao H; Zhang Y; Li Y; Vasdev N; Gao Y; Chen Y; Zhang QJ Hematol. Oncol 2016, 9 (1), 93. 10.1186/s13045-016-0327-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bhedi DN; Vishwakarma RA; Deka V; More DM; Ramalingam M; Suthar A; Dalal R; Parikh S; Tannu AA Tricyclic Compounds for the Treatment of Inflammatory Disorder. International Patent WO 2009/016565 Al, 2011.
- (5).Crooks PA; Jordan CT; Wei X Use of Parthenolide Derivatives as Antileukemic and Cytotoxic Agents. U.S. Patent US 9,447,112 B2, 2016.
- (6).Li X; Wang B; Zhang J; Yan M Org. Lett 2011, 11, 374–377. 10.1021/ol102570b. [DOI] [PubMed] [Google Scholar]
- (7).Rowland AT; Gill BC; J. Org. Chem 1988, 53, 434–437. 10.1021/jo00237a042. [DOI] [Google Scholar]
- (8).Pinner A; Klein F Berichte der Dtsch. Chem. Gesellschaft 1877, 10, 1889–1897. 10.1002/cber.187701002154. [DOI] [Google Scholar]
- (9).Watanabe K; Kogoshi N; Miki H; Torisawa Y Synth. Commun 2009, 39, 2008–2013. 10.1080/00397910802632548. [DOI] [Google Scholar]
- (10).Pfaff D; Nemecek G; Podlech JA Helv. Chim. Acta 2012, 95, 1851–1856. 10.1002/hlca.201200435. [DOI] [Google Scholar]
- (11).Aplander K; Hidestål O; Katebzadeh K; Lindström UM Green Chem. 2006, 8, 22–24. 10.1039/b513656c. [DOI] [Google Scholar]
- (12).Trinh Thi Thanh V; Cuong Pham V; Doan Thi Mai H; Litaudon M; Guéritte F; Retailleau P; Nguyen VH; Chau VM J. Nat. Prod 2012, 75, 1578–1583. 10.1021/np3003832. [DOI] [PubMed] [Google Scholar]
- (13).Gupta V; Sahu D; Jain S; Vanka K; Singh RP Org. Biomol. Chem 2019, 17, 8853–8857. 10.1039/c9ob01345h. [DOI] [PubMed] [Google Scholar]
- (14).Kelley AM; Minerali E; Wilent JE; Chambers NJ; Stingley KJ; Wilson GT; Petersen KS Tetrahedron Lett. 2019, 60, 1262–1264. 10.1016/j.tetlet.2019.03.074. [DOI] [Google Scholar]
- (15).Kelley AM; Haywood RD; White JC; Petersen KS ChemistrySelect 2020, 5, 3018–3022. 10.1002/slct.202000312. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.