

Abstract
There is increasing evidence regarding the prevalence of genetic cardiomyopathies, for which arrhythmias may be the first presentation. Ventricular and atrial arrhythmias presenting in the absence of known myocardial disease are often labelled as “idiopathic”, or “lone”. While ventricular arrhythmias are well-recognized as presentation for arrhythmogenic cardiomyopathy in the right ventricle, the scope of arrhythmogenic cardiomyopathy has broadened to include those with dominant left ventricular involvement, usually with a phenotype of dilated cardiomyopathy. In addition, careful evaluation for genetic cardiomyopathy is also warranted for patients presenting with frequent PVC’s, conduction system disease, and early onset atrial fibrillation, in which most detected genes are in the cardiomyopathy panels. Sudden death can occur early in the course of these genetic cardiomyopathies, for which risk is not adequately tracked by left ventricular ejection fraction. Only a few of the cardiomyopathy genotypes implicated in early sudden death are recognized in current indications for implantable cardioverter defibrillators which otherwise rely upon a left ventricular ejection fraction ≤ 0.35 in dilated cardiomyopathy. The genetic diagnoses impact other aspects of clinical management such as exercise prescription and pharmacologic therapy of arrhythmias, and new therapies are coming into clinical investigation for specific genetic cardiomyopathies. The expansion of available genetic information and implications raises new challenges for genetic counseling, particularly with the family member who has no evidence of a cardiomyopathy phenotype and may face a potentially negative impact of a genetic diagnosis. Discussions of risk for both probands and relatives need to be tailored to their numeric literacy during shared decision-making. For patients presenting with arrhythmias or cardiomyopathy, extension of genetic testing and its implications will enable cascade screening, intervention to change the trajectory for specific genotype-phenotype profiles, and enable further development and evaluation of emerging targeted therapies.
Introduction
There is increasing recognition of the prevalence of genetic cardiomyopathies. Genetic cardiomyopathies often present with cardiac arrhythmias, which, in the absence of clinical heart failure, may too readily be labelled as “idiopathic,” “isolated,” or “lone.” Many of these disorders are now considered arrhythmogenic cardiomyopathies (ACM). This term reflects evolution from the initial recognition of families with sudden death, ventricular arrhythmias, and morphologic disease of the right ventricle (RV), defined first as arrhythmogenic RV dysplasia and then arrhythmogenic RV cardiomyopathy (ARVC), linked to genetic desmosomal disease.1 As described in the 2019 Heart Rhythm Society Expert Consensus Statement,2 the scope of ACM has expanded to include those predominantly expressed in the left ventricle (LV), most commonly with a dilated cardiomyopathy phenotype. In this review, the broad term ACM will encompass both the desmosomal and other genetic diseases primarily affecting the right ventricle (ACM-RV) and those primarily affecting the left ventricle (ACM-LV), with recognition that both ventricles are often involved together. The distinct phenotype of hypertrophic cardiomyopathy, which is notorious for its presentation as sudden death in athletes, receives separate consideration. Consideration of genetic cardiomyopathy is increasingly relevant, however, also for patients presenting with early onset atrial fibrillation, conduction system disease, or frequent PVCs, any of which can be the presenting symptoms in the absence of HF.
When evaluating a patient with new arrhythmias and possible CM, it is useful to consider three contexts according to the relative timing and severity of arrhythmias and myocardial dysfunction:
Arrhythmias dominate the clinical picture with mild or no apparent impairment of left ventricular function.
Arrhythmias appear in parallel with other evidence of CM not previously recognized.
Arrhythmias have developed only after CM and HF are advanced.
This review will focus on evaluation and recognition of patients in the first two settings with atrial fibrillation, conduction system disease, or ventricular arrhythmias (VA) as the presentation of genetic cardiomyopathy. The major morphologic phenotypes of genetic CM will then be reviewed, including examples of genetic variants associated with arrhythmias, recognizing that the reported prevalence and prognosis of pathogenic variants vary among different regions, and continue to evolve with new information.
Initial Evaluation in Patients Presenting with Arrhythmias
Historical features to be elicited should focus on evidence of other cardiac disease. Abnormal ECGs may have been reported in the past. Prior cardiac evaluation may have been triggered by atypical chest pain, which can occur for multiple reasons with cardiomyopathies. Comparison of current to past activity level, such as during previous family holidays, can reveal unrecognized decline in exercise tolerance. Some pathogenic myocardial variants also affect skeletal muscle, so patterns of muscle weakness can suggest genetic heart disease. The history should note other features specific to some cardiomyopathies, such as familial partial lipodystrophy with LMNA disease.
A 3-generation family history should be obtained,2,3 to include not only heart failure, CM, and “enlarged heart”, but also skeletal muscle disease, early onset atrial fibrillation (AF), pacemaker implantation, or sudden cardiac death (SCD), sometimes hidden within a diagnosis of “massive heart attack”, drowning, single motor vehicle accident, or occurring in infants. CM can also be part of recognized genetic syndromes with prominent non-cardiac features (which will not be reviewed here).
Electrocardiography
ECG documentation of the arrhythmia can suggest the nature of underlying cardiomyopathy. Baseline abnormalities of repolarization, hypertrophy, or conduction favor the presence of underlying disease.4 The morphology of PVCs and other VA can be helpful as discussed below. High voltage of left ventricular hypertrophy suggests hypertrophic cardiomyopathy (HCM), often with prominent septal forces, while voltage can decrease progressively with some genetic CM, such as with LMNA or desmoplakin diseases. Ambulatory cardiac monitoring is useful for identifying arrhythmias and quantifying whether frequency of AF or PVCs is sufficiently high to contribute to ventricular dysfunction.
Echocardiography
Transthoracic echocardiography is widely available, inexpensive, and remains the standard first-line imaging for evaluation of patients presenting with ventricular arrhythmia. It can help to exclude underlying coronary artery or valvular disease, and may identify features such as ventricular dilation, global and/or regional ventricular systolic dysfunction, atrial enlargement or abnormal myocardial wall thickness that indicate CM phenotypes. Assessment of global longitudinal strain enhances recognition of early disease and of serial changes in systolic function during the course of genetic cardiomyopathies.5
Cardiac Magnetic Resonance Imaging
Cardiac magnetic resonance imaging (CMR) is the recognized gold standard for quantitative measurement of ventricular size, function, morphology, and myocardial tissue characteristics. It allows visualization of myocardial edema, inflammation, and fibrosis. Gadolinium contrast accumulates in areas of expanded extracellular matrix, allowing recognition of myocardial fibrosis as regions of late gadolinium enhancement (LGE). When trying to determine whether VA are “benign”, the presence of LGE is a relatively reliable indicator of myocardial disease and a potential substrate for scar-related arrhythmias (Figure 1).6 The morphology and location of LGE allows for the discrimination of non-ischemic fibrosis from scarring related to myocardial infarction. The extent of LGE has been linked to risk of VA/SCD in dilated cardiomyopathy (DCM)7–9 and some signature LGE patterns have been associated with specific genotypes.10
Figure 1.
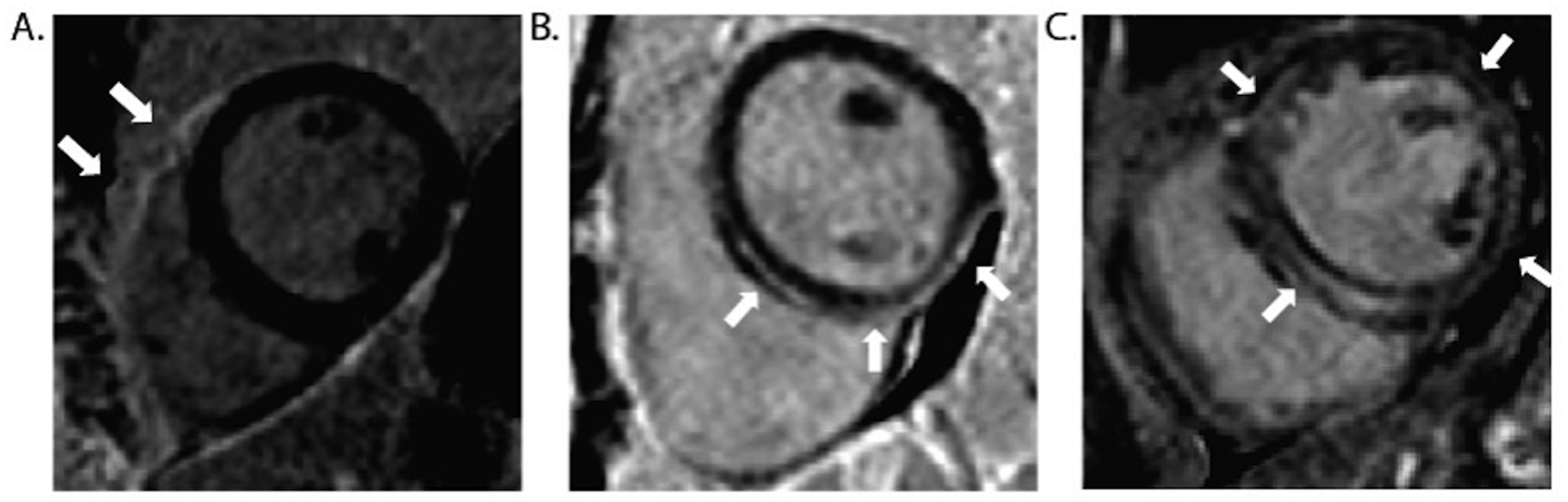
Cardiac magnetic resonance (CMR) images of genetic CM showing typical patterns of late gadolinium enhancement (LGE). A. 39M with family history of arrhythmogenic right ventricular cardiomyopathy with pathogenic PKP2 variant. CMR showed normal function of the left ventricle (LV) and right ventricle (RV), with LGE of the anterior RV free wall (arrows) but no LGE in the LV. B. 21F pathogenic DSP mutation and family history of dilated cardiomyopathy, ventricular arrhythmias, and sudden death. CMR showed normal LVEF 0.58 with sub-epicardial LGE of the inferior LV wall, extending into the septum without separate RV involvement. C. 45M with atrial fibrillation and family history of dilated cardiomyopathy, CMR showed a ring-like pattern of intramural LGE in the LV. Genetic testing revealed a pathogenic LMNA variant.
Diffuse interstitial fibrosis may not result in visible LGE but can be appreciated by novel imaging techniques.11 Parametric mapping (T1, T2, and extra-cellular volume, ECV) allows for further discrimination of myocardial edema, inflammation, infiltration, and fibrosis. Global elevation of the native myocardial T1 and the ECV are indicators of diffuse fibrosis and associated with increased arrhythmia risk, even when focal LGE is not apparent.12,13
Imaging of the right ventricle (RV) presents challenges, as CMR may not detect thin areas of fibrosis that can nonetheless cause ventricular tachycardia (VT).14 Discrimination of LGE in the thin RV free wall is further complicated by bordering epicardial fat, which can be distinguished using pre-contrast T1 mapping. Co-registration of the native T1 map with LGE images may increase diagnostic sensitivity for detection of fibro-fatty infiltration that is a hallmark of desmosomal disease.
Overlap Between Inflammatory and Genetic Cardiomyopathies
Inflammatory cardiomyopathies, most commonly sarcoidosis and Chagas disease in endemic areas, need to be distinguished from genetic cardiomyopathies. Cardiac sarcoid with predominant RV involvement can be hard to distinguish from ACM-RV.15 In the absence of confirmed extracardiac sarcoid, the diagnosis of active cardiac sarcoidosis may depend upon focal cardiac areas of increased fluoro-deoxy-glucose uptake on positron emission tomography after myocardial glucose uptake is suppressed during a no-carbohydrate diet. However, this distinction is complex because some genetic cardiomyopathies, notably those due to DSP and other desmosomal mutations, can also cause areas of inflammation.16
The distinction between inflammatory and genetic CM is further blurred by evidence that they can occur together. An early pathologic series of ARVC showed lymphocytic myocarditis in 2/3 of hearts.17 Case series document patients who presented with acute chest pain, troponin elevations, and imaging typical of myocarditis, but who were later found to have desmoplakin CM.18,19 Some genes implicated in familial CM may increase susceptibility to viral myocarditis.20 In vitro and murine models of desmosomal variants have shown activation of nuclear factor kappaB signaling with increased levels of inflammatory cytokines.21 In recent small studies, auto-antibodies to components of the intercalated disks have been described in families with ACM-RV and proposed for diagnosis of desmosomal disease.22,23
Current Selection of Patients for Genetic Testing
Beyond recommendations for the 3-generation family history, formal indications are evolving for genetic testing in the evaluation of arrhythmias and cardiomyopathies. The family history should not be considered a reliable proxy for the presence of genetic disease, which can occur with a negative family history. A suggestive family history may increase the likelihood of finding a pathogenic variant by up to 2-fold, but its absence does not signify absence of genetic CM. Clinical penetrance is highly variable and many family histories are incomplete. In a recent study of genetic CM with recurrent VA, family history was negative for sudden cardiac death (SCD) in 65% patients and for CM in 73%.24 In a large Maastricht registry, the presence of an arrhythmia phenotype of AF, conduction disease, or VA at the time of CM diagnosis doubled the rate of positive genetic tests for CM.25
Underlying genetic CM should remain a consideration even in the presence of an acquired cause of CM. Compared to a control population without CM, patients with CM in the settings of excess alcohol consumption, cardiotoxicity from anthracycline chemotherapy, or peri-partum cardiomyopathy all had higher prevalence of genetic CM variants, often truncating TTN variants.26–28
The AHA Scientific Statement on Inherited Cardiac Disease indicated, based on a moderate level of consensus, that genetic testing can be useful in patients with familial or “idiopathic” cardiomyopathy.29 Genetic testing was also recommended by the AHA/ACC guidelines for HCM and by the Heart Failure Society of America for HCM, DCM, and ACM, with lower strength of recommendation for RCM.30,31 The Heart Rhythm Society emphasizes the importance of genetic testing for possible ACM,2 which has major implications for consideration of ICDs (Table 1). All these recommendations emphasize the importance of genetic counseling and a multidisciplinary team experienced in the care of genetic CM.
Table 1.
Recommendations for ICDs as Primary Prevention in Familial Cardiomyopathy
| Phenotype | Recommended consideration for ICD | Level | Comments |
|---|---|---|---|
| Hypertrophic Cardiomyopathy | Single risk factor: Family history of sudden death Massive LVH (≥ 30 mm) Unexplained syncope LVEF < 0.50 LV apical aneurysm | IIa | From 2020 AHA/ACC Guideline for HCM31 |
| NSVT or LGE ≥ 15% | IIb | ||
| Dilated Cardiomyopathy | Non-ischemic CM with EF ≤ 0.35, despite GDMT**, and NYHA II-III symptoms of HF | I | From 2017 AHA/ACC/HRS Guideline for Management of Patients with Ventricular Arrhythmias and Prevention of Sudden Cardiac Death32 |
| LVEF≤ 0.30 despite GDMT**, NYHA Class I | IIb | ||
| Specific genetic DCM when LVEF ≥ 0.35 | Lamin A/C mutation with ≥ 2 risk factors: Male, LVEF<0.45, NSVT, Non missense variant | IIa | |
| Lamin A/C ≥ 2 risk factors Male sex, LVEF < 0.45, NSVT | IIa | From 2019 HRS Expert Consensus Statement on Evaluation, Risk stratification, and Management of Arrhythmogenic Cardiomyopathy2 | |
| Lamin A/C and pacing indication | IIa | ||
| Phospholamban LVEF < 0.45 or NSVT | IIa | ||
| Filamin C LVEF < 0.45 | IIa | ||
| Arrhythmogenic RV CM (ARVC) | (Resuscitated SCA, sustained VT*) or significant ventricular dysfunction with RVEF or LVEF ≤ 0.35 | I | From 2017 AHA/ACC/HRS Guideline32 |
| 3 major criteria, 2 major+ 2 minor, 1 major+ 4 minor criteria | IIa | From 2019 HRS Expert Consensus Statement2 Major criteria: NSVT, inducible VT, LVEF ≤ 0.49 Minor criteria: Male sex, > 1000 PVCs/24 hours, RV dysfunction by 2010 criteria33, proband status, ≥ 2 variants (cannot count both NSVT and PVCs.) | |
| 2 major, 1 major+2 minor criteria, or 4 minor criteria | IIb | ||
| LV Non- Compaction | NSVT associated with reduced LVEF | IIa | From 2019 HRS Expert Consensus Statement2 |
| Neuromuscular disorders | Emery-Dreifuss and limb-girdle type IB dystrophies with progressive cardiac involvement | IIa | From 2017 AHA/ACC/HRS Guideline32 |
|
Examples of variants that can cause ACM for which there is no recommendation when LVEF ≥ 0.35
RNA-binding motif 20 (RBM20) Type V voltage-gated cardiac sodium channel (SCN5A) Desmoplakin (DSP) ACM not meeting criteria for ACM-RV | |||
| Familial CM unspecified | Familial cardiomyopathy associated with sudden death | IIb | From ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities34, and 2013 Appropriate Use Criteria for ICDs and CRTs35 |
Syncope and sustained ventricular tachycardia are considered to be secondary prevention
GDMT = guideline-directed medical therapy for at least 3 months
Arrhythmia Mechanisms and Phenotypes in Genetic CM
Atrial Fibrillation
The association between atrial fibrillation and atrial flutter (here abbreviated together as AF) and CM is well-recognized, occurring in up to 30% of patients with inherited cardiomyopathies, increasing to 40–50% in severe HF.36,37 AF can also lead to the tachycardia-induced CM phenotype, which does not preclude an underlying genetic CM, as described below. This has been attributed to rapid ventricular response, but also to the irregularity of AF, which may cause impairment in cardiomyocyte excitation-contraction coupling.38
AF may often be the first presentation of genetic CM with other clinical evidence of disease developing years later.39,40 Familial AF has also been reported in families with pathogenic variants in genes associated with inherited CM.40–42 Such cases appeared to be rare until data from population-based studies began to emerge, in which loss-of-function variants in TTN were associated with AF,43 raising questions of when patients with unexplained AF should be screened for disease-associated variants in both CM and arrhythmia genes.44,45 A recent study sequenced 1,293 patients with early-onset AF (diagnosed before age 66) for genes included on commercial cardiomyopathy and arrhythmia panels (Figure 2A). A disease-associated variant was found in 10.1% patients overall, and in 16.7% with AF onset before 30 years (Figure 2B).40 Most implicated variants were in genes associated with DCM, followed by HCM and ACM-RV, with fewer in genes associated with channelopathies. In early onset AF without clinical CM, the CM genes most often implicated were TTN, MYH7, MYH6, and LMNA. In these patients presenting with AF before age 66, presence of a pathogenic or likely pathogenic variant conferred a 1.5-fold higher risk of death during median 10-year follow-up, an effect that was most marked in younger patients.46
Figure 2.
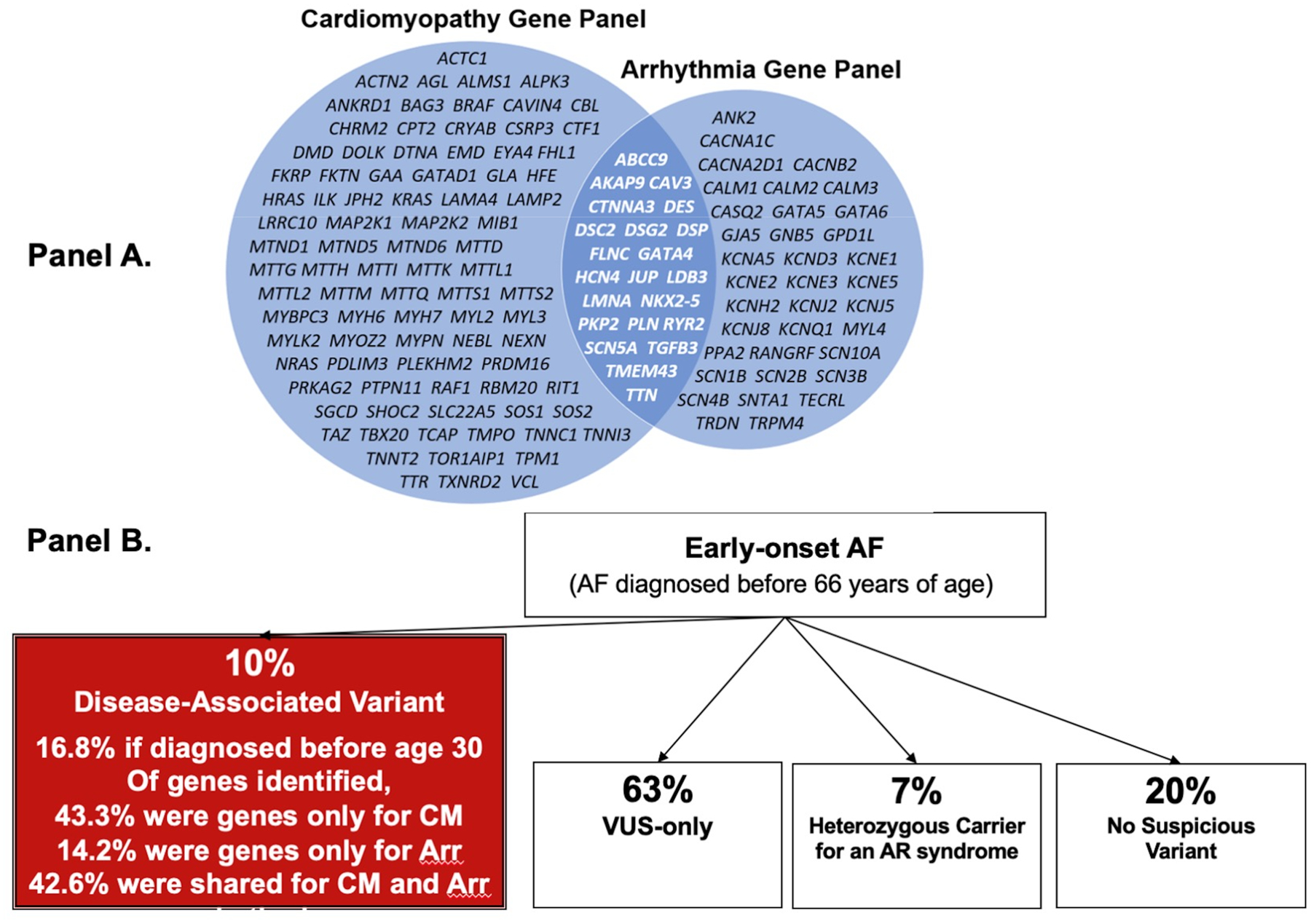
Panel A presents the genes often included on comprehensive commercial panels for cardiomyopathy and arrhythmia genes, showing the overlap of genes most commonly implicated in both. Panel B presents the results from sequencing 1,293 participants with early-onset AF defined as AF diagnosed before age 65.40 According to the genes listed in Panel A, 43% of disease-associated variants occurred in genes commonly included on cardiomyopathy panels, 14% in genes on arrhythmia panels, and 43% in genes included on both panels.
CM = cardiomyopathy, Arr = arrythmia, AR = autosomal recessive
Multiple mechanisms for AF have been described. Both automaticity and reentry may play roles. Paroxysmal AF often appears to be initiated by rapid activity originating in the posterior left atrium in musculature that includes sleeves of myocardium along the pulmonary veins, and this rapid focal activity may perpetuate AF or initiate reentrant waves. Atrial fibrosis and oxidative stress have been proposed as key mediators of AF initiation and maintenance. Atrial fibrosis can facilitate both automaticity and reentry by creating areas of slow conduction and intra-atrial reentry paths and diminishing coupling between myocyte bundles.47 Fibrosis is common in HF as progressively elevated filling pressures cause left atrial distention, myocyte stretch, and fibrosis, creating areas of slow conduction and intra-atrial re-entry.48 The extent to which underlying genetic disease contributes to the initiating electrophysiologic perturbations and the fibrosis that sustains AF is not well defined.
When AF and a pathogenic CM gene are identified, a next step is to evaluate for other evidence of the CM phenotype (Table 2). Finding a decreased LVEF in the setting of a new AF diagnosis could reflect limitations to imaging during an irregular and/or fast heart rhythm, reversible LV dysfunction secondary to AF, and/or an underlying DCM phenotype, so re-assessment is indicated after AF therapy. The frequency of AF is increased in association with any CM genotype, with truncating TTN variants and LMNA variants being commonly implicated for DCM (Figure 3).25 When evaluation of new AF instead reveals a hypercontractile LV with LVH meeting criteria for HCM, the most likely genetic variants are in sarcomeric proteins, particularly MYH7.49
Table 2.
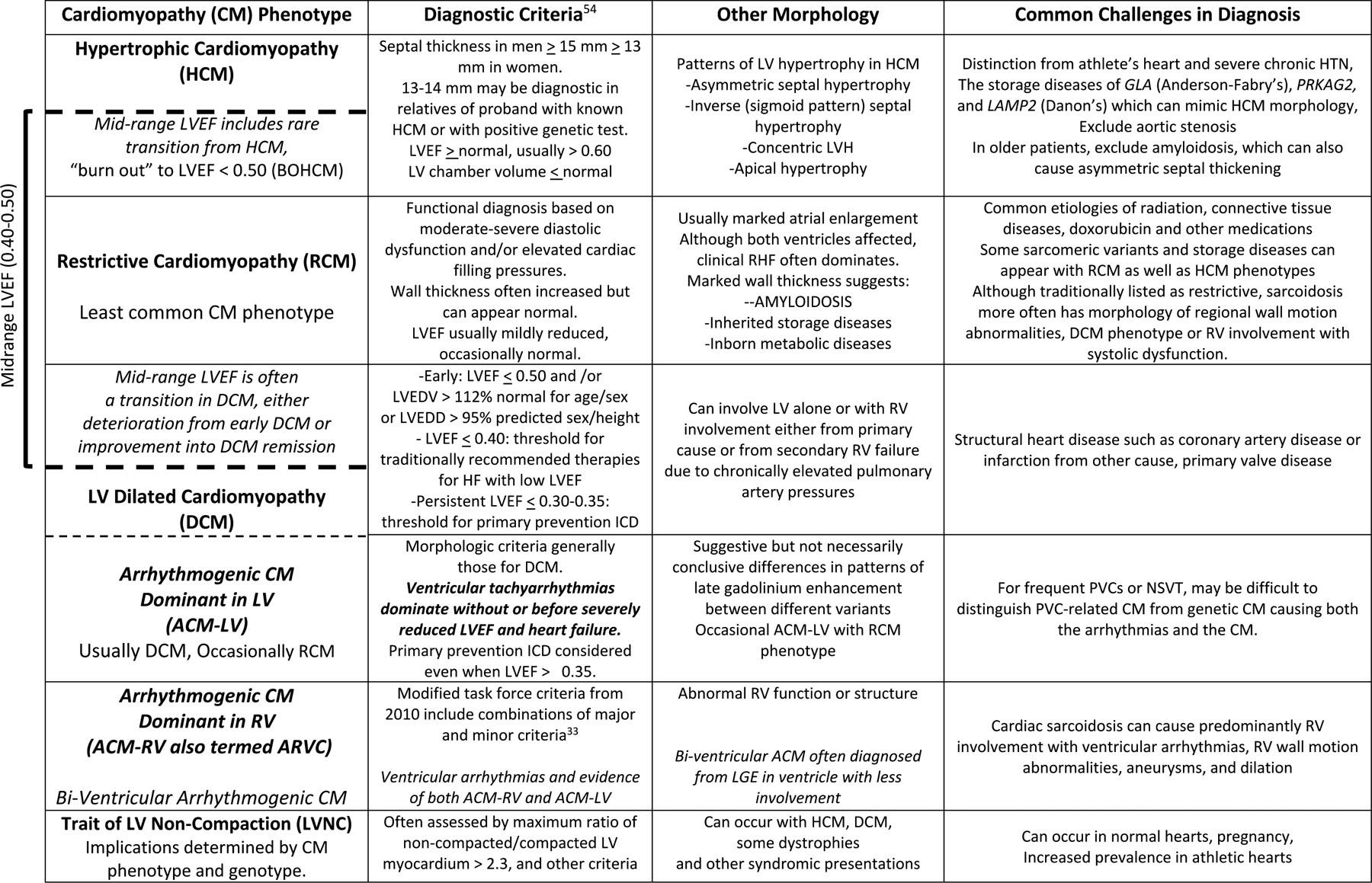
Phenotypes and Morphologies of Cardiomyopathies
|
ACM-LV Arrhythmogenic cardiomyopathy with dominant LV involvement, ACM-RV with dominant RV involvement
Shown within the bracket are the overlapping phenotypes contributing to “Mid-Range LVEF 0.40–0.50”, a term which describes a heterogeneous group of patients rather than a phenotype with specific implications (see text).
Abbreviations: BOHCM = ”burned out” HCM, LV = left ventricle, RV = right ventricle , LDEDV = LV end diastolic volume, LVEDD = LV end diastolic dimension, LVEF = LV ejection fraction
Figure 3.
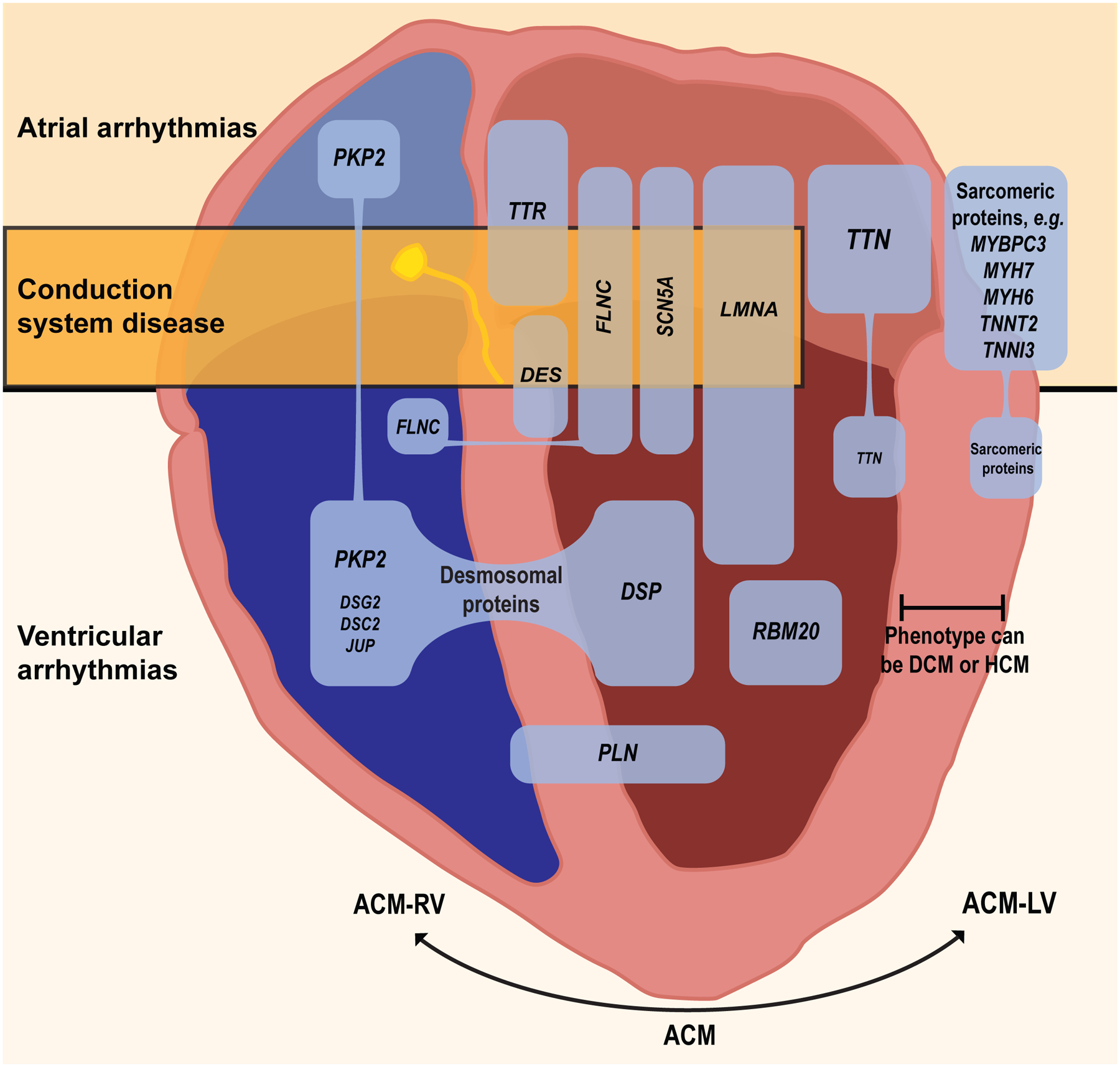
Examples of selected cardiomyopathy genes commonly associated with atrial or ventricular arrhythmias or conduction disease. The common genes coding for titin and the sarcomeric proteins are associated with a large number of variants, many of which are implicated in atrial fibrillation, but a minority of which are associated with ventricular arrhythmias, and conduction disease is rare. Variants recognized in less common genes such as LMNA and FLNC are more consistently associated with ventricular arrhythmias and sudden cardiac death. Variants in genes for desmosomal proteins, phospholamban, and filamin-C can be associated with both RV and LV involvement. ACM= general term for arrhythmogenic cardiomyopathy, ACM-RV = arrhythmogenic cardiomyopathy with dominant RV involvement; ACM-LV= arrhythmogenic cardiomyopathy with dominant LV involvement, DCM = dilated cardiomyopathy, HCM = hypertrophic cardiomyopathy. (Illustration credit: Ben Smith).
AF also occurs in restrictive cardiomyopathy, which is less common than HCM and DCM (Table 2). Most RCM results from acquired or systemic disease, but can be due to amyloidosis, for which the common genetic cause is mutant transthyretin (TTR). AF may be the initial presentation of ATTR-CM (transthyretin amyloid cardiomyopathy) when other signs have been overlooked.50 In an international ATTR-CM registry, AF occurred in half of the subjects with the VAL122Ile mutation.51 Pathogenic variants in DES and FLNC have been associated with an increased incidence of AF in either DCM or RCM phenotype.
Anticoagulation and Pacing for AF with Genetic CM
Regardless of the CHADS2-VASC score, anticoagulation for AF is generally recommended for all patients with a clinical diagnosis of HCM, all women and most men with DCM, and more recently for ACM-RV as well.2,52,53 Anticoagulation is indicated for all patients with AF and cardiac amyloidosis regardless of LVEF or HF severity. Therapy for AF in ATTR-CM is most effective early after diagnosis, as only 17% of patients with advanced disease can maintain sinus rhythm.55 ATTR-CM amyloidosis has been associated with frequent development of a left atrial thrombus despite anticoagulation.56
While CM variants are well-described in early onset AF, it is not clear how identification of a CM variant should influence care for a patient with early-onset AF as the first presentation.44,45 For instance, it is not known whether the stronger recommendations for anticoagulation with CM should extend to patients with AF and positive genotype in the absence of CM phenotype. Should patients with AF as their only manifestation of an ACM phenotype restrict activity or be evaluated for risk of VA/SCD? Although these questions remain unanswered, for a patient with AF and an arrhythmogenic genotype who then develops a need for permanent pacing, implantation of a defibrillator device should be considered.
Conduction Disease
Right bundle branch block (RBBB) is the most common conduction delay encountered in the absence of known heart disease. The incidence of complete RBBB without known heart disease varies from < 1% in young airmen57 to 1.3% in the Women’s Health Initiative,58 and increases with age. There is no consistent association with mortality after adjustment for known cardiovascular risk factors.59 During cardiac evaluation, however, RBBB can be an early clue to RV desmosomal disease and is also common with cardiac sarcoidosis and Chagas’ disease.54
Left bundle branch block (LBBB) is seen in 0.1 – 1% of the population without known cardiac disease. In the Framingham Heart Study, 28% of patients developing LBBB without prior cardiac history had a new diagnosis of HF within a mean 3 years,60 a rate 7-fold higher than without LBBB. The association of new LBBB with future heart failure may reflect subclinical CM or ischemic heart disease, and/or impaired LV function from dyssynchrony that can improve with biventricular pacing.61 LBBB occurs in approximately 25–30% of all DCM but was present in over 50% of patients with LMNA variants in one series.25
LMNA variants are associated with progressive conduction system disease as well as with AF and VA (Figure 3). This may relate in part to its predilection to cause fibrosis in the septum. Some degree of atrioventricular block is present at diagnosis in 38% to 78% of LMNA CM.62,63 By the time VA appear, most patients have PR prolongation and up to 1/3 have complete heart block. Complete heart block requiring a pacemaker may occur several years before evidence of DCM, which develops in almost all patients who reach their seventh decade with cardiac LMNA disease.62
Primary cardiac conduction defects also occur due to genetic variants that may not cause CM. Loss of function mutations in SCN5A, encoding the cardiac sodium channel, are a well-recognized cause of progressive conduction system disease (Lenègre disease) in humans and in mice, but DCM is not a common feature of these mutations.64,65 Other SCN5A mutations have been associated with atrioventricular block and DCM,66 and with PVC’s and related CM with subsequent VA as discussed below.
Conduction disease is also common in ATTR-CM. Prolonged PR interval and prolonged QRS are present in almost half of patients at the time of presentation with cardiac transthyretin deposits.56 About one in 10 patients with ATTR-CM presents with high degree AV block and one of the remaining 9 will progress to complete heart block.
In addition to LMNA, SCN5A, and transthyretin, other genes associated with both conduction disease and CM include FLNC and DES. Conduction disease also commonly accompanies the muscular dystrophies (see below). Atrial standstill can occur with Emery-Dreifuss dystrophy and Friedreich’s ataxia. An important implication of conduction disease in a patient with a CM is for consideration of an ICD, especially if pacing is contemplated. It is now increasingly well-recognized that sudden cardiac death (SCD) may occur early in the course of the disease as well as after progression to HF with LVEF ≤ 0.35, for which ICDs are generally recommended regardless of the need for pacing or etiology of HF (Table 1).
The mechanisms underlying conduction disease in CM are not well-defined. Lenègre disease (which does not usually progress to CM) can be caused by SCN5A mutations that have been shown to cause ventricular fibrosis in mice.64 Amyloid infiltration is similarly thought to cause conduction block.67 Multiple mechanisms, including altered transcriptional regulation of multiple downstream genes, have been proposed in LMNA cardiomyopathy.68
Ventricular Arrhythmias
PVC’s, non-sustained VT (NSVT), sustained monomorphic VT, and polymorphic VT/ventricular fibrillation can all occur in any CM later in the course of disease, associated with myocardial fibrosis, electrical remodeling, neurohormonal activation, depletion of energy stores and relative ischemia, as well as from proarrhythmic effects of drugs and electrolyte abnormalities.69 In arrhythmogenic cardiomyopathies, VA may emerge early, sometimes as the first presentation of CM.70 Initial evaluation seeks to distinguish ACM from idiopathic VA that occur in the absence of structural heart disease and are usually benign.71 Fibrosis, seen with LGE on CMR imaging, strongly favors myocardial disease rather than an idiopathic arrhythmia, and often corresponds to the site of origin of VA (Figure 1).
VA can be due to automaticity, triggered activity, or reentry. Fibrosis is commonly associated with VA and sudden death risk in CM. Fibrosis can diminish cellular coupling which can slow conduction and facilitate automaticity or propagation of wavefronts initiated by automatic foci. LMNA, DSP, and FLNC mutations are associated with prominent fibrosis (often in specific locations such as the septum in LMNA-associated CM) and it is possible that the extent and distribution of fibrosis contributes to their arrhythmic propensity. The extent to which mutations also disrupt cellular electrophysiology varies and is not well defined for most genes.
One exception is SCN5A mutations which cause multiple arrhythmia syndromes. Some SCN5A mutations are clearly associated with CM, while others (type 3 Long QT syndrome, Lenègre disease, Brugada syndrome) generally are not.72,73 However, there is growing recognition that Brugada syndrome can be associated with abnormal contractile function74, and DCM related to bundle-branch block has been described with other SCN5A mutations.75 Mutant sodium channels may alter intracellular sodium and calcium homeostasis, leading to frequent ectopy and DCM.76,77 Sodium channel mis-localization and dysfunction are also seen with mutations in other DCM-associated genes,78 consistent with an emerging appreciation of a role of the sodium channel in interacting with macromolecular complexes to regulate cell structure, distinct from its role in ion conduction, as discussed below.79 The unusual syndrome of Multifocal ectopic Purkinje-related premature contractions (MEPPC) is discussed below. In addition, HF itself, regardless of cause, has been associated with sodium channel dysfunction in animal models.80,81
There is also increasing appreciation that CM due to genes that do not code for ion channels may nevertheless lead to impaired ion channel function. Patients with ACM-RV due to PKP2 mutations can develop arrhythmias when there is little structural abnormality. These mutations reduce expression or localization of multiple genes whose protein products are involved in intracellular calcium homeostasis (type 2 ryanodine receptors, ankyrin 2, L-type calcium channels, triadin, and calsequestrin-2), disrupting intracellular calcium handling and increasing susceptibility to ventricular arrhythmias caused by triggered activity, consistent with the exercise induced arrhythmias and sudden death observed in ACM-RV.73,82 Similarly, ACM-RV mutations have also been shown to directly affect cardiac sodium channel abundance and intracellular localization, effects that likely perturb normal conduction and increase susceptibility to VA. Re-entrant VT in ACM-RV, however, is often associated with areas of fibro-fatty replacement that develop during the disease.
Sites of VA Origin: ECG Correlations
The QRS morphology of PVCs or VT can point to their sites of origin and provides clues to the presence and type of CM (Figure 4). VA with a dominant S wave in V1 are designated as having LBBB morphology and usually originate from the RV or interventricular septum. An LBBB pattern with an inferior axis (tall R waves in the inferior leads) is typical of RV outflow or periaortic LV outflow tract origin, both common locations for idiopathic arrhythmias as well as VA in ACM. VA of RV origin are usually present in ACM-RV (Figure 4A). VA with a dominant R wave in V1 are designated as having an RBBB morphology, usually originating from the LV (Figure 4B) and associated with myocardial disease.
Figure 4.
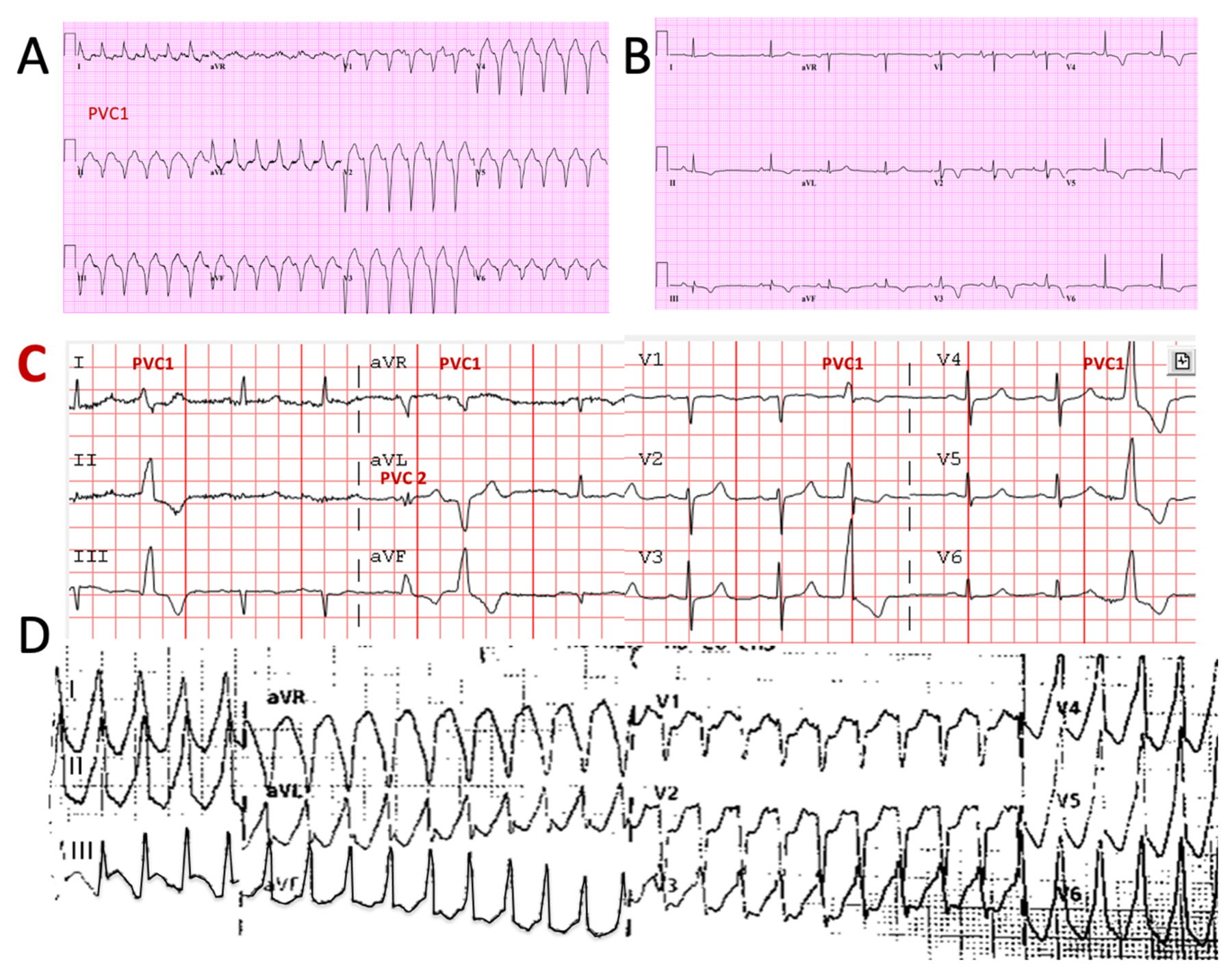
Examples of 12-lead electrocardiograms (ECG) from 2 patients with arrhythmogenic cardiomyopathies Panels A and B are from a 53-year-old man with ACM-RV. Panel A shows SMVT with atrio-ventricular dissociation. V1 has a left bundle branch block-type configuration and the frontal plane is directed superiorly, consistent with a VT origin in the inferior RV. Panel B shows sinus rhythm ECG findings typical for RV disease with T-wave inversion in leads V1–V6 and inferiorly, and a slurred and prolonged terminal s-wave in V2. Panel C is from a 77-year old man with a long history of PVCs that had been considered benign in the absence of either known disease or concerning family history. The most frequent PVC (PVC1) has a right bundle branch configuration in V1 with dominant R waves across the precordium and inferiorly directed frontal plan axis, consistent with an origin in the periaortic LV. Panel D in the same patient shows the tracing during ventricular tachycardia, taken by paramedics after he presented with syncope. Genetic testing revealed a pathogenic TTN mutation.
ACM-RV=arrhythmogenic right ventricular dysplasia, LV= left ventricle, RV=right ventricle, SMVT = sustained monomorphic ventricular tachycardia
Premature Ventricular Contractions and Non-sustained VT
PVCs are common in CM, sometimes as the initial presentation (Figure 4C), but can also be idiopathic or from structural heart disease. PVCs are noted on routine 12-lead ECG in 1.5 to 6% of adults and are associated with a greater than 2 fold increased risk of sudden death, heart failure, and cardiac death.83–85 NSVT in DCM is associated with a greater likelihood of identifying a pathogenic variant and consistently linked to risk of adverse outcomes that include HF events as well as VA/SCD.25
The likelihood of underlying CM increases with multifocal PVCs and PVCs with RBBB pattern (LV origin). Fibrosis detected as LGE on CMR imaging generally indicates the presence of CM and increased risk of sustained VA.10 In a multicenter registry of 686 patients who had ≥1000 PVCs/24 hours considered to be idiopathic with LVEF > 0.50, MRI showed LGE in 15% of patients, of whom 27% experienced VT/VF, cardiac arrest or death during median follow up of 5 years.86 A ring-like pattern of 3 contiguous segments of LGE in the same short axis slice was associated with the greatest risk (Figure 1C), with 50% of these patients experiencing a VA event, compared to 19% of those with other LGE patterns, and 0.3% of patients with no LGE.13
A high density of PVCs over prolonged time can itself depress LVEF, which may normalize when they are suppressed. This PVC-induced CM is reported in 7 to 26% of patients referred for ablation of PVCs and LVEF improves in about two-thirds of them following successful PVC suppression.87,88 Most patients with PVC-CM have a PVC burden of more than 15%, but daily frequency can vary substantially, and depressed LVEF has also been seen with fewer PVCs. Of note, interstitial fibrosis occurs in some animal models of PVC induced myopathy89 and LV function can recover after successful suppression of PVCs despite the presence of LGE.90
Notably, the majority of patients with very frequent PVCs do not have LV dysfunction, suggesting heterogeneity in susceptibility, for which genetic factors could play a role. Thus, a patient who has PVCs with reduced LVEF may have idiopathic PVCs depressing LV function, an underlying cardiomyopathic process, or a combination of primary CM with ectopy contributing to depressed ventricular function. Assessment may be further complicated by difficulty measuring ventricular function in the presence of frequent PVCs.
Multifocal ectopic Purkinje-related premature contractions (MEPPC)
Multifocal ectopic Purkinje-related premature contractions (MEPPC) is an example of a genetic syndrome with specific therapeutic implications. It was described near-simultaneously by four groups in 2012.76,91,92 MEPPC is characterized by multifocal PVCs that have a relatively narrow QRS, consistent with an origin from the Purkinje system, and can produce ventricular dysfunction. It is due to specific variants in SCN5A that confer an unusual “gain of function” in the cardiac sodium channel. It has high penetrance in affected families. Recognition is important because pharmacologic therapy with sodium-channel blockers can suppress the PVCs with recovery of ventricular function if initiated sufficiently early in the disease.93
Sustained Ventricular Arrhythmias
Sustained monomorphic ventricular tachycardia (SMVT)
Sustained monomorphic ventricular tachycardia (SMVT) is relatively uncommon among the general population of DCM, occurring in fewer than 1–3% of patients annually.94,95 Much of our understanding of SMVT derives from the small number of patients with recurrent episodes referred for VT ablation. More than 90% of these VTs are due to scar-related reentry and fewer than 10% are due to bundle branch reentry involving the Purkinje system or a focal mechanism.96 Ebert et al performed genetic testing on 98 consecutive patients with DCM, referred for catheter ablation of sustained monomorphic VT in the LV, finding pathogenic mutations in 38%, dominated by variants in LMNA and TTN, but also including PLN, SCN5A, RBM20, and DSP.24 Genotype-positive patients had worse outcomes, with 81% having recurrent VT and 51% dying or requiring heart transplant during median follow-up of 28 months.
VA have been a major component of the traditional ARVC criteria, with SMVT occurring in more than 40% of patients.97 SMVT typically originates from areas of RV subepicardial fibro-fatty myocardial replacement giving rise to VA with a LBBB-like QRS (Figure 4A). LBBB-like VT with a superior frontal plane axis favors ACM over idiopathic VT, as it indicates an origin outside the outflow tract.98
Polymorphic VT/Ventricular Fibrillation
Polymorphic VT/Ventricular Fibrillation occurs in ACM regardless of which ventricle is most affected, and in HCM. A variety of mechanisms associated with electrophysiologic remodeling in hypertrophy and heart failure may be involved. Genetic cardiomyopathies are associated with arrhythmogenic mechanisms linked to calcium handling and other electrophysiologic perturbations.99 Some appear to be due to scar-related reentry100 or associated with rapid activity in the Purkinje system driving the ventricle into fibrillation.101 The polymorphic VT torsade de pointes associated with QT prolongation also occurs, typically in association with other factors prolonging the QT interval such as hypokalemia from diuresis, bradycardia, and QT-prolonging drugs. Polymorphic VT or VF with HCM cardiomyopathy may result not only from the myofibrillar disarray but also from ischemia, particularly with exercise, related to the excess myocardial oxygen demand and the abnormal coronary vasculature that commonly accompanies HCM.
Phenotypes of Genetic CM
The diagnosis and treatment of cardiomyopathies have traditionally focused on their classification as hypertrophic (HCM), dilated (DCM), and restrictive (RCM), based on left ventricular morphology and function (Table 2). In contrast, arrhythmogenic cardiomyopathies (ACM) are instead defined by the prominence of VA in the clinical presentation and further classified by the ventricle most affected.102 (Figure 3). Arrhythmogenic cardiomyopathy with dominant LV involvement (ACM-LV) is usually associated with the phenotype of DCM (Table 2) and occasionally with RCM, while structural RV abnormalities of ACM with dominant RV involvement are more variable. Some degree of involvement of both ventricles is common, particularly with desmosomal disease.102
The rapid acceleration of genetic testing has outpaced our techniques to identify and track the associated phenotypes to inform prognosis and management. The layering of phenotype and genotype is further complicated by overlaps between the major types of CM (Table 2).
Hypertrophic CM Phenotype
Hypertrophic cardiomyopathy (HCM) can present with AF, syncope, or resuscitated SCD. The morphologic phenotype, as described in Table 2, hinges on LV wall thicknesses and patterns of hypertrophy.103 The prevalence of genetic HCM, typically an autosomal dominant disease, is currently estimated at 1/200–250 adults, about twice as high as phenotypic prevalence.104 Multi-center data in almost 6000 patients confirms the impression of higher clinical penetrance in men.105 However, once diagnosed, women appear to have similar age-adjusted rates of VA and higher rates of progressive HF and mortality than men.106
HCM Genotypes
HCM was the one of the first cardiomyopathies to be linked to a specific mutation, the missense variant R403G in MYH7 in a large French-Canadian family.107 Pathogenic variants are identified in about 60% of HCM, with about 1500 mutations in at least 11 genes.108 MYH7 and MYBPC3 account for about 80% of those identified, with another 10% in other proteins of the sarcomeric complex. A second causal gene is present in about 5% of patients, who typically have more severe disease. However, more data is needed in non-white populations, where variant frequencies and interpretation differ. For example, benign variants were misclassified among Americans of African ancestry,109 and variants of unknown significance predicted to be deleterious were enriched among Singaporeans of Chinese ancestry with HCM.110
Implications of HCM Genotype
Genetic testing can help to distinguish HCM from the athletic heart, long-standing hypertensive heart disease, and the look-alike phenocopies (Table 2). In a meta-analysis and in a large prospective registry, the presence of sarcomeric variants in HCM was associated with earlier onset of disease and up to 2-fold higher rates of AF and VA/SCD than sarcomere-negative HCM.108,111 Risks were intermediate for patients with variants of unknown significance (VUS).111 Higher event rates were seen with MYH7 variants than MYBPC3 variants. A study of 80 patients with thin filament mutations showed less LV hypertrophy and a lower prevalence of outflow tract obstruction, but similar rates of VA and SCD, and higher progression to late- stage heart failure compared to 150 patients with thick-filament mutations.112 It is increasingly apparent, however, that the implications will often depend more on the specific variants than on the genes affected. Recent work with mouse models of two different cTNNT mutations causing HCM revealed opposite responses to reducing CAMKII activation, with potential variant-specific implications for prevention of both disease progression and SCD in human HCM.113
Particularly in young athletes. syncope or cardiac arrest can be the presentation of HCM in asymptomatic patients, which can result from primary polymorphic VT/VF, ischemic arrhythmias, or from hemodynamic consequences of dynamic outflow obstruction. Recommendations for ICDs are guided by risk factors for sudden death as noted in Table 1, and also by a multi-component risk score that yields an estimated annual risk for discussion with patients.114
A new therapy targeted to decrease hypercontractility in HCM, mavacamten inhibits myosin ATPase activity and will soon be available to treat symptoms of outflow tract obstruction and improve exercise capacity in obstructive HCM.115 It is not yet known how the specific genetic variants will influence clinical responses to mavacamten, but studies with human induced pluripotent stem cells demonstrated more response to mavacamten with an ACTC1 variant than with a variant of the more commonly implicated MYH7.116
Sarcomeric variants causing the HCM phenotype generally share effects leading to hypercontractility, impaired relaxation, and high energy expenditure, typically with hypersensitivity to calcium. Some of the same variants can cause a restrictive CM (see below) with features overlapping with HCM. Sarcomeric variants associated with DCM generally affect different sections of the sarcomeric genes, causing decreased calcium sensitivity, decreased contractility, and eventually the dilated LV with low LVEF.117
Dilated Cardiomyopathy Phenotype
Dilated cardiomyopathy (DCM) can present with AF or VA, less commonly conduction disease. The phenotype is generally defined by decrease in LVEF and/or increase in LV volume or dimension (Table 2),104,118 and usually excludes structural causes such as myocardial infarction and primary valve disease. Historical estimates of 1/2500–10,000 prevalence have been revised, currently up to 1/250–1/400 adults.104 The U.S. DCM Consortium of 25 U.S. HF programs studied 1220 patients meeting criteria of both LVEF < 0.50 and LV end-diastolic dimension of ≥ 95th percentile, and their first-degree relatives available for study,119 revealing absolute rates of 11% incidence of familial DCM (defined as LV with both criteria in a first-degree relative), and a higher rate of 24.1% using an expanded definition including either criterion.) Models extrapolating to complete families yielded estimates of 29.7% with familial DCM meeting both criteria and 56.9% by either criterion. Prevalence of genetic cause of DCM by the narrow definition was higher in Black probands, 39.4% versus 28%, without difference between Hispanic and non-Hispanic ethnicity.
DCM Genotypes
Pathogenic variants have been found in 20–50% of genotyped populations with DCM, usually with rates 1.5 to 2-fold higher in subgroups with a positive family history.120,121 Positive genotypes are also more common in patients who have arrhythmias at presentation (Figure 5). Unrecognized viral myocarditis has likely been over-implicated as a cause of DCM, as the path of post-viral DCM has been difficult to follow in human studies, although extensively investigated in animal models. LGE on CMR that was at one time considered evidence of myocarditis is now increasingly recognized as typical of genetic CM, as discussed above.
Figure 5.
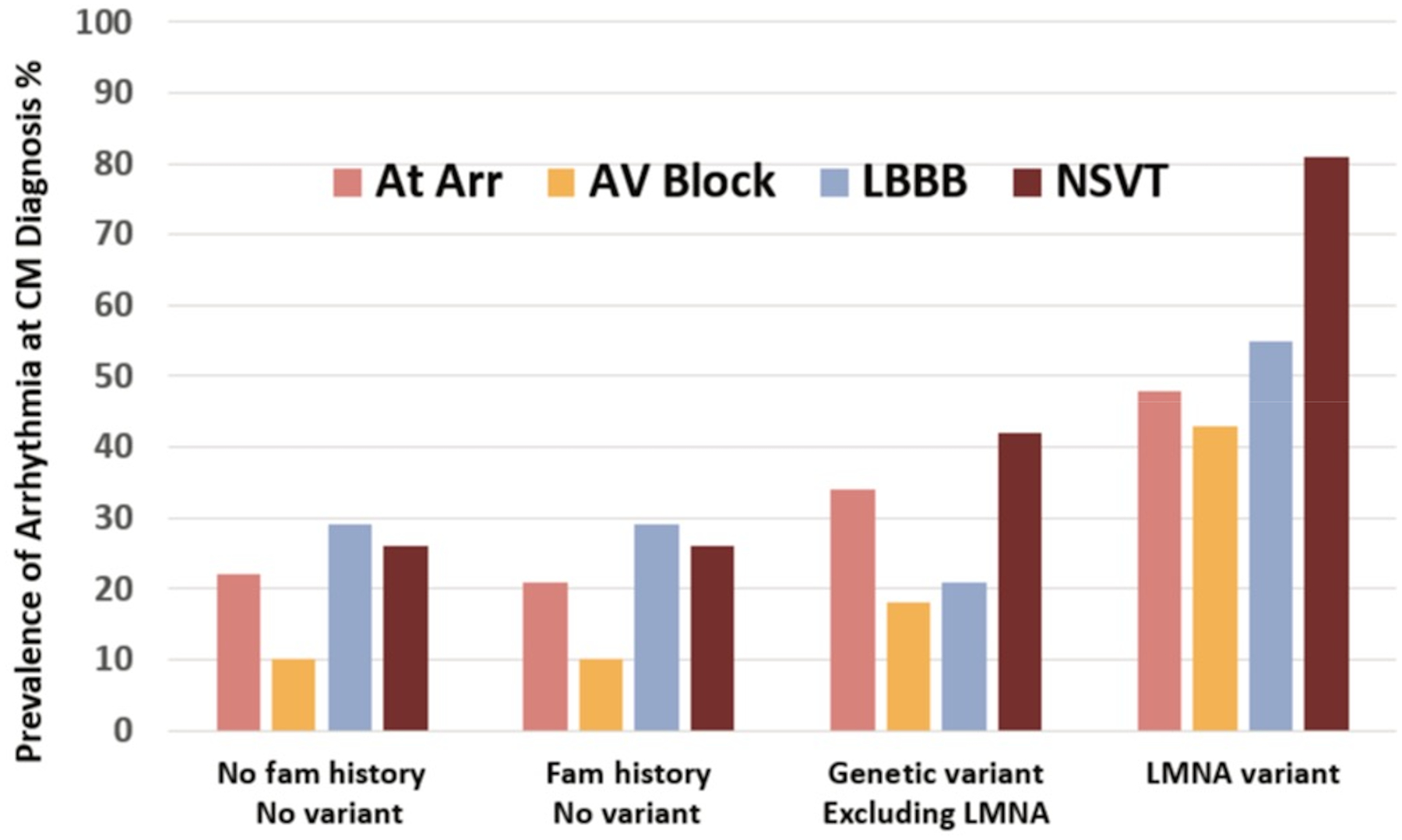
Arrhythmias at the time of presentation for 689 patients with dilated cardiomyopathy in relation to the presence or absence of family history and identified pathogenic variants on genetic testing. At Arr = atrial arrhythmias, AV = atrioventricular, LBBB = left bundle branch block, NSVT = non-sustained ventricular tachycardia. Figure drawn from published data from the Maastricht Cardiomyopathy Registry, with permission from Verdonschot and Hunner.25
DCM is associated with the largest sets of implicated genes, often 40–50, among which the prevalence of specific variants varies across regions.122 Many variants are now being re-scrutinized as too common in the unaffected population or insufficiently linked to CM by accumulating data. Two recent large series suggest that fewer than 20 genes are clearly implicated.25,123 Variants in TTN and the sarcomeric proteins together account for up to half of positive testing in patients with DCM. Examples of other variants commonly cited as causing DCM are shown in Table 3.
Table 3:
Genes Commonly Implicated in Genetic Dilated Cardiomyopathy
| Pathogenic Variant with Moderate-Strong Association with DCM | Gene | Approximate prevalence in DCM | Other phenotypes | Association with ACM |
|---|---|---|---|---|
| Titin | TTN | 12–25% | HCM ( |
In small proportion of truncating variants |
| Lamin A/C | LMNA | 4–8% |
|
High |
| Myosin heavy chain 7 | MYH7 | 3–4% | HCM ( |
Present |
| Troponin T2 | TNNT2 | 2–4% | HCM | Present |
| Filamin C | FLNC | 2–4% ** | HCM, RCM, ACM-RV, |
High |
| RNA binding motif 20 | RBM20 | 2% | High | |
| Type V voltage-gated cardiac sodium channel | SCN5A | 2–3% | Brugada syndrome | High for some variants |
| Desmoplakin | DSP | 2% | ACM-RV, |
High |
| Phospholamban | PLN | < 1% * | ACM-RV | High |
| Bcl-associated athanogene 3 | BAG-3 | 0.1–3%** |
|
Present** |
| Desmin | DES | < 1% | More often RCM |
High |
|
Other Sarcomeric Proteins, e.g.: Myosin heavy chain 6 Tropomyosin Troponin C1 Myosin binding protein C3 |
MYH6
TPM1 TNNC1 MYBPC3 |
Variable representation in DCM populations | HCM, |
Occasional variants implicated |
ACM = arrhythmogenic cardiomyopathy, characterized by early and severe ventricular arrhythmias
ACM-RV = arrhythmogenic cardiomyopathy with dominant RV involvement
DCM = dilated cardiomyopathy phenotype, HCM = hypertrophic cardiomyopathy, RCM = restrictive cardiomyopathy
![]() = associated with skeletal myopathy
= associated with skeletal myopathy
Associations between genotype and arrhythmia phenotypes vary due to relatively small series, referral center bias, and geographic heterogeneity.
Higher prevalence of a founder mutation in Netherlands/Germany
Implications of Genotype for DCM
In contrast to variants causing HCM, some of the specific genes identified for DCM are consistently linked to higher likelihood of VA, disease progression, and death. In the large series from Maastricht assessing arrhythmias at the time of first CM evaluation, the adverse impact of genetic etiology on DCM outcomes was mediated primarily through increased risk associated with arrhythmia phenotypes, defined as AF, conduction disease, or VA at initial evaluation.25 In another recent series of 487 patients with DCM, pathogenic variants in LMNA and desmosomal proteins were associated with increased risk of ventricular arrhythmias and sudden death, while other variants were associated with outcomes similar to DCM without an identified pathogenic variant.121
Many Titin Variants
Although they are not among the most frequent causes of ACM, attention is focused on TTN variants due to their frequency in DCM. TTN, the largest gene in the body, encodes titin, the largest protein in the body and an integral part of the sarcomeric complex. Extending from the Z disc to the M band, titin is involved in force transmission, cell organization, and signaling. While variants in titin are much more common than for other CM genes, the variant frequency is similar when normalized for size.122 Because the gene is so large, and therefore missense variants so common, genetic studies to date have focused on truncating variants. Truncating TTN variants account for 12–25% of pathogenic variants implicated in DCM, up to 25% in familial CM and 18% of patients without family history.
Truncating TTN variants considered pathogenic have also been described in 2–3% of normal subjects without known cardiac disease, raising the likelihood that CM attributed to some variants may in fact be coincidental to another cause or a different pathogenic variant.104 In a multi-national study of 639 patients with DCM, 44% of patients with TTN variants implicated as causal also had pathogenic variants in another DCM gene.122
TTN is commonly implicated in a “two-hit” cardiomyopathy, developing in the setting of an acquired insult (or another CM pathogenic variant); common clinical settings in which TTN variants appear to increase CM risk include postpartum, heavy alcohol use, and cancer chemotherapy. Favorable reverse remodeling with major LVEF improvement after initiation of recommended HF medications occurred in 69% of patients in the large study above, a higher rate than the 30–40% expected in general DCM.
The TTN variants exemplify a DCM phenotype in which systolic dysfunction usually dominates the clinical picture, with mean LVEF already reduced to 0.30 at diagnosis.125 The most common presenting symptom in 537 patients was dyspnea, 21% presenting with palpitations and about 10% of patients with AF.126 By the end of almost 10 years, 30% with TTN variants had developed AF, 45% had NSVT, and 9% had experienced sustained VT.127 VA occurs rarely in the absence of marked left ventricular dysfunction, but LVEF, LGE, and NSVT all predict both HF and arrhythmic events, as does family history. Although not common, there are TTN variants with a high VA/SCD rate consistent with ACM.
Arrhythmogenic Cardiomyopathy With Dominant LV Involvement
VA occur commonly during progression of any dilated LV cardiomyopathy. However, pathogenic variants causing VA early in the course of CM are increasingly recognized. Current estimates are that about 30% of genetic DCM is associated with high risk for VA that can occur before prominent LV dilation or dysfunction,121 but estimating the prevalence of ACM with dominant LV involvement has been hindered by lack of a consistent definition.102 Most commonly implicated are LMNA and desmosomal proteins (Table 2), which together account for about 10% of all DCM patients; RBM20 and SCN5A occur in about 2% each, and FLNC and DES occur in about 1% each.128
Lamin A/C
Pathogenic LMNA variants are identified in 4–8% of DCM, with “laminopathies” considered among the most malignant and highly penetrant ACM.24,129 Differential splicing of the single lamin gene produces lamin A and lamin C, which are nuclear envelope proteins involved in regulation of DNA replication, gene expression, replication, mechanosignaling, and cytoplasmic transport. LMNA mutations causing cardiomyopathies can also involve skeletal muscle, such as with Emory-Dreifuss or limb-girdle dystrophy, amd are also associated with familial partial lipodystrophy.
Arrhythmias are frequently the first presentation of LMNA cardiomyopathy (Figure 3), often preceding the diagnosis of DCM. The most common initial rhythms are AV block or AF, but life-threatening VA can be the presentation. At first VA, mean LVEF was 0.42±0.15, while 25% of patients still had LVEF greater than 0.50. By 7 years after diagnosis, over half of patients had AV block, over half had AF, and a third had sustained VA.63
The risk of developing sustained VA was 3-fold higher for male sex or LVEF < 0.50 at presentation, 2.5-fold higher with non-missense mutations. Due to the high incidence of VA, the recommended indication for primary prevention ICD in the presence of LMNA mutation is the presence of at least 2 risk factors: male sex, non-missense variant, NSVT or LVEF < 0.45 (Table 1).32 LVEF < 0.50 at presentation was associated with almost 5-fold risk of end-stage HF and many patients undergo heart transplantation.63 Mitogen-activated protein kinase signaling has been implicated in the pathogenesis of LMNA disease (and perhaps other DCM), and a clinical trial of p38 MAPK inhibition is currently ongoing in patients with LVEF < 0.50.130
Desmoplakin And LV Desmosomal CM
DSP accounts for about 4% of variants causing ACM-LV and is traditionally grouped with other proteins of the desmosomal complex, which connects the myocytes with each other and also contributes to structure and function within the cell, discussed below. However, unlike the other desmosomal proteins, the VA, fibrosis, and depressed ventricular function with DSP variants are often dominated by LV involvement.102
A recent multi-center series reported 107 subjects with pathogenic desmoplakin variants.131 Of those with decreased ventricular function, 79% had predominant LV involvement, among whom 31% also had decreased RVEF. The Arrhythmogenic Task Force criteria from 2010 for ARVC33 were met in only 42% of desmoplakin patients with LV dominance and 57% of those with RV dominance of DSP disease. LGE was seen in 74% of patients with LV involvement (Figure 1B), usually sub-epicardial in the inferior wall and occasionally associated with intramural fat. Cutaneous findings of palmoplantar hyperkeratosis and curly hair seen in the autosomal recessive Naxos disease, as described below, are also prominent with the autosomal dominant desmoplakin disease of the LV and may be overlooked in patients who use hair-straighteners.131
As discussed above, presentations consistent with acute myocarditis, which can include VA, chest pain and troponin elevations, are described with other desmosomal disease18,19 but may be particularly common with desmoplakin.19,131 These patients often have sub-epicardial LGE initially, but may not return with VA or the DCM phenotype until years later.
Patients with LV dominant disease often present in their thirties. Half of VA/SCD events occur in patients with LVEF > 0.35 and 15% with LVEF > 0.55.131 Current indications for primary prevention ICD with desmosomal disease have focused on traditional ARVC criteria, and exclude many patients with ACM-LV with DSP variants, who nonetheless merit special consideration to prevent sudden death before the LVEF is less than 0.35 (Table 1). Although exercise restriction is recommended for desmosomal disease with predominantly RV involvement, as described below, the association of strenuous exercise with earlier and more severe disease has not been established for desmoplakin disease in the LV, perhaps because the LV is less distended than the RV during exercise.132
Filamin C
FLNC is a recent addition to genetic CM panels, so the 1% prevalence described is likely to increase. Considered a cytoskeletal protein, filamin-C stabilizes polymerized actin and anchors membrane proteins to the cytoskeleton in both skeletal and cardiac muscle. Strong evidence associates variants in FLNC with DCM and the skeletal myofibrillar myopathies, but CM often occurs without recognized skeletal myopathy.133 This ACM usually has an LV dominant phenotype but occasional biventricular and RV dominant phenotypes can occur.134 Missense variants in FLNC have also been associated with both HCM and RCM phenotypes, suggesting a wide spectrum of cardiac disease.135
FLNC may present with AF, conduction disease, or VA prior to a diagnosis of CM.134 Both the arrhythmogenic phenotype and the potential for bi-ventricular involvement are shared by FLNC and desmosomal cardiomyopathies (Figure 3), although the pathogenesis appears distinct. Both are also characterized by a sub-epicardial location of LGE, sometimes in a ring-like pattern.
Patients with truncating variants in FLNC have a cumulative 15–27% incidence of major VA or SCD events over 5-year follow-up,121,136 which is similar to rates for LMNA and DSP for both probands and carriers. The risk of VA correlated with frequent PVCs and NSVT on ambulatory monitoring, and with the degree of LGE but not with LVEF.137,138 ICD recommendations in the HRS Expert Consensus Statement2 include FLNC with LVEF ≤ 0.45 (Table 1). Although LVEF remained stable during 5-year follow-up in most patients, 22% progressed to end-stage HF, most of whom had lower LVEF at presentation.
RNA binding-motif 20
Variants in this RNA-splicing factor gene RBM20 are implicated in 2–3% of familial DCM.139 This protein modulates RNA splicing for over 30 genes,140 including those coding for titin, sarcomeric proteins, and proteins involved in calcium handling. The resulting variants of these gene products have been implicated in systolic dysfunction but other pathways are likely involved also, particularly with regard to calcium handling and VA.
RMB20 variants have shown high penetrance rates of 66%−96% and earlier age at presentation than for general DCM populations or for titin CM. Among index cases in a multicenter series of 74 patients, family histories of CM were present in 72% and of SCD in 51%.141 VA/SCD events have been reported in 20%−44% of patients with RBM20 variants99,141,142 compared to 2.2% in an unselected CM registry.138,141 VA event rates with RBM20 are similar to those with LMNA disease63 and also often occur with EF > 0.45, as in 1/3 of an international series of 74 RBM20 patients reported by Parikh et al. Special consideration for ICD implantation may thus be warranted for RBM20, as it is for LMNA (Table 1).
Data emerging from studies in pluripotent stem cells and from RBM20 knock-out mice suggests that abnormal calcium handling may contribute to intracellular calcium overload and the VA phenotype. This might eventually provide a target for therapy of ACM with RBM20 variants, and potentially for other CM, through modulation of calcium homeostasis.143
Phospholamban
Phospholamban (PLN) is a transmembrane protein of the sarcoplasmic reticulum (SR) membrane. Until phosphorylated, PLN inhibits the SR ATPase (SERCA2) from transporting calcium back into the SR, with greater inhibition by the DCM-associated variants.144 The R14del PLN founder mutation accounted for 13% of 240 Dutch patients with DCM and also 12% of Dutch patients with arrhythmogenic RV cardiomyopathy145, but occurs in less than 1% of DCM in other regions. PLN cardiomyopathy is associated with marked fibrosis in the LV posterolateral wall and occasionally with fibro-fatty infiltration of the RV that qualifies as ACM-RV.146 Malignant VA can occur before LVEF is severely reduced and progression to heart transplantation is frequent. A pathogenic PLN variant is a current indication to consider early ICD placement.
The complex role of SCN5A variants in multiple arrhythmias and CM is discussed above with the specific arrhythmias.
Arrhythmogenic Cardiomyopathy With Dominant RV Involvement
The major hallmark of arrhythmogenic CM with dominant RV involvement is the predominance of VA events rather than the variable abnormalities in RV morphology or function. ARVC was initially described as a cause of sudden death in athletes in Italy.147 The first gene was identified in families on the Greek island of Naxos, in whom SCD was frequent and associated with “woolly hair” and hyperkeratosis of the hands and feet. The RV was dilated with areas of thinning and replacement by fat. The Naxos syndrome, which is autosomal recessive, is caused by mutations in JUP, encoding for plakoglobin, a protein of the desmosomal complex. Variants in genes encoding other components (PKP2, DSP, DSG2, DSC2) are also well-recognized causes of desmosomal disease affecting predominantly the RV. Variants in other genes such as RYR2 have occasionally been associated with similar phenotypes.148
The prevalence of CM by ARVC criteria is currently estimated at 1/2000–5000 adults.149 In a transatlantic study of 439 probands meeting this criteria, pathogenic variants were identified in 63%, up to 89% with positive family history. Plakophilin (encoded by PKP2) was the most common desmosomal protein implicated, accounting for 74% of identified variants; others were desmoplakin (DSP) 4%, desmoglein (DSG2) 6%, and desmocollin (DSC2) 2%, and only 2 patients had variants in plakoglobin (JUP), which are rare outside Mediterranean countries. Coexistence of multiple variants, identified in 4–16% of desmosomal cardiomyopathies, have been associated with earlier and more severe disease.150
Desmosomal Disease of the Right Ventricle
The desmosome is a structure that bridges the cell membrane and connects the myocytes with each other. It also contributes to the structure and integration of functions within the cell. Disruption of the desmosomal structure can lead to cell separation and death. Because desmosomes are also central to integrity of connective tissue, some pathogenic variants are associated also with the distinctive findings of the hair and skin, as discussed above. The progressive fibro-fatty deposits within the myocardium may be replacement for lost myocardial cells but have also been proposed to result from effects of displaced plakoglobin to alter nuclear signaling for stem cells,151 resulting in differentiation into adipocytes and fibroblasts in areas that may represent substrate for frequent PVCs and reentrant VT.
RV dilation and dysfunction are often present but visualization can be difficult. Abnormalities of the RV on the ECG and echo imaging are a prominent part of the original criteria for ARVC, later revised to also include genotype.33 Although myocyte disconnection and damage are seen on pathologic examination, myocyte loss may be limited, and few patients progress to end-stage HF of either ventricle. The reason for predominant RV involvement with desmosomal disease is uncertain, but greater vulnerability of the thin RV myocardium to mechanical and hemodynamic stress, particularly during the ventricular dilation that occurs with exercise, has been postulated.132 Models of desmosomal disease show early and more severe RV dysfunction and fibro-fatty replacement when the animals perform frequent exercise.152 Clinical presentation in affected families with the RV phenotype occurs earlier with greater severity for relatives who engage in high-level exercise.153
Patients with desmosomal ACM affecting the RV often present in their 30s. In a series of 1001 patients and family members, 61% of probands presented with sustained VA.149 Some patients present first with very frequent PVCs. Sustained VA occurred in 72% of patients during mean 7-year follow-up; sudden death occurred in 16% of patients without ICDs compared to < 1% in those with ICDs. Indications for ICD are based on the ARVC disease criteria105 and specific risk factors for VA4 (Table 1).154 The course is usually dominated by VA, as symptomatic HF developed in only 13% of index cases in this large series, with only 4% going to heart transplantation.
Although most patients with arrhythmogenic CM have a phenotype dominated by one ventricle, disease is often present in both ventricles, particularly with the desmosomal variants. Early pathologic studies demonstrated some fibro-fatty involvement of the LV in 76% of patients with dominant RV disease whose hearts were examined after death or transplant.155 Using CMR, a more recent study in 89 patients meeting criteria for ARVC showed LV LGE in 67% of patients, of whom most have detectable decrease in LVEF.4 Conversely, evidence of RV involvement was found in 31% of patients with predominant LV involvement with DSP mutations.156 Bi-ventricular involvement has also been found with variants in other ACM genes such as PLN and FLNC.
Restrictive Cardiomyopathy Phenotype
Unlike HCM and DCM, the phenotype of restrictive cardiomyopathy (RCM) has variable morphology and is defined instead primarily by diastolic dysfunction on echocardiography and elevated filling pressures during hemodynamic evaluation. RCM typically presents with mildly reduced LVEF, which is encompassed within the newly categorized “Mid-range LVEF” from 0.40–0.50, but this EF range should not be considered a specific phenotype. The LVEF of 0.40–0.50 is instead the intersection of many conditions including DCM which is deteriorating or improving into remission, and HCM with “burn out”, as well as RCM and mildly decreased LVEF from structural heart disease (Table 2).
Aside from systemic metabolic diseases and hemochromatosis, the major genetic cause of RCM arises from variants in transthyretin (TTR) causing amyloid cardiomyopathy (ATTR-CM). Three ATTR-CM variants, Val122Ile associated with African descent, Thr60Ala often with concomitant neuropathy, and Val30Met with early neuropathy have been best characterized.51 Both AF and conduction disease may present prior to recognition of amyloidosis as previously discussed. VA are relatively uncommon and small series have not demonstrated benefit of ICDs. Tafamadis, a stabilizer of the TTR tetramer, has slowed progression of both cardiac disease and neuropathy, and decreased mortality by 30%.157 Patisiran, an siRNA agent and inotersen, an antisense oligonucleotide, target hepatic production and reduce circulating TTR by over 85%.158 Early diagnosis directly impacts treatment benefit, as benefits of these therapies have been limited to those with few cardiac symptoms at presentation.
Although most RCM is due to acquired causes or to amyloidosis, genetic disease is increasingly recognized in the remaining cases. Sarcomeric genes are most commonly implicated, particularly those of the thin filaments and associated proteins.159 HCM and RCM can exist with the same sarcomeric variant in different members of the same family.160 The RCM phenotype is seen with DES mutations and associated skeletal muscle involvement, and has occasionally been reported with FLN-C variants.
Skeletal Myopathies
Heritable myopathies are a heterogeneous group of disorders that can be accompanied by CM, arrhythmias, and conduction disease. Cardiac involvement varies in onset and severity, but is occasionally the early or dominant presentation when neuromuscular manifestations are mild. Multiple genes causing CM are associated with variable degrees of skeletal myopathy (Table 3).
The most common myopathy is myotonic dystrophy type I (MD1), due to expansion of an unstable trinucleotide repeat in the DMPK gene, in which the number of repeats and clinical severity increases from generation to generation.161,162 Severe conduction defects, VA, and SCD may appear early in disease, but late sudden deaths are usually attributed to respiratory events163,164 and only 10% of patients with adult-onset disease progress to reduced LVEF.
Emery-Dreifuss muscular dystrophy is due to mutations in one of several genes encoding nuclear envelope proteins. X-linked disease due to mutations in EDMD1, is associated with atrioventricular conduction defects, AF, and atrial standstill that can precede overt CM and neuromuscular symptoms.165–167 Autosomal dominant Emery-Dreifuss disease and limb-girdle muscular dystrophy can both occur with LMNA variants, conferring high risk of arrhythmias as for other laminopathies (Figure 3).168 Desmin (DES)-associated myopathy is an autosomal dominant myofibrillar myopathy that may present with prominent CM or arrhythmias, with AF and conduction disease common and SCD also described.169
In the Duchenne’s and Becker’s dystrophinopathies, the prevalence of arrhythmias generally parallels later progression of HF, which is commonly the mode of death.120 Arrhythmias and conduction abnormalities are common with congenital syndromic myopathies and those associated with mitochondrial genetic syndromes, but rarely present early in disease.170
The Trait of Left Ventricular Non-Compaction
LV non-compaction (LVNC) is a morphologic description of prominent trabeculation of the LV above a thin compacted layer of myocardium, creating crypts that communicate with the LV cavity but not with the epicardial coronary arteries.171,172 (Table 3) Areas of non-compaction can occur with abnormal embryologic development, but can appear later in life with CM and neuromuscular disorders, as well as in athletes, normal pregnant women, and up to 8% of normal subjects.171 The trabeculation could represent a dynamic response to the intensity and regionality of wall stress, such as in HCM, where it is may function to prevent apical aneurysms.173 Recent work has implicated variants in a member of the PR domain-containing family of transcriptional regulators (PRDM16) in development of both LVNC and DCM.174 It has been suggested that this protein may direct myocardial adaptation to improve oxygenation in settings of chronic wall stress in athletes and pregnant women.175 While LVNC is often listed as a separate CM phenotype, it may instead be considered a dynamic morphologic trait. The presence of LVNC in a patient with arrhythmias or CM may increase suspicion for underlying genetic CM, particularly with a positive family history. Current management of patients meeting LVNC criteria, however, is largely guided by the presence or absence of accompanying CM or arrhythmia phenotypes.
Implications of Identifying a Genetic Cardiomyopathy
The implications of identifying a pathogenic variant for CM after presentation with an arrhythmia depend upon the nature of the arrhythmia, the variant, and the current phenotypic expression of CM. With the goal of improving prognosis, the most common current consideration is the level of risk for SCD and whether to recommend implantation of a defibrillator. However, earlier identification of CM may also help to decrease CM disease progression. While the most effective therapies to stabilize CM are currently guided only by phenotype, gene-specific treatments targeting molecular etiologies are becoming available or in development.
Decisions Regarding Primary Prevention ICD
Patients presenting with life-threatening VA/SCD should be evaluated for an ICD as secondary prevention of SCD, regardless of genotype. For patients presenting with unexplained syncope, genetic variants associated with VA will lower the threshold for EP study or ICD implantation. For patients requiring pacemakers, adding defibrillation capability should be strongly influenced by genotype and phenotype.
For primary prevention of SCD with a DCM phenotype, an LVEF ≤ 0.35 is often the indication regardless of CM etiology (Table 1). However, genetic diagnosis and/or family history may trigger consideration before CM has progressed to an LVEF ≤ 0.35, and/or before waiting 3–6 months to determine the effect of HF medications on LVEF. Special consideration for early placement of a primary prevention ICD is currently an important consideration for pathogenic variants in LMNA, FLNC or PLN, and should be extended to other types of ACM, as discussed above. There are no randomized trials establishing ICD benefit in these subgroups; recommendations derive from scientific statements and expert consensus.2,32 The presence and extent of LGE on CMR are increasingly linked with the risk of VA/SCD in DCM and HCM. However, CMR may be less sensitive for assessing risk in those genetic diseases in which VA may arise from other mechanisms independent of the extent of myocardial fibrosis. ICD indications will continue to evolve both for implicated genetic variants and for other indicators of increased risk.
Although the family history cannot be relied upon to identify the presence of a known pathogenic variant, the risk of SCD with either DCM or HCM is higher in patients with a strong family history, even when the genetic variant has not been identified.114,128 This is reflected in the guideline risk factors for HCM and as a Class IIb indication for a primary prevention ICD in patients with other CM phenotypes and family history of SCD associated with CM.34 This reflects the likelihood that sudden death risk is influenced by as yet unspecified genetic background beyond the implicated causative variant.
All consideration of ICD implantation requires discussion of risk/benefit discussion as part of shared decision-making. Decisions regarding risk of sudden death are strongly influenced by age and trajectory of both cardiac disease and general health. Women with CM have lower rates of sudden death, and patients reaching older age without arrhythmic events are generally at lower risk of life-threatening VA, particularly with genetic CM. Patients with advanced left ventricular dysfunction, a heavy HF symptom burden, and frequent HF hospitalizations are increasingly likely to die from end-stage HF rather than from untimely VA. Indications for ICD are predicated on the anticipation that patients otherwise will live at least one year with good quality of life.32,34
Current Treatments to Modify Genetic CM Progression
Fibrosis and ventricular remodeling herald disease progression and worsening outcome, including arrhythmia burden, with all HF. For genetic variants associated with DCM, there is thus good rationale, without specific confirmatory data, to support early initiation of HF therapies before the LVEF falls to <0.40. A current trial in Netherlands is testing whether the mineralocorticoid receptor antagonist eplerenone will decrease progression of phospholamban CM.176 Specific implications for therapy determined by genetic causes of DCM are currently limited, but include sodium channel blockers for the SCN5A variants associated with MEPPC.
For patients with HCM, a goal is to prevent progression of hypertrophy. A recent trial in asymptomatic carriers of sarcomeric mutations in HCM families showed that early initiation of valsartan diminished a composite score of disease progression.177 As discussed above, mavacamtem is an allosteric modulator developed to inhibit cardiac myosin and diminish hypercontractility, shown to improve symptoms and cause reverse remodeling with decreases in LVH and left atrial size.115,178,179 Intravenous enzyme replacement is routine for the HCM look-alike phenocopy, Anderson-Fabry’s disease, with genetic defect in alpha-galactosidase A, which is now also treated with an oral chaperone protein, migalastat.180 Genetic information can also help distinguish the HCM look-alike ATTR-CM, that has multiple targeted treatments as described above.
Counseling About Genetic Testing for Genetic Cardiomyopathy
The expansion of genetic testing further increases the need for qualified genetic counseling for probands and family members. Large gaps remain in the diagnostic yield for most genetic CM. Particularly with positive family history, a negative genetic test cannot rule out a genetic cause and serial screening remains important for first-degree relatives.2,31,181 While a variant of uncertain significance (VUS) should usually not be used for clinical decision-making,182 its identification challenges patient understanding of the risk, as it might later be re-classified. The American College of Medical Genetics and Genomics (ACMG) has promulgated criteria under which a VUS can be reclassified as pathogenic.182. Importantly, the identification of a rare variant in a CM-associated gene in a patient with a CM does not establish pathogenicity of that variant.
The impact of a positive test will vary according to the genetic results and implications for therapy, lifestyle, and future expectations. Discussion of potential risks will often be influenced by sex, as the penetrance and severity of genetic CM are generally lower for women than for men, as shown in a recent meta-analysis of genotype-phenotype association in DCM for most implicated genes.183
Genetic testing is recommended for first-degree relatives of a proband with a pathogenic variant for CM. A positive test can potentially have negative implications for asymptomatic individuals who do not yet carry a cardiac diagnosis. The Genetic Information Nondiscrimination Act of 2008 and the Patient Protection and Affordable Care Act of 2010 provide some protection from genetic discrimination for employment and health insurance in the United States, but in most states there are gaps in protection and the laws do not offer protection from discrimination regarding life, disability, or long-term care insurance.184 A phenotype-negative patient may elect not to be tested, but should recognize that he/she may still carry a mutation and may still transmit a pathogenic variant to children.
Patient understanding of disease risk and the need for intervention reflects their risk perceptions, which are influenced by health literacy and numeracy. Words describing possible events as “common” and “rare” are interpreted differently by providers than by patients.185 Numeracy may be low even in highly educated populations.186 People have greater difficulty comprehending relative risk, so absolute risk should be used.187 Fractions are confusing, so rates using rounded denominators such as X in 100 or X in 1000 may be understood better than proportions centered on a numerator of one, such as 1 in 384.185 Strategies such as teach-back can encourage patients to share their perceptions of risks and benefits, identifying misconceptions that can be addressed.188
Expanding Implications of Genetic Testing to Guide Therapy
As new therapies emerge for genetic CM, potential benefits of genetic testing for patients and families will increase. The definition of mutation-associated disease mechanisms at the molecular level is increasingly enabling the development of mechanism-based therapies. One approach is to “repurpose” existing drugs for the new indication of DCM: examples include an in vitro study suggesting efficacy of lovastatin in LMNA cardiomyopathy.189 Another is to develop novel “first in class” agents that target specific molecular mechanisms in CM subtypes. Examples of new agents that were developed using this strategy and are now becoming available include the myosin inhibitor mavacamtem for patients with hypertrophic cardiomyopathy,178 the myosin activator omecamtiv for advanced systolic dysfunction,190 and tafamidis to stabilize mutant transthyretin in amyloid cardiomyopathy.157 Inhibition of multiple kinases (JNK, ERK1/2, p38α MAP kinase) has been used to correct the DCM phenotype in mice with LMNA DCM191 and a clinical trial is underway in humans using a p38 MAP kinase inhibitor for LMNA DCM.192 In vivo gene editing using adenoviral delivery of CRISPR/cas9 constructs corrected the molecular defect in mice with dystrophin-related DCM.193 In DCM caused by titin truncating mutations, correction of the truncation by CRISPR/cas9, or proteasome inhibition that reduced the amount of mutant protein, corrected the contractile defect in cardiomyocytes derived from induced pluripotent stem cells (iPSC-CMs).194 An intron-mediated TTN enhancer that promoted cardiac-specific TTN expression in similarly-derived cardiomyocytes promoted normal TTN expression in mice.195 Similarly, adenoviral mediated modulation of RNA splicing in iPSC-CMs with an HCM-associated MYBPC3 mutation corrected the hypertrophic phenotype.196 The first-in-human gene transfection with the LAMP2 gene has been reported for 3 patients with Danon disease, a HCM phenocopy.197
Tools are now becoming available to better understand and address arrhythmias in genetic CM, such as better animal and cellular models coupled to gene editing, that raise the prospect that therapies specific to mechanism, gene, and mutation are on the horizon. Development of these therapies will be enabled by increased identification of mutation carriers and their phenotypic journeys, and further advances beyond the one gene - one disease paradigm.73 The rationale to expand genetic testing thus extends from current management of patients and families to acceleration of an exciting future that holds promise for correction of their underlying molecular defects.
KEY POINTS.
Genetic cardiomyopathy should be considered in patients who present with “idiopathic” arrhythmias, including atrial fibrillation, conduction disease, frequent PVCs and ventricular tachycardia.
The presence of an apparently acquired cause of cardiomyopathy or the absence of family history does not rule out genetic cardiomyopathy.
The 12-lead electrocardiogram, echocardiogram, enhanced cardiac magnetic resonance imaging, and sequencing can help to layer the clinical phenotype with the genotype for arrhythmogenic cardiomyopathies.
Sudden death can occur early in arrhythmogenic cardiomyopathy, before the ejection fraction is low.
The increase in genetic information and implications raises new challenges for genetic counseling and shared decision-making.
Genetic testing enables cascade screening, specific treatments for some subtypes, and evaluation of emerging targeted therapies.
Sources of Funding
This work has been supported by the National Institutes of Health R01HL155197 - MBS, LWS; R01HL149826 - DR; R01HL140074 - QSW; American Heart Association: Atrial Fibrillation Strategically Focused Research Network (SFRN) award 18SFRN34110369 - DR, MBS, QW; Ventricular Arrhythmia/Sudden Cardiac Death Strategically Focused Research Network (SFRN) award 19SFRN34830019 - DR, MBS, WGS; Atrial Fibrillation Collaborative Grant Award 20SCG35540037 - MBS, LWS; NHGRI Vanderbilt Genomic Medicine T32 HG008341 - MCL; and the generous support from the Wilds family for the VUMC Genetic Cardiomyopathy Program-LWS, KG, MBS, and JLL.
Abbreviations
- ACM
Arrhythmogenic cardiomyopathy
- ACM-LV
Arrhythmogenic cardiomyopathy dominant in LV
- ACM-RV
Arrhythmogenic cardiomyopathy dominant in RV
- ARVC
traditional term for arrhythmogenic cardiomyopathy as originally linked to desmosomal disease
- AF
atrial fibrillation/flutter
- ATTR-CM
transthyretin amyloid cardiomyopathy
- CM
cardiomyopathy
- CMR
cardiac magnetic resonance imaging
- DCM
dilated cardiomyopathy
- HCM
hypertrophic cardiomyopathy
- HF
heart failure
- LGE
late gadolinium enhancement
- LVEF
left ventricular ejection fraction
- NSVT
non-sustained ventricular tachycardia
- PVC
Premature ventricular contraction
- RCM
restrictive cardiomyopathy
- SCD
sudden cardiac death
- SMVT
sustained monomorphic ventricular tachycardia
- VA
ventricular arrhythmias
- VT
ventricular tachycardia
- VUS
variant of unknown significance
Footnotes
Disclosures
RRH has received honoraria from Akcea Therapeutics and Alnylam Pharmaceuticals
REFERENCES:
- 1.Corrado D, van Tintelen PJ, McKenna WJ, Hauer RNW, Anastastakis A, Asimaki A, Basso C, Bauce B, Brunckhorst C, Bucciarelli-Ducci C, et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2020;41:1414–1429. doi: 10.1093/eurheartj/ehz669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16:e301–e372. doi: 10.1016/j.hrthm.2019.05.007 [DOI] [PubMed] [Google Scholar]
- 3.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr., Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147–239. doi: 10.1016/j.jacc.2013.05.019 [DOI] [PubMed] [Google Scholar]
- 4.Cipriani A, Bauce B, De Lazzari M, Rigato I, Bariani R, Meneghin S, Pilichou K, Motta R, Aliberti C, Thiene G, et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis With Dilated Cardiomyopathy. Journal of the American Heart Association. 2020;9. doi: 10.1161/jaha.119.014628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bui QM, Hong KN, Kraushaar M, Ma GS, Brambatti M, Kahn AM, Battiha CE, Boynton K, Storm G, Mestroni L, et al. Myocardial Strain and Association With Clinical Outcomes in Danon Disease: A Model for Monitoring Progression of Genetic Cardiomyopathies. Journal of the American Heart Association. 2021;10. doi: 10.1161/jaha.121.022544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark DE, Parikh A, Dendy JM, Diamond AB, George-Durrett K, Fish FA, Slaughter JC, Fitch W, Hughes SG, Soslow JH. COVID-19 Myocardial Pathology Evaluation in Athletes With Cardiac Magnetic Resonance (COMPETE CMR). Circulation. 2021;143:609–612. doi: 10.1161/circulationaha.120.052573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muser D, Santangeli P, Castro SA, Casado Arroyo R, Maeda S, Benhayon DA, Liuba I, Liang JJ, Sadek MM, Chahal A, et al. Risk Stratification of Patients With Apparently Idiopathic Premature Ventricular Contractions: A Multicenter International CMR Registry. JACC Clin Electrophysiol. 2020;6:722–735. doi: 10.1016/j.jacep.2019.10.015 [DOI] [PubMed] [Google Scholar]
- 8.Alba AC, Gaztañaga J, Foroutan F, Thavendiranathan P, Merlo M, Alonso-Rodriguez D, Vallejo-García V, Vidal-Perez R, Corros-Vicente C, Barreiro-Pérez M, et al. Prognostic Value of Late Gadolinium Enhancement for the Prediction of Cardiovascular Outcomes in Dilated Cardiomyopathy. Circulation: Cardiovascular Imaging. 2020;13. doi: 10.1161/circimaging.119.010105 [DOI] [PubMed] [Google Scholar]
- 9.Piers SR, Everaerts K, van der Geest RJ, Hazebroek MR, Siebelink HM, Pison LA, Schalij MJ, Bekkers SC, Heymans S, Zeppenfeld K. Myocardial scar predicts monomorphic ventricular tachycardia but not polymorphic ventricular tachycardia or ventricular fibrillation in nonischemic dilated cardiomyopathy. Heart Rhythm. 2015;12:2106–2114. doi: 10.1016/j.hrthm.2015.05.026 [DOI] [PubMed] [Google Scholar]
- 10.Augusto JB, Eiros R, Nakou E, Moura-Ferreira S, Treibel TA, Captur G, Akhtar MM, Protonotarios A, Gossios TD, Savvatis K, et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study. European Heart Journal - Cardiovascular Imaging. 2019. doi: 10.1093/ehjci/jez188 [DOI] [PubMed] [Google Scholar]
- 11.Glashan CA, Androulakis AFA, Tao Q, Glashan RN, Wisse LJ, Ebert M, De Ruiter MC, Van Meer BJ, Brouwer C, Dekkers OM, et al. Whole human heart histology to validate electroanatomical voltage mapping in patients with non-ischaemic cardiomyopathy and ventricular tachycardia. European Heart Journal. 2018;39:2867–2875. doi: 10.1093/eurheartj/ehy168 [DOI] [PubMed] [Google Scholar]
- 12.Nakamori S, Ngo LH, Rodriguez J, Neisius U, Manning WJ, Nezafat R. T1 Mapping Tissue Heterogeneity Provides Improved Risk Stratification for ICDs Without Needing Gadolinium in Patients With Dilated Cardiomyopathy. JACC Cardiovasc Imaging. 2020;13:1917–1930. doi: 10.1016/j.jcmg.2020.03.014 [DOI] [PubMed] [Google Scholar]
- 13.Muser D, Nucifora G, Castro SA, Enriquez A, Chahal CAA, Magnani S, Kumareswaran R, Arkles J, Supple G, Schaller R, et al. Myocardial Substrate Characterization by CMR T1 Mapping in Patients With NICM and No LGE Undergoing Catheter Ablation of VT. JACC Clin Electrophysiol. 2021;7:831–840. doi: 10.1016/j.jacep.2020.10.002 [DOI] [PubMed] [Google Scholar]
- 14.Tschabrunn CM, Zado ES, Schaller R, Garcia FC, Kumareswaran R, Hsue W, Santangeli P, Marchlinski FE. Isolated critical epicardial arrhythmogenic substrate abnormalities in patients with arrhythmogenic right ventricular cardiomyopathy and ventricular tachycardia. Heart Rhythm. 2021. doi: 10.1016/j.hrthm.2021.11.035 [DOI] [PubMed] [Google Scholar]
- 15.Philips B, Madhavan S, James CA, Te Riele ASJM, Murray B, Tichnell C, Bhonsale A, Nazarian S, Judge DP, Calkins H, et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy and Cardiac Sarcoidosis. Circulation: Arrhythmia and Electrophysiology. 2014;7:230–236. doi: 10.1161/circep.113.000932 [DOI] [PubMed] [Google Scholar]
- 16.Protonotarios A, Wicks E, Ashworth M, Stephenson E, Guttmann O, Savvatis K, Sekhri N, Mohiddin SA, Syrris P, Menezes L, et al. Prevalence of 18F-fluorodeoxyglucose positron emission tomography abnormalities in patients with arrhythmogenic right ventricular cardiomyopathy. International Journal of Cardiology. 2019;284:99–104. doi: 10.1016/j.ijcard.2018.10.083 [DOI] [PubMed] [Google Scholar]
- 17.Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation. 1996;94:983–991. doi: 10.1161/01.cir.94.5.983 [DOI] [PubMed] [Google Scholar]
- 18.Ader F, Surget E, Charron P, Redheuil A, Zouaghi A, Maltret A, Marijon E, Denjoy I, Hermida A, Fressart V, et al. Inherited Cardiomyopathies Revealed by Clinically Suspected Myocarditis. Circulation: Genomic and Precision Medicine. 2020;13. doi: 10.1161/circgen.119.002744 [DOI] [PubMed] [Google Scholar]
- 19.Scheel PJ 3rd, Murray B, Tichnell C, James CA, Tandri H, Calkins H, Chelko SP, Gilotra NA. Arrhythmogenic Right Ventricular Cardiomyopathy Presenting as Clinical Myocarditis in Women. Am J Cardiol. 2021;145:128–134. doi: 10.1016/j.amjcard.2020.12.090 [DOI] [PubMed] [Google Scholar]
- 20.Kontorovich AR, Patel N, Moscati A, Richter F, Peter I, Purevjav E, Selejan SR, Kindermann I, Towbin JA, Bohm M, et al. Myopathic Cardiac Genotypes Increase Risk for Myocarditis. JACC Basic Transl Sci. 2021;6:584–592. doi: 10.1016/j.jacbts.2021.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chelko SP, Asimaki A, Lowenthal J, Bueno-Beti C, Bedja D, Scalco A, Amat-Alarcon N, Andersen P, Judge DP, Tung L, et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation. 2019;140:1491–1505. doi: 10.1161/circulationaha.119.040676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caforio ALP, Re F, Avella A, Marcolongo R, Baratta P, Seguso M, Gallo N, Plebani M, Izquierdo-Bajo A, Cheng C-Y, et al. Evidence From Family Studies for Autoimmunity in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation. 2020;141:1238–1248. doi: 10.1161/circulationaha.119.043931 [DOI] [PubMed] [Google Scholar]
- 23.Chatterjee D, Fatah M, Akdis D, Spears DA, Koopmann TT, Mittal K, Rafiq MA, Cattanach BM, Zhao Q, Healey JS, et al. An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis. European Heart Journal. 2018;39:3932–3944. doi: 10.1093/eurheartj/ehy567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebert M, Wijnmaalen AP, de Riva M, Trines SA, Androulakis AFA, Glashan CA, Schalij MJ, Peter van Tintelen J, Jongbloed JDH, Zeppenfeld K. Prevalence and Prognostic Impact of Pathogenic Variants in Patients With Dilated Cardiomyopathy Referred for Ventricular Tachycardia Ablation. JACC Clin Electrophysiol. 2020;6:1103–1114. doi: 10.1016/j.jacep.2020.04.025 [DOI] [PubMed] [Google Scholar]
- 25.Verdonschot JAJ, Hazebroek MR, Krapels IPC, Henkens MTHM, Raafs A, Wang P, Merken JJ, Claes GRF, Vanhoutte EK, Van Den Wijngaard A, et al. Implications of Genetic Testing in Dilated Cardiomyopathy. Circulation: Genomic and Precision Medicine. 2020;13:476–487. doi: 10.1161/circgen.120.003031 [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Pavia P, Kim Y, Restrepo-Cordoba MA, Lunde IG, Wakimoto H, Smith AM, Toepfer CN, Getz K, Gorham J, Patel P, et al. Genetic Variants Associated With Cancer Therapy–Induced Cardiomyopathy. Circulation. 2019;140:31–41. doi: 10.1161/circulationaha.118.037934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ware JS, Amor-Salamanca A, Tayal U, Govind R, Serrano I, Salazar-Mendiguchia J, Garcia-Pinilla JM, Pascual-Figal DA, Nunez J, Guzzo-Merello G, et al. Genetic Etiology for Alcohol-Induced Cardiac Toxicity. J Am Coll Cardiol. 2018;71:2293–2302. doi: 10.1016/j.jacc.2018.03.462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goli R, Li J, Brandimarto J, Levine LD, Riis V, Mcafee Q, Depalma S, Haghighi A, Seidman JG, Seidman CE, et al. Genetic and Phenotypic Landscape of Peripartum Cardiomyopathy. Circulation. 2021;143:1852–1862. doi: 10.1161/circulationaha.120.052395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Musunuru K, Hershberger RE, Day SM, Klinedinst NJ, Landstrom AP, Parikh VN, Prakash S, Semsarian C, Sturm AC. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circulation: Genomic and Precision Medicine. 2020;13. doi: 10.1161/hcg.0000000000000067 [DOI] [PubMed] [Google Scholar]
- 30.Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, Mcbride KL, Morales A, Taylor MRG, Vatta M, Ware SM. Genetic Evaluation of Cardiomyopathy—A Heart Failure Society of America Practice Guideline. Journal of Cardiac Failure. 2018;24:281–302. doi: 10.1016/j.cardfail.2018.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, Evanovich LL, Hung J, Joglar JA, Kantor P, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy. Circulation. 2020;142. doi: 10.1161/cir.0000000000000937 [DOI] [PubMed] [Google Scholar]
- 32.Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, Deal BJ, Dickfeld T, Field ME, Fonarow GC, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Heart Rhythm. 2018;15:e73–e189. doi: 10.1016/j.hrthm.2017.10.036 [DOI] [PubMed] [Google Scholar]
- 33.Marcus FI, Mckenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MGPJ, Daubert JP, et al. Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Circulation. 2010;121:1533–1541. doi: 10.1161/circulationaha.108.840827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Epstein AE, Dimarco JP, Ellenbogen KA, Estes NA 3rd, Freedman RA, Gettes LS, Gillinov AM, Gregoratos G, Hammill SC, Hayes DL, et al. ACC/AHA/HRS 2008 Guidelines for device-based therapy of cardiac rhythm abnormalities. Heart Rhythm. 2008;5:e1–62. doi: 10.1016/j.hrthm.2008.04.014 [DOI] [PubMed] [Google Scholar]
- 35.Russo AM, Stainback RF, Bailey SR, Epstein AE, Heidenreich PA, Jessup M, Kapa S, Kremers MS, Lindsay BD, Stevenson LW. ACCF/HRS/AHA/ASE/HFSA/SCAI/SCCT/SCMR 2013 appropriate use criteria for implantable cardioverter-defibrillators and cardiac resynchronization therapy: a report of the American College of Cardiology Foundation appropriate use criteria task force, Heart Rhythm Society, American Heart Association, American Society of Echocardiography, Heart Failure Society of America, Society for Cardiovascular Angiography and Interventions, Society of Cardiovascular Computed Tomography, and Society for Cardiovascular Magnetic Resonance. Heart Rhythm. 2013;10:e11–58. doi: 10.1016/j.hrthm.2013.01.008 [DOI] [PubMed] [Google Scholar]
- 36.Grunig E, Tasman JA, Kucherer H, Franz W, Kubler W, Katus HA. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol. 1998;31:186–194. doi: 10.1016/s0735-1097(97)00434-8 [DOI] [PubMed] [Google Scholar]
- 37.Yeung C, Enriquez A, Suarez-Fuster L, Baranchuk A. Atrial fibrillation in patients with inherited cardiomyopathies. Europace. 2019;21:22–32. doi: 10.1093/europace/euy064 [DOI] [PubMed] [Google Scholar]
- 38.Pabel S, Knierim M, Stehle T, Alebrand F, Paulus M, Sieme M, Herwig M, Barsch F, Körtl T, Pöppl A, et al. Effects of Atrial Fibrillation on the Human Ventricle. Circulation Research. 2022. doi: 10.1161/circresaha.121.319718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodyer WR, Dunn K, Caleshu C, Jackson M, Wylie J, Moscarello T, Platt J, Reuter C, Smith A, Trela A, et al. Broad Genetic Testing in a Clinical Setting Uncovers a High Prevalence of Titin Loss-of-Function Variants in Very Early Onset Atrial Fibrillation. Circ Genom Precis Med. 2019;12:e002713. doi: 10.1161/CIRCGEN.119.002713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoneda ZT, Anderson KC, Quintana JA, O’Neill MJ, Sims RA, Glazer AM, Shaffer CM, Crawford DM, Stricker T, Ye F, et al. Early-Onset Atrial Fibrillation and the Prevalence of Rare Variants in Cardiomyopathy and Arrhythmia Genes. JAMA Cardiology. 2021;6:1371. doi: 10.1001/jamacardio.2021.3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pan H, Richards AA, Zhu X, Joglar JA, Yin HL, Garg V. A novel mutation in LAMIN A/C is associated with isolated early-onset atrial fibrillation and progressive atrioventricular block followed by cardiomyopathy and sudden cardiac death. Heart Rhythm. 2009;6:707–710. doi: 10.1016/j.hrthm.2009.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi SH, Weng LC, Roselli C, Lin H, Haggerty CM, Shoemaker MB, Barnard J, Arking DE, Chasman DI, Albert CM, et al. Association Between Titin Loss-of-Function Variants and Early-Onset Atrial Fibrillation. JAMA. 2018;320:2354–2364. doi: 10.1001/jama.2018.18179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shoemaker MB, Shah RL, Roden DM, Perez MV. How Will Genetics Inform the Clinical Care of Atrial Fibrillation? Circ Res. 2020;127:111–127. doi: 10.1161/CIRCRESAHA.120.316365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomaselli GF, Roy-Puckelwartz MJ. Early-Onset Atrial Fibrillation and Heritable Heart Disease-To Test or Not to Test? JAMA Cardiol. 2021;6:1359–1361. doi: 10.1001/jamacardio.2021.3367 [DOI] [PubMed] [Google Scholar]
- 46.Yoneda ZT, Anderson KC, Fei Y, Quintana JA, O’Neill MJ, Sims RA, Sun L, Glazer AM, Davogustto G, El-Harasis M, et al. Mortality in Early-onset Atrial Fibrillation and Rare Variants in Cardiomyopathy and Arrhythmia Genes. 2022. doi: [DOI] [PMC free article] [PubMed]
- 47.Mikhailov AV, Kalyanasundaram A, Li N, Scott SS, Artiga EJ, Subr MM, Zhao J, Hansen BJ, Hummel JD, Fedorov VV. Comprehensive evaluation of electrophysiological and 3D structural features of human atrial myocardium with insights on atrial fibrillation maintenance mechanisms. J Mol Cell Cardiol. 2021;151:56–71. doi: 10.1016/j.yjmcc.2020.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jansen HJ, Bohne LJ, Gillis AM, Rose RA. Atrial remodeling and atrial fibrillation in acquired forms of cardiovascular disease. Heart Rhythm O2. 2020;1:147–159. doi: 10.1016/j.hroo.2020.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee S-P, Ashley EA, Homburger J, Caleshu C, Green EM, Jacoby D, Colan SD, Arteaga-Fernández E, Day SM, Girolami F, et al. Incident Atrial Fibrillation Is Associated With MYH7 Sarcomeric Gene Variation in Hypertrophic Cardiomyopathy. Circulation: Heart Failure. 2018;11. doi: 10.1161/circheartfailure.118.005191 [DOI] [PubMed] [Google Scholar]
- 50.Bishop E, Brown EE, Fajardo J, Barouch LA, Judge DP, Halushka MK. Seven factors predict a delayed diagnosis of cardiac amyloidosis. Amyloid. 2018;25:174–179. doi: 10.1080/13506129.2018.1498782 [DOI] [PubMed] [Google Scholar]
- 51.Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, Judge DP, Lenihan DJ, Gottlieb SS, Shah SJ, et al. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68:161–172. doi: 10.1016/j.jacc.2016.03.596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, et al. ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary. J Am Coll Cardiol. 2011;58:2703–2738. doi: 10.1016/j.jacc.2011.10.825 [DOI] [PubMed] [Google Scholar]
- 53.Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, Evanovich LL, Hung J, Joglar JA, Kantor P, et al. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: Executive Summary. Circulation. 2020;142:e533–e557. doi: 10.1161/CIR.0000000000000938 [DOI] [PubMed] [Google Scholar]
- 54.Lakdawala NK, Stevenson LW, Loscalzo J. Cardiomyopathies and Myocarditis. Jameson JL, Fauci AS, Kasper DL, Hauser SL, Longo DL, Loscalzo J, eds. In: Harrison’s Manual of Medicine, 20e. McGraw-Hill Education; 2020:1779–1796. [Google Scholar]
- 55.Donnellan E, Wazni OM, Saliba WI, Hanna M, Kanj M, Patel DR, Wilner B, Kochar A, Jaber WA. Prevalence, Incidence, and Impact on Mortality of Conduction System Disease in Transthyretin Cardiac Amyloidosis. Am J Cardiol. 2020;128:140–146. doi: 10.1016/j.amjcard.2020.05.021 [DOI] [PubMed] [Google Scholar]
- 56.Donnellan E, Wazni OM, Saliba WI, Baranowski B, Hanna M, Martyn M, Patel D, Trulock K, Menon V, Hussein A, et al. Cardiac devices in patients with transthyretin amyloidosis: Impact on functional class, left ventricular function, mitral regurgitation, and mortality. J Cardiovasc Electrophysiol. 2019;30:2427–2432. doi: 10.1111/jce.14180 [DOI] [PubMed] [Google Scholar]
- 57.Rotman M, Triebwasser JH. A clinical and follow-up study of right and left bundle branch block. Circulation. 1975;51:477–484. doi: 10.1161/01.cir.51.3.477 [DOI] [PubMed] [Google Scholar]
- 58.Zhang ZM, Rautaharju PM, Soliman EZ, Manson JE, Cain ME, Martin LW, Bavry AA, Mehta L, Vitolins M, Prineas RJ. Mortality risk associated with bundle branch blocks and related repolarization abnormalities. Am J Cardiol. 2012;110:1489–1495. doi: 10.1016/j.amjcard.2012.06.060 [DOI] [PubMed] [Google Scholar]
- 59.Xiong Y, Wang L, Liu W, Hankey GJ, Xu B, Wang S. The Prognostic Significance of Right Bundle Branch Block: A Meta-analysis of Prospective Cohort Studies. Clinical Cardiology. 2015;38:604–613. doi: 10.1002/clc.22454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schneider JF, Thomas HE Jr., Kreger BE, McNamara PM, Kannel WB. Newly acquired left bundle-branch block: the Framingham study. Ann Intern Med. 1979;90:303–310. doi: 10.7326/0003-4819-90-3-303 [DOI] [PubMed] [Google Scholar]
- 61.Zannad F, Huvelle E, Dickstein K, Van Veldhuisen DJ, Stellbrink C, Køber L, Cazeau S, Ritter P, Maggioni AP, Ferrari R, et al. Left bundle branch block as a risk factor for progression to heart failure. European Journal of Heart Failure. 2007;9:7–14. doi: 10.1016/j.ejheart.2006.04.011 [DOI] [PubMed] [Google Scholar]
- 62.Pasotti M, Klersy C, Pilotto A, Marziliano N, Rapezzi C, Serio A, Mannarino S, Gambarin F, Favalli V, Grasso M, et al. Long-Term Outcome and Risk Stratification in Dilated Cardiolaminopathies. Journal of the American College of Cardiology. 2008;52:1250–1260. doi: 10.1016/j.jacc.2008.06.044 [DOI] [PubMed] [Google Scholar]
- 63.Kumar S, Baldinger SH, Gandjbakhch E, Maury P, Sellal J-M, Androulakis AFA, Waintraub X, Charron P, Rollin A, Richard P, et al. Long-Term Arrhythmic and Nonarrhythmic Outcomes of Lamin A/C Mutation Carriers. Journal of the American College of Cardiology. 2016;68:2299–2307. doi: 10.1016/j.jacc.2016.08.058 [DOI] [PubMed] [Google Scholar]
- 64.Royer A, Van Veen TAB, Le Bouter S, Marionneau CL, Griol-Charhbili V, LéOni A-L, Steenman M, Van Rijen HVM, Demolombe S, Goddard CA, et al. Mouse Model of SCN5A-Linked Hereditary Leneégre’s Disease. Circulation. 2005;111:1738–1746. doi: 10.1161/01.cir.0000160853.19867.61 [DOI] [PubMed] [Google Scholar]
- 65.Probst V, Kyndt F, Potet F, Trochu J-N, Mialet G, Demolombe S, Schott J-J, Baró I, Escande D, Le Marec H. Haploinsufficiency in combination with aging causes SCN5A-linked hereditary Lenègre disease. Journal of the American College of Cardiology. 2003;41:643–652. doi: 10.1016/s0735-1097(02)02864-4 [DOI] [PubMed] [Google Scholar]
- 66.Ge J, Sun A, Paajanen V, Wang S, Su C, Yang Z, Li Y, Wang S, Jia J, Wang K, et al. Molecular and Clinical Characterization of a Novel SCN5A Mutation Associated With Atrioventricular Block and Dilated Cardiomyopathy. Circulation: Arrhythmia and Electrophysiology. 2008;1:83–92. doi: 10.1161/circep.107.750752 [DOI] [PubMed] [Google Scholar]
- 67.John R Arrhythmias in Cardiac Amyloidosis. The Journal of Innovations in Cardiac Rhythm Management. 2018:3051–3057. doi: 10.19102/icrm.2018.090301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Crasto S, My I, Di Pasquale E. The Broad Spectrum of LMNA Cardiac Diseases: From Molecular Mechanisms to Clinical Phenotype. Front Physiol. 2020;11:761. doi: 10.3389/fphys.2020.00761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang KC, Kyle JW, Makielski JC, Dudley SC Jr. Mechanisms of sudden cardiac death: oxidants and metabolism. Circ Res. 2015;116:1937–1955. doi: 10.1161/CIRCRESAHA.116.304691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marian AJ, Asatryan B, Wehrens XHT. Genetic basis and molecular biology of cardiac arrhythmias in cardiomyopathies. Cardiovascular Research. 2020;116:1600–1619. doi: 10.1093/cvr/cvaa116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharma A, Assis F, James CA, Murray B, Tichnell C, Tandri H, Calkins H. Misdiagnosis of ARVC leading to inappropriate ICD implant and subsequent ICD removal – lessons learned. Journal of Cardiovascular Electrophysiology. 2019;30:2020–2026. doi: 10.1111/jce.14088 [DOI] [PubMed] [Google Scholar]
- 72.Peters S, Thompson BA, Perrin M, James P, Zentner D, Kalman JM, Vandenberg JI, Fatkin D. Arrhythmic Phenotypes Are a Defining Feature of Dilated Cardiomyopathy-Associated SCN5A Variants: A Systematic Review. Circulation: Genomic and Precision Medicine. 2021. doi: 10.1161/circgen.121.003432 [DOI] [PubMed] [Google Scholar]
- 73.Cerrone M, Remme CA, Tadros R, Bezzina CR, Delmar M. Beyond the One Gene–One Disease Paradigm. Circulation. 2019;140:595–610. doi: 10.1161/circulationaha.118.035954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ben-Haim Y, Asimaki A, Behr ER. Brugada syndrome and arrhythmogenic cardiomyopathy: overlapping disorders of the connexome? EP Europace. 2021;23:653–664. doi: 10.1093/europace/euaa277 [DOI] [PubMed] [Google Scholar]
- 75.Watanabe H, Yang T, Stroud DM, Lowe JS, Harris L, Atack TC, Wang DW, Hipkens SB, Leake B, Hall L, et al. Striking In Vivo Phenotype of a Disease-Associated Human SCN5A Mutation Producing Minimal Changes in Vitro. Circulation. 2011;124:1001–1011. doi: 10.1161/circulationaha.110.987248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gosselin-Badaroudine P, Keller DI, Huang H, Pouliot V, Chatelier A, Osswald S, Brink M, Chahine M. A Proton Leak Current through the Cardiac Sodium Channel Is Linked to Mixed Arrhythmia and the Dilated Cardiomyopathy Phenotype. PLoS ONE. 2012;7:e38331. doi: 10.1371/journal.pone.0038331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moreau A, Gosselin-Badaroudine P, Boutjdir M, Chahine M. Mutations in the Voltage Sensors of Domains I and II of Nav1.5 that are Associated with Arrhythmias and Dilated Cardiomyopathy Generate Gating Pore Currents. Front Pharmacol. 2015;6:301. doi: 10.3389/fphar.2015.00301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Chkourko Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, et al. Missense Mutations in Plakophilin-2 Cause Sodium Current Deficit and Associate With a Brugada Syndrome Phenotype. Circulation. 2014;129:1092–1103. doi: 10.1161/circulationaha.113.003077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rivaud MR, Delmar M, Remme CA. Heritable arrhythmia syndromes associated with abnormal cardiac sodium channel function: ionic and non-ionic mechanisms. Cardiovascular Research. 2020;116:1557–1570. doi: 10.1093/cvr/cvaa082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rivaud MR, Agullo‐Pascual E, Lin X, Leo‐Macias A, Zhang M, Rothenberg E, Bezzina CR, Delmar M, Remme CA. Sodium Channel Remodeling in Subcellular Microdomains of Murine Failing Cardiomyocytes. Journal of the American Heart Association. 2017;6. doi: 10.1161/jaha.117.007622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ke HY, Yang HY, Francis AJ, Collins TP, Surendran H, Alvarez‐Laviada A, Firth JM, Macleod KT. Changes in cellular Ca 2+ and Na+ regulation during the progression towards heart failure in the guinea pig. The Journal of Physiology. 2020;598:1339–1359. doi: 10.1113/jp277038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cerrone M, Montnach J, Lin X, Zhao Y-T, Zhang M, Agullo-Pascual E, Leo-Macias A, Alvarado FJ, Dolgalev I, Karathanos TV, et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nature Communications. 2017;8. doi: 10.1038/s41467-017-00127-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ataklte F, Erqou S, Laukkanen J, Kaptoge S. Meta-analysis of ventricular premature complexes and their relation to cardiac mortality in general populations. Am J Cardiol. 2013;112:1263–1270. doi: 10.1016/j.amjcard.2013.05.065 [DOI] [PubMed] [Google Scholar]
- 84.Simpson RJ Jr., Cascio WE, Schreiner PJ, Crow RS, Rautaharju PM, Heiss G. Prevalence of premature ventricular contractions in a population of African American and white men and women: the Atherosclerosis Risk in Communities (ARIC) study. Am Heart J. 2002;143:535–540. doi: 10.1067/mhj.2002.120298 [DOI] [PubMed] [Google Scholar]
- 85.Nguyen KT, Vittinghoff E, Dewland TA, Dukes JW, Soliman EZ, Stein PK, Gottdiener JS, Alonso A, Chen LY, Psaty BM, et al. Ectopy on a Single 12-Lead ECG, Incident Cardiac Myopathy, and Death in the Community. J Am Heart Assoc. 2017;6. doi: 10.1161/JAHA.117.006028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Muser D, Nucifora G, Pieroni M, Castro SA, Casado Arroyo R, Maeda S, Benhayon DA, Liuba I, Sadek M, Magnani S, et al. Prognostic Value of Non-Ischemic Ring-Like Left Ventricular Scar in Patients with Apparently Idiopathic Non-Sustained Ventricular Arrhythmias. Circulation. 2021. doi: 10.1161/circulationaha.120.047640 [DOI] [PubMed] [Google Scholar]
- 87.Latchamsetty R, Yokokawa M, Morady F, Kim HM, Mathew S, Tilz R, Kuck KH, Nagashima K, Tedrow U, Stevenson WG, et al. Multicenter Outcomes for Catheter Ablation of Idiopathic Premature Ventricular Complexes. JACC Clin Electrophysiol. 2015;1:116–123. doi: 10.1016/j.jacep.2015.04.005 [DOI] [PubMed] [Google Scholar]
- 88.Huizar JF, Tan AY, Kaszala K, Ellenbogen KA. Clinical and translational insights on premature ventricular contractions and PVC-induced cardiomyopathy. Prog Cardiovasc Dis. 2021;66:17–27. doi: 10.1016/j.pcad.2021.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Walters TE, Szilagyi J, Alhede C, Sievers R, Fang Q, Olgin J, Gerstenfeld EP. Dyssynchrony and Fibrosis Persist After Resolution of Cardiomyopathy in a Swine Premature Ventricular Contraction Model. JACC Clin Electrophysiol. 2020;6:1367–1376. doi: 10.1016/j.jacep.2020.06.020 [DOI] [PubMed] [Google Scholar]
- 90.Ghannam M, Siontis KC, Kim HM, Cochet H, Jais P, Eng MJ, Attili A, Sharaf-Dabbagh G, Latchamsetty R, Jongnarangsin K, et al. Factors predictive for delayed enhancement in cardiac resonance imaging in patients undergoing catheter ablation of premature ventricular complexes. Heart Rhythm O2. 2021;2:64–72. doi: 10.1016/j.hroo.2020.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Laurent G, Saal S, Amarouch MY, Beziau DM, Marsman RF, Faivre L, Barc J, Dina C, Bertaux G, Barthez O, et al. Multifocal ectopic Purkinje-related premature contractions: a new SCN5A-related cardiac channelopathy. J Am Coll Cardiol. 2012;60:144–156. doi: 10.1016/j.jacc.2012.02.052 [DOI] [PubMed] [Google Scholar]
- 92.Nair K, Pekhletski R, Harris L, Care M, Morel C, Farid T, Backx PH, Szabo E, Nanthakumar K. Escape capture bigeminy: phenotypic marker of cardiac sodium channel voltage sensor mutation R222Q. Heart Rhythm. 2012;9:1681–1688 e1681. doi: 10.1016/j.hrthm.2012.06.029 [DOI] [PubMed] [Google Scholar]
- 93.Mann SA, Castro ML, Ohanian M, Guo G, Zodgekar P, Sheu A, Stockhammer K, Thompson T, Playford D, Subbiah R, et al. R222Q SCN5A mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. J Am Coll Cardiol. 2012;60:1566–1573. doi: 10.1016/j.jacc.2012.05.050 [DOI] [PubMed] [Google Scholar]
- 94.Ellenbogen KA, Levine JH, Berger RD, Daubert JP, Winters SL, Greenstein E, Shalaby A, Schaechter A, Subacius H, Kadish A. Are Implantable Cardioverter Defibrillator Shocks a Surrogate for Sudden Cardiac Death in Patients With Nonischemic Cardiomyopathy? Circulation. 2006;113:776–782. doi: 10.1161/circulationaha.105.561571 [DOI] [PubMed] [Google Scholar]
- 95.Køber L, Thune JJ, Nielsen JC, Haarbo J, Videbæk L, Korup E, Jensen G, Hildebrandt P, Steffensen FH, Bruun NE, et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. New England Journal of Medicine. 2016;375:1221–1230. doi: 10.1056/nejmoa1608029 [DOI] [PubMed] [Google Scholar]
- 96.Lopera G, Stevenson WG, Soejima K, Maisel WH, Koplan B, Sapp JL, Satt SD, Epstein LM. Identification and Ablation of Three Types of Ventricular Tachycardia Involving the His-Purkinje System in Patients with Heart Disease. Journal of Cardiovascular Electrophysiology. 2004;15:52–58. doi: 10.1046/j.1540-8167.2004.03189.x [DOI] [PubMed] [Google Scholar]
- 97.Cadrin-Tourigny J, Bosman LP, Wang W, Tadros R, Bhonsale A, Bourfiss M, Lie ØH, Saguner AM, Svensson A, Andorin A, et al. Sudden Cardiac Death Prediction in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation: Arrhythmia and Electrophysiology. 2021;14. doi: 10.1161/circep.120.008509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wijnmaalen AP, Stevenson WG, Schalij MJ, Field ME, Stephenson K, Tedrow UB, Koplan BA, Putter H, Epstein LM, Zeppenfeld K. ECG Identification of Scar-Related Ventricular Tachycardia With a Left Bundle-Branch Block Configuration. Circulation: Arrhythmia and Electrophysiology. 2011;4:486–493. doi: 10.1161/circep.110.959338 [DOI] [PubMed] [Google Scholar]
- 99.Van Den Hoogenhof MMG, Beqqali A, Amin AS, Van Der Made I, Aufiero S, Khan MAF, Schumacher CA, Jansweijer JA, Van Spaendonck-Zwarts KY, Remme CA, et al. RBM20 Mutations Induce an Arrhythmogenic Dilated Cardiomyopathy Related to Disturbed Calcium Handling. Circulation. 2018;138:1330–1342. doi: 10.1161/circulationaha.117.031947 [DOI] [PubMed] [Google Scholar]
- 100.Nakamura T, Schaeffer B, Tanigawa S, Muthalaly RG, John RM, Michaud GF, Tedrow UB, Stevenson WG. Catheter ablation of polymorphic ventricular tachycardia/fibrillation in patients with and without structural heart disease. Heart Rhythm. 2019;16:1021–1027. doi: 10.1016/j.hrthm.2019.01.032 [DOI] [PubMed] [Google Scholar]
- 101.Hocini M, Ramirez FD, Szumowski Ł, Maury P, Cheniti G, Duchateau J, Pambrun T, Derval N, Sacher F, Cochet H, et al. Purkinje triggers of ventricular fibrillation in patients with hypertrophic cardiomyopathy. Journal of Cardiovascular Electrophysiology. 2021;32:2987–2994. doi: 10.1111/jce.15231 [DOI] [PubMed] [Google Scholar]
- 102.Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–2187. doi: 10.1016/j.jacc.2008.09.019 [DOI] [PubMed] [Google Scholar]
- 103.Marian AJ, Braunwald E. Hypertrophic Cardiomyopathy. Circulation Research. 2017;121:749–770. doi: 10.1161/circresaha.117.311059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mckenna WJ, Judge DP. Epidemiology of the inherited cardiomyopathies. Nature Reviews Cardiology. 2021;18:22–36. doi: 10.1038/s41569-020-0428-2 [DOI] [PubMed] [Google Scholar]
- 105.Olivotto I, Maron MS, Adabag AS, Casey SA, Vargiu D, Link MS, Udelson JE, Cecchi F, Maron BJ. Gender-related differences in the clinical presentation and outcome of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;46:480–487. doi: 10.1016/j.jacc.2005.04.043 [DOI] [PubMed] [Google Scholar]
- 106.Lakdawala NK, Olivotto I, Day SM, Han L, Ashley EA, Michels M, Ingles J, Semsarian C, Jacoby D, Jefferies JL, et al. Associations Between Female Sex, Sarcomere Variants, and Clinical Outcomes in Hypertrophic Cardiomyopathy. Circ Genom Precis Med. 2021;14:e003062. doi: 10.1161/circgen.120.003062 [DOI] [PubMed] [Google Scholar]
- 107.Pare JA, Fraser RG, Pirozynski WJ, Shanks JA, Stubington D. Hereditary cardiovascular dysplasia. A form of familial cardiomyopathy. Am J Med. 1961;31:37–62. doi: 10.1016/0002-9343(61)90222-4 [DOI] [PubMed] [Google Scholar]
- 108.Sedaghat-Hamedani F, Kayvanpour E, Tugrul OF, Lai A, Amr A, Haas J, Proctor T, Ehlermann P, Jensen K, Katus HA, et al. Clinical outcomes associated with sarcomere mutations in hypertrophic cardiomyopathy: a meta-analysis on 7675 individuals. Clinical Research in Cardiology. 2018;107:30–41. doi: 10.1007/s00392-017-1155-5 [DOI] [PubMed] [Google Scholar]
- 109.Manrai AK, Funke BH, Rehm HL, Olesen MS, Maron BA, Szolovits P, Margulies DM, Loscalzo J, Kohane IS. Genetic Misdiagnoses and the Potential for Health Disparities. N Engl J Med. 2016;375:655–665. doi: 10.1056/NEJMsa1507092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pua CJ, Tham N, Chin CWL, Walsh R, Khor CC, Toepfer CN, Repetti GG, Garfinkel AC, Ewoldt JF, Cloonan P, et al. Genetic Studies of Hypertrophic Cardiomyopathy in Singaporeans Identify Variants in TNNI3 and TNNT2 That Are Common in Chinese Patients. Circ Genom Precis Med. 2020;13:424–434. doi: 10.1161/CIRCGEN.119.002823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138:1387–1398. doi: 10.1161/CIRCULATIONAHA.117.033200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Coppini R, Ho CY, Ashley E, Day S, Ferrantini C, Girolami F, Tomberli B, Bardi S, Torricelli F, Cecchi F, et al. Clinical Phenotype and Outcome of Hypertrophic Cardiomyopathy Associated With Thin-Filament Gene Mutations. Journal of the American College of Cardiology. 2014;64:2589–2600. doi: 10.1016/j.jacc.2014.09.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lehman SJ, Tal-Grinspan L, Lynn ML, Strom J, Benitez GE, Anderson ME, Tardiff JC. Chronic Calmodulin-Kinase II Activation Drives Disease Progression in Mutation-Specific Hypertrophic Cardiomyopathy. Circulation. 2019;139:1517–1529. doi: 10.1161/circulationaha.118.034549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.O’Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, Biagini E, Gimeno JR, Limongelli G, Mckenna WJ, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD). European Heart Journal. 2014;35:2010–2020. doi: 10.1093/eurheartj/eht439 [DOI] [PubMed] [Google Scholar]
- 115.Olivotto I, Oreziak A, Barriales-Villa R, Abraham TP, Masri A, Garcia-Pavia P, Saberi S, Lakdawala NK, Wheeler MT, Owens A, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet. 2020;396:759–769. doi: 10.1016/s0140-6736(20)31792-x [DOI] [PubMed] [Google Scholar]
- 116.Bhagwan JR, Mosqueira D, Chairez-Cantu K, Mannhardt I, Bodbin SE, Bakar M, Smith JGW, Denning C. Isogenic models of hypertrophic cardiomyopathy unveil differential phenotypes and mechanism-driven therapeutics. Journal of Molecular and Cellular Cardiology. 2020;145:43–53. doi: 10.1016/j.yjmcc.2020.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Garfinkel AC, Seidman JG, Seidman CE. Genetic Pathogenesis of Hypertrophic and Dilated Cardiomyopathy. Heart Fail Clin. 2018;14:139–146. doi: 10.1016/j.hfc.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mestroni L Guidelines for the study of familial dilated cardiomyopathies. European Heart Journal. 1999;20:93–102. doi: 10.1053/euhj.1998.1145 [DOI] [PubMed] [Google Scholar]
- 119.Huggins GS, Kinnamon DD, Haas GJ, Jordan E, Hofmeyer M, Kransdorf E, Ewald GA, Morris AA, Owens A, Lowes B, et al. Prevalence and Cumulative Risk of Familial Idiopathic Dilated Cardiomyopathy. JAMA. 2022;327:454. doi: 10.1001/jama.2021.24674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McNally EM, Mestroni L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circulation Research. 2017;121:731–748. doi: 10.1161/circresaha.116.309396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, Stolfo D, Haywood ME, Dal Ferro M, Altinier A, et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J Am Coll Cardiol. 2019;74:1480–1490. doi: 10.1016/j.jacc.2019.06.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Müller S, Kayvanpour E, Vogel B, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. European Heart Journal. 2015;36:1123–1135. doi: 10.1093/eurheartj/ehu301 [DOI] [PubMed] [Google Scholar]
- 123.Jordan E, Peterson L, Ai T, Asatryan B, Bronicki L, Brown E, Celeghin R, Edwards M, Fan J, Ingles J, et al. An Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation. 2021;144:7–19. doi: 10.1161/circulationaha.120.053033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hershberger RE, Jordan E. Dilated Cardiomyopathy Overview. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, eds. In: GeneReviews(®). University of Washington, Seattle; 1993. [PubMed] [Google Scholar]
- 125.Haggerty CM, Damrauer SM, Levin MG, Birtwell D, Carey DJ, Golden AM, Hartzel DN, Hu Y, Judy R, Kelly MA, et al. Genomics-First Evaluation of Heart Disease Associated With Titin-Truncating Variants. Circulation. 2019;140:42–54. doi: 10.1161/circulationaha.119.039573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Akhtar MM, Lorenzini M, Cicerchia M, Ochoa JP, Hey TM, Sabater Molina M, Restrepo-Cordoba MA, Dal Ferro M, Stolfo D, Johnson R, et al. Clinical Phenotypes and Prognosis of Dilated Cardiomyopathy Caused by Truncating Variants in the TTN Gene. Circulation: Heart Failure. 2020;13. doi: 10.1161/circheartfailure.119.006832 [DOI] [PubMed] [Google Scholar]
- 127.Tayal U, Newsome S, Buchan R, Whiffin N, Halliday B, Lota A, Roberts A, Baksi AJ, Voges I, Midwinter W, et al. Phenotype and Clinical Outcomes of Titin Cardiomyopathy. Journal of the American College of Cardiology. 2017;70:2264–2274. doi: 10.1016/j.jacc.2017.08.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Spezzacatene A, Sinagra G, Merlo M, Barbati G, Graw SL, Brun F, Slavov D, Di Lenarda A, Salcedo EE, Towbin JA, et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life‐Threatening Arrhythmias. Journal of the American Heart Association. 2015;4:e002149. doi: 10.1161/jaha.115.002149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Van Berlo JH, De Voogt WG, Van Der Kooi AJ, Van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbüchel H, De Visser M, et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? Journal of Molecular Medicine. 2005;83:79–83. doi: 10.1007/s00109-004-0589-1 [DOI] [PubMed] [Google Scholar]
- 130.Fatkin D, Huttner IG, Kovacic JC, Seidman JG, Seidman CE. Precision Medicine in the Management of Dilated Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;74:2921–2938. doi: 10.1016/j.jacc.2019.10.011 [DOI] [PubMed] [Google Scholar]
- 131.Smith ED, Lakdawala NK, Papoutsidakis N, Aubert G, Mazzanti A, Mccanta AC, Agarwal PP, Arscott P, Dellefave-Castillo LM, Vorovich EE, et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation. 2020;141:1872–1884. doi: 10.1161/circulationaha.119.044934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.La Gerche A, Heidbuchel H, Burns AT, Mooney DJ, Taylor AJ, Pfluger HB, Inder WJ, Macisaac AI, Prior DL. Disproportionate exercise load and remodeling of the athlete’s right ventricle. Med Sci Sports Exerc. 2011;43:974–981. doi: 10.1249/MSS.0b013e31820607a3 [DOI] [PubMed] [Google Scholar]
- 133.Verdonschot JAJ, Vanhoutte EK, Claes GRF, Helderman‐Van Den Enden ATJM, Hoeijmakers JGJ, Hellebrekers DMEI, Haan A, Christiaans I, Lekanne Deprez RH, Boen HM, et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. Human Mutation. 2020;41:1091–1111. doi: 10.1002/humu.24004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gigli M, Stolfo D, Graw SL, Merlo M, Gregorio C, Nee Chen S, Dal Ferro M, Paldinomd A, De Angelis G, Brun F, et al. Phenotypic Expression, Natural History, and Risk Stratification of Cardiomyopathy Caused by Filamin C Truncating Variants. Circulation. 2021;144:1600–1611. doi: 10.1161/circulationaha.121.053521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Valdés-Mas R, Gutiérrez-Fernández A, Gómez J, Coto E, Astudillo A, Puente DA, Reguero JR, Álvarez V, Morís C, León D, et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nature Communications. 2014;5:5326. doi: 10.1038/ncomms6326 [DOI] [PubMed] [Google Scholar]
- 136.Begay RL, Graw SL, Sinagra G, Asimaki A, Rowland TJ, Slavov DB, Gowan K, Jones KL, Brun F, Merlo M, et al. Filamin C Truncation Mutations Are Associated With Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell–Cell Adhesion Structures. JACC: Clinical Electrophysiology. 2018;4:504–514. doi: 10.1016/j.jacep.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Celeghin R, Cipriani A, Bariani R, Bueno Marinas M, Cason M, Bevilacqua M, De Gaspari M, Rizzo S, Rigato I, Da Pozzo S, et al. Filamin-C variant-associated cardiomyopathy: A pooled analysis of individual patient data to evaluate the clinical profile and risk of sudden cardiac death. Heart Rhythm. 2021. doi: 10.1016/j.hrthm.2021.09.029 [DOI] [PubMed] [Google Scholar]
- 138.Akhtar MM, Lorenzini M, Pavlou M, Ochoa JP, O’Mahony C, Restrepo-Cordoba MA, Segura-Rodriguez D, Bermudez-Jimenez F, Molina P, Cuenca S, et al. Association of Left Ventricular Systolic Dysfunction Among Carriers of Truncating Variants in Filamin C With Frequent Ventricular Arrhythmia and End-stage Heart Failure. JAMA Cardiol. 2021;6:891–901. doi: 10.1001/jamacardio.2021.1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kayvanpour E, Sedaghat-Hamedani F, Amr A, Lai A, Haas J, Holzer DB, Frese KS, Keller A, Jensen K, Katus HA, et al. Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin Res Cardiol. 2017;106:127–139. doi: 10.1007/s00392-016-1033-6 [DOI] [PubMed] [Google Scholar]
- 140.Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med. 2012;18:766–773. doi: 10.1038/nm.2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Parikh VN, Caleshu C, Reuter C, Lazzeroni LC, Ingles J, Garcia J, Mccaleb K, Adesiyun T, Sedaghat-Hamedani F, Kumar S, et al. Regional Variation in RBM20 Causes a Highly Penetrant Arrhythmogenic Cardiomyopathy. Circulation: Heart Failure. 2019;12. doi: 10.1161/circheartfailure.118.005371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hey TM, Rasmussen TB, Madsen T, Aagaard MM, Harbo M, Mølgaard H, Møller JE, Eiskjær H, Mogensen J. Pathogenic RBM20 Variants Are Associated With a Severe Disease Expression in Male Patients With Dilated Cardiomyopathy. Circulation: Heart Failure. 2019;12. doi: 10.1161/circheartfailure.118.005700 [DOI] [PubMed] [Google Scholar]
- 143.Lennermann D, Backs J, Van Den Hoogenhof MMG. New Insights in RBM20 Cardiomyopathy. Current Heart Failure Reports. 2020;17:234–246. doi: 10.1007/s11897-020-00475-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Young HS, Ceholski DK, Trieber CA. Deception in simplicity: hereditary phospholamban mutations in dilated cardiomyopathy. Biochem Cell Biol. 2015;93:1–7. doi: 10.1139/bcb-2014-0080 [DOI] [PubMed] [Google Scholar]
- 145.Hof IE, Van Der Heijden JF, Kranias EG, Sanoudou D, De Boer RA, Van Tintelen JP, Van Der Zwaag PA, Doevendans PA. Prevalence and cardiac phenotype of patients with a phospholamban mutation. Netherlands Heart Journal. 2019;27:64–69. doi: 10.1007/s12471-018-1211-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Te Rijdt WP, Ten Sande JN, Gorter TM, Van Der Zwaag PA, Van Rijsingen IA, Boekholdt SM, Van Tintelen JP, Van Haelst PL, Planken RN, De Boer RA, et al. Myocardial fibrosis as an early feature in phospholamban p.Arg14del mutation carriers: phenotypic insights from cardiovascular magnetic resonance imaging. European Heart Journal - Cardiovascular Imaging. 2019;20:92–100. doi: 10.1093/ehjci/jey047 [DOI] [PubMed] [Google Scholar]
- 147.Mazzanti A, Ng K, Faragli A, Maragna R, Chiodaroli E, Orphanou N, Monteforte N, Memmi M, Gambelli P, Novelli V, et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Clinical Course and Predictors of Arrhythmic Risk. J Am Coll Cardiol. 2016;68:2540–2550. doi: 10.1016/j.jacc.2016.09.951 [DOI] [PubMed] [Google Scholar]
- 148.McNally E, MacLeod H, Dellefave-Castillo L. Arrhythmogenic Right Ventricular Cardiomyopathy. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, eds. In: GeneReviews(®). University of Washington, Seattle: Copyright © 1993–2022, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 1993. [Google Scholar]
- 149.Groeneweg JA, Bhonsale A, James CA, Te Riele AS, Dooijes D, Tichnell C, Murray B, Wiesfeld ACP, Sawant AC, Kassamali B, et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circulation: Cardiovascular Genetics. 2015;8:437–446. doi: 10.1161/circgenetics.114.001003 [DOI] [PubMed] [Google Scholar]
- 150.Corrado D, Basso C, Judge DP. Arrhythmogenic Cardiomyopathy. Circulation Research. 2017;121:784–802. doi: 10.1161/circresaha.117.309345 [DOI] [PubMed] [Google Scholar]
- 151.Garcia-Gras E Suppression of canonical Wnt/ -catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. Journal of Clinical Investigation. 2006;116:2012–2021. doi: 10.1172/jci27751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kirchhof P, Fabritz L, Zwiener M, Witt H, SchaäFers M, Zellerhoff S, Paul M, Athai T, Hiller K-H, Baba HA, et al. Age- and Training-Dependent Development of Arrhythmogenic Right Ventricular Cardiomyopathy in Heterozygous Plakoglobin-Deficient Mice. Circulation. 2006;114:1799–1806. doi: 10.1161/circulationaha.106.624502 [DOI] [PubMed] [Google Scholar]
- 153.James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1290–1297. doi: 10.1016/j.jacc.2013.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bosman LP, Nielsen Gerlach CL, Cadrin-Tourigny J, Orgeron G, Tichnell C, Murray B, Bourfiss M, Van Der Heijden JF, Yap S-C, Zeppenfeld K, et al. Comparing clinical performance of current implantable cardioverter-defibrillator implantation recommendations in arrhythmogenic right ventricular cardiomyopathy. EP Europace. 2022;24:296–305. doi: 10.1093/europace/euab162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 1997;30:1512–1520. doi: 10.1016/s0735-1097(97)00332-x [DOI] [PubMed] [Google Scholar]
- 156.Hylind R, Beauséjour-Ladouceur V, Plovanich ME, Helms A, Smith E, Joyce E, Granter S, Stevenson LW, Cirino AL, Mcdonough BA, et al. Cardiocutaneous Features of Autosomal Dominant Desmoplakin-Associated Arrhythmogenic Cardiomyopathy. Circulation: Genomic and Precision Medicine. 2020;13. doi: 10.1161/circgen.120.003081 [DOI] [PubMed] [Google Scholar]
- 157.Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. New England Journal of Medicine. 2018;379:1007–1016. doi: 10.1056/nejmoa1805689 [DOI] [PubMed] [Google Scholar]
- 158.Kittleson MM, Maurer MS, Ambardekar AV, Bullock-Palmer RP, Chang PP, Eisen HJ, Nair AP, Nativi-Nicolau J, Ruberg FL. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement From the American Heart Association. Circulation. 2020;142. doi: 10.1161/cir.0000000000000792 [DOI] [PubMed] [Google Scholar]
- 159.Cimiotti D, Budde H, Hassoun R, Jaquet K. Genetic Restrictive Cardiomyopathy: Causes and Consequences—An Integrative Approach. International Journal of Molecular Sciences. 2021;22:558. doi: 10.3390/ijms22020558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vio R, Angelini A, Basso C, Cipriani A, Zorzi A, Melacini P, Thiene G, Rampazzo A, Corrado D, Calore C. Hypertrophic Cardiomyopathy and Primary Restrictive Cardiomyopathy: Similarities, Differences and Phenocopies. Journal of Clinical Medicine. 2021;10:1954. doi: 10.3390/jcm10091954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wahbi K, Furling D. Cardiovascular manifestations of myotonic dystrophy. Trends Cardiovasc Med. 2020;30:232–238. doi: 10.1016/j.tcm.2019.06.001 [DOI] [PubMed] [Google Scholar]
- 162.Tanawuttiwat T, Wagner KR, Tomaselli G, Nazarian S. Left Ventricular Dysfunction and Conduction Disturbances in Patients With Myotonic Muscular Dystrophy Type I and II. JAMA Cardiology. 2017;2:225. doi: 10.1001/jamacardio.2016.4145 [DOI] [PubMed] [Google Scholar]
- 163.Mathieu J, Allard P, Potvin L, Prévost C, Bégin P. A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology. 1999;52:1658–1658. doi: 10.1212/wnl.52.8.1658 [DOI] [PubMed] [Google Scholar]
- 164.Denicourt M, Pham MT, Mathieu J, Breton R. DM1 Patients with Small CTG Expansions are also at Risk of Severe Conduction Abnormalities. J Neuromuscul Dis. 2015;2:99–105. doi: [PubMed] [Google Scholar]
- 165.Marchel M, Madej-Pilarczyk A, Tymińska A, Steckiewicz R, Ostrowska E, Wysińska J, Russo V, Grabowski M, Opolski G. Cardiac Arrhythmias in Muscular Dystrophies Associated with Emerinopathy and Laminopathy: A Cohort Study. Journal of Clinical Medicine. 2021;10:732. doi: 10.3390/jcm10040732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Stoyanov N, Winterfield J, Varma N, Gollob MH. Atrial arrhythmias in the young: early onset atrial arrhythmias preceding a diagnosis of a primary muscular dystrophy. EP Europace. 2014;16:1814–1820. doi: 10.1093/europace/euu141 [DOI] [PubMed] [Google Scholar]
- 167.Yoneda ZT, Anderson KC, Estrada JC, Quintana JA, Strickland T, Montgomery JA, Michaud GF, Roden DM, Shoemaker MB. Genetic Testing for Early Onset Atrial Arrhythmias Changes Clinical Management: 2 Cases of Cardiac Emerinopathy. JACC Clin Electrophysiol. 2021;7:410–412. doi: 10.1016/j.jacep.2020.11.006 [DOI] [PubMed] [Google Scholar]
- 168.Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. European Heart Journal. 2015;36:2793–2867. doi: 10.1093/eurheartj/ehv316 [DOI] [PubMed] [Google Scholar]
- 169.van Spaendonck-Zwarts KY, van Hessem L, Jongbloed JD, de Walle HE, Capetanaki Y, van der Kooi AJ, van Langen IM, van den Berg MP, van Tintelen JP. Desmin-related myopathy. Clin Genet. 2011;80:354–366. doi: 10.1111/j.1399-0004.2010.01512.x [DOI] [PubMed] [Google Scholar]
- 170.Feingold B, Mahle WT, Auerbach S, Clemens P, Domenighetti AA, Jefferies JL, Judge DP, Lal AK, Markham LW, Parks WJ, et al. Management of Cardiac Involvement Associated With Neuromuscular Diseases: A Scientific Statement From the American Heart Association. Circulation. 2017;136. doi: 10.1161/cir.0000000000000526 [DOI] [PubMed] [Google Scholar]
- 171.Arbustini E, Favalli V, Narula N, Serio A, Grasso M. Left Ventricular Noncompaction: A Distinct Genetic Cardiomyopathy? J Am Coll Cardiol. 2016;68:949–966. doi: 10.1016/j.jacc.2016.05.096 [DOI] [PubMed] [Google Scholar]
- 172.Hoedemaekers YM, Germans T. Left Ventricular Noncompaction. Baars HF, Doevendans PA, Houweling AC, van Tintelen JP, eds. In: Clinical Cardiogenetics. Springer Nature Switzerland; 2020:115–138. [Google Scholar]
- 173.Abela M, D’Silva A. Left Ventricular Trabeculations in Athletes: Epiphenomenon or Phenotype of Disease? Current Treatment Options in Cardiovascular Medicine. 2018;20. doi: 10.1007/s11936-018-0698-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wu T, Liang Z, Zhang Z, Liu C, Zhang L, Gu Y, Peterson KL, Evans SM, Fu XD, Chen J. PRDM16 Is a Compact Myocardium-Enriched Transcription Factor Required to Maintain Compact Myocardial Cardiomyocyte Identity in Left Ventricle. Circulation. 2022;145:586–602. doi: 10.1161/CIRCULATIONAHA.121.056666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mably JD, Wu JC, Wang D-Z. New Insights Into the Molecular Underpinnings of LVNC. Circulation. 2022;145:603–605. doi: 10.1161/circulationaha.121.058371 [DOI] [PubMed] [Google Scholar]
- 176.Te Rijdt WP, Hoorntje ET, De Brouwer R, Oomen A, Amin A, Van Der Heijden JF, Karper JC, Westenbrink BD, Silljé HHW, Te Riele ASJM, et al. Rationale and design of the PHOspholamban RElated CArdiomyopathy intervention STudy (i-PHORECAST). Netherlands Heart Journal. 2021. doi: 10.1007/s12471-021-01584-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ho CY, Day SM, Axelsson A, Russell MW, Zahka K, Lever HM, Pereira AC, Colan SD, Margossian R, Murphy AM, et al. Valsartan in early-stage hypertrophic cardiomyopathy: a randomized phase 2 trial. Nature Medicine. 2021;27:1818–1824. doi: 10.1038/s41591-021-01505-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ho CY, Mealiffe ME, Bach RG, Bhattacharya M, Choudhury L, Edelberg JM, Hegde SM, Jacoby D, Lakdawala NK, Lester SJ, et al. Evaluation of Mavacamten in Symptomatic Patients With Nonobstructive Hypertrophic Cardiomyopathy. J Am Coll Cardiol. 2020;75:2649–2660. doi: 10.1016/j.jacc.2020.03.064 [DOI] [PubMed] [Google Scholar]
- 179.Saberi S, Cardim N, Yamani M, Schulz-Menger J, Li W, Florea V, Sehnert AJ, Kwong RY, Jerosch-Herold M, Masri A, et al. Mavacamten Favorably Impacts Cardiac Structure in Obstructive Hypertrophic Cardiomyopathy: EXPLORER-HCM Cardiac Magnetic Resonance Substudy Analysis. Circulation. 2021;143:606–608. doi: 10.1161/circulationaha.120.052359 [DOI] [PubMed] [Google Scholar]
- 180.Müntze J, Gensler D, Maniuc O, Liu D, Cairns T, Oder D, Hu K, Lorenz K, Frantz S, Wanner C, et al. Oral Chaperone Therapy Migalastat for Treating Fabry Disease: Enzymatic Response and Serum Biomarker Changes After 1 Year. Clinical Pharmacology & Therapeutics. 2019;105:1224–1233. doi: 10.1002/cpt.1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, Mcbride KL, Morales A, Taylor MRG, Vatta M, Ware SM. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine. 2018;20:899–909. doi: 10.1038/s41436-018-0039-z [DOI] [PubMed] [Google Scholar]
- 182.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kayvanpour E, Sedaghat-Hamedani F, Amr A, Lai A, Haas J, Holzer DB, Frese KS, Keller A, Jensen K, Katus HA, et al. Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clinical Research in Cardiology. 2017;106:127–139. doi: 10.1007/s00392-016-1033-6 [DOI] [PubMed] [Google Scholar]
- 184.Seaver LH, Khushf G, King NMP, Matalon D, Sanghavi K, Vatta M, Wees K. Points to consider to avoid unfair discrimination and the misuse of genetic information: A statement of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine. 2021. doi: 10.1016/j.gim.2021.11.002 [DOI] [PubMed] [Google Scholar]
- 185.Carey M, Herrmann A, Hall A, Mansfield E, Fakes K. Exploring health literacy and preferences for risk communication among medical oncology patients. PLOS ONE. 2018;13:e0203988. doi: 10.1371/journal.pone.0203988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lipkus IM, Samsa G, Rimer BK. General Performance on a Numeracy Scale among Highly Educated Samples. Medical Decision Making. 2001;21:37–44. doi: [DOI] [PubMed] [Google Scholar]
- 187.Zipkin DA, Umscheid CA, Keating NL, Allen E, Aung K, Beyth R, Kaatz S, Mann DM, Sussman JB, Korenstein D, et al. Evidence-based risk communication: a systematic review. Ann Intern Med. 2014;161:270–280. doi: 10.7326/M14-0295 [DOI] [PubMed] [Google Scholar]
- 188.Talevski J, Wong Shee A, Rasmussen B, Kemp G, Beauchamp A. Teach-back: A systematic review of implementation and impacts. PLOS ONE. 2020;15:e0231350. doi: 10.1371/journal.pone.0231350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sayed N, Liu C, Ameen M, Himmati F, Zhang JZ, Khanamiri S, Moonen JR, Wnorowski A, Cheng L, Rhee JW, et al. Clinical trial in a dish using iPSCs shows lovastatin improves endothelial dysfunction and cellular cross-talk in LMNA cardiomyopathy. Sci Transl Med. 2020;12. doi: 10.1126/scitranslmed.aax9276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Teerlink JR, Diaz R, Felker GM, Mcmurray JJV, Metra M, Solomon SD, Adams KF, Anand I, Arias-Mendoza A, Biering-Sørensen T, et al. Cardiac Myosin Activation with Omecamtiv Mecarbil in Systolic Heart Failure. New England Journal of Medicine. 2021;384:105–116. doi: 10.1056/nejmoa2025797 [DOI] [PubMed] [Google Scholar]
- 191.Muchir A, Worman HJ. Targeting Mitogen-Activated Protein Kinase Signaling in Mouse Models of Cardiomyopathy Caused by Lamin A/C Gene Mutations. In: Methods in Enzymology. Elsevier; 2016:557–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Judge DP, Lakdawala NK, Taylor MR, Mestroni L, Li H, Oliver C, Angeli FS, Lee PA, Macrae C. Abstract 12210: Long-Term Efficacy and Safety of ARRY-371797 (PF-0765803) in an Open-Label Rollover Study in Patients With Dilated Cardiomyopathy Due to a Lamin A/C Gene Mutation. Circulation. 2021;144:A12210–A12210. doi: doi: 10.1161/circ.144.suppl_1.12210 [DOI] [Google Scholar]
- 193.El Refaey M, Xu L, Gao Y, Canan BD, Adesanya TMA, Warner SC, Akagi K, Symer DE, Mohler PJ, Ma J, et al. In Vivo Genome Editing Restores Dystrophin Expression and Cardiac Function in Dystrophic Mice. Circulation Research. 2017;121:923–929. doi: 10.1161/circresaha.117.310996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fomin A, Gärtner A, Cyganek L, Tiburcy M, Tuleta I, Wellers L, Folsche L, Hobbach AJ, von Frieling-Salewsky M, Unger A, et al. Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci Transl Med. 2021;13:eabd3079. doi: 10.1126/scitranslmed.abd3079 [DOI] [PubMed] [Google Scholar]
- 195.Kim Y, Kim S, Neyazi M, Schmid M, Letendre J, Ewoldt J, Xiao F, Cloonan P, Sharma A, Wasson L, et al. Abstract 12802: Intron-Mediated Enhancement of TTN Regulates Sarcomere Formation and Function. Circulation. 2021;144:A12802–A12802. doi: doi: 10.1161/circ.144.suppl_1.12802 [DOI] [Google Scholar]
- 196.Prondzynski M, Krämer E, Laufer SD, Shibamiya A, Pless O, Flenner F, Müller OJ, Münch J, Redwood C, Hansen A, et al. Evaluation of MYBPC3 trans -Splicing and Gene Replacement as Therapeutic Options in Human iPSC-Derived Cardiomyocytes. Molecular Therapy - Nucleic Acids. 2017;7:475–486. doi: 10.1016/j.omtn.2017.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Greenberg B, Eshraghian E, Battiprolu P, Ricks D, Yarabe P, Schwartz J, Patel K, Shah G, Trevejo J. Abstract 10727: Results from First-in-Human Clinical Trial of RP-A501 (AAV9:LAMP2B) Gene Therapy Treatment for Danon Disease. Circulation. 2021;144:A10727–A10727. doi: doi: 10.1161/circ.144.suppl_1.10727 [DOI] [Google Scholar]