

Abstract
Apolipoprotein L1 (APOL1)-associated focal segmental glomerulosclerosis (FSGS) is the dominant form of FSGS in Black individuals. There are no targeted therapies for this condition, in part because the molecular mechanisms underlying APOL1’s pathogenic contribution to FSGS are incompletely understood. Studying the transcriptomic landscape of APOL1 FSGS in patient kidneys is an important way to discover genes and molecular behaviors that are unique or most relevant to the human disease. With the hypothesis that the pathology driven by the high-risk APOL1 genotype is reflected in alteration of gene expression across the glomerular transcriptome, we compared expression and co-expression profiles of 15,703 genes in 16 Black patients with FSGS at high-risk vs 14 Black patients with a low-risk APOL1 genotype. Expression data from APOL1-inducible HEK293 cells and normal human glomeruli were used to pursue genes and molecular pathways uncovered in these studies. We discovered increased expression of APOL1 and nine other significant differentially expressed genes in high-risk patients. This included stanniocalcin, which has a role in mitochondrial and calcium-related processes along with differential correlations between high- and low-risk APOL1 and metabolism pathway genes. There were similar correlations with extracellular matrix- and immune-related genes, but significant loss of co-expression of mitochondrial genes in high-risk FSGS, and an NF-κB-down regulating gene, NKIRAS1, as the most significant hub gene with strong differential correlations with NDUF family (mitochondrial respiratory genes) and immune-related (JAK-STAT) genes. Thus, differences in mitochondrial gene regulation appear to underlie many differences observed between high- and low-risk Black patients with FSGS.
Keywords: Focal segmental glomerulosclerosis, gene expression, nephrotic syndrome, glomerulus, mitochondria
Graphical Abstract
Introduction
Positive selection events can result in common, population-specific variants. This is the case for the two exonic variants of apolipoprotein L1 (APOL1), G1 - comprised of two missense variants, and G2, a six base-pair deletion1. One copy of risk variant (RV) APOL1 protects against trypanosomiasis. Two copies (a high-risk [“HR”] genotype) are associated with increased risk of multiple kidney diseases, including focal segmental glomerulosclerosis (FSGS) (17x increased odds)1, 2 3. 13% of Black Americans carry the HR APOL1 genotype, but this rises to 60–70% in those with FSGS3, 4. These RVs are rare in individuals without recent African ancestry5. Thus, the added kidney disease risk from APOL1 uniquely affects Black individuals, who already face disproportionate health burdens due to structural racism and other negative social drivers6.
To ultimately develop treatments and cures for APOL1-associated FSGS, we must discover the mechanisms by which its RVs cause/contribute to it. While APOL1 is absent outside of humans and some primates, transgenic animals and cell lines have provided insights into disease pathogenesis7–9. Insights include; (1) increased APOL1 expression by immune activation10 (2) toxicity associated with the RV and increased expression8, 11, (3) APOL1 ion channel formation and function dependent on complex intracellular trafficking and pH changes12, 13, and (4) multiple pathways implicated in cellular damage including mitochondrial dysfunction14–16, inflammasome activation17, ER stress18, 19, increased cation transport13, 20, and global protein synthesis inhibition21, 22.
However, a gap remains in understanding the molecular components driving the APOL1associated FSGS phenotype in patients. Dissecting the transcriptomic landscape of kidney tissue from affected patients – where APOL1’s regulation and expression are within its native genomic context – can help address this gap. APOL1-associated FSGS genes, pathways, or transcriptional behaviors discovered this way would arguably have increased specificity and relevancy towards understanding the human disease.
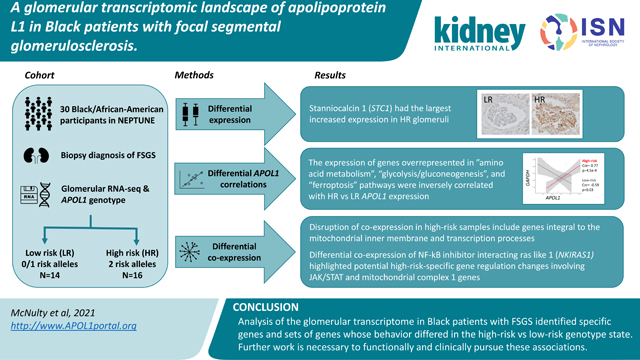
Our conceptual model of APOL1’s role in FSGS was: (1) a HR genotype primarily exerts its effects via a change in APOL1 function (given that the RV are protein-altering variants) rather than massive changes in its own expression and (2) this HR-specific APOL1 function results in differences in cellular programs which are potentially revealed through changes in gene expression and co-expression of glomerular genes. To test this model, we performed glomerular transcriptome-wide differential expression and differential co-expression studies comparing Black patients with biopsy-proven FSGS23 with a low-risk (“LR”; 0 or 1 risk alleles) or HR genotype (Figure 1).
Figure 1 -. Analysis schematic:

APOL1 risk variants were genotyped for Black participants with FSGS from the NEPTUNE cohort. There were 16 high-risk (“HR,” 2 risk variants) and 14 low-risk (“LR,” 0 or 1 risk variant) participants. Three glomerular transcriptomic analyses were used to illuminate mRNA expression differences as a function of risk genotype and APOL1 expression. (1) differential expression to identify genes with varying expression between the HR and LR FSGS group. (2) transcriptome-wide correlation of single genes with APOL1 to identify similarities and differences between genes coexpressed with APOL1 specifically in HR versus LR state. (3) differential co-expression to go beyond solely APOL1 correlations and identify groups of genes whose co-expression differs with each other in the HR and LR APOL1 state. The latter two analyses empowered identification of potential molecular perturbations between groups that are not reflected through differential expression.
Methods
The work described has been carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki).
NEPTUNE
Samples were from the Nephrotic Syndrome Study Network (NEPTUNE)23, a biopsy-based, longitudinal study of patients with proteinuric kidney diseases. We selected FSGS patients with a self-reported race/ancestry ‘Black/African American’ or a genotype-based African ancestry from Peddy24.
APOL1 Genotyping
Twenty-four samples were genotyped through previously described whole-genome sequencing (WGS) and Sanger sequencing25, 26, five through WGS alone, and one using RNA-seq.
Glomerular RNA-seq
Using standard methods, glomerular mRNA underwent sequencing and quality control followed by alignment, gene expression counting of protein-coding genes, and normalization27–32. We used sampleNetwork32 to identify samples with outlying expression profiles, standardized connectivity < −3. This process was repeated until no outliers remained (see Supplement for details).
Cell-Type-Specific Gene Expression
Using the genes unique to a cell type33, we approximated the cell-type abundances as the mean of the cell-type-specific quantile normalized gene expression in each sample. The difference in mean cell-type expression between groups was tested with a Wilcoxon test and a Bonferroni adjusted significance threshold of 0.05/24=0.002.
Differential Expression
We conducted the differential expression analysis with DESeq230 using the filtered gene count matrix from StringTie, adjusting for age, sex, and RNAseq batch. Cook’s distance was used to flag associations influenced by outliers. Results were plotted with EnhancedVolcano [https://github.com/kevinblighe/EnhancedVolcano].
Immunohistochemical staining of renal biopsies
The Brigham and Women’s Hospital Institutional Review Board approved collection and immunostaining of archival human renal biopsies. For analysis, we searched for sections from patients with (1) normal kidney histology, (2) FSGS and a low risk APOL1 genotype, and (3) FSGS and a high risk APOL1 genotype. Furthermore, we sought FSGS patients with similar stages of disease. 4 mm paraffin-processed, formalin-fixed kidney sections from eligible patients were subjected to antigen-retrieval (pressure cooker or microwave in citrate buffer, pH 6), stained with antibodies against NFkB (1:2500; ab32360), STC1 (1:500; Abcam ab229477) or NKIRas1 (1:100; Santa Cruz sc-271169), followed by horseradish peroxidase–conjugated secondary antibodies (Dako), and counterstained with hematoxylin. Care was taken that glomeruli chosen for imaging were well-preserved with either no or only segmental collapse or scarring, in order to ensure preserved glomerular cellular architecture.Images were taken with an Olympus BX53 microscope with DP73 camera and processed using Olympus CellSens standard software.
APOL1 Genome-Wide Correlations
In each group, we calculated Spearman correlations between APOL1 and normalized expression for each gene. The ranked correlations were assessed by Webgestalt (WEB-based Gene SeT AnaLysis Toolkit) 34 for analysis of enrichment of “annotation terms”. Enriched annotation terms with FDR < 0.05 in both HR and LR analyses were considered overlapping; the sign of the normalized enrichment score was used to determine if the enrichments were concordant or discordant. A threshold of FDR < 0.2 was used to compare enrichments with normal tissue.
Differential Co-expression
Differential co-expression gene modules were built using methods outlined by DiffCoEx35 (see Supplement for full details). Briefly, we calculated the adjacency matrix of Spearman correlations for HR and LR separately and computed the matrix of adjacency differences, ranging from 0 to 1 (where 1 indicates the strongest difference in correlations). This created a topology overlap matrix which was then clustered into modules with Weighted Gene Correlation Network Analysis (WGCNA). Intra-modular connectivity for each gene was defined as the sum of adjacencies (from the matrix of adjacency differences) for each gene normalized by the largest connectivity value. Hub genes were defined as genes with intra-modular connectivity greater than 0.7. Submodules were identified by comparing the genes’ k-means clustering within a DiffCoEx module in HR and LR separately. Submodule eigengenes (1st principal component) were used to compare patterns between submodules. We used Cytoscape (v3.7.2) to visualize the correlation networks.
Enrichment Analyses – Webgestalt, DAVID
We used Webgestalt for gene set enrichment analyses. Correlations were ranked by decreasing magnitude in each group separately. We tested enrichment in all Gene Ontology (GO) categories, KEGG, Panther, Wikipathway, and Reactome databases. 2,000 simulations were used to generate p-values. FDR was from reference dataset-specific adjustments. DAVID (v6.8)36, 37 was used for over-representation analyses of gene modules from DiffCoEx, using all our analysis genes as background.
CMAP
To test enrichment in the Broad Connectivity Map (CMAP)38, we selected genes with |log2 fold change| >0.75 and an unadjusted p-value < 0.05, excluding genes flagged by Cook’s distance or low counts, and reported results from their HA1E cell-line only. CMAP connectivity scores, ranging from −100 to 100, quantify the relationship between the query signatures and our differentially expressed genes. A score of 95 indicates that only 5% of the reference datasets have shown stronger connectivity than the query.
HEK293 Analyses
We used the same carefully characterized lines for the RNAseq experiment here as previously published20, 39. Briefly, HEK293 cells were stably transfected with APOL1 on a TET ON promoter and harvested nine hours after tetracycline induction. As shown in those publications, Western blotting for APOL1 after induction with the same dose of tetracycline demonstrated similar expression levels. DESeq2 was used to test for differential expression between four G1 replicates and two G0 replicates. Genes included were those with greater than ten normalized counts in all samples. Cook’s distance filtered was not applied due to the small sample size. Only replicated genes are reported.
Statistical Analyses
R Studio Version 1.2.5033 was used for all analyses and figures created with ggplot, ggcorplot, and ggpairs.
Results
Participants were from NEPTUNE23, a biopsy-based cohort of patients with proteinuric kidney diseases. Inclusion criteria for this study were a histologic diagnosis of FSGS, glomerular RNA-seq data, APOL1 genotyping, and self-identified ‘Black/African-American’ race or genotype-predicted African continental ancestry. This resulted in 14 LR and 16 HR samples.
There were no significant demographic or clinical differences at the time of biopsy (Table S1, Table 1). There were no significant differences in cell-type distribution between HR and LR groups based on a comparison of the mean expression of cell type-specific genes33 in the glomerular transcriptome data (Figure S1). These data support the validity of comparing these two groups as a function of APOL1 genotype with limited confounding by phenotypic or cell type differences.
Table 1:
Participant Demographics
| Low-Risk | High-Risk | Pa | |
|---|---|---|---|
|
| |||
| n | 14 | 16 | |
| Sex = Male (%) | 9(64.3) | 9 (56.2) | 0.72 |
| Age of Biopsy (median [IQR]) | 29.5 [13.3, 57.0] | 16.5 [15.8, 26.8] | 0.38 |
| Age of Onset (median [IQR]) | 20.0 [4.8, 56.3] | 16.0 [15.0, 23.0] | 0.98 |
| Race (%) | 0.49 | ||
| Black/African American | 13 (92.9) | 13 (81.2) | |
| Multi-Racial | 1 (7.1) | 0 (0.0) | |
| White/Caucasian | 0 (0.0) | 1 (6.2) | |
| Unknown | 0 (0.0) | 2 (12.5) | |
| Black (%) | 0.49 | ||
| Yes | 14 (100.0) | 13 (81.2) | |
| No | 0 (0.0) | 2 (12.5) | |
| NA | 0 (0.0) | 1 (6.2) | |
| Genotype-predicted Ancestry (%) | 0.49 | ||
| African | 13 (92.9) | 13 (81.2) | |
| Admixed American | 0 (0.0) | 2 (12.5) | |
| Unknown | 1 (7.1) | 0 (0.0) | |
| NA | 0 (0.0) | 1 (6.2) | |
| Immunosuppression at Biopsy = yes (%) | 5 (35.7) | 4 (25.0) | 0.69 |
| eGFR at Biopsy (median [IQR]) | 66.7 [47.7, 89.4] | 62.9 [45.9, 71.1] | 0.28 |
| UPCR at Biopsy (median [IQR]) | 2.8 [1.4, 5.5] | 2.3 [1.1, 6.1] | 0.90 |
| APOL1 Risk Alleles (%) | <0.01 | ||
| 0 | 3 (21.4) | 0 (0.0) | |
| 1 | 11 (78.6) | 0 (0.0) | |
| 2 | 0 (0.0) | 16 (100.0) | |
| APOL1 Haplotype (%) | <0.01 | ||
| G0_G0 | 3 (21.4) | 0 (0.0) | |
| G0_G1 | 6 (42.9) | 0 (0.0) | |
| G0_G2 | 5 (35.7) | 0 (0.0) | |
| G1_G1 | 0 (0.0) | 5 (31.2) | |
| G1_G2 | 0 (0.0) | 7 (43.8) | |
| G2_G2 | 0 (0.0) | 4 (25.0) | |
Continuous variables were tested with non-parametric Wilcoxon rank-sum tests, categorical variables were tested with Exact tests. IQR= interquartile range, eGFR = estimated glomerular filtration rate, UPCR = urine protein/creatinine ratio,
Differential expression identified increased HR APOL1 expression and nine genome-wide significant genes
Here, in contrast to a previous study that included non-FSGS patients and used array-based expression40, glomerular expression of APOL1 was significantly higher in the HR state (log2FC =0.57, p=0.02, Figure 2a). Among 15,703 genes, we discovered nine genes with significant differential glomerular expression (FDR < 0.05) between HR and LR FSGS (Table 2, Figure 2a, Table S2). Stanniocalcin 1 (STC1) had the largest up-regulation (log2FC = 3.50, padj=0.02) in HR, and C-C motif chemokine ligand 18 (CCL18) had the largest down-regulation (log2FC = −4.94, padj=0.02).
Figure 2 -. Differential expression volcano plot:

a) Genes (represented as dots) up-regulated in high-risk samples, as compared to low-risk samples, are indicated with positive log2 fold change. Vertical hashed bars indicate log2 fold change of −0.5 and 0.5, which we define as the threshold for differential expression. The horizontal hashed bar indicates the adjusted significant p-value threshold 0.05 (Punadjusted ~ 2.5×10–5). Red dots identify the nine genes that passed the multiple testing correction with absolute fold change > 0.5. Green dots are differentially expressed genes that do not pass our significance threshold. Insert - Differential expression of APOL1 in the low risk (gray) and high risk (red) states. b) Differences in glomerular immunohistochemical staining of STC1 in the normal human kidney compared to biopsies of patients with Collapsing FSGS and zero or two confirmed APOL1 risk alleles. Most-preserved glomeruli are captured. STC1 is only weakly expressed in normal glomeruli and in glomeruli of patients with collapsing FSGS and wildtype APOL1 risk alleles. In contrast, patients with FSGS with collapsing features and two APOL1 risk alleles show increased glomerular staining, diffusely detected in the glomerular endothelium. No definitive staining was seen in podocytes. Scale bars: 50mm.
Table 2:
Differentially Expressed Genes
| Gene | Log2 FC | P-value | P-adj |
|---|---|---|---|
|
| |||
| STCl | 3.50 | 8.07 × 10−6 | 0.02 |
| CCN3 | 1.89 | 6.63 × 10−6 | 0.02 |
| CCN6 | 1.42 | 7.83 × 10−6 | 0.02 |
| HIPKl | 1.23 | 2.21 × 10−6 | 0.01 |
| ANO5 | 1.07 | 1.96 × 10−6 | 0.01 |
| ALDOC | 0.72 | 1.42 × 10−5 | 0.02 |
| DGKD | −0.90 | 5.84 × 10−7 | 0.0l |
| NOS2 | −1.76 | 2.40 × 10−5 | 0.04 |
| CCL18 | −4.94 | 8.40 × 10−6 | 0.02 |
Log2 FC = log2 fold change, positive values are upregulated in HR, compared to LR.
P-adj = FDR adjusted p-value
We followed-up this result using an independent dataset of HEK293 cell gene expression data measured after induction of either wild-type (G0) or G1 APOL1 transgenes20. Four of the six upregulated genes (STC1, ALDOC, CCN3, HIPK1) replicated differential expression in this system (padj < 0.05; Table S3). These in vitro results from non-immune cells suggest that increased HR APOL1 expression in FSGS patients may be causally related to these genes’ glomerular expression changes and that their expression in FSGS patients does not solely come from non-resident immune/inflammatory cells.
We next performed immunohistochemical staining of STC1 from kidney biopsies of one patient without kidney pathology and four patients with similar stages of biopsy-proven FSGS with collapsing features and no other glomerular pathology. FSGS with collapsing features is a common histologic finding among patients with a high risk APOL1 genotype. The four patients were tested for APOL1 risk genotypes (Table S4). Two patients were wildtype for the risk genotypes (G0G0) and two patients had a high-risk genotype (G1G1 and G1G2). The presence of active, florid collapse in some glomeruli, together with more advanced segmental sclerosis and unremarkable glomeruli with evidence of podocyte injury, made these cases the best available candidates for comparatively studying the glomerular expression of the three target proteins in question. STC1 was weakly expressed in the healthy and LR glomeruli. In glomeruli of HR patients however, there was increased STC1 staining, specifically detected in the glomerular endothelium (Figure 2b).
Connectivity Map highlights the role of calcium channels upstream differential expression
To infer potential drivers of the differential expression signature observed between HR and LR FSGS, we used an in silico method, the Connectivity Map (CMAP)38, 41. CMAP is a catalog of gene expression signatures of cell lines after being directly perturbed via chemical administration, gene knockdown, or gene overexpression. The CMAP database includes an immortalized human kidney epithelial cell line (HA1E). We hypothesized that an HAE1 differential expression signature observed between unexposed cells and those exposed to a subset of chemical or transcriptomic perturbations would either match or be the opposite of the differential expression signature in our FSGS samples (comprised of 235 genes with an |FC| > 0.75 between HR and LR and punadj < 0.05).
The expression signatures for the top perturbagens that match or oppose the APOL1 FSGS signature are presented in Figure 3. For chemicals, a strong positive match was synephrine (connectivity score=99.61), an adrenergic receptor agonist and L-type calcium channel activator; and a strong opposing match was levetiracetam (connectivity score=−99.23), an N-type calcium channel inhibitor. Together, these results support a difference in calcium channel regulation between HR and LR FSGS patients that is contributing to the differentially expressed glomerular genes, with higher calcium channel activation in HR patients. Full CMAP results are in Table S5.
Figure 3 -. Connectivity Map (CMAP):

a) Schematic of CMAP analysis. Differentially expressed glomerular genes in the high risk (HR) versus low risk (LR) state with absolute log2 fold change > 0.75 and unadjusted p-value < 0.05 were selected for the CMAP query. Red dots indicate genes with higher expression in the HR group, and gray dots indicate genes with higher expression in LR group. This gene set pattern was then compared to CMAP analyses of the impact of perturbagens - including compounds, gene knockdowns, and gene overexpression – on gene expression of the HAE1 kidney cell. Perturbagens with a positive score match our analysis, i.e., genes increased/decreased in HR are also increased/decreased in response to the perturbagen. Perturbagens with a negative score inversely match our analysis, i.e., genes increased/decreased in HR are decreased/increased in response to the perturbagen. b) Heatmap of CMAP HAE1 z-scores from the top three positive and negatively enriched perturbations for each category (KD=knockdown, OE=overexpression, CP=compound). Genes (y-axis) are sorted by the log2 fold change in the high-risk versus low-risk FSGS differential expression analysis. Numbers in parentheses indicate percentile of match among all perturbagens applied to HAE1 cells.
Gene set enrichment analyses of APOL1 correlations highlight enriched pathways unique to and shared by high-risk and low-risk groups
We then aimed to test our model that the HR genotype substantially alters APOL1’s function, leading to altered co-expression with other glomerular genes and ultimately to differences in cellular biology between risk states. To do this, we first computed expression correlations of 15,703 genes with APOL1 (HR vs LR separately), ranked them by correlation magnitude, and performed gene set enrichment analysis(GSEA)34 to identify biologic pathways that were shared and different between the APOL1 risk states (Tables S6, S7).
First, most of the significantly enriched pathways (FDR < 0.05) were unique to LR and HR - 94% and 87%, respectively. The vast majority of HR enriched pathways comprised genes positively correlated with APOL1, whereas enriched pathways in the LR group derived from positively and negatively correlated groups of genes (Figure 4a).
Figure 4 -. APOL1 genome-wide correlations:

a) Histogram of gene set enrichment analysis (GSEA) normalized enrichment scores (NES) with FDR < 0.05, stratified by risk genotype. Enrichments among genes negatively correlated with APOL1 are indicated by a negative NES, and enrichments among genes positively correlated with APOL1 are indicated by a positive NES. In the low risk (LR) group, there was enrichment among genes positively and negatively correlated with APOL1. In high risk (HR), most enrichments were among genes positively correlated with APOL1 expression. b) Comparison of enrichment terms common to both HR and LR FSGS colored by gene annotation dataset. Annotations to the right of the hashed line indicate gene sets enriched among genes positively correlated with both HR and LR APOL1 expression. Annotations to the left of the hashed line indicate gene sets negatively correlated with LR APOL1 expression and positively correlated with HR APOL1 expression.
Twenty-three pathways were significantly correlated with both HR and LR APOL1 expression (FDR < 0.05). Fourteen of these were comprised of genes positively correlated with both HR and LR APOL1 expression. These included gene sets related to the extracellular matrix (e.g., collagen formation, laminin interactions) and the immune system (Figure 4b). The role of APOL1 in innate immunity has been established10. However, the shared positive correlation of HR and LR APOL1 with extracellular matrix genes (e.g., laminins, integrins, collagen) in FSGS has not been reported in either risk state. These results suggest that targeted study of APOL1’s potential coregulation and/or biological interactions with the extracellular matrix and these specific genes could be fruitful for further mechanistic understanding (Data File S1).
The component genes of 9 of 23 gene sets were significantly correlated in a positive direction with HR APOL1 but negatively correlated with LR APOL1; many of them related to metabolism (Figure 4b). The strongest of these discordant annotations were the Wikipathways term “Amino Acid Metabolism” and the KEGG Pathway “Glycolysis/Gluconeogenesis.” An additional discordant term was “Ferroptosis,” a form of programmed cell death caused by iron-dependent phospholipid peroxidation42. In HR APOL1 glomeruli of FSGS patients, expression of genes in the ferroptosis pathway increase as well. Autophagy has been implicated as one potential mechanism participating in APOL1 associated glomerular disease11, 43, 44, and the APOL1 family of proteins, in general, are known to function in cell death pathways45, 46. The extent to which any of these pathways has a role in APOL1 kidney disease requires further experimental clarification.
To triangulate which APOL1 correlation signature may be aberrant, we compared APOL1 correlation enrichment of the nine discordantly-correlated pathways from normal-appearing glomeruli microdissected from nephrectomies. With this data, we could more confidently assign the “disease state” to the FSGS subtype whose signature was discordant with the normal glomerular expression signature. In the normal tissue, there was an enrichment of three of these pathways (FDR< 0.2); “pteridine-containing compound,” “nucleoside monophosphate,” and “organic acid transporter activity” (Table S8). In all three, the patterns in the normal tissue matched LR FSGS. This suggests that for these pathways, HR APOL1’s correlation patterns with its co-expression partners represent the pathologic state.
Differential co-expression identifies gene modules and submodules with changes in correlation patterns between APOL1 states
Independent studies have demonstrated that differentially co-expressed genes are often not differentially expressed47. Furthermore, even without differential expression, groups of genes comprising regulatory networks can differentially co-express (“re-wire”) between biologic conditions48. As such, we hypothesized that biological pathways disrupted in APOL1-associated FSGS might be reflected by these types of changes in transcriptomic network relationships and not limited to differentially expressed genes or those only correlated with APOL1. To test this, we turned to the differential co-expression method35 (“DiffCoEx”). Using the same 30 samples and 15,703 genes from previous analyses, we used DiffCoEx to identify and cluster genes whose correlation patterns changed between HR and LR FSGS patients’ glomeruli.
1,273 genes showed varying degrees of differentially co-expression with each other, clustering into four modules (“Green”−137 genes, “Purple”- 40 genes, “Pink” – 847 genes, “Brown” – 249 genes; Figure 5a). These modules were defined based on their component genes’ magnitudes and direction of correlation within themselves - and in relation to other modules - in the HR vs LR state. By clustering each module’s genes separately in the HR and LR state, we further refined them into smaller, putatively more specific, “submodules” (Figure 5b, 5c). Once the DiffCoEx modules and submodules were defined, we sought to more clearly understand relationships within and between them and to identify enriched gene sets and key genes – “hub genes” (Figure 5d, Table S2, Figure S2, S3).
Figure 5 -. Differential co-expression analysis of 15,703 protein-coding genes in high-risk versus low-risk FSGS:
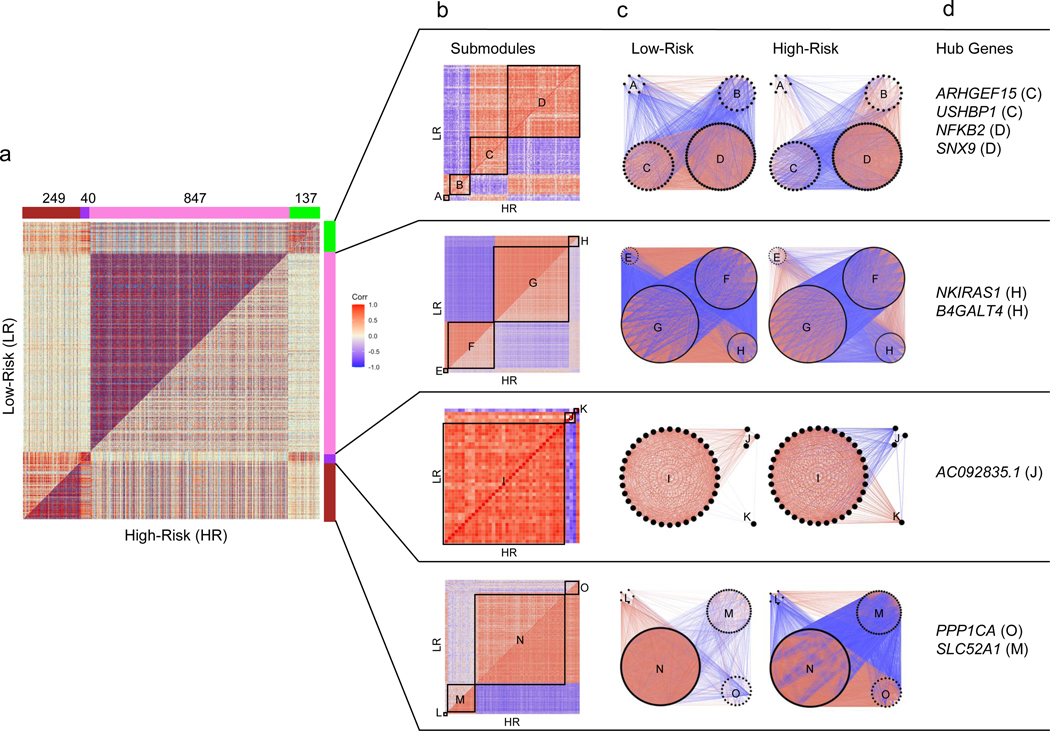
(Positive correlations are red and negative are blue throughout all figures). a) Symmetric correlation heatmap of differentially co-expressed genes grouped into four modules (“Brown,” “Purple,” “Pink,” “Green”). Each row and column is a single gene. The upper triangle (above the diagonal) reflects correlations in the low risk (LR) samples, and the lower triangle (below the diagonal) the high risk (HR) samples. The number of genes in each module is indicated above the module color. b) Submodule correlation heatmaps for each differentially co-expressed gene module, identified by hierarchical clustering of module genes in each group. LR correlations are reflected on the upper triangle and HR on the bottom. Submodules are labeled alphabetically. c) Gene network view of correlations within and between gene submodules stratified by risk genotype. d) Hub genes (most differentially co-expressed genes), for each module with the corresponding submodule label. Genes with a normalized connectivity > 0.7 were considered hub genes.
Disruption of co-expression in high-risk samples include genes integral to the mitochondrial inner membrane and transcription processes
The Pink module was defined by stronger co-expression of its 847 genes in the LR state, with 91% of gene pairs having a stronger absolute correlation in LR. There was significant enrichment for terms such as “mitochondrial inner membrane,” “oxidative phosphorylation,” “mitochondrial respiratory chain complex I,” “mRNA splicing,” and “ribonucleoprotein” (Table S9). Previous studies have implicated mitochondrial dysfunction, abnormal energetics and reduced transcription in APOL1 associated FSGS14–16, 21, 49, 50. Given its large size and its enrichment for genes, subcellular structures, and pathways previously implicated in APOL1 FSGS, we focused on more deeply characterizing the Pink module and its submodules. In particular, given the weaker co-expression in the HR state, we hypothesized that genes in the Pink module might harbor sets of genes with aberrant coregulation that could contribute to, or be the result of, the inciting pathobiology of APOL1-associated FSGS.
There were four Pink submodules (“Pink_E,” “Pink_F,” “Pink_G,” “Pink_H”). We performed an over-representation analysis of each submodule using DAVID (Table S10). We also computed eigengene correlations between them in the HR and LR FSGS state and in normal tissue (Figure 6a). Enriched terms in submodules Pink_E and Pink_F were UniProt terms “spliceosome” and “ATP binding,”and “nucleoplasm.” Genes in submodules Pink_G and Pink_H were mitochondrially enriched and included terms such as GO term “oxidative phosphorylation.” Notably, the Pink_H submodule was enriched for the mitochondrial inner membrane translocase subunit (TIM complex – TIMM17A, TIMM23, TIMM23B). A previous study found that the TIM complex was required for APOL1 import into the mitochondrial matrix and was a precursor of APOL1-induced cytotoxicity16.
Figure 6 -. Characterization of the ‘Pink’ differentially co-expressed module:

a) Spearman correlation of submodule eigengenes in high risk (HR), low risk (LR), and normal glomerular tissue (HR – red; LR – black, normal – blue). Eigengenes summarize overall gene expression behavior in each submodule and were defined as the first principal component of the group- specific gene expression matrices. Submodules are labeled along the x and y-axis. Diagonal – eigengene density plots. Below diagonal – scatter plot comparing submodule eigengenes (each point represents a sample). Above diagonal – Spearman correlations stratified by group (“Corr” – all groups combined). Correlation significance - “***” p < 0.01, “**” p<0.01, “*” p<0.05, “.” p<0.1. b) HR and LR correlation schematic of the four ‘Pink’ submodules with selected enriched biology. c) We identified the top 10% of Pink module genes differentially co-expression with NKIRAS1, resulting in 30 and 55 genes from F and G submodules, respectively. Presented here are HR, LR, and healthy NKIRAS1 correlations with other genes from the corresponding enrichment analysis. These genes are among the strongest differentially correlated genes with NKIRAS also enriched for known biology. Absolute correlations are stronger in the LR group compared to the HR group and correlation patterns in the healthy tissue are more similar to LR, compared to HR group.
We present a summary of the enriched biology and the overall co-expression patterns in the Pink submodules in Figure 6b. Overall there were opposite directions of co-expression between each submodule by risk genotype, and the magnitude of correlation was always stronger in the LR state. The normal tissue co-expression patterns were more similar to LR than HR (Figure 6a). From this, we deduce that the co-expression patterns of these glomerular genes in HR APOL1 represent the diseased state.
Differential co-expression of NKIRAS1 highlights potential high-risk-specific gene regulation changes involving JAK/STAT and mitochondrial complex 1 genes
In differential co-expression networks, hub genes are those with the most substantial differences in correlations with other genes between two comparative groups48. Here, the strongest hub gene was NF-κB inhibitor interacting ras like 1 (NKIRAS1), located within submodule Pink_H (Table S2). NKIRAS1 down-regulates NF-κB by inhibiting the degradation of one of its inhibitors, NF-κB inhibitor beta (NFKBIB)51 52. In LR, NKIRAS1 has strong positive correlations with genes in submodule Pink_G, a relationship that becomes non-significant or negative in HR. Conversely, NKIRAS1 has strong negative correlations with submodule Pink_F in LR, a relationship that becomes non-significant or positive in HR
We hypothesized that genes showing the strongest differences in co-expression with NKIRAS1 might illuminate regulatory pathways unique to, or dysregulated in, APOL1-associated FSGS. We focused our enrichment analysis on the top 10% of genes from the Pink module with the greatest difference in NKIRAS1 correlations (Tables S2, S11). Figure 6c lists correlations between NKIRAS1 and its top differentially co-expressed, biologically enriched genes. The Pink_F submodule genes had the strongest enrichment for the type I interferon signaling pathway (padj=7.3×10−4), including STAT2, which can bind to APOL1’s transcription start site10. Of note, five of the 7 JAK/STAT genes were clustered to submodule Pink_F; comparison of JAK/STAT correlations with NKIRAS1 are in Figure S4. The Pink_G module genes were enriched in the mitochondria (padj=5.7 ×10−3), and more specifically, mitochondrial respiratory chain complex I, (padj=2.1 ×10−2). This includes four NADH:ubiquinone oxidoreductase (NDUF) genes composing complex 1 of the respiratory chain. Given the stronger correlation strengths of NKIRAS1 in the LR groups and shared correlation patterns between normal kidney and LR glomeruli, we hypothesize that a regulation pathway, with inverse regulation of immune and mitochondrial genes, is being disrupted in HR.
Given the central role of NKIRAS1 in our differential co-expression results and the fact that it is novel in the glomerular biology, FSGS, and APOL1 literature, we sought to further characterize it, and its inhibition target NF-κB, with immunostaining of kidney biopsy sections from the same five patients described above. We posited that the differential co-expression could reflect differing regulation or localization of this gene in the glomeruli. Thus, we hypothesized that we would see differential cellular localization of NKIRAS1 within glomeruli of HR vs LR patients, but not differential expression. Immunohistochemical analysis of NKIRAS1 showed weak expression of NKIRAS1 in healthy and LR glomeruli and a noticeable shift of NKIRAS1 from the nucleus to the cytoplasm of HR glomeruli. There was no significant difference in staining intensity by risk genotype (Figure S5).
NKIRAS1 is a known inhibitor of NFkB. Given that NKIRAS1 is differentially co-expressed between HR and LR, we posited that downstream regulation of NFkB may be affected. The NFKB1 antibody used targets p50 and p105 in both active (nucleus) and inactive (cytoplasm) states. Thus, we hypothesized that the expression and/or localization of NFkB might be affected by differential co-expression of NKIRAS1. We found that NFkB shows a nuclear staining pattern in the kidneys of normal and low-risk genotype patients. High risk genotype individuals appeared to show increased staining both in the nucleus and cytoplasm of podocytes (Figure S5).
Discussion
Given that the majority of Black patients with FSGS have APOL1-associated disease, developing treatments and cures targeting it can make a major health impact in this population. To this end, we used three complementary glomerular transcriptomic analyses to illuminate genes, pathways, and networks that may be contributing to, or driving, the pathobiology of APOL1-associated FSGS. We focused exclusively on Black FSGS patients to increase confidence that differences observed were more likely to be specific to APOL1 risk genotype and not confounded by genetic ancestry, structural racism, or other NS histologies. And by comparing two groups of FSGS patients differing significantly only by APOL1 status (rather than a case-control study), we could be more certain that changes we discovered were related to APOL1-associated FSGS rather than FSGS non-specifically.
A major distinction between HR and LR FSGS related to differences in expression and/or co-expression of genes localized to mitochondrial compartments and/or processes. In the past decade, many experimental studies using inducible cell lines have linked increased HR APOL1 expression to mitochondrial dysfunction 14–16, 21, 49, 50. Here, we extend these in vitro findings to patients with FSGS, increasing the relevancy and importance of dissecting these pathways. Furthermore, there was a closer match of the mitochondrial co-expression signatures of normal tissue with the LR state. Thus, we are more confident in classifying the mitochondrial co-expression patterns in HR FSGS glomeruli as pathologic.
STC1 was the most significantly increased transcript in HR glomeruli, a finding supported on the protein level via immunohistochemistry. Interestingly, this increased STC1 protein expression appeared localized to the glomerular endothelium. It is notable that in a previous genetic study of Black individuals with ESKD, a variant at the STC1 locus had a weak statistical interaction with APOL153. STC1, along with two of the other five most up-regulated genes - HIPK1 and CCN6 – have been shown to regulate mitochondrial function through cytoprotective, antioxidant pathways54–56. In mesenchymal stromal cells, the up-regulation of STC1 uncouples oxidative phosphorylation, reduces reactive oxygen species, and switches metabolism towards glycolysis54. Supporting this, we discovered significant enrichment for glycolysis genes among positive APOL1 correlations specifically in the HR state. We found that induction of the APOL1 RV in the HEK293 cells resulted in increased STC1 expression. Future work is needed to test the hypothesis that increased STC1 expression is triggered to help rescue cells from APOL1-induced cytotoxicity.
Our differential co-expression data further supported disruption of mitochondrial function in HR FSGS, with co-expression changes between TIM complex genes TIMM17A, TIMM23, and TIMM23B and mitochondrial inner membrane genes integral to the respiratory chain complex 1. Notably, two mitochondrial genes in these modules, SLC25A4 (Pink_H), a component of the inner membrane, and ATP5F1C (Pink_G), an ATP synthase subunit, have been previously identified as APOL1 binding partners that play a role in the mitochondrial permeability transition pore (mPTP) function16. Additionally, via CMAP, we identified NDUFA8, a component of respiratory chain complex I, as a potential upstream target differentiating HR vs LR FSGS. Previous work has shown that APOL1 RVs activate opening of the mPTP which depolarizes the mitochondrial membrane leading to cytotoxicity16. Overall, the mitochondria appear to behave differently in the setting of HR vs LR gene expression, and some of the mitochondrial components of this response may contribute to driving the biology of APOL1 kidney injury. Results from a number of analyses also pointed to differences in calcium regulation between HR and LR FSGS. STC1 plays a role in calcium regulation, an observation that has been linked to mitochondrial function57, 58. The CMAP analysis of kidney cells found that a calcium channel agonist (synephrine) and inhibitor (levetiracetam) closely matched and opposed, respectively, the differential expression pattern in the HR vs LR group. Furthermore, it is recognized that levetiracetam has an impact on mitochondrial function59–61. Future analyses in relevant model systems are needed to directly determine whether calcium modulating drugs can alter APOL1’s cellular impact, the potential role of STC1 in HR FSGS pathology vis-à-vis calcium dynamics, and the overall relationship between calcium regulation and mitochondrial function in glomerular cells.
The largest hub-gene in the entire analysis was NF-κB inhibitor interacting ras like 1 (NKIRAS1). NKIRAS1’s known function is to inhibit degradation of the NF-κB inhibitor beta (NFKBIB), which ultimately prevents NF-kB from traveling into the nucleus and initiation transcription51 52. There were no previous reports of NKIRAS1 in relation to glomerular disease or APOL1. And while transcriptomic- and protein-based analysis did not allow us to conclusively define the role of these genes in APOL1-associated FSGS in this study, we found that NKIRAS1 had strong differential co-expression with NDUF and JAK-STAT family genes. A subset of JAK-STAT genes showed strong negative correlations with NKIRAS1 in the LR group but strong positive correlations in HR. JAK-STAT genes have previously been implicated in the pathology of FSGS and APOL1 regulation20. NF-κB and the STAT genes also play a role in mitochondrial regulation62 63, raising interesting questions about potentially pleiotropic roles for these genes in APOL1-attributed FSGS and how NKIRAS1 may fit in mechanistically. While the underlying biology remains to be discovered, our data suggests that the NF-κB pathway is differentially regulated in APOL1associated FSGS.
There are a number of aspects to take into account when interpreting this study. First, mainly due to available sample sizes, we were agnostic to G1 vs G2 when classifying HR alleles and used the recessive model in all of our analyses, with 11 of 14 LR samples having one risk allele. Second, because we compared two diseased groups, it is challenging to determine which signatures represent the potential contributory disease state. Using expression from normal glomerular tissue helped to discern the disease state. Future experimental work in controlled, manipulable systems will be necessary to more precisely define disease signatures from glomerular cells picked up in this study and more definitively dissect regulatory hierarchies and causal mechanisms. In addition, given that gene expression changes in the glomeruli are almost certainly not the sole contributor to this disease, studying the transcriptome/proteome from other cell-types (e.g. from kidney tubulointerstitium or the immune system) would also be valuable in defining drivers and modifiers of APOL1-associated FSGS. Finally, while our protein-based studies were intriguing, more comprehensive immunohistochemical analyses of APOL1 genotyped patients with FSGS are needed. For instance, because clinical testing for APOL1 is rare, we had limited ability to match larger numbers of cases with controls. Thus, the generalizability of the cases shown here is unclear. In addition, due to the absence of frozen tissue in these archival samples, we were unable to perform dual immunostaining to label glomerular cell types. There are also many other candidate proteins that could be analyzed in the future. Finally, there is likely heterogeneity in environmental and genetic factors contributing to LR FSGS and in the “second hits” in the HR group. These factors may influence the glomerular transcriptome for each person, with a downstream impact on the between-group comparative gene expression analyses. While this does not alter the validity of the results presented here, it may impact their generalizability to APOL1 FSGS transcriptomic studies in which samples have different environmental and genetic factors contributing to their disease.
In conclusion, this glomerular transcriptomic analysis of APOL1-associated FSGS in people extended mitochondrially-related discoveries from model systems, replicated immune-related findings from previous human analyses, and identified new genes and pathways for hypothesisdriven, mechanistic pursuit. Multiple lines of inquiry converged on the concepts that mitochondrial dysfunction, metabolic dysregulation, and a loss of gene coregulation in the HR state have a substantial and specific role in APOL1-associated FSGS. We highlighted the most impactful, cohesive findings from our analyses in the body of this paper, but there were many other significant relationships between APOL1, genes, and modules that we could not include here. Thus, we’ve created the “APOL1 Portal” (http://APOL1portal.org) to make all of these analyses and all of the underlying expression data publicly available for browsing, download, and secondary analysis.
Supplementary Material
Acknowledgments:
The authors would like to thank Dr. Seongkyu Han and Dr. Christopher Benway for their discussions and contributions to Figures 1 and 3, and Terri Woo for their contribution to immunohistochemical staining of kidney biopsies.
MS is supported by NIH RO1 DK108805, DK119380, and RC2 DK122397. DJ, NB, and JF are supported by 2 UM1 DK105554-06. The Nephrotic Syndrome Study Network Consortium (NEPTUNE), U54- DK- 083912, is a part of the National Institutes of Health (NIH) Rare Disease Clinical Research Network (RDCRN), supported through a collaboration between the Office of Rare Diseases Research, National Center for Advancing Translational Sciences and the National Institute of Diabetes, Digestive, and Kidney Diseases. Additional funding and/or programmatic support for this project has also been provided by the University of Michigan, the NephCure Kidney International and the Halpin Foundation.
Disclosure Statement:
MGS has consulted for Maze Therapeutics and Janssen and is on the Scientific Advisory Board of Natera. DJF and MRP are inventors on patents related to APOL1, own equity in Apolo1bio, and receive research support and have consulted for Vertex.
Appendix
Members of the Nephrotic Syndrome Study Network (NEPTUNE)
NEPTUNE Enrolling Centers
Cleveland Clinic, Cleveland, OH: K Dell*, J Sedor**, M Schachere#, J Negrey#
Children’s Hospital, Los Angeles, CA: K Lemley*, B Silesky#
Children’s Mercy Hospital, Kansas City, MO: T Srivastava*, A Garrett#
Cohen Children’s Hospital, New Hyde Park, NY: C Sethna*, K Laurent #
Columbia University, New York, NY: P Canetta*, A Pradhan#
Emory University, Atlanta, GA: L Greenbaum*, C Wang**, C Kang#
Harbor-University of California Los Angeles Medical Center: S Adler*, J LaPage#
John H. Stroger Jr. Hospital of Cook County, Chicago, IL: A Athavale*, M Itteera
Johns Hopkins Medicine, Baltimore, MD: M Atkinson*, T Dell#
Mayo Clinic, Rochester, MN: F Fervenza*, M Hogan**, J Lieske*, V Chernitskiy#
Montefiore Medical Center, Bronx, NY: F Kaskel*, M Ross*, P Flynn#
NIDDK Intramural, Bethesda MD: J Kopp*, J Blake#
New York University Medical Center, New York, NY: H Trachtman*, O Zhdanova**, F Modersitzki#, S Vento#
Stanford University, Stanford, CA: R Lafayette*, K Mehta#
Temple University, Philadelphia, PA: C Gadegbeku*, S Quinn-Boyle#
University Health Network Toronto: M Hladunewich**, H Reich**, P Ling#, M Romano#
University of Miami, Miami, FL: A Fornoni*, C Bidot#
University of Michigan, Ann Arbor, MI: M Kretzler*, D Gipson*, A Williams#, C Klida#
University of North Carolina, Chapel Hill, NC: V Derebail*, K Gibson*, E Cole#, J Ormond-Foster#
University of Pennsylvania, Philadelphia, PA: L Holzman*, K Meyers**, K Kallem#, A Swenson#
University of Texas Southwestern, Dallas, TX: K Sambandam*, Z Wang#, M Rogers#
University of Washington, Seattle, WA: A Jefferson*, S Hingorani**, K Tuttle**§, M Bray #, E Pao#, A Cooper#§
Wake Forest University Baptist Health, Winston-Salem, NC: JJ Lin*, Stefanie Baker#
Data Analysis and Coordinating Center: M Kretzler*, L Barisoni**, J Bixler, H Desmond, S Eddy, D Fermin, C Gadegbeku**, B Gillespie**, D Gipson**, L Holzman**, V Kurtz, M Larkina, S Li, S Li, CC Lienczewski, J Liu, T Mainieri, L Mariani**, M Sampson**, J Sedor**, A Smith, A Williams, J Zee.
Digital Pathology Committee: Carmen Avila-Casado (University Health Network, Toronto), Serena Bagnasco (Johns Hopkins University), Joseph Gaut (Washington University in St Louis), Stephen Hewitt (National Cancer Institute), Jeff Hodgin (University of Michigan), Kevin Lemley (Children’s Hospital of Los Angeles), Laura Mariani (University of Michigan), Matthew Palmer (University of Pennsylvania), Avi Rosenberg (Johns Hopkins University), Virginie Royal (University of Montreal), David Thomas (University of Miami), Jarcy Zee (University of Pennsylvania) Co-Chairs: Laura Barisoni (Duke University) and Cynthia Nast (Cedar Sinai).
*Principal Investigator; **Co-investigator; #Study Coordinator
§Providence Medical Research Center, Spokane, WA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tzur S, Rosset S, Shemer R, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 2010; 128: 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol 2011; 22: 21292137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng DK, Robertson CC, Woroniecki RP, et al. APOL1-associated glomerular disease among African-American children: a collaboration of the Chronic Kidney Disease in Children (CKiD) and Nephrotic Syndrome Study Network (NEPTUNE) cohorts. Nephrol Dial Transplant 2017; 32: 983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foster MC, Coresh J, Fornage M, et al. APOL1 variants associate with increased risk of CKD among African Americans. J Am Soc Nephrol 2013; 24: 1484–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bignall ONR 2nd, Crews DC. Stony the road we trod: towards racial justice in kidney care. Nat Rev Nephrol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar V, Singhal PC. APOL1 and kidney cell function. Am J Physiol Renal Physiol 2019; 317: F463–F477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Datta S, Kataria R, Zhang JY, et al. Kidney Disease-Associated APOL1 Variants Have Dose-Dependent, Dominant Toxic Gain-of-Function. J Am Soc Nephrol 2020; 31: 20832096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Toole JF, Schilling W, Kunze D, et al. ApoL1 Overexpression Drives Variant-Independent Cytotoxicity. J Am Soc Nephrol 2018; 29: 869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nichols B, Jog P, Lee JH, et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int 2015; 87: 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beckerman P, Bi-Karchin J, Park AS, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat Med 2017; 23: 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schaub C, Verdi J, Lee P, et al. Cation channel conductance and pH gating of the innate immunity factor APOL1 is governed by pore lining residues within the C-terminal domain. J Biol Chem 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giovinazzo JA, Thomson RP, Khalizova N, et al. Apolipoprotein L-1 renal risk variants form active channels at the plasma membrane driving cytotoxicity. Elife 2020; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Granado D, Muller D, Krausel V, et al. Intracellular APOL1 Risk Variants Cause Cytotoxicity Accompanied by Energy Depletion. J Am Soc Nephrol 2017; 28: 3227–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma L, Chou JW, Snipes JA, et al. APOL1 Renal-Risk Variants Induce Mitochondrial Dysfunction. J Am Soc Nephrol 2017; 28: 1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah SS, Lannon H, Dias L, et al. APOL1 Kidney Risk Variants Induce Cell Death via Mitochondrial Translocation and Opening of the Mitochondrial Permeability Transition Pore. J Am Soc Nephrol 2019; 30: 2355–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wakashin H, Heymann J, Roshanravan H, et al. APOL1 renal risk variants exacerbate podocyte injury by increasing inflammatory stress. BMC nephrology 2020; 21: 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lan X, Jhaveri A, Cheng K, et al. APOL1 risk variants enhance podocyte necrosis through compromising lysosomal membrane permeability. Am J Physiol Renal Physiol 2014; 307: F326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chun J, Zhang JY, Wilkins MS, et al. Recruitment of APOL1 kidney disease risk variants to lipid droplets attenuates cell toxicity. Proc Natl Acad Sci U S A 2019; 116: 3712–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olabisi OA, Zhang JY, VerPlank L, et al. APOL1 kidney disease risk variants cause cytotoxicity by depleting cellular potassium and inducing stress-activated protein kinases. Proc Natl Acad Sci U S A 2016; 113: 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Datta S, Kataria R, Zhang JY, et al. Kidney Disease-Associated APOL1 Variants Have Dose-Dependent, Dominant Toxic Gain-of-Function. J Am Soc Nephrol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okamoto K, Rausch JW, Wakashin H, et al. APOL1 risk allele RNA contributes to renal toxicity by activating protein kinase R. Commun Biol 2018; 1: 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gadegbeku CA, Gipson DS, Holzman LB, et al. Design of the Nephrotic Syndrome Study Network (NEPTUNE) to evaluate primary glomerular nephropathy by a multidisciplinary approach. Kidney Int 2013; 83: 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pedersen BS, Quinlan AR. Who’s Who? Detecting and Resolving Sample Anomalies in Human DNA Sequencing Studies with Peddy. Am J Hum Genet 2017; 100: 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sampson MG, Robertson CC, Martini S, et al. Integrative Genomics Identifies Novel Associations with APOL1 Risk Genotypes in Black NEPTUNE Subjects. J Am Soc Nephrol 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gillies CE, Putler R, Menon R, et al. An eQTL Landscape of Kidney Tissue in Human Nephrotic Syndrome. Am J Hum Genet 2018; 103: 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wingett SW, Andrews S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res 2018; 7: 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pertea M, Pertea GM, Antonescu CM, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 2015; 33: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Jenkins DF, Manimaran S, et al. Alternative empirical Bayes models for adjusting for batch effects in genomic studies. BMC Bioinformatics 2018; 19: 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oldham MC, Langfelder P, Horvath S. Network methods for describing sample relationships in genomic datasets: application to Huntington’s disease. BMC Syst Biol 2012; 6: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menon R, Otto EA, Hoover P, et al. Single cell transcriptomics identifies focal segmental glomerulosclerosis remission endothelial biomarker. JCI Insight 2020; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liao Y, Wang J, Jaehnig EJ, et al. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 2019; 47: W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tesson BM, Breitling R, Jansen RC. DiffCoEx: a simple and sensitive method to find differentially coexpressed gene modules. BMC Bioinformatics 2010; 11: 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 37.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subramanian A, Narayan R, Corsello SM, et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017; 171: 1437–1452 e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang JY, Wang M, Tian L, et al. UBD modifies APOL1-induced kidney disease risk. Proc Natl Acad Sci U S A 2018; 115: 3446–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sampson MG, Robertson CC, Martini S, et al. Integrative Genomics Identifies Novel Associations with APOL1 Risk Genotypes in Black NEPTUNE Subjects. J Am Soc Nephrol 2016; 27: 814–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 2006; 313: 1929–1935. [DOI] [PubMed] [Google Scholar]
- 42.Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar V, Ayasolla K, Jha A, et al. Disrupted apolipoprotein L1-miR193a axis dedifferentiates podocytes through autophagy blockade in an APOL1 risk milieu. American journal of physiology Cell physiology 2019; 317: C209–C225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wan G, Zhaorigetu S, Liu Z, et al. Apolipoprotein L1, a novel Bcl-2 homology domain 3only lipid-binding protein, induces autophagic cell death. J Biol Chem 2008; 283: 2154021549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith EE, Malik HS. The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res 2009; 19: 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vanhollebeke B, Pays E. The function of apolipoproteins L. Cellular and molecular life sciences : CMLS 2006; 63: 1937–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hudson NJ, Reverter A, Dalrymple BP. A differential wiring analysis of expression data correctly identifies the gene containing the causal mutation. PLoS Comput Biol 2009; 5: e1000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhuva DD, Cursons J, Smyth GK, et al. Differential co-expression-based detection of conditional relationships in transcriptional data: comparative analysis and application to breast cancer. Genome Biol 2019; 20: 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma L, Ainsworth HC, Snipes JA, et al. APOL1 Kidney-Risk Variants Induce Mitochondrial Fission. Kidney Int Rep 2020; 5: 891–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li D, Snipes JA, Murea M, et al. An Acidic Environment Induces APOL1-Associated Mitochondrial Fragmentation. Am J Nephrol 2020; 51: 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fenwick C, Na SY, Voll RE, et al. A subclass of Ras proteins that regulate the degradation of IkappaB. Science 2000; 287: 869–873. [DOI] [PubMed] [Google Scholar]
- 52.Oeckinghaus A, Postler TS, Rao P, et al. kappaB-Ras proteins regulate both NF-kappaBdependent inflammation and Ral-dependent proliferation. Cell Rep 2014; 8: 1793–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Divers J, Palmer ND, Lu L, et al. Gene-gene interactions in APOL1-associated nephropathy. Nephrol Dial Transplant 2014; 29: 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Shan P, Srivastava A, et al. Endothelial Stanniocalcin 1 Maintains Mitochondrial Bioenergetics and Prevents Oxidant-Induced Lung Injury via Toll-Like Receptor 4. Antioxid Redox Signal 2019; 30: 1775–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meng L, Zhao X, Zhang H. HIPK1 Interference Attenuates Inflammation and Oxidative Stress of Acute Lung Injury via Autophagy. Med Sci Monit 2019; 25: 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patra M, Mahata SK, Padhan DK, et al. CCN6 regulates mitochondrial function. J Cell Sci 2016; 129: 2841–2851. [DOI] [PubMed] [Google Scholar]
- 57.Guan J, Mishra S, Shi J, et al. Stanniocalcin1 is a key mediator of amyloidogenic light chain induced cardiotoxicity. Basic Res Cardiol 2013; 108: 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ellard JP, McCudden CR, Tanega C, et al. The respiratory effects of stanniocalcin-1 (STC1) on intact mitochondria and cells: STC-1 uncouples oxidative phosphorylation and its actions are modulated by nucleotide triphosphates. Mol Cell Endocrinol 2007; 264: 90101. [DOI] [PubMed] [Google Scholar]
- 59.Ando S, Funato M, Ohuchi K, et al. The Protective Effects of Levetiracetam on a Human iPSCs-Derived Spinal Muscular Atrophy Model. Neurochem Res 2019; 44: 1773–1779. [DOI] [PubMed] [Google Scholar]
- 60.Rogers SK, Shapiro LA, Tobin RP, et al. Levetiracetam Differentially Alters CD95 Expression of Neuronal Cells and the Mitochondrial Membrane Potential of Immune and Neuronal Cells in vitro. Front Neurol 2014; 5: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gibbs JE, Walker MC, Cock HR. Levetiracetam: antiepileptic properties and protective effects on mitochondrial dysfunction in experimental status epilepticus. Epilepsia 2006; 47: 469–478. [DOI] [PubMed] [Google Scholar]
- 62.Albensi BC. What Is Nuclear Factor Kappa B (NF-kappaB) Doing in and to the Mitochondrion? Front Cell Dev Biol 2019; 7: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meier JA, Larner AC. Toward a new STATe: the role of STATs in mitochondrial function. Semin Immunol 2014; 26: 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.