

Abstract
It is becoming increasingly clear that the intravenous administration of nanoparticles elicits an immune response that compromises delivery efficiency and can be life threatening. This study investigated both the systemic and tissue-level cytokine response to repeat administration of lipoplexes coated with either lactose or PEG. We report that blood cytokine levels differ significantly from that observed in individual tissues. While we consistently observed a reduced cytokine response to lactosylated particles, this did not result in enhanced delivery or expression as compared to PEGylated formulations. We also document that repeat injection did not increase plasmid levels in the liver, lung, or spleen, but delivery to the tumor was enhanced under these conditions. In addition, we show that changes in neither blood nor tissue cytokines correlated strongly with reporter gene expression, and we observed relatively constant expression efficiencies (RLU/ng plasmid) across all tissues despite a considerably reduced cytokine response in the tumor. Together, these results indicate that both biodistribution and cytokine responses are dramatically altered by a repeat intravenous injection of lipoplexes, and that the mechanisms regulating reporter gene expression are not straightforward.
Keywords: DNA delivery, Lipoplex, Lipid nanoparticle, Pegylation, Immune Response, Cancer, Non-viral gene delivery, Transfection, Plasmid DNA, Cationic lipid
INTRODUCTION
Many studies have documented the biodistribution and clearance of both synthetic (e.g., liposomes, polymeric nanoparticles, lipoplexes) and “natural” (e.g., viruses, exosomes) nanoparticles1–5. The vast majority of this work has focused on applications for cancer treatment and therefore utilized tumor-bearing mouse models6–9. As a method of improving cancer treatment, researchers have attempted to develop delivery systems that preferentially target tumors9–13. In this regard, it is generally considered advantageous to design particles for prolonged circulation in order to maximize accumulation in the tumor1, 7, 8, 14. The predominant strategy for extending circulation times involves utilizing a polyethyleneglycol (PEG) coating to prevent uptake by phagocytic cells involved in clearance. However, a significant drawback of utilizing PEGylated components is the development of anti-PEG antibodies that dramatically accelerate clearance of repeat intravenous doses of PEGylated nanoparticles15–20. This phenomenon is known as “accelerated blood clearance” (ABC) and greatly limits the effectiveness of subsequent injections21–24. These effects not only compromise delivery to tumors but can also elicit life-threatening immune reactions that have forced the termination of clinical trials16, 17. In fact, high levels of pre-existing anti-PEG antibodies have been correlated with adverse events during the infusion of therapeutic nanoparticles, and clinical trials in the U.S. are now required to monitor these antibodies before and after treatment16, 17, 20, 25–27. Accordingly, we and others have investigated alternative coatings that are less immunogenic7, 21, 28–30.
Despite the predominant focus on the production of antibodies in response to nanoparticles, it is important to recognize that accelerated blood clearance is also observed in immunocompromised mice that are incapable of mounting an adaptive immune response24. It follows that systemic innate immune responses must also contribute to the accelerated blood clearance phenomenon in addition to their role in the adverse infusion reactions noted above. In the context of evaluating the potential for adverse immune responses during infusion, studies have monitored the levels of blood cytokines at relatively early timepoints, i.e., 1–4 h31–33. It is worth noting that the inclusion of nucleic acids appears to enhance the immunogenicity of PEG which further complicates the development of delivery vehicles for siRNA, mRNA, and genes24, 32. Our previous work has characterized the effects of different formulation variables (e.g., CpG, lipids, coatings) on the early cytokine response after repeated intravenous administration, and we have demonstrated that PEGylation elicits a potent, wide-ranging elevation of blood cytokines33.
In addition to the heightened immunogenicity of PEG and the potential for systemic cytokine responses, it should also be recognized that mammalian cells have an extensive system for detecting the presence of foreign nucleic acids within the cytoplasm34–37. This intricate detection system has evolved as a defense against viral infection and includes dozens of distinct mechanisms for preventing the expression of viral genes. Not surprisingly, this compromises the expression of plasmids containing a viral promoter (e.g., CMV), and it can also complicate the prolonged expression of therapeutic mRNA38–43. Although these antiviral mechanisms are intracellular, infected cells produce cytokines that are secreted locally to inhibit viral replication in neighboring cells34, 37, 44. It follows that cytokines involved in an antiviral response that limits gene expression are more localized and prolonged than those associated with a systemic infusion reaction. Therefore, the tissue-level antiviral response may not be accurately reflected by blood cytokine levels, especially when measured at early time points. Accordingly, we monitored both blood and tissue cytokines 24 and 72 hours after repeated intravenous administration of lipoplexes formulated with different coatings in an attempt to gain insight into the systemic and tissue-level immune responses elicited by lipoplexes and their effects on gene expression. Importantly, these experiments were conducted in a fully immunocompetent mouse strain bearing a murine colon tumor (CT26).
MATERIALS AND METHODS
Materials
Cholesterol, diarachidoyl-sn-glycero-3-phosphocholine (DAPC), sphingosine, lactosylceramide (C-16), and PEG750-ceramide (C-16) were obtained from Avanti Polar Lipids (Alabaster, AL).
Lipoplex preparation
Lipids were dissolved in chloroform and used to prepare liposomes comprised of sphingosine:cholesterol:DAPC at a mole ratio of 3:2:5 in water as previously described33, 45–48. Briefly, the lipid mixture was dried with nitrogen gas to achieve a thin film that was incubated under vacuum overnight before rehydration with distilled water and sonication to achieve clarity (1 – 2 min). Lactosylceramide or PEG-ceramide were included in the chloroform-lipid mixture and incorporated into liposomes at a 5% mole ratio as previously described21. Lipoplexes were prepared by combining liposomes at an N/P ratio of 0.5 with an equal volume of an altered (CMV removed, ROSA26 added) pSelect-LucSh (Invivogen, San Diego, CA) plasmid encoding luciferase (purified and eluted in water via the “salt-sensitive” protocol, and stored at −20 °C)45, 49. The diameters of these preparations were 237.9 ± 7.5 (lactose) and 273.4 ± 10.4 (PEG) nm as reported previously21.
Animal studies
For in vivo experiments, lipoplex preparations were concentrated by centrifugation with a centricon filter as previously described4, 50, and they were then diluted 1:1 (v/v) with 12% hydroxyl ethyl starch (MW 250,000, Fresenius; Linz, Austria) immediately prior to intravenous administration. Prior to treatment with lipoplexes, female immunocompetent balb/c mice 6–8 weeks old were purchased from Jackson labs (Bar Harbor, ME) and inoculated in the flank with 1 × 106 CT26.WT cells (murine colon carcinoma, ATCC® CRL-2638). When tumor volumes reached approximately 80–100 mm3 (≈ 7 days), each mouse received an intravenous tail vein injection of lipoplexes, and the animals were sacrificed 24 or 72 h after the first or second injection. In an attempt to simulate clinical infusion, suspensions containing 50 μg plasmid in 200 μl were slowly injected (over approximately 10 seconds) via tail vein as previously described48, 51. We have demonstrated that this approach sharply reduces expression in tumors and organs (> 100-fold) by avoiding hydrodynamic effects of a rapid bolus injection that are known to enhance delivery (data not shown)21, 52. Luciferase expression was monitored in extracted tissues with Promega Luciferase Assay Reagents (Madison, WI) as previously described53. All animal procedures were approved by the IACUC committee of The University of Colorado Anschutz Medical Campus and conformed to the guidelines established by the National Institutes of Health.
Measurement of Plasmid Blood Levels
To monitor plasmid levels in the blood, mice were bled (15–50 μl) at 60 minutes using their submandibular veins as previously described33, 46, 48. Whole blood was collected and centrifuged (2,000 × g for 10 minutes) to separate plasma from the blood cell fraction, and each sample was prepared with a Qiagen DNeasy Blood and Tissue kit (Qiagen, Germantown, MD). Both the plasma and cell pellet were treated with the included lysis buffer per manufacturer’s instructions. The samples were then subject to quantitative PCR using a QuantiTech RTPCR Kit (Qiagen, Germantown, MD) on an Applied Biosystems 7500 RTPCR instrument (Grand Island, NY). A standard curve of pure plasmid was used to determine quantity. Although our PCR measurements do not directly determine whether the plasmid is associated with the lipoplex, previous studies have shown that free plasmid composed of naturally-occurring nucleotides is completely degraded (i.e., not detectable) within 10 min of intravenous administration in a mouse54. While some studies have employed chemically-modified nucleotides that impart nuclease resistance, our study utilized unmodified DNA and thus we assume that the reported levels predominantly represent plasmid that is protected from degradation by its association with lipoplexes.
Determination of plasmid levels in tissues
To determine delivery of plasmid DNA to mouse tissues, animals were sacrificed 24 or 72 h after each lipoplex administration, and extracted organs were harvested and flash frozen in liquid nitrogen. Organs were subsequently thawed, weighed, and DNA was extracted using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Germantown, MD) as described above33, 45, 46, 48. Quantitative PCR (qPCR) was performed on tissue samples using QuantiTech RTPCR Kit (Qiagen, Germantown, MD) on an Applied Biosystems 7500 RTPCR instrument (Grand Island, NY)55. A standard curve of pure plasmid was used for quantification in addition to amplicon efficiency factors that account for different efficiencies of amplification33, 46, 48, 55. We have previously determined the extraction efficiencies of plasmid from isolated organs, and these values were used to calculate the plasmid levels depicted in the results46. It should be noted that the amplified PCR fragment is 1072 base pairs representing approximately one-sixth of the plasmid sequence. Therefore, our methods cannot distinguish between intact plasmid and large fragments that encode both primer sites. Previous studies have shown that free plasmid is rapidly degraded in tissues54, 56, and thus the presence of large fragments would be unlikely at the time points (24 and 72 h) used in our experiments.
Cytokine Response
A separate set of tumor-bearing mice was used to quantify cytokine levels after repetitive injection of lipoplexes. In addition to the coated lipoplex formulations described above, mice were treated with phosphate buffered saline as a negative control33. In these studies, triplicate mice were treated with lipoplex formulations as described above, and blood was collected 24 and 72 h after both the first and second injection of lipoplexes (3-day interval). Serum samples were allowed to clot for 30 minutes and spun at 2,000 × g for 15 minutes per manufacturer’s instructions (R&D Systems, Minneapolis MN). For solid tissues (liver, lung, spleen, tumor), extracted tissues were homogenized in buffer and cytokines extracted/quantified as described by the manufacturer. Samples were assayed in triplicate for interferon gamma (IFN γ), interleukin 6 (IL-6), and tumor necrosis factor alpha (TNF α) and using ELISA kits purchased from R&D Systems (Minneapolis MN). The limits of detection of IFN γ, IL-6, and TNF α are 2, 1, and 7 pg/mL for blood and 20, 40, 140 pg/g for tissues, respectively.
In vitro Experiments
Murine colon carcinoma (CT26.WT) cells were cultured at 37 °C, 5% carbon dioxide with 100% humidity in Minimum Essential Media (MEM), 10% fetal bovine serum (FBS), 50 U/ml penicillin, 50 μg/ml streptomycin (all media from Cellgro MediaTech Inc., a Corning Acquisition, Manassas, VA) as previously described45, 57. Cells were seeded at 20,000 cells/well in 96-well plates 24 hours prior to treatment. Lipoplexes were pre-incubated 1:1 v/v in FBS (i.e., 50% FBS to mimic in vivo serum protein conditions) for 30 minutes prior to dilution to 10% FBS with 100% MEM and then administered to cells for transfection. Formulations were applied to the center of each well and allowed to incubate for 4 hours as described previously45, 57. After 4 hours, the transfection media was carefully removed, cells were washed with PBS, and then returned to 10% FBS growth media. At the indicated times, cells were lysed with 30 μl Promega lysis buffer in the −80 °C freezer according to manufacturer’s instructions (Promega, Madison, WI). The lysates were divided into three fractions; one fraction (40 uL) was used for quantitative PCR measurements as described above, and the other fraction (80 uL) was assayed for protein content with a Bio-Rad protein assay (Bio-Rad, Hercules, CA) on a 96-well THERMOmax plate reader (Molecular Devices, Sunnyvale, CA). The third fraction (40 uL) was used for luminescence quantification on a Monolight Luminometer according to manufacturer’s instructions (BD Biosciences, San Jose, CA) as previously described45.
Statistics
Significance was determined using a two-tailed unpaired t test and Taylor Series was used to calculate error for the in vitro PCR and luminescence measurements as different replicates were used including additional technical replicates on qPCR. All statistics and calculations were done with GraphPad Prism Software (San Diego, CA).
RESULTS
Lipoplexes were formulated with either 5% lactosylceramide or 5% PEG-ceramide and repeat administered (3-day interval) to tumor-bearing mice via tail vein injection. This dosing interval is consistent with our previous studies and is sufficient to avoid any potential saturation of uptake mechanisms that could affect repeat injections5, 33. To assess the role of coatings on accelerated blood clearance, blood was collected 1 h after each injection, and plasmid levels were assessed in both the plasma and cell fraction. As shown in figure 1, both coatings exhibited comparable levels (60–80% of injected dose) of plasmid in the blood (plasma + cell fractions) 1 h after the first injection, with the majority associated with the plasma. In contrast, both formulations were predominantly found in the cell fraction 1 h after the second injection with greater levels of plasmid from the lactose formulation in the plasma and higher amounts of PEGylated lipoplexes associated with blood cells. These results are consistent with our previous studies showing longer plasma circulation times provided by PEG as compared to lactose after a single injection21 and also with the accelerated clearance of PEGylated liposomes after repeat injections described in prior studies22–24, 58. However, we also observe accelerated plasma clearance of lactosylated lipoplexes after repeat injection (Fig. 1A). Taken together these results suggest that accelerated plasma clearance may be a more general phenomenon, at least for lipoplexes, consistent with the potent immune responses that are known to be elicited in response to nucleic acid-containing nanoparticles32, 33, 59.
Fig. 1.
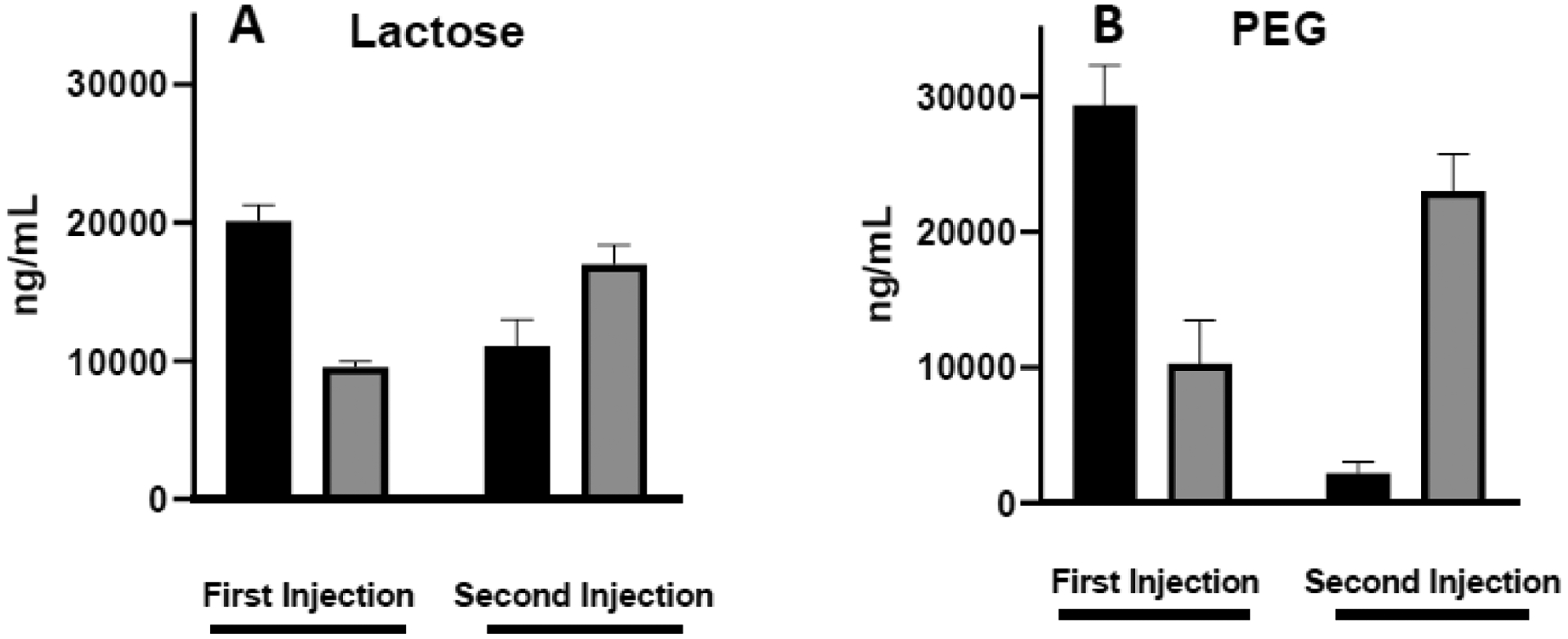
Lipoplex levels in blood 1 h after repeated intravenous injections. Plasmid levels were quantified in the plasma (black bars) and cell fraction (shaded bars) after repeat administration of lipoplexes coated with lactose (A) or PEG (B). Each bar represents the mean ± one standard error of measurements on triplicate mice.
Previous studies have established that clearance by the liver, spleen, and lungs accounts for virtually all of the injected dose of lipoplexes after a single injection21, 46, 55. In this study we monitored the accumulation of plasmid in these organs as well as the tumor 24 and 72 h after the first and second injections. After accounting for tissue weights, accumulation in the liver (≈ 1 g) was dramatically greater than other tissues (≈ 0.1 g) with significant accumulation also occurring in the spleen regardless of the coating (Fig. 2). Surprisingly, the second dose did not result in additional distribution to either of these organs or the lungs. In contrast to the absence of additional accumulation of the second dose in the liver, spleen, and lungs, deposition in the tumor approximately doubled after the second dose. Considering the accelerated clearance of the second dose, one might expect diminished accumulation. However, the absence of deposition after the second dose, especially in the liver and spleen, raises questions as to an alternative fate of repeat doses. The fact that the tumor exhibited supplemental accumulation after repeat dosing suggests that the accelerated blood clearance does not prevent additional deposition, consistent with our previous study demonstrating progressive increases in tumor accumulation after four doses of lipoplexes33.
Fig. 2.
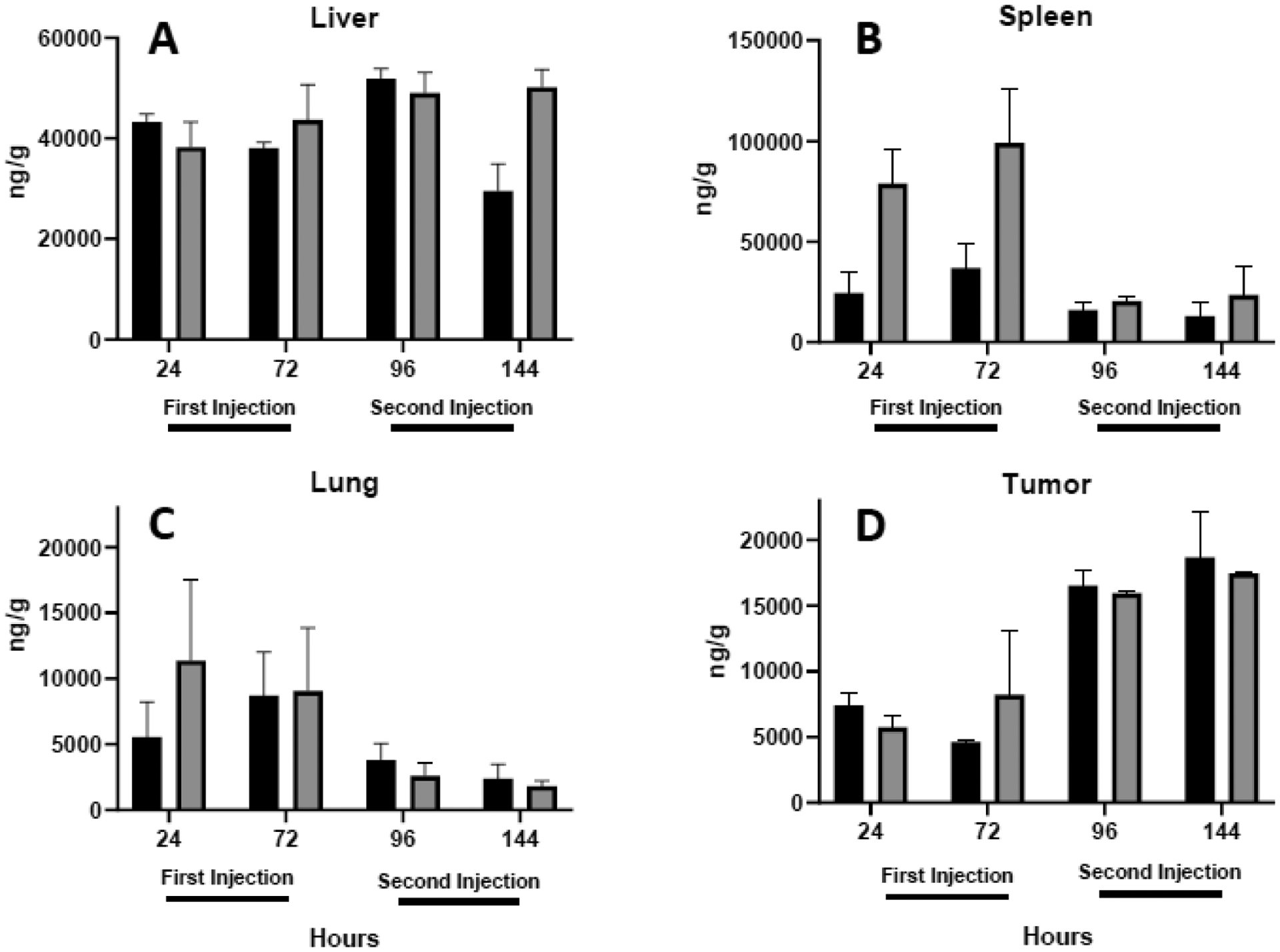
Total plasmid accumulation in tissues. Quantitative PCR was used to monitor the accumulation of plasmid per gram tissue in the liver (A), spleen (B), lung (C), and tumor (D) 24 and 72 h after each intravenous injection of lactosylated (black bars) or PEGylated (shaded bars) lipoplexes. Note the different scales used in the y-axes. Each bar represents the mean ± one standard error of measurements on organs from triplicate mice.
Changes in luciferase expression between the first and second injection generally corresponded with the observed changes in accumulation, i.e., expression in the liver, spleen, and lungs was not consistently enhanced after the second injection while that in the tumor maintained an approximately two-fold increase (Fig. 3). A closer look at the data reveals many cases where expression does not correlate with the amount of plasmid. For example, accumulation of plasmid in the liver was comparable for the two formulations (Fig. 2A), but expression was markedly higher for PEGylated lipoplexes at all time points (Fig. 3A). Similarly, luciferase expression in the spleen initially increased after the second injection despite sharp decreases in plasmid levels (compare figs. 2B+3B). Overall, the data indicate that expression is not well correlated with delivery, and that tissue type, coatings, timing, and repeat injection can each affect the levels of reporter gene expression that are observed in delivery studies.
Fig. 3.
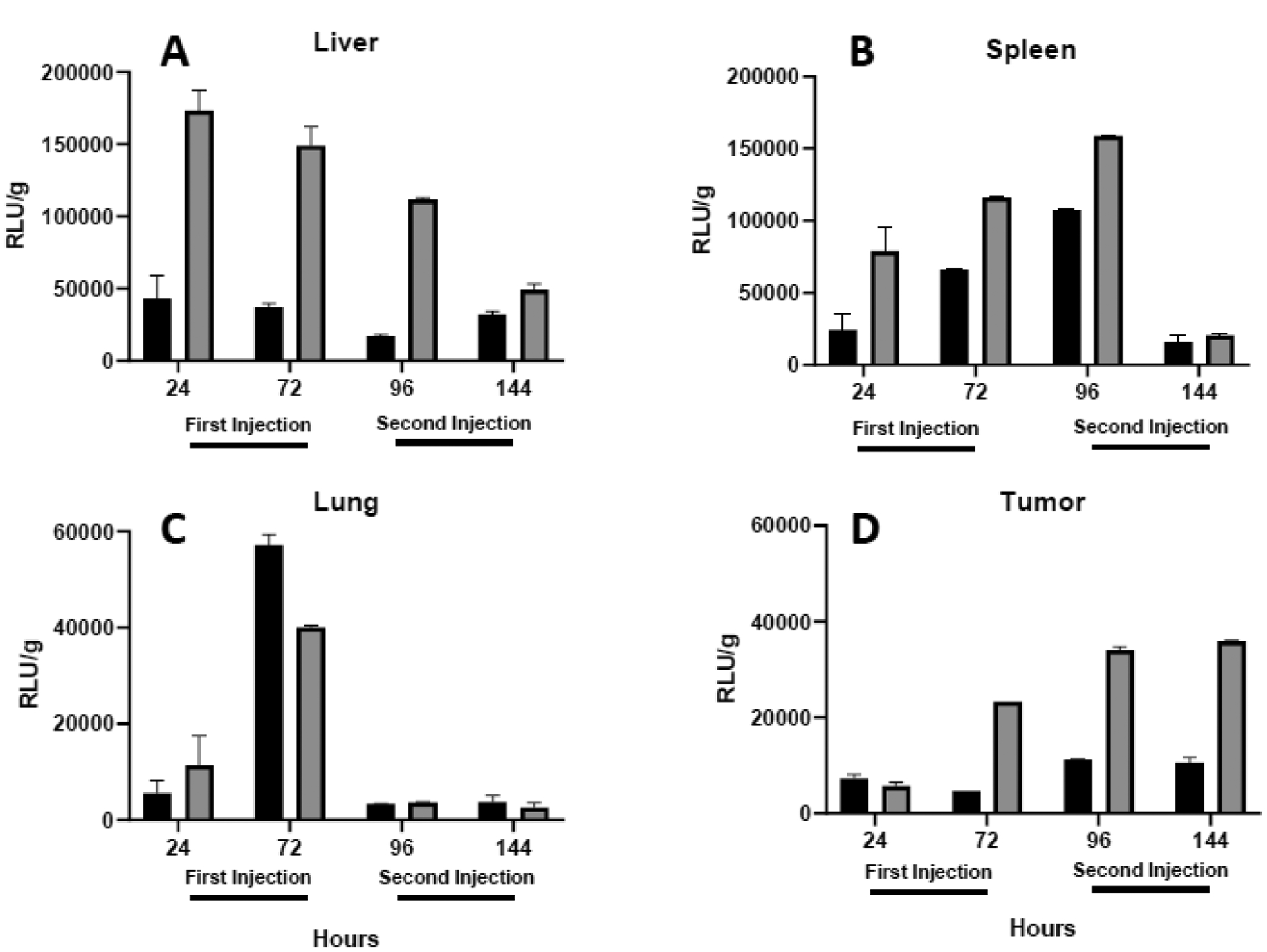
Reporter gene expression. Luciferase expression per gram tissue is depicted for the liver (A), spleen (B), lung (C), and tumor (D) 24 and 72 h after each intravenous injection of lactosylated (black bars) or PEGylated (shaded bars) lipoplexes. Note the different scales used in the y-axes. Each bar represents the mean ± one standard error of measurements on organs from triplicate mice.
Cytokine levels in the blood are often used as a measure of the innate immune response, and excessive cytokines (“storm”) can be problematic32, 60. As mentioned above, cytokines are also secreted in response to microbial invasion and can silence gene expression34, 37, 44. To evaluate the effect of lactose and PEG coatings on the cytokine response after repeat injection, we monitored blood levels of IFN γ, IL-6, and TNF α at 24 and 72 h after each injection. As shown in figure 4, each cytokine was elevated 24 h after the initial injection of lipoplexes and decreased to lower but still elevated, levels after 72 h. The second injection of lipoplexes elicited an increase in blood cytokines comparable to that seen 24 h after the first injection and again levels subsided after 72 h. In most cases, cytokines were more elevated after administration of PEGylated lipoplexes as compared to those coated with lactose, consistent with our previous findings21.
Fig. 4.
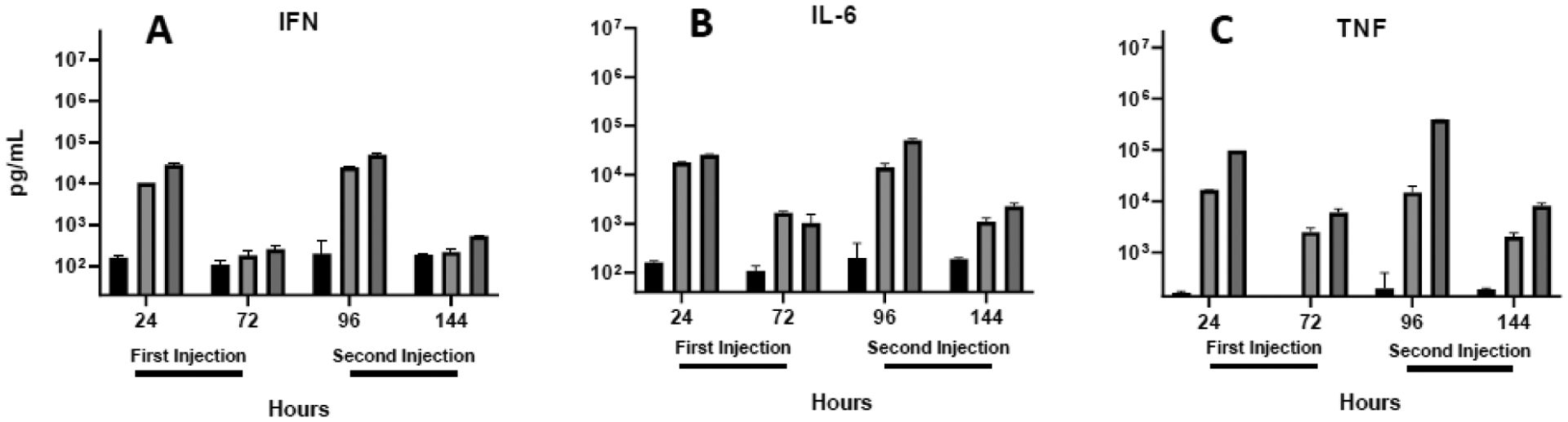
Cytokine levels in the blood. IFN γ (A), IL-6 (B), and TNFα (C) were quantified in plasma 24 and 72 h after each intravenous injection. Each bar represents the mean ± one standard error of measurements (per mL plasma) on triplicate mice injected with saline (PBS; black bars) or lipoplexes coated with either lactose (light shading) or PEG (dark shading).
As compared to changes observed in the blood, all cytokines were significantly more elevated in the liver, spleen, and lung 24 h after lipoplex injection (compare Figs. 4 to 5–7). As seen in the blood, cytokines decreased 72 h after each injection, and PEGylated lipoplexes generally elicited greater levels of cytokines than that seen with lactose-coated particles. In contrast to blood, all liver, spleen, and lung cytokines were elevated to a greater extent after the second injection. In sharp contrast to the striking increases observed in the liver, spleen, and lung, cytokine levels remained relatively low in the tumor and exhibited concentrations after the second injection that were comparable to that seen after the initial injection (Figs. 5D,6D,7D). The overall lower tumor cytokine levels and notable absence of a further elevation after the second injection is distinctly different from that observed in all other tissues. Consistent with the well-established immunosuppression that is characteristic of the tumor microenvironment61–63, tumor cytokine levels were significantly below that measured in organs and blood in most cases, regardless of coating(Table 1).
Fig. 5.
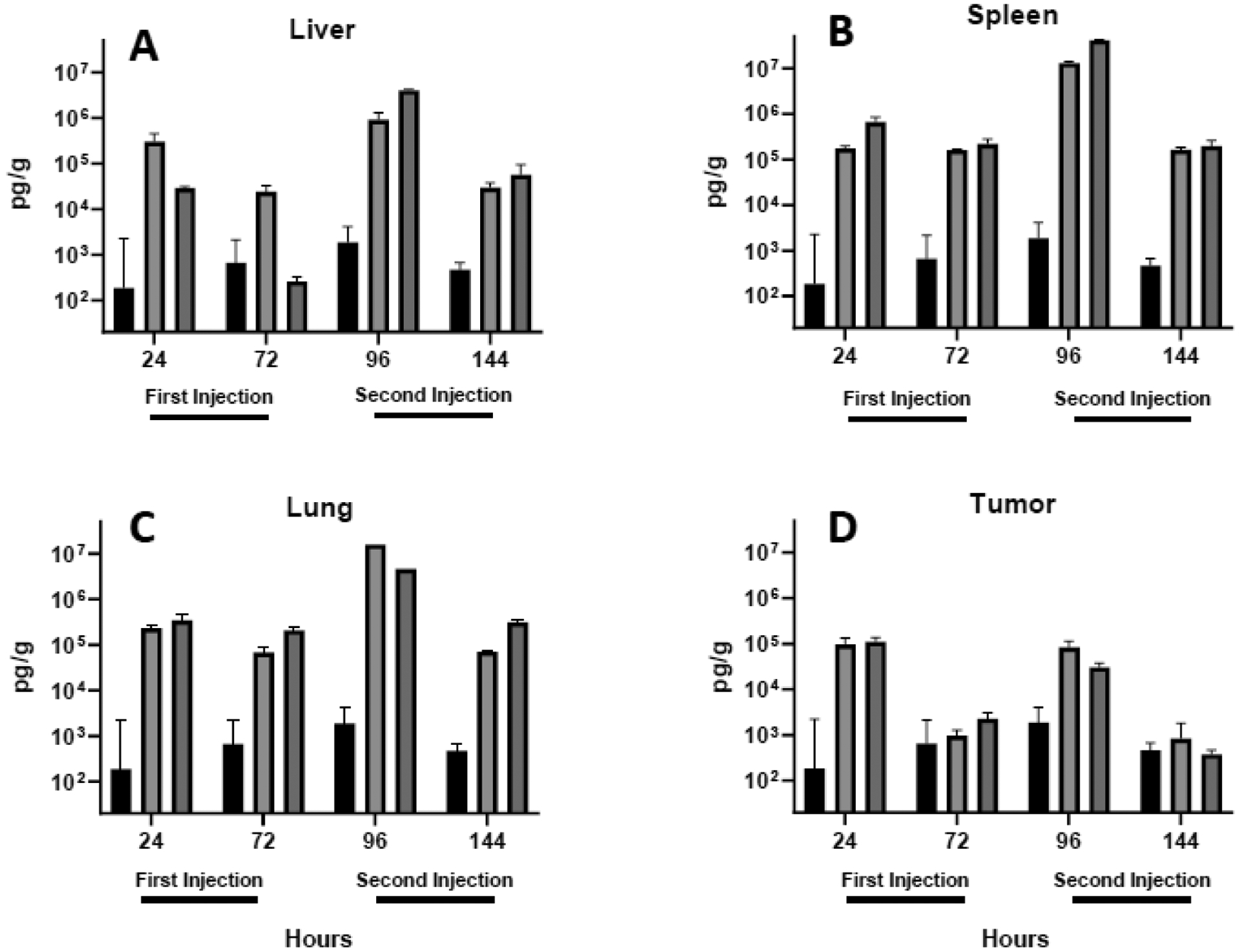
IFN γ levels in the liver (A), spleen (B), lung (C) and tumor (D) were quantified in tissues extracted 24 and 72 h after each intravenous injection. Each bar represents the mean ± one standard error of measurements (per gram tissue) on triplicate mice injected with saline (PBS; black bars) or lipoplexes coated with either lactose (light shading) or PEG (dark shading).
Fig. 7.
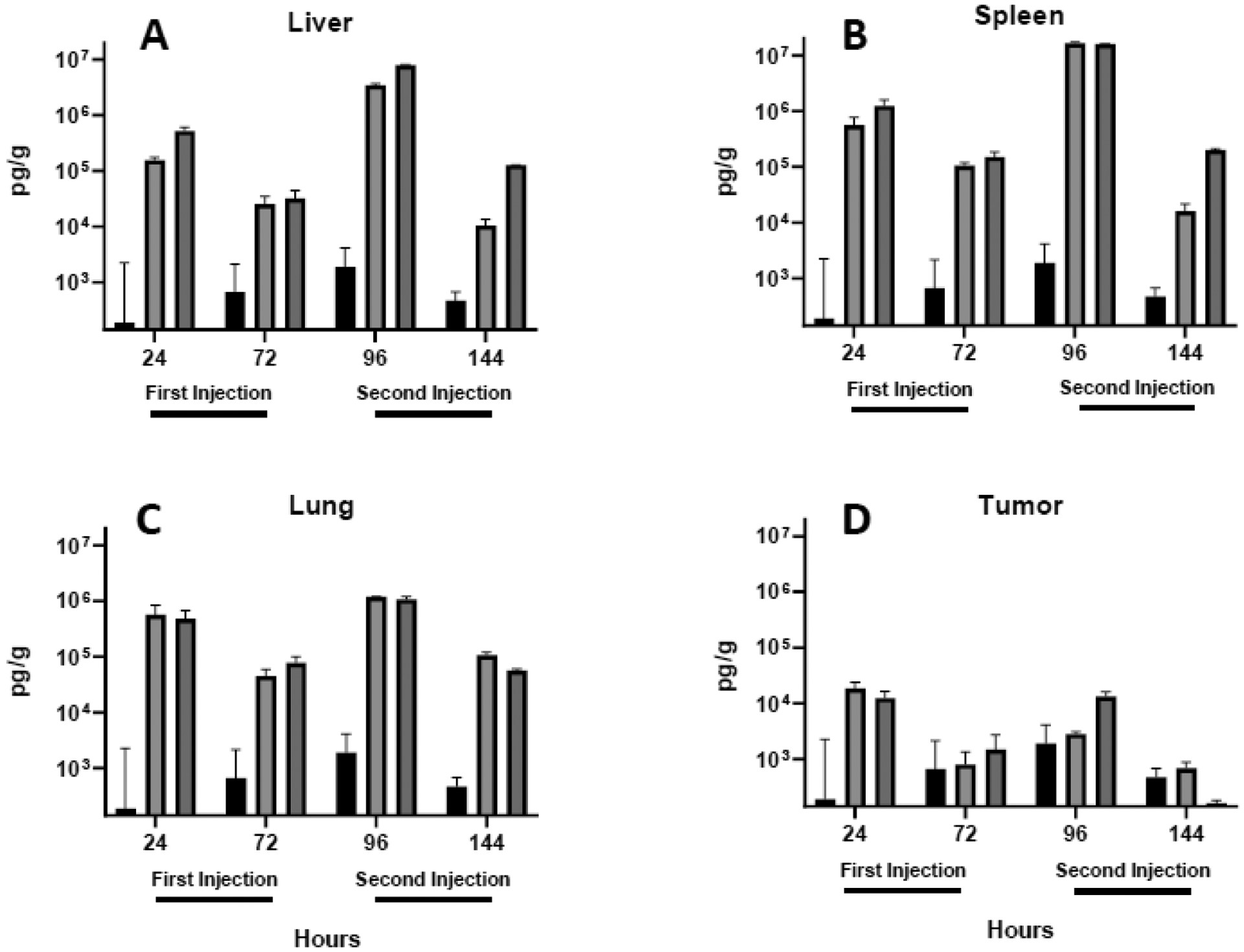
TNFα levels in the liver (A), spleen (B), lung (C) and tumor (D) were quantified in lungs extracted 24 and 72 h after each intravenous injection. Each bar represents the mean ± one standard error of measurements (per gram tissue) on triplicate mice injected with saline (PBS; black bars) or lipoplexes coated with either lactose (light shading) or PEG (dark shading).
Fig. 6.
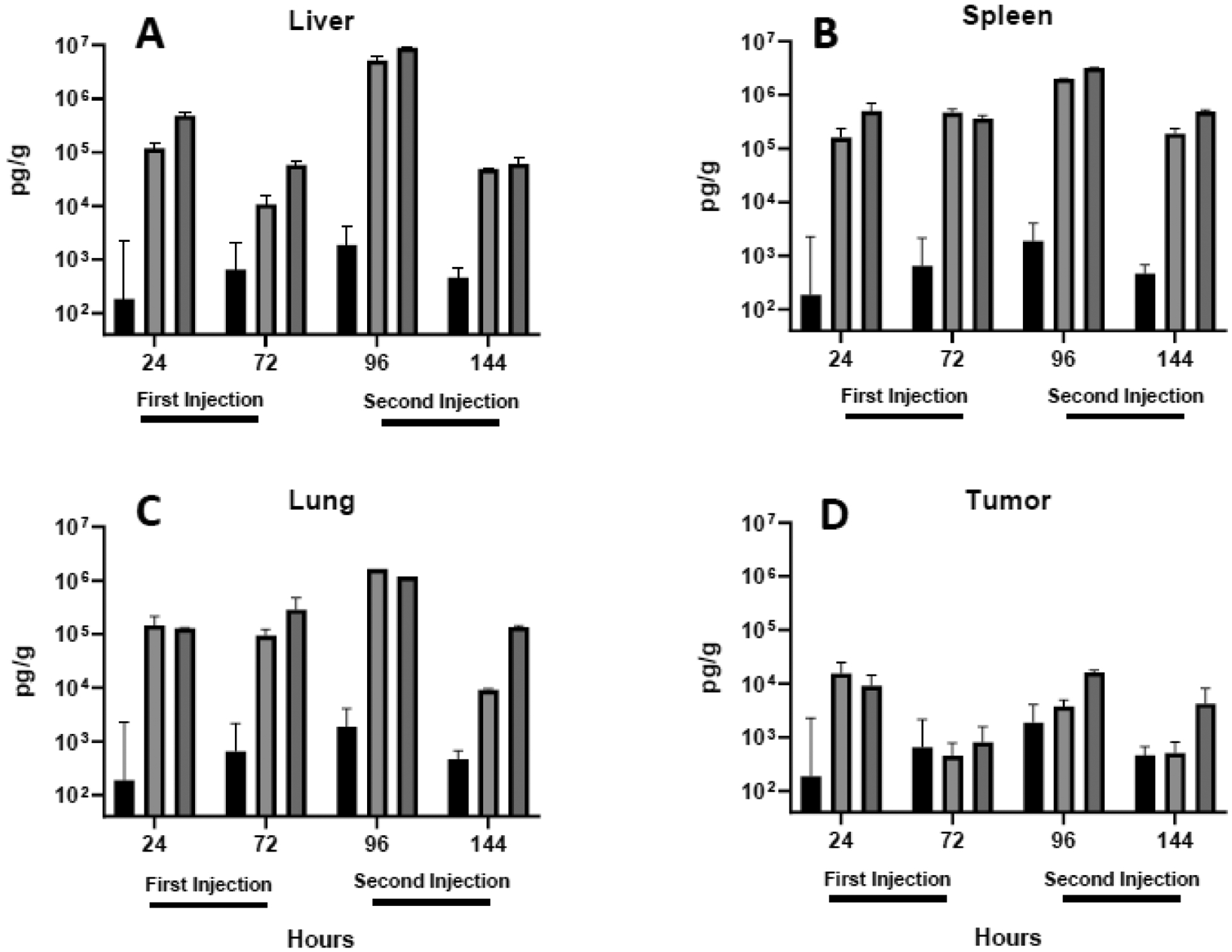
IL-6 levels in the liver (A), spleen (B), lung (C) and tumor (D) were quantified in tissues extracted 24 and 72 h after each intravenous injection. Each bar represents the mean ± one standard error of measurements (per gram tissue) on triplicate mice injected with saline (PBS; black bars) or lipoplexes coated with either lactose (light shading) or PEG (dark shading).
Table 1:
p values for comparing tumor to organ cytokine levels.
| IFN-γ | Lactose | PEG | ||||||
|---|---|---|---|---|---|---|---|---|
| 1st Injection | 2nd Injection | 1st Injection | 2nd Injection | |||||
| Tumor vs. | 24 | 72 | 96 | 144 | 24 | 72 | 96 | 144 |
| Liver | 0.1402 | 0.0450 | 0.0734 | 0.0237 | 0.0336 | 0.0505 | 0.0011 | 0.1311 |
| Spleen | 0.0511 | 0.0007 | 0.0020 | 0.0030 | 0.0100 | 0.0508 | 0.0004 | < 0.0001 |
| Lung | 00108 | 0.0226 | < 0.0001 | 0.0063 | 0.0715 | 0.0113 | < 0.0001 | 0.0068 |
| Blood | 0.0612 | 0.0489 | 0.0820 | 0.4008 | 0.0336 | 0.0505 | 0.0273 | 0.0830 |
| IL-6 | Lactose | PEG | ||||||
| 1st Injection | 2nd Injection | 1st Injection | 2nd Injection | |||||
| Tumor vs. | 24 | 72 | 96 | 144 | 24 | 72 | 96 | 144 |
| Liver | 0.0217 | 0.0661 | 0.0108 | 0.0099 | 0.0126 | 0.0153 | 0.0004 | 0.0412 |
| Spleen | 0.0822 | 0.0064 | < 0.0001 | 0.0211 | 0.0473 | 0.0073 | 0.0003 | 0.0019 |
| Lung | 0.0805 | 0.0301 | < 0.0001 | 0.0001 | < 0.0001 | 0.1216 | < 0.0001 | 0.0004 |
| Blood | 0.7189 | 0.0161 | 0.0144 | 0.0637 | 0.0311 | 0.7247 | 0.0008 | 0.4888 |
| TNF-α | Lactose | PEG | ||||||
| 1st Injection | 2nd Injectioin | 1st Injection | 2nd Injection | |||||
| Tumor vs. | 24 | 72 | 96 | 144 | 24 | 72 | 96 | 144 |
| Liver | 0.0049 | 0.0512 | 0.0010 | 0.0328 | 0.0098 | 0.0477 | 0.0004 | < 0.0001 |
| Spleen | 0.0532 | 0.0063 | 0.0011 | 0.0428 | 0.0268 | 0.0168 | < 0.0001 | 0.0007 |
| Lung | 0.0724 | 0.0313 | < 0.0001 | 0.0053 | 0.0565 | 0.0304 | 0.0050 | 0.0012 |
| Blood | 0.6052 | 0.0156 | 0.0523 | 0.0175 | 0.0008 | 0.0082 | < 0.0001 | 0.0055 |
Shaded values are statistically significant, p < 0.05.
One striking observation is that the levels of blood cytokines do not accurately reflect those observed in tissues. In fact, we observed significant differences between blood and tissue cytokines in all organs at all timepoints (see Table 2 for statistical analysis). Furthermore, this difference was noted for all cytokines suggesting that the measurement of blood cytokines that is typically conducted do not accurately reflect innate immune responses at the tissue level (compare Fig. 4 to Figs. 5–7).
Table 2:
p values for comparing blood to tissue cytokine levels.
| IFN-γ | Lactose | PEG | ||||||
|---|---|---|---|---|---|---|---|---|
| 1st Injection | 2nd Injection | 1st Injection | 2nd Injection | |||||
| Blood vs. | 24 | 72 | 96 | 144 | 24 | 72 | 96 | 144 |
| Liver | 0.0822 | 0.0424 | 0.0657 | 0.0241 | 0.0060 | 0.0106 | < 0.0001 | < 0.0001 |
| Spleen | 0.0058 | 0.0007 | 0.0020 | 0.0063 | 0.0284 | 0.0262 | 0.0001 | 0.0315 |
| Lung | 0.0073 | 0.0222 | < 0.0001 | 0.0035 | 0.0448 | 0.0111 | < 0.0001 | 0.0068 |
| Tumor | 0.0612 | 0.0489 | 0.0820 | 0.4008 | 0.0336 | 0.0505 | 0.0273 | 0.0830 |
| IL-6 | Lactose | PEG | ||||||
| 1st Injection | 2nd Injection | 1st Injection | 2nd Injection | |||||
| Blood vs. | 24 | 72 | 96 | 144 | 24 | 72 | 96 | 144 |
| Liver | 0.0300 | 0.0834 | 0.0108 | 0.0011 | 0.0138 | 0.0156 | 0.0004 | 0.0424 |
| Spleen | 0.0865 | 0.0064 | < 0.0001 | 0.0213 | 0.0506 | 0.0073 | 0.0003 | 0.0021 |
| Lung | 0.0854 | 0.0308 | < 0.0001 | 0.0005 | 0.0005 | 0.1218 | < 0.0001 | 0.0018 |
| Tumor | 0.7189 | 0.0161 | 0.0144 | 0.0637 | 0.0311 | 0.7247 | 0.0008 | 0.4888 |
| TNF-α | Lactose | PEG | ||||||
| 1st Injection | 2nd Injection | 1st Injection | 2nd Injection | |||||
| Blood vs. | 24 | 72 | 96 | 144 | 24 | 72 | 96 | 144 |
| Liver | 0.0070 | 0.0586 | 0.0010 | 0.0418 | 0.0142 | 0.0649 | 0.0005 | < 0.0001 |
| Spleen | 0.0530 | 0.0066 | 0.0011 | 0.0503 | 0.0308 | 0.0179 | < 0.0001 | 0.0006 |
| Lung | 0.0720 | 0.0338 | <0.0001 | 0.0054 | 0.0818 | 0.0343 | 0.0120 | 0.0007 |
| Tumor | 0.6052 | 0.0156 | 0.0523 | 0.0175 | 0.0008 | 0.0082 | < 0.0001 | 0.0055 |
Shaded values are statistically significant, p < 0.05.
As mentioned above, cytokines (especially interferons) are secreted as part of an antiviral response that functions to combat infection by suppressing viral gene expression35, 37, 40–42. In this context, it is important to note that the plasmid used in our studies contains a mammalian promoter and has been altered to minimize the CpG sequences and reduce immunogenicity33, 49. In order to more clearly determine whether reporter gene expression of our plasmid is being affected by changes in tissue cytokines, we utilized our qPCR measurements to calculate the efficiency of expression in individual tissues, i.e., RLU/ng plasmid. As shown in Figure 8, we observe differences in expression efficiency among tissues, coatings, and injections. More specifically, expression efficiency increased sharply after the second injection in the spleen but decreased in the lung. Surprisingly, the distinct increase in expression efficiency observed in the spleen after the second injection was maintained despite large increases in all three cytokines at 96 h and lower levels by 144 h (compare figs. 5–7+8B). While expression efficiency was relatively constant in the liver and tumor, PEGylated lipoplexes consistently resulted in more efficient expression in those organs whereas lactosylated formulations exhibited higher efficiency in the spleen (Fig. 8). In contrast to what might be expected if cytokines were involved in suppressing reporter gene expression, expression efficiency in the spleen was highest after the second injection and remained high while all cytokines peaked 24 h after the second injection and subsequently decreased. Taken together, our data are not consistent with a role for tissue cytokines in directly modulating reporter gene expression.
Fig. 8.
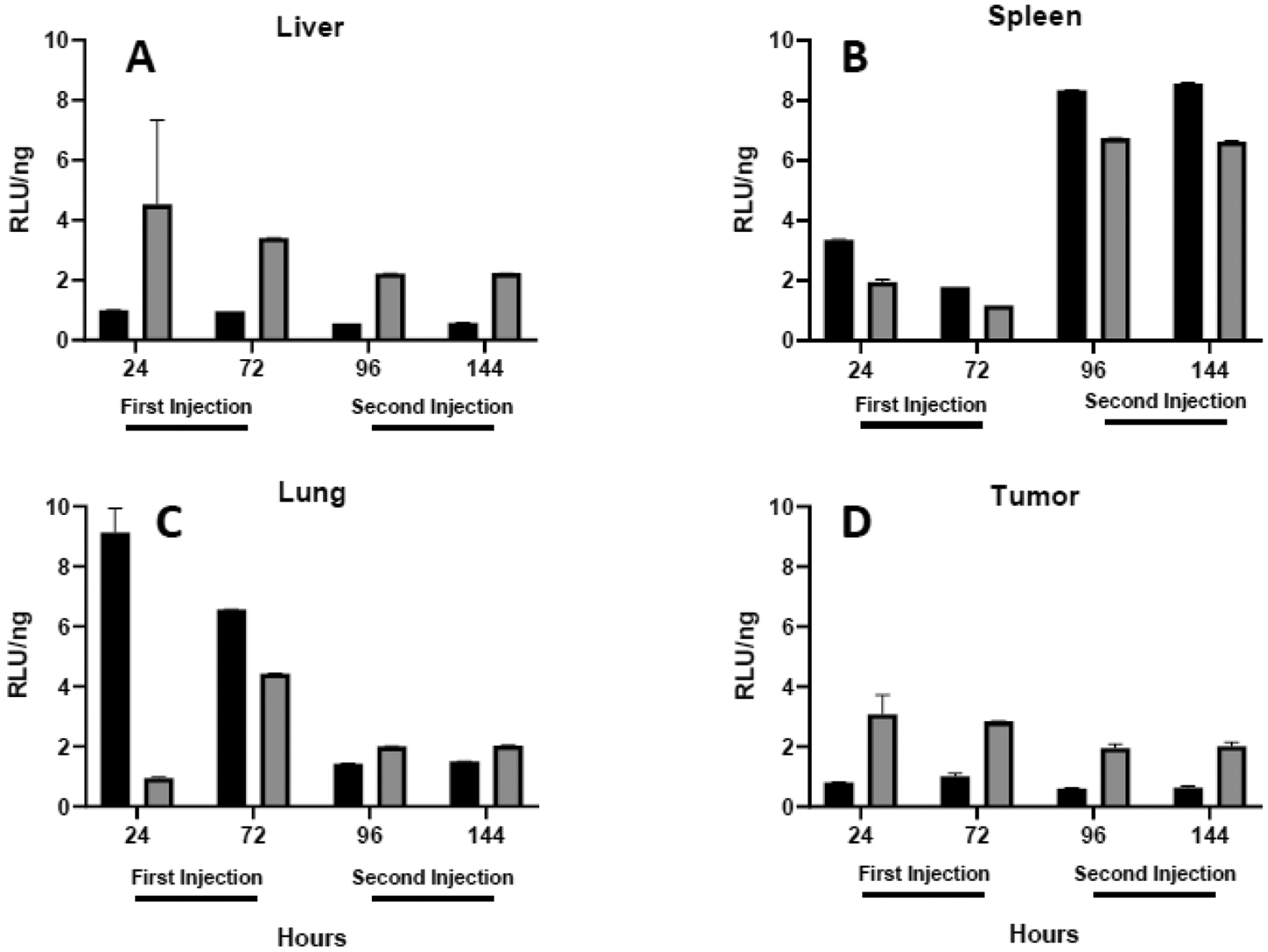
Expression efficiency in tissues. Luciferase expression in liver (A), spleen (B), lungs (C) or tumor (D) is standardized against the amount of plasmid to depict the efficiency with which plasmids are expressed 24 and 72 h after each intravenous injection. Each bar represents the mean ± one standard error of measurements on triplicate mice injected with lipoplexes coated with either 5% lactose (black) or PEG (shaded).
As seen in many gene delivery studies, we observed a poor correlation of plasmid levels with expression in vivo (Figs. 2 + 3). This poor correlation was even observed within individual tissues (Fig. 8). In an attempt to better understand these results, we performed in vitro transfection experiments in the same tumor cell line (CT26) that was implanted in our mouse experiments. To be consistent with our in vivo experiments, RTPCR was used to quantify plasmid levels in our cell preparations in which luciferase expression was also quantified at different times after a single treatment with lipoplexes (Fig. 9). It is interesting to note that < 10% of the plasmid applied to the cultured cells was present in the cells (after 4 h), and the amount of plasmid (per mg cellular protein) progressively decreased over 96 h (data not shown). In addition, our data are consistent with the well established abilities of PEG to inhibit interactions with cells and reduce transfection rates19, 64. When considered as expression efficiency (i.e., RLU/ng plasmid), it is evident that in vitro expression efficiency increases after 24 h in contrast to the relatively stable tumor values observed in vivo. Another striking difference is that the in vitro values are >1000-fold higher than those observed for the tumor in vivo (compare y axes in Figs. 8 + 9). These stark disparities between in vitro and in vivo expression efficiency suggest that the plasmid delivered to a tumor in a mouse exhibits fundamentally different behavior from that within cancer cells in culture.
Fig. 9.
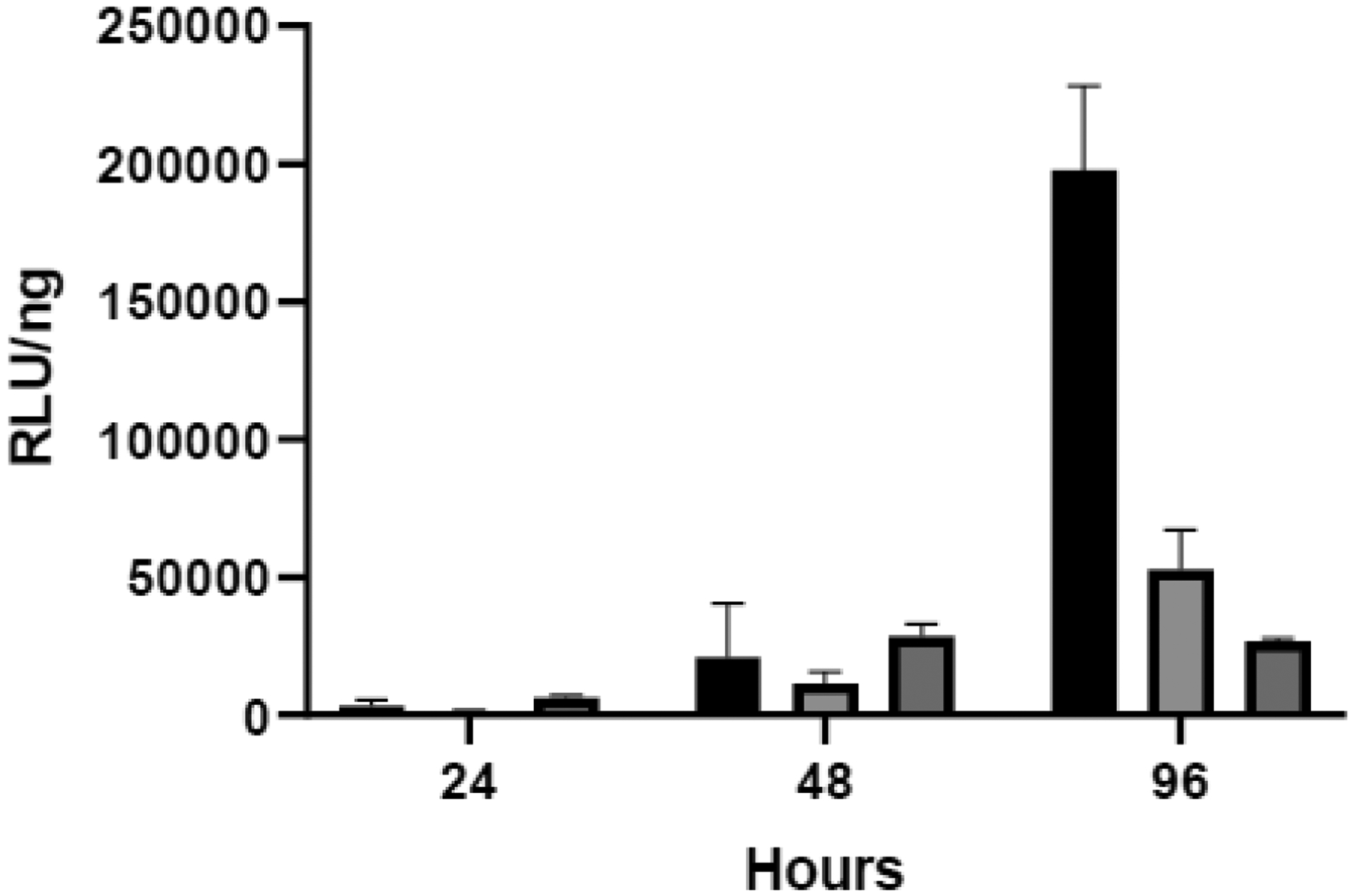
In vitro expression efficiency in CT26 cells. Luciferase expression in CT26 cells in culture was standardized against the amount of plasmid (assessed by qPCR by using 3 technical replicates per each of the three triplicate samples as is recommended by the manufacturer) to depict the efficiency after a single lipoplex treatment. Error was estimated by using the Taylor series expansion on both the qPCR and luminescence measurements. Each bar represents the mean ± one error of measurements on triplicate wells of cells incubated with uncoated lipoplexes (black bars) or lipoplexes coated with either 5% lactose (light shading) or PEG (dark shading).
DISCUSSION
It is well established that PEGylated nanoparticles can elicit an immune response that can cause repeat injections to exhibit accelerated blood clearance (ABC)22–24, 58. In addition to the potential for ABC to compromise delivery, high levels of pre-existing anti-PEG antibodies have been correlated with life-threatening infusion reactions16, 17. However, studies have established that ABC also occurs in immunocompromised animals that are incapable of producing antibodies, demonstrating that the innate immune response is involved in accelerated clearance24. Previous work has documented the extensive uptake of naked lipoplexes by circulating immune cells48, 54, 65, and our recent work has demonstrated that a lactose coating provides an equivalent reduction in leukocyte uptake and superior tumor accumulation to that observed with PEGylated lipoplexes21. We have previously shown that lactosylated lipoplexes elicit less of a cytokine response as compared to PEG, but the results in Figure 1 clearly indicate that ABC is also observed when lipoplexes are coated with lactose. Because both coatings elicited an elevated systemic cytokine response (Fig. 4), these results are consistent with previous studies showing a role for innate immunity in the ABC phenomenon24.
Considering the accelerated clearance discussed above, it would be expected that organ accumulation after the second injection would be limited to the relatively rapid deposition that occurs prior to clearance21, 46, 66. Not only did we observe reduced accumulation relative to the initial dose, our data indicate no additional deposition after the second injection in any of the clearance organs suggesting that repeat administration triggers very rapid processing and/or an alternative mechanism that bypasses these organs (Fig. 2). The stimulation of rapid processing by the organs is consistent with the sharp reduction in plasmid accumulation observed in the spleen and lung after repeat administration, but a similar reduction was not evident in the liver. This difference among these organs could potentially be explained by previous studies showing that prolonged presence in the liver indicates delivery to hepatocytes as opposed to macrophages67. It is important to note that we have previously observed considerable accumulation in each of these organs with uncoated lipoplexes that are cleared much more rapidly than the coated formulations used in this study21, 46, 48. Therefore, we conclude that the circulation time is sufficient to allow organ accumulation even under accelerated clearance conditions, yet we do not observe additional plasmid in liver, lung, or spleen after the second dose. In contrast to that observed in the clearance organs, additional accumulation is definitely evident in the tumor after repeat injection, further demonstrating that circulation times after the second injection are sufficient to allow supplemental deposition (Fig. 2). Accordingly, it is unclear if particle deposition after the second injection is preferentially cleared and/or if lipoplex uptake by these organs is inhibited during repeat administration.
Changes in reporter gene expression correlated poorly with changes in plasmid accumulation in the clearance organs (liver, spleen, and lung), but a better correlation was observed in the tumor (compare Figs 2+3). Considering previous work showing that cytokines can suppress gene expression, we performed ELISA assays to monitor the three main cytokines that have been correlated to changes in reporter gene expression32, 39. Although levels of specific cytokines can vary in response to foreign material, the levels of IFNγ, IL-6, and TNFα exhibited very similar trends and thereby serve as general indicators of the cytokine response. Unlike most prior studies, we measured both blood and tissue cytokines, and we observed striking differences, i.e., blood levels were generally 10–1000-fold lower than that seen in tissues and exhibited different trends (compare Figs. 4–7; see Table 2 for statistical analysis). These sharp differences between blood and tissue cytokines suggest that nanoparticle administration elicits disparate responses at the systemic and tissue level. However, the levels of cytokines in the tissue did not correlate with expression even after accounting for changing plasmid levels (Fig. 8), suggesting that other mechanisms are more directly involved in controlling reporter gene expression. This is best illustrated by the fact that expression efficiency in the tumor was comparable to or below that in other organs despite tumor cytokines being maintained at much lower levels throughout our study. Because cytokine production by infected cells is thought to be indicative of an antiviral response, our data may indicate that the plasmid alterations used in our study (mammalian promoter, minimal CpG) may be sufficient to circumvent these silencing mechanisms34, 37.
Finally, we conducted in vitro transfection experiments to determine how expression patterns within the same cancer cells differed in the absence of the complicated stromal environment and immune cells that are present in vivo. Although our cell culture experiments involved only a single treatment with lipoplexes, the trends in expression were markedly different from that observed in vivo. More specifically, expression per plasmid in vitro clearly increased after 24 h as would be expected from a rapidly dividing population of cells. It is important to point out that cell division allows plasmids to access the nucleus, and previous studies have demonstrated that gene expression from plasmids strongly correlates with mitotic activity33, 68, 69. Because we observe progressive increases in tumor size in vivo indicating that implanted CT26 cancer cells were rapidly dividing throughout our experiments, it would be expected that plasmid internalized by cancer cells is readily expressed. Even more striking than the time-dependent differences is the approximately 1000-fold higher level of expression efficiency observed in vitro as compared to in vivo (compare Figs. 8D + 9). It is important to recognize that our cell culture experiments involved removing unbound lipoplexes after 4 h and washing cells prior to assessing expression and plasmid levels. Accordingly, we assume that most, if not all, of the remaining lipoplexes have been internalized under in vitro conditions. It follows that the expression efficiency (RLU/ng plasmid) measured in our cell culture experiments represents that which would be observed if plasmid were internalized by rapidly dividing cancer cells. Therefore, the dramatically lower levels of expression we observe in vivo may indicate that a large majority of the plasmid measured in tissues may be extracellular, at least at the timepoints used in our experiments. Considering the strong association of lipoplexes with the negatively charged molecules present in the extracellular matrix70, we suggest that the extensive network of hyaluronic acid and glucosaminoglycans in vivo may hinder internalization of nanoparticles after extravasation from the blood. Another contributing factor could be that the vast majority of particles present in tumors after intravenous administration become associated with stromal cells that are not rapidly dividing11, 71. Therefore, plasmid expression from lipoplexes internalized by stromal cells would be minimal.
In conclusion, our results showing a distinct cytokine response to intravenous lipoplex administration are consistent with previous studies suggesting a role for an innate immune response in the accelerated blood clearance phenomenon observed after repeat administration24. In addition to reduced circulation times, this study documents a dramatic difference between the first and second intravenous injection in terms of biodistribution, expression, and cytokine response, and this was observed with lipoplexes coated with either lactose or PEG. The inability of the second injection to increase plasmid levels in the liver, lung, and spleen differs from our previous studies using uncoated lipoplexes, indicating that a response to the hydrophilic coating may contribute to these effects33. The fact that additional accumulation was evident in the tumor after repeat injection suggests that the immune response to our lipoplexes includes mechanisms that reduce uptake in clearance organs. This may be an important consideration when developing nucleic acid-based therapeutics for treating these organs (e.g., liver diseases), and prolonged dosing intervals may allow this response to dissipate. In this context, it is noteworthy that Onpattro® (an approved siRNA product targeting the liver) is dosed at 3-week intervals25. Conversely, the persistent ability of the tumor to accumulate lipoplexes may selectively permit higher expression to be achieved by repeat injection (relative to organs), consistent with our previous findings33. While the use of a lactose coating generally reduces the cytokine response as compared to PEGylated lipoplexes, changes in expression did not correlate with either blood or tissue cytokine levels. Surprisingly, the levels of blood cytokines do not correlate with tissue cytokines and are generally 100-fold lower. This suggests that the systemic immune response can differ markedly from that in individual tissues, which may be an important consideration for tissue-specific therapies. A comparison of in vivo and in vitro expression from the same cancer cells suggests that much of the plasmid in tumors is extracellular and/or internalized by slowly dividing cells in which expression is delayed/prevented. It is worth noting that we observe relatively constant expression efficiency in all tissues in contrast to previous studies that reported up to 100-fold greater efficiency in the tumor68, 72, 73.
Taken together, our results demonstrate that repeat injection of lipoplexes elicits an immune response that dramatically alters delivery, consistent with the well-established refractory period described in other studies39, 74, 75. These prior studies attributed the inability of repeat dosing to enhance luciferase activity to the production of inflammatory cytokines76, and our results indicate that mechanisms that directly restrict particle uptake contribute to the refractory period. Considering the findings from those prior studies, it is possible that the consistently lower levels of inflammatory cytokines in the tumor may contribute to the unimpeded delivery to that tissue after repeat injection (Table 2). We propose that studies utilizing repeat administration of nanoparticles should account for the stark differences as compared to an initial intravenous injection and be aware of the significantly-enhanced cytokine response at the tissue level.
Acknowledgments
This work was supported by grants from the ALSAM Foundation and the National Institutes of Health (RO1 GM129046).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors have no competing interests to declare.
References
- 1.Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal doxorubicin - Review of animal and human studies. Clin Pharmacokinet. 2003;42(5):419–436. [DOI] [PubMed] [Google Scholar]
- 2.Gabizon A, Price DC, Huberty J, Bresalier RS, Papahadjopoulos D. Effect of Liposome Composition and Other Factors on the Targeting of Liposomes to Experimental-Tumors - Biodistribution and Imaging Studies. Cancer Res. Oct 1 1990;50(19):6371–6378. [PubMed] [Google Scholar]
- 3.Smyth T, Kullberg M, Malik N, Smith-Jones P, Graner MW, Anchordoquy TJ. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. Journal of Controlled Release. Feb 10 2015;199:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Bradshaw-Pierce EL, Delille A, Gustafson DL, Anchordoquy TJ. In vivo comparative study of lipid/DNA complexes with different in vitro serum stability: effects on biodistribution and tumor accumulation. J Pharm Sci. Jan 2008;97(1):237–50. doi: 10.1002/jps.21076 [DOI] [PubMed] [Google Scholar]
- 5.Mates JM, Yao ZL, Cheplowitz AM, et al. Mouse Liver Sinusoidal Endothelium Eliminates HIV-Like Particles from Blood at a Rate of 100 Million per Minute by a Second-Order Kinetic Process. Front Immunol. Jan 24 2017;8doi:ARTN 35 10.3389/fimmu.2017.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukumori Y, Ichikawa H. Nanoparticles for cancer therapy and diagnosis. Adv Powder Technol. 2006;17(1):1–28. [Google Scholar]
- 7.Gabizon A, Papahadjopoulos D. Liposome Formulations with Prolonged Circulation Time in Blood and Enhanced Uptake by Tumors. Proceedings of the National Academy of Sciences of the United States of America. Sep 1988;85(18):6949–6953. doi:DOI 10.1073/pnas.85.18.6949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabizon AA. Selective Tumor-Localization and Improved Therapeutic Index of Anthracyclines Encapsulated in Long-Circulating Liposomes. Cancer Res. Feb 15 1992;52(4):891–896. [PubMed] [Google Scholar]
- 9.Park K The drug delivery field needs a well-diversified technology portfolio. J Control Release. Jan 10 2017;245:177. doi: 10.1016/j.jconrel.2016.12.010 [DOI] [PubMed] [Google Scholar]
- 10.Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nature Reviews. 2016;1(May 2016):1–12. [Google Scholar]
- 11.Dai Q, Wilhelm S, Ding D, et al. Quantifying the Ligand-Coated Nanoparticle Delivery to Cancer Cells in Solid Tumors. Acs Nano. Aug 2018;12(8):8423–8435. doi: 10.1021/acsnano.8b03900 [DOI] [PubMed] [Google Scholar]
- 12.Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WC. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. May 2009;9(5):1909–15. doi: 10.1021/nl900031y [DOI] [PubMed] [Google Scholar]
- 13.Sykes EA, Chen J, Zheng G, Chan WCW. Investigating the Impact of Nanoparticle Size on Active and Passive Tumor Targeting Efficiency. Acs Nano. Jun 2014;8(6):5696–5706. [DOI] [PubMed] [Google Scholar]
- 14.Dos Santos N, Allen C, Doppen AM, et al. Influence of poly(ethylene glycol) grafting density and polymer length on liposomes: Relating plasma circulation lifetimes to protein binding. Biochimica Et Biophysica Acta-Biomembranes. Jun 2007;1768(6):1367–1377. [DOI] [PubMed] [Google Scholar]
- 15.Anchordoquy TJ, Simberg D. Watching the gorilla and questioning delivery dogma. Journal of Controlled Release. Sep 28 2017;262:87–90. doi: 10.1016/j.jconrel.2017.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ganson NJ, Povsic TJ, Sullenger BA, et al. Pre-existing anti-polyethylene glycol antibody linked to first-exposure allergic reactions to pegnivacogin, a PEGylated RNA aptamer. Journal of Allergy and Clinical Immunology. May 2016;137(5):1610–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Povsic TJ, Lawrence MG, Lincoff AM, et al. Pre-existing anti-PEG antibodies are associated with severe immediate allergic reactions to pegnivacogin, a PEGylated aptamer. Journal of Allergy and Clinical Immunology. Dec 2016;138(6):1712–1715. [DOI] [PubMed] [Google Scholar]
- 18.Tillmann H, Ganson NJ, Patel K, et al. High Prevalence of Pre-Existing Antibodies against Polyethylene Glycol (Peg) in Hepatitis C (Hcv) Patients Which Is Not Associated with Impaired Response to Peg-Interferon. J Hepatol. 2010;52:S129–S129. [Google Scholar]
- 19.Verhoef JJF, Anchordoquy TJ. Questioning the use of PEGylation for drug delivery. Drug Delivery and Translational Research. Dec 2013;3(6):499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verhoef JJF, Carpenter JF, Anchordoquy TJ, Schellekens H. Potential induction of anti-PEG antibodies and complement activation toward PEGylated therapeutics. Drug Discovery Today. Dec 2014;19(12):1945–1952. [DOI] [PubMed] [Google Scholar]
- 21.Betker JL, Anchordoquy TJ. The Use of Lactose as an Alternative Coating for Nanoparticles. J Pharm Sci. Apr 2020;109(4):1573–1580. doi: 10.1016/j.xphs.2020.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dams ET, Laverman P, Oyen WJ, et al. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J Pharmacol Exp Ther. Mar 2000;292(3):1071–9. [PubMed] [Google Scholar]
- 23.Ishida T, Kiwada H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int J Pharm. Apr 16 2008;354(1–2):56–62. [DOI] [PubMed] [Google Scholar]
- 24.Semple SC, Harasym TO, Clow KA, Ansell SM, Klimuk SK, Hope MJ. Immunogenicity and rapid blood clearance of liposomes containing polyethylene glycol-lipid conjugates and nucleic acid. Journal of Pharmacology and Experimental Therapeutics. Mar 2005;312(3):1020–1026. [DOI] [PubMed] [Google Scholar]
- 25.Onpattro PI. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210922s000lbl.pdf
- 26.Yang Q, Jacobs TM, McCallen JD, et al. Analysis of Pre-existing IgG and IgM Antibodies against Polyethylene Glycol (PEG) in the General Population. Anal Chem. Dec 6 2016;88(23):11804–11812. doi: 10.1021/acs.analchem.6b03437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang P, Sun F, Liu SJ, Jiang SY. Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation. Journal of Controlled Release. Dec 28 2016;244:184–193. doi: 10.1016/j.jconrel.2016.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abu Lila AS, Nawata K, Shimizu T, Ishida T, Kiwada H. Use of polyglycerol (PG), instead of polyethylene glycol (PEG), prevents induction of the accelerated blood clearance phenomenon against long-circulating liposomes upon repeated administration. Int J Pharm. Nov 1 2013;456(1):235–242. doi: 10.1016/j.ijpharm.2013.07.059 [DOI] [PubMed] [Google Scholar]
- 29.Park K Attenuating the immunogenicity of PEGylated liposomes by gangliosides. Journal of Controlled Release. Mar 28 2017;250:116–116. doi: 10.1016/j.jconrel.2017.03.002 [DOI] [PubMed] [Google Scholar]
- 30.Zhang P, Sun F, Tsao C, et al. Zwitterionic gel encapsulation promotes protein stability, enhances pharmacokinetics, and reduces immunogenicity. Proceedings of the National Academy of Sciences of the United States of America. Sep 29 2015;112(39):12046–12051. doi: 10.1073/pnas.1512465112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abrams MT, Koser ML, Seitzer J, et al. Evaluation of efficacy, biodistribution, and inflammation for a potent siRNA nanoparticle: effect of dexamethasone co-treatment. Mol Ther. Jan 2010;18(1):171–80. doi: 10.1038/mt.2009.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alaaeldin E, Abu Lila AS, Moriyoshi N, et al. The Co-Delivery of Oxaliplatin Abrogates the Immunogenic Response to PEGylated siRNA-Lipoplex. Pharm Res. Sep 2013;30(9):2344–2354. [DOI] [PubMed] [Google Scholar]
- 33.Betker JL, Anchordoquy TJ. Nonadditive Effects of Repetitive Administration of Lipoplexes in Immunocompetent Mice. J Pharm Sci. Mar 2017;106(3):872–881. doi: 10.1016/j.xphs.2016.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan YK, Gack MU. Viral evasion of intracellular DNA and RNA sensing. Nature Reviews Microbiology. Jun 2016;14(6):360–373. doi: 10.1038/nrmicro.2016.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan YK, Gack MU. RIG-I-like receptor regulation in virus infection and immunity. Curr Opin Virol. Jun 2015;12:7–14. doi: 10.1016/j.coviro.2015.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiang JJ, Davis ME, Gack MU. Regulation of RIG-I-like receptor signaling by host and viral proteins. Cytokine Growth Factor Rev. Oct 2014;25(5):491–505. doi: 10.1016/j.cytogfr.2014.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nature Reviews Immunology. Sep 2020;20(9):537–551. doi: 10.1038/s41577-020-0288-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hassett KJ, Benenato KE, Jacquinet E, et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol Ther-Nucl Acids. Apr 15 2019;15:1–11. doi: 10.1016/j.omtn.2019.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindberg MF, Le Gall T, Carmoy N, et al. Efficient in vivo transfection and safety profile of a CpG-free and codon optimized luciferase plasmid using a cationic lipophosphoramidate in a multiple intravenous administration procedure. Biomaterials. Aug 2015;59:1–11. [DOI] [PubMed] [Google Scholar]
- 40.Morrissey D, van Pijkeren JP, Rajendran S, et al. Control and Augmentation of Long-Term Plasmid Transgene Expression In Vivo in Murine Muscle Tissue and Ex Vivo in Patient Mesenchymal Tissue. J Biomed Biotechnol. 2012;doi:Artn 379845 10.1155/2012/379845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morrissey D, Collins S, Casey G, O’Sullivan GC, Tangney M. Variables affecting duration of plasmid transgene transcription in vivo. Hum Gene Ther. Apr 2008;19(4):413–413. [Google Scholar]
- 42.Brito LA, Chan M, Shaw CA, et al. A Cationic Nanoemulsion for the Delivery of Next-generation RNA Vaccines. Mol Ther. Dec 2014;22(12):2118–2129. doi: 10.1038/mt.2014.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamamoto A, Kormann M, Rosenecker J, Rudolph C. Current prospects for mRNA gene delivery. Eur J Pharm Biopharm. Mar 2009;71(3):484–489. doi: 10.1016/j.ejpb.2008.09.016 [DOI] [PubMed] [Google Scholar]
- 44.Sellins K, Fradkin L, Liggitt D, Dow S. Type I Interferons potently suppress gene expression following gene delivery using liposome-DNA complexes. Mol Ther. Sep 2005;12(3):451–459. doi: 10.1016/j.ymthe.2005.04.008 [DOI] [PubMed] [Google Scholar]
- 45.Betker JL, Anchordoquy TJ. Relating toxicity to transfection: using sphingosine to maintain prolonged expression in vitro. Mol Pharm. Jan 5 2015;12(1):264–73. doi: 10.1021/mp500604r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Betker JL, Anchordoquy TJ. Assessing the effect of a nude mouse model on nanoparticle-mediated gene delivery. Drug Deliv Transl Res. Feb 2017;7(1):162–167. doi: 10.1007/s13346-016-0327-6 [DOI] [PubMed] [Google Scholar]
- 47.Betker JL, Gomez J, Anchordoquy TJ. The effects of lipoplex formulation variables on the protein corona and comparisons with in vitro transfection efficiency. J Control Release. Nov 10 2013;171(3):261–8. doi: 10.1016/j.jconrel.2013.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Betker JL, Jones D, Childs CR, et al. Nanoparticle uptake by circulating leukocytes: A major barrier to tumor delivery. Journal of Controlled Release. Sep 28 2018;286:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watcharanurak K, Nishikawa M, Takahashi Y, Takakura Y. Controlling the kinetics of interferon transgene expression for improved gene therapy. J Drug Target. Nov 2012;20(9):764–9. doi: 10.3109/1061186X.2012.716848 [DOI] [PubMed] [Google Scholar]
- 50.Xu L, Betker J, Yin H, Anchordoquy TJ. Ligands located within a cholesterol domain enhance gene delivery to the target tissue. J Control Release. May 30 2012;160(1):57–63. doi: 10.1016/j.jconrel.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fete MG, Betker JL, Shoemaker RK, Anchordoquy TJ. A Novel Method for Conjugating the Terminal Amine of Peptide Ligands to Cholesterol: Synthesis iRGD-Cholesterol. Ther Deliv. 2019;10(1):11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. Jul 1999;6(7):1258–66. doi: 10.1038/sj.gt.3300947 [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Anchordoquy TJ. The role of lipid charge density in the serum stability of cationic lipid/DNA complexes. Biochim Biophys Acta. May 27 2004;1663(1–2):143–57. doi: 10.1016/j.bbamem.2004.03.004 [DOI] [PubMed] [Google Scholar]
- 54.Thierry AR, Rabinovich P, Peng B, Mahan LC, Bryant JL, Gallo RC. Characterization of liposome-mediated gene delivery: expression, stability and pharmacokinetics of plasmid DNA. Gene Ther. Mar 1997;4(3):226–37. doi: 10.1038/sj.gt.3300350 [DOI] [PubMed] [Google Scholar]
- 55.Betker JL, Anchordoquy TJ. Effect of charge ratio on lipoplex-mediated gene delivery and liver toxicity. Ther Deliv. 2015;6(11):1243–1253. [DOI] [PubMed] [Google Scholar]
- 56.Kawabata K, Takakura Y, Hashida M. The Fate of Plasmid DNA after Intravenous-Injection in Mice - Involvement of Scavenger Receptors in Its Hepatic-Uptake. Pharm Res. Jun 1995;12(6):825–830. doi:Doi 10.1023/A:1016248701505 [DOI] [PubMed] [Google Scholar]
- 57.Betker JL, Kullberg M, Gomez J, Anchordoquy TJ. Cholesterol domains enhance transfection. Ther Deliv. Apr 2013;4(4):453–62. doi: 10.4155/tde.13.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ishida T, Ichihara M, Wang X, et al. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J Control Release. May 1 2006;112(1):15–25. doi: 10.1016/j.jconrel.2006.01.005 [DOI] [PubMed] [Google Scholar]
- 59.Judge A, McClintock K, Phelps JR, MacLachlan I. Hypersensitivity and loss of disease site targeting caused by antibody responses to PEGylated liposomes. Mol Ther. Feb 2006;13(2):328–337. doi: 10.1016/j.ymthe.2005.09.014 [DOI] [PubMed] [Google Scholar]
- 60.Fajgenbaum DC, June CH. Cytokine Storm. New England Journal of Medicine. Dec 3 2020;383(23):2255–2273. doi: 10.1056/NEJMra2026131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Q, Yu SN, Li AP, Xu HX, Han XW, Wu KM. Targeting interlukin-6 to relieve immunosuppression in tumor microenvironment. Tumor Biol. Jun 22 2017;39(6) [DOI] [PubMed] [Google Scholar]
- 62.Schlosser HA, Theurich S, Shimabukuro-Vornhagen A, Holtick U, Stippel DL, von Bergwelt-Baildon M. Overcoming tumor-mediated immunosuppression. Immunotherapy. 2014;6(9):973–988. [DOI] [PubMed] [Google Scholar]
- 63.Wang DZ, DuBois RN. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis. Oct 2015;36(10):1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harvie P, Wong FMP, Bally MB. Use of poly(ethylene glycol)-lipid conjugates to regulate the surface attributes and transfection activity of lipid-DNA particles. J Pharm Sci. May 2000;89(5):652–663. [DOI] [PubMed] [Google Scholar]
- 65.McLean JW, Fox EA, Baluk P, et al. Organ-specific endothelial cell uptake of cationic liposome-DNA complexes in mice. American Journal of Physiology-Heart and Circulatory Physiology. Jul 1997;273(1):H387–H404. [DOI] [PubMed] [Google Scholar]
- 66.Barron LG, Gagne L, Szoka FC. Lipoplex-mediated gene delivery to the lung occurs within 60 minutes of intravenous administration. Hum Gene Ther. Jul 1 1999;10(10):1683–1694. [DOI] [PubMed] [Google Scholar]
- 67.Scherphof G, Roerdink F, Dijkstra J, Ellens H, Dezanger R, Wisse E. Uptake of Liposomes by Rat and Mouse Hepatocytes and Kupffer Cells. Biol Cell. 1983;47(1):47–57. [Google Scholar]
- 68.Ambegia E, Ansell S, Cullis P, Heyes J, Palmer L, MacLachlan I. Stabilized plasmid-lipid particles containing PEG-diacylglycerols exhibit extended circulation lifetimes and tumor selective gene expression. Biochimica Et Biophysica Acta-Biomembranes. May 20 2005;1669(2):155–163. [DOI] [PubMed] [Google Scholar]
- 69.Wiethoff CM, Middaugh CR. Barriers to nonviral gene delivery. J Pharm Sci. Feb 2003;92(2):203–217. [DOI] [PubMed] [Google Scholar]
- 70.Wiethoff CM, Smith JG, Koe GS, Middaugh CR. The potential role of proteoglycans in cationic lipid-mediated gene delivery - Studies of the interaction of cationic lipid-DNA complexes with model glycosaminoglycans. J Biol Chem. Aug 31 2001;276(35):32806–32813. doi:DOI 10.1074/jbc.M007940200 [DOI] [PubMed] [Google Scholar]
- 71.Miao L, Newby JM, Lin CM, et al. The Binding Site Barrier Elicited by Tumor Associated Fibroblasts Interferes Disposition of Nanoparticles in Stroma-Vessel Type Tumors. Acs Nano. Oct 2016;10(10):9243–9258. doi: 10.1021/acsnano.6b02776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heyes J, Palmer L, Chan K, Giesbrecht C, Jeffs L, MacLachlan I. Lipid encapsulation enables the effective systemic delivery of polyplex plasmid DNA. Mol Ther. Apr 2007;15(4):713–720. [DOI] [PubMed] [Google Scholar]
- 73.Hatakeyama H, Akita H, Harashima H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: A strategy for overcoming the PEG dilemma. Adv Drug Del Rev. Mar 18 2011;63(3):152–160. doi: 10.1016/j.addr.2010.09.001 [DOI] [PubMed] [Google Scholar]
- 74.Li S, Wu SP, Whitmore M, et al. Effect of immune response on gene transfer to the lung via systemic administration of cationic lipidic vectors. Am J Physiol-Lung C. May 1999;276(5):L796–L804. [DOI] [PubMed] [Google Scholar]
- 75.Song YK, Liu F, Chu SY, Liu DX. Characterization of cationic liposome-mediated gene transfer in vivo by intravenous administration. Hum Gene Ther. Sep 1 1997;8(13):1585–1594. [DOI] [PubMed] [Google Scholar]
- 76.Tan YD, Li S, Pitt BR, Huang L. The inhibitory role of CpG immunostimulatory motifs in cationic lipid vector-mediated transgene expression in vivo. Hum Gene Ther. Sep 1 1999;10(13):2153–2161. [DOI] [PubMed] [Google Scholar]