

Abstract
Autophagy is a well-conserved self-eating mechanism of cell survival during periods of nutrient deprivation, stress and injury. Autophagy is implicated in many pathophysiological conditions across all organ systems. The cornea is an avascular transparent tissue that is prone to damage by trauma, injury and infection. Following insult, the cornea undergoes a complex wound healing process, which is regulated by multiple factors including autophagy. The involvement of autophagy in keratoconus and HSV-1 infection has been demonstrated, underlining the importance of this mechanism in corneal disorders. However, the role of autophagy in corneal wound repair, fibrosis and angiogenesis is still unclear. Recently, we characterized the expression of autophagy-related genes in cornea and are studying their role in the modulation of corneal conditions including fibrosis and dystrophies. Preliminary results presented within this review article support further investigation of the dynamic modulation of autophagy-related genes in corneal health and disease. This article provides an overview of how autophagy modulates corneal function.
Keywords: Autophagy, Cornea, Stroma, Corneal fibrosis, Keratoconus, Angiogenesis, Autophagy dysregulation, Atg
1. Introduction
Autophagy (also known as autophagocytosis) is a well-orchestrated, self-regulated intracellular degradative mechanism of cytoplasmic proteins and cellular organelles through the lysosomal machinery. Autophagy (“auto” -self and “phagein” -to eat), a self-eating cellular machinery, was initially identified in the 1960s. Recent studies have found autophagy crucial for normal homeostasis and healthy functioning of cells. The primary homeostatic roles of autophagy include expelling and recycling of obsolete organelles and unessential, harmful and misfolded proteins [1]. Autophagy involves bulk as well as selective degradation of cytoplasmic proteins and cellular organelles into peptides and amino acids for favorable use by the cell [2].
Dysregulated autophagy has been implicated in many biological and pathological conditions with either increased or decreased autophagic events. For example, autophagy pathways are stimulated in cells during nutrient deprivation and stress conditions including growth factor deprivation, glucose deprivation, reduced oxygen availability and compromised normal homeostasis (Fig. 1) [3,4]. It is important to note that cultivated cells in which autophagy had been increased by nutrient deprivation can reestablish normal cellular volume, proliferative capacity and basal levels of autophagy following nutrient replenishment. Basal autophagy is imperative for cell recovery in the face of adversity [4]. Moreover, lysosomal storage disorders result from inhibited autophagy (accumulation of intracellular proteins) whereas stimulated autophagy is important for the growth of some neoplastic tumors (maintenance of cancer cell viability) [5,6]. Autophagy is also important as a defense against intracellular pathogens [7]. In this regard, autophagy leads to the production of autophagosomes by engulfing invading intracellular pathogens (xenophagy) and fusing with lysosomes to facilitate their destruction and eventual elimination. Nevertheless, some viruses and bacteria are able to resist degradation by lysosomes and employ autophagosomes and/or lysosomes as sites for replication [7].
Fig. 1. Schematic showing the mechanism of action of autophagy after cell injury.
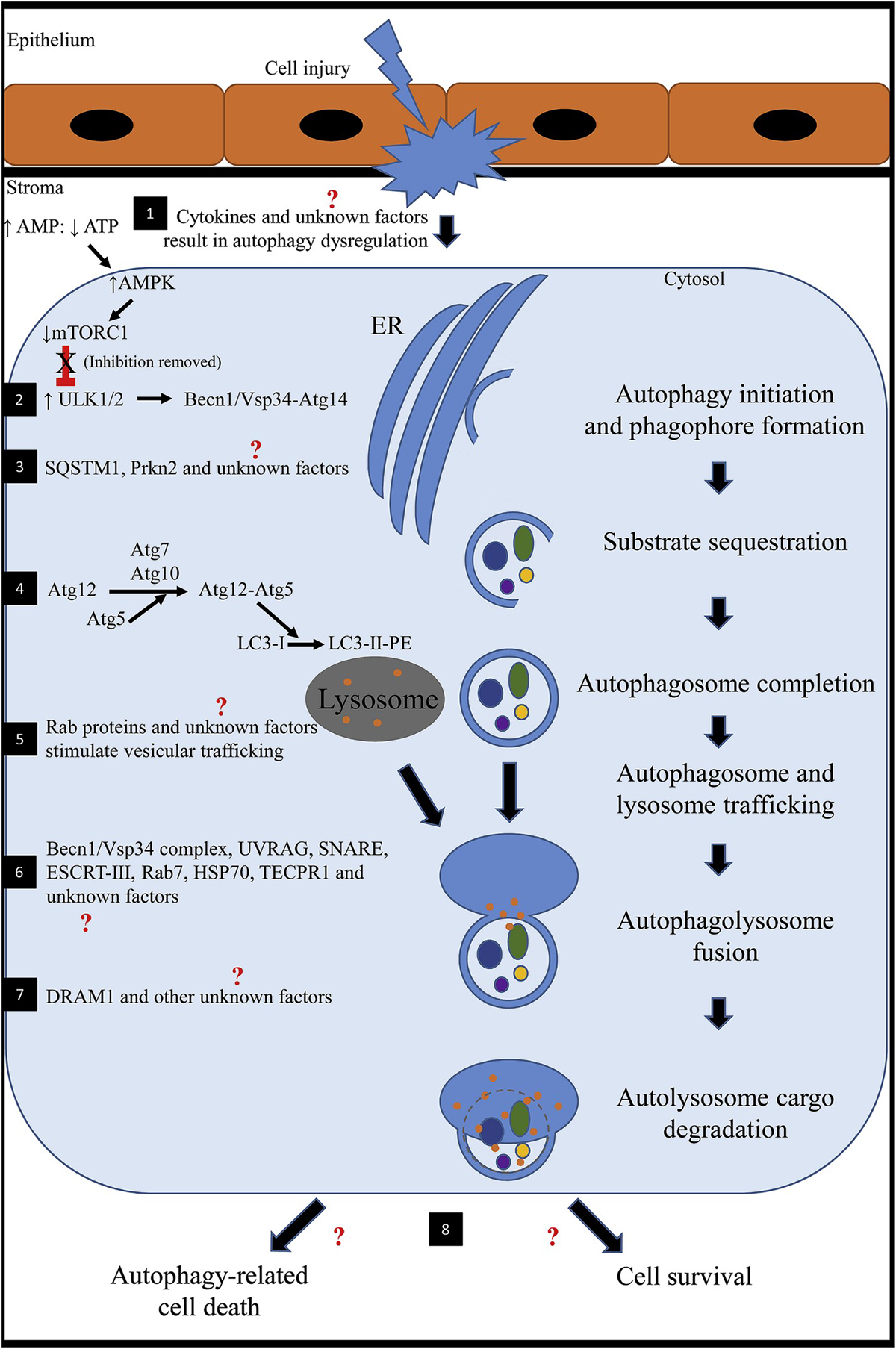
1) Following stress (injury to the corneal epithelium and underlying stroma), nutrient deprivation or hypoxia, cytokines (such as TNFα, IL-1, TGFβ) are released and initiate a cascade of signaling pathways. It is unknown how these factors specifically affect autophagy in the cornea. An altered ratio of AMP to ATP results in upregulation of AMPK. Increased AMPK inhibits mTOR1. In health, mTOR1 represses autophagy because there are sufficient nutrients for the cell to survive. AMPK-inhibition of mTOR1 activates ULK1 (mammalian homologue of Atg1). 2) ULK1 stimulates BECN1 to complex with Vsp34-Atg14 to regulate and initiate formation of the nascent autophagosome (phagophore) at the ER in response to stress signaling pathways. 3) SQSTM1, PRKN2 and other unknown factors selectively target obsolete organelles, misfolded proteins and fragmented/damaged organelles for degradation. 4) Atg7 and Atg10 facilitates Atg5 to conjugate with Atg12. The Atg12-Atg5 complex stimulates LC3-I (cytosolic form) conjugation to PE to form an LC3-II-PE complex that binds the surface of the expanding phagophore. Lipids are continually supplied until the autophagosomal membrane is a double-membraned enclosed structure. 5) The newly formed autophagosome and lysosome traffic toward one another for fusion. In general, Rab GTPases have been found to be involved with intracellular vesicular trafficking. Transport typically involves three steps including 1) budding of a vesicle from the donor membrane, 2) selective identification of the vesicle to the acceptor membrane and 3) docking and fusion with the selected membrane. Rab proteins have not been investigated concerning autophagy in the cornea. 6) A number of factors have been identified to be involved in autophagosome-lysosome fusion; however, many have not been identified in the cornea. Known factors include BECN1/Vsp34 complex, UVRAG, SNARE, ESCRT-III, Rab 7, HSP70 and TECPR1. 7) After fusion, the autolysosome undergoes a series of changes including acidification of the encircled environment and release of degradative enzymes. DRAM1 has been implicated and promotes the afore mentioned processes. 8) Ultimately, the degraded protein products are predominantly recycled into peptides and amino acids for use by the cell or the cell might undergo autophagy-related cell death. The latter is a somewhat similar process to apoptosis; however, autophagy-related cell death is not well understood and might be implicated in disease development.
(AMP: adenosine monophosphate; AMPK: 5′ AMP-activated protein kinase; Atg: autophagy-related protein; ATP: adenosine triphosphate; BECN1: Beclin 1; DRAM1: DNA damage regulated autophagy modulator 1; ER: endoplasmic reticulum; ESCRT: endosomal sorting complex required for transport; HSP70: heat shock protein 70; IL-1: interleukin 1; LC3: microtubule associated protein 1 light chain 3 alpha; mTORC1: mammalian target of rapamycin complex 1; PE: phosphatidylethanolamine; PRKN2: E3 ubiquitin-protein ligase parkin; Rab: ras-related GTP-binding protein; SNARE: soluble N-ethylmaleimide-sensitive fusion attachment protein receptors; SQSTM1: sequestosome 1; TECPR1: tectonin beta-propeller repeat containing 1; TGFβ: transforming growth factor beta; TNFα: tumor necrosis factor alpha; ULK1: unc-51-like kinase 1; UVRAG: ultraviolet irradiation resistance-associated gene; Vsp34: vesicular protein sorting 34).
Properly regulated autophagy is critical for the health and survival of mammalian cells [1,5]. Novel therapeutic approaches are being developed that serve to either control autophagy or to restore homeostasis in the face of dysregulated autophagy, as occurs in various disease states, including those that affect the mammalian cornea. Therefore, improved understanding regarding how autophagy regulates external stressors is needed before therapeutic approaches to autophagy modulation in ocular disease can be investigated and harnessed.
In addition to the oxidative and endoplasmic reticulum (ER) stress, many signaling molecules including mTOR1, 5′ adenosine monophosphate-activated protein kinase (AMPK) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) have been found to be involved in the regulation of autophagy [8,9]. The role of these signaling pathways and their interactions in the normal and diseased corneas is still largely unknown at this time.
Though the role of autophagy in ocular health has been documented by few studies [1,2,8,10–14]; little has been published regarding its role in corneal function specifically. The cornea provides two thirds of the refractive power to the eye, and corneal diseases are a major cause of blindness worldwide [15]. Injury (physical, chemical, infectious or iatrogenic) to the corneal epithelium and underlying stroma typically triggers a highly orchestrated wound healing process and results in regeneration of normal corneal structure and function. After an insult, a battery of cytokines released from the corneal epithelium and lacrimal glands arrive at the site of corneal damage and activate transparent quiescent stromal keratocytes to become fibroblasts. Under the influence of transforming growth factor β (TGFβ), fibroblasts transdifferentiate into metabolically active, opaque, light-scattering corneal myofibroblasts to facilitate wound repair by synthesizing and secreting large amounts of extracellular matrix components (ECM), collagens, and α-smooth muscle actin (αSMA) stress fibers. The excessive and prolonged production of ECM components by myofibroblasts leads to the development of fibrosis and neovascularization in the cornea, and resultant compromised vision [16–18].
Recent studies have shown the potential therapeutic applications of autophagy in controlling specific ophthalmic conditions [8–10,19–21]. For example, rapamycin, a well-known autophagy inducer and mTOR inhibitor, has been successfully used for the treatment of age-related macular degeneration (AMD), Fuchs dystrophy and keratoconus [8–10,19,20]. However, further studies are required to elucidate the role of pharmacological manipulation of autophagy dynamics in a wider variety of ocular diseases. Results of only a few studies have uncovered the importance of autophagy and its dynamics in diseases of the cornea, thus the discovery of novel regulators and pharmacological agents that target autophagy machinery might have greater clinical applicability [8–10,21].
In light of the fact that autophagy plays an important role in ocular diseases, a literature review was conducted using the keywords: “autophagy” and “cornea” from the PubMed Central and Google Scholar. This article presents an overview of the role of autophagy in corneal health maintenance and disease modulation.
2. Autophagy
2.1. Autophagy mechanisms
Autophagy is classified into three distinct categories: macro-autophagy, micro-autophagy and chaperone-mediated autophagy (Fig. 2). Macro-autophagy is a process whereby cellular material is either non-selectively or selectively enclosed into double-membraned vesicles (autophagosomes) that fuse with lysosomes creating autolysosomes (Fig. 3). Formation of autophagosomes is the result of a highly conserved process involving at least 31 autophagy-related proteins (Atg). The generation of autophagosomes entails three major steps including, 1) vesicle nucleation (formation), 2) vesicle elongation (expansion) and 3) vesicle completion (fusion). Dedicated forms of macro-autophagy include mitophagy (mitochondria), xenophagy (microbe) and lipophagy (lipid droplet) [1,2,22]. For the purposes of this paper, macro-autophagy will be referred to as autophagy. Micro-autophagy results in the sequestration of cellular structures by way of direct invagination of the lysosomal membrane. Both macro- and micro-autophagy are able to facilitate the transport of large structures, such as organelles. Chaperone-mediated autophagy involves specifically targeted proteins that are complexed with a chaperone protein. The lysosomal membrane receptor lysosomal-associated membrane protein 2A (LAMP2A) recognizes the chaperone protein complex, and the targeted protein is unfolded and translocated across the lysosomal membrane. The need for protein unfolding precludes the transport of large structures via chaperone-mediated autophagy [1,2,23–25].
Fig. 2. The three major degradative autophagy pathways.
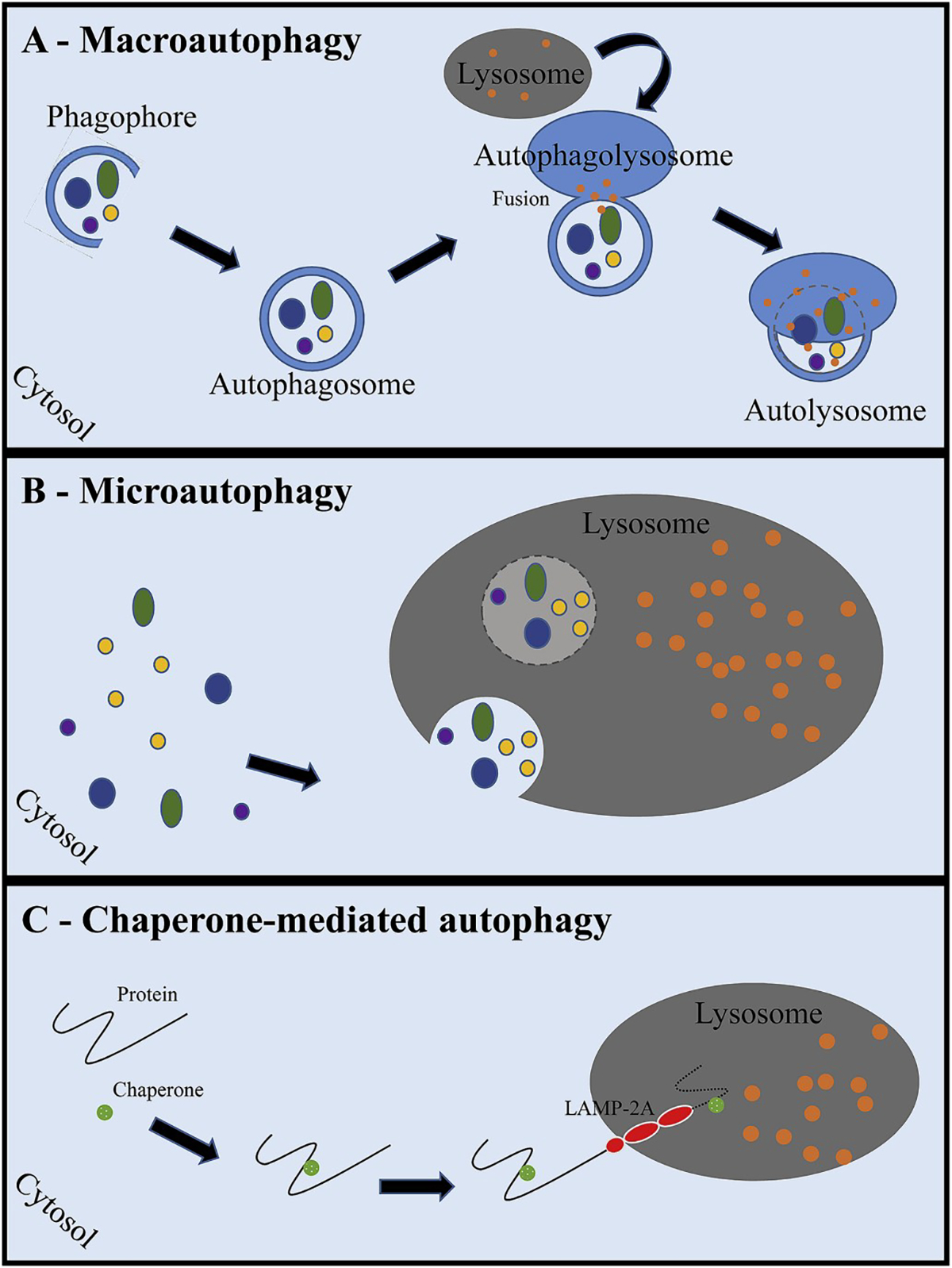
In macro-autophagy (A), cytosolic components are engulfed into a double-membraned phagophore. Completion of the double-membrane with sequestered cargo is called an autophagosome. The autophagosome then fuses with the lysosome to create an autophagolysosome. Lysosomal degradative granules then break down the cargo within the autolysosome. Eventually, the degraded protein products are released into the cytosol for use by the cell. Microautophagy (B) is a process by which the lysosome directly engulfs cytosolic components by way of lysosomal membrane invagination, similar to endocytosis. A chaperone binds to proteins within the cytosol that have been targeted for degradation (C), and the lysosomal-associated membrane protein-2A (LAMP-2A) recognizes and binds the protein complex. The protein is then unfolded and translocated into the lysosome.
(LAMP-2A: lysosomal associated membrane protein-2A).
Fig. 3. Basic steps of autolysosome formation.
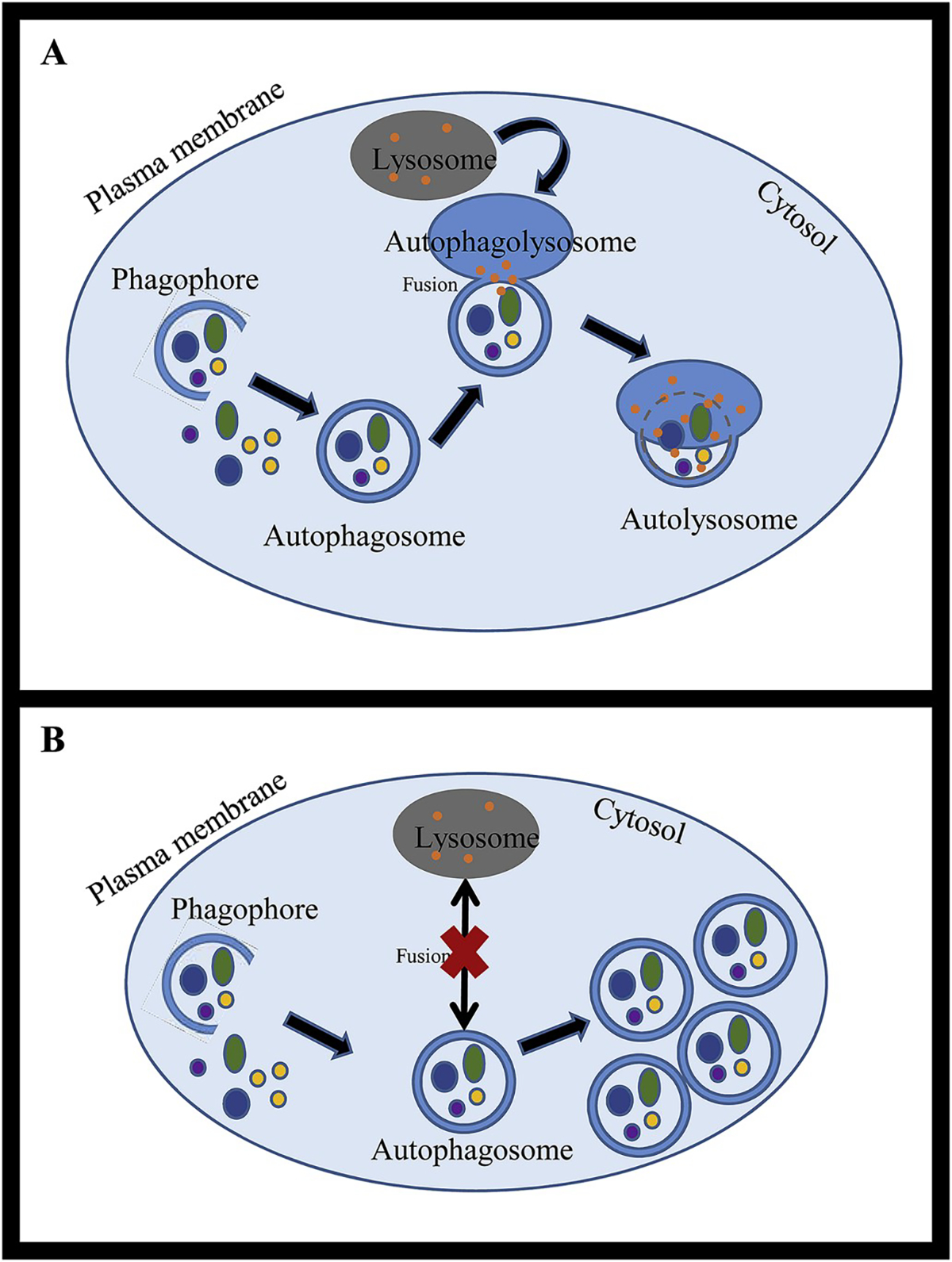
Within the cytoplasm, a double-membraned phagophore begins to form and encircles obsolete organelles and misfolded proteins for autophagy degradation. Once the double-membrane structure is complete, it is called an autophagosome. The autophagosome fuses with the lysosome (autophagolysosome) and degradative lysosomal granules break down the cargo (autolysosome). This complete degradative process is called autophagy flux (A). Autophagy dysregulation results in impaired autophagy flux. This might result in accumulation of autophagosomes due to lack of lysosomal fusion (B), slowed autophagy flux or impaired function downstream to autophagolysosomal formation.
2.2. Selective autophagy
Initially, autophagy was described as a non-selective degradative mechanism considering the fact that it allowed elimination of large cytoplasmic protein cargoes with no specific target protein or organelles. Subsequent studies revealed the presence of specific cellular receptors for autophagy. The selective autophagosomal degradation of mitochondria and other specific organelles, proteins or pathogens led scientists to coin a new term “selective autophagy”. Succeeding research classified selective autophagy into two broad classes: ubiquitin-dependent and ubiquitin-independent selective autophagy based on the interactions of autophagy receptors with cellular targets. The process was considered ubiquitin-dependent autophagy if an autophagy receptor interacted with ubiquitinated cargo via their ubiquitin binding domain. Conversely, the process in which autophagy receptors interacted directly with a cargo of different proteins or organelles was referred as ubiquitin-independent autophagy.
2.3. Autophagy assays
Autophagy assays measure either flux or steady state [26]. It is important to understand the differences between these two states. Autophagy flux encompasses the entire functional autophagy pathway from phagophore formation to degradation within the autolysosome [26]. The use of various inducers or inhibitors of lysosomal fusion aid in measures of autophagy flux. Key proteins in this process are sequestosome 1 (SQSTM1/p62), microtubule-associated protein 1A/1B-light chain 3 (LC3), lysosomal-associated membrane protein 1 (LAMP1), Beclin 1 (BECN1), vacuolar adenosine triphosphatase (V-ATPase) and many others [26]. Assays that measure steady state assess the form, level and location of pathway proteins at a single point in time of the autophagy pathway (Fig. 4) [26]. Steady state assays do not provide information about autophagy pathway conclusion [26,27]. Note that others have described autophagy flux as “complete autophagy” and autophagy steady state as “incomplete autophagy”. Terminology continues to evolve [26].
Fig. 4. Schematic of normal autophagy function (center) and the various points of failure that might result in autophagy dysfunction.
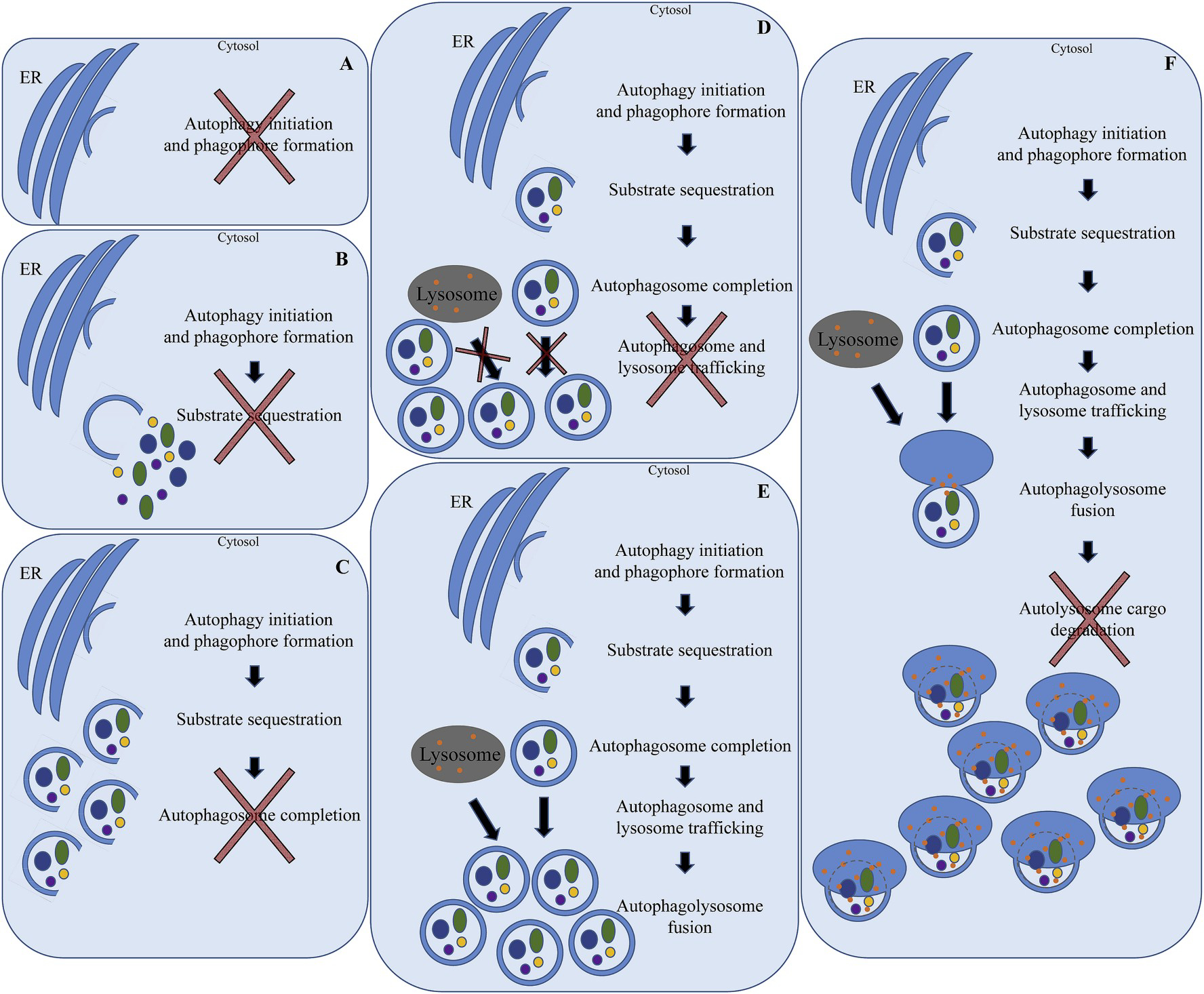
A) Failure of initiation cell signaling pathways to start phagofore formation. This might also occur due to lack of substrate from the endoplasmic reticulum. B) Failure of sequestration of cargo into the forming autophagosome. This might occur due to failed targeting of selected substrates or failed signaling and trafficking of selected substrates. C) Failure of complete double-membrane synthesis (elongation) of the developing autophagosome. This might result in either accumulation of partially formed autophagosomes or failed synthesis of the initial forming autophagosome. D) Failure of lysosome and autophagosome vesicular trafficking, which results in accumulation of autophagosomes. E) Failure to fuse the lysosome and autophagosome, which results in accumulation of autophagosomes. F) Failure to degrade autolysosomal sequestered cargo, which results in accumulation of autolysosomes.
(ER: endoplasmic reticulum).
Knowing the difference between autophagy flux and steady state is critical to understanding the role of autophagy in both health and disease and in developing either therapeutic targets or methods to stage or monitor disease progression. Klionsky et al. provide a thorough review of specific assays that are beneficial for determining steady state and flux, including commonly encountered challenges [26]. To monitor the steady state of autophagy, the following assays were recommended by those authors: 1) electron microscopy, 2) LC3A/B western blotting and ubiquitin-like protein conjugation systems, 3) fluorescence microscopy, 4) mTOR1 activation and Atg1 kinase activity and 5) transcriptional regulation (assessing levels of LC3, Rab proteins and Atg messenger ribonucleic acid [mRNA] by northern blot or quantitative real-time polymerase chain reaction [qRT-PCR]). To monitor flux states, the following assays were recommended: 1) autophagy protein degradation, 2) turnover of LC3-II (LC3B), 3) green fluorescent protein (GFP)-Atg8/LC3 lysosomal delivery and proteolysis, 4) p62/SQSTM1 western blot, 5) autophagy sequestration assays, 6) turnover of autophagy compartments, 7) autophagosome-lysosome colocalization and dequenching assay, 8) sequestration and processing assays in plants, 9) tandem red fluorescent protein (RFP)-GFP fluorescence microscopy, 10) tissue fractionation and 11) analysis in vivo (for complete details, see Ref. [26]).
All Atgs are not specific to autophagy alone and might be implicated in other cellular pathways in health or disease. Further guidance for the use and interpretation of assays tracking autophagy can be found here [26]. These authors suggest that the term ‘autophagy dysregulation’ be used to describe any generalized autophagy aberration. The term encompasses both alterations in autophagy flux and steady state.
3. Cornea
The cornea is an avascular and transparent tissue that is primarily comprised of three layers, the epithelium, stroma and endothelium. The epithelium is the outermost component of the cornea made of non-keratinized, stratified squamous epithelial cells. Corneal epithelial cells have an average lifespan of 7–10 days and repeatedly turnover through a process of involution, apoptosis and desquamation [16]. The apex of the outermost epithelial cells has microvilli and microplicae covered by a glycocalyx. The villi increase the overall surface area to promote adherence of the tear film. Cells of the epithelium maintain tight junctional complexes, and the deepest cellular layer (basal layer) adheres to the basement membrane via hemidesmosomes, thus securing the epithelium to underlying layers. Epithelial stem cells are located at the limbal region of the cornea [16,28]. The stroma is the thickest layer of the cornea (85%) and provides structural integrity. The stroma is comprised of type I collagen complexed with type V collagen fibers arranged in parallel to create fibrils. The highly organized fibrils reduce forward light scatter and provide transparency. Keratocytes are the major cell type of the stroma and maintain the extracellular matrix (ECM). These cells produce collagen components, glycosaminoglycans and matrix metalloproteases (MMPs) [16]. The deepest layer is the endothelium, which is a single layer of squamous cells. Adjacent endothelial cells are connected by lateral interdigitations. Endothelial cell Na/K-ATPase pumps maintain corneal clarity by ensuring that the cornea is in a state of persistent deturgescence. Similar to the epithelium, endothelial cells are attached to Descemet membrane by hemidesmosomes [28].
Interestingly, the avascular nature of the cornea results from an active inhibitory process, thus maintaining its transparency, which is essential for normal visual function. The tears and aqueous humor provide nutrients to the cornea. In the face of injury, adventitious microvascular incursions from the limbal region and from end branches of the ophthalmic and facial arteries may invade the cornea (referred to as neovascularization) [17,28]. A number of antiangiogenic factors are normally present in the cornea. Examples include soluble vascular endothelial growth factor (sVEGF) receptors −1, −2, −3, pigment epithelium-derived factor, angiostatin, endostatin and thrombospondin-1 [17,29]. The combined effects of these antiangiogenic factors include inhibiting growth factor availability and blocking vascular endothelial cell proliferation and/or migration. Excessive and prolonged neovascularization of the cornea due to severe injury can result in impaired vision. Corneal blood vessels might regress at a very slow rate or persist indefinitely [17,29,30].
4. Ocular implications of dysregulated autophagy
4.1. The corneal epithelium
The corneal epithelium represents that part of the visual axis that is also an interface with the external environment and must therefore be responsible for dynamic responses required to protect the ocular surface from various stimuli and insults. Corneal exposure to atmospheric oxygen and other environmental stressors leads to the production of free radicals including reactive oxygen species (ROS), resulting in corneal epithelial damage or disrupted corneal physiology and function [31]. Biomarkers of oxidative stress, such as malondialdehyde (MDA; for lipid peroxidation) and 3-nitrotyrosine (NT; for peroxynitrite formation) are elevated in the corneal epithelium in a variety of corneal diseases [31]. Oxidative damage also results in autophagy induction and defective lysosomal clearance, which we demonstrated in human corneal epithelial (HCE) cells [8]. These findings suggest that autophagy is an inherent response of HCE cells similar to other mammalian cell types subjected to oxidative damage and that prolonged oxidative insult results in autophagy dysregulation. Oxidatively stressed HCE cells also showed mTOR-mediated regulation of autophagy [8], which might pave the way to the use of autophagy-modulating drugs (e.g. rapamycin, trehalose and other functional analogues) for the therapeutic management of oxidative stress-associated corneal diseases [32]. Concerning the epithelial stem cells that are located at the peripheral region of the cornea, Park et al. investigated two stages of autophagy, vesicular biogenesis (early-stage) and turnover (end-stage). The authors found that preservation of end-stage autophagy aided in maintenance of limbal epithelial proliferation [33], which might prove critical for corneal wound healing.
4.1.1. Disease perspective: keratoconus
Keratoconus (KC) is a corneal degenerative condition characterized by restricted corneal thinning and corneal protrusion causing irregular astigmatism and eventual visual loss. KC is the most common primary ectasia and appears in the second decade of life [34]. The condition has no sex or ethnicity predilection and can be bilateral or asymmetric. The etiology is not fully understood [35]. When compared to controls, corneal buttons from KC patients have degraded ECM (with subsequent thinning of the corneal stroma), reduced expression of antioxidant enzymes and increased levels of free radicals due to oxidative damage. This suggests that oxidative stress plays a role in the pathogenesis of KC [31,36–38]. In consideration of the fact that induction of autophagy has been proposed as a cellular response to oxidative stress [39], we evaluated expression of autophagosomal (LC3-II) and lysosomal markers (LAMP1) in cone and periphery regions of KC patient epithelium of different clinical grades based on the Amsler-Krumeich classification [8]. Atg expressions were significantly reduced in the cone region of KC Grades II– and III-affected corneas (more severe disease) compared to the peripheral region [8]. This observation implies that there is insufficient autophagy activity in the cone (diseased) region compared to the matched peripheral (non-diseased) region of the same eyes with increasing disease severity. Altered expression of Atg levels is also observed in non-ocular progressive disorders including Alzheimer’s disease, amyotrophic lateral sclerosis and familial Parkinson’s disease [40]. Iqbal et al. investigated the expression of LC3 in healthy corneal tissue and in corneal tissue affected by keratoconus. Immunohistochemistry (IHC) demonstrated that the expression of LC3 was increased in keratoconus (epithelium, stroma and endothelium) and that LC3 increased in parallel with disease severity. The highest level of expression was seen in advanced keratoconus. The authors suggested that the level of expression of LC3 might be used as a marker for staging disease and monitoring progression [21]. The study design did not ascertain whether the presence of autophagy was desirable/undesirable for disease management. Another study found reduced fold change expression of Atgs involved with synthesis of autophagosomes in the ectatic cone regions compared to peripheral regions [41]. Additionally, KC corneas showed increased levels of lysosomal enzymes such as cathepsins B, G and V/L, which might indicate abnormal lysosomal function and defective autophagic flux [37]. It has been reported that damage to the lysosomal membranes due to oxidative stress results in excessive release of proteolytic enzymes, which might trigger corneal thinning characteristic to KC [37]. A high level of autofluorescence was seen in KC corneal epithelium, which might be the result of failure of autophagy activity leading to an accumulation of non-degradable cellular materials or organelle deposits in the lysosomal compartment [21]. KC epithelium showed dysregulation of Atgs that might be associated with the abnormal apoptosis in KC cornea because of autophagy mediated cell death [42]. Taken together, these results suggest that an oxidative stress-induced defect in the autophagy-lysosomal pathway might be involved in the progression and pathogenesis of KC.
4.1.2. Disease perspective: dry eye disease
Dry eye disease (DED) is a common condition affecting 5.5–33.7% of the population worldwide [43] and is typically characterized by ocular surface discomfort, increased tear osmolarity, tear-film instability and inflammation [44]. Studies have demonstrated that inflammation plays a key role in DED [44], leading to stimulated expression of pro-inflammatory factors such as interleukins 6, 8, 17 [45–47] and MMPs [48]. However, chronic inflammatory conditions and the associated innate immune responses work in tandem with autophagy dysregulation to cause cellular dysfunction [49,50]. Therefore, with improved understanding of the role of autophagy regarding the regulation of corneal inflammation, treatments targeting autophagy and inflammation together might find clinical applications. To that end, autophagy activators (e.g. trehalose) and inhibitors (e.g. chloroquine) might be useful for modulation of the inflammatory process in corneal diseases. Using in vitro dry eye models, we observed that HCE cells under desiccation stress secreted inflammatory factors and had increased expression levels of LC3 and LAMP1, which indicates that the upregulation of autophagy is an inherent response [51]. We observed that chloroquine was able to rescue the phenotype of HCE cells from desiccation stress-induced inflammation without altering autophagy flux [51]. This suggests that the mechanisms underlying such rescue are independent of classical autophagy pathways, necessitating additional studies to unravel regulatory signaling networks. In support of this observation, topical trehalose treatment has been shown to alleviate symptoms of DED [52].
4.1.3. Disease perspective: ocular infections
Bacteria.
Invading microorganisms are recognized by the cells of the immune system using pattern recognition receptors (PRRs). Once recognized, the cells initiate microbial autophagy, which might occur in any of the stages of microbial adhesion, endocytosis or intracellular escape [53]. There are two types of antimicrobial autophagy. The first type is LC3-associated phagocytosis (LAP) whereby bacteria are engulfed in a phagosome [53]. The second is xenophagy whereby double-membraned autophagosomes directly engulf intracellular microbes [54]. Reports of studies regarding antimicrobial autophagy in the context of ocular health are sparse. However, it is evident from literature in other fields (reviewed here [55]) that acquisition of common infections might be associated with autophagy dysregulation.
Virus.
In the cornea, a common infection is that of herpes simplex viruses 1 and 2 (HSV-1; HSV-2). HSV-1 enters epithelial cells (primary target for infection) in a pH-dependent manner by way of the receptors nectin-1, herpes viral entry mediator (HVEM) and paired immunoglobulin-like 2 receptor alpha (PILR-α) [56]. HSV-1 enters the stroma through 3-O-sulfated heparin sulfate (3-OS HS) receptors on corneal fibroblasts, causing viral stromal keratitis [57]. In one study, HSV-1 was suppressed in corneal cells by autophagy stimulation [58], while in another, active HSV-1 infection suppressed autophagy by interacting directly with Atg BECN1 [2]. The authors concluded that HSV-1 infection caused a more rapid stimulation of the immune response and activation of inflammasome (NLRP3), but it is not clear how activation of inflammasome occurs through autophagy-dependent mechanisms [2]. Yakoub and Shukla demonstrated a distinction between basal autophagy and induced autophagy. Basal autophagy supported HSV-2 infection, while induced autophagy negated HSV-2 infection [59]. In another study, infection with both HSV-1 and HSV-2 affected both autophagy and apoptosis in a coordinated manner, with autophagy appearing to play a cytoprotective role in HSV-infected cells through inhibition of apoptosis. This phenomenon was shown by enhanced autophagosome formation, triggered cytoplasmic acidification, increased LC3B lipidation and elevated ratio of apoptotic cells. Bafilomycin A1 (autophagy inhibitor) triggered a significant increase in the apoptotic responses of HSV-1- and HSV-2-infected cells [14].
Protozoa.
Toxoplasma gondii also enters endothelial cells by endocytosis. Toxoplasma infections occur in immunocompromised and immune-incompetent patients and generally result in posterior uveitis [1]. Yu et al. found that CCAAT/enhancer-binding protein β (C/EBP β) mediates the killing of T. gondii by inducing autophagy in nonhematopoietic cells. Increased T. gondii killing induced by C/EBP β overexpression was blocked by the mTOR activator phosphatidic acid and was increased by the inhibitor AZD8055 [60]. Engagement of CD40 on a mouse endothelial cell line also resulted in killing of T. gondii. CD40 stimulation increased expression of the autophagy proteins BECN1 and LC3-II, enhanced autophagy flux and led to recruitment of LC3 around the parasite [61].
4.2. The corneal stroma
The corneal stroma is the thickest layer of the cornea, characterized by orderly arranged collagen fibrils that provide refractive power and quiescent keratocytes that help maintain this unique layer. Loss of homeostasis in keratocytes or their malfunction results in loss of corneal clarity and visual impairment. To date, there have been few reports focusing on the specific role of autophagy in the corneal stroma [21,62,63]. Corneal fibrosis occurs within the stroma and is a complex repair process that serves to maintain tissue integrity in the face of injury. Dynamic and intricate interactions between cells, growth factors, components of the ECM and cytokines are involved (a complete review of the subject is beyond the scope of this paper, see Ref. [16]). Injury to the epithelium and stroma cause the underlying collagen fibrils to become disorganized and initiate the following cascade of events: inflammation, myofibroblast differentiation, ECM deposition and fibrosis. Cytokines (tumor necrosis factor alpha [TNFα], interleukin 1 [IL-1]) released by the injured tissue cause keratocytes to proliferate into fibroblasts and migrate to the wound. TGFβ 1 and 2, produced by the injured epithelial cells, transdifferentiates fibroblasts into myofibroblasts. Under optimal conditions (physiological healing), there is no scar formation and myofibroblasts undergo apoptosis. With pathological healing (chronic or severe), excessive ECM deposition results and a corneal scar forms, resulting in loss of vision [17,30].
Schnyder corneal dystrophy (SCCD), a rare hereditary form of corneal dystrophy, occurs in the stroma and is the result of an abnormal accumulation of cholesterol and phospholipids [62]. Multilamellar bodies (MLB) are concentric cytoplasmic membranes that develop through an autophagy-dependent mechanism and are associated with SCCD [62]. Autophagy biomarkers, LC3/Atg8 and p62/SQSTM1, were used to demonstrate the presence of autophagy by IHC. Serum starvation or rapamycin treatment upregulated autophagy, while 3-methyladenine was used to inhibit autophagy. The researchers concluded that 3-methyladenine-mediated inhibition of autophagy was beneficial and resulted in decreased multilamellar body formation [62]. Granular corneal dystrophy type 2 is a genetic condition that results in progressive accumulation of granular and lattice deposits with age [63]. Using a transgenic mouse model for granular corneal dystrophy type 2, autophagy biomarker LC3 was identified in affected subepithelial stroma by IHC. Autophagosomes were also identified by transmission electron microscopy (TEM) in proximity to the affected cornea in homozygous mice. No definitive conclusion was ascertained as to whether the presence of autophagy was beneficial or detrimental in granular corneal dystrophy type 2 diseased cornea in that study [63]. Similar to research in other corneal tissue layers, autophagy dysregulation appears to be present with stromal disease. In the latter study, it is possible that increased autophagosomes are beneficial (similarly to in SCCD) and might result in reduced stromal granular deposition. We predict that autophagy might play a role in reducing ECM deposition in the development of corneal fibrosis.
4.2.1. Disease perspective: corneal wound healing and fibrosis
Although the role of autophagy in corneal fibrosis has not been well-defined, specific ECM constituents have been previously shown to modulate autophagy-signaling pathways [27]. Constituents of the ECM that have been shown to influence autophagy include decorin, collagen VI, laminin alpha-2, endostatin, endorepellin and krigle V [27]. All the aforementioned proteins are considered activators of autophagy other than laminin alpha −2, which is an inhibitor. Moreover, these constituents exert their influence independently of nutrient availability [27]. Using rapamycin to induce autophagy in a rat model of peritendinous fibrosis (assessed by LC3 and p62 biomarkers) alleviated the severity of peritendinous fibrosis in vivo [64]. This observation suggests that there might be autophagy dysregulation, either decreased autophagy flux or increased accumulation of autophagosomes, in the development of corneal fibrosis. Pulmonary fibrosis investigations have determined that the loss of autophagy-related protein Atg7 in endothelial cells leads to loss of endothelium, upregulation of TGFβ (pro-fibrotic gene) signaling and impaired autophagy flux with significant changes in endothelial cell structure [65]. TGFβ plays a major role in myofibroblast transdifferentiation and has been implicated in numerous fibrotic ocular diseases. Ding et al. reported that autophagy processes regulate TGFβ expression and suppress renal fibrosis induced by a unilateral ureteral obstruction mouse model [66]. Further investigation of autophagy’s role in corneal fibrosis is warranted.
In our pilot experiments with primary human donor corneal stromal fibroblast (HSF) cultures, we determined that certain autophagy protein levels were elevated after the cells had been wounded by scratch (Fig. 5). These results showed that both LC3A/B and LAMP1 levels were elevated in the wounded cells compared to control cells. We also determined that at this time point (6 h post-wound), p62 levels were not appreciably altered. Results of this experiment provide proof of the concept that autophagy alterations occur during corneal wound healing and should therefore be further investigated for clinical application.
Fig. 5. Expression of autophagy-related proteins in wounded HSF cells.
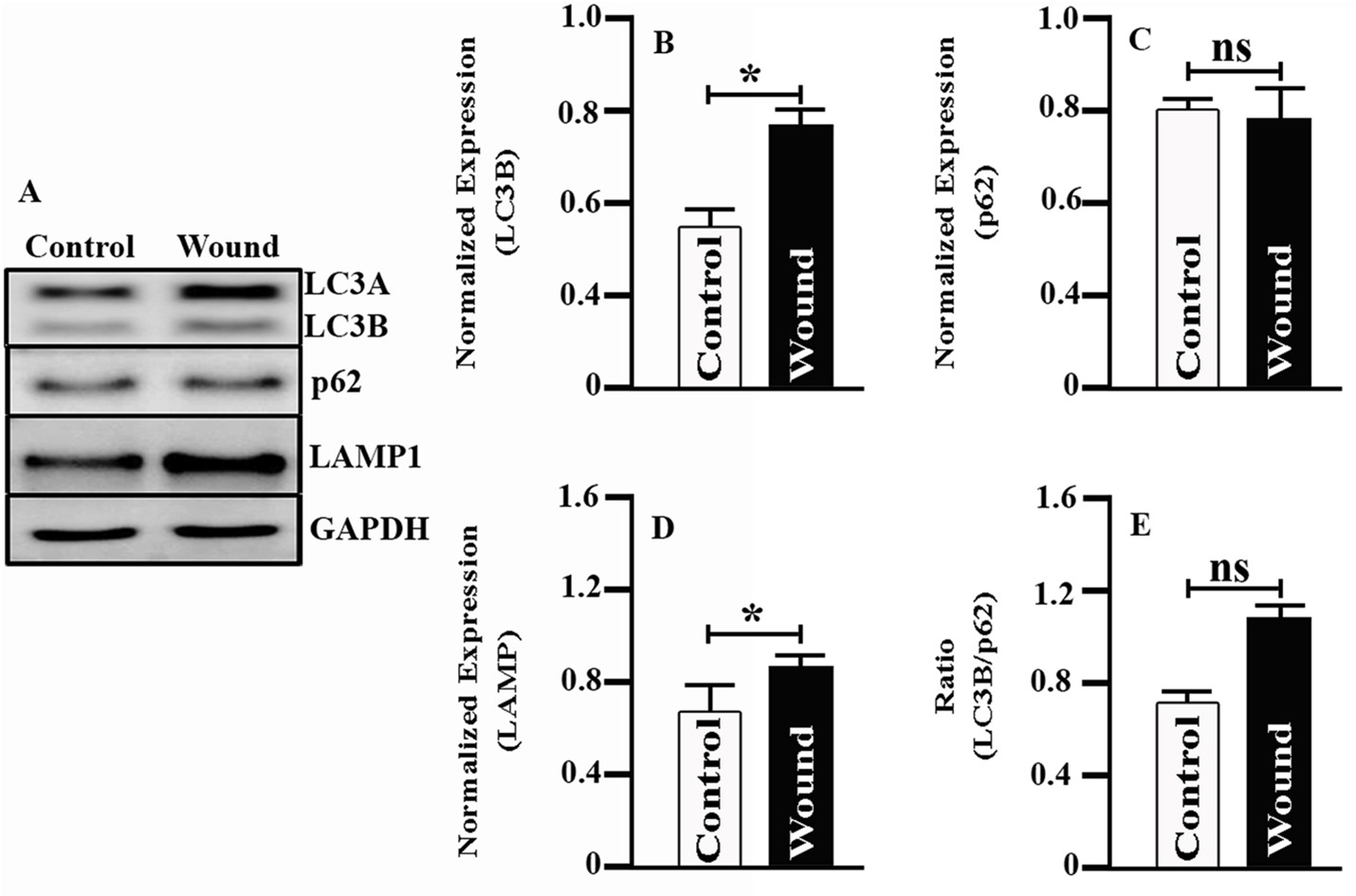
A) Western blot analysis shows expression of LC3A/B, p62 and LAMP1 in normal (control) and wounded (scratch) HSF cells. B) Densitometric analysis of LC3B, p62 and LAMP1 proteins. GAPDH served as the loading control and was used for densitometric analyses. C) Ratio of LC3B and p62. Data are expressed as mean ± SD, n = 3, statistical significance was denoted by * (p < 0.05) and ns (not significant as compared to control).
(GAPDH: glyceraldehyde 3-phosphate dehydrogenase; HSF: human stromal fibroblast. LAMP1: lysosomal-associated membrane protein 1; LC3: microtubule associated protein 1 light chain 3 alpha).
4.2.2. Disease perspective: corneal angiogenesis
Although the cornea is normally avascular, neovascularization might be initiated following an injury. There are three primary types of neovascularization: superficial vascularization, vascular pannus and deep stromal vascularization. Neovascularization is characterized by the incursion of vessels that have relatively poor structural integrity that might result in increased leakage of blood components into the local tissue milieu [67]. For example, VEGF is a major cytokine that stimulates endothelial cells and promotes neovascularization. Investigations have shown that inhibition of VEGF can inhibit vessel formation and represents a new therapeutic target [68]. Inhibitory antibodies targeting VEGF-A signaling pathways, such as bevacizumab and ranibizumab, are currently used in clinical settings [17]. VEGF has also been shown to increase the expression of the Atgs BECN1 and LC3 and to inhibit inflammation through activation of autophagy in a spinal cord injury model [69]. Chemerin is a recently identified adipokine associated with metabolic patients that has been shown to act as a proangiogenic factor. Shen et al. reported that in vitro, chemerin treated endothelial cells increased mitochondrial ROS, LC3-II, BECN1, Atg7 and Atg12-Atg5 complex suggesting an upregulation of autophagy flux. In a follow up study, the knockdown of chemerin receptor 23 resulted in inhibition of autophagy [70]. Taken together, these studies provide a functional link between VEGF and autophagy modulation suggesting that the pharmacological manipulation of autophagy could be useful in the management of corneal repair when neovascularization is problematic.
4.3. The corneal endothelium
Integrity loss of the endothelium in association with epithelial and stromal injury might result in corneal perforation (iris prolapse) or failed deturgescence (corneal edema) [28]. Dysfunctional autophagy has been found to play a role in corneal dystrophies (described previously and more later) [10,12,13]. In a study investigating brain microvascular endothelial cells and their role in blood-brain barrier (BBB) integrity during cerebral ischemia-reperfusion injury, rapamycin and lithium carbonate (mTOR-dependent and mTOR-independent autophagy inducers, respectively) and 3-methyladenine (autophagy inhibitor) were used to modulate autophagy. Investigators determined that enhanced autophagy exerted a beneficial effect on BBB integrity during ischemic-reperfusion injury [71]. There are a number of substances that have been found to activate autophagy and might be considered as therapeutic agents. These include curcumin (induces autophagy to protect vascular endothelial cell survival from oxidative stress damage), decorin (promotes endothelial cell autophagy and inhibits angiogenesis), endostatin (induces autophagy in endothelial cells by modulating BECN1 and β-catenin levels) and resveratrol (induces autophagy through the cAMP signaling pathway to dampen vascular endothelial inflammation) [72–75]. By inhibiting superoxide anion generation, inducing NO production, activating survival kinases and maintaining mitochondrial function and cell viability, vitamin D has also been shown to prevent endothelial cell death through alterations in the crosstalk between apoptosis and autophagy [76].
4.3.1. Disease perspective: corneal dystrophy
Fuchs endothelial corneal dystrophy.
Fuchs endothelial corneal dystrophy (FECD) is a progressive disease resulting in bilateral loss of corneal endothelial cells (CEnCs). It is characterized by decreased CEnC density, ECM excrescences (guttae) and thickening of Descemet membrane [12,13]. Meng et al. used two α2 collagen VIII (Col8a2) knock-in mouse models and human FECD tissues to show upregulation of autophagy by demonstrating increased DRAM1 compared to controls [13]. Using lithium treatment of cultured CEnCs and a mouse model of FECD, increased autophagy was further demonstrated in this condition through the employment of autophagy biomarkers p62, Tmem74, Tm9sf1 and Tmem166 by real time PCR and of Atg5–12 conjugate by western blotting [12]. Recent studies have shown that mitochondrial dysfunction (diminished mitochondrial antioxidant abilities, fewer mitochondria, damaged mitochondrial DNA and fragmented mitochondria) plays a role in FECD. Dysfunctional mitochondria are a source of ROS and therefore increased oxidative damage to the cell [77,78]. The disease causes activation of ER stress and the unfolded protein response (UPR) that stimulate autophagy [13,79]. Using the autophagy biomarker LC3, Benischke et al. concluded that upregulation of mitochondrial autophagy (mitophagy) resulted in decreased mitochondrial mass and depletion of functional mitochondria in FECD [79]. Furthermore, inhibition of autophagy flux led to increased mitochondrial mass in FECD. TEM showed an increased number of autophagolysosomes with engulfed damaged mitochondria [80]. Based on these results, treatments to decrease autophagy flux should be investigated to help FECD patients.
Granular corneal dystrophy type 2.
Granular corneal dystrophy (GCD; Avellino corneal dystrophy) is slowly progressive and results in degeneration of the corneal endothelium. There are two types; type 1 and type 2. GCD type 2 (GCD2) is an autosomal dominant disease due to a mutation (R124H) in the TGFβ1 gene (chromosome 5q31) and results in corneal stromal opacities [2,81]. Transforming growth factor β-induced protein (TGFβIp) accumulates in corneal dystrophies and the affected corneal fibroblasts exhibit morphologic changes, increased sensitivity to oxidative stress, fragmented and dysfunctional mitochondria and autophagy dysregulation [10,82]. Choi et al. found that mutant-TGFβIp co-localized with LC3-enriched cytosolic vesicles and that LC3-II (indicates active autophagy) and SQSTM1/p62 levels were significantly increased in GCD2 corneal fibroblasts, all indicating an upregulation of autophagy. Bafilomycin A was used to inhibit autophagy by blocking fusion of autophagosomes and lysosomes, and rapamycin was used to stimulate autophagy [10]. Nie et al. found that lithium chloride treatment of corneal fibroblasts in vitro reduced the expression of TGFβIp and increased autophagy and autophagy flux in mutant-TGFβIp-overexpressing cells. Results of that study indicated that lithium chloride might be considered as a therapeutic agent for GCD [83]. In addition, enhanced autophagy clearance of mutant-TGFβ1 protein was seen in GCD corneal fibroblast cells with a treatment of melatonin and rapamycin [84].
5. Autophagy paradigms for application in corneal diseases
To investigate the presence of autophagy in normal corneal stroma, our laboratory focused on autophagy biomarkers that have previously been usefully employed in other organs (Table 1), in addition to some already identified in the cornea. The proteins include LC3/Atg8, BECN1/Atg6, Atg5, DRAM1, SQSTM1/p62 and PRKN2 (Table 2). PCR performed by using cDNA from HCE, HSF and human corneal endothelium (HCN) primary culture cells showed that all Atgs were present in all cell types tested (Fig. 6). In addition, we demonstrated that altered levels of LC3 and LAMP1 were present in the epithelium of keratoconus subjects compared to healthy corneas (Fig. 7), substantiating the role of autophagy in corneal disease. We also showed that HCE cells could respond to oxidative stress and the autophagy inhibitor chloroquine by modulating autophagy markers LC3 and LAMP1 as well as lysosomal content (Fig. 8). In addition, we observed that the process of wound healing in HSF cells was associated with alterations to LC3B and LAMP1 (Fig. 5), suggesting that autophagy could be an important pathway influencing corneal wound healing in various wound repair conditions and/or stages. It is further likely that both stromal and endothelial cells will also respond in a similar fashion when encountering stressful stimuli. We predict that the modulation of autophagy by pharmacological agents (such as rapamycin, chloroquine and trehalose) may be possible in corneal disease but must be contextualized to the specific disease process present. This approach also provides the opportunity for possibly co-regulating multiple pathways, such as inflammation and autophagy, using dual-action modulators or formulations containing synergistic drugs.
Table 1. Autophagy-related genes that have been identified and implicated in various disease processes within the body.
The currently known roles/functions of each autophagy-related gene are listed, as well as the known pathological disorder.
| Autophagy genes | Role/function | Tissue(s) with implicated genes | Reference(s) |
|---|---|---|---|
|
| |||
| LC3 | LC3-I (cytosolic form) conjugates to phosphatidylethanolamine (PE) to form an LC3-II-PE complex on the surface of the expanding phagophore. Lipids are continually supplied until the autophagosomal membrane is a double-membraned enclosed structure. | - Neurodegenerative disease (Ex: amyotrophic lateral sclerosis) - Cancer (Ex: breast, ovarian, prostate, colorectal) - Renal (Ex: Fabry’s Disease) - Corneal endothelium (Fuchs endothelial corneal dystrophy) - Corneal stroma (Granular corneal dystrophy) - Congenital cataracts - Primary open angle glaucoma - Age-related macular degeneration - Pulmonary epithelia (Ex: Chronic obstructive pulmonary disease) |
[10,13,85–95] |
| BECN1 | Becn1 complexes with Vsp34-Atg14 to regulate and initiate formation of the nascent autophagosome (phagophore). | - Neurodegenerative disease (Ex: amyotrophic lateral sclerosis) - Cancer (Ex: breast, ovarian, prostate) - Corneal stroma (Granular corneal dystrophy) - Primary open angle glaucoma - Pulmonary epithelia (Ex: Chronic obstructive pulmonary disease) |
[10,85,87,90,91,94,96] |
| Atg5 | Atg7 activates Atg5 to conjugate with Atg12. The Atg12-Atg5 complex stimulates lipidation of LC3, which ultimately promotes extension and growth of the double-membraned phagophore to become an autophagosome. | - Neurodegenerative disease - Corneal endothelium (Fuchs endothelial corneal dystrophy) - Congenital cataracts - Primary open angle glaucoma - Age-related macular degeneration |
[13,88,92,93,97,98] |
| DRAM1 | DRAM1 is a part of the p53 tumor suppressor pathway that aids in autophagosome clearance through promotion of lysosomal acidification and upregulation of lysosomal enzymes. | - Corneal endothelium (Fuchs endothelial corneal dystrophy) | [13] |
| SQSTM1 | Also known as p62, SQSTM1 is an autophagosomal membrane protein that selectively targets other proteins for autophagy. The protein is integral to autophagy of the mitochondria (mitophagy). | - Skeletal disease (Ex: Page’s Disease) - Prion disease |
[99,100] |
| PRKN2 | As part of a multiprotein E3 ubiquitin ligase complex, Prkn2 regulates the selective targeting of proteins for proteasomal degradation. | -Neurodegenerative disease (Ex: Parkinson’s disease) | [89,96,97] |
(Atg: autophagy-related protein; BECN1: Beclin 1; DRAM1: DNA damage regulated autophagy modulator 1; LC3: microtubule associated protein 1 light chain 3 alpha; PE: phosphatidylethanolamine; PRKN2: E3 ubiquitin-protein ligase parkin; SQSTM1: sequestosome 1; Vsp34: vesicular protein sorting 34).
Table 2.
Primers used for PCR analysis.
| Autophagy-related gene | Gene sequence (5’–3’) |
|---|---|
|
| |
| LC3 | F: TCCTGTACATGGTCTATGCC R: CGTTTACCCTGCGTTTGT |
| BECN1 | F: CTGGACAAGCTGAAGAAGAC R: TGTGAGGATACCCAAGCA |
| Atg5 | F: GAAGGCACACCACTGAAA R: AGCTCAGATGCTCACTCA |
| DRAM1 | F: GTCAGCCGCCTTCATTAT R: TCCACTCCAGCTTGGTTA |
| SQSTM1 | F: GGTGAAACACGGACACTT R: GACTGGAGTTCACCTGTAGA |
| PRKN2 | F: GCACCTGATCGCAACAAA R: CTCTGTAGGCCTGAGTAGTT |
(Atg5: autophagy-related protein 5; BECN1: Beclin 1; DRAM1: DNA damage regulated autophagy modulator 1; LC3: microtubule associated protein 1 light chain 3 alpha; PRKN2: E3 ubiquitin-protein ligase parkin; SQSTM1: sequestosome 1).
Fig. 6. Gel electrophoresis results of autophagy-related proteins in corneal tissues.
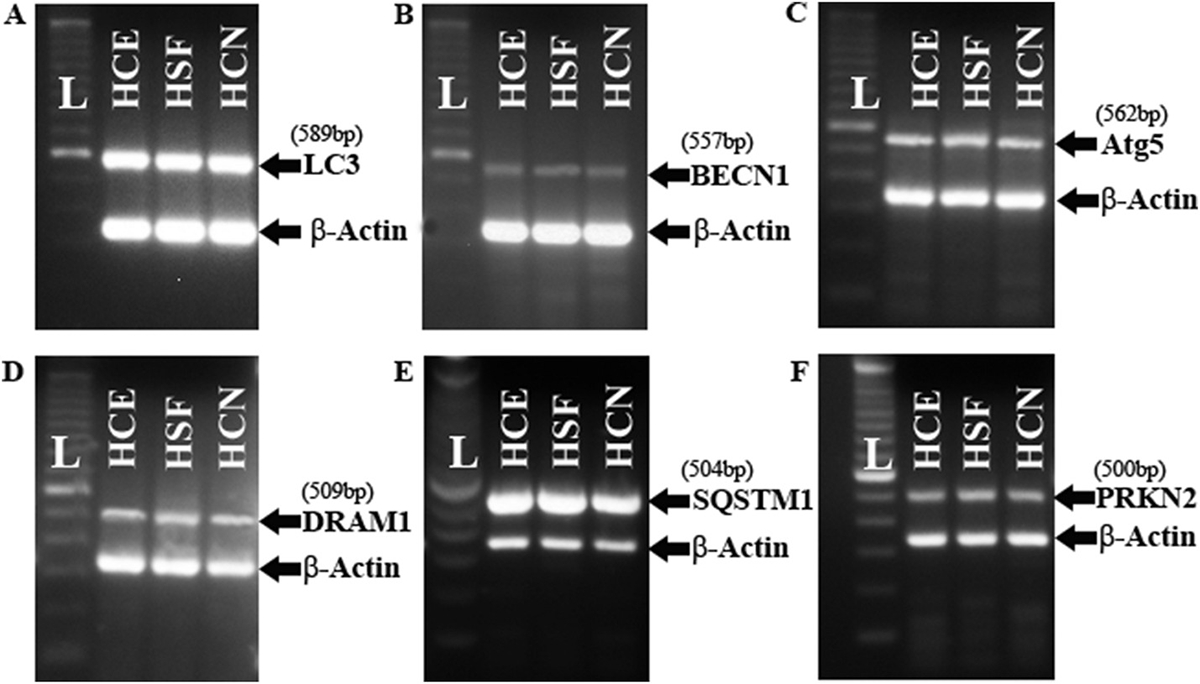
LC3 (A), BECN1 (B), Atg5 (C), DRAM1 (D), SQSTM1 (E) and PRKN2 (F) were present in human corneal epithelium (HCE), human stromal fibroblast (HSF) and human corneal endothelium (HCN). β-Actin served as the control.
(Atg5: autophagy-related protein 5; BECN1: Beclin 1; DRAM1: DNA damage regulated autophagy modulator 1; HCE: human corneal epithelium; HCN: human corneal endothelium; HSF: human stromal fibroblast; L: ladder; LC3: microtubule associated protein 1 light chain 3 alpha; PRKN2: E3 ubiquitin-protein ligase parkin; SQSTM1: sequestosome 1).
Fig. 7. Expression of autophagy-related proteins in KC epithelium.
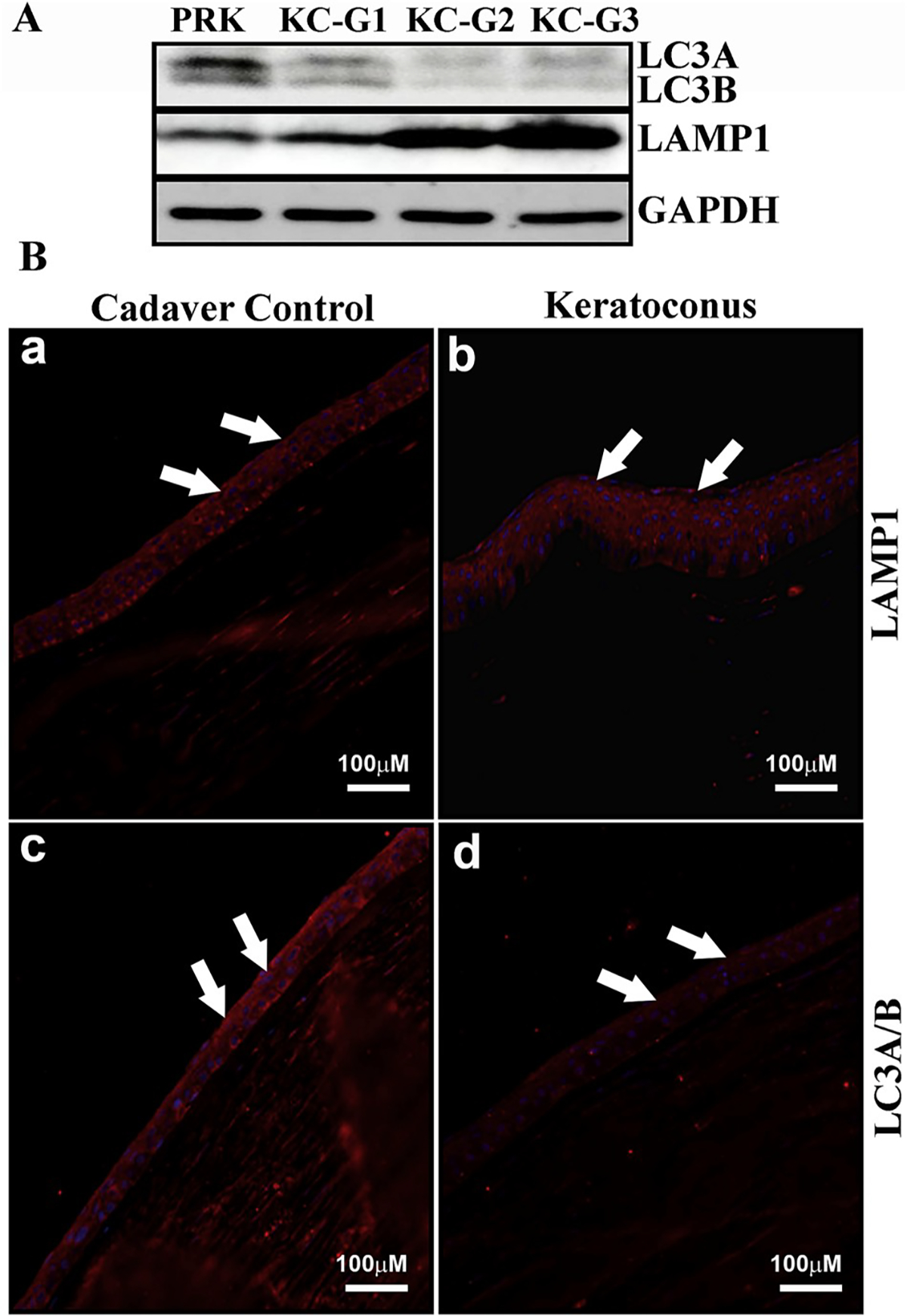
A) Western blot analysis of LC3A/B and LAMP1 expression in different clinical grades of KC epithelium compared to healthy cornea collected prior to PRK (control). GAPDH served as the loading control. B) Immunofluorescence of LC3A/B and LAMP1 in KC epithelium compared to healthy donor corneal epithelium (arrows).
(GAPDH: glyceraldehyde 3-phosphate dehydrogenase; KC-G: keratoconus-grade; LAMP1: lysosomal-associated membrane protein 1; LC3: microtubule associated protein 1 light chain 3 alpha; PRK: photorefractive keratectomy).
Fig. 8. Expression of autophagy-related proteins in oxidatively stressed HCE cells.
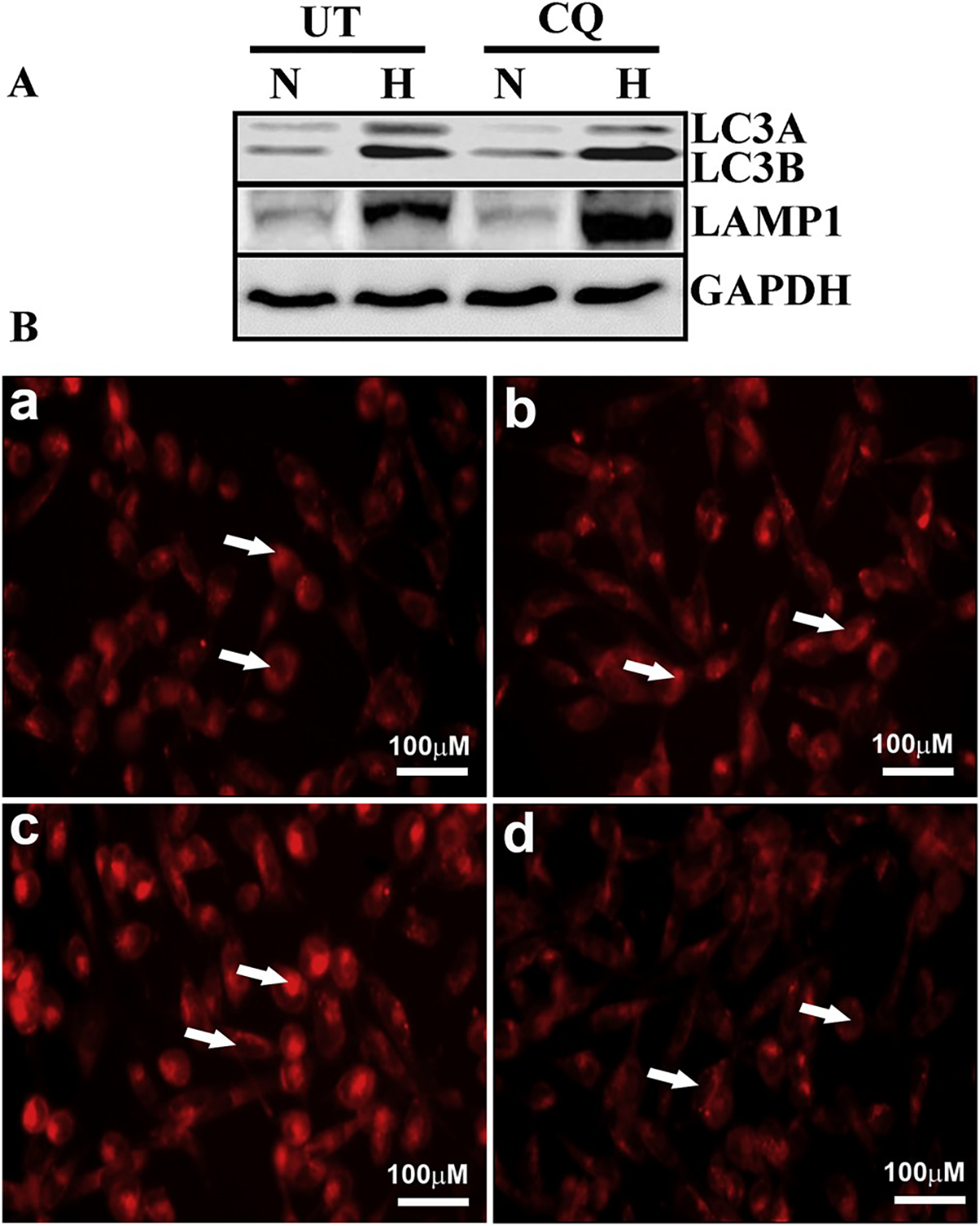
A) Western blot analysis shows expression of LC3A/B and LAMP1 in HCE cells under normoxic (N) and hyperoxic (H) conditions with chloroquine treatment (CQ) or sham treatment control (UT). GAPDH served as the loading control. B) Immunofluorescence of lysosomal content in oxidatively stressed HCE cells. Panels A–D show lysotracker staining (arrows) of normoxic cells (a), normoxic cells treated with chloroquine (b), hyperoxic cells (c) and hyperoxic cells treated with chloroquine (d).
(CQ: chloroquine; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; H: hyperoxia; HCE: human corneal epithelium; LAMP1: lysosomal-associated membrane protein 1; LC3: microtubule associated protein 1 light chain 3 alpha; N: normoxia).
In light of the fact that one of the most rudimentary roles of autophagy is to remove obsolete or dysfunctional organelles and proteins, autophagy dysregulation might play a role in the net accumulation of ECM that results in corneal haze. It is well recognized that lysosomal storage disorders, characterized by progressive accumulation of intracellular undigested material, are considered primary autophagy disorders. Impaired autophagy flux represents the underlying commonality among different lysosomal storage disorders [14]. Our laboratory aims to investigate in detail whether the development of corneal haze and other corneal diseases might also be considered a primary autophagy disorder.
Other roles of autophagy that are not well understood in the context of the cornea are those of providing nutrients via recycling, which would be especially important for this avascular tissue and in responding to invading pathogens. There might be unidentified autophagy mechanisms that provide signaling or cell behavior modulatory functions, which could be useful for the administration of therapeutic drugs to prevent or treat pathologic conditions. Understanding how autophagy reacts to various infectious pathogens might aid our clinical treatment arsenal.
Identification of Atgs and characterization of their specific alterations during response to injury and consequent healing processes in each one of the primary corneal layers might provide novel therapeutic targets to prevent loss of visual function. Wound healing in the corneal stroma is a multi-phasic process involving a number of cell signaling mechanisms and the production of various signaling factors and ECM components, which require the cells to transform in a spatio-temporal manner for optimal outcome. Therefore, we hypothesize that by activating both the induction of autophagy as well as autophagy flux, aberrant function of keratocytes might be prevented. Further, in cases where oxidative stress plays a role in disease pathology, increased autophagy activity will likely lessen damage to both stromal and epithelial cells resulting from ROS. Pursuant to this, in cases where inflammation is the primary culprit, modulating autophagy might have the dual effect of returning cells to homeostasis as well as regulating the feedback activation of chronic inflammatory signaling. Further investigation is therefore underway.
Acknowledgements
Thanks are due to undergraduate students, Mr. Prashant R. Sinha and Ms. Sally Heil, for their assistance in western blotting, electrophoresis and cryo-sectioning.
Sources of funding
This work was primarily supported by the University of Missouri Ruth M. Kraeuchi Missouri Endowed Chair Ophthalmology Fund (RRM) and partially by the NIH/NEI R01EY017294 grant (RRM) and the Veterans Health Affairs Merit 1I01BX00357 grant (RRM).
Abbreviations:
- 3-OS HS
3-O-sulfated heparin sulfate
- AMPK
5′ adenosine monophosphate-activated protein kinase
- Atg
autophagy-related proteins
- BBB
blood-brain barrier
- BECN1
beclin 1
- c-Met/Met
mesenchymal-epithelial transition factor
- C/EBP β
CCAAT/enhancer-binding protein β
- DED
dry eye disease
- ECM
extracellular matrix
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- HCE
human corneal epithelium
- HCN
human corneal endothelium
- HGF
hepatocyte growth factor
- HSF
human stromal fibroblast
- HSV-1
herpes simplex virus 1
- HVEM
herpes viral entry mediator
- IHC
immunohistochemistry
- IL-1
interleukin 1
- LAMP1
lysosomal-associated membrane protein 1
- LAMP2A
lysosomal-associated membrane protein 2A
- LAP
LC3-associated phagocytosis
- LC3
microtubule-associated protein 1A/1B-light chain 3
- MDA
malondialdehyde
- MMP
matrix metalloprotease
- MPK8
mitogen-activated protein kinase 8
- mRNA
messenger ribonucleic acid
- mTOR1
mammalian target of rapamycin complex 1
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NLRP3
inflammasome
- NT
3-nitrotyrosine
- PILR-α
paired immunoglobulin-like 2 receptor alpha
- PRKN2
E3 ubiquitin-protein ligase parkin
- PRR
pattern recognition receptor
- qRT-PCR
quantitative real-time polymerase chain reaction
- RFP
red fluorescent protein
- ROS
reactive oxygen species
- SQSTM1
sequestosome 1
- sVEGF
soluble vascular endothelial growth factor
- TEM
transmission electron microscopy
- TGFβ
transforming growth factor beta
- TGFβIp
transforming growth factor beta-induced protein
- TNFα
tumor necrosis factor alpha
- UPR
unfolded protein response
- V-ATPase
vacuolar adenosine triphosphatase
Footnotes
Declaration of interests
None.
References
- [1].Chai P, Ni H, Zhang H, Fan X. The evolving functions of autophagy in ocular health: a double-edged sword. Int J Biol Sci 2016;12:1332–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Frost LS, Mitchell CH, Boesze-Battaglia K. Autophagy in the eye: implications for ocular cell health. Exp Eye Res 2014;124:56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006;10:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005;120:237–48. [DOI] [PubMed] [Google Scholar]
- [5].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol 2010;221:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wileman T. Autophagy as a defense against intracellular pathogens. Essays Biochem 2013;55:153–63. [DOI] [PubMed] [Google Scholar]
- [8].Shetty R, Sharma A, Pahuja N, Chevour P, Padmajan N, Dhamodaran K, et al. Oxidative stress induces dysregulated autophagy in corneal epithelium of keratoconus patients. PLoS One 2017;12:e0184628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Matthaei M, Hu J, Meng H, Lackner EM, Eberhart CG, Qian J, et al. Endothelial cell whole genome expression analysis in a mouse model of early-onset Fuchs’ endothelial corneal dystrophy. Invest Ophthalmol Vis Sci 2013;54:1931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Choi SI, Kim BY, Dadakhujaev S, Oh JY, Kim TI, Kim JY, et al. Impaired autophagy and delayed autophagic clearance of transforming growth factor beta-induced protein (TGFBI) in granular corneal dystrophy type 2. Autophagy 2012;8:1782–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jiang Y, Yin X, Stuart PM, Leib DA. Dendritic cell autophagy contributes to herpes simplex virus-driven stromal keratitis and immunopathology. mBio 2015;6. e01426–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kim EC, Meng H, Jun AS. Lithium treatment increases endothelial cell survival and autophagy in a mouse model of Fuchs endothelial corneal dystrophy. Br J Ophthalmol 2013;97:1068–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Meng H, Matthaei M, Ramanan N, Grebe R, Chakravarti S, Speck CL, et al. L450W and Q455K Col8a2 knock-in mouse models of Fuchs endothelial corneal dystrophy show distinct phenotypes and evidence for altered autophagy. Invest Ophthalmol Vis Sci 2013;54:1887–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Petrovski G, Pasztor K, Orosz L, Albert R, Mencel E, Moe MC, et al. Herpes simplex virus types 1 and 2 modulate autophagy in SIRC corneal cells. J Biosci 2014;39:683–92. [DOI] [PubMed] [Google Scholar]
- [15].Whitcher JP, Srinivasan M, Upadhyay MP. Corneal blindness: a global perspective. Bull World Health Organ 2001;79:214–21. [PMC free article] [PubMed] [Google Scholar]
- [16].Bukowiecki A, Hos D, Cursiefen C, Eming SA. Wound-healing studies in cornea and skin: parallels, differences and opportunities. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Korntner S, Lehner C, Gehwolf R, Wagner A, Grutz M, Kunkel N, et al. Limiting angiogenesis to modulate scar formation. Adv Drug Deliv Rev 2018. 10.1016/j.addr.2018.02.010.S0169-409X(18)30038-3. [DOI] [PubMed] [Google Scholar]
- [18].Lim M, Goldstein MH, Tuli S, Schultz GS. Growth factor, cytokine and protease interactions during corneal wound healing. Ocul Surf 2003;1:53–65. [DOI] [PubMed] [Google Scholar]
- [19].Kaarniranta K, Kauppinen A, Blasiak J, Salminen A. Autophagy regulating kinases as potential therapeutic targets for age-related macular degeneration. Future Med Chem 2012;4:2153–61. [DOI] [PubMed] [Google Scholar]
- [20].Kaarniranta K, Sinha D, Blasiak J, Kauppinen A, Vereb Z, Salminen A, et al. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013;9:973–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Iqbal O, Fisher G, Vira S, Syed D, Sadeghi N, Freeman D, et al. Increased expression of secreted frizzled-related protein-1 and microtubule-associated protein light chain 3 in keratoconus. Cornea 2013;32:702–7. [DOI] [PubMed] [Google Scholar]
- [22].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011;27:107–32. [DOI] [PubMed] [Google Scholar]
- [23].Gump JM, Thorburn A. Autophagy and apoptosis: what is the connection? Trends Cell Biol 2011;21:387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mallat A, Lodder J, Teixeira-Clerc F, Moreau R, Codogno P, Lotersztajn S. Autophagy: a multifaceted partner in liver fibrosis. BioMed Res Int 2014;2014:869390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mao S, Zhang J. Role of autophagy in chronic kidney diseases. Int J Clin Exp Med 2015;8:22022–9. [PMC free article] [PubMed] [Google Scholar]
- [26].Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2014;4:151–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Neill T, Schaefer L, Iozzo RV. Instructive roles of extracellular matrix on autophagy. Am J Pathol 2014;184:2146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Richardson A, Wakefield D, Di Girolamo N. Fate mapping mammalian corneal epithelia. Ocul Surf 2016;14:82–99. [DOI] [PubMed] [Google Scholar]
- [29].Roshandel D, Eslani M, Baradaran-Rafii A, Cheung AY, Kurji K, Jabbehdari S, et al. Current and upcoming therapies for corneal neovascularization. Ocul Surf 2018;16:398–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chaurasia SS, Lim RR, Lakshminarayanan R, Mohan RR. Nanomedicine approaches for corneal diseases. J Funct Biomater 2015;6:277–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Buddi R, Lin B, Atilano SR, Zorapapel NC, Kenney MC, Brown DJ. Evidence of oxidative stress in human corneal diseases. J Histochem Cytochem: Off J Histochem Soc 2002;50:341–51. [DOI] [PubMed] [Google Scholar]
- [32].Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 2015;125:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Park JK, Peng H, Katsnelson J, Yang W, Kaplan N, Dong Y, et al. MicroRNAs-103/107 coordinately regulate macropinocytosis and autophagy. J Cell Biol 2016;215:667–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Romero-Jimenez M, Santodomingo-Rubido J, Wolffsohn JS. Keratoconus: a review. Contact Lens Anterior Eye 2010;33:157–66. quiz 205. [DOI] [PubMed] [Google Scholar]
- [35].Mas Tur V, MacGregor C, Jayaswal R, O’Brart D, Maycock N. A review of keratoconus: diagnosis, pathophysiology, and genetics. Surv Ophthalmol 2017;62:770–83. [DOI] [PubMed] [Google Scholar]
- [36].Chwa M, Atilano SR, Reddy V, Jordan N, Kim DW, Kenney MC. Increased stress-induced generation of reactive oxygen species and apoptosis in human keratoconus fibroblasts. Invest Ophthalmol Vis Sci 2006;47:1902–10. [DOI] [PubMed] [Google Scholar]
- [37].Kenney MC, Brown DJ, Rajeev B. The elusive causes of keratoconus: a working hypothesis. CLAO J 2000;26:10–3. American journal of ophthalmology. 2000;130:263. [PubMed] [Google Scholar]
- [38].Arnal E, Peris-Martinez C, Menezo JL, Johnsen-Soriano S, Romero FJ. Oxidative stress in keratoconus? Invest Ophthalmol Vis Sci 2011;52:8592–7. [DOI] [PubMed] [Google Scholar]
- [39].Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxidants Redox Signal 2006;8:152–62. [DOI] [PubMed] [Google Scholar]
- [40].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013;19:983–97. [DOI] [PubMed] [Google Scholar]
- [41].Pahuja N, Kumar NR, Shroff R, Shetty R, Nuijts RM, Ghosh A, et al. Differential molecular expression of extracellular matrix and inflammatory genes at the corneal cone apex drives focal weakening in keratoconus. Invest Ophthalmol Vis Sci 2016;57:5372–82. [DOI] [PubMed] [Google Scholar]
- [42].You J, Wen L, Roufas A, Hodge C, Sutton G, Madigan MC. Expression of HGF and c-met proteins in human keratoconus corneas. Journal of ophthalmology 2015;2015:852986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].The epidemiology of dry eye disease: report of the epidemiology subcommittee of the international dry eye workshop 2007 Ocul Surf 2007;5:93–107. [DOI] [PubMed] [Google Scholar]
- [44].Ogawa Y, Tsubota K. Dry eye disease and inflammation. Inflamm Regen 2013;33:238–48. [Google Scholar]
- [45].Kang MH, Kim MK, Lee HJ, Lee HI, Wee WR, Lee JH. Interleukin-17 in various ocular surface inflammatory diseases. J Kor Med Sci 2011;26:938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Massingale ML, Li X, Vallabhajosyula M, Chen D, Wei Y, Asbell PA. Analysis of inflammatory cytokines in the tears of dry eye patients. Cornea 2009;28:1023–7. [DOI] [PubMed] [Google Scholar]
- [47].Turner K, Pflugfelder SC, Ji Z, Feuer WJ, Stern M, Reis BL. Interleukin-6 levels in the conjunctival epithelium of patients with dry eye disease treated with cyclosporine ophthalmic emulsion. Cornea 2000;19:492–6. [DOI] [PubMed] [Google Scholar]
- [48].Pflugfelder S, Fang B, De Paiva C. Matrix metalloproteinase-9 in the pathophysiology and diagnosis of dry eye syndrome. Metalloproteinases Med 2017;4:37–46. [Google Scholar]
- [49].Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med 2017;6:ss. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Saitoh T, Akira S. Regulation of innate immune responses by autophagy-related proteins. J Cell Biol 2010;189:925–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shivakumar S, Panigrahi T, Shetty R, Subramani M, Ghosh A, Jeyabalan N. Chloroquine protects human corneal epithelial cells from desiccation stress induced inflammation without altering the autophagy flux. BioMed Res Int 2018;2018:7627329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Matsuo T, Tsuchida Y, Morimoto N. Trehalose eye drops in the treatment of dry eye syndrome. Ophthalmology 2002;109:2024–9. [DOI] [PubMed] [Google Scholar]
- [53].Cemma M, Brumell JH. Interactions of pathogenic bacteria with autophagy systems. Curr Biol: CB 2012;22:R540–5. [DOI] [PubMed] [Google Scholar]
- [54].Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011;469:323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013;13:722–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shah A, Farooq AV, Tiwari V, Kim MJ, Shukla D. HSV-1 infection of human corneal epithelial cells: receptor-mediated entry and trends of re-infection. Mol Vis 2010;16:2476–86. [PMC free article] [PubMed] [Google Scholar]
- [57].Tiwari V, Clement C, Xu D, Valyi-Nagy T, Yue BY, Liu J, et al. Role for 3-O-sulfated heparan sulfate as the receptor for herpes simplex virus type 1 entry into primary human corneal fibroblasts. J Virol 2006;80:8970–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yakoub AM, Shukla D. Autophagy stimulation abrogates herpes simplex virus-1 infection. Sci Rep 2015;5:9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yakoub AM, Shukla D. Basal autophagy is required for herpes simplex virus-2 infection. Sci Rep 2015;5:12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yu Y, Zhao N, An J, Zhang X. CCAAT/Enhancer-Binding protein beta mediates the killing of toxoplasma gondii by inducing autophagy in nonhematopoietic cells. DNA Cell Biol 2017;36:212–8. [DOI] [PubMed] [Google Scholar]
- [61].Van Grol J, Muniz-Feliciano L, Portillo JA, Bonilha VL, Subauste CS. CD40 induces anti-Toxoplasma gondii activity in nonhematopoietic cells dependent on autophagy proteins. Infect Immun 2013;81:2002–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Szabo DJ, Nagymihaly R, Vereb Z, Josifovska N, Noer A, Liskova P, et al. Ex vivo 3D human corneal stroma model for Schnyder corneal dystrophy - role of autophagy in its pathogenesis and resolution. Histol Histopathol 2018;33:455–62. [DOI] [PubMed] [Google Scholar]
- [63].Yamazoe K, Yoshida S, Yasuda M, Hatou S, Inagaki E, Ogawa Y, et al. Development of a transgenic mouse with R124H human TGFBI mutation associated with granular corneal dystrophy type 2. PLoS One 2015;10:e0133397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zheng W, Qian Y, Chen S, Ruan H, Fan C. Rapamycin protects against peritendinous fibrosis through activation of autophagy. Front Pharmacol 2018;9:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Singh KK, Lovren F, Pan Y, Quan A, Ramadan A, Matkar PN, et al. The essential autophagy gene ATG7 modulates organ fibrosis via regulation of endothelial-to-mesenchymal transition. J Biol Chem 2015;290:2547–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ding Y, Kim S, Lee SY, Koo JK, Wang Z, Choi ME. Autophagy regulates TGF-beta expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J Am Soc Nephrol 2014;25:2835–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Stevenson W, Cheng SF, Dastjerdi MH, Ferrari G, Dana R. Corneal neovascularization and the utility of topical VEGF inhibition: ranibizumab (Lucentis) vs bevacizumab (Avastin). Ocul Surf 2012;10:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Zhu XR, Du JH. Autophagy: a potential target for the treatment of intraocular neovascularization. Int J Ophthalmol 2018;11:695–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wang H, Wang Y, Li D, Liu Z, Zhao Z, Han D, et al. VEGF inhibits the inflammation in spinal cord injury through activation of autophagy. Biochem Biophys Res Commun 2015;464:453–8. [DOI] [PubMed] [Google Scholar]
- [70].Shen W, Tian C, Chen H, Yang Y, Zhu D, Gao P, et al. Oxidative stress mediates chemerin-induced autophagy in endothelial cells. Free Radic Biol Med 2013;55:73–82. [DOI] [PubMed] [Google Scholar]
- [71].Li H, Gao A, Feng D, Wang Y, Zhang L, Cui Y, et al. Evaluation of the protective potential of brain microvascular endothelial cell autophagy on blood-brain barrier integrity during experimental cerebral ischemia-reperfusion injury. Translat stroke Res 2014;5:618–26. [DOI] [PubMed] [Google Scholar]
- [72].Han J, Pan XY, Xu Y, Xiao Y, An Y, Tie L, et al. Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy 2012;8:812–25. [DOI] [PubMed] [Google Scholar]
- [73].Buraschi S, Neill T, Goyal A, Poluzzi C, Smythies J, Owens RT, et al. Decorin causes autophagy in endothelial cells via Peg 3. Proc Natl Acad Sci U S A 2013;110:E2582–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Nguyen TM, Subramanian IV, Xiao X, Ghosh G, Nguyen P, Kelekar A, et al. Endostatin induces autophagy in endothelial cells by modulating Beclin 1 and beta-catenin levels. J Cell Mol Med 2009;13:3687–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Chen ML, Yi L, Jin X, Liang XY, Zhou Y, Zhang T, et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy 2013;9:2033–45. [DOI] [PubMed] [Google Scholar]
- [76].Uberti F, Lattuada D, Morsanuto V, Nava U, Bolis G, Vacca G, et al. Vitamin D protects human endothelial cells from oxidative stress through the autophagic and survival pathways. J Clin Endocrinol Metab 2014;99:1367–74. [DOI] [PubMed] [Google Scholar]
- [77].Halilovic A, Schmedt T, Benischke AS, Hamill C, Chen Y, Santos JH, et al. Menadione-induced DNA damage leads to mitochondrial dysfunction and fragmentation during rosette formation in Fuchs endothelial corneal dystrophy. Antioxidants Redox Signal 2016;24:1072–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Twig G, Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxidants Redox Signal 2011;14:1939–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Benischke AS, Vasanth S, Miyai T, Katikireddy KR, White T, Chen Y, et al. Activation of mitophagy leads to decline in Mfn2 and loss of mitochondrial mass in Fuchs endothelial corneal dystrophy. Sci Rep 2017;7:6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Jalimarada SS, Ogando DG, Bonanno JA. Loss of ion transporters and increased unfolded protein response in Fuchs’ dystrophy. Mol Vis 2014;20:1668–79. [PMC free article] [PubMed] [Google Scholar]
- [81].Stone EM, Mathers WD, Rosenwasser GO, Holland EJ, Folberg R, Krachmer JH, et al. Three autosomal dominant corneal dystrophies map to chromosome 5q. Nat Genet 1994;6:47–51. [DOI] [PubMed] [Google Scholar]
- [82].Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Prog Retin Eye Res 2016;50:67–88. [DOI] [PubMed] [Google Scholar]
- [83].Nie D, Peng Y, Li M, Liu X, Zhu M, Ye L. Lithium chloride (LiCl) induced autophagy and downregulated expression of transforming growth factor beta-induced protein (TGFBI) in granular corneal dystrophy. Exp Eye Res 2018;173:44–50. [DOI] [PubMed] [Google Scholar]
- [84].Choi SI, Kim KS, Oh JY, Jin JY, Lee GH, Kim EK. Melatonin induces autophagy via an mTOR-dependent pathway and enhances clearance of mutant-TGFBIp. J Pineal Res 2013;54:361–72. [DOI] [PubMed] [Google Scholar]
- [85].Berardi DE, Campodonico PB, Diaz Bessone MI, Urtreger AJ, Todaro LB. Autophagy: friend or foe in breast cancer development, progression, and treatment. Int J Breast Cancer 2011;2011:595092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Brennan LA, Kantorow WL, Chauss D, McGreal R, He S, Mattucci L, et al. Spatial expression patterns of autophagy genes in the eye lens and induction of autophagy in lens cells. Mol Vis 2012;18:1773–86. [PMC free article] [PubMed] [Google Scholar]
- [87].Choi SI, Kim BY, Dadakhujaev S, Jester JV, Ryu H, Kim TI, et al. Inhibition of TGFBIp expression by lithium: implications for TGFBI-linked corneal dystrophy therapy. Invest Ophthalmol Vis Sci 2011;52:3293–300. [DOI] [PubMed] [Google Scholar]
- [88].Lichtenstein A, Minogue PJ, Beyer EC, Berthoud VM. Autophagy: a pathway that contributes to connexin degradation. J Cell Sci 2011;124:910–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Liebau MC, Braun F, Hopker K, Weitbrecht C, Bartels V, Muller RU, et al. Dysregulated autophagy contributes to podocyte damage in Fabry’s disease. PLoS One 2013;8:e63506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Mizumura K, Cloonan S, Choi ME, Hashimoto S, Nakahira K, Ryter SW, et al. Autophagy: friend or foe in lung disease? Ann Am Thorac Soc 2016;13(Suppl 1):S40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Park HY, Kim JH, Park CK. Activation of autophagy induces retinal ganglion cell death in a chronic hypertensive glaucoma model. Cell Death Dis 2012;3. e290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Rodriguez-Muela N, Germain F, Marino G, Fitze PS, Boya P. Autophagy promotes survival of retinal ganglion cells after optic nerve axotomy in mice. Cell Death Differ 2012;19:162–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, Neufeld AH. Autophagy, exosomes and drusen formation in age-related macular degeneration. Autophagy 2014;5:563–4. [DOI] [PubMed] [Google Scholar]
- [94].Wu S, Sun C, Tian D, Li Y, Gao X, He S, et al. Expression and clinical significances of Beclin1, LC3 and mTOR in colorectal cancer. Int J Clin Exp Pathol 2015;8:3882–91. [PMC free article] [PubMed] [Google Scholar]
- [95].Zhao Z, Xue J, Zhao X, Lu J, Liu P. Prognostic role of autophagy-related proteins in epithelial ovarian cancer: a meta-analysis of observational studies. Minerva Med 2017;108:277–86. [DOI] [PubMed] [Google Scholar]
- [96].Frake RA, Ricketts T, Menzies FM, Rubinsztein DC. Autophagy and neurodegeneration. J Clin Invest 2015;125:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Liang P, Le W. Role of autophagy in the pathogenesis of multiple sclerosis. Neurosci Bull 2015;31:435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Yoshii SR, Kuma A, Akashi T, Hara T, Yamamoto A, Kurikawa Y, et al. Systemic analysis of atg5-null mice rescued from neonatal lethality by transgenic ATG5 expression in neurons. Dev Cell 2016;39:116–30. [DOI] [PubMed] [Google Scholar]
- [99].Garner TP, Long J, Layfield R, Searle MS. Impact of p62/SQSTM1 UBA domain mutations linked to Paget’s disease of bone on ubiquitin recognition. Biochemistry 2011;50:4665–74. [DOI] [PubMed] [Google Scholar]
- [100].Homma T, Ishibashi D, Nakagaki T, Satoh K, Sano K, Atarashi R, et al. Increased expression of p62/SQSTM1 in prion diseases and its association with pathogenic prion protein. Sci Rep 2014;4:4504. [DOI] [PMC free article] [PubMed] [Google Scholar]