

Abstract
Genomic alterations underlying chemotherapy resistance remains poorly characterized in pediatric acute myeloid leukemia (AML). In this study, we used whole exome sequencing to identify gene mutations associated with chemo-resistance in 44 pediatric AML patients. We identified previously unreported mutations involving epigenetic regulators such as KDM5C, SRIT6, CHD4, and PRPF6 in pediatric AML patients. Despite low prevalence in general pediatric AML, mutations involving epigenetic regulators including splicing factors, were collectively enriched as a group in primary chemo-resistance AML patients. In addition, clonal evolution analysis of secondary chemo-resistance AML patients reveals dominant clone at diagnosis could survive several course of intensified chemotherapy. And gain of new mutations in genes such as MVP, TCF3, SS18, and BCL10, may contribute to chemo-resistance at relapse. These results provide novel insights into the genetic basis of treatment failure in pediatric AML.
Keywords: Primary chemo-resistance, Epigenetic regulator mutations, Pediatric leukemia, Whole exome sequencing
Chemotherapy resistance remains a major concern in pediatric acute myeloid leukemia (AML) despite substantial improvement of treatment outcomes [1,2]. While the molecular basis of therapeutic failure was intensely interrogated, genomic alterations of chemotherapy resistance remains poorly characterized in pediatric AML. We have performed whole exome sequencing (WES) to identify gene mutations associated with primary and secondary chemo-resistance in pediatric AML.
Specimens were collected from patients treated at the Hematology and Blood Diseases Hospital of Tianjin and Children’s Hospital of Soochow University in China. All of the patients, including 18 cases of primary chemo-resistance (including 1 case of both primary and secondary chemoresistance), 3 cases of secondary chemo-resistance, 23 cases of primary complete remission, provided written consent and were enrolled in institutional protocols (Supplementary Table 1 and Data I). Primary and secondary chemo-resistance were defined based on criteria of failure to achieve complete remission (CR) of 1 cycle or 2 cycles of induction chemotherapy after the initial diagnosis or after relapse [3]. Bone marrow aspirate samples were obtained at diagnosis (pretreatment) and relapse. Leukemia cells were sorted by a surface marker selection of CD13, CD33, CD34, and CD117 using flow cytometry, and T or B cells were purified as a germline control. Detailed sample preparation and sequencing procedures are provided in the supplementary methods.
Whole exome sequence were performed on collected specimens from 44 pediatric AML patients. Mutation analysis, including single nucleotide variants (SNVs) and small indels, revealed a total of approximately 378 nonsynonymous mutations with an average of 8 protein-coding mutations per case (Supplementary Data II). The sequencing coverage of WES was approximately 150–200X, which has the power to detect variants with a mutation allele frequency (MAF) of at least above 0.1. Detailed methods and mutation analysis results are provided in the supplementary methods.
We first compared the mutation frequency of 44 total pediatric AML cases, to that of a cohort of 200 adult AML cases from the The Cancer Genome Atlas (TCGA) dataset. We observed that the mutation frequency of signaling molecules was much higher in pediatric AML than in adult AML. The most frequently mutated genes were NRAS (27%, 12/44) and KIT (20%, 9/44) in pediatric AML, which have significantly higher mutation frequencies than in adult AML (NRAS, P < 0.001; KIT, P < 0.001, Chi-Squared Test) (Supplementary Fig. 1). Other recurrent mutations in signaling pathways were also highly enriched in pediatric AML, such as JAK2 (7% vs 0.5%, P < 0.01), CSF3R (7% vs 0.5%, P < 0.01), and MPL (4.4% vs 0.5%, P = 0.03, Chi-Squared Test). In contrast to a high prevalence of mutations in signaling molecules, epigenetic regulators such as DNMT3A (0%, 0/44), TET2 (0%, 0/44), and IDH1/2 (2%, 1/44) are rarely mutated in pediatric AML patients, while they are commonly found mutated in adult AML (Supplementary Fig. 1). Our results are in consistence with previous findings from targeted sequencing regarding mutation frequencies of DNMT3A, TET2, and IDH1/2 genes in pediatric and adult AML(Supplementary Table 4) [4]. It is worth noting that we uncovered several novel mutations of epigenetic regulators that were previously unreported in AML, such as KDM5C, SIRT6, CHD4, PRPF6, and SS18 (Supplementary Table 2). Taken together, the pathogenesis and mutation spectrum of pediatric AML is different from adult AML, as mutations of signaling molecules are commonly found in pediatric AML, whereas epigenetic regulators are more frequently mutated in adult AML.
We assessed whether any mutated genes were associated with primary chemo-resistance in our pediatric cohort. The chemo-sensitive group contained 23 primary complete remission samples and 3 primary chemo-sensitive diagnosed samples of secondary chemo-resistant patients. In addition to a high recurrence of FLT3-ITD (44%, 8/18), we observed that none of the signaling molecule mutations were significantly enriched in the primary chemo-resistant group. It has been reported that KIT mutations are frequently mutated in pediatric AML and associated with adverse outcomes [5]. In our studied cohort, KIT mutations were found in relapsed (57%, 4/7) and primary chemo-resistant (28%, 2/7) AML patients (Supplementary Table 3).
Notably, mutations of epigenetic regulators were collectively enriched in the primary chemo-resistant group (7/18 vs 2/26, P < 0.01, Chi-Squared Test), including the histone modification enzymes KDM5C, SIRT6, and SETD8, splicesomal factors PRPF6 and PRPF8, and other epigenetic regulators CHD4 and IDH2 (Fig. 1, Supplementary Table 2). We found a high allele frequency of the KDM5C point mutation (35%) and SIRT6 frameshift mutation (42%) in one chemorefractory t(8:21) translocated AML patient (Fig. 2, Supplementary Fig. 2). KDM5C is a histone demethylase that specifically demethylates di- and trimethylated lysine 4 on histone 3 (H3K4), which is frequently mutated in clear cell renal cell carcinomas (ccRCCs) [6]. Interestingly, MLL2 as a H3K4 methyltransferase, were found recurrently mutated in AML [7]. SIRT6, a histone H3 lysine 9 (H3K9) deacetylase, were found dysregulated in AML patients and implicated in regulating DNA damage repair [8]. CHD4 as a chromodomain helicase DNA-binding protein, was also implicated in determining sensitivity of chemo-drugs to AML cells [9]. In addition, we also identified splicing factor mutations PRPF6 and PRPF8 in chemorefractory AML patients. PRPF6 and PRPF8, two evolutionarily conserved spliceosomal proteins, are the key components in the U4/U6.U5 tri-snRNP spliceosome complex, which is critical for the exon excision and ligation process from pre-mRNA to mature mRNA. PRPF6 and PRPF8 may promote leukemogenesis and chemotherapy resistance by aberrant splicing [10]. In light of the recent discovery of epigenetic alteration mediating chemo-resistance in adult AML [11,12], we speculated that these mutations of epigenetic regulators found in our cohort play a critical role in chemorefractory pediatric AML.
Fig. 1.
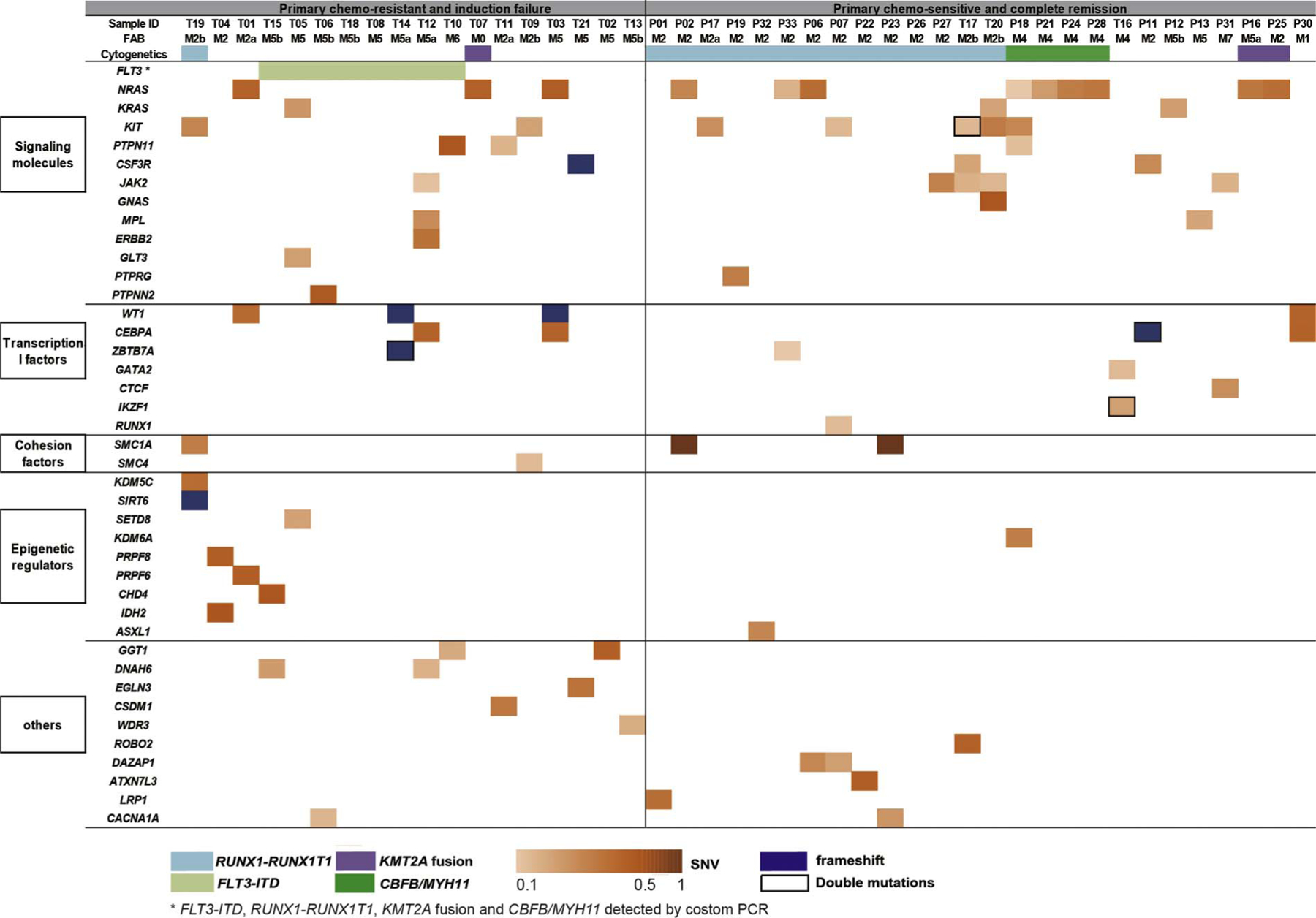
Mutation spectrum of 44 pediatric AML patients. Mutations present in primary chemo-resistance (n = 18) and primary chemo-sensitive (n = 26) pediatric AML patients are shown in two separated groups. Cytogenetic subtypes, SNVs and frameshift mutations are shown in colored labels as indicated at the bottom of the figure.
Fig. 2.
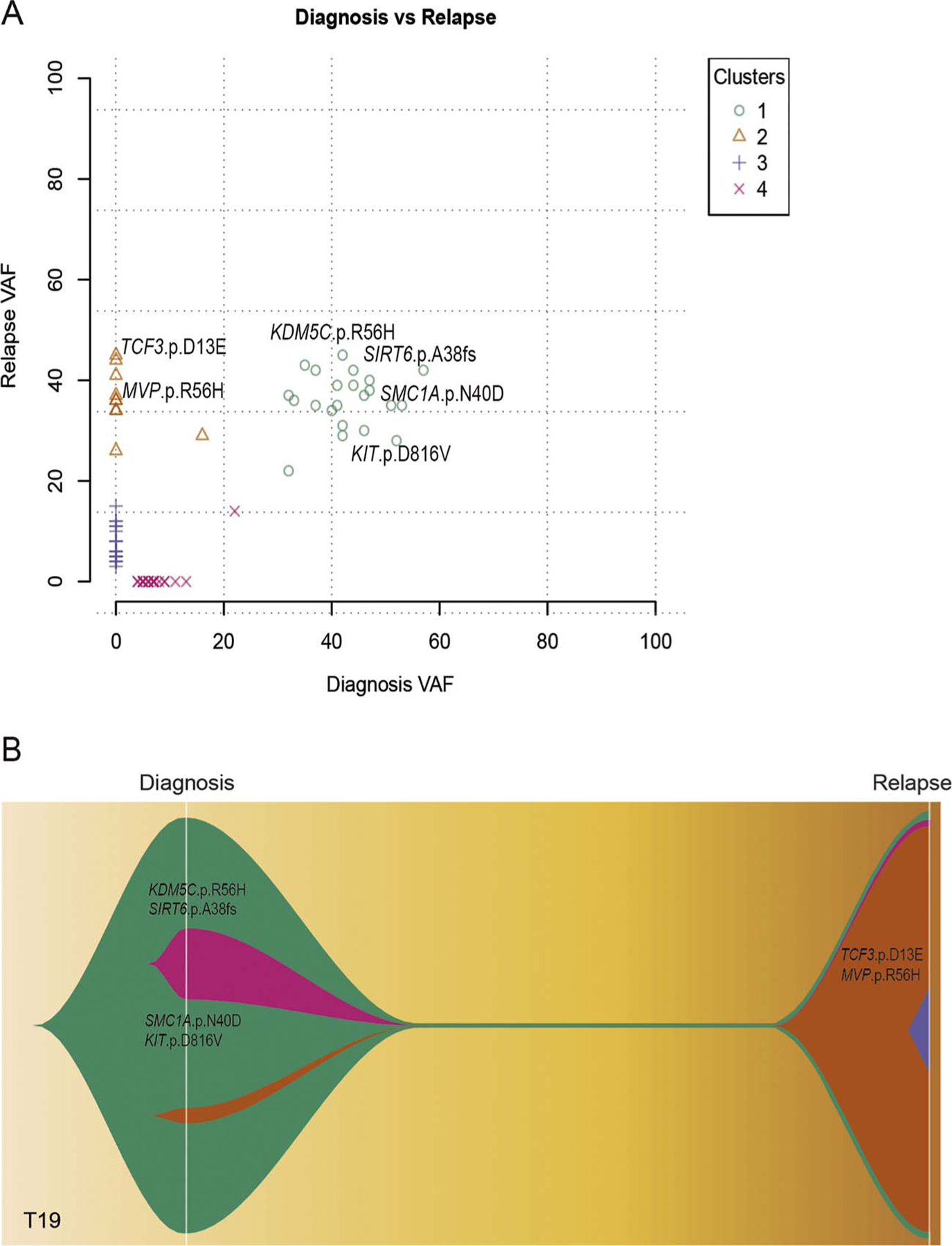
Clonal architecture and evolution patterns from diagnosis to relapse of one representative chemorefractory pediatric AML case, T19. A. Plot graph for four mutational clusters (1–4) as constructed by mutational allele frequency at diagnosis and relapse. B. Clonal evolution graph created by fishplot to show clonal lineage changes from diagnosis to relapse.
To investigate the molecular basis and clonal evolution underlying secondary chemo-resistant AML, we performed whole exome sequencing of diagnosis-relapse paired samples from 4 pediatric AML patients, who underwent chemotherapy induction failure at relapse. We analyzed clonal composition inferred from variant allele frequency changes from diagnosis to relapse. Besides from T16, which showed lack of mutations in dominant clone at diagnosis (Supplementary Fig. 2D), we identified the same mutation turnover pattern in other 3 chemo-resistant cases (T17, T19, T20) containing a t(8;21) translocation. The dominant clone with t(8;21) translocation persists even after several courses of intensified chemotherapy. Gain of new mutations in genes such as MVP, TCF3, SS18, and BCL10, may contribute to chemo-resistance in relapsed AML (Fig. 2, Supplementary Fig. A, B and 2C). One particular case is T19, who showed early-relapse (approximately 6 months) and was resistant to chemotherapy. By mutation frequency and clonal evolution analysis, we uncovered a high frequency of mutations in KDM5C and SIRT6, which coexisted with KIT and SMC1A mutations from diagnosis to relapse (Fig. 2A and B, Cluster 1). And a minor clone with the acquisition of additional mutations in TCF3 and MVP was expanded at relapse (Fig. 2A and B, Cluster 2). These epigenetics-related and relapse-specific mutations may contribute to the aggressiveness and chemo-resistance phenotype of t (8: 21) translocated AML.
The development of new treatment strategies for pediatric AML needs to be based on an improved understanding of the molecular mechanisms underlying chemotherapy resistance. Previously, Brown, et al. had shown that SETBP1, ASXL1, and RELN mutations were associated with primary chemo-resistance in adult and pediatric AML patients by utilizing targeted sequencing of 670 genes [12]. We have, for the first time, performed whole exome sequencing on a Chinese cohort of chemorefractory pediatric AML patients. We identified novel mutations in epigenetic regulators and spliceosomal components, such as KDM5C, SIRT6, CHD4, PRPF6, and SS18, that may play a critical role in leukemogenesis and chemo-resistance in pediatric AML patients. Analysis of clonal evolution in chemorefractory AML suggests that the persistence of epigenetic factor mutations may contribute to chemo-resistance and relapse. Taken together, our study sheds light on genetic lesions and evolutionary patterns underlying chemotherapy resistance and failure in pediatric AML.
Supplementary Material
Acknowledgements
This work was supported by the National Key Basic Research Program of China (2014CB542001 to Q.W.), the National Nature Science Foundation of China (81425003 to Q.W.; 81421002 to X.Z.; 81470339 to X.Z.; 81400137 to Y.Z.), CAMS innovation fund for Medical Science (2016-12M-1-002 to X.Z.), the PUMC Youth Fund(3332016091 to Y.Z.) and Social Development Project of Jiangsu Province (CXTDA2017014 to S.H.)
Footnotes
Conflict of interest
The authors declare no potential conflicts of interest.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.leukres.2017.12.001.
Contributor Information
Di Zhan, Key Laboratory of Genomic and Precision Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Yingchi Zhang, State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China; Division of Pediatric Blood Diseases Center, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China.
Peifang Xiao, The Children’s Hospital of Soochow University, Suzhou 215003, China.
Xinchang Zheng, Key Laboratory of Genomic and Precision Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Min Ruan, State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China; Division of Pediatric Blood Diseases Center, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China.
Jingliao Zhang, State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China; Division of Pediatric Blood Diseases Center, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China.
Aili Chen, Key Laboratory of Genomic and Precision Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Yao Zou, State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China; Division of Pediatric Blood Diseases Center, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China.
Yumei Chen, State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China; Division of Pediatric Blood Diseases Center, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China.
Gang Huang, Divisions of Pathology and Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, Ohio 45229, USA.
Shaoyan Hu, The Children’s Hospital of Soochow University, Suzhou 215003, China.
Qian-fei Wang, Key Laboratory of Genomic and Precision Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Xiaofan Zhu, State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China; Division of Pediatric Blood Diseases Center, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin, China.
References
- [1].Pession A, Masetti R, Rizzari C, Putti MC, Casale F, Fagioli F, et al. , Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia, Blood 122 (2013) 170–178. [DOI] [PubMed] [Google Scholar]
- [2].Quarello P, Fagioli F, Basso G, Putti MC, Berger M, Luciani M, et al. , Outcome of children with acute myeloid leukaemia (AML) experiencing primary induction failure in the AIEOP AML 2002/01 clinical trial, British Journal of Haematology 171 (2015) 566–573. [DOI] [PubMed] [Google Scholar]
- [3].Cheson BD, Bennett JM, Kopecky KJ, Buchner T, Willman CL, Estey EH, et al. , Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia, Journal of Clinical Oncology 21 (2003) 4642–4649. [DOI] [PubMed] [Google Scholar]
- [4].Liang DC, Liu HC, Yang CP, Jaing TH, Hung IJ, Yeh TC, et al. , Cooperating gene mutations in childhood acute myeloid leukemia with special reference on mutations of ASXL1, TET2, IDH1, IDH2, and DNMT3A, Blood 121 (2013) 2988–2995. [DOI] [PubMed] [Google Scholar]
- [5].Tokumasu M, Murata C, Shimada A, Ohki K, Hayashi Y, Saito AM, et al. , Adverse prognostic impact of KIT mutations in childhood CBF-AML: the results of the Japanese Pediatric Leukemia/Lymphoma Study Group AML-05 trial, Leukemia 29 (2015) 2438–2441. [DOI] [PubMed] [Google Scholar]
- [6].Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, et al. , Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes, Nature 463 (2010) 360–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mar BG, Bullinger L, Basu E, Schlis K, Silverman LB, Dohner K, et al. , Sequencing histone-modifying enzymes identifies UTX mutations in acute lymphoblastic leukemia, Leukemia 26 (2012) 1881–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cea M, Cagnetta A, Lovera D, Grasso R, Colombo N, Bergamaschi M, et al. , A Novel Synthetic Lethal Approach Targeting SIRT6 in Acute Myeloid Leukemia, Blood 126 (2015) 1375. [Google Scholar]
- [9].Sperlazza J, Rahmani M, Beckta J, Aust M, Hawkins E, Wang SZ, et al. , Depletion of the chromatin remodeler CHD4 sensitizes AML blasts to genotoxic agents and reduces tumor formation, Blood 126 (2015) 1462–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kurtovic-Kozaric A, Przychodzen B, Singh J, Konarska MM, Clemente MJ, Otrock ZK, et al. , PRPF8 defects cause missplicing in myeloid malignancies, Leukemia 29 (2015) 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, et al. , DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling, Nature Medicine 22 (2016) 1488–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Brown FC, Cifani P, Drill E, He J, Still E, Zhong S, et al. , Genomics of primary chemoresistance and remission induction failure in paediatric and adult acute myeloid leukaemia, British Journal of Haematology 176 (2017) 86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.