

Abstract
Protein arginine (R) methylation is a post-translational modification involved in various biological processes, such as RNA splicing, DNA repair, immune response, signal transduction, and tumor development. Although several advancements were made in the study of this modification by mass spectrometry, researchers still face the problem of a high false discovery rate. We present a dataset of high-quality methylations obtained from several different heavy methyl stable isotope labeling with amino acids in cell culture experiments analyzed with a machine learning–based tool and show that this model allows for improved high-confidence identification of real methyl-peptides. Overall, our results are consistent with the notion that protein R methylation modulates protein–RNA interactions and suggest a role in rewiring protein–protein interactions, for which we provide experimental evidence for a representative case (i.e., NONO [non-POU domain–containing octamer-binding protein]–paraspeckle component 1 [PSPC1]). Upon intersecting our R-methyl-sites dataset with the PhosphoSitePlus phosphorylation dataset, we observed that R methylation correlates differently with S/T-Y phosphorylation in response to various stimuli. Finally, we explored the application of heavy methyl stable isotope labeling with amino acids in cell culture to identify unconventional methylated residues and successfully identified novel histone methylation marks on serine 28 and threonine 32 of H3. The database generated, named ProMetheusDB, is freely accessible at https://bioserver.ieo.it/shiny/app/prometheusdb.
Keywords: heavy methyl SILAC, machine learning, mass spectrometry, protein methylation, PTM crosstalk, protein–protein interactions
Abbreviations: ADMA, asymmetric dimethyl-arginine; dRT, retention time difference; FDR, false discovery rate; GO, Gene Ontology; hmSILAC, heavy methyl SILAC; IP, immunoprecipitation; K, lysine; LLPS, liquid–liquid phase separation; LogRatio, log2 H/L intensity ratio; ME, mass error; ML, machine learning; MMA, monomethyl-arginine; MS, mass spectrometry; NONO, non-POU domain–containing octamer-binding protein; PPARα, peroxisome proliferator–activated receptor alpha; PPI, protein–protein interaction; PRMT, protein arginine methyltransferase; ProMetheusDB, ProMetheus database; PSPC1, paraspeckle component 1; PTM, post-translational modification; R, arginine; RBP, RNA-binding protein; RNP, ribonucleoprotein; RT, room temperature; SDMA, symmetric dimethyl-arginine; SILAC, stable isotope labeling with amino acids in cell culture; WCE, whole-cell extract
Graphical Abstract
Highlights
-
•
hmSEEKER 2.0 identifies methyl-peptides from hmSILAC data through machine learning.
-
•
Arginine methylation plays a role in modulating protein–protein interactions.
-
•
Arginine methylations occur more frequently in proximity of phosphorylation sites.
-
•
hmSEEKER 2.0 was used to identify methylations occurring on nonstandard amino acids.
In Brief
Protein arginine methylation is an important post-translational modification whose high-throughput identification by mass spectrometry is hampered by a high false discovery rate. We developed hmSEEKER 2.0 to identify methyl-sites from heavy methyl SILAC data through a machine learning–based algorithm. We generated ProMetheusDB, the largest comprehensive database of high-confidence protein methylations. The functional analysis of ProMetheusDB suggests a role of arginine methylation in processes such as metabolism, immunity, protein–protein interactions, and functional crosstalk with phosphorylation.
Protein methylation is a widespread post-translational modification (PTM) that consists of the addition of one or more methyl (CH3) groups to a residue, most frequently an arginine (R) or lysine (K). R can be monomethylated (monomethyl-arginine [MMA]) or dimethylated, and the dimethylation can be symmetrical (symmetric dimethyl-arginine [SDMA]) if the two methyl groups are bound to different nitrogen atoms of the guanidino group or asymmetrical (asymmetric dimethyl-arginine [ADMA]) if they are bound to the same atom. Overall, methylation does not change the charge state of R but alters their steric hindrance and reduces their ability to form hydrogen bonds with other molecules, thus affecting their interactions with other biomolecules, both positively and negatively (1). Along this line, the distinction between ADMA and SDMA is crucial, because the two modifications are recognized by distinct readers and can lead to completely different functional outcomes, as exemplified by asymmetrical and symmetrical dimethylation on R3 of histone H4 (H4R3) that are associated to transcriptional activation and repression, respectively (2).
In mammals, the transfer of methyl groups from the biological donor SAM to R is carried out by the nine members of the protein arginine methyltransferase (PRMT) family that are classified into three groups: type I PRMTs catalyze the formation of MMA and ADMA; type II PRMTs catalyze the formation of MMA and SDMA; and PRMT7, which is the only type III PRMT, leads to MMA only (3). PRMT1 is the most active enzyme among type I PRMTs and PRMTs in general, being overall responsible for ∼85% of the methylation events in the human cell (4), whereas PRMT5 is the most active type II PRMT (5).
While methylation of histones and its implication in transcriptional regulation has been extensively studied in several model systems and functional states (6), the concept that R methylation is widespread on non-histone proteins is supported by more recent studies (7, 8, 9, 10, 11). PRMT1, PRMT3, PRMT4, PRMT5, and PRMT7 have all been reported to catalyze methylation on RNA-binding proteins (RBPs), modulating their interactions with RNAs and thus impacting on biological processes, such as mRNA splicing, miRNA maturation, translation, and ribonucleoprotein (RNP) granules assembly (8, 12, 13, 14, 15, 16, 17). Importantly, PRMTs are often aberrantly expressed in cancer (18, 19) and in neurodegenerative and metabolic disorders (20, 21). Hence, there is a great interest in developing therapies based on the pharmacological modulation of these enzymes, with some PRMT inhibitors already in clinical trials (identifiers NCT03573310, NCT02783300, NCT03614728, and NCT03666988) (19, 22).
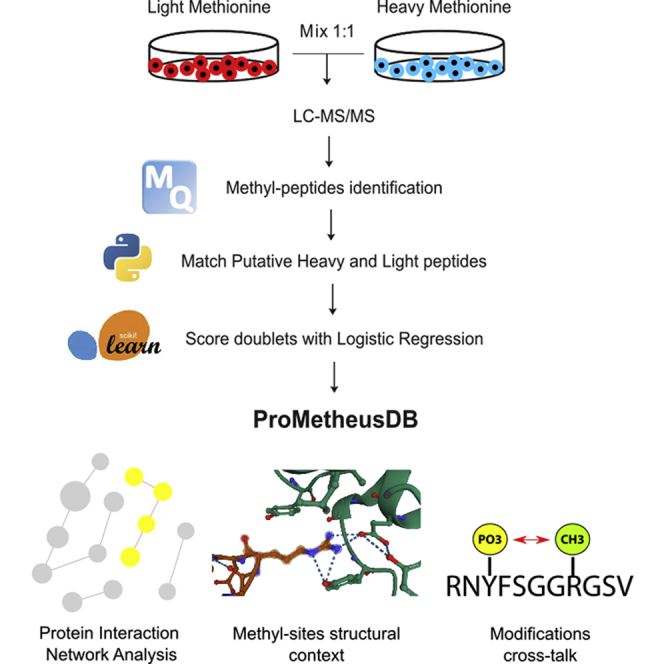
Recent studies on protein methylation took advantage of the progress made by LC–MS/MS–based proteomics technology. Several mass spectrometry (MS)–based studies have annotated progressively larger methyl-proteomes, yet it was shown that the proteome-wide identification of methyl-sites through LC–MS/MS is prone to a high false discovery rate (FDR) (23). In fact, the chemical methylesterification of an acidic residue (aspartate and glutamate) and some conservative amino acid substitutions (alanine to valine and aspartate to glutamate) can be erroneously identified as in vivo methylations on nearby R residues by peptide search engines. To address this issue, Ong and Mann (24) proposed the heavy methyl stable isotope labeling with amino acids in cell culture (hmSILAC) metabolic labeling strategy, whereby cells are grown in the presence of either natural methionine (M0) or [13CD3]-methionine (M4) to isotopically label the methyl groups. Upon mixing the heavy and light samples, methylated peptides can be detected by MS as pairs of MS1 peaks separated by a specific mass difference. The identification of these doublets allows distinguishing naturally occurring methyl-peptides from false positives, which cannot exist in heavy form since the isotopically heavy methyl groups can derive only from in vivo enzymatic methylation; hence, hmSILAC has been proposed as a strategy to orthogonally validate true methyl-site annotations by MS.
Until recently, the application of hmSILAC was held back by the lack of computational tools for the data analysis, given that common peptide search engines cannot handle isotopic labeling encoded by a PTM. To overcome this limitation, Tay et al. (25) developed MethylQuant, and we published hmSEEKER (26), two computational tools that operate downstream of a traditional peptide search engines [MaxQuant, (Max Planck Institute of Biochemistry) (27) in the case of hmSEEKER] by matching heavy- and light-labeled methyl-peptides to their counterparts. Since the release of the first version of hmSEEKER, we undertook an effort to improve its performance: following a strategy similar to that described in (26), we trained a machine learning (ML) model to recognize hmSILAC doublets generated by M-containing peptides, which was then used to reanalyze all the hmSILAC experiments collected in our laboratory so far in order to generate a comprehensive dataset of high-confidence protein methylations.
While the majority of MS-based methyl-proteomics studies have traditionally focused only on R and K methylation, growing evidence suggests that methylation can naturally occur also on aspartate (D), glutamate (E), asparagine (N), glutamine (Q), serine (S), threonine (T), histidine (H), cysteine (C), and protein N and C termini (28). This bias is mainly because of the lack of methods for the affinity enrichment of these unconventional methylations and the limited knowledge about the enzymes that set or remove them.
Overall, our MS-proteomics workflow and the improved computational analysis of the MS data have produced what, at the time of writing, is the largest orthogonally validated MS-based methyl-proteomics dataset. We then integrated this dataset with a list of R-methyl-sites that are significantly regulated in perturbed SILAC experiments, thus generating the ProMetheus database (ProMetheusDB) of high-confidence protein methylation sites.
The in-depth analysis of ProMetheusDB not only confirmed the role of R methylation in different aspects of RNA metabolism but also suggested its involvement in novel cellular processes, such as antigen processing and presentation, macrophage metabolism, and immunity. We also investigated the potential crosstalk between R methylation and S/T-Y phosphorylation and carried out a structural analysis of R-methyl-sites at protein–protein interaction (PPI) interfaces. In this context, we confirmed the computational prediction that R methylation of the protein non-POU domain–containing octamer-binding protein (NONO) positively affects its binding to paraspeckle component 1 (PSPC1).
ProMetheusDB and the functional analysis associated can serve as a precious resource for all scientists focusing on this important PTM and on the enzymes responsible of their dynamic regulation, many of which represent promising pharmacological targets for treatment of various diseases, from neurological disorders to cancer (19, 22).
Experimental Procedures
Experimental Design and Statistical Rationale
The annotation of ProMetheusDB was obtained by combining datasets derived from two distinct proteomics strategies. The majority of the methylations were annotated from hmSILAC experiments that allow this PTM to be orthogonally validated, as described (24). This initial list was then integrated with a second list of regulated methyl-peptides derived from MS-proteomics experiments where the biochemical protocol to separate and affinity-enrich methyl-peptides was coupled to metabolic labeling with standard SILAC amino acids (K/R) and stimulation of cells with different perturbations. Each SILAC experiment was done in duplicates (forward and reverse), and only peptides whose regulation was reproducible in at least one pair of experiments were considered significantly regulated. Significantly upregulated or downregulated were determined as described previously (13, 16, 29, 30): briefly, for each SILAC experiment, the mean and standard deviation of the unmodified peptide SILAC log2 ratios distribution were calculated; then, these values were used to calculate the z-scores of the methyl-peptide SILAC ratios; finally, a peptide was classified as significantly regulated in a given experiment if its z-score was >2 or <−2 in both the forward and reverse replicates of that experiment. The analyzed experiments are described in supplemental Table S1.
Extraction and Protease Digestion of hmSILAC-Labeled Histones Before LC–MS Analysis
hmSILAC-labeled HeLa S3 cells (L and H channels mixed in 1:1 ratio) were resuspended in lysis buffer (10% sucrose, 0.5 mM EGTA, 60 mM KCl, 15 mM NaCl, 15 mM Hepes, 0.5 mM PMSF, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 1 mM DTT, 5 mM sodium butyrate, 5 mM NaF, 30 μg/ml spermine, 30 μg/ml spermidine, and 0.5% Triton X-100), and nuclei were separated from cytoplasm by centrifugation on a sucrose cushion (20 ml lysis buffer, 10% sucrose) for 30 min at 3695g, 4 °C. Histones were then extracted through 0.4 M hydrochloric acid for 5 h at 4 °C and dialyzed overnight in 100 mM CH3COOH. Dialyzed histones were then lyophilized and either kept at −80 °C until use or directly resuspended in milliQ water before sample processing prior to MS analysis. To maximize the protein sequence coverage, four different aliquots of 5 μg histones each were in-solution digested overnight using different proteases, such as ArgC, trypsin, LysargiNase, and LysC. Proteolytic peptides were then desalted and concentrated by microchromatography onto SCX and C18 Stage Tips microcolumn, prior to LC–MS/MS analysis.
LC–MS/MS Analysis
Peptide mixtures were analyzed by online nano-flow LC–MS/MS using an EASY-nLC 1000 or 1200 (Thermo Fisher Scientific) connected to a Q Exactive instrument (Thermo Fisher Scientific) through a nanoelectrospray ion source. The nano-LC system was operated in one column set up with a 50 cm analytical column (75 μm inner diameter) packed with C18 resin (EASY-Spray PEPMAP RSLC C18 2 μm 50 cm × 75 μm; Thermo Fisher Scientific). Solvent A was 0.1% formic acid, and solvent B was 0.1% formic acid in 80% acetonitrile. Samples were injected in an aqueous 0.1% TFA solution at a flow rate of 500 nl/min. Peptides were separated with a gradient of 5 to 40% solvent B over 90 min followed by a gradient of 40 to 60% for 10 min and 60 to 80% over 5 min at a flow rate of 250 nl/min in the EASY-nLC system. Histone peptides were separated with a gradient of 5 to 35% solvent B over 100 min followed by a gradient of 35 to 95% for 10 min and 60 to 80% over 8 min at a flow rate of 250 nl/min in the EASY-nLC system. The Q Exactive was operated in the data-dependent acquisition mode to automatically switch between full-scan MS and MS/MS acquisition. Survey full-scan MS spectra (from m/z 300 to 1150) were analyzed in the Orbitrap detector with resolution R = 35,000 at m/z 400. The 10 most intense peptide ions with charge states ≥2 were sequentially isolated to a target value of 3 × 106 and fragmented by higher energy collision dissociation with a normalized collision energy setting of 25%. The maximum allowed ion accumulation times were 20 ms for full scans and 50 ms for MS/MS, and the target value for MS/MS was set to 106. The dynamic exclusion time was set to 20 s.
MS Raw Data Processing with MaxQuant
MS raw data were analyzed with the integrated MaxQuant software, version 1.3.0.5 or version 1.5.2.8 (for SILAC) or version 1.6.2.10 (for hmSILAC), using the Andromeda search engine (27, 31). In all MaxQuant searches, the estimated FDR of all peptide identifications was set to a maximum of 1%. The mass tolerance for precursor ions was set to 20 ppm during the first search and to 4.5 ppm during the main search. The mass tolerance for fragment ions was set to 20 ppm. A maximum of three missed cleavages was permitted, and the minimum peptide length was fixed at six amino acids. The minimum modified peptide score was set to 1 (identifications were subsequently filtered during hmSEEKER analysis). The 2021_01 version of the Human UniProt–SwissProt reference proteome (UP000005640; 42,371 entries) was used for peptide identification. Other parameters were different, according to the experiment and the sample to be analyzed.
For the hmSILAC experiments, each MS raw data file was analyzed twice to identify heavy and light methyl-peptides separately. In the “light” analysis, monomethylation of K/R (+14.016 Da), dimethylation of K/R (+28.031 Da), trimethylation of K (+42.047 Da), and oxidation of M (+15.995 Da) were specified as variable modifications; carbamidomethylation of Cys (+57.021 Da) was specified as fixed modification. In the “heavy” analysis, heavy monomethylation of K/R (+18.038 Da), heavy dimethylation of K/R (+36.076 Da), heavy trimethylation of K (+54.114 Da), and oxidation of M were specified as variable modifications; carbamydomethylation of cysteine (C) and isotope-labeled methionine (MS) (+4.022 Da) were specified as fixed modifications. Enzyme specificity was set to either trypsin/P or LysargiNase. For the standard SILAC methyl-proteomics experiments, we indicated K8 + R10 and/or K4 + R6 as SILAC labels. N-terminal acetylation (+42.010 Da), M oxidation, monomethyl-K/R and dimethyl-K/R were set as variable modifications; carbamidomethylation of C was set as fixed modification. Enzyme specificity was set to trypsin/P. For histone hmSILAC experiments, MS raw data were also analyzed twice. Monomethylation (light or heavy) was allowed on K, R, D, E, H, Q, N, S, and T, and we included K acetylation (+42.011 Da) as a variable modification. Enzyme specificity was set to trypsin/P, ArgC, LysC, or LysargiNase. A database containing only human histone sequences was used as reference.
Validation of Methyl-peptides with hmSEEKER 2.0
Identification of light and heavy methyl-peptide doublets was carried out with hmSEEKER (26). The input of hmSEEKER consists of the “msms.txt” and “allPeptides.txt” generated by MaxQuant. Methyl-peptide identifications reported in “msms.txt” were filtered to only retain peptides with Andromeda score >25, Delta score >12 and localization probability of the methylation sites >0.9. hmSILAC doublets were then reconstructed based on the MS1 peaks reported in “allPeptides.txt.” Initially, hmSILAC v1.0 called a methyl-peptide doublet when two peaks had the same charge, |ME| <2 ppm, |dRT| <0.5 min, and |LogRatio| <1. The ML model developed in this study within hmSEEKER, v2.0 is a logistic regression model trained with Python package Scikit-learn, v0.23.1, as described in the Results section. To avoid biases in the model, the true positive and true negative doublets were obtained by re-analyzing a subset of MS raw data that was representative of all hmSILAC experiments we performed (supplemental Table S1).
Functional and Structural Analysis of the ProMetheusDB
Motif analysis was done using pLogo (University of Connecticut) (32). Sequence windows centered on regulated (or unchanging) R-methyl-sites were submitted as foreground; sequence windows centered on all identified R-methyl-sites (including nonquantified ones) were submitted as background; the resulting position weight matrices were then downloaded and visualized with the Logomaker Python package (33). Functional enrichment analysis was performed using the “gprofiler2” R package (34); terms from Gene Ontology (GO): Biological Processes (GO:BP), cellular component (GO:CC), molecular function (GO:MF), Reactome (REAC), Kyoto Encyclopedia of Genes and Genomes pathways, and CORUM complexes were used as data sources; only the most significant nonredundant terms are reported in the figures (full lists of enriched terms are reported in supplemental Table S2). Overlap of the annotated modification sites with InterPro domains (35) was performed with an in-house Python script; the InterPro database was filtered to include only regions classified as “domain” or “homologous superfamily.” Overlap of modification sites with disordered regions predicted by AlphaFold (36) was performed as previously described (37). Mapping of modification sites on interaction surfaces was performed with the Mechismo (University of Heidelberg) web application (38), with the stringency threshold set to “medium.” The database of currently annotated phosphorylation sites was downloaded on May 28, 2019 from PhosphoSitePlus (39). Protein–protein functional interaction networks were generated within Cytoscape (Institute for Systems Biology) (40) using the ReactomeFI PlugIn plugin (41), the same plugin was also used to perform the clustering; protein–protein physical interactions were downloaded from the IMEx database (42); network analysis was performed with Cytoscape and the Python package Pyntacle (43). The dataset of random R-sites was extracted from the 2021_01 version of the Human UniProt–SwissProt reference proteome. Fisher’s exact tests were performed with the Scipy package in Python. The ProMetheusDB web interface was implemented in R using the Shiny, ShinyFiles, and DataTable libraries.
Protein Immunoprecipitation of NONO and Western blot Analysis of its R Methylation State and Co-immunoprecipitation of PSPC1
Immunoprecipitation (IP) of NONO was performed starting from 1 mg of HeLa whole-cell extract (WCE). Briefly, 30 × 106 HeLa cells were harvested, washed twice with cold PBS, and resuspended in two volumes of radioimmunoprecipitation assay buffer (10 mM Tris [pH 8], 150 nM NaCl, 0.1% SDS, 1% Triton, 1 mM EDTA, 0.1% sodium deoxycholate, 1 mM PMSF, 1 mM DTT, and 1× Protease and Phosphatase Inhibitor Cocktails [Roche], supplemented with 10 kU of benzonase [Merck Life Science]). The suspension was rotated on a wheel for 45 min at room temperature (RT) (vortex every 10 min), centrifuged at 12,000g for 1 h at 4 °C, and the supernatant was transferred into a new Eppendorf tube. Proteins were quantified by bicinchoninic acid colorimetric assay (Pierce Bicinchoninic Acid Protein assay kit), and 1 mg of WCE was used for the IP, 8% of which was saved as input (80 μg to be divided into four SDS-PAGE gels). The WCE used for the IP was rotated at 4 °C overnight with 4 μg of anti-NONO/p54 (catalog no.: sc-376865; Santa Cruz). G protein–coupled magnetic beads (Dynabeads; Thermo Fisher Scientific) were saturated with a blocking solution (0.5% bovine serum albumin) and rotated at 4 °C overnight on a wheel. The following day, the beads were added to the lysate in 1:100 proportion with the primary antibodies and incubated for 3 h at 4 °C on the wheel; the captured complexes were washed four times with the radioimmunoprecipitation assay buffer and then incubated 10 min at 95° with LDS sample buffer (2×) supplemented with 100 mM DTT to elute the immunoprecipitated proteins. Equal protein amounts were separated by SDS-PAGE electrophoresis (NuPAGE Novex 4–12% Bis–Tris Gel 1.5 mm; Thermo Fisher Scientific) and transferred on transfer membrane (Immobilon-P; Merck Millipore) by wet-transfer method. Membrane blocking was performed with 10% bovine serum albumin/Tris-buffered saline and 0.1% Tween-20 for 1 h at RT and followed by overnight incubation with the selected primary antibodies and subsequent incubation with the horseradish peroxidase–conjugated secondary antibodies (Cell Signaling Technology) for 1 h at RT. Proteins were detected by enhanced chemiluminescence (Bio-Rad). For the Western blot analysis, the following primary antibodies were used: (i) anti-NONO (catalog no.: SC-376865; 1:500 dilution) purchased from Santa Cruz; (ii) anti-ADMA (catalog no.: ASYM24 07-414; 1:1000 dilution) and anti-SDMA (catalog no.: SYM10 07-412; 1:2000 dilution) purchased from Millipore; (iii) anti-MMA (catalog no.: D5A12; 1:1000 dilution) purchased from Cell Signaling Technology; (iv) anti-PSPC1 (catalog no.: A302-461; 1:5000 dilution) purchased from Bethyl Laboratories; and (v) anti-GAPDH (catalog no.: Ab9484; 1:3000 dilution) purchased from Abcam. Quantification of the signal intensity for each band was performed by Fiji software (44), and signal intensity was normalized at four different levels: (i) quantification of NONO in the input was normalized on GAPDH (as loading control); (ii) quantification of NONO in its IP was normalized on the previous normalized input for each condition; (iii) R methylation (MMA, ADMA, or SDMA) and PSPC1 signals were normalized on the amount of normalized NONO in the IP; and (iv) signal intensity in MS023 condition was normalized on dimethyl sulfoxide (untreated).
Results
Implementation of ML in hmSEEKER v2.0 to Improve the Detection of Methyl-peptide Doublets from hmSILAC Data
During the first implementation of hmSEEKER (26), we defined empirical cutoff values to distinguish true hmSILAC doublets from false positives. However, these criteria were not only conservative and resulted in a very low FDR but also a suboptimal sensitivity. We addressed this point by training a logistic regression ML model to discriminate between putative true and putative false hmSILAC doublets (Fig. 1A). This model allowed us to increase the number of hmSILAC-validated methyl-peptides in our dataset without compromising the FDR and provides more rigorous criteria for the identification of hmSILAC doublets compared with the original ones, which were estimated empirically. To obtain a dataset for model training, we followed the same strategy employed (26) to determine optimal cutoffs. Specifically, we analyzed MS raw files from a subset of hmSILAC experiments that were representative of all the cell lines and enrichment methods employed, then we used MaxQuant to search nonmethylated peptides that contained one or more methionine (M) residues: in fact, the labeling with M0 (“light”) or M4 (“heavy”) causes M-containing peptides to generate hmSILAC doublets that have the same properties as those generated by methyl-peptides. Thus, we used M-containing peptides as “mock methyl-peptides.” To model true negatives, we also included in the dataset peptides without M and randomly assigned methylations to them to mimic an erroneous identification by MaxQuant (Fig. 1B). The peptides in the dataset were processed with hmSEEKER using the following cutoffs: |mass error (ME)| <100 ppm, |retention time difference (dRT)| <5 min, and |log2 H/L intensity ratio (LogRatio)| <25. These cutoffs allowed us to retrieve as many putative doublets as possible, to have enough data for the training of the model. Doublets generated by a “mock methyl-peptide” were then filtered to only include “matched” doublets (i.e., those where both the heavy and light peptides are identified) and labeled “1” (n = 4434); doublets generated by a “true negative” peptide were labeled “0” (n = 3618). In total, the dataset used to train the predictor consisted of 8052 doublets (supplemental Fig. S1A).
Fig. 1.

Rationale of hmSEEKER, development of the machine learning (ML) model and analysis of hmSILAC data.A, schematic representation of the hmSEEKER workflow upon metabolic labeling with stable isotope–encoded methionine (M). Cells grown in “light” (M0) and “heavy” (M4) media are mixed in 1:1 proportion, and then proteins are extracted, digested, and analyzed by LC–MS/MS. MS spectra are analyzed with MaxQuant to obtain peptide and PTM identifications. The hmSEEKER software reads MaxQuant peptide identifications and, for each methyl-peptide that passes the quality filters, finds its corresponding MS1 peak, then searches the corresponding heavy/light counterpart. A peak doublet is defined by the difference in the retention time (dRT), the log-transformed intensity ratio (LogRatio), and the deviation between expected and observed m/z delta (mass error [ME]); these parameters are used to predict if the peak pair is a true hmSILAC doublet or a false positive. B, schematic examples of true positive and true negative doublets. C, receiving operator characteristic (ROC) curves obtained by testing the models trained by using either the raw or absolute value of the features. D, representation of the ML model weights with (blue) and without (red) taking the absolute values of the features. E, comparison of the performance of hmSEEKER before and after the introduction of the ML predictor (∗MCC = Matthew's correlation coefficient). F, summary of hmSILAC experiments analyzed to produce the orthogonally validated methyl-proteome. A detailed description of these experiments is available in supplemental Table S1. G, composition of the high-confidence hmSILAC R-methyl-proteome before (left) and after (right) the implementation of the ML model. hmSILAC, heavy methyl stable isotope labeling with amino acids in cell culture; MS, mass spectrometry; PTM, post-translational modification.
The independent variables (features) used within the logistic regression model were the ME, dRT, and LogRatio; the dependent variable was whether the doublet was a true hmSILAC doublet (“1”) or a random peak pair (“0”). The model was trained on 6000 doublets (3000 true + 3000 false; supplemental Fig. S1A) using stratified fivefold crossvalidation. To reduce the impact of potential outliers, we applied a quantile transformation to the features, which were then normalized based on their median and interquartile range values. Moreover, we observed that taking the absolute values of the features improved the performance of the model, allowing it to reach an area under the receiving operator characteristic curve >0.99 (Fig. 1C). Upon validation of the model on the remaining 2052 doublets, we found that less than 2% of the doublets were incorrectly labeled by the logistic regression (supplemental Fig. S1B). Inspection of the logistic regression coefficients revealed that ME was the most important feature as it had the largest absolute weight (−5.393), followed by LogRatio (weight = −3.067), whereas dRT (weight = −0.927) appeared to be the least important (Fig. 1D).
We then tested whether the inclusion of the ML model improved hmSEEKER v1.0 performance by comparing it to the default cutoffs previously defined (26): the entire set of 8052 doublets used for the training of the model was reanalyzed twice, first with the default cutoffs (i.e., |ME| <2 ppm, |dRT| <0.5 min, and |LogRatio| <1) and then by using the model predictions. The logistic regression showed an increase in sensitivity and accuracy; the metrics F1 score and Matthew’s correlation coefficient were also improved upon applying the model (Fig. 1E). We also checked in detail how each doublet in the training dataset was classified by applying the original cutoffs or the ML model. Indeed, we observed that the original cutoffs were biased toward “negative” doublets (supplemental Fig. S1C) and that the ML model, by defining less stringent criteria to call a positive doublet, allows hmSEEKER to recover 768 true positive doublets from those previously classified as false negative (corresponding to 90% of the total false negatives found with the default cutoffs). Because of the less stringent criteria, the number of false positives increased by 100, a relatively low amount compared with the 768 new true positives. In conclusion, we believe that the unbiased and data-driven nature of ML was able to find the optimal compromise between sensitivity and specificity, whereas the human-defined cutoffs tended to be conservative.
Reannotation of the Largest High-Quality Human Methyl-proteome
We first applied hmSEEKER v1.0 as described (26) to our hmSILAC data. Briefly, the data consisted of samples from four different cell lines (HeLa, SK-OV-3, NB4, and U2OS), which were subjected to different biochemical pipelines prior to LC–MS/MS, such as immunoenrichment of methyl-peptides with CST PTMScan kits or immunoenrichment of proteins with antibodies against methyl-R, methyl-K, or components of the Large Drosha Complex. These enrichment methods were combined with separation techniques such as PAGE, isoelectric focusing, and high-pH reversed-phase liquid chromatography to reduce sample complexity and further boost the identification of methyl-peptides (Fig. 1F and supplemental Table S1). Samples were then acquired on a Q Exactive mass spectrometer, and the resulting MS raw data were processed with MaxQuant to identify R-methyl-peptides and K-methyl-peptides.
The high-confidence methyl-proteome obtained from hmSEEKER v1.0 contained 1724 methyl-peptides, mapping to 476 different proteins, which carried a total of 1198 methyl-sites and 1491 methylation events (Fig. 1G, left). By reanalyzing the hmSILAC dataset with hmSEEKER v2.0, we annotated 2154 methyl-peptides (+25% compared with hmSEEKER v1.0) mapping on 660 proteins and 1882 methylation events (1551 on R and 331 on K; +26% compared with v1.0) distributed on 1561 methyl-sites (1266 R-sites and 295 K-sites; +30% compared with v1.0) (Fig. 1G, right).
We then set to expand further this experimental dataset by including the methyl-peptides that resulted as upregulated or downregulated in response to a certain biological stimulus (Fig. 2A and supplemental Table S1). The inclusion of these peptides in the repository was performed under the assumption that methyl-peptides displaying significant changes upon a stimulus are likely to be enzymatic, whereas amino acid substitutions and chemical artifacts should remain unaffected by any kind of perturbation. The significantly regulated methyl-peptides were extrapolated from previous studies carried out by our group, whereby R methylation changes were profiled through standard SILAC labeling in response to: (i) PRMT1 overexpression or knockdown (13); (ii) cell treatment with the PRMT5 inhibitor GSK591 or with the PRMT type I inhibitor MS023 (16, 29); and (iii) cell treatment with cisplatin (CDDP) (30). We thus obtained a final dataset consisting of 2246 methyl-peptides, 672 methyl-proteins, 1599 methyl-sites, and 1933 methylation events (Fig. 2B, left), which we named ProMetheusDB, a repository of high-quality K/R-methyl-peptides from human cancer cell lines (https://bioserver.ieo.it/shiny/app/prometheusdb).
Fig. 2.

Integration of the hmSILAC and SILAC data.A, summary of SILAC experiments used to expand the high-confidence methyl-proteome. A detailed description of these datasets and the linked experiments is available in supplemental Table S1. B, composition of the R-methyl-proteome upon the inclusion of dynamically modulated SILAC methyl-peptides, representing the ProMetheusDB. C, Venn diagram comparing R-methyl-peptides in ProMetheusDB and annotated in PhosphoSitePlus. D, Venn diagram comparing R-methyl-sites identified as monomethylated or dimethylated. hmSILAC, heavy methyl stable isotope labeling with amino acids in cell culture.
To facilitate the accessibility and dissemination of our data, we set ProMetheusDB as an open access repository that can be browsed via a web interface. This interface consists of an interactive modification-centric table displaying all the unique methylation events within ProMetheusDB, which can be filtered by protein, cell line, modified residue, methylation degree, or by searching a motif of interest within the site sequence windows. For each modification row, it is possible to visualize a peptide-centric child table that contains the list of identified peptides bearing it; within these child tables, the source of the methyl-peptide identification (hmSILAC or normal SILAC) is indicated, as well as their regulation state in the different quantitative experiments, where applicable.
Analysis of the Composition of ProMetheusDB
To characterize the features of ProMetheusDB, we first checked its composition in terms of K and R methylation events. Despite enriching for R methylation in most of our samples, we identified proteins bearing K-methyl-sites or combinations of K- and R-methyl-sites (supplemental Fig. S2A), although peptides on which K and R methylation coexist were overall rare (only 53, supplemental Fig. S2B), suggesting that K- and R-methyl-sites occur in distinct protein regions. While K methylation was included in the MS data analysis to avoid erroneous assignment of R methylation events to K and vice versa, the coverage of the R-methyl-peptides in ProMetheusDB is much larger than that of K-methyl-peptides, because of the employment of anti–pan-methyl-R antibodies in most of our experiments. Hence, we subsequently focused on the R-methyl-proteome, which comprises 1909 R-methyl-peptides, 548 proteins, 1303 R-sites, and 1599 R-modifications (Fig. 2B, right).
We first compared the R-methyl-sites in ProMetheusDB to the hmSILAC-validated R-methyl-sites in PhosphoSitePlus (Fig. 2C) and found that 716 (55%) of the sites present in ProMetheusDB are already annotated, whereas the remaining 587 (45%) are novel. So, our repository significantly expands the annotation of this PTM in the human proteome.
We then compared monomethyl-sites and dimethyl-sites and observed that, of the total 1303 R sites, 753 are only monomethylated, 254 are only dimethylated, and 296 are identified in both forms (Fig. 2D). Since previous studies reported ADMA as the most abundant methylation mark (45), we sought to verify if these counts of monomethylated and dimethylated R residues reflect the natural situation or a bias in the sample preparation procedure. We grouped our experiments based on the presence or not of the antibody-based affinity-enrichment step with the PTMScan Motif antibody kits (CST) in the protocol and observed that, in the datasets deriving from experiments where no affinity enrichment was performed, the number of dimethyl-R-sites tends to be greater than (supplemental Fig. S2C, R-methyl-protein IP and protein IP) or similar to the number of monomethyl-R-sites (supplemental Fig. S2C, input and K-methyl-protein IP). This result confirmed the possible bias in the antibody specificity and suggests caution when extrapolating conclusions on the different methylation degree representativeness.
Functional Analysis of the R-methyl-proteome Suggests New Roles of R Methylation in Inflammation and Immunity
To explore the possible impact of protein R methylation on cellular processes, we generated a network of functional PPIs, starting from the full list of R-methyl-proteins included in ProMetheusDB. This analysis highlighted the presence of five subnetworks enriched for distinct biological pathways (Fig. 3, A and B). Cluster 1 is enriched for DNA repair, Fc gamma receptor–dependent phagocytosis, and peroxisome proliferator–activated receptor (PPARα)–mediated gene activation; cluster 2 is enriched for functional terms related to mitosis; cluster 3 contains several components of the spliceosome; cluster 4 displays an enrichment for the term Antigen processing; and finally, cluster 5 contains proteins related to translational regulation.
Fig. 3.

Functional analysis of the R-methyl-protein network.A, protein clusters identified in the Reactome functional interaction network of R-methyl-proteins. Node size is proportional to the number of methyl-sites annotated on that protein; red borders highlight proteins bearing regulated methyl-sites. B, top five most enriched functional GO terms per each protein cluster. C, topology analysis performed on the Reactome interaction network displaying the increase in degree and centrality of proteins with increasing numbers of R-methyl-sites (p values are calculated with Mann–Whitney test). GO, Gene Ontology.
Since our group previously observed a strong trend of methylated proteins to be part of multisubunit complexes (9), we asked whether this observation still held true in this expanded dataset: we found that hypermethylated proteins (bearing five or more R-methyl-sites) presented significantly higher node degree and network centrality compared with nonmethylated proteins detected in the input samples (Fig. 3C). Several of the high-centrality and hypermethylated proteins are known RBPs, such as RBMX, various ribosomal proteins, nucleolin, and heterogeneous nuclear RNPs (46). However, the presence of R-methyl-proteins in cellular pathways related to PPARα (a regulator of lipid metabolism) (47), cytoskeleton organization, and phagocytosis was particularly interesting. Among these proteins we found G3BP1, which is on the one hand linked to stress granule formation and on the other hand has been shown to participate in DNA/RNA-sensing pathways implicated in the regulation of innate immunity (48, 49); the cytoskeletal protein actin (ACTB); the nuclear receptors NCOA2 and NCOA3, which are involved in metabolism, inflammation, and adipocyte differentiation (50); CUL1, a component of several E3 ubiquitin–protein ligase complexes (51); SMARCA5, a helicase with nucleosome-remodeling activity that has a role in transcription, phosphorylation of H2AX, and maintenance of chromatin structures during DNA replication (52). Taken together, these proteins and pathways suggest a role of R methylation in innate and adaptive immunity that is not limited to PRMT1-mediated transcriptional regulation (53, 54, 55). Moreover, our analysis revealed novel R-methyl-sites on protein already annotated as methylated on other residues, such as NONO (R142), SMARCA5 (R616), HSP90B1 (R51, R557), and ACTB (R206, R312).
As a further investigation, we integrated the network of protein interactions with the quantitative data on methylation dynamics obtained from the SILAC experiments to identify which proteins present at least one R-methyl-site that is significantly regulated in at least one SILAC experiment (i.e., regulated in the same way in one pair of matched forward and reverse replicates). Proteins featuring one or more regulated R-sites (circled in red in Fig. 3A) showed higher node degree and centrality in the network (Fig. 4A). Moreover, we observed that proteins not bearing regulated methylations were not enriched for any specific functional term, whereas proteins whose sites were regulated were mainly associated to RNA binding and splicing (Fig. 4B). This suggests the existence of two categories of R-methyl-sites: “dynamic” methyl-sites, which are predominant on protein binding to or modifying RNA and whose changes may exert effects on protein–RNA interactions, and “structural” or “constitutive” methyl-sites, which are overall refractory to modulation and occur on a more heterogenous set of proteins.
Fig. 4.

Functional analysis of dynamically regulated R methylations.A, topology analysis performed on the Reactome network reveals that proteins bearing at least one significantly regulated R-methyl-site have significantly higher degree and network centrality than those without regulated R-methyl-sites (p values are calculated with Mann–Whitney test). B, functional enrichment performed on proteins bearing at least one regulated R-methyl-site (top) or no regulated R-methyl-sites (bottom). C, intersection of PhaSepDB and ProMetheusDB shows that 60% of R-methyl-proteins are involved in the process of liquid–liquid phase separation (LLPS). D, motif analysis performed on significantly regulated (top) and unchanging (bottom) R-sites. Logos were generated using the full list of identified methyl-sites as background.
Since RBPs are frequently involved in the process of liquid–liquid phase separation (LLPS), we asked whether a link could exist between R methylation and LLPS. We intersected ProMetheusDB with PhaSepDB, a database of proteins involved in LLPS, and found that 60% of the proteins in our dataset were annotated as part of at least one membrane-less organelle (Fig. 4C). This observation was corroborated by a very recent study published by our group showing that R methylation is involved in the assembly of membrane-less organelles through the modulation of RBP–RNA interaction (17).
Finally, a motif analysis showed that, on the one hand, the regulated sites are surrounded by Gs (Fig. 4D, top), an observation consistent with the notion that the major type I and II PRMTs (PRMT1 and PRMT5, respectively) preferentially target sites in glycine- and arginine-rich motifs; on the other hand, the unchanging R-methyl-sites did not produce a significant enrichment for any consensus motif (Fig. 4D, bottom): this result suggest that unchanging methylations are catalyzed by methyltransferases other than the best-known ones possibly recognizing different sequence motifs, still uncharacterized.
Structural Survey of R Methylation Highlights Novel Biological Mechanisms
In the last 2 decades, it has been reported that intrinsically disordered regions of proteins play a role in different processes (such as PPI and protein–nucleic acid interactions and LLPS) and are often R-methylated (9, 56, 57). In addition, methylated R can serve as docking sites for the recruitment of other proteins and the formation of multiprotein complexes; two examples of such “readers” are represented by the Tudor domain, present in several proteins linked to RNA processing (58), and the WD40 repeat domain, found on a wide range of scaffolding proteins (59).
To study the link between protein structure and R methylation, we intersected the R-methyl-sites in the ProMetheusDB with disordered regions and domains annotated in MobiDB (60) and confirmed that the vast majority (77%) of the R-methyl-sites are indeed located in disordered regions (Fig. 5A). When we carried out the same analysis on a subset of 1500 nonmethylated R sites randomly extracted from the human proteome, we observed a more even distribution between structured domains and disordered regions. As a matter of fact, when performing a Fisher's exact test on the counts of methylated or unmodified R located in structured or disordered regions, we found that the distribution was not random (Fig. 5A, top), which corroborates previous evidence that R-methyl-sites tend to occur much more frequently within low complexity and disordered regions. Interestingly, when we performed the same analysis on methylated and unmethylated K, we found that K methylation is instead more frequent on structured domains (Fig. 5A, bottom). This result was further corroborated by two additional analyses: first, we expanded our strategy to K ubiquitination, K sumoylation, K acetylation, and S/T-Y phosphorylation sites that were downloaded from PhosphoSitePlus (supplemental Fig. S3A); second, we applied a recently described approach that identifies disordered regions based on AlphaFold predictions (36, 37) (supplemental Fig. S3B). The results showed that the occurrence of R methylation in disordered protein regions is not a feature generically common to all PTMs (supplemental Fig. S3).
Fig. 5.

Structural analysis of R-methylated protein regions.A, counts of R-methyl-sites (top) and K-methyl-sites (bottom) that occur in regions that are annotated as either domains or disordered in the MobiDB database. Counts of random R and K sites are also included as a comparison. p Value between methyl-site counts and random site counts was calculated with Fisher's exact test. B, counts of R-methyl-sites occurring on specific protein domains, as annotated in the InterPro database. Domains linked to RNA-binding proteins such as RNA-binding domain and K homology domain are the most represented. C, network obtained from Mechismo, showing the methylated proteins in ProMetheusDB and their interaction partners. DNA/RNA and chemicals are highlighted in orange and blue, respectively. R-methylated proteins are indicated by a green border. Self-loops indicated proteins that form a dimer and present a methylation site at the interface of the two monomers (i.e., MAT2A). D, crystal structure of NONO (orange) and PSPC1 (green) showing how R256 of NONO interacts with L222 of PSPC1, which suggest a possible role in the interaction between the two proteins. E, IP and WB profiling of NONO, its R methylation state, and PSPC1 coenrichment, in HeLa cells treated with either DMSO (negative control) or 10 μM MS023 for 48 h. Quantification of signal intensity for each band was performed as described in the Experimental Procedures section. DMSO, dimethyl sulfoxide; IP, immunopreipitation; NONO, non-POU domain–containing octamer-binding protein; PSPC1, paraspeckle component 1; WB, Western blot.
Despite being a minority, we focused on the 308 R-methyl-sites located in structured regions and mapped them onto InterPro (35) domains to investigate their possible association to specific protein domains: most R-methyl-sites localize within RNA-binding domains, K homology domains, like-Sm (LSM) domains, and P-loop NTPase domains (Fig. 5B). In addition to the “RBD,” whose enrichment is expected, the K homology domain is also mainly located in RBPs (specifically heterogeneous RNPs (61)), the domains belonging to the LSM family are often found on protein related to RNA biogenesis (62), and the P-loop NTPase fold is one of the most common nucleotide-binding domains; hence, these results are consistent with the notion that RNA binding and processing are mechanisms highly dependent on this PTM.
We then investigated the possible role of R methylation in regulating the interaction of proteins with other types of biomolecules. To do so, we analyzed ProMetheusDB using Mechismo, a web application that maps alterations of amino acid residues (induced by mutations or PTMs) onto protein crystal structures, to identify modified sites that are putatively involved in physical interactions with different kinds of biomolecules (38, 63). We found that 32 methylated R residues were involved in a total of 46 interactions with nucleic acid (n = 14), chemical compounds (n = 15), copies of the same protein (n = 4), and other proteins (n = 13) (Fig. 5C). The presence of few interactions with nucleic acids is somewhat unexpected, but it can be explained by the fact that many RBPs bind RNA through low-complexity regions, for which crystal structures are currently unavailable. Still, these data support the hypothesis that R methylation can also modulate interactions with other biomolecules, such as protein–protein and protein–chemical interactions.
We followed up the R-methyl-sites indicated by Mechismo analysis as putatively involved in PPIs. Based on reports that methylation of the guanidine group of R can exert distinct effects on PPIs by reducing the number of hydrogen bond donors on this amino acid, while at the same time increasing its hydrophobicity (1), we hypothesized that methylation could on the one hand enhance interactions between Rs and hydrophobic residues and on the other hand inhibit the interactions between Rs and negatively charged amino acids.
An interesting case study highlighted by this structural analysis was represented by the protein pair NONO–PSPC1, where R256 of NONO is located at the interface of interaction with PSPC1 (Fig. 5D). We hypothesized that methylation of R256 would increase its hydrophobicity, thus stabilizing the NONO–PSPC1 interaction interface, which also entails apolar residues, such as L222 of PSPC1. To verify this prediction, we set up the NONO IP in HeLa cells and profiled PSCP1 co-IP efficiency, both in basal conditions and upon treatment with the PRMT type I inhibitor MS023 (64), which typically leads to ADMA reduction and MMA/SDMA increase (30, 65, 66). When we profiled both NONO methylation and its interaction with PSPC1, we observed that MS023 induced the decrease of ADMA on NONO, with a parallel increase of MMA and, to a minor extent, of SDMA; in parallel, a mild increase of the amount of PSPC1 coimmunoprecipitated was also observed, confirming our prediction (Fig. 5E). In contrast, we suggest that methylation of R154 of SRSF1 disrupts the hydrogen bonds that this residue can form with E543 and Y549 of SRPK1, leading to a reduced binding of SRSF1 with SRPK1 (supplemental Fig. S5A). This could have important implications on the splicing of various gene transcripts and the consequent production of alternative protein isoforms, including, for example, vascular endothelial growth factor (67). Finally, a third interesting interaction suggested by Mechismo involves R264 of SAM synthase (MAT2A); this modification is located at the interface between the two subunits forming the active dimer of the enzyme, where it contacts E57 of the other MAT2A monomer, ATP, and metal ions serving as cofactors (supplemental Fig. S5B) (68). Based on structural modeling, it is possible to hypothesize that R264 methylation of MAT2A could impact on the catalytic activity of the enzyme, serving as a mechanism of regulation of overall SAM levels in the cell.
R-Methyl-Sites Located Near to and Distant from Phosphorylation Sites Show Distinct Responses to Stimuli
The crosstalk between R methylation and S/T-Y phosphorylation has already been described in the literature and linked to subcellular localization of proteins (69) and the promotion of stem-like properties in cancer (70). Interestingly, these two PTMs share some features like their preferential localization in intrinsically disordered regions (supplemental Fig. S3A) and their role in modulating protein–RNA interactions and LLPS (14, 71, 72). To elaborate on this aspect, we determined 15-amino acid windows centered on each R-methyl-site annotated in ProMetheusDB and assessed if phosphorylated residues are preferentially enriched within these windows. We found that 764 (49.0%) of the 1559 R-methyl-sites present an S, T, or Y phosphorylation site within their 15-amino acid window; by repeating this analysis on 1500 R residues randomly selected from the human proteome, we observed that the percentage of sites featuring a phosphorylation site in the same amino acid window was more than halved (328 sites, 21.9%; Fig. 6A), indicating a statistically significant co-occurrence of these PTMs.
Fig. 6.

Crosstalk of R methylation and S/T-Y phosphorylation.A, counts of R-methyl-sites that occur in proximity of a phosphorylated site. As a control, counts of randomly sampled R sites from the human proteome are shown, and the comparison indicates a significant correlation between R methylation and S/T-Y phosphorylation (p values are calculated by Fisher’s exact test). B, functional terms enriched from proteins bearing proximal R methylation and S/T-Y phosphorylation sites. C–F, counts of R-methyl-sites classified based on their regulation state upon external stimuli and their proximity to S/T-Y phosphorylation sites (p values are calculated by Fisher’s exact test).
We then asked whether this co-occurrence was linked to a specific biological process: by performing GO analysis on the protein displaying such PTM, we found an even stronger enrichment of terms linked to RNA binding and post-transcriptional regulation compared with the proteins in ProMetheusDB (Fig. 6B). To assess whether R-methyl-sites co-occurring with phosphosites are more likely subject to regulation in response to external cues, we intersected the results of the PTM crosstalk analysis with the dynamic information from the SILAC experiments. Notably, we found that the MS023-regulated methyl-sites occur significantly more frequently in the proximity of phospho-Y and phospho-S than the nonregulated ones (Fig. 6, C and D); the opposite is true for other stimuli, with R-methyl-sites regulated by CDDP or PRMT1 depletion significantly more distant from phospho-Y and phospho-T sites (Fig. 6, E and F). The opposite responses to these stimuli suggest that these PTMs can functionally interact in multiple ways and, more in general, hints toward the widespread existence of a molecular barcode of PTMs across the cellular proteome, similar to the one extensively described on histones.
We then expanded the analysis of possible crosstalk of R methylation with other PTMs and found that R-methyl-sites seem to significantly anticorrelate with nearby K ubiquitination sites, when compared with the dataset of randomly selected R sites (supplemental Fig. S4A). Instead, the associations between R methylation and K acetylation or K sumoylation are not statistically significant (supplemental Fig. S4, B and C). Because the numbers of K acetylation, K ubiquitination, and K sumoylation sites annotated in PhosphoSitePlus are at least one order of magnitude lower than that of phosphosites, these results should be taken with caution; nevertheless, they seem to corroborate the specificity of crosstalk between R methylation and S/T-Y phosphorylation, which is not generically applicable to every annotated PTM.
Methylation Beyond Arginine: hmSILAC-based Detection of Noncanonical Methylation Sites for High-Confidence Annotation of Novel Histone H3 Methylation Marks
Protein methylation has been observed not only on K and R but also on aspartate (D), glutamate (E), asparagine (N), glutamine (Q), serine (S), threonine (T) and histidine (H). However, the systematic study on these noncanonical methylated residues by MS is hindered, not only by the high FDR that already plagues R methylation analysis but also by the difficulty in pinpointing the PTM to the exact residue when multiple putative methyl-sites are present on the same peptide. In this context, we set to assess to what extent the hmSILAC strategy could help filling this gap in knowledge, with the rationale that all enzyme-driven methylations should use SAM as the universal methyl-group donor. We thus reanalyzed all the MS-raw data from hmSILAC-labeled non-affinity-enriched samples with the Andromeda search engine of MaxQuant, allowing monomethylation to occur on R, K, D, E, N, Q, S, T, and H. The MaxQuant output data were then processed with hmSEEKER v2.0 using the same criteria applied for high-quality R-methyl-sites identification, to produce a list of 86 orthogonally validated methylation sites in total, 22 of which occurring on noncanonical residues (Fig. 7A), located on 16 individual proteins. These novel modifications mainly occur on proteins functionally linked to mRNA splicing and RNPs (Fig. 7B). Most proteins only present one type of methylated residue (most commonly R), with very few proteins bearing combinations of up to four different methylated residues (Fig. 7C), such as EEF1A1, PRPF8, and HNRNPU.
Fig. 7.

Explorative analysis of noncanonical methylation sites.A, number of methyl-sites detected upon the reanalysis of the input MS data, grouped by residue. B, Gene Ontology terms, pathways, and complexes significantly enriched among proteins bearing methylations on non-K/R residues. C, UpSets plot representation of the co-occurrence of methylated residues on different proteins. D, MS/MS spectrum of the histone H3 27 to 40 peptide methylated on S28 and T32. MS, mass spectrometry.
This exploratory analysis also revealed the presence of two unconventional novel methyl-sites on H3, on S28 (H3S28me) and T32 (H3T32me). We were prompted to focus on noncanonical methylation on histones by this initial experimental evidence, together with two considerations: first, histone methylation is widely regarded as a core component of the histone code, hence the detection of novel methyl-sites may help dissecting this molecular language; second, since standard database search engines produce suboptimal results when multiple (>5) PTMs are searched in a large protein database (73), the strategy of limiting the analysis to histones could be a reasonable trade-off between expanding our methylation search to noncanonical methyl-residues and handling the explosion of false positives and negatives, linked to the expansion of the search space.
We thus applied an optimized biochemical and analytical pipeline to a set of hmSILAC-labeled histone samples: from a biochemical point of view, we took advantage of multiple proteases (i.e., trypsin, LysC, ArgC, and LysargiNase) digestion of histones to generate overlapping peptides and maximize protein coverage; from the computational point of view, the MS raw data were searched with MaxQuant using a filtered version of the UniProt Human protein database that included only histone sequences, using the same search parameters selected for the global methylation search. The subsequent analysis with hmSEEKER v2.0 for methyl-peptides pair matching and orthogonal validation of novel methyl-sites allowed us to unambiguously reidentify monomethylation on H3S28 and H3T32 (Figs. 7D and S6).
Discussion
In this work, we describe ProMetheusDB, our current hmSILAC-validated repository of protein methylation sites, annotated upon the reanalysis of previous hmSILAC experiments with an updated version of the hmSEEKER bioinformatics tool. Doublets of light and heavy methyl-peptides were evaluated by an ML model that was trained on doublets generated by M-containing peptides, which, unlike proper methyl-peptides, can be easily identified by database search engines and therefore provide a reliable positive control to our tool. After training the model, we observed that the dRT parameter of the doublets was the least important for discriminating true and false peptide pairs; this is in contrast with the first iteration of hmSEEKER, where dRT and ME were the main predictors of methyl-peptide doublets, and the H/L ratio parameter was introduced later. This choice of features also differentiates our ML model from the one adopted by MethylQuant (25), which scores methyl-peptide pairs based on the isotope distribution and elution profile correlation of the two peaks. Moreover, while we set the initial cutoffs with the aim of minimizing the number of false positives, the predictions produced by the ML model allowed us to find a compromise between false positives and false negatives, by recovering 768 false negatives versus the introduction of only 100 new false positives, thus keeping the FDR of hmSEEKER at 2.4% (precision = 0.976).
Functional analysis performed on R-methyl-sites revealed that proteins carrying multiple R-methyl-sites and/or dynamically regulated ones are more strongly interconnected in protein interaction networks, thus representing potential hubs of PPIs. In line with the current literature, we found that most of the regulated R-methyl-sites occurs in glycine and arginine–rich regions and on RBPs. However, when we analyzed the subset of methyl-sites that did not respond to the biological stimuli, we observed no significant enrichment; we therefore argue that these unchanging (“structural”) methyl-sites could represent a heterogeneous set of sites that is methylated by currently uncharacterized PRMTs with different motif preferences.
Interestingly, the protein clusters emerging from the network analysis suggest a potential role of R methylation in the immune response, as exemplified by the “FCGR-dependent phagocytosis” cluster. While it is known that PRMT1-mediated H4R3me2a promotes the expression of PPARα, a transcription factor regulating monocyte differentiation (53), our results expand the role of R methylation in immunity beyond mere transcriptional regulation, showing that several methyl-proteins are involved in antigen processing and exogenous DNA/RNA-sensing pathways (such as CUL1 and G3BP1, respectively). Other clusters that might be connected to this biological process are the ones related to cytoskeleton dynamics and lipid metabolism. In fact, anti-inflammatory macrophages metabolize lipids as a source of energy, whereas inflammatory ones use fatty acids to produce signal molecules like prostaglandins and leukotrienes (74, 75). The impact of R methylation on proteins of the cytoskeleton has been already described in neuronal development, where PRMTs regulate the formation of both the axon and the dendrites; it is possible PRMTs control cytoskeleton dynamics also in macrophages and, potentially, other cell types.
By mapping the R-methyl-sites annotated in ProMetheusDB onto protein structures, we confirmed that this PTM mostly occurs in unstructured low-complexity regions, characterized by short linear motifs that can mediate PPIs (76). This observation, together with the notion that R-methyl-proteins are central nodes of PPI networks, suggests a functional link between R methylation and PPI modulation. Indeed, by inspecting available 3D complexes, we also found a small number of sites where R methylation could modulate PPIs by increasing or reducing the hydrophobicity and hydrogen-bonding capability of R situated at protein interfaces. In particular, our data show that NONO can be methylated at R256; moreover, recent study by Xiang-Bo Wan et al. (77) identified R251 of NONO as a methylation target of PRMT1. Both residues are predicted by Mechismo to interact with apolar residues of PSPC1 (L222 and L171, respectively). Moreover, the amino acid substitution of NONO R256 with isoleucine (I), an apolar residue, is a mutation associated with colorectal cancer in the ActiveDriverDB database (78) and is predicted by Mechismo to enhance NONO–PSPC1 interaction; similarly, a hypothetical R251I substitution is also predicted to strengthen the binding. Overall, these observations suggest a model where an increase in the hydrophobicity of NONO, caused by either an amino acid substitution or by PRMT1-mediated hypermethylation, can elicit an oncogenic effect (at least in colorectal cancer) by modulating the interaction between NONO and PSPC1.
The observation that an R-methyl-site is involved in SRSF1–SRPK1 interaction is also intriguing, as Ngo et al. (67) have recently shown that disrupting this interaction promotes alternative splicing events triggering the translation of a vascular endothelial growth factor isoform that displays antiangiogenic properties. In this study, the authors used a PPI inhibitor to modulate SRSF1–SRPK1 interaction; along this line, based on our result, it will be interesting to assess whether pharmacological modulation of PRMTs can elicit the same functional effect.
Mechismo suggested another interesting interaction potentially targeted by R methylation that involves R264 of SAM synthase (MAT2A). The observation that the enzyme that synthesizes SAM, the methyl-group donor, can also be methylated in a pocket relevant for its catalytic activity is remarkable, because it suggests the possible existence of a regulatory feedback loop controlling SAM levels in the cell. It has already been reported that the RNA methyltransferase METTL16 binds and methylates the 3′-UTR region of MAT2A mRNA to prevent its translation. When SAM levels are low, the mRNA of MAT2A cannot be methylated because of lack of the methyl donor and the protein is translated (79). Thus, MAT2A protein methylation at R264 could represent an additional layer of regulation of the enzyme activity.
Phosphorylation and methylation sites colocalize on disordered SRGG motifs of some proteins, where the two PTMs are mutually exclusive because the presence of negative charges inhibits the binding of PRMTs to their recognition motifs (69). By overlapping ProMetheusDB with the PhosphoSitePlus phosphoproteomics dataset, we confirmed that methylated R are significantly more likely to not only occur in the proximity of a phospho-S but also expand this observation to phospho-T and phospho-Y residues. Furthermore, the analysis of co-occurrence of these two PTMs in the context of dynamic regulation suggests the existence of a link between the regulation of an R-methyl-site and its proximity to a phosphosite. Stimuli such as the use of type I PRMTs inhibitor MS023 cause a change in the methylation of R-sites that are close to phosphosites; instead, depletion of PRMT1 by RNA interference and the treatment with cisplatin affect R-methyl-sites that are distant from phosphosites. This result highlights the importance of developing, in the future, experimental pipelines enabling the simultaneous profiling of these two (or even more) PTMs, a strategy that is currently still at a pioneering stage (71). Our analysis, for instance, focused exclusively on methyl-sites, whereas the datasets of phosphorylation, acetylation, ubiquitination, and sumoylation sites were mostly derived from other studies focused on one individual PTM at a time; as such, we cannot be sure which modifications truly coexist and which are mutually exclusive.
The application of the hmSILAC-based biochemical and analytical pipeline led to the annotation of methyl-sites also on amino acids that are traditionally excluded from proteome with MS-based analysis, such as D, E, N, Q, S, T, and H, which also included the identification of a few putative novel methylation marks on histones (i.e., H3S28, H3T32). These two PTMs occurring on the peptide 27 to 40 of histone H3 are particularly interesting for their potential crosstalk with functionally relevant modifications on K27 and K36. For instance, methylation of H3K27 is a repressive mark (80), whereas H3K36me2 is found in transcribed regions (81). Our experimental data showed that the S28me mark could coexist with K27me and T32me, whereas peptides carrying simultaneously K27me and T32me, or bearing S28me/T32me in combination with K36me, were not detected (supplemental Fig. S5). A more in-depth analysis of this peptide and its numerous differentially modified isoforms is needed to corroborate this observation: measuring the relative abundance of the H3K27me/S28me and H3S28me/T32me peptide isoforms could allow developing some hypotheses about their biological role and possible crosstalk with other epigenetic marks. Similarly, it would be interesting to confirm whether H3S28me/T32me are mutually exclusive with K36 methylation or to find how these novel methylation marks are associated with neighboring acetylation and phosphorylation.
We argue that our analysis of noncanonical methylation sites could be linked to our previous analysis on PTM crosstalk. A recent article from the Eyers’ group (82) explored the field of noncanonical phosphorylations and identified several of these modifications on R, K, D, E, H, and C. Interestingly, these residues overlap with the putative noncanonical methyl-residues we have investigated here: it is, therefore, possible to envisage that the crosstalk of methylation and phosphorylation is not limited to proximal sites but could also occur through the physical competition for the same substrates. An intriguing case study emerging from our study is H3S28, a known phosphosite that we found methylated in the hmSILAC histone dataset. Phosphorylation of H3S28 can occur in response to extracellular stress and is proposed to override repressive epigenetic marks to temporarily express genes that would normally be silenced (83). We hypothesize that methylation of H3S28 may cooperate with methylation of H3K27 in silencing genes (80), which would explain why we did not detect H3S28 in combination with the aforementioned active transcription marks H3K36me2/me3 and H3K27ac (84). As a future perspective, since H3S28ph is also necessary for chromatin condensation (85), we could investigate how this residue is modified specifically during mitosis.
Some major points remain to be addressed in the methyl-proteomics field. First, there is a need for more efficient workflows that allow to annotate R-methyl-proteomes through sensibly smaller scale experiments, as the prerequisite for the investigation of this PTM in more relevant model systems, such as primary cells, tissues, organoid, similarly to what was made possible in the phosphoproteomics field (86). In fact, while this work describes how the R-methyl-proteomics can be improved at the level of MS-data processing, yet the sample preparation and LC–MS/MS acquisition steps are still prone to relevant limitations because methyl-peptides tend to be less abundant than unmodified peptides and thus less likely to be selected for MS/MS analysis. One solution to reduce the identification bias toward more abundant peptides and thus increase the coverage of the methyl-proteome could be the incorporation of data-independent acquisition methods into methyl-proteomics workflows (87). Along the same line, and more urgently, better strategies for the biochemical or affinity-enrichment of methylated peptides are needed; in particular, more efficient affinity-reagents recognizing dimethyl-R-peptides would allow addressing the bias toward R monomethylation introduced by the use of the CST anti-MMA kit currently available, as suggested by the evidence that monomethyl- and dimethyl-R-sites are more evenly represented in the datasets deriving from experiments when no affinity-enrichment steps through the CST kits were performed (i.e., fractionated WCE, protein IPs; supplemental Fig. S2C). Similarly, efficient antibodies for the enrichment of K-methyl-peptides are also essential to map the non-histone K-methyl-proteome, which remains essentially unexplored.
Besides the annotation of the steady-state methyl-proteome, we recognize a need to accelerate the acquisition of dynamic data. In the near future, it would be useful to apply isobaric mass tags to MS-based methyl-proteomics profiling to overcome the limited throughput of SILAC.
From the analytical point of view, some future developments can also be conceived: first, spectral library searching (88) is faster than conventional database searching; however its application to methyl-proteomics was limited by the low number of high-confidence methyl-peptide spectra available. The MS/MS spectra of methyl-peptides that were orthogonally validated by hmSILAC could be used to build a spectral library to be used as reference, thus speeding up the analysis of new data. Second, it is crucial to separately analyze ADMA and SDMA, which can be distinguished by searching for their diagnostic neutral loss ions within the MS/MS spectra (29, 89). While tools to detect neutral losses are available (90), they are hard to apply successfully because of the aforementioned scarcity of high-confidence spectra. Thus, it is recommended to visually inspect the MS/MS spectra of dimethyl-R-peptides, which is impractical for large datasets such as ProMetheusDB.
Data Availability
Raw data are available via ProteomeXchange (91) with identifier PXD027949.
MS/MS spectra are available via MS-Viewer with the following search keys: (1) traditional methyl-K/R search: 7w3qxdupp7; (2) global methyl-K/R/D/E/N/Q/S/T/H: p78hhhjj52; and (3) histone methyl-K/R/D/E/N/Q/S/T/H: 9arjmvlxfp.
ProMetheusDB can be accessed and browsed via a web interface at: https://bioserver.ieo.it/shiny/app/prometheusdb.
hmSEEKER 2.0 is freely available at: https://bitbucket.org/EMassi/hmseeker/src/master/.
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
We thank all members of the Bonaldi group for their support, suggestions, and critical discussion, in particular the ex-members of the group D. Musiani and V. Spadotto for producing part of the methyl-proteomics data that have been used to generate the ProMetheusDB. This project is supported by the Italian Association for Cancer Research (IG grant: 2018-15741).
Author contributions
E. M., R. G., and T. B. conceptualization; E. M., A. Y., and F. R. software; E. M., A. Y., and F. R. formal analysis; M. M., L. N., and A. C. investigation; T. B. resources; E. M., A. Y., and F. R. data curation; E. M. writing–original draft; R. G., F. R., and T. B. writing–review & editing; E. M. visualization; T. B. supervision; T. B. project administration; T. B. funding acquisition.
Funding and additional information
E.M. is supported by a fellowship of the Italian Foundation for Cancer Research (FIRC fellowship: 2018-22506). L.N. was supported by a fellowship from the Fondazione Istituto Europeo di Oncologia- Centro Cardiologico Monzino (FIEO-CCM). E.M. and M.M. are PhD students at the European School of Molecular Medicine (SEMM). L.N. was a PhD student at the European School of Molecular Medicine (SEMM).
Footnotes
Present address for Luciano Nicosia: Leukaemia Biology Laboratory, Cancer Research UK Manchester Institute, The University of Manchester, Oglesby Cancer Research Centre Building, Manchester, M20 4GJ, United Kingdom (UK).
Present address for Avinash Yadav: GSK Vaccines Srl, Siena, Italy.
Supplemental Data
References
- 1.Fulton M.D., Brown T., Zheng Y.G. The biological Axis of protein arginine methylation and asymmetric dimethylarginine. Int. J. Mol. Sci. 2019;20:3322. doi: 10.3390/ijms20133322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wysocka J., Allis C.D., Coonrod S. Histone arginine methylation and its dynamic regulation. Front. Biosci. 2006;11:344–355. doi: 10.2741/1802. [DOI] [PubMed] [Google Scholar]
- 3.Lorton B.M., Shechter D. Cellular consequences of arginine methylation. Cell Mol. Life Sci. 2019;76:2933–2956. doi: 10.1007/s00018-019-03140-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamaguchi A., Kitajo K. The effect of PRMT1-mediated arginine methylation on the subcellular localization, stress granules, and detergent-insoluble aggregates of FUS/TLS. PLoS One. 2012;7 doi: 10.1371/journal.pone.0049267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stopa N., Krebs J.E., Shechter D. The PRMT5 arginine methyltransferase: Many roles in development, cancer and beyond. Cell Mol. Life Sci. 2015;72:2041–2059. doi: 10.1007/s00018-015-1847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Lorenzo A., Bedford M.T. Histone arginine methylation. FEBS Lett. 2011;585:2024–2031. doi: 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu Q., Schapira M., Arrowsmith C.H., Barsyte-Lovejoy D. Protein arginine methylation: From enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 2021;20:509–530. doi: 10.1038/s41573-021-00159-8. [DOI] [PubMed] [Google Scholar]
- 8.Guccione E., Richard S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 2019;20:642–657. doi: 10.1038/s41580-019-0155-x. [DOI] [PubMed] [Google Scholar]
- 9.Bremang M., Cuomo A., Agresta A.M., Stugiewicz M., Spadotto V., Bonaldi T. Mass spectrometry-based identification and characterisation of lysine and arginine methylation in the human proteome. Mol. Biosyst. 2013;9:2231–2247. doi: 10.1039/c3mb00009e. [DOI] [PubMed] [Google Scholar]
- 10.Geoghegan V., Guo A., Trudgian D., Thomas B., Acuto O. Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling. Nat. Commun. 2015;6:6758. doi: 10.1038/ncomms7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsen S.C., Sylvestersen K.B., Mund A., Lyon D., Mullari M., Madsen M.V., Daniel J.A., Jensen L.J., Nielsen M.L. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signal. 2016;9:rs9. doi: 10.1126/scisignal.aaf7329. [DOI] [PubMed] [Google Scholar]
- 12.Szewczyk M.M., Ishikawa Y., Organ S., Sakai N., Li F., Halabelian L., Ackloo S., Couzens A.L., Eram M., Dilworth D., Fukushi H., Harding R., Dela Seña C.C., Sugo T., Hayashi K., et al. Pharmacological inhibition of PRMT7 links arginine monomethylation to the cellular stress response. Nat. Commun. 2020;11:2396. doi: 10.1038/s41467-020-16271-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spadotto V., Giambruno R., Massignani E., Mihailovich M., Maniaci M., Patuzzo F., Ghini F., Nicassio F., Bonaldi T. PRMT1-mediated methylation of the microprocessor-associated proteins regulates microRNA biogenesis. Nucl. Acids Res. 2020;48:96–115. doi: 10.1093/nar/gkz1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schisa J.A., Elaswad M.T. An emerging role for post-translational modifications in regulating RNP condensates in the germ line. Front. Mol. Biosci. 2021;8:658020. doi: 10.3389/fmolb.2021.658020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng D., Côté J., Shaaban S., Bedford M.T. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol. Cell. 2007;25:71–83. doi: 10.1016/j.molcel.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 16.Fong J.Y., Pignata L., Goy P.A., Kawabata K.C., Lee S.C., Koh C.M., Musiani D., Massignani E., Kotini A.G., Penson A., Wun C.M., Shen Y., Schwarz M., Low D.H., Rialdi A. Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell. 2019;36:194–209.e9. doi: 10.1016/j.ccell.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maniaci M., Boffo F.L., Massignani E., Bonaldi T. Systematic analysis of the impact of R-methylation on RBPs-RNA interactions: a proteomic approach. Front. Mol. Biosci. 2021;8:688973. doi: 10.3389/fmolb.2021.688973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bao X., Zhao S., Liu T., Liu Y., Liu Y., Yang X. Overexpression of PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J. Histochem. Cytochem. 2013;61:206–217. doi: 10.1369/0022155413475452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang J.W., Cho Y., Bae G.U., Kim S.N., Kim Y.K. Protein arginine methyltransferases: Promising targets for cancer therapy. Exp. Mol. Med. 2021;53:788–808. doi: 10.1038/s12276-021-00613-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Couto E.S.A., Wu C.Y., Citadin C.T., Clemons G.A., Possoit H.E., Grames M.S., Lien C.F., Minagar A., Lee R.H., Frankel A., Lin H.W. Protein arginine methyltransferases in cardiovascular and neuronal function. Mol. Neurobiol. 2020;57:1716–1732. doi: 10.1007/s12035-019-01850-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.vanLieshout T.L., Ljubicic V. The emergence of protein arginine methyltransferases in skeletal muscle and metabolic disease. Am. J. Physiol. Endocrinol. Metab. 2019;317:E1070–E1080. doi: 10.1152/ajpendo.00251.2019. [DOI] [PubMed] [Google Scholar]
- 22.Smith E., Zhou W., Shindiapina P., Sif S., Li C., Baiocchi R.A. Recent advances in targeting protein arginine methyltransferase enzymes in cancer therapy. Expert Opin. Ther. Targets. 2018;22:527–545. doi: 10.1080/14728222.2018.1474203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hart-Smith G., Yagoub D., Tay A.P., Pickford R., Wilkins M.R. Large scale mass spectrometry-based identifications of enzyme-mediated protein methylation are subject to high false discovery rates. Mol. Cell Proteomics. 2016;15:989–1006. doi: 10.1074/mcp.M115.055384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ong S.E., Mann M. Identifying and quantifying sites of protein methylation by heavy methyl SILAC. Curr. Protoc. Protein Sci. 2006 doi: 10.1002/0471140864.ps1409s46. [DOI] [PubMed] [Google Scholar]
- 25.Tay A.P., Geoghegan V., Yagoub D., Wilkins M.R., Hart-Smith G. MethylQuant: A tool for sensitive validation of enzyme-mediated protein methylation sites from heavy-methyl SILAC data. J. Proteome Res. 2018;17:359–373. doi: 10.1021/acs.jproteome.7b00601. [DOI] [PubMed] [Google Scholar]
- 26.Massignani E., Cuomo A., Musiani D., Jammula S., Pavesi G., Bonaldi T. hmSEEKER: Identification of hmSILAC doublets in MaxQuant output data. Proteomics. 2019;19 doi: 10.1002/pmic.201800300. [DOI] [PubMed] [Google Scholar]
- 27.Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 28.Afjehi-Sadat L., Garcia B.A. Comprehending dynamic protein methylation with mass spectrometry. Curr. Opin. Chem. Biol. 2013;17:12–19. doi: 10.1016/j.cbpa.2012.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Musiani D., Bok J., Massignani E., Wu L., Tabaglio T., Ippolito M.R., Cuomo A., Ozbek U., Zorgati H., Ghoshdastider U., Robinson R.C., Guccione E., Bonaldi T. Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci. Signal. 2019;12 doi: 10.1126/scisignal.aat8388. [DOI] [PubMed] [Google Scholar]
- 30.Musiani D., Giambruno R., Massignani E., Ippolito M.R., Maniaci M., Jammula S., Manganaro D., Cuomo A., Nicosia L., Pasini D., Bonaldi T. PRMT1 is recruited via DNA-PK to chromatin where it sustains the senescence-associated secretory phenotype in response to cisplatin. Cell Rep. 2020;30:1208–1222.e9. doi: 10.1016/j.celrep.2019.12.061. [DOI] [PubMed] [Google Scholar]
- 31.Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 32.O'Shea J.P., Chou M.F., Quader S.A., Ryan J.K., Church G.M., Schwartz D. pLogo: A probabilistic approach to visualizing sequence motifs. Nat. Methods. 2013;10:1211–1212. doi: 10.1038/nmeth.2646. [DOI] [PubMed] [Google Scholar]
- 33.Tareen A., Kinney J.B. Logomaker: Beautiful sequence logos in Python. Bioinformatics. 2020;36:2272–2274. doi: 10.1093/bioinformatics/btz921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolberg L., Raudvere U., Kuzmin I., Vilo J., Peterson H. Gprofiler2 -- R. package gene list functional enrichment analysis namespace conversion toolset G:profiler. F1000Res. 2020 doi: 10.12688/f1000research.24956.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blum M., Chang H.Y., Chuguransky S., Grego T., Kandasaamy S., Mitchell A., Nuka G., Paysan-Lafosse T., Qureshi M., Raj S., Richardson L., Salazar G.A., Williams L., Bork P., Bridge A., et al. The InterPro protein families and domains database: 20 years on. Nucl. Acids Res. 2021;49:D344–D354. doi: 10.1093/nar/gkaa977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., Bridgland A., Meyer C., Kohl S.A.A., Ballard A.J., Cowie A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bludau I., Willems S., Zeng W.F., Strauss M.T., Hansen F.M., Tanzer M.C., Karayel O., Schulman B.A., Mann M. The structural context of PTMs at a proteome wide scale. bioRxiv. 2022 doi: 10.1101/2022.02.23.481596. preprint. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Betts M.J., Lu Q., Jiang Y., Drusko A., Wichmann O., Utz M., Valtierra-Gutiérrez I.A., Schlesner M., Jaeger N., Jones D.T., Pfister S., Lichter P., Eils R., Siebert R., Bork P., et al. Mechismo: Predicting the mechanistic impact of mutations and modifications on molecular interactions. Nucl. Acids Res. 2015;43:e10. doi: 10.1093/nar/gku1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucl. Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Croft D., O'Kelly G., Wu G., Haw R., Gillespie M., Matthews L., Caudy M., Garapati P., Gopinath G., Jassal B., Jupe S., Kalatskaya I., Mahajan S., May B., Ndegwa N., et al. Reactome: A database of reactions, pathways and biological processes. Nucl. Acids Res. 2011;39:D691–D697. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Orchard S., Kerrien S., Abbani S., Aranda B., Bhate J., Bidwell S., Bridge A., Briganti L., Brinkman F.S., Cesareni G., Chatr-aryamontri A., Chautard E., Chen C., Dumousseau M., Goll J., et al. Protein interaction data curation: The international molecular exchange (IMEx) consortium. Nat. Methods. 2012;9:345–350. doi: 10.1038/nmeth.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parca L., Truglio M., Biagini T., Castellana S., Petrizzelli F., Capocefalo D., Jordán F., Carella M., Mazza T. Pyntacle: A parallel computing-enabled framework for large-scale network biology analysis. Gigascience. 2020;9 doi: 10.1093/gigascience/giaa115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.Y., White D.J., Hartenstein V., Eliceiri K., Tomancak P., et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davids M., Teerlink T. Plasma concentrations of arginine and asymmetric dimethylarginine do not reflect their intracellular concentrations in peripheral blood mononuclear cells. Metabolism. 2013;62:1455–1461. doi: 10.1016/j.metabol.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 46.Geuens T., Bouhy D., Timmerman V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016;135:851–867. doi: 10.1007/s00439-016-1683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoon M. The role of PPARalpha in lipid metabolism and obesity: Focusing on the effects of estrogen on PPARalpha actions. Pharmacol. Res. 2009;60:151–159. doi: 10.1016/j.phrs.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 48.Liu Z.S., Cai H., Xue W., Wang M., Xia T., Li W.J., Xing J.Q., Zhao M., Huang Y.J., Chen S., Wu S.M., Wang X., Liu X., Pang X., Zhang Z.Y., et al. G3BP1 promotes DNA binding and activation of cGAS. Nat. Immunol. 2019;20:18–28. doi: 10.1038/s41590-018-0262-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reineke L.C., Lloyd R.E. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J. Virol. 2015;89:2575–2589. doi: 10.1128/JVI.02791-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rollins D.A., Coppo M., Rogatsky I. Minireview: Nuclear receptor coregulators of the p160 family: Insights into inflammation and metabolism. Mol. Endocrinol. 2015;29:502–517. doi: 10.1210/me.2015-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Michel J.J., Xiong Y. Human CUL-1, but not other cullin family members, selectively interacts with SKP1 to form a complex with SKP2 and cyclin A. Cell Growth Differ. 1998;9:435–449. [PubMed] [Google Scholar]
- 52.Oppikofer M., Bai T., Gan Y., Haley B., Liu P., Sandoval W., Ciferri C., Cochran A.G. Expansion of the ISWI chromatin remodeler family with new active complexes. EMBO Rep. 2017;18:1697–1706. doi: 10.15252/embr.201744011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tikhanovich I., Zhao J., Olson J., Adams A., Taylor R., Bridges B., Marshall L., Roberts B., Weinman S.A. Protein arginine methyltransferase 1 modulates innate immune responses through regulation of peroxisome proliferator-activated receptor gamma-dependent macrophage differentiation. J. Biol. Chem. 2017;292:6882–6894. doi: 10.1074/jbc.M117.778761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cho J.H., Lee R., Kim E., Choi Y.E., Choi E.J. PRMT1 negatively regulates activation-induced cell death in macrophages by arginine methylation of GAPDH. Exp. Cell Res. 2018;368:50–58. doi: 10.1016/j.yexcr.2018.04.012. [DOI] [PubMed] [Google Scholar]
- 55.Zhao J., O'Neil M., Vittal A., Weinman S.A., Tikhanovich I. PRMT1-Dependent macrophage IL-6 production is required for alcohol-induced HCC progression. Gene Expr. 2019;19:137–150. doi: 10.3727/105221618X15372014086197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Musselman C.A., Kutateladze T.G. Characterization of functional disordered regions within chromatin-associated proteins. iScience. 2021;24:102070. doi: 10.1016/j.isci.2021.102070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Darling A.L., Uversky V.N. Intrinsic disorder and posttranslational modifications: The darker side of the biological dark matter. Front. Genet. 2018;9:158. doi: 10.3389/fgene.2018.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ying M., Chen D. Tudor domain-containing proteins of Drosophila melanogaster. Dev. Growth Differ. 2012;54:32–43. doi: 10.1111/j.1440-169x.2011.01308.x. [DOI] [PubMed] [Google Scholar]
- 59.Jain B.P., Pandey S. WD40 repeat proteins: signalling scaffold with diverse functions. Protein J. 2018;37:391–406. doi: 10.1007/s10930-018-9785-7. [DOI] [PubMed] [Google Scholar]
- 60.Piovesan D., Necci M., Escobedo N., Monzon A.M., Hatos A., Mičetić I., Quaglia F., Paladin L., Ramasamy P., Dosztányi Z., Vranken W.F., Davey N.E., Parisi G., Fuxreiter M., Tosatto S.C.E. MobiDB: Intrinsically disordered proteins in 2021. Nucl. Acids Res. 2021;49:D361–D367. doi: 10.1093/nar/gkaa1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Valverde R., Edwards L., Regan L. Structure and function of KH domains. FEBS J. 2008;275:2712–2726. doi: 10.1111/j.1742-4658.2008.06411.x. [DOI] [PubMed] [Google Scholar]
- 62.He W., Parker R. Functions of Lsm proteins in mRNA degradation and splicing. Curr. Opin. Cell Biol. 2000;12:346–350. doi: 10.1016/s0955-0674(00)00098-3. [DOI] [PubMed] [Google Scholar]
- 63.Fic W., Bastock R., Raimondi F., Los E., Inoue Y., Gallop J.L., Russell R.B., St Johnston D. RhoGAP19D inhibits Cdc42 laterally to control epithelial cell shape and prevent invasion. J. Cell Biol. 2021;220 doi: 10.1083/jcb.202009116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eram M.S., Shen Y., Szewczyk M., Wu H., Senisterra G., Li F., Butler K.V., Kaniskan H.Ü., Speed B.A., Dela Seña C., Dong A., Zeng H., Schapira M., Brown P.J., Arrowsmith C.H., et al. A potent, selective, and cell-active inhibitor of human type I protein arginine methyltransferases. ACS Chem. Biol. 2016;11:772–781. doi: 10.1021/acschembio.5b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dhar S., Vemulapalli V., Patananan A.N., Huang G.L., Di Lorenzo A., Richard S., Comb M.J., Guo A., Clarke S.G., Bedford M.T. Loss of the major Type I arginine methyltransferase PRMT1 causes substrate scavenging by other PRMTs. Sci. Rep. 2013;3:1311. doi: 10.1038/srep01311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hartel N.G., Chew B., Qin J., Xu J., Graham N.A. Deep protein methylation profiling by combined chemical and immunoaffinity approaches reveals novel PRMT1 targets. Mol. Cell Proteomics. 2019;18:2149–2164. doi: 10.1074/mcp.RA119.001625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Q., Zeng C., Liu H., Yung K.W.Y., Chen C., Xie Q., Zhang Y., Wan S.W.C., Mak B.S.W., Xia J., Xiong S., Ngo J.C.K. Protein-protein interaction inhibitor of SRPKs alters the splicing isoforms of VEGF and inhibits angiogenesis. iScience. 2021;24:102423. doi: 10.1016/j.isci.2021.102423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Quinlan C.L., Kaiser S.E., Bolaños B., Nowlin D., Grantner R., Karlicek-Bryant S., Feng J.L., Jenkinson S., Freeman-Cook K., Dann S.G., Wang X., Wells P.A., Fantin V.R., Stewart A.E., Grant S.K., et al. Targeting S-adenosylmethionine biosynthesis with a novel allosteric inhibitor of Mat2A. Nat. Chem. Biol. 2017;13:785–792. doi: 10.1038/nchembio.2384. [DOI] [PubMed] [Google Scholar]
- 69.Smith D.L., Erce M.A., Lai Y.W., Tomasetig F., Hart-Smith G., Hamey J.J., Wilkins M.R. Crosstalk of phosphorylation and arginine methylation in disordered SRGG repeats of saccharomycescerevisiae fibrillarin and its association with nucleolar localization. J. Mol. Biol. 2020;432:448–466. doi: 10.1016/j.jmb.2019.11.006. [DOI] [PubMed] [Google Scholar]
- 70.Liu N., Yang R., Shi Y., Chen L., Liu Y., Wang Z., Liu S., Ouyang L., Wang H., Lai W., Mao C., Wang M., Cheng Y., Liu S., Wang X., et al. The cross-talk between methylation and phosphorylation in lymphoid-specific helicase drives cancer stem-like properties. Signal Transduct. Target. Ther. 2020;5:197. doi: 10.1038/s41392-020-00249-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hamey J.J., Nguyen A., Wilkins M.R. Discovery of arginine methylation, phosphorylation, and their Co-occurrence in condensate-associated proteins in Saccharomyces cerevisiae. J. Proteome Res. 2021;20:2420–2434. doi: 10.1021/acs.jproteome.0c00927. [DOI] [PubMed] [Google Scholar]
- 72.Owen I., Shewmaker F. The role of post-translational modifications in the phase transitions of intrinsically disordered proteins. Int. J. Mol. Sci. 2019;20:5501. doi: 10.3390/ijms20215501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verheggen K., Raeder H., Berven F.S., Martens L., Barsnes H., Vaudel M. Anatomy and evolution of database search engines-a central component of mass spectrometry based proteomic workflows. Mass Spectrom. Rev. 2020;39:292–306. doi: 10.1002/mas.21543. [DOI] [PubMed] [Google Scholar]
- 74.Yan J., Horng T. Lipid metabolism in regulation of macrophage functions. Trends Cell Biol. 2020;30:979–989. doi: 10.1016/j.tcb.2020.09.006. [DOI] [PubMed] [Google Scholar]
- 75.Batista-Gonzalez A., Vidal R., Criollo A., Carreño L.J. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front. Immunol. 2019;10:2993. doi: 10.3389/fimmu.2019.02993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tompa P., Davey N.E., Gibson T.J., Babu M.M. A million peptide motifs for the molecular biologist. Mol. Cell. 2014;55:161–169. doi: 10.1016/j.molcel.2014.05.032. [DOI] [PubMed] [Google Scholar]
- 77.Yin X.K., Wang Y.L., Wang F., Feng W.X., Bai S.M., Zhao W.W., Feng L.L., Wei M.B., Qin C.L., Wang F., Chen Z.L., Yi H.J., Huang Y., Xie P.Y., Kim T., et al. PRMT1 enhances oncogenic arginine methylation of NONO in colorectal cancer. Oncogene. 2021;40:1375–1389. doi: 10.1038/s41388-020-01617-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krassowski M., Paczkowska M., Cullion K., Huang T., Dzneladze I., Ouellette B.F.F., Yamada J.T., Fradet-Turcotte A., Reimand J. ActiveDriverDB: Human disease mutations and genome variation in post-translational modification sites of proteins. Nucl. Acids Res. 2018;46:D901–D910. doi: 10.1093/nar/gkx973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pendleton K.E., Chen B., Liu K., Hunter O.V., Xie Y., Tu B.P., Conrad N.K. The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. 2017;169:824–835.e14. doi: 10.1016/j.cell.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wiles E.T., Selker E.U. H3K27 methylation: A promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev. 2017;43:31–37. doi: 10.1016/j.gde.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang C., Zhu B. Roles of H3K36-specific histone methyltransferases in transcription: Antagonizing silencing and safeguarding transcription fidelity. Biophys. Rep. 2018;4:170–177. doi: 10.1007/s41048-018-0063-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hardman G., Eyers C.E. High-throughput characterization of histidine phosphorylation sites using UPAX and tandem mass spectrometry. Methods Mol. Biol. 2020;2077:225–235. doi: 10.1007/978-1-4939-9884-5_15. [DOI] [PubMed] [Google Scholar]
- 83.Sawicka A., Seiser C. Histone H3 phosphorylation - a versatile chromatin modification for different occasions. Biochimie. 2012;94:2193–2201. doi: 10.1016/j.biochi.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang T., Zhang Z., Dong Q., Xiong J., Zhu B. Histone H3K27 acetylation is dispensable for enhancer activity in mouse embryonic stem cells. Genome Biol. 2020;21:45. doi: 10.1186/s13059-020-01957-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sawicka A., Hartl D., Goiser M., Pusch O., Stocsits R.R., Tamir I.M., Mechtler K., Seiser C. H3S28 phosphorylation is a hallmark of the transcriptional response to cellular stress. Genome Res. 2014;24:1808–1820. doi: 10.1101/gr.176255.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lindhorst P.H., Hummon A.B. Proteomics of colorectal cancer: Tumors, organoids, and cell cultures-A minireview. Front. Mol. Biosci. 2020;7:604492. doi: 10.3389/fmolb.2020.604492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bekker-Jensen D.B., Bernhardt O.M., Hogrebe A., Martinez-Val A., Verbeke L., Gandhi T., Kelstrup C.D., Reiter L., Olsen J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020;11:787. doi: 10.1038/s41467-020-14609-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lam H. Building and searching tandem mass spectral libraries for peptide identification. Mol. Cell Proteomics. 2011;10 doi: 10.1074/mcp.R111.008565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brame C.J., Moran M.F., McBroom-Cerajewski L.D. A mass spectrometry based method for distinguishing between symmetrically and asymmetrically dimethylated arginine residues. Rapid Commun. Mass Spectrom. 2004;18:877–881. doi: 10.1002/rcm.1421. [DOI] [PubMed] [Google Scholar]
- 90.Kelstrup C.D., Frese C., Heck A.J., Olsen J.V., Nielsen M.L. Analytical utility of mass spectral binning in proteomic experiments by SPectral immonium Ion detection (SPIID) Mol. Cell Proteomics. 2014;13:1914–1924. doi: 10.1074/mcp.O113.035915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucl. Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are available via ProteomeXchange (91) with identifier PXD027949.
MS/MS spectra are available via MS-Viewer with the following search keys: (1) traditional methyl-K/R search: 7w3qxdupp7; (2) global methyl-K/R/D/E/N/Q/S/T/H: p78hhhjj52; and (3) histone methyl-K/R/D/E/N/Q/S/T/H: 9arjmvlxfp.
ProMetheusDB can be accessed and browsed via a web interface at: https://bioserver.ieo.it/shiny/app/prometheusdb.
hmSEEKER 2.0 is freely available at: https://bitbucket.org/EMassi/hmseeker/src/master/.