

Abstract
Cells detached from the extracellular matrix (ECM) can trigger different modes of cell death, and the survival of ECM‐detached cells is one of the prerequisites for the metastatic cascade. Ferroptosis, a form of iron‐dependent programmed cell death, has recently been found to be involved in matrix‐detached cancer cells. However, the molecular mechanisms by which ECM‐detached cells escape ferroptosis are not fully understood. Here, we observed that cell migration‐inducing protein (CEMIP) upregulation facilitates ferroptosis resistance during ECM detachment by promoting cystine uptake in prostate cancer (PCa) cells. Meanwhile, silencing CEMIP causes it to lose its ability to promote cystine uptake and inhibit ferroptosis. Mechanistically, the interaction of CEMIP with inositol 1,4,5‐trisphosphate receptor type 3 (ITPR3) modulates calcium ion (Ca2+) leakage from the endoplasmic reticulum, activating calcium/calmodulin‐dependent protein kinase II (CaMKII), which further facilitates nuclear factor erythroid 2‐related factor 2 (NRF2) phosphorylation and nuclear localization, leading to elevated transcription of solute carrier family 7 member 11 (SLC7A11), a glutamate/cystine antiporter, in PCa cells. Our findings delineate a novel role of CEMIP in ferroptosis resistance during ECM detachment and provide new insights into therapeutic strategies for metastatic PCa.
Keywords: CEMIP, ferroptosis, NRF2, prostate cancer, SLC7A11
Ferroptosis was triggered by ECM detachment in prostate cancer cells, while the survival cells presented a ferroptosis‐resistant phenotype. Suppressing CEMIP expression led to detached cell death by boosting ferroptosis through inhibiting the ITPR3/CaMKII/NRF2/SLC7A11 pathway.
Abbreviations
- ARE
antioxidant response element
- Ca2+
calcium ion
- CaMKII
calcium/calmodulin‐dependent protein kinase II
- CCK‐8
cell counting kit‐8
- CEMIP
cell migration‐inducing protein
- Co‐IP
co‐immunoprecipitation
- DR
detachment‐resistant
- ECM
extracellular matrix
- ER
endoplasmic reticulum
- Fe2+
ferrous ion
- GSH/GSSG
the ratio of reduced glutathione to oxidized glutathione
- IHC
immunohistochemistry
- ITPR3
inositol 1,4,5‐trisphosphate receptor type 3
- KEAP1
Kelch‐like ECH‐associated protein 1
- MDA
malondialdehyde
- NC
negative control
- NRF2
nuclear factor erythroid 2‐related factor 2
- PCa
prostate cancer
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- ROS
reactive oxygen species
- SLC7A11
solute carrier family 7 member 11
- TEM
transmission electron microscope
1. INTRODUCTION
Prostate cancer (PCa), with an estimated 1.4 million new cases and 375,000 deaths occurring in 2020, is the second most diagnosed malignant neoplasm in men worldwide. 1 Metastasis was found in 5% of PCa cases at first diagnosis, with a 5‐year survival rate of <30%. 2 , 3 PCa cells in primary tumors often metastasize to the lymph nodes and distant organs, such as the bones, the lung, and the liver. These sites generally display terribly low responsiveness to initial treatment and have a high risk of post‐treatment relapse.
Cancer cell survival after detachment from the extracellular matrix (ECM) is one of the prerequisites for the metastatic cascade. Further, resistance to anoikis, a form of apoptosis induced by cell detachment, is crucial for the survival of ECM‐detached cancer cells. 4 Growing evidence has shown that different modus of cellular changes in response to ECM detachment may also be involved in this procedure and induce cell death independent of anoikis. 5 , 6 , 7 Consequently, understanding the survival mechanisms in addition to anoikis resistance in ECM‐detached cancer cells is essential for the development of effective chemotherapeutic strategies to inhibit tumor progression and metastasis.
Ferroptosis, a form of nonapoptotic cell death based on iron‐dependent accumulation of reactive oxygen species (ROS), was first discovered by Dixon in 2012. 8 ECM detachment can cause a series of deleterious metabolic alterations, including a remarkable increase in ROS levels. 9 Therefore, ferroptosis may be activated when cells are detached from the ECM. Recent studies have shown that ECM detachment can trigger ferroptosis and that upregulation of the integrin α6β4 enables cancer cells to evade ferroptosis. 10 The cooperation of ΔNp63 with the B‐cell lymphoma 2 protein inhibits oxidative stress‐induced ferroptosis and confers clonogenic survival against matrix detachment. 11 However, whether matrix detachment triggers ferroptosis and how ECM‐detached cells escape ferroptosis in PCa remains unclear.
Here, we demonstrate that ECM‐detached PCa cells undergo ferroptosis, and detachment‐resistant (DR) PCa cell models induced by continuous suspension culture exhibit tolerance to ferroptosis. Cell migration‐inducing protein (CEMIP) is significantly overexpressed in DR PCa cell models and protects detached PCa cells against ferroptosis. Accumulating evidence suggests that CEMIP upregulation is related to cancer metastasis, cancer progression, and poor prognosis. 12 , 13 , 14 , 15 However, the effect of CEMIP on ferroptosis in metastatic PCa is unknown. In the present study, we aimed to discover the underlying regulatory mechanism of CEMIP in ferroptosis resistance during ECM detachment in PCa cells and to investigate the mechanisms involved.
2. MATERIALS AND METHODS
The detailed procedures for the cell viability assay and measurement of intracellular ROS, malondialdehyde (MDA) and the ratio of reduced glutathione to oxidized glutathione (GSH/GSSG), patient tissue specimens, western blotting, nuclear and cytosolic protein extraction, quantitative real‐time polymerase chain reaction (qRT‐PCR), measurement of intracellular ferrous ion (Fe2+) and calcium ion (Ca2+), immunofluorescence staining, transmission electron microscope (TEM), plasmid transfection, soft agar assay, invasion and migration assays, luciferase reporter assay, co‐immunoprecipitation (Co‐IP), and animal studies are described in the Supplementary Materials and Methods.
2.1. Cell culture and construction of the detachment‐resistant cells model
PC‐3, DU145, and LNCaP cell lines were purchased from the Cell Bank of the Chinese Academy of Sciences. All cell lines were cultured in RPMI‐1640 medium (Hyclone, GE Healthcare Life Sciences) containing 10% FBS (Biologic Industries) and 1% penicillin/streptomycin (Servicebio) under standard conditions (5% CO2, 37°C). The method used to construct the DR cells model was consistent that used in our previous studies. 16 In general, parental cells were suspension cultured in ultra‐low‐attachment six‐well plates (Corning Life Sciences) for 7 days and then transferred to normal plates for continued adherent culturing for 2 days; the cells that re‐adhered were considered DR cells.
2.2. Measurement of cystine uptake
Uptake of cystine was measured with BioTracker Cystine‐FITC Live Cell Dye (Sigma‐Aldrich) using a previously reported method. 17 The pretreated cells were seeded on coverslips and incubated with complete medium for 24 h. After washing three times with choline chloride buffer, cells were starved at 37°C for 30 min in the same buffer. Then, cells were incubated with cystine‐FITC (5 μM) in the dark for 60 min. Finally, cells were washed with choline chloride buffer three times and FITC fluorescence was detected using a Nikon A1Si Laser Scanning Confocal Microscope (Nikon Instruments).
2.3. Immunohistochemistry
Immunohistochemical staining was undertaken as described previously, 16 with antibodies specific for SLC7A11 (ProteinTech, 1:200 dilution). Four categories of staining intensity and their corresponding scores as follows: negative (0), weak (1), moderate (2), strong (3). H‐scores were calculated to quantify the immunoreactivity of tumor cells according to the following formula: (1× percentage of weak) + (2× percentage of moderate) + (3× percentage of strong).
2.4. Statistical analysis
All statistical analyses were performed using GraphPad Prism 9.0 software and were presented as the mean ± SD. All in vitro experiments were repeated three times. The differences between two groups were compared using Student’s t‐test. Comparisons among multiple sets of data were performed using ANOVA. Survival analysis were conducted with the log‐rank test. p < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001).
3. RESULTS
3.1. Resistance to ferroptosis is required for survival of extracellular matrix‐detached prostate cancer cells
To clarify whether ferroptosis is involved in the death mechanism of PCa cells after detachment from the ECM, we seeded parental PC‐3 and DU145 cells in ultra‐low‐attachment 96‐well plates. Cells from both cell lines were treated with the apoptosis inhibitor Z‐VAD‐FMK (10 µM), the ferroptosis inhibitor Ferrostatin‐1 (5 µM), or both. Both Z‐VAD‐FMK and Ferrostatin‐1 were able to improve the cell viability rate separately. However, Z‐VAD‐FMK combined with Ferrostatin‐1 showed the most significant increase in cell viability (p < 0.001) (Figure 1A). This indicates that the death mechanism of PCa cells after ECM detachment involves both apoptosis and ferroptosis.
FIGURE 1.
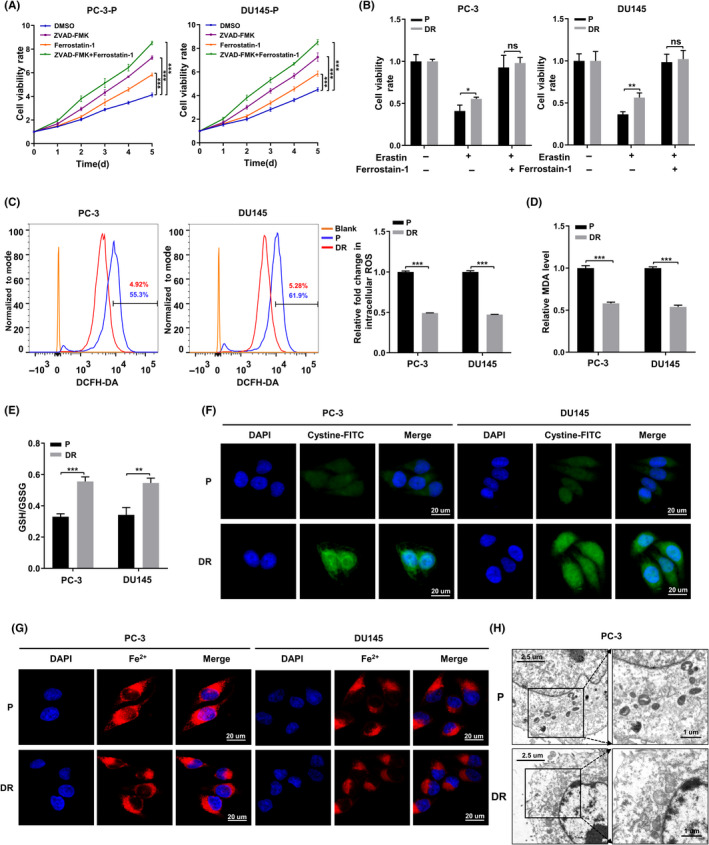
Death mechanism of PCa cells after detaching from extracellular matrix (ECM) involves ferroptosis. (A) CCK‐8 assays depicting the change in cell viability of parental prostate cancer (PCa) cells were treated with DMSO or ZVAD‐FMK (10 µM) or Ferrostatin‐1 (5 µM) or ZVAD‐FMK in combination with Ferrostatin‐1 after suspended culture for 5 days. (B) Cell viability in parental and DR PCa cells were treated with DMSO or erastin (10 µM) or erastin in combination with Ferrostatin‐1 (5 µM) for 24 h. (C) Intracellular ROS of parental and DR PCa cells were stained by DCFH‐DA and determined by flow cytometry. (D, E) Concentrations of MDA and GSH/GSSH ratio were measured in parental and DR PCa cells. (F, G) Intracellular cystine and Fe2+ of parental and DR PCa cells were detected by using cystine‐FITC and FeRhoNox‐1 fluorescent probe, respectively; confocal microscopy was used to record the fluorescence signal. Scale bars, 20 μm. (H) The morphological changes of mitochondria were detected by TEM in parental and detachment‐resistant (DR) PCa cells. Scale bars represent 2.5 and 1 µm, respectively. Data are presented as representative images or as mean ± SD from three independent repeats. *p < 0.05, **p < 0.01, ***p < 0.001
To further investigate the influence of ferroptosis resistance during ECM detachment in PCa cells, we established DR cell models using PC‐3 and DU145 cell lines, as described previously. 16 The CCK‐8 assay showed that the ferroptosis inducer erastin (10 µM) reduced cell viability in both parental and DR cells. However, DR cells were significantly more resistant to erastin than parental cells. In addition, Ferrostatin‐1 was able to reverse the effect of erastin on cell viability (Figure 1B).
Next, we observed ferroptosis‐related changes under nonadherent conditions in both parental and DR cells. ROS and MDA levels in DR cells were significantly lower than those in the parental cells (Figure 1C,D). The ratio of GSH/GSSG and the uptake levels of cystine in DR cells were notably higher than those in their parental counterparts (Figure 1E,F). No significant difference was observed in the levels of Fe2+ accumulation among two groups (Figure 1G). In addition, the differences in ferroptosis among parental and DR cells were also confirmed by transmission electron microscopy (TEM). As shown in Figure 1H, compared with DR cells, the mitochondria in parental cells were significantly smaller, while the mitochondrial cristae were shorter or had completely disappeared. These findings indicate that detachment from the ECM can lead to ferroptosis in PCa cells. Further, resistance to ferroptosis is required for the survival of PCa cells detached from the ECM.
3.2. Cell migration‐inducing protein mediates resistance to ferroptosis in DR prostate cancer cells
Using genome microarray assays, we previously found that CEMIP was significantly overexpressed in DR PCa cells compared to their parental cells. 16 Western blotting analysis and quantitative real‐time polymerase chain reaction (qRT‐PCR) analysis further verified that the transcription level and protein level of CEMIP in DR PCa cells were significantly higher than those of their parental cells (Figure S1A,B). To evaluate whether CEMIP affects ferroptosis in PCa cells after ECM detachment, we downregulated CEMIP by short hairpin RNA in DR PC‐3 and DU145 cells (Figure 2A). The CCK‐8 assay revealed that, when treated with erastin, the cell viability of the CEMIP‐silenced (shCEMIP) group was dramatically lower than that of the negative control (NC) group. Ferrostatin‐1 was able to restore the cell viability of the shCEMIP group (Figure 2B). We also observed that CEMIP downregulation markedly improved the levels of ROS and MDA (Figure 2C,D) and decreased the ratio of GSH/GSSG and the cystine uptake levels (Figure 2E,F) in DR cells. There were no significant differences in the intracellular Fe2+ levels between groups (Figure 2G). Consistent with these results, TEM showed that, compared to the NC group, CEMIP downregulation resulted in increased mitochondrial membrane density and smaller mitochondria (Figure 2H). To fully understand the influence of CEMIP on ferroptosis in PCa cells, we also stably overexpressed CEMIP in parental PC‐3 and DU145 cells. The results obtained were opposite to those obtained via CEMIP downregulation in DR cells, as shown in Figure S2. Collectively, downregulation of CEMIP attenuates resistance to ferroptosis in DR PCa cells. However, overexpression of CEMIP facilitates resistance to ferroptosis in parental PCa cells.
FIGURE 2.
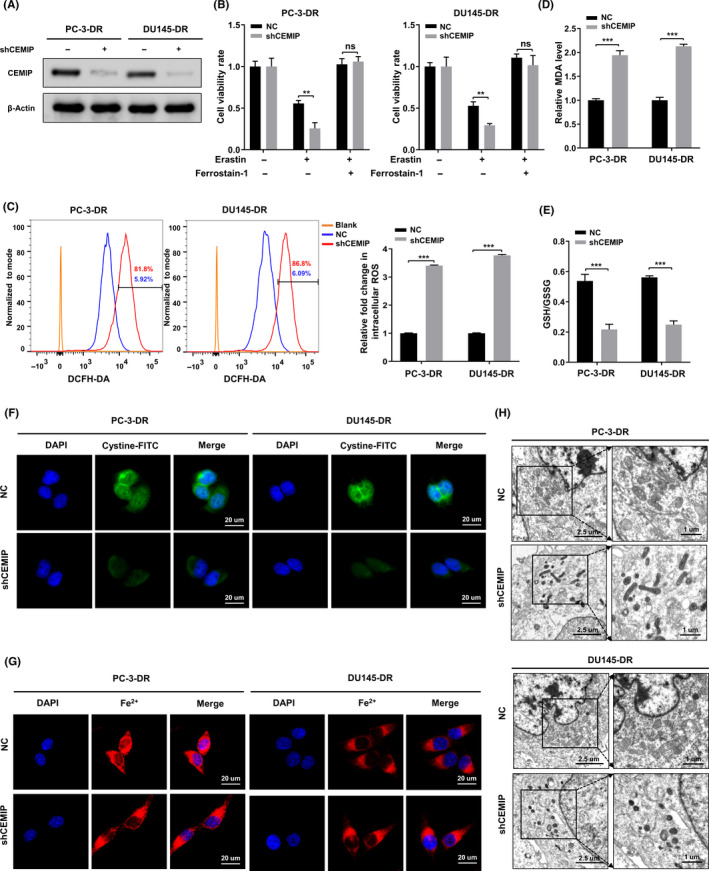
Downregulation of cell migration‐inducing protein (CEMIP) attenuates resistance to ferroptosis in detachment‐resistant (DR) prostate cancer (PCa) cells. (A) Western blotting analysis of CEMIP expression in the indicated DR PCa cells. (B) Indicated DR PCa cells were treated with DMSO or erastin (10 µM) or erastin in combination with Ferrostatin‐1 (5 µM) for 24 h, and cell viability was assayed by CCK‐8 assay. (C) Intracellular ROS of the indicated DR PCa cells were stained by DCFH‐DA and determined by flow cytometry. (D, E) Concentrations of malondialdehyde (MDA) and GSH/GSSH ratio were measured in the indicated DR PCa cells. (F, G) Intracellular cystine and Fe2+ of the indicated DR PCa cells were detected by using a cystine‐FITC and FeRhoNox‐1 fluorescent probe, respectively; confocal microscopy was used to recorded the fluorescence signal. Scale bars, 20 µm. (H) The morphological changes of mitochondria were detected by transmission electron microscope (TEM) in the indicated DR PCa cells. Scale bars represent 2.5 and 1 µm, respectively. Data are presented as representative images or as mean ± SD from three independent repeats. **p < 0.01, ***p < 0.001
3.3. Cell migration‐inducing protein knockdown impairs antioxidant capacity of detachment‐resistant prostate cancer cells by downregulating SLC7A11
As shown in Figure 3A,B, western blotting analysis and qRT‐PCR suggested that, consistent with CEMIP, the expression of solute carrier family 7 member 11 (SLC7A11) in DR PCa cells was obviously higher than that in parental cells. Downregulation of CEMIP by short hairpin RNA in DR PCa cells led to a remarkable decrease of SLC7A11 expression (Figure 3C,D). Expression data and corresponding clinical information of PCa patients (Table S3) extracted from The Cancer Genome Atlas database (https://portal.gdc.cancer.gov/) showed that SLC7A11 was notably upregulated in PCa tissues compared with normal tissues, both in the expression analysis of unpaired samples and paired samples (Figure 3E,F). The expression of SLC7A11 was positively associated with the Gleason score of patients with PCa (Figure 3G). Finally, the influence of SLC7A11 expression on the progress‐free interval in PCa patients from TCGA was evaluated by univariate and multivariate Cox regression analysis. We found SLC7A11 to be significant in univariate Cox analysis but not significant in the multivariate Cox model, including SLC7A11, Gleason score, T stage, N stage, and M stage (Figure 3H, Table S4). The baseline characteristics for PCa patients in the high and low SLC7A11 groups are shown in Table S5. Western blotting analysis also verified that SLC7A11 was upregulated in PCa tissues compared to matched normal tissues (Figure 3I). Consistent with recent reports, 18 immunohistochemistry (IHC) analysis of PCa specimens also confirmed that SLC7A11 expression in PCa tissues was higher than that in corresponding pericarcinous specimens and that SLC7A11 H‐scores were positively correlated with the Gleason scores in carcinoma tissues (Figure 3J). These results suggested that SLC7A11 may be a major downstream effector of CEMIP. To test this hypothesis, we performed SLC7A11 rescue experiments in shCEMIP DR PCa and control cells. As shown in Figure S3, overexpression of SLC7A11 in DR PCa cells reversed the reduction in SLC7A11 expression caused by CEMIP knockdown. Further experiments confirmed that overexpression of SLC7A11 could significantly rescue the uptake level of cystine and the ratio of GSH/GSSG induced by CEMIP knockdown (Figure 3K,L). The CCK‐8 assay revealed that overexpression of SLC7A11 was able to reverse the decrease in cell viability caused by CEMIP knockdown (Figure 3M). Silencing SLC7A11 reversed the increased antioxidant capacity and cell viability induced by CEMIP overexpression in LNCaP cells (Figure S4). Overall, these results indicate that SLC7A11 mediates CEMIP‐induced resistance to ferroptosis in PCa cells.
FIGURE 3.
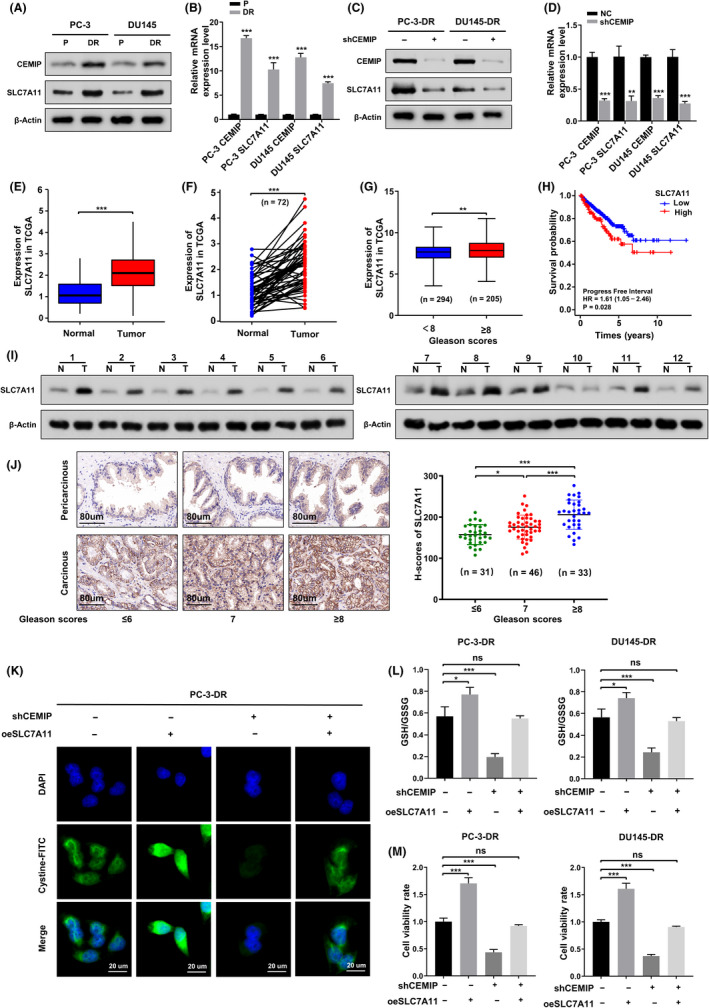
Silence of cell migration‐inducing protein (CEMIP) attenuates antioxidant capacity of detachment‐resistant (DR) prostate cancer (PCa) cells by downregulating SLC7A11. (A) Western blotting analysis of SLC7A11 expression in the indicated parental and DR PCa cells. (B) Quantitative real‐time polymerase chain reaction (qRT‐PCR) analysis of the transcription levels of SLC7A11 in indicated parental and DR PCa cells. (C) Western blotting to analyze the change of SLC7A11 caused by CEMIP knockdown in the indicated DR PCa cells. (D) qRT‐PCR to analyze changes in the transcription level of SLC7A11 caused by CEMIP knockdown in the indicated DR PCa cells. (E, F) Expression levels of SLC7A11 comparison between prostate tumor samples and normal tissues from the TCGA; (E) unpaired; (F) paired. (G) Expression levels of SLC7A11 in prostate cancer patients with different Gleason scores from the TCGA. (H) Univariate Cox regression analysis of the progress‐free interval (PFI) in prostate cancer patients with different expression levels of SLC7A11 from the TCGA. (I) Western blotting analysis of SLC7A11 expression in PCa tissues (N, normal tissues; T, tumor tissues). (J) Left panel, immunohistochemistry (IHC) analysis of SLC7A11 expression difference between PCa tissues and corresponding pericarcinous specimens. Right panel, H‐scores of SLC7A11 among PCa tissues with different Gleason scores. (K) Intracellular cystine were detected by using a cystine‐FITC fluorescent probe in the presence of shCEMIP plasmids or oeSLC7A11 plasmids or both shCEMIP plasmids and oeSLC7A11 plasmids in the indicated DR PCa cells; confocal microscopy was used to record the fluorescence signal. Scale bars, 20 μm. (L) GSH/GSSH ratio was measured in the presence of shCEMIP plasmids or oeSLC7A11 plasmids or both shCEMIP plasmids, and oeSLC7A11 plasmids in the indicated DR PCa cells. (M) Cell viability was detected by CCK‐8 assay in the indicated group. Data are presented as representative images or as mean ± SD from three independent repeats. *p < 0.05, **p < 0.01, ***p < 0.001
3.4. SLC7A11 rescues the progression of shCEMIP detachment‐resistant prostate cancer cells in vitro and in vivo
To investigate the function of SLC7A11 on proliferation, invasion, and migration of shCEMIP DR PCa cells, we performed rescue experiments in vitro. The soft agar assay indicated that the anchorage‐independent growth ability was weakened by CEMIP knockdown. However, it could be rescued by SLC7A11 overexpression (Figure 4A). The transwell assay showed that overexpression of SLC7A11 led to an increased capacity for migration and invasion. Further, SLC7A11 overexpression could rescue the decreased capacity of migration and invasion induced by CEMIP knockdown (Figure 4B).
FIGURE 4.
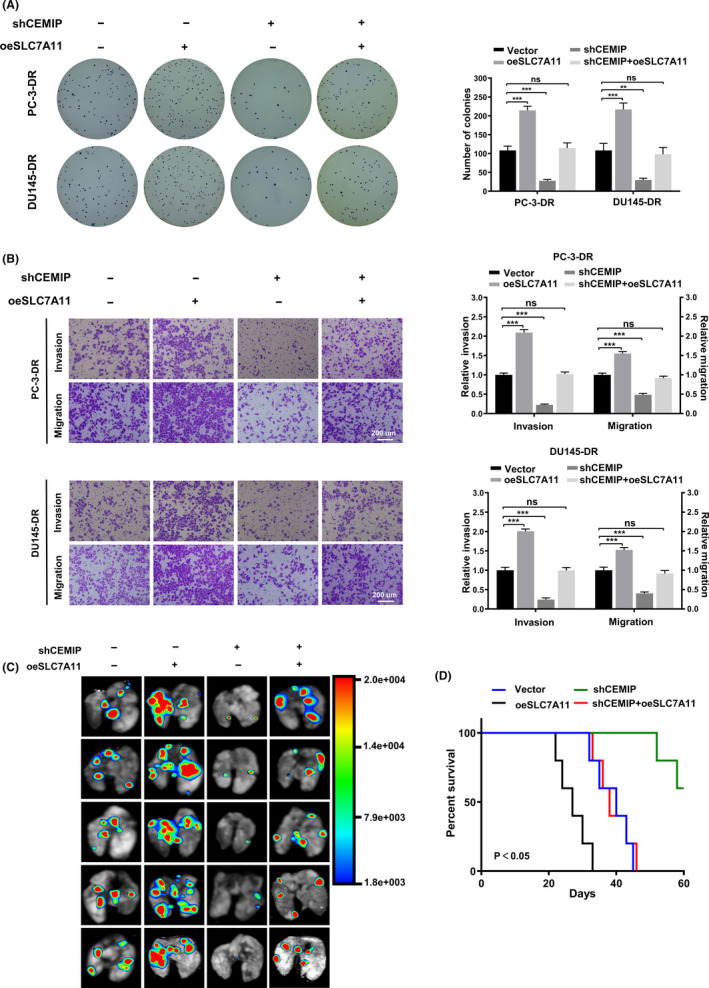
SLC7A11 rescues the invasion and metastasis of shCEMIP detachment‐resistant (DR) prostate cancer (PCa) cells in vitro and in vivo. (A) Representative images (left panel) and quantification (right panel) of soft agar plates indicating anchorage‐independent growth of the DR PCa cells stably transfected as indicated. (B) Transwell analysis of the invasion and migration capability of DR PCa cells stably transfected as indicated. Scale bars, 200 μm. (C) Representative image of metastatic lung colonization in nude mice (n = 5 per group) treated with tail vein injection of DR PC‐3 cells stably transfected as indicated. (D) Kaplan–Meier curves for nude mice. Data are presented as representative images or as mean ± SD from three independent repeats. **p < 0.01, ***p < 0.001
To further investigate the effects of CEMIP‐mediated SLC7A11 on PCa metastasis in vivo, we established an athymic nude mouse lung metastasis model by tail vein injection of fluorescent DR PC‐3 cells. Lung tumor metastasis was monitored two months after tail vein injection of the DR PC‐3 cells. Bioluminescence imaging showed that knockdown of CEMIP significantly decreased the number of pulmonary metastasis foci, and overexpression of SLC7A11 increased the number of pulmonary metastasis foci. In addition, overexpression of SLC7A11 markedly reversed the decrease in the number of pulmonary metastasis foci caused by CEMIP knockdown (Figure 4C). Consistent with the above results, athymic nude mice treated with a tail vein injection of DR PC‐3 cells stably transfected with a shCEMIP plasmid displayed better survival than the vector group. Further, overexpression of SLC7A11 significantly reversed the ascendant survival caused by CEMIP knockdown (Figure 4D). Collectively, these results suggest that SLC7A11 mediates CEMIP‐induced promotion of proliferation, invasion, migration, and metastasis in PCa.
3.5. Cell migration‐inducing protein mediates the expression of SLC7A11 by altering the subcellular localization of nuclear factor erythroid 2‐related factor 2
To further explore the regulation of SLC7A11 by CEMIP, we focused on the transcription factor nuclear factor erythroid 2‐related factor 2 (NRF2), as it is the primary transcription factor that promotes SLC7A11 transcription in response to oxidative stress. 19 , 20 , 21 NRF2 is bound with Kelch‐like ECH‐associated protein 1 (KEAP1) and persists in an inactivated state through ubiquitination and degradation in the proteasome under basal conditions. 22 , 23 However, during increased oxidative stress, NRF2 is phosphorylated at serine 40 (S40) and is no longer degraded by ubiquitination. 24 Once phosphorylated, NRF2 translocates into the nucleus and activates the transcription of antioxidant response element (ARE)‐containing genes. 25 , 26 As shown in Figure 5A,B, although there was no difference in the transcription levels of NRF2 between DR PCa cells and parental cells, the protein levels and the phosphorylation levels of NRF2 were remarkably higher in DR PCa cells than in parental cells. Meanwhile, the protein levels of KEAP1 were not significantly altered. Both cell lines were fractionated into cytosolic and nuclear fractions. In DR PCa cells, NRF2 was more concentrated in the nucleus than in parental PCa cells (Figure 5C). Next, we silenced CEMIP in DR PCa cells and found that, compared with control group cells, the protein levels and the phosphorylation levels of NRF2 in shCEMIP DR PCa cells were significantly reduced, and NRF2 was more translocated into the cytoplasm. The transcriptional levels of NRF2 and the protein levels of KEAP1 in PCa cells remained unchanged (Figure 5D–F). Furthermore, the translocation of NRF2 between the cytoplasm and nucleus was confirmed by immunofluorescence staining (Figure 5G).
FIGURE 5.
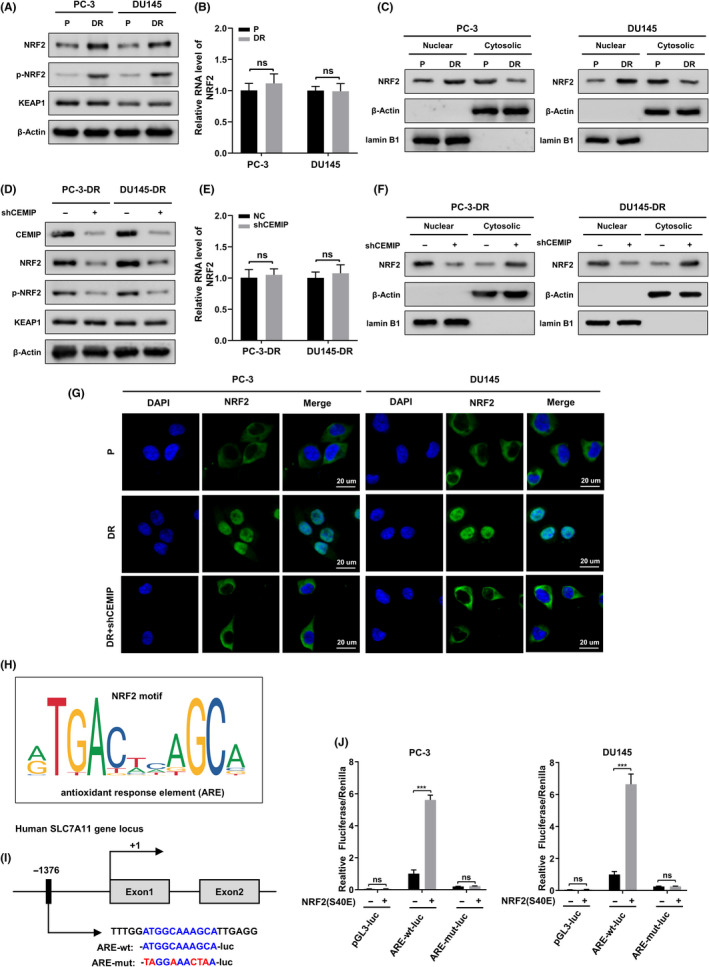
Cell migration‐inducing protein (CEMIP) drives NRF2 translocate from cytoplasm to nucleus and furthermore promotes the expression of SLC7A11. (A) KEAP1 protein, nuclear factor erythroid 2‐related factor 2 (NRF2) protein, and its phosphorylation levels (p‐NRF2) in the whole cell lysate from the indicated parental and detachment‐resistant (DR) prostate cancer (PCa) cells were assessed using western blotting. (B) Quantitative real‐time polymerase chain reaction (qRT‐PCR) analysis of the transcription levels of NRF2 in indicated parental and DR PCa cells. (C) Indicated parental PCa cells were subjected to nuclear and cytosolic fractionation, and NRF2 protein levels were determined by western blotting. Lamin B1 and β‐Actin were used as nuclear and cytoplasmic markers, respectively. (D) The protein levels of NRF2, p‐NRF2, and KEAP1 were assayed by western blotting in the indicated DR PCa cells transfected with empty plasmids or shCEMIP plasmids. (E) qRT‐PCR to analyze changes in the transcription level of NRF2 caused by CEMIP knockdown in the indicated DR PCa cells. (F) The NRF2 protein levels in cytoplasmic fraction and nuclear fraction from the indicated cells were analyzed using western blotting. (G) NRF2 location in the indicated cells was observed using fluorescent microscopy. Scale bars, 20 μm. (H) The positional matrices for the human ARE sequences, JASPAR ID MA0150.1, which have been generated by the frequency at which these sites have been found experimentally to be occupied by NRF2. (I) Schematic representation of the ARE in the SLC7A11 promoter and its mutant (AREmut). The +1 denotes the transcription initiation site. (J) Luciferase assays performed using the control pGL3, ARE‐wt, and ARE‐mut constructs in the indicated parental PCa cells transfected with empty vector or NRF(S40E) plasmids. Data are presented as representative images or as mean ± SD from three independent repeats. ***p < 0.001
To verify the transcriptional regulation of SLC7A11 by NRF2, we used JASPAR 27 (http://jaspar.genereg.net) to identify the binding profile of NRF2 in Homo sapiens (Figure 5H). Next, as shown in Figure 5I, we identified a canonical ARE upstream of the SLC7A11 transcription start site and cloned luciferase reporter constructs into a pGL3 plasmid containing the putative ARE (ARE‐wt‐luc) or its mutant (ARE‐mut‐luc). We also constructed NRF2(S40E) mutant plasmids, in which the S40 site was replaced with glutamic acid to mimic continual phosphorylation. 28 Subsequently, we performed a dual‐luciferase assay to analyze promoter activity. As expected, ectopic NRF2(S40E) expression significantly activated ARE‐wt‐luc activity. However, no activation of ARE‐mut‐luc activity was observed upon overexpression of NRF2(S40E) (Figure 5J). These results suggest that CEMIP promotes NRF2 phosphorylation and localization to the nucleus, which in turn promotes transcription of SLC7A11.
3.6. Activation of calcium/calmodulin‐dependent protein kinase II result in cell migration‐inducing protein‐mediated nuclear factor erythroid 2‐related factor 2 phosphorylation at serine 40
The decreased phosphorylation level of NRF2 is accompanied by downregulation of CEMIP. However, CEMIP is a hyaluronidase rather than a classic protein kinase. 29 Studies have reported that detachment from the ECM activates endoplasmic reticulum (ER) stress, which leads to increased cytosolic Ca2+ levels. 14 , 30 Recently, it was confirmed that CEMIP can interact with the ER receptor inositol 1,4,5‐trisphosphate receptor type 3 (ITPR3), a key player in Ca2+ signaling that mediates Ca2+ leakage from the ER, leading to elevated cytosolic Ca2+ levels. 31 , 32 To verify the interaction of CEMIP and ITPR3 in parental PCa cells and DR PCa cells, we performed a Co‐IP experiment. As shown in Figure 6A,B, the interaction between CEMIP and ITPR3 in DR PCa cells was increased compared to that in parental cells. The cytosolic Ca2+ levels in DR PCa cells were significantly higher than those in parental PCa cells (Figure 6C). Furthermore, knockdown of CEMIP significantly decreased both the interaction between CEMIP and ITPR3 and the cytosolic Ca2+ levels in DR PCa cells (Figure 6D–F). Double immunofluorescent staining indicated that CEMIP and ITPR3 were colocalized in the cytoplasm of DR PC‐3 cells. However, this colocalization relationship could be significantly weakened by knockdown of CEMIP (Figure 6G).
FIGURE 6.
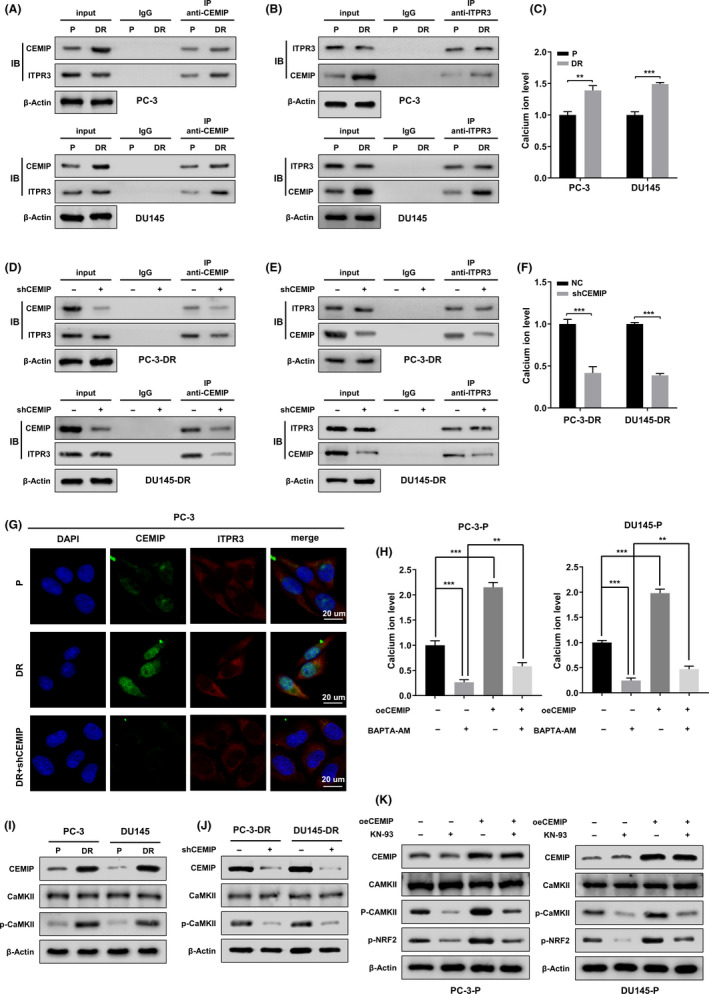
Cell migration‐inducing protein (CEMIP) mediates nuclear factor erythroid 2‐related factor 2 (NRF2) phosphorylation at serine 40 (S40) by activating CaMKII. (A, B) Western blotting showing Co‐IP of ITPR3 with anti‐CEMIP antibody and reciprocal co‐immunoprecipitation of CEMIP with anti‐ITPR3 antibody in the indicated parental and detachment‐resistant (DR) prostate cancer (PCa) cells (upper panel, PC‐3 cells, lower panel, DU145 cells). The lysate immunoprecipitated with anti‐immunoglobulin G antibody served as negative control. (C) Cytosolic Ca2+ levels of the indicated parental and DR PCa cells were detected by Fluo‐4 AM fluorescent probe. (D, E) After the indicated DR PCa cells were transfected with empty plasmids or shCEMIP plasmids, the whole cell lysates were subjected to immunoprecipitation using anti‐CEMIP antibody, followed by immunoblotting (IB) with anti‐CEMIP and anti‐ITPR3 antibody. Reciprocal IP was performed using anti‐ITPR3 antibody, followed by IB with anti‐CEMIP and anti‐ITPR3 antibody. The lysate immunoprecipitated with anti‐immunoglobulin G antibody was served as negative control. (F) Cytosolic Ca2+ levels of the indicated DR PCa cells transfected with empty plasmids or shCEMIP plasmids were detected by Fluo‐4 AM fluorescent probe. (G) Double immunofluorescent staining revealed the changes of colocation relationships between CEMIP and ITPR3 proteins in the indicated PCa cells. Scale bars, 20 μm. (H) The indicated parental PCa cells were transfected with empty plasmids or oeCEMIP plasmids, then treated with calcium chelator BAPTA‐AM (1 μM) or DMSO for 24 h, and respective cytosolic Ca2+ levels were detected by Fluo‐4 AM fluorescent probe. (I) CaMKII expression and its phosphorylation levels (p‐CaMKII) in the whole cell lysate from the indicated parental and DR PCa cells were assessed using western blotting. (J) The expression of CaMKII and p‐CaMKII were assayed by western blotting in the indicated DR PCa cells transfected with empty plasmids or shCEMIP plasmids. (K) The indicated parental PCa cells were transfected with empty plasmids or oeCEMIP plasmids, then treated with CaMKII inhibitor KN‐93 (10 μM) or DMSO for 24 h. Western blotting analysis the expression of CEMIP, CaMKII, p‐CaMKII, and p‐NRF2 in different groups. Data are presented as representative images or as mean ± SD from three independent repeats. **p < 0.01, ***p < 0.001
To further address the effect of CEMIP on cytosolic Ca2+ levels, we performed rescue experiments in parental PCa cells. As shown in Figure 6H, overexpression of CEMIP significantly rescued the inhibition of cytosolic Ca2+ levels caused by the calcium chelator BAPTA‐AM (1 µM). Based on the above research results, we further tested calcium/calmodulin‐dependent kinase II (CaMKII) and its phosphorylation level at threonine 286 (T286), as CaMKII is a family of multifunctional serine/threonine protein kinases, and phosphorylated activation of CaMKII requires both Ca2+ and calmodulin. 33 , 34 Western blotting analysis revealed that the phosphorylation level of CaMKII at T286 in DR PCa cells was remarkably higher than that in parental cells. However, there was no obvious difference in the expression of CaMKII between the two groups (Figure 6I). Knockdown of CEMIP in DR PCa cells did not affect the expression of CaMKII but did reduce its activity (Figure 6J).
Next, to elucidate the regulation of CEMIP on CaMKII and to determine whether NRF2 was the major downstream effector of CaMKII, we conducted a rescue experiment in parental PCa cells using the CaMKII inhibitor KN‐93 (10 µM) and CEMIP‐overexpression plasmids. Western blot analysis showed that the depressed phosphorylation level of CaMKII at T286 caused by KN‐93 could be restored by CEMIP overexpression (Figure 6K). Furthermore, the inhibition of CaMKII activity by KN‐93 was accompanied by a decrease in the phosphorylation level of NRF2 (Figure 6K). These results indicate that CEMIP activates CaMKII by increasing the level of cytosolic Ca2+, thereby promoting NRF2 phosphorylation in PCa cells.
4. DISCUSSION
Previous studies have shown that cells under detached conditions may undergo anoikis 35 and non‐apoptotic cell death induced by rectifying metabolic deficiencies. 4 , 36 Escape from anoikis and other non‐apoptotic cell death forms is a prerequisite for the metastatic cascade of cancer cells. 37 Although the underlying mechanism of anoikis resistance acquisition in cancer cells has been extensively researched, the mechanisms by which detached cancer cells escape from non‐apoptotic cell death, especially ferroptosis, remain unclear. In this study, we identified the role and potential molecular basis of CEMIP on ferroptosis in ECM‐detached PCa cells. Compared with parental cells, DR PCa cells were less susceptible to ferroptosis inducers. Knockdown of CEMIP decreases cystine uptake and increases intracellular ROS, thereby triggering ferroptosis in DR PCa cells under detachment conditions. In contrast, overexpression of CEMIP compromises erastin‐induced cell death in parental cells under detachment conditions. Collectively, our results delineate a novel role of CEMIP in ferroptosis resistance during ECM detachment, defining CEMIP as a promising therapeutic target for metastatic PCa.
Our study links CEMIP expression to ferroptosis resistance based on the induction of ferroptosis upon silencing of endogenous CEMIP in ECM‐detached DR PCa cells. Our results show that overexpression of CEMIP protects parental PC‐3 and DU145 cells from ferroptosis under detachment conditions. Considering that CEMIP can reduce ROS levels in ECM‐detached PCa cells, we wondered whether CEMIP regulates the expression of antioxidant‐related genes. System Xc − is a membrane sodium‐independent cystine/glutamate antiporter. It is responsible for maintaining redox homeostasis by exporting intracellular glutamate and importing extracellular cystine at a 1:1 ratio, cystine is subsequently reduced to cysteine and used to synthesize the major antioxidant GSH. 38 , 39 Given the difference in the ratio of GSH/GSSG and the uptake level of cystine between DR PCa cells and their shCEMIP group cells, we speculated that CEMIP might regulate the expression level of SLC7A11, which is the functional subunit of system Xc −. 40 We found that CEMIP promotes ferroptosis resistance by facilitating NRF2 phosphorylation and nuclear localization, leading to elevated SLC7A11 transcription in PCa cells. Furthermore, in vivo analysis confirmed that silencing of CEMIP inhibits metastasis formation in a tail vein cancer metastasis mouse model; overexpression of SLC7A11 reversed this effect. Previous studies have also found that CEMIP can promote metastasis of hepatocellular carcinomas and brain tumors. 14 , 41 NRF2 is a key oxidant defense transcription factor. 42 Several studies have revealed that phosphorylation and nuclear translocation of the NRF2 protein may enhance the transcription of AREs of target genes under oxidative stress conditions. 43 , 44 , 45 The antioxidant effect of NRF2 can prevent carcinogenesis in normal lung tissue but may accelerate malignant cell growth after tumor initiation. 46 NRF2 inducers, such as sulforaphane, can protect against prostate carcinogenesis in genetic models. 47 NRF2 activation is responsible for the increased resistance to ER stress‐induced apoptosis in PCa cells. 48 The role of NRF2 in tumor cell detachment from the ECM has not been extensively studied. A few studies have found that NRF2 protein levels increase after epithelial ovarian cancer cell detachment from the ECM. 49 AMP‐activated protein kinase (AMPK) and activating transcription factor 4 (ATF4) may induce the expression of NRF2 and its target proteins, thereby sustaining cell survival following ECM detachment. 37 , 50 Our study is the first to demonstrate that NRF2 activation protects ECM‐detached PCa cells from ferroptosis by promoting SLC7A11 transcription.
Cell migration‐inducing protein localized in the ER is found to mediate ER Ca2+ leakage and ultimately increase cytosolic Ca2+ levels. 13 , 14 Ca2+, an important intracellular messenger, is involved in different cellular processes linked to cancer, such as the cell cycle, apoptosis, and autophagy. 51 Increased intracellular Ca2+ levels can activate different kinases, including MAPK, 52 PKC, 14 and CaMKII. 53 A previous study identified ITPR3 as an CEMIP‐interacting protein in colon cancer by immunoprecipitation and mass spectrometry. 32 ITPR3 is a Ca2+ release channel that resides in the membranes of the ER, which is extremely important for intracellular Ca2+ homeostasis. 54 The present study revealed that the interaction of CEMIP and ITPR3 led to ER Ca2+ leakage and increased cytosolic Ca2+ levels in ECM‐detached PCa cells, subsequently activating CaMKII. Furthermore, functional studies revealed that CaMKII facilitates NRF2 phosphorylation at S40 and nuclear localization, leading to elevated SLC7A11 transcription in PCa cells.
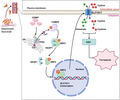
In summary, our results highlight that ferroptosis is involved in the detachment of PCa cells from the ECM. Overexpression of CEMIP promoted ferroptosis resistance by activating the ITPR3/CaMKII/NRF2/SLC7A11 pathway (Figure 7), thereby providing new insights into the development of therapeutic strategies for metastatic PCa. However, our understanding of other potential mechanisms responsible for CEMIP‐mediated ferroptosis resistance under detachment conditions is far from complete. Further research is required to understand the complete mechanistic basis of the observed effects.
FIGURE 7.
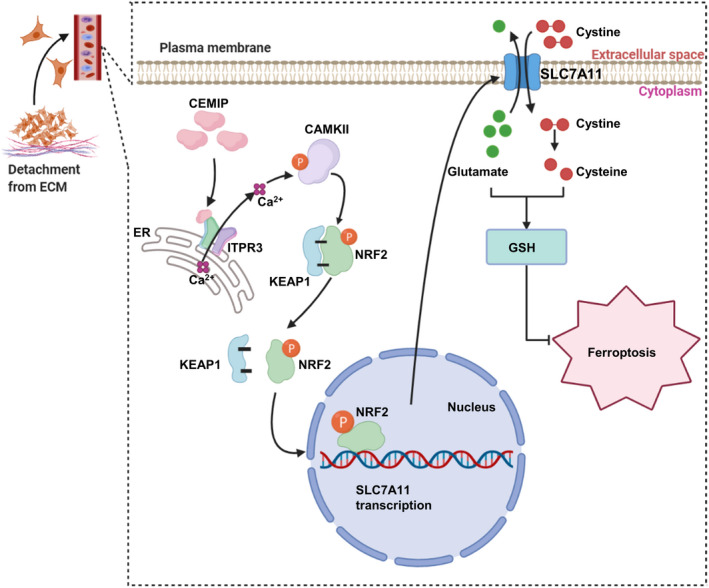
A scheme for cell migration‐inducing protein (CEMIP)‐mediated ITPR3/CaMKII/NRF2/SLC7A11 pathway inhibits ferroptosis in detachment‐resistant prostate cancer cells. CEMIP interacted with ITPR3 and promoted Ca2+ leakage from the endoplasmic reticulum and thereby activated CaMKII. Activated CaMKII (p‐CaMKII) further promoted the phosphorylation of NRF2, which led to NRF2 dissociated with KEAP1 and translocated from cytoplasm to nucleus. NRF2 facilitated SLC7A11 transcription and led to an increase in GSH levels, thereby inhibits ferroptosis
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported by the National Natural Science Foundation of China (grant numbers 81974399, 81772751, and 82103610).
Liu B, Li X, Wang D, et al. CEMIP promotes extracellular matrix‐detached prostate cancer cell survival by inhibiting ferroptosis. Cancer Sci. 2022;113:2056–2070. doi: 10.1111/cas.15356
Bing Liu, Xuexiang Li and Decai Wang contributed equally to this work.
Contributor Information
Yarong Song, Email: tjmusong@126.com.
Yifei Xing, Email: yfxing@hust.edu.cn.
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209‐249. [DOI] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7‐30. [DOI] [PubMed] [Google Scholar]
- 3. Tan PS, Aguiar P Jr, Haaland B, Lopes G. Addition of abiraterone, docetaxel, bisphosphonate, celecoxib or combinations to androgen‐deprivation therapy (ADT) for metastatic hormone‐sensitive prostate cancer (mHSPC): a network meta‐analysis. Prostate Cancer Prostatic Dis. 2018;21:516‐523. [DOI] [PubMed] [Google Scholar]
- 4. Hawk MA, Gorsuch CL, Fagan P, et al. RIPK1‐mediated induction of mitophagy compromises the viability of extracellular‐matrix‐detached cells. Nat Cell Biol. 2018;20:272‐284. [DOI] [PubMed] [Google Scholar]
- 5. Mason JA, Cockfield JA, Pape DJ, et al. SGK1 signaling promotes glucose metabolism and survival in extracellular matrix detached cells. Cell Rep. 2021;34:108821. [DOI] [PubMed] [Google Scholar]
- 6. Wang D, Zhang L, Hu A, et al. Loss of 4.1N in epithelial ovarian cancer results in EMT and matrix‐detached cell death resistance. Protein Cell. 2021;12:107‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buchheit CL, Weigel KJ, Schafer ZT. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer. 2014;14:632‐641. [DOI] [PubMed] [Google Scholar]
- 8. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149:1060‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hawk MA, Schafer ZT. Mechanisms of redox metabolism and cancer cell survival during extracellular matrix detachment. J Biol Chem. 2018;293:7531‐7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brown CW, Amante JJ, Goel HL, Mercurio AM. The alpha6beta4 integrin promotes resistance to ferroptosis. J Cell Biol. 2017;216:4287‐4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang GX, Tu HC, Dong Y, et al. DeltaNp63 inhibits oxidative stress‐induced cell death, including ferroptosis, and cooperates with the BCL‐2 family to promote clonogenic survival. Cell Rep. 2017;21:2926‐2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang L, Yu T, Li W, et al. The miR‐29c‐KIAA1199 axis regulates gastric cancer migration by binding with WBP11 and PTP4A3. Oncogene. 2019;38:3134‐3150. [DOI] [PubMed] [Google Scholar]
- 13. Li L, Yan LH, Manoj S, Li Y, Lu L. Central role of CEMIP in tumorigenesis and its potential as therapeutic target. J Cancer. 2017;8:2238‐2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Evensen NA, Kuscu C, Nguyen HL, et al. Unraveling the role of KIAA1199, a novel endoplasmic reticulum protein, in cancer cell migration. J Natl Cancer Inst. 2013;105:1402‐1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Michishita E, Garces G, Barrett JC, Horikawa I. Upregulation of the KIAA1199 gene is associated with cellular mortality. Cancer Lett. 2006;239:71‐77. [DOI] [PubMed] [Google Scholar]
- 16. Zhang P, Song Y, Sun Y, et al. AMPK/GSK3beta/beta‐catenin cascade‐triggered overexpression of CEMIP promotes migration and invasion in anoikis‐resistant prostate cancer cells by enhancing metabolic reprogramming. FASEB J. 2018;32:3924‐3935. [DOI] [PubMed] [Google Scholar]
- 17. Taylor WR, Fedorka SR, Gad I, et al. Small‐molecule ferroptotic agents with potential to selectively target cancer stem cells. Sci Rep. 2019;9:5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhong W, Weiss HL, Jayswal RD, et al. Extracellular redox state shift: a novel approach to target prostate cancer invasion. Free Radic Biol Med. 2018;117:99‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34:21‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tebay LE, Robertson H, Durant ST, et al. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic Biol Med. 2015;88:108‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dodson M, Castro‐Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu K, Alcivar AL, Ma J, et al. NRF2 induction supporting breast cancer cell survival is enabled by oxidative stress‐induced DPP3‐KEAP1 interaction. Cancer Res. 2017;77:2881‐2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal. 2018;29:1756‐1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anandhan A, Dodson M, Schmidlin CJ, Liu P, Zhang DD. Breakdown of an ironclad defense system: the critical role of NRF2 in mediating ferroptosis. Cell Chem Biol. 2020;27:436‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Y, Zhao S, Fu Y, et al. Computational repositioning of dimethyl fumarate for treating alcoholic liver disease. Cell Death Dis. 2020;11:641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cho MK, Kim WD, Ki SH, et al. Role of Galpha12 and Galpha13 as novel switches for the activity of Nrf2, a key antioxidative transcription factor. Mol Cell Biol. 2007;27:6195‐6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fornes O, Castro‐Mondragon JA, Khan A, et al. JASPAR 2020: update of the open‐access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87‐D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Numazawa S, Ishikawa M, Yoshida A, Tanaka S, Yoshida T. Atypical protein kinase C mediates activation of NF‐E2‐related factor 2 in response to oxidative stress. Am J Physiol Cell Physiol. 2003;285:C334‐342. [DOI] [PubMed] [Google Scholar]
- 29. Tammi MI, Oikari S, Pasonen‐Seppanen S, Rilla K, Auvinen P, Tammi RH. Activated hyaluronan metabolism in the tumor matrix – causes and consequences. Matrix Biol. 2019;78–79:147‐164. [DOI] [PubMed] [Google Scholar]
- 30. Cubillos‐Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168:692‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Y, Jia S, Jiang WG. KIAA1199 and its biological role in human cancer and cancer cells (review). Oncol Rep. 2014;31:1503‐1508. [DOI] [PubMed] [Google Scholar]
- 32. Tiwari A, Schneider M, Fiorino A, et al. Early insights into the function of KIAA1199, a markedly overexpressed protein in human colorectal tumors. PLoS One. 2013;8:e69473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhattacharyya M, Karandur D, Kuriyan J. Structural insights into the regulation of Ca(2+)/calmodulin‐dependent protein kinase II (CaMKII). Cold Spring Harb Perspect Biol. 2020;12:a035147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu G, Cheng CJ, Lin SC, et al. Organelle‐derived acetyl‐CoA promotes prostate cancer cell survival, migration, and metastasis via activation of calmodulin kinase II. Cancer Res. 2018;78:2490‐2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim YS, Gupta Vallur P, Jones VM, et al. Context‐dependent activation of SIRT3 is necessary for anchorage‐independent survival and metastasis of ovarian cancer cells. Oncogene. 2020;39:1619‐1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ishikawa F, Ushida K, Mori K, Shibanuma M. Loss of anchorage primarily induces non‐apoptotic cell death in a human mammary epithelial cell line under atypical focal adhesion kinase signaling. Cell Death Dis. 2015;6:e1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Endo H, Owada S, Inagaki Y, Shida Y, Tatemichi M. Metabolic reprogramming sustains cancer cell survival following extracellular matrix detachment. Redox Biol. 2020;36:101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu J, Xia X, Huang P. xCT: a critical molecule that links cancer metabolism to redox signaling. Mol Ther. 2020;28:2358‐2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12:599‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rodrigues G, Hoshino A, Kenific CM, et al. Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nat Cell Biol. 2019;21:1403‐1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sun X, Ou Z, Chen R, et al. Activation of the p62‐Keap1‐NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1‐Nrf2 pathway. Trends Pharmacol Sci. 2013;34:340‐346. [DOI] [PubMed] [Google Scholar]
- 44. Zhang J, Anshul F, Malhotra DK, Jaume J, Dworkin LD, Gong R. Microdose lithium protects against pancreatic islet destruction and renal impairment in streptozotocin‐elicited diabetes. Antioxidants (Basel). 2021;10:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chaiprasongsuk A, Janjetovic Z, Kim TK, et al. Protective effects of novel derivatives of vitamin D3 and lumisterol against UVB‐induced damage in human keratinocytes involve activation of Nrf2 and p53 defense mechanisms. Redox Biol. 2019;24:101206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Satoh H, Moriguchi T, Saigusa D, et al. NRF2 intensifies host defense systems to prevent lung carcinogenesis, but after tumor initiation accelerates malignant cell growth. Cancer Res. 2016;76:3088‐3096. [DOI] [PubMed] [Google Scholar]
- 47. Dinkova‐Kostova AT, Fahey JW, Kostov RV, Kensler TW. KEAP1 and done? Targeting the NRF2 pathway with sulforaphane. Trends Food Sci Technol. 2017;69:257‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bellezza I, Scarpelli P, Pizzo SV, Grottelli S, Costanzi E, Minelli A. ROS‐independent Nrf2 activation in prostate cancer. Oncotarget. 2017;8:67506‐67518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Q, Gu T, Ma L, et al. Efficient iron utilization compensates for loss of extracellular matrix of ovarian cancer spheroids. Free Radic Biol Med. 2021;164:369‐380. [DOI] [PubMed] [Google Scholar]
- 50. Dey S, Sayers CM, Verginadis II, et al. ATF4‐dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J Clin Invest. 2015;125:2592‐2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Patergnani S, Danese A, Bouhamida E, et al. Various aspects of calcium signaling in the regulation of apoptosis, autophagy, cell proliferation, and cancer. Int J Mol Sci. 2020;21:8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Song N, Ma J, Meng XW, et al. Heat shock protein 70 protects the heart from ischemia/reperfusion injury through inhibition of p38 MAPK signaling. Oxid Med Cell Longev. 2020;2020:3908641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yan T, Zhao Y. Acetaldehyde induces phosphorylation of dynamin‐related protein 1 and mitochondrial dysfunction via elevating intracellular ROS and Ca(2+) levels. Redox Biol. 2020;28:101381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yue L, Wang L, Du Y, et al. Type 3 inositol 1,4,5‐Trisphosphate receptor is a crucial regulator of calcium dynamics mediated by endoplasmic reticulum in HEK cells. Cells. 2020;9:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Supplementary Material