

Abstract
Cholesterol is an essential plasma membrane lipid for the maintenance of cellular homeostasis and cancer cell proliferation. Free cholesterol is harmful to cells; therefore, excessive free cholesterol must be quickly esterified by acetyl‐coenzyme A:cholesterol acetyltransferase (ACAT) and exported by scavenger receptor class B member I (SR‐BI) or ATP‐binding cassette protein A1 from specific cells such as macrophage foam cells, which contain cholesteryl ester‐derived vacuoles. Many vacuoles are present in the cytoplasm of Burkitt lymphoma cells. In this study, we observed that these vacuoles are often seen in high‐grade lymphomas. Cell culture study using lymphoma cell lines found that esterified cholesterol is the main component of these vacuoles and the expression of cholesterol metabolism‐related molecules was significantly upregulated in lymphoma cell lines, with SR‐BI and ACAT inhibitors (BLT‐1 and CI‐976, respectively) impeding lymphoma cell proliferation. Cytoplasmic free cholesterol was increased by ACAT and SR‐BI inhibitors, and the accumulation of free cholesterol induced lymphoma cell apoptosis by inducing endoplasmic reticulum stress. Furthermore, synergistic effects of SR‐BI and ACAT inhibitors were observed in a preclinical study. Treatment with SR‐BI inhibitor suppressed lymphoma progression in a tumor‐bearing mouse model, whereas ACAT inhibitor did not. Therefore, SR‐BI inhibitors are potential new antilymphoma therapeutics that target cholesterol metabolism.
Keywords: ACAT, cholesterol, ER stress, lymphoma, SR‐BI
This study revealed that the main component of vacuoles seen in high‐grade lymphomas is esterified cholesterol. The expression of cholesterol metabolism‐related molecules such as scavenger receptor class B member I and acetyl‐coenzyme A:cholesterol acetyltransferase is significantly upregulated in lymphoma cells. Scavenger receptor class B member I inhibitor has potential as a new therapy for targeting the cholesterol metabolism pathway in lymphomas.

Abbreviations
- ABCA1
ATP‐binding cassette protein A1
- ACAT
acetyl‐coenzyme A:cholesterol acetyltransferase
- ATF6
activating transcription factor 6
- ATLL
adult T‐cell leukemia/lymphoma
- CBDCA
carboplatin
- CE
cholesteryl ester
- CHOP
C/EBP homologous protein
- DLBCL
diffuse large B‐cell lymphoma
- eIF2α
eukaryotic translation initiation factor 2α
- EBV
Epstein–Barr virus
- ER
endoplasmic reticulum
- GCB
germinal center B‐cell‐like subtype
- HDL
high‐density lipoprotein
- HD‐MTX
high‐dose methotrexate
- HMG‐CoA reductase
3‐hydroxy‐3‐methylglutaryl‐CoA reductase
- IRE1
inositol‐requiring enzyme 1
- LDL
low‐density lipoprotein
- LDLR
LDL receptor
- LPDS
lipoprotein‐deficient serum
- LXR
liver X receptor
- PCNSL
primary central nervous system lymphoma
- RT‐qPCR
real‐time quantitative PCR
- sIL‐2R
soluble interleukin‐2 receptor
- SR‐BI
scavenger receptor class B member I
- SREBP2
sterol response element‐binding protein 2
- UPR
unfolded protein response
- WBRT
whole‐brain radiation therapy
1. INTRODUCTION
Non‐Hodgkin lymphoma, including Burkitt lymphoma, DLBCL, and ATLL, are highly aggressive lymphomas that have difficult clinical courses. 1 The overall survival of patients with Burkitt lymphoma and DLBCL is significantly improved by additional treatment with rituximab, 2 , 3 although relapsed leukemia/lymphoma is resistant to this treatment, and novel therapeutic strategies are now in clinical trials. Adult T‐cell leukemia/lymphoma is associated with retroviral infections, such as human T‐cell leukemia virus type I or human T‐cell lymphotropic virus type I. Outcomes from aggressive types of ATLL are poor, even when new anti‐CCR4 Abs are used as an additional treatment. 4 In addition, most cases of PCNSL are high‐grade DLBCL and standard therapeutic protocol is chemotherapy with HD‐MTX, combination therapy with rituximab, and WBRT. However, the 5‐year overall survival with combined HD‐MTX and WBRT is approximately 20%. 5 , 6 Therefore, the detailed mechanisms underlying the resistance of aggressive lymphomas to antilymphoma therapies should be identified to guide the treatment of lymphoma.
Cholesterol is one of the main components of cell membranes and is a precursor of bile acid and steroidogenic hormones. 7 It is transported around the body in its bound form: LDL, or HDL. Low‐density lipoprotein is delivered into cells through receptor‐mediated endocytosis by the LDLR, while ABCA1 and SR‐BI are involved in cholesterol transport mediated by HDL binding. Cholesterol uptake and its metabolic upregulation are important factors in cancer cell growth and proliferation. 8 Low‐density lipoprotein receptor and the cholesterol synthesis enzyme HMG‐CoA reductase are overexpressed in cancer cells, including leukemia/lymphoma cells. 9 Excess free cholesterol is harmful to cells 10 , 11 ; therefore, free cholesterol needs to be quickly esterified to CE by ACAT. The efflux of accumulated free cholesterol is a mechanism for regulating cholesterol homeostasis. Many researchers have attempted to treat cancer cells with ACAT or HMG‐CoA reductase inhibitors. In brain cancers, an LXR agonist promotes tumor cell death by inducing LDLR downregulation and increasing ABCA1 expression, and the antitumor effect of the LXR agonist is observed in a preclinical model. 12 , 13 Therefore, targeting cholesterol metabolism could be a promising approach to cancer therapy; however, the effects of these drugs in treating cancers, including leukemia/lymphoma, are currently limited.
Many vacuoles are detected in the cytoplasm of Burkitt lymphoma cells, which are important characteristics in morphological diagnosis. 14 Similarly, intracytoplasmic vacuoles are also observed in some circulating ATLL cells 15 ; however, their components are uncharacterized. Comparable vacuoles composed of accumulated CE are observed in foamy macrophages, which are the primary indicators of atherosclerosis. 16 Therefore, the lymphoma vacuoles might contain large amounts of cholesterol and play an important role in cell proliferation. Targeting cholesterol metabolism could be an effective antilymphoma therapy.
In the present study, we observed vacuoles in high‐grade lymphomas and hypothesized that these vacuoles reflected excessive cholesterol uptake with a high accumulation of CE. Molecules related to cholesterol homeostasis were overexpressed in several T‐cell and B‐cell lymphoma cell lines, including PCNSL. Interestingly, an SR‐BI inhibitor induced lymphoma cell apoptosis and prolonged survival in a preclinical model. The SR‐BI inhibitor also synergistically increased the cytotoxic effects of the ACAT inhibitor. The present study indicated that blocking cholesterol efflux could be a novel antilymphoma strategy.
2. MATERIALS AND METHODS
2.1. Patients and samples
Touch imprint cytology samples were obtained from lymph node biopsies from 92 patients diagnosed with follicular lymphoma (n = 15), ATLL (n = 23, lymphomatous type), or DLBCL (n = 54) from 2005 to 2014 at Kumamoto University Hospital and Saitama Medical Center of Saitama Medical University. Touch imprint cytology samples were obtained from the brains of 11 patients diagnosed with PCNSL from 2001 to 2012 at Kumamoto University Hospital. Written informed consent was obtained from all the patients. The study design and protocols were approved by the Kumamoto University Review Board (#1174) and Saitama Medical University Review Board (#1965). Intracytoplasmic vacuoles were evaluated in 10 randomly selected areas of high‐power field of a microscope by two pathologists who were blinded to information about the patients’ backgrounds or their prognosis.
2.2. Cells and cell culture conditions
Human ATLL cell lines (ATN‐1, ED, ATL‐T, ATL‐2s, ATL‐35T, and MT‐1), EBV‐infected B cell lines (103‐LCL and 141‐LCL), B cell lymphoma cell lines (DAUDI, BALL1, TL‐1, RAJI, and SLVL) and PCNSL cell lines (HKBML and TK) were maintained in RPMI supplemented with 10% FBS. ED, ATL‐T, ATL‐2s, ATL‐35T, and MT‐1 cells were previously established by Matsuoka and Wakutani, 17 , 18 and other cell lines were purchased from the RIKEN Cell Bank.
Peripheral blood mononuclear cells were acquired from healthy volunteer donors. Written informed consent for sample collection and subsequent analysis was obtained. All protocols using human macrophages were approved by the Kumamoto University Hospital Review Board (No. 486) and carried out in accordance with the approved guidelines.
2.3. Cell staining
May–Grünwald–Giemsa and Sudan black B were purchased from Wako Chemicals, while Filipin III was obtained from Sigma‐Aldrich. Staining was carried out according to the manufacturer’s protocol.
2.4. Electron microscopy
The following cells were cultured with or without 40 μg/ml LDL for 24 h, cells were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer for 1 h and postfixed in 1% osmium tetroxide. After the dehydration in a graded series of ethanol solutions with propylene oxide and embedding in Epon 812, ultrathin sections were cut with an ultratome, stained with uranyl acetate and lead citrate, and observed with a Hitachi H‐7700 electron microscope (Hitachi).
2.5. Chemicals
The ACAT inhibitor CI‐976 was purchased from Santa Cruz Biotechnology. The SR‐BI inhibitor BLT‐1 was purchased from Sigma‐Aldrich. Lipoprotein‐deficient serum was purchased from Johnson Matthey. Human LDL and HDL were purchased from Biomedical Technology.
2.6. Cytotoxicity assay
Cell viability was determined with a water‐soluble tetrazolium salt (WST‐8) colorimetric assay using a CCK‐8 (Dojindo). Cells were cultured in a 96‐well plate (approximately 10,000–20,000 cells/well) for 1–2 days and then subjected to the indicated treatments. After treatment, 10 µl WST‐8 solution was added to each well and incubated for 30–60 min. Absorbance at 450 nm was measured using a microplate reader (SpectraMax i3x system; Molecular Devices). The culture medium containing WST‐8 without cells was used to set the background threshold, while culture medium containing WST‐8 with cells was used as a control. Cell viability was determined with the following formula: Cell viability (%) = (sample − background) / (control − background) × 100.
2.7. Apoptosis assay
Activation of caspase‐3/7 was measured by the Amplite Fluorimetric Caspase 3/7 Assay Kit (AAT Bioquest), according to the manufacturer’s protocol.
2.8. Intracellular lipid analysis
Intracellular total cholesterol, free cholesterol, and triglyceride analyses were carried out using the cholesterol E test, the free cholesterol E test, and the triglyceride E test, respectively (Wako Chemicals), according to the manufacturer’s protocol.
2.9. Real‐time quantitative PCR
Total RNA was isolated using RNAiso Plus reagent, and reverse‐transcribed using the PrimeScript RT reagent kit (Takara Bio). Real‐time quantitative PCR was carried out using TaqMan polymerase with SYBR green fluorescence (Takara Bio) and an ABI PRISM 7300 sequence detector (Applied Biosystems). The primer sequences are described in Table S1.
2.10. Western blot analysis
Cells were lysed in ice‐cold lysis buffer supplemented with protease inhibitor cocktail (Sigma‐Aldrich). Polyvinylidene fluoride membranes were incubated with primary Abs such as anti‐ACAT1 (GTX102637; Cene Tex), anti‐SREBP2 (ab30682; Abcam), anti‐lamin a/c (4777; Cell Signaling Technology), anti‐GRP78 Bip (3177; Cell Signaling Technology), anti‐ATF6 (65880; Cell Signaling Technology), anti‐eIF2α (9722; Cell Signaling Technology), anti‐phospho‐eIF2α (9721; Cell Signaling Technology), anti‐IRE1α (Ab48187; Abcam), anti‐JNK (9258; Cell Signaling Technology), anti‐phospho‐JNK (9251; Cell Signaling Technology), anti‐p38 MAPK (8690; Cell Signaling Technology), anti‐phospho‐p38 MAPK (4511; Cell Signaling Technology), anti‐SR‐BI (NB400‐104; Novus Biologicals), anti‐ABCA1 (NB400‐105; Novus Biologicals), anti‐LDLR (ab52818; Abcam), anti‐phospho‐IRE1 (ab48187; Abcam), and anti‐β‐actin (sc‐47778; Santa Cruz Biotechnology). Horseradish peroxidase‐conjugated goat anti‐mouse IgG (7076; Cell Signaling Technology) or HRP‐conjugated goat anti‐rabbit IgG (7074; Cell Signaling Technology) was used as the secondary Ab. Immunoreactive bands were visualized using a Pierce western blotting substrate plus kit (Thermo Fisher Scientific) and an ImageQuant LAS‐4000 mini (Fujifilm).
2.11. Lymphoma tumor‐bearing mouse model
SCID mice (6–8 weeks old) were purchased from Charles River Japan. ED cells (2 × 106 cells/mouse) were suspended in 100 µl RPMI and injected s.c. into SCID mice. CI‐976 and BLT‐1 were intraperitoneally administered on days 14–32 or on days 10–36 after the injection of ED cells. Nude mice (6–8 weeks old) were purchased from Charles River Japan. HKBML cells (5 × 105 cells/mouse) were suspended in 5 µL RPMI and injected into the brain. BLT‐1 was intraperitoneally administered on days 7–29 after the injection of HKBML cells. All animal experiments were approved by the Ethics Committee for Animal Experiments of Kumamoto University (Permission Number: B24‐125) and carried out in accordance with the Guidelines for Animal Experiments of Kumamoto University.
2.12. Statistics
All data are representative of two or three independent experiments. Data are expressed as mean ± SD. Differences between the groups were examined for statistical significance using the Mann–Whitney U‐test and nonrepeated measures ANOVA with Bonferroni’s test. Overall murine survival was assessed using Kaplan–Meier analysis and compared using the log–rank test. Statistical significance was set at p < 0.05.
3. RESULTS
3.1. Intracytoplasmic vacuoles observed in lymphoma cells have high malignant potential
We first examined the cytoplasmic vacuoles in the touch imprint cytology samples; we divided the cases into two groups according to the percentage of vacuole‐positive lymphoma cells (negative, none or <5%; positive, ≥5%). Follicular lymphoma is low‐grade/indolent, whereas ATLL and DLBCL are aggressive subtypes of lymphoma. Lymphoma cells with intracytoplasmic vacuoles were detected in 57.4%, 69.5%, and 13.3% of DLBCL, ATLL, and follicular lymphoma cases, respectively (Figures 1A and S1A). Cytoplasmic vacuoles were also observed in 6 of 11 PCNS lymphoma patients (Figure 1A). Those cytoplasmic vacuoles were positively stained with Sudan black, indicating that lipid was contained in the vacuoles (Figure 1B). Patients with vacuole‐positive DLBCL had significantly shorter survival times than those without vacuoles (Figure 1C). However, the presence of vacuoles was not associated with the GCB/non‐GCB subtype, serum sIL‐2R, or serum LDH (Figure S1). These observations suggested that intracytoplasmic vacuoles were often present in high‐grade lymphomas.
FIGURE 1.
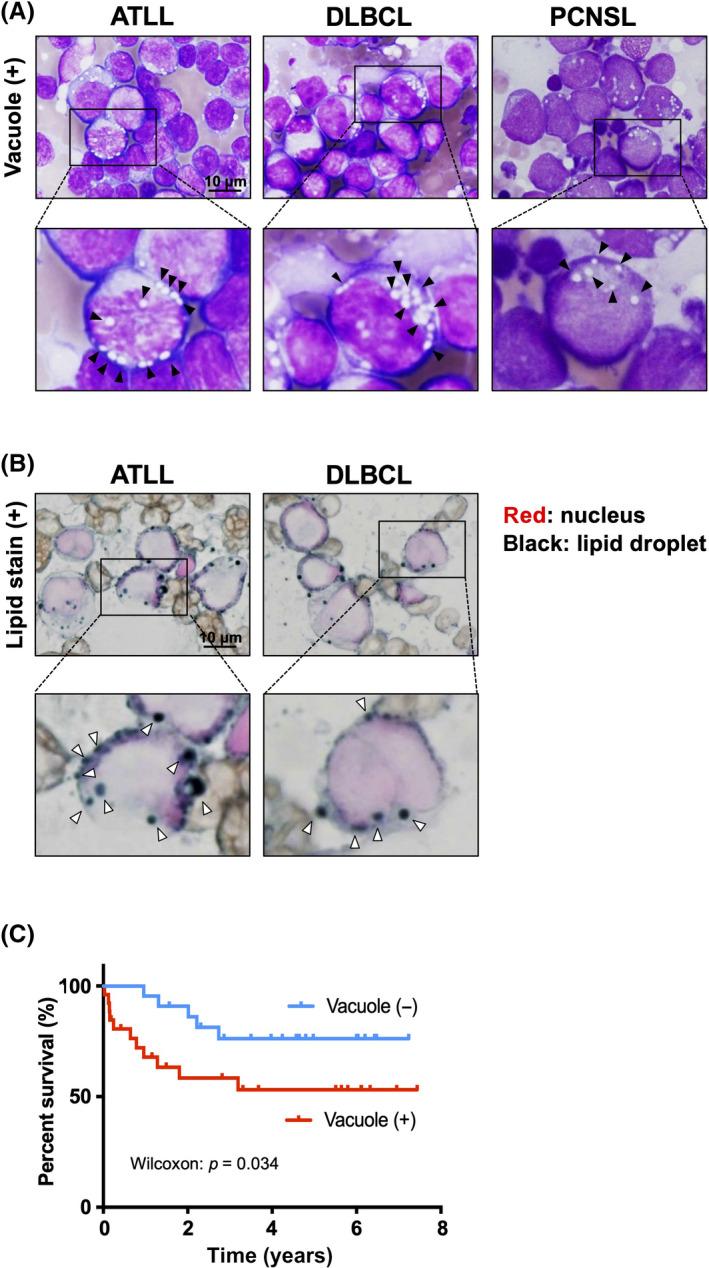
Lipid accumulation in lymphoma cells. (A) Cytoplasmic vacuoles in adult T‐cell leukemia/lymphoma (ATLL), diffuse large B‐cell lymphoma (DLBCL), and primary central nervous system lymphoma (PCNSL) cells. (B) Detection of lipid accumulation in lymphocytes by Sudan black B staining. (C) Kaplan–Meier estimates comparing the overall survival rate between vacuole‐positive (+) DLBCL and vacuole‐negative (−) DLBCL
3.2. Excessive LDL uptake induces vacuole formation in lymphoma cells
Next we examined whether intracytoplasmic vacuoles were detected in lymphoma cell lines. The cytoplasmic vacuoles were observed in all ATLL cell lines and PCNSL cell lines, and in two of five B‐cell lymphoma cell lines (Figure 2A,B and Table 1). Cytoplasmic vacuoles were also positively stained with Sudan black and Filipin III (Figures 2C and S2A). In this study, T lymphocytes stimulated by the anti‐CD3 Ab and anti‐CD28 Ab (activated T cells), B lymphocytes (B cells), and B lymphocytes transformed by EBV (141‐ and 103‐LCL cells) were used as nonlymphomatous control cells. No positive‐stained vacuoles were seen in normal T lymphocytes, normal B lymphocytes, or one of the two EBV‐transformed B cell lines (Figure 2C).
FIGURE 2.
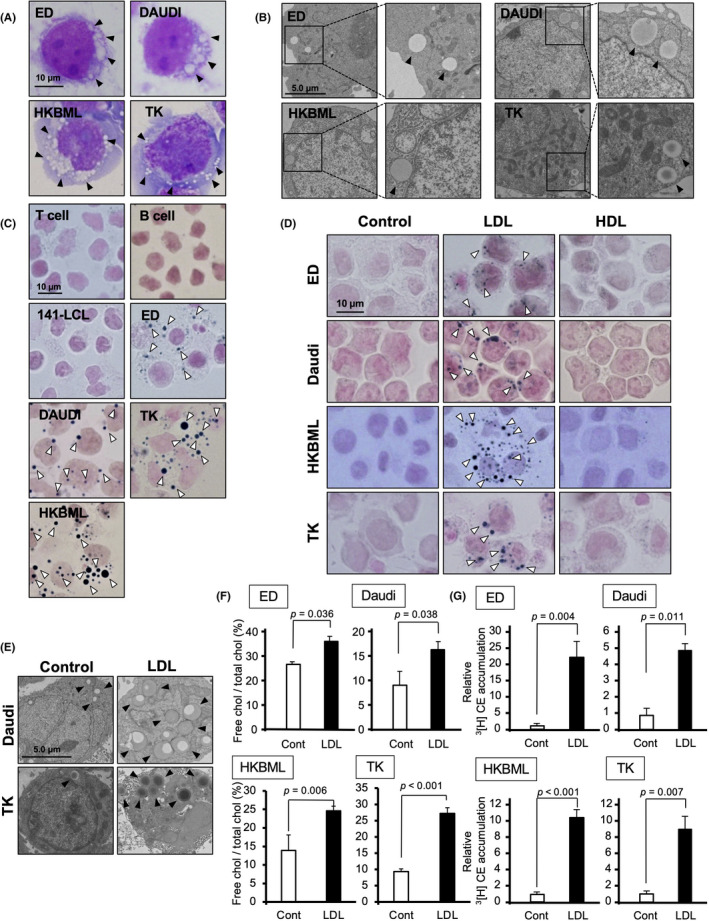
Low‐density lipoprotein (LDL) induces cholesterol accumulation in lymphoma cells. (A) Giemsa staining of human adult T‐cell leukemia/lymphoma cell line (ED), human B cell lymphoma cell line (DAUDI), and human primary central nervous system lymphoma cell lines (HKBML and TK). (B) Electron microscopy images of lymphoma cell lines. (C) Detection of lipid accumulation in lymphoma cell lines by Sudan black B staining. (D) Microscopy of Sudan black B stained cells to determine the lipid accumulation of lymphoma cells incubated with LDL (40 μg/ml) or high‐density lipoprotein (HDL; 40 μg/ml) for 24 h under lipoprotein‐deficient serum (LPDS, 2.5%) conditions. (E) Electron microscopy of DAUDI and TK cells treated with LDL (40 μg/ml) for 24 h. (F) Total cholesterol (chol) and free cholesterol of lymphoma cells following incubation with LDL (40 μg/ml) for 24 h. (G) 3[H] cholesteryl ester (CE) in lymphoma cells following incubation with LDL (40 μg/ml) in the presence of 3[H]oleate for 24 h
TABLE 1.
Cholesterol accumulation in lymphoma cells
| Lipid stain | 3[H]‐cholesteryl ester | ||
|---|---|---|---|
| T lymphocyte | Activated T cells | − | − |
| ATLL | ATN‐1 | + | + |
| ED | + | + | |
| ATL‐T | + | − | |
| ATL‐2s | + | + | |
| ATL‐35T | + | + | |
| EBV‐infected B cell | 103‐LCL | − | − |
| 141‐LCL | − | − | |
| B cell lymphoma | DAUDI | + | + |
| BALL1 | + | ND | |
| TL‐1 | − | − | |
| RAJI | − | + | |
| SLVL | − | − | |
| PCNS lymphoma | TK | + | + |
| HKBML | + | + |
Abbreviations: ATLL, adult T‐cell leukemia/lymphoma; EBV, Epstein–Barr virus; ND, Not detected; PCNS lymphoma, primary central nervous system lymphoma.
We then investigated whether similar vacuoles could be induced by treatment with lipoproteins such as LDL and HDL under LPDS conditions. We found that treatment with LDL induced lipid accumulation and the appearance of vacuoles in lymphoma cell lines, but HDL did not induce lipid accumulation (Figure 2D,E). As summarized in Table 1, lipid accumulation was observed in all of the ATLL cell lines, one of the two EBV‐transformed B cell lines, two of the five B‐cell lymphoma cell lines and two of the two PCNS lymphoma cell lines. Furthermore, the uptake of LDL induced the accumulation of both free cholesterol and CE in lymphoma cells (Figure 2F,G and Table 1), whereas the content of triglyceride in lymphoma cells was not changed by the uptake of LDL (Figure S2B). This indicated that intracytoplasmic vacuoles were associated with cholesterol accumulation in lymphoma cells.
3.3. Upregulation of cholesterol metabolism‐related molecules in lymphoma cells
Significant upregulation of LDLR mRNA and protein was detected in all ATLL cell lines, one B cell lymphoma cell line, and two PCNS lymphoma cell lines compared with the following nonlymphomatous control cells: T lymphocytes stimulated by the anti‐CD3 Ab and anti‐CD28 Ab (activated T cells), B lymphocytes (B cells), and B lymphocytes transformed by EBV (103‐LCL cells) (Figures 3A,B and S3). This suggested that LDLR played an important role in cell homeostasis in these lymphoma cell lines.
FIGURE 3.
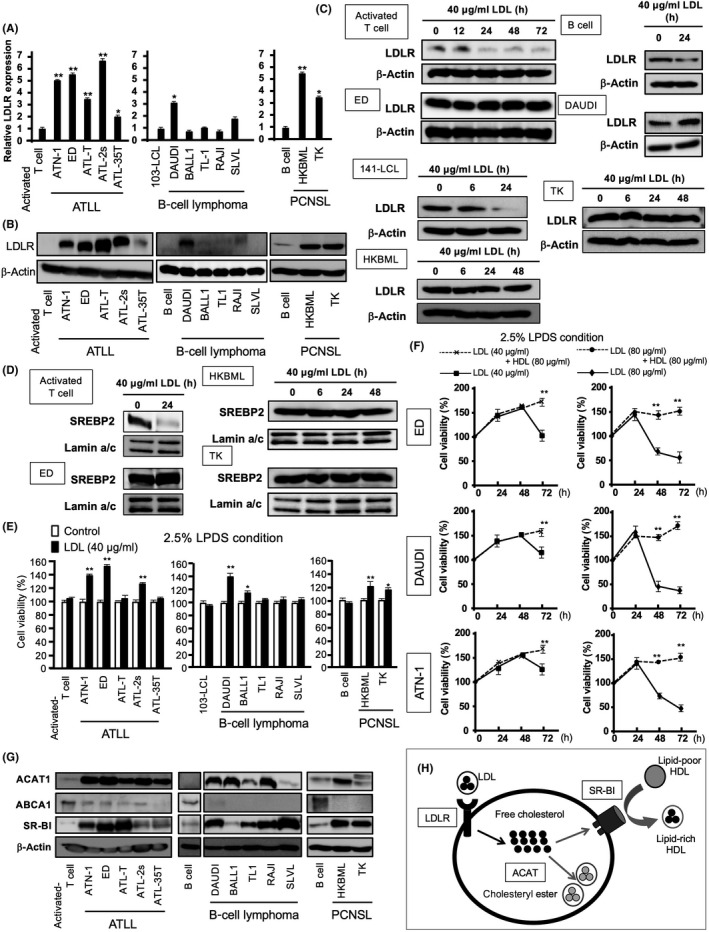
Upregulation of cholesterol metabolism‐related factors in lymphoma cells. (A,B) mRNA and protein levels of low‐density lipoprotein receptor (LDLR) detected by real‐time quantitative PCR (A), and western blotting (B), respectively. (C) Activated T cells, Epstein–Barr virus‐infected 141‐LCL cells, and B cells were used as controls. LDLR expression of activated T cells, B cells, and lymphoma cells following incubation with LDL (40 μg/ml) for 12, 24, 48 or 72 h measured by western blotting. (D) Sterol response element‐binding protein 2 (SREBP2) expression of activated T cells and lymphoma cells following incubation with LDL (40 μg/ml) for 24 h measured by western blotting. (E) Cell proliferation of lymphocytes and lymphoma cells following incubation with LDL (40 μg/ml) for 24 h using LPDS. (F) Cell proliferation of lymphoma cells following incubation with LDL (80 μg/ml) (circles) or LDL (80 μg/ml) + high‐density lipoprotein (HDL) (80 μg/ml) (diamonds) for 48 h using LPDS. (G) Cholesterol metabolism‐related proteins detected by western blotting. (H) Formation of intracellular vacuoles or cholesteryl ester droplets reflected the excessive uptake of cholesterol and increased acetyl‐coenzyme A:cholesterol acetyltransferase (ACAT) activity. Accumulated cholesterol was exported through SR‐B1 and converted to HDL. ACAT and scavenger receptor class B member I (SR‐BI) inhibitors induced the accumulation of free cholesterol, which subsequently induced cell apoptosis due to the cytotoxic effects of cholesterol. Data are presented as mean ± SD. * p‐value <0.05, ** p‐value <0.01 compared with activated T cells, 103‐LCL cells, or LDL (40 or 80 µg/ml) treatment. ABCA1, ATP‐binding cassette protein A1; ATLL, adult T‐cell leukemia/lymphoma; PCNSL, primary central nervous system lymphoma
The LDL uptake induces the downregulation of LDLR expression in several cell types, including both normal and tumor cells. Low‐density lipoprotein receptor is regulated by SREBP2, which is sensitive to cholesterol content in the ER at the transcriptional level. 19 Interestingly, the expression of LDLR and SREBP2 was unchanged in lymphoma cells following LDL stimulation, whereas nonlymphomatous T cells, the EBV‐infected B cell line (141‐LCL), and B cells showed the downregulation of the expression of both genes (Figure 3C,D). This indicated that the abrogation of LDLR downregulation was caused by SREBP2 overexpression in lymphoma cells and suggested that LDL metabolism was needed for homeostasis processes, such as cell survival and proliferation in lymphomas.
Low‐density lipoprotein (40–80 µg/ml) enhanced cell proliferation in LDLR‐overexpressing lymphoma cell lines using LPDS after 24 h (Figure 3E,F). In contrast, long‐term (48 or 72 h) incubation using LDL (40–80 µg/ml) with LPDS decreased lymphoma cell viability, and the cytotoxicity of excessive LDL was abrogated by HDL (80 µg/ml) (Figure 3F). This suggested that HDL inhibited free cholesterol accumulation by inducing cholesterol efflux through SR‐BI or ABCA1. There was increased SR‐BI and ACAT1 expression, and decreased ABCA1 expression in lymphoma cell lines (Figure 3G). Acetyl‐coenzyme A:cholesterol acetyltransferase 1, SR‐BI, and SREBP2 increased in some lymphoma cell lines, whereas HMGCR did not substantially change (Figure S4). This suggested that LDLR, ACAT1, and SR‐BI played important roles in cholesterol metabolism in lymphoma cells, and excessive free cholesterol was cleared by esterification and the efflux system (Figure 3H).
3.4. Acetyl‐coenzyme A:cholesterol acetyltransferase and SR‐BI inhibitors suppressed lymphoma cell proliferation in vitro
The ACAT inhibitor (CI‐976) and SR‐BI inhibitor (BLT‐1) inhibited lymphoma cell proliferation in a dose‐dependent manner (Figure 4A). In contrast, these inhibitors had no effect on the viability of activated T cells, 141‐LCL cells, or B cells (Figure 4B). The knockdown of SR‐BI and ACAT in lymphoma cells also induced cell death and reduced lymphoma proliferation (Figure S5). Caspase‐3/7 activation in ATN‐1 and ED cells was induced by treatment with either CI‐976 or BLT‐1 (Figure 4C). The caspase inhibitor z‐VAD‐fmk protected against cell death induced by treatment with BLT‐1 or CI‐976 (Figure S6). Furthermore, an additive effect was observed after combined treatment with both CI‐976 and BLT‐1 in ATN‐1 and ED cells (Figure 4C,D). Similar results were observed in B‐cell lymphomas, represented by DAUDI, BALL‐1, and TK cells (Figure 4E,F). In addition, CI‐976 and BLT‐1 synergistically increased the cytotoxic activity of CBDCA (Figure S7). These findings suggest that cholesterol metabolism regulation using ACAT and SR‐BI inhibitors could be a promising therapy for reducing lymphoma proliferation.
FIGURE 4.
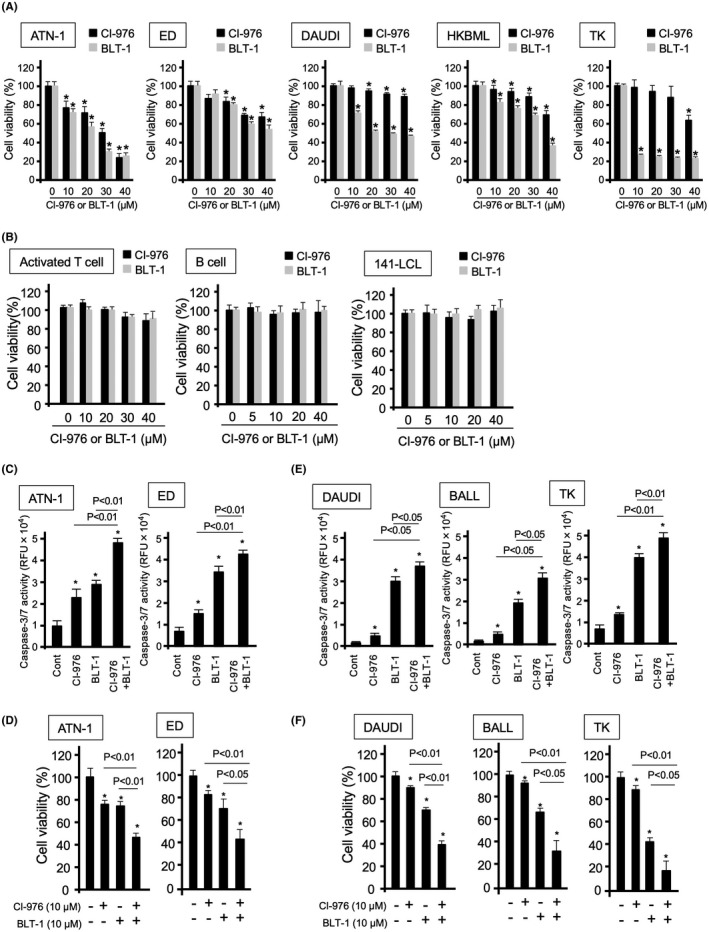
Effects of acetyl‐coenzyme A:cholesterol acetyltransferase and scavenger receptor class B member I inhibitors on lymphoma cell proliferation. (A) Proliferation of lymphoma cells following incubation with the indicated concentrations of CI‐976 or BLT‐1 for 24 h. (B) Proliferation of activated T cells, B cells, and Epstein–Barr virus‐infected 141‐LCL cells following incubation with the indicated concentrations of CI‐976 or BLT‐1 for 24 h. (C) Caspase‐3/7 activation of lymphoma cells following incubation with the indicated concentrations of CI‐976 (10 µM) and/or BLT‐1 (10 µM) for 24 h. (D) Proliferation of lymphoma cells following incubation with the indicated concentrations of CI‐976 (10 µM) and/or BLT‐1 (10 µM) for 24 h. (E) Caspase‐3/7 activation of lymphoma cells following incubation with the indicated concentrations of CI‐976 (10 µM) and/or BLT‐1 (10 µM) for 24 h. (F) Proliferation of lymphoma cells following incubation with the indicated concentrations of CI‐976 (10 µM) and/or BLT‐1 (10 µM) for 24 h. Data are presented as mean ± SD. *p‐value <0.05 compared with the control (Cont). RFU, relative fluorescence unit
3.5. Excessive LDL uptake enhanced the antilymphoma effect of ACAT and SR‐BI inhibitors
Low‐density lipoprotein uptake enhanced CI‐976‐ and BLT‐1‐induced cell death in lymphoma cells, whereas there was no effect on activated T cell proliferation (Figure 5A). Treatment with both CI‐976 and BLT‐1 resulted in a greater accumulation of free cholesterol than that observed in the single‐inhibitor treatments (Figure 5B), and significantly inhibited cell proliferation in the presence of LDL (Figure 5C). This showed that the enhanced accumulation of LDL‐derived free cholesterol by ACAT inhibitor blocking free cholesterol esterification, and SR‐BI inhibitor blocking cholesterol efflux (Figure S8), selectively induced cell death in lymphoma cells. In contrast, HDL significantly attenuated the suppressive effects of CI‐976 on lymphoma cell proliferation (Figure 5D), while having no effect on activated T cell and lymphoma cell proliferation (Figure 5E). The suppressive effects of BLT‐1 on lymphoma proliferation were not influenced by HDL addition (Figure 5F). These data indicated that free cholesterol efflux through the SR‐BI‐HDL pathway is involved in lymphoma cell viability.
FIGURE 5.
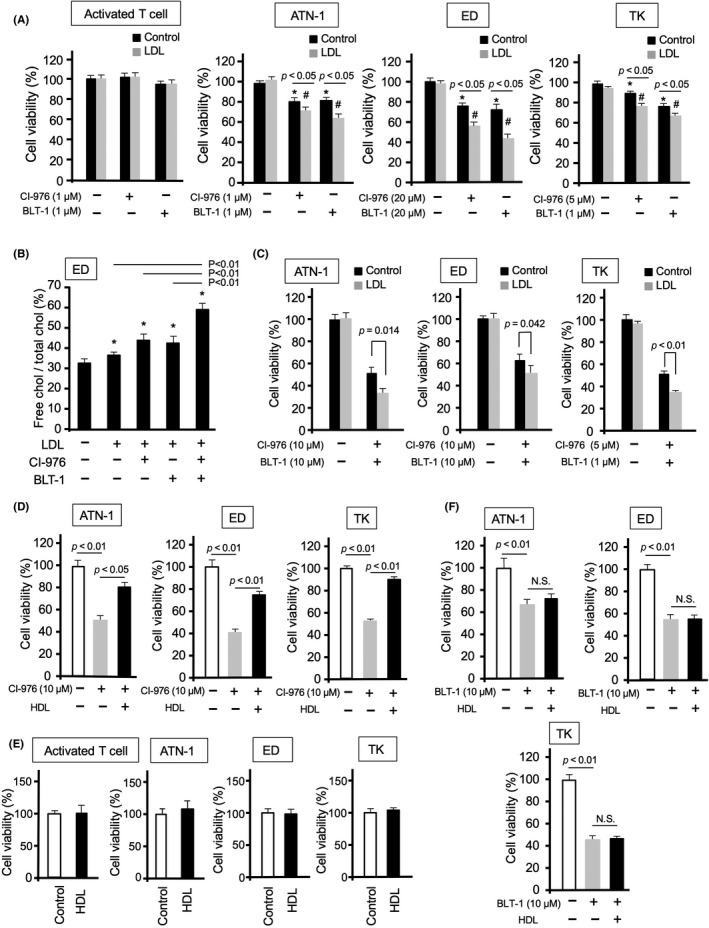
Effects of acetyl‐coenzyme A:cholesterol acetyltransferase and scavenger receptor class B member I inhibitors on lymphoma cell proliferation during incubation with lipoproteins. (A) Proliferation of activated T cells and lymphoma cells following incubation with or without low‐density lipoprotein (LDL) (40 µg/ml) for 24 h. (B) Total cholesterol (chol) and free cholesterol of ED cells following incubation with LDL (40 μg/ml) in the presence of CI‐976 and/or BLT‐1 for 24 h. (C) Proliferation of lymphoma cells following incubation with both CI‐976 (10 µM) and BLT‐1 (10 µM) in the presence of LDL for 24 h. (D) Proliferation of lymphoma cells following incubation with CI‐976 (10 µM) in the presence of high‐density lipoprotein (HDL) (40 µg/ml) for 48 h. (E) Proliferation of activated T cells and lymphoma cells following incubation with HDL (40 µg/ml) for 48 h. (F) Proliferation of lymphoma cells following incubation with BLT‐1 (10 µM) in the presence of HDL (40 µg/ml) for 48 h. Data are presented as mean ± SD. #p‐value <0.05, *p‐value <0.05 compared with untreated controls
3.6. Scavenger receptor class B member I inhibitor induces ER stress through cholesterol accumulation in lymphoma cells
Excess cholesterol accumulation induces ER stress in macrophages. 20 The expression of CHOP, a major ER stress response protein, and other ER stress response‐related proteins, including Bip, ATF6, p‐eIF2α, and p‐IRE1α, was induced in lymphoma cells following BLT‐1 treatment (Figure 6A,B). In addition, the MAPK kinases p38 and JNK were activated in lymphoma cells following BLT‐1 treatment (Figure 6C), which correlated with previous results showing that IRE1α induces apoptosis through the activation of p38 and JNK pathways. 21 Furthermore, BLT‐1‐induced cell death in lymphoma cells was suppressed by incubation with p38 and JNK inhibitors (Figure [Link], [Link]). These data indicated that BLT‐1‐induced cholesterol accumulation is involved in apoptosis by promoting ER stress responses.
FIGURE 6.
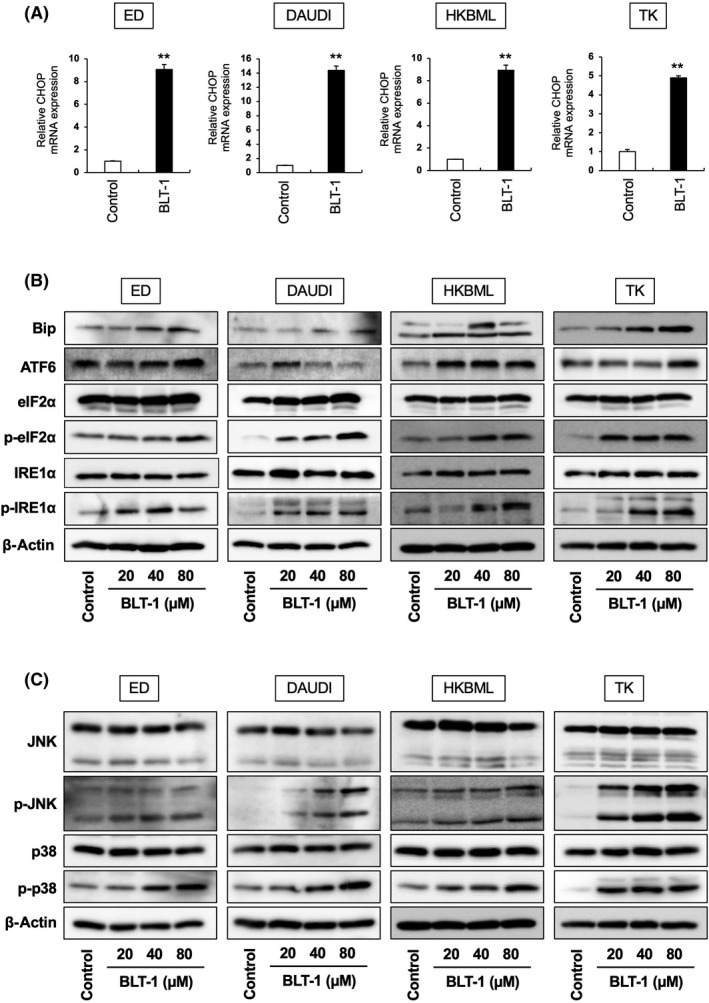
Effects of scavenger receptor class B member I inhibitors on endoplasmic reticulum stress‐related protein expression in lymphoma cells. (A) C/EBP homologous protein (CHOP) expression of lymphoma cells following incubation with BLT‐1 (40 μM) for 3 h, measured by real‐time quantitative PCR. (B) Western blot analysis of Bip, ATF6, eIF2α, p‐eIF2α, IRE1α, p‐eIF2α, and β‐actin expression of lymphoma cells following incubation with BLT‐1 for 3 h. (C) Western blot analysis of JNK, p‐JNK, p38, p‐p38, and β‐actin expression of lymphoma cells following incubation with BLT‐1 for 3 h
3.7. Scavenger receptor class B member I inhibitor abrogated lymphoma progression and prolonged survival in vivo
The effects of i.p. administration of CI‐976 and BLT‐1 after ED cells were injected s.c. into SCID mice showed that BLT‐1 significantly suppressed tumor development, whereas CI‐976 displayed no antilymphoma effects (Figure 7A). Cleaved caspase 3‐positive cells were increased in s.c. tumor tissues treated with BLT‐1 (Figure 7B). BLT‐1 significantly prolonged the survival in the lymphoma‐bearing mouse model (Figure 7C). In addition, BLT‐1 significantly prolonged the survival in the PCNS lymphoma‐bearing mouse model (Figure 7D). Cleaved caspase 3‐positive cells were also increased in intracerebral tumor tissues treated with BLT‐1 (Figure 7E). These results suggest that the SR‐BI inhibitor could be a promising candidate agent for antilymphoma therapy.
FIGURE 7.
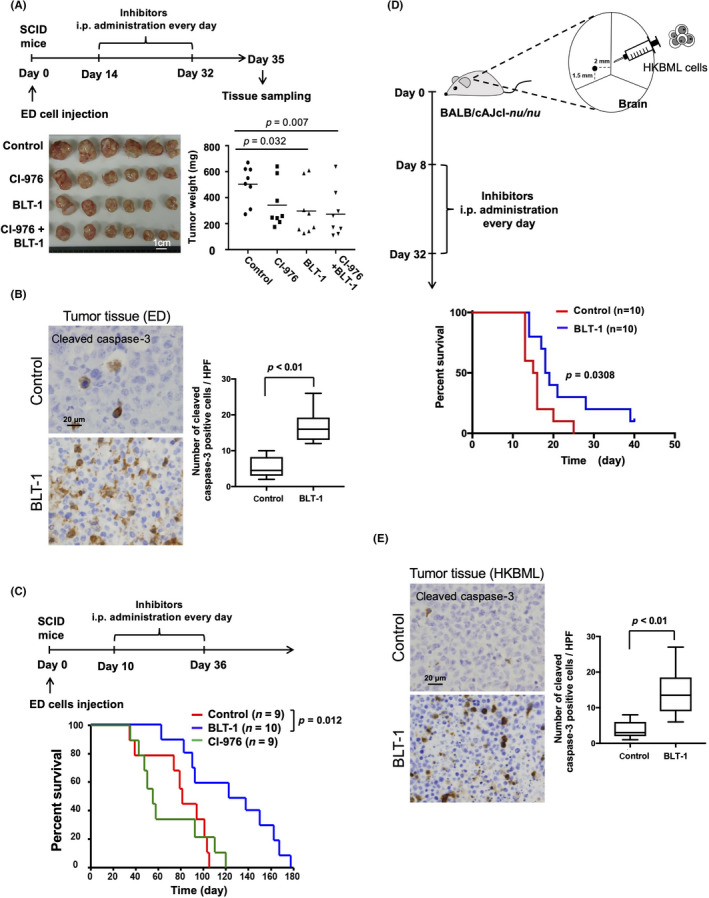
Effects of the acetyl‐coenzyme A:cholesterol acetyltransferase and scavenger receptor class B member I inhibitors on tumor progression in tumor‐bearing mice. (A) SCID mice were injected s.c. with ED cells and treated with CI‐976 (20 mg/kg) and BLT‐1 (20 mg/kg), followed by the measurement of tumor weights (scale bar = 1 cm). (B) Cleaved caspase 3‐positive cells in s.c. tumor tissues were evaluated by immunostaining. (C) SCID mice were injected s.c. with ED cells and treated with CI‐976 (20 mg/kg) and BLT‐1 (20 mg/kg), followed by the measurement of survival rates. (D) Nude mice were injected with HKBML cells into the brain and treated with BLT‐1 (20 mg/kg), followed by the measurement of the survival rate. (E) Cleaved caspase 3‐positive cells in intracerebral tumor tissues were evaluated by immunostaining
4. DISCUSSION
Formation of cholesterol‐associated intracytoplasmic vacuoles was observed in ATLL and PCNSL cell lines compared with B‐cell lymphoma cell lines. These intracytoplasmic vacuoles could be associated with the clinical course of patients with the more aggressive ATLL and PCNSL compared with patients with the comparatively milder DLBCL, or follicular lymphoma. Furthermore, significantly upregulated cholesterol metabolism‐related factors ACAT1 and SR‐BI were found in lymphoma cells. The inhibition of SR‐BI and ACAT caused the accumulation of free cholesterol and suppressed lymphoma growth by inhibiting free cholesterol efflux and CE formation (Figure S8). The effect of SR‐BI and ACAT inhibitors in high‐grade lymphomas was stronger than that in low‐grade lymphoma, indicating that targeting cholesterol metabolism is an effective therapy for high‐grade lymphomas (Table S2 and Figure S10). The ACAT inhibitors have anticancer effects in some cancer cell lines 22 , 23 , 24 ; however, no significant therapeutic effects were observed in either the present study or in another study using a murine model. Recent in vitro and in vivo studies showed that ACAT inhibition abrogates pancreatic cancer growth and progression by increasing intracellular free cholesterol accumulation, which increases ER stress and apoptosis. 25 Cholesterol ester droplet formation and ACAT1 overexpression are observed in clear cell renal cell carcinoma, although the effects of ACAT inhibitors has never been investigated. 26 Overexpression of ACAT1 and SR‐BI is observed in nasopharyngeal, breast, and pancreatic cancers 27 , 28 , 29 ; however, few studies have described the upregulation or functional significance of cholesterol metabolism in lymphoma. Our data provided the first evidence that targeting SR‐BI and ACAT could be an effective strategy for treating lymphomas by regulating cholesterol metabolism.
Cholesterol homeostasis is tightly regulated by a complex protein network that monitors cholesterol import, synthesis, export, metabolism, and esterification. 7 Both SREBF2 and LXR are key regulators of cholesterol homeostasis. 30 The ER cholesterol level is a sensor for intracellular cholesterol homeostasis. A decrease in ER cholesterol triggers the translocation of SREBF2 from the ER to the Golgi, to the nucleus, where it activates the transcription of genes involved in cholesterol synthesis, such as HMGCR, and cholesterol import into cells (LDLR). Overexpression of LDLR is observed in several cancer cell types, although its significance is only elucidated in pancreatic cancer. Silencing of LDLR abrogates cell proliferation, tumorigenic capacity, and ERK1/2 activation, and enhances the cytotoxic effects of chemotherapy drugs. 30 Consequently, pancreatic cancer patients with high LDLR expression have poor clinical prognoses. 30 These results indicate that upregulated cholesterol uptake and metabolism are closely associated with cancer cell chemoresistance.
Lymphomas are difficult to cure, with the prognosis of PCNSL in particular being especially poor. Methotrexate‐based chemotherapy is selected for PCNSL because the standard therapy for general lymphoma is ineffective. 31 , 32 Recently, treatment with tirabrutinib, a Bruton tyrosine kinase inhibitor, and chemotherapy with autologous stem cell transplantation have shown efficacy in PCNSL; however, the prognosis remains poor. 33 , 34 , 35 Metabolically active tumors show elevated glycolysis and lipogenesis, resulting in increased lipid levels in tumor cells, which induces tumor proliferation. 36 In normal cells, cholesterol is strictly maintained at stable levels. 19 However, when ER cholesterol levels increase, a negative feedback loop is triggered that inhibits de novo cholesterol synthesis. 37 , 38 Excess free cholesterol is esterified (detoxified) to CE by intracellular ACAT, which is located in the ER of normal cells such as macrophages. In contrast, the CD36 scavenger receptor protein SR‐BI is abundantly expressed in the liver where it functions in the reverse cholesterol transport pathway, and in steroidogenic tissues, where it mediates cholesterol delivery. 39 Furthermore, SR‐BI is an HDL receptor functioning as a bidirectional cholesterol transporter. In normal cells such as macrophages, this process involves the selective transfer of CE/free cholesterol in an HDL particle into the cell in the absence of endocytic uptake or HDL degradation. 40 , 41 In prostate cancer, LDLR and SR‐BI overexpression is suggested to be related to castration‐resistant prostate cancer progression. 42 In contrast, SR‐BI silencing abrogated cell proliferation. 43 Overexpression of SR‐BI in breast cancer cells is closely associated with poorer clinical outcomes. 28 Expression of SR‐BI is significantly higher in Burkitt lymphoma and DLBCL cells than in normal B lymphocytes and targeting of SR‐BI by HDL nanoparticles has an antilymphoma effect in combination with cholesterol starvation. 44 These researchers described the potential antilymphoma effect of an SR‐BI inhibitor, which is consistent with the results in this study. The SR‐BI inhibitor increased the cytotoxic effects of CBDCA on lymphoma cells, indicating that cholesterol efflux through SR‐BI was associated with chemoresistance (Figure S7). These results indicated that targeting cholesterol metabolism could be a promising approach for treating aggressive chemoresistant lymphomas.
Endoplasmic reticulum stress is a condition in which unfolded proteins possess an incorrect high‐order structure and accumulate in the ER. As ER stress interferes with cell function, normal cells have an avoidance system called the UPR. 45 , 46 The CHOP transcription factor is involved in ER stress‐dependent apoptosis if UPR does not work properly or is overstressed. 47 , 48 , 49 The accumulation of UPR in mammals is mainly mediated by stress sensor proteins including PERK, IRE1, and ATF6. 48 , 50 , 51 , 52 The dissociation of a molecular chaperone (Bip) from stress sensor proteins is involved in the activation of UPR signaling by binding to UPR. 53 , 54 Endoplasmic reticulum stress is suggested to be a therapeutic target for cancer. Toyocamycin is an IRE1 RNase domain inhibitor that induces apoptosis in myeloma cells. 55 In the present study, SR‐BI inhibitor induced apoptosis through the ER stress signaling pathway in lymphoma cells (Figure 6).
In conclusion, SR‐BI and ACAT were overexpressed in lymphoma cells with LDLR expression maintained after LDL stimulation. There was no negative feedback system and cholesterol efflux was activated through the HDL‐SR‐BI pathway. This suggested that the activation of LDL‐derived cholesterol uptake, and cholesterol efflux through the HDL‐SR‐BI pathway played critical roles in lymphoma cell survival and proliferation. The changes in cholesterol metabolism due to treatment with SR‐BI and ACAT inhibitors suppressed lymphoma cell proliferation through cholesterol accumulation (Figure S8) and the efficacy of the SR‐BI inhibitor was verified in a tumor‐bearing mouse model. These findings indicated that SR‐BI and ACAT inhibitors have therapeutic potential in lymphoma treatment by targeting the cholesterol metabolism pathway.
DISCLOSURE
The authors declare no competing interests. The authors A.M. and Y.K. are editorial board members.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Fig S10
Table S1
Table S2
ACKNOWLEDGMENTS
We thank Ms. Ikuko Miyagawa, Ms. Michiko Tokunaga, and Mr. Takenobu Nakagawa for their technical assistance. This work was supported by Japan Society for the Promotion Science KAKENHI (Grant numbers: 16H05162, 16K09247, 16K15503, and 20H03459). We would like to thank Editage for English language editing.
Yano H, Fujiwara Y, Horlad H, et al. Blocking cholesterol efflux mechanism is a potential target for antilymphoma therapy. Cancer Sci. 2022;113:2129–2143. doi: 10.1111/cas.15349
Funding information
Japan Society for the Promotion of Science, KAKENHI Grant/Award Number: 16H05162, 16K09247, 16K15503, and 20H03459.
Contributor Information
Yukio Fujiwara, Email: fuji-y@kumamoto-u.ac.jp.
Yoshihiro Komohara, Email: ycomo@kumamoto-u.ac.jp.
DATA AVAILABILITY STATEMENT
The datasets analyzed during the study are available from the corresponding authors on reasonable request.
REFERENCES
- 1. Giulino‐Roth L, Goldman S. Recent molecular and therapeutic advances in B‐ cell non‐Hodgkin lymphoma in children. Br J Haematol. 2016;173(4):531‐544. [DOI] [PubMed] [Google Scholar]
- 2. Hoelzer D, Walewski J, Döhner H, et al. Improved outcome of adult Burkitt lymphoma/leukemia with rituximab and chemotherapy: report of a large prospective multicenter trial. Blood. 2014;124(26):3870‐3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jaeger U, Trneny M, Melzer H, et al. Rituximab maintenance for patients with aggressive B cell lymphoma in first remission: results of the randomized NHL13 trial. Haematologica. 2015;100(7):955‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tsukasaki K, Tobinai K. Human T cell lymphotropic virus type I‐associated adult T cell leukemia‐lymphoma: new directions in clinical research. Clin Cancer Res. 2014;20(20):5217‐5225. [DOI] [PubMed] [Google Scholar]
- 5. Abrey LE, DeAngelis LM, Yahalom J. Long‐term survival in primary CNS lymphoma. J Clin Oncol. 1998;16:859‐863. [DOI] [PubMed] [Google Scholar]
- 6. Ferreri AJ, Reni M, Foppoli M, et al. High‐dose cytarabine plus high‐dose methotrexate versus high‐dose methotrexate alone in patients with primary CNS lymphoma: a randomised phase 2 trial. Lancet. 2009;374(9700):1512‐1520. [DOI] [PubMed] [Google Scholar]
- 7. Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. 2008;9(2):125‐138. [DOI] [PubMed] [Google Scholar]
- 8. Tosi MR, Tugnoli V. Cholesteryl esters in malignancy. Clin Chim Acta. 2005;359(1–2):27‐45. [DOI] [PubMed] [Google Scholar]
- 9. Vitols S, Norgren S, Juliusson G, Tatidis L, Luthman H. Multilevel regulation of low‐density lipoprotein receptor and 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase gene expression in normal and leukemic cells. Blood. 1994;84(8):2689‐2698. [PubMed] [Google Scholar]
- 10. Brown MS, Ho YK, Goldstein JL. The cholesteryl ester cycle in macrophage foam cells: continual hydrolysis and re‐esterification of cytoplasmic cholesteryl esters. J Biol Chem. 1980;255:9344‐9352. [PubMed] [Google Scholar]
- 11. Warner GJ, Stoudt G, Bamberger M, Johnson WJ, Rothblat GH. Cell toxicity induced by inhibition of acyl coenzyme A cholesterol acyltransferase and accumulation of unesterified cholesterol. J Biol Chem. 1995;270(11):5772‐5778. [DOI] [PubMed] [Google Scholar]
- 12. Villa GR, Hulce JJ, Zanca C, et al. An LXR‐cholesterol axis creates a metabolic co‐dependency for brain cancers. Cancer Cell. 2016;30(5):683‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guo D, Reinitz F, Youssef M, et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP‐1/LDLR‐dependent pathway. Cancer Discov. 2011;1(5):442‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Said JW. Aggressive B cell lymphomas: how many categories do we need? Mod Pathol. 2013;26(Suppl 1):S42‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsukasaki K, Imaizumi Y, Tawara M, et al. Diversity of leukaemic cell morphology in ATL correlates with prognostic factors, aberrant immunophenotype and defective HTLV‐1 genotype. Br J Haematol. 1999;105(2):369‐375. [DOI] [PubMed] [Google Scholar]
- 16. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118(4):653‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Satou Y, Matsuoka M. Implication of the HTLV‐I bZIP factor gene in the leukemogenesis of adult T‐cell leukemia. Int J Hematol. 2007;86(2):107‐112. [DOI] [PubMed] [Google Scholar]
- 18. Katoh T, Harada T, Morikawa S, Wakutani T. IL‐2‐ and IL‐2‐R‐ independent proliferation of T‐cell lines from adult T‐cell leukemia/lymphoma patients. Int J Cancer. 1986;38(2):265‐274. [DOI] [PubMed] [Google Scholar]
- 19. Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. Switch‐like control of SREBP‐2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 2008;8(6):512‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsukano H, Gotoh T, Endo M, et al. The endoplasmic reticulum stress‐C/EBP homologous protein pathway‐mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2010;30(10):1925‐1932. [DOI] [PubMed] [Google Scholar]
- 21. Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic reticulum stress‐induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16(11):1345‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paillasse MR, de Medina P, Amouroux G, Mhamdi L, Poirot M, Silvente‐Poirot S. Signaling through cholesterol esterification: a new pathway for the cholecystokinin 2 receptor involved in cell growth and invasion. J Lipid Res. 2009;50(11):2203‐2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bemlih S, Poirier MD, El Andaloussi A. Acyl‐coenzyme A: cholesterol acyltransferase inhibitor Avasimibe affect survival and proliferation of glioma tumor cell lines. Cancer Biol Ther. 2010;9(12):1025‐1032. [DOI] [PubMed] [Google Scholar]
- 24. Antalis CJ, Arnold T, Rasool T, Lee B, Buhman KK, Siddiqui RA. High ACAT1 expression in estrogen receptor negative basal‐like breast cancer cells is associated with LDL‐induced proliferation. Breast Cancer Res Treat. 2010;122(3):661‐670. [DOI] [PubMed] [Google Scholar]
- 25. Li J, Gu D, Lee SS, et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene. 2016;35(50):6378‐6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matsumoto K, Fujiwara Y, Nagai R, Yoshida M, Ueda S. Expression of two isozymes of acyl‐coenzyme A: cholesterol acyltransferase‐1 and ‐2 in clear cell type renal cell carcinoma. Int J Urol. 2008;15(2):166‐170. [DOI] [PubMed] [Google Scholar]
- 27. Liu YY, Lin SJ, Chen YY, et al. High‐density lipoprotein cholesterol as a predictor of poor survival in patients with nasopharyngeal carcinoma. Oncotarget. 2016;7(28):42978‐42987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J, Wang J, Li M, Yin L, Li XA, Zhang TG. Up‐regulated expression of scavenger receptor class B type 1 (SR‐B1) is associated with malignant behaviors and poor prognosis of breast cancer. Pathol Res Pract. 2016;212(6):555‐559. [DOI] [PubMed] [Google Scholar]
- 29. Julovi SM, Xue A, LE Thanh TN, et al. Apolipoprotein A‐II Plus lipid emulsion enhance cell growth via SR‐B1 and target pancreatic cancer in vitro and in vivo. PLoS One. 2016;11(3):e0151475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guillaumond F, Bidaut G, Ouaissi M, et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci USA. 2015;112(8):2473‐2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hiraga S, Arita N, Ohnishi T, et al. Rapid infusion of high‐dose methotrexate resulting in enhanced penetration into cerebrospinal fluid and intensified tumor response in primary central nervous system lymphomas. J Neurosurg. 1999;91(2):221‐230. [DOI] [PubMed] [Google Scholar]
- 32. Gregory G, Arumugaswamy A, Leung T, et al. Rituximab is associated with improved survival for aggressive B cell CNS lymphoma. Neuro Oncol. 2013;15(8):1068‐1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Narita Y, Nagane M, Mishima K, et al. Phase 1/2 study of tirabrutinib, a second‐generation Bruton's tyro‐sine kinase inhibitor, in relapsed/refractory primary central nervous system lymphoma. Neuro Oncol. 2021;23(1):122‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Illerhaus G, Kasenda B, Ihorst G, et al. High‐dose chemotherapy with autologous haemopoietic stem cell transplantation for newly diagnosed primary CNS lymphoma: a prospective, single‐arm, phase 2 trial. Lancet Haematol. 2016;3(8):e388‐e397. [DOI] [PubMed] [Google Scholar]
- 35. Omuro A, Correa DD, DeAngelis LM, et al. R‐MPV followed by high‐dose chemotherapy with TBC and autologous stem‐cell transplant for newly diagnosed primary CNS lymphoma. Blood. 2015;125(9):1403‐1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhatt AP, Jacobs SR, Freemerman AJ, et al. Dysregulation of fatty acid synthesis and glycolysis in non‐Hodgkin lymphoma. Proc Natl Acad Sci USA. 2012;109(29):11818‐11823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Olkkonen VM, Johansson M, Suchanek M, et al. The OSBP‐related proteins (ORPs): global sterol sensors for co‐ordination of cellular lipid metabolism, membrane trafficking and signalling processes? Biochem Soc Trans. 2006;34(Pt 3):389‐391. [DOI] [PubMed] [Google Scholar]
- 38. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zannis VI, Chroni A, Krieger M. Role of apoA‐I, ABCA1, LCAT, and SR‐BI in the biogenesis of HDL. J Mol Med. 2006;84(4):276‐294. [DOI] [PubMed] [Google Scholar]
- 40. Ji Y, Jian B, Wang N, et al. Scavenger receptor BI promotes high density lipoprotein‐mediated cellular cholesterol efflux. J Biol Chem. 1997;272(34):20982‐20985. [DOI] [PubMed] [Google Scholar]
- 41. Jian B, de la Llera‐Moya M, Ji Y, et al. Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem. 1998;273(10):5599‐5606. [DOI] [PubMed] [Google Scholar]
- 42. Leon CG, Locke JA, Adomat HH, et al. Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration‐resistant prostate cancer in a mouse xenograft model. Prostate. 2010;70(4):390‐400. [DOI] [PubMed] [Google Scholar]
- 43. Twiddy AL, Cox ME, Wasan KM. Knockdown of scavenger receptor class B type I reduces prostate specific antigen secretion and viability of prostate cancer cells. Prostate. 2012;72(9):955‐965. [DOI] [PubMed] [Google Scholar]
- 44. Yang S, Damiano MG, Zhang H, et al. Biomimetic, synthetic HDL nanostructures for lymphoma. Proc Natl Acad Sci USA. 2013;110(7):2511‐2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569(1–2):29‐63. [DOI] [PubMed] [Google Scholar]
- 46. Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110(10):1383‐1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nakagawa T, Zhu H, Morishima N, et al. Caspase‐12 mediates endoplasmic‐reticulum‐specific apoptosis and cytotoxicity by amyloid‐beta. Nature. 2000;403(6765):98‐103. [DOI] [PubMed] [Google Scholar]
- 48. Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664‐666. [DOI] [PubMed] [Google Scholar]
- 49. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11(4):381‐389. [DOI] [PubMed] [Google Scholar]
- 50. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic‐reticulum‐resident kinase. Nature. 1999;397(6716):271‐274. [DOI] [PubMed] [Google Scholar]
- 51. Tirasophon W, Lee K, Callaghan B, Welihinda A, Kaufman RJ. The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response. Genes Dev. 2000;14(21):2725‐2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li M, Baumeister P, Roy BI, et al. ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress‐induced changes and synergistic interactions with NF‐Y and YY1. Mol Cell Biol. 2000;20(14):5096‐5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of Bip and ER stress transducers in the unfolded‐protein response. Nat Cell Biol. 2000;2(6):326‐332. [DOI] [PubMed] [Google Scholar]
- 54. Ma K, Vattem KM, Wek RC. Dimerization and release of molecular chaperone inhibition facilitate activation of eukaryotic initiation factor‐2 kinase in response to endoplasmic reticulum stress. J Biol Chem 2002;277(21):18728‐18735. [DOI] [PubMed] [Google Scholar]
- 55. Ri M, Tashiro E, Oikawa D, et al. Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress‐induced XBP1 mRNA splicing. Blood Cancer J. 2012;2(7):e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Fig S5
Fig S6
Fig S7
Fig S8
Fig S9
Fig S10
Table S1
Table S2
Data Availability Statement
The datasets analyzed during the study are available from the corresponding authors on reasonable request.