

Abstract
Neutrophils are the first defenders of the innate system for injury and infection. They have gradually been recognized as important participants in tumor initiation and development due to their heterogeneity and plasticity. In the tumor microenvironment (TME), neutrophils can exert antitumor and protumor functions, depending on the surroundings. Tumor cells systemically alter intracellular amino acid (AA) metabolism and extracellular AA distribution to meet their proliferation need, leading to metabolic reprogramming and TME reshaping. However, the underlying mechanisms that determine how altered AAs affect neutrophils in TME are less‐explored. Here, we identified that abundant glutamate releasing from tumor cells blunted neutrophils’ cell‐killing effects toward tumor cells in vitro and in vivo. Mass spectrometric detection, flow cytometry, and western blot experiments proved that increased levels of pSTAT3/RAB10/ARF4, mediated by glutamate, were accompanied with immunosuppressive phenotypes of neutrophils in TME. We also discovered that riluzole, an FDA‐approved glutamate release inhibitor, significantly inhibited tumor growth by restoring neutrophils’ cell‐killing effects and decreasing glutamate secretion from tumor cells. These findings highlight the importance of tumor‐released glutamate on neutrophil transformation in TME, providing new possible cancer treatments targeting altered glutamate metabolism.
Keywords: glutamate, neutrophil, riluzole, STAT3, tumor microenvironment
Tumor cell‐released glutamate could transform neutrophils from tumor‐cell killing to immunosuppressive phenotype, which is presumed to be the switch from N1 to N2. Specifically, glutamate might induce this switch by promoting pSTAT3 levels in neutrophils. Treatment with riluzole, a glutamate‐release inhibitor, significantly ameliorated this switch in tumor microenvironment by inhibiting glutamate release from tumor cell.
Abbreviations
- AA
amino acid
- ARF4
ADP‐ribosylation factor 4
- ARG
arginase
- FATP2
fatty acid transport protein 2
- GLS
glutaminase
- GluR
metabotropic glutamate receptor
- HIF‐1α
hypoxia inducible factor‐1α
- iGluR
ionotropic glutamate receptor
- LOX‐1
lectin type oxidized LDL receptor 1
- MDSC
myeloid‐derived suppressor cell
- MPO
myeloperoxidase
- NSCLC
non‐small cell lung cancer
- RAB10
Ras‐related GTP‐binding protein 10
- STAT3
signal transducer and activator of transcription 3
- TGF‐β
transforming growth factοr‐β
- TME
tumor microenvironment
1. INTRODUCTION
Abundant AA supply and production are important factors that maintain the high proliferative rate of tumor cells. 1 It has been reported that AA metabolism alterations (including glutamate, alanine, methionine, and cysteine), either intracellular or extracellular, are related with tumor development and progression, 2 , 3 , 4 in which oncogene activation and tumor suppressor gene mutation generally drive the alteration of catalytic enzyme expression and activity. 5 Several AA‐related receptors and transporters are also associated with cancer progression in tumor cell‐autonomous and nonautonomous ways. 2 , 6 , 7 In addition to their direct role of protein synthesis in tumor cells as materials, AAs in TME are also involved in energy generation and tumor‐infiltrated immune cell training, providing new ways for cancer treatment targeting AA metabolism.
The TME is tightly sculpted by tumor cells through the release of various chemokines and metabolites. 8 This leads to systemic reprogramming of surrounding cells, including fibroblast, endothelial, innate, and adaptive immune cells. 9 Neutrophils are the first defenders of the innate system for injury and infection. Neutrophils have been gradually recognized as important participants in tumor initiation and development due to their heterogeneity and plasticity. 10 Neutrophil‐contained granules and proteins are shown to kill tumor cells and facilitate tumor cell migration simultaneously. 11 , 12 , 13 The role of neutrophils in TME remains controversial, with evidence for antitumor and protumor functions depending on the surroundings. 14 Understanding the molecular mechanisms that drive neutrophils’ plasticity and the switch between pro‐ and antitumor effects is needed urgently. Based on previous evidence that AA metabolism was tightly associated with neutrophil functionality, 15 , 16 we hypothesized that altered AAs in TME might be one of the factors that influence neutrophil phenotype.
In this study, we found that tumor cells secreted glutamate in vitro and in vivo. High‐dose glutamate stimulation on neutrophils dampened cytotoxicity toward tumor cells. Specifically, glutamate upregulates the expression of pSTAT3, ARF4, and RAB10 in neutrophils, along with decreased cytotoxicity and inhibitory effects on T cell proliferation. Moreover, riluzole (glutamate release inhibitor) effectively restored neutrophil cytotoxicity toward tumor cells by reducing glutamate secretion in TME. These results indicate that glutamate was important tumor‐released AAs for neutrophil transformation in TME, which provided the possible mechanisms for neutrophil plasticity in tumors.
2. MATERIALS AND METHODS
2.1. Mice and treatments
Male C57/BL6 mice were purchased from TengXin Experimental Animal Company. All mice were fed and maintained at the animal facility of Army Medical University. All experiments were carried out with weight‐matched (18–22 g) male mice and approved by the Ethics Committee of Army Medical University.
2.2. Tumor cell line culture
Three murine tumor cell lines, melanoma (B16), Lewis lung carcinoma (LLC), and colon adenocarcinoma (MC48), were purchased from ATCC. Dulbecco’s modified Eagle’s medium (11965) and FBS (10100) were purchased from Thermo Fisher Scientific. All murine tumor cell lines (LLC, MC38, and B16) were cultured with DMEM (10% FBS supplemented). Luciferase‐containing lentiviral vectors (79943; BPS Bioscience) were coated and transfected with tumor cell lines LLC, MC38, and B16. The cells that stably expressed luciferase were selected by ampicillin and passaged for the following in vitro cytotoxicity effects.
2.3. Tumor model
Tumor cells (106 cells) were subcutaneously injected into the left armpit of male C57/BL6 mice and tumor sizes were monitored every 2 days. Tumor volume was calculated by the formula 0.5 × length × width2. To study the contribution of neutrophils/CD8 T cells to the riluzole‐mediated antitumor effect, five mice in each group were i.p. injected with 200 μg anti‐LY6G (Clone 1Α8, BP0075‐1; BioXCell) and 200 μg anti‐CD8α (Clone:53–6.7, ΒΕ0004‐1; BioXCell) four times before and after tumor inoculation (days −2, 1, 4, and 7). To test the cell‐killing effect of neutrophils from riluzole‐treated tumor tissues, five mice in each group were treated with riluzole (18 mg/kg, 0768; Tocris) through i.p. injection four times after tumor inoculation (days 1, 4, 7, and 10) according to a previous study. 17 Tumor tissues were then harvested and ground into single cell suspension by a Tissue Digestion Kit (05401020001; Sigma Aldrich) for the following studies.
2.4. Neutrophil isolation
Neutrophils from blood were purified by using LY6G MACS Microbeads (130120337; Miltenyi Biotec). Briefly, red blood cells were removed by ACK lysis buffer (BL5031; Biosharp). Dead cells were cleared up by a Dead Cell Removal Kit (130090101; Miltenyi Biotec). Single cell suspensions were then incubated with LY6G‐labeling beads (10 μl beads for 107 cells) at 4°C for 15 min. For neutrophils from tumors, single cell suspensions were incubated with LY6G MACS Microbeads and the Dead Cell Removal Kit on ice for 15 min. These LY6G‐labeled cells were separated by LS column separators. Isolated neutrophils were confirmed by FACS that 95% were murine neutrophils (CD45+CD11b+LY6G+) and the viability rate was more than 90%.
2.5. In vitro cytotoxicity assay
The cytotoxicity effects of neutrophils were determined as previously described. 18 Briefly, 5 × 104 tumor cells were seeded into 96‐well plates. After 6 h, normal DMEM culture medium was replaced with FBS‐free DMEM. Neutrophils isolated from blood or tumor tissues were harvested as described above. Isolated neutrophils were put into tumor cells (Luciferase expressing) and seeded into 96‐well plates at tumor cell : neutrophil ratios of 1:1, 1:10, and 1:50. After 24 h of coculturing, neutrophils and dead cells were washed away by PBS. The number of surviving tumor cells was evaluated using the firefly luciferase detection kit (16177; Thermo Fisher) with luminometer. The RLU values of DMEM without cells (Blank), tumor cell medium with/without neutrophils (control and coculture) were recorded, tumor cell viability (%) was presented as ([RLUcoculture − RLUblank]/[RLUcontrol − RLUblank] × 100%). L‐Glutamic acid (1.00291) and GLS inhibitor CB‐839 (S7655) were purchased from Sigma‐Aldrich and Selleckchem, respectively.
2.6. Medium AA analysis
The AA component and concentration of cultured cell supernatants were analyzed by AminoSAAYA (LA8080; HITACHI). Blank medium (DMEM without FBS) was used as control. Standard protein sample (VWD1E) and control sample (VWD1F) were used to define the specific protein signals. Tumor cell lines (106 cells) were cultured with DMEM (2 ml, FBS‐free) for 24 h. The cell cultured mediums were then centrifuged at 6800 g for 15 min and diluted with 5% trichloroacetic acid. Supernatants were collected for the following AA analysis. Relative AA concentration in cell cultured medium was calculated by comparing to blank medium. The curves of standard proteins sample, blank FBS‐free DMEM medium, and cultured medium were shown in Figure S1.
2.7. Glutamate detection
The glutamate concentration was determined by a fluorometric glutamate assay kit (STA‐674; Cell Biolabs). For cell culture medium (106 cells in 2 ml culture medium), supernatants were centrifuged at 6800 g for 10 min and diluted with assay buffer. For tumors, the tissues were homogenized and sonicated with hydrolysis buffer (STA‐674, 200 μl for 20 mg tissue; Cell Biolabs). The glutamate concentration was calculated by comparing the sample RFU value to the standard curve.
2.8. Flow cytometry
For the flow cytometry study, C57/BL6 mice bearing LLC tumors of approximately 150 mm3 were treated with riluzole by i.p. injection once every 2 days for a total of five times. Tumors and peripheral blood were harvested 1 day following the last treatment. Single cell suspensions were prepared and costained for CD4 (Clone:GK1.5, 100421; BioLegend), CD8 (Clone:YTS156.7.7, 126606 and Clone:YTS156.7.7, 126613; BioLegend), CD45 (Clone:30‐F11, 103130; BioLegend), FOXP3(Clone:MF‐14, 126409; BioLegend), CD11b (Clone:M1/70, 101201; BioLegend), and Gr‐1 (Clone:RB6‐8C5, 108407; BioLegend) for FACS analysis. Gating schematics for specific cell populations are shown in Figure S2.
2.9. Immunofluorescence
Immunofluorescence analysis was carried out as previously described. 19 Briefly, isolated neutrophils were smeared on glass slides and incubated with primary Abs diluted with 5% BSA containing 0.1% Triton‐100X at 4℃ overnight. LY6G Ab (ab238132) was purchased from Abcam, ARF4 (bs‐8750R) from Bioss, and Abs of RAB10 (8127) and pSTAT3 (Tyr705, 9145) were purchased from Cell Signaling Technology. The slices were subsequently washed with PBST three times and incubated with Alexa Fluor‐488/555 conjugated secondary Ab (1:800) for 2 h at 37℃. The slices were incubated with DAPI for nucleus staining and observed under a fluorescence microscope (IX‐81; Olympus).
2.10. Western blot analysis
After 24 h, neutrophils treated with gradient glutamate were lysed in 1× RIPA lysis buffer (P0013C; Beyotime) and Protease Inhibitor Cocktail (78429; Thermo Fisher Scientific). Antibodies of ARF4 (bs‐8750R; Bioss), RAB10 (8127; Cell Signaling Technology), pSTAT3 (Tyr705, 9145; Cell Signaling Technology), and β‐actin (3700S; Cell Signaling Technology) were incubated overnight. Goat anti‐rabbit/mouse HRP tagged secondary antibody (A0208 and A0216; Beyotime) were subsequently incubated at room temperature for 1 h. Finally, protein blots were visualized western ECL substrate (1705060; Bio‐Rad).
2.11. CD8+ T cell proliferation experiment
Murine CD8+ T cells were isolated from spleen by using CD8 MACS Microbeads (130120337; Miltenyi Biotec). The cells were harvested and resuspended with 5 μM CellTrace CFSE staining solution (C34554; Thermo Fisher Scientific). Cells were incubated for 20 min in a 37°C water bath. Aliquots of stained cells were distributed into anti‐CD3 (5 μg/ml, 11003081; eBioscience) and anti‐CD28 (2.5 μg/ml,16028981; eBioscience) coated 24‐well plates. These CD8+ T cells were cocultured with isolated neutrophils with RMPI‐1640 medium at different coculture ratios (neutrophil number : CD8+ T cell number) for 24 h. After gating out dead cells, the proliferated CD8+ T cells were analyzed by FACS in FITC channel.
2.12. Mass spectrometry
Murine neutrophils were isolated from blood using the Percoll gradient method 20 (P4937, pH 8.5–9.5; Sigma) and treated with/without glutamate (100 μM, G0355000, CAS 56‐86‐0; Sigma) for 24 h. Then 107 neutrophils were lysed in 200μl lysis buffer combined with sonication on ice. After digestion with trypsin (V5280; Promega), samples were shifted to Luming Biological Technology for the following steps, according to a previous study 21 : iTRAQ labeling and hydrophilic interaction chromatography fractionation, LC‐MS/MS, and data analysis. Proteins with changes greater than 1.5‐fold and p < 0.05 were considered differentially expressed.
2.13. Tumor Immune Single‐cell Hub analysis
Tumor Immune Single‐cell Hub provides detailed cell‐type annotation at the single‐cell level, enabling the exploration of TME. 22 In this study, single cell‐sequencing data of human NSCLC were retrieved and compared (GSE127465 and GSE99254), and mRNA relative expression of intended genes in different cell types from the GSE127465 dataset are presented. The mRNA expression levels are shown as log2 values of TPM.
2.14. Gene Expression Profiling Interactive Analysis database
We tested the mRNA correlation between GLS and human neutrophil marker genes (CD11b/ITGAM and CD66b/CEACAM8) from human LUAD and LUSC samples in the GEPIA database, as well as comparable normal tissues. The Pearson correlation coefficient values (R) and p values were analyzed and presented according to a previous study. 23
2.15. Statistical analysis
All experiments were repeated at least three times in duplicate. Results are presented as mean ± SEM. When comparing two groups, p values were calculated using two‐tailed Student’s t‐tests. For multiple treatment group comparisons, significance was determined by one‐way ANOVA, followed by Tukey’s post hoc multiple comparisons test. All statistical analyses were undertaken with GraphPad Prism software.
3. RESULTS
3.1. Glutamate is released from tumor cells in TME
We analyzed the AA release from three murine tumor cell lines (LLC, MC38, and B16) in vitro using the High‐Speed Amino Acid Analyzer AminoSAAYA. According to the standardized AA sample curve, 20 AAs/metabolites were detected in cell medium (Figure S1). After 48 h, supernatants were collected for the AA detection and cells were harvested for cell recounting. The standardized data (Figure 1A) showed that Glu and Ala were concordantly increased after culturing. We then measured Glu levels from tumor tissues and serum of tumor‐bearing mice (LLC, MC38, and B16). Using the fluorometric glutamate assay kit assay, we analyzed the Glu concentration in tumors and paratumor tissues (control). As shown in Figure 1B, glutamate levels in tumors were significantly increased compared to control groups, indicating that tumor tissues were glutamate‐abundant. We did not see statistical differences in Glu levels in serum (Figure 1C), whereas the glutamate concentration in tumor tissue was increased approximately 2–7‐fold compared to serum.
FIGURE 1.
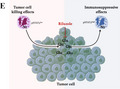
Analysis of glutamate production and glutaminase (GLS) expression in tumor. (A) Fourteen amino acids (AAs) in culture medium from three tumor cell lines (LLC, MC38, and B16). n = 3. (B) Glutamate concentration in tumor tissue was detected when tumor size reached 150 mm3. Control represents paratumor tissues. (C) Glutamate concentration in serum. Control represents tumor‐free mice. n = 5 for B and C. (D) Schematic diagram of glutamate synthesis and release. (E) Tumor Immune Single‐cell Hub database analysis of GLS expression comparison in human tumor tissue. Left panel presents different cell types. Right panel shows the relative GLS expression log2(transcripts per million [TPM]) in different cell types. (F, G) Gene Expression Profiling Interactive Analysis correlation analysis between GLS and ITGAM (CD11b)/CEACAM8 (CD66b) in The Cancer Genome Atlas human non‐small‐cell lung cancer (NSCLC) database (lung adenocarcinoma and lung squamous cell carcinoma, n = 481) and related normal tissues (n = 126). Correlation coefficients (r) were calculated by Pearson analysis with p value included
L‐glutamate is converted from L‐glutamine by GLS. 24 The increased L‐glutamine consumption and GLS expression driven by oncogenes (cMyc) are hallmarks of cancer cells, 25 leading to excessive glutamate production (Figure 1D). Tumor‐released glutamate was shown to promote tumor cell proliferation in an autocrine‐dependent manner. 26 , 27 However, the effect of extracellular glutamate on neutrophils in TME is still unknown. Using TISCH (http://tisch.comp‐genomics.org), we analyzed the mRNA levels of GLS in different cell types from human NSCLC samples. Compared to tumor cells, T cells, and macrophages, neutrophils expressed relatively low levels of GLS, indicating that tumor cells (Figure 1E), T cells, and macrophages were possibly the main source for glutamate release in TME. Using GEPIA (http://gepia.cancer‐pku.cn ), we tested the mRNA correlation between GLS and human neutrophil marker genes (CD11b/ITGAM and CD66b/CEACAM8) from human LUAD and LUSC samples. GLS expression was positively related to ITGAM (CD11b, r = 0.26, p < 0.0001; Figure 1F) and CEACAM8 (CD66, r = 0.26, p < 0.0001; Figure 1G) in tumor tissues, which were not observed in normal tissues. Here, we proved that tumor cells were capable of secreting glutamate in vitro and in vivo, making TME into a glutamate‐abundant environment.
3.2. Tumor cell‐killing effect by neutrophils is significantly blunted in TME, which could be restored by glutamate release inhibitor riluzole
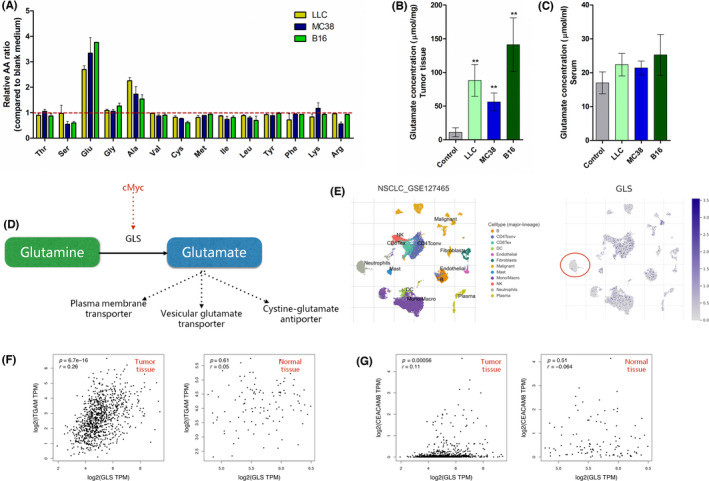
Neutrophils show both protumor and antitumor effects, depending on various surroundings. 10 , 11 , 27 , 28 , 29 A previous study and our data both showed that glutamate was abundant in tumor tissues. 2 , 28 We hypothesized whether glutamate could modulate neutrophil functions in tumor. Hence, we compared the cytotoxic effects between blood‐derived neutrophils and tumor‐derived neutrophils. The isolated neutrophils were cocultured at varying ratios with luciferase‐labeled tumor cells. The blood‐derived neutrophils presented obvious cytotoxic potential in a ratio‐dependent manner, which was not observed in intratumoral neutrophils (Figure 2A–C). This phenomenon was also observed previously, 29 , 30 , 31 indicating TME possibly switched the neutrophil phenotype. In order to explore glutamate effects on neutrophils, we used glutamate release inhibitor riluzole 32 to treat LLC‐tumor bearing mice. As shown in Figure 2D–F, riluzole treatment significantly inhibited glutamate release from tumor cells in vitro and in vivo. LY6G+ intratumoral neutrophil cells from control mice were found to be noncytotoxic up to a ratio of 1:10, whereas neutrophils isolated from the tumors of riluzole‐treated mice showed dose‐dependent cytotoxicity, with more significantly killing effects than control groups at ratios of 1:10 and 1:50 (Figure 2G). These data preliminarily showed that glutamate release blocked by riluzole increased neutrophil cytotoxic effects.
FIGURE 2.
Cytotoxic effect of neutrophils from blood and tumor tissue. (A–C) Viability of luciferase‐tagged tumor cells after 24 h of coculture with isolated neutrophils from blood and tumor tissues (three types). n = 5 (D) Glutamate release from three tumor cell lines after treating with gradient riluzole for 24 h. *Means compared to control. (E, F) Glutamate concentration in tumor tissues and serum from tumor‐free mice (control), LLC and riluzole‐treated LLC‐bearing mice. *Means compared to LLC. (G) Intratumoral neutrophil cytotoxic effects on LLC at different coculture ratios. */**Differences between riluzole group and blank group. n = 5. Data are presented as mean ± SEM
3.3. Glutamate decreases neutrophil cytotoxicity toward tumor cells in vitro
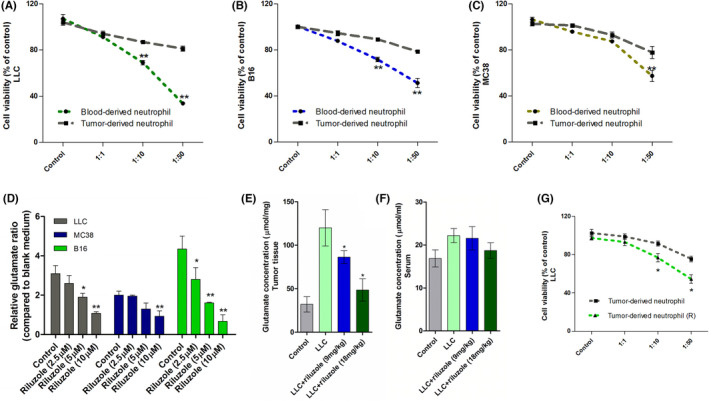
We next used glutamate‐pretreated neutrophils to coculture with tumor cell line LLC. As shown in Figure 3A, glutamate pretreatment decreased neutrophil cytotoxicity toward tumor cells in dose‐dependent manner. The cell‐killing effects were almost abolished at treatment doses above 50 μM glutamate. Riluzole treatment on neutrophils did not directly influence neutrophils’ cytotoxic functions to tumor cell line LLC (Figure 3B), but it directly inhibited tumor cell growth in a dose‐dependent manner (Figure 3C) in vitro. Previous studies have shown that targeting glutamate synthesis and release in tumor was effective to inhibiting tumor cell proliferation. 17 , 33 , 34 , 35 , 36 Our data showed that GLS inhibitor (CB‐839) killed tumor cells directly and sensitized tumor cells to neutrophil cytotoxicity (Figure 3D–F). Tumor cells incubated with glutamate release inhibitor riluzole (5 μM) were more vulnerable to neutrophil cytotoxicity (Figure 3G–I). Glutamate‐pretreated neutrophils showed weak cell‐killing effects toward tumor cell lines compared to the control group (Figure 3G–I). These data confirmed that glutamate synthesis and release from tumor cells were responsible for blunting neutrophil cytotoxicity in vitro and in vivo.
FIGURE 3.
Cytotoxic effect of neutrophils on tumor cells mediated by riluzole and glutamate. (A) Cell viability of LLC after coculturing with glutamate treated neutrophils at different ratios. (B) Cell viability of LLC after coculturing with riluzole pretreated neutrophils at different ratios. */**Significant differences between glutamate (100 μM) and blank neutrophil groups. (C) Cell viability of three tumor cell types after riluzole treatment for 24 h. (D–F) Cell viability of LLC/B16/MC38 cocultured with/without neutrophils at 1:10 ratio combined with gradient doses of CB‐839 treatments (0–2.5 μM) for 24 h. **Significant differences between tumor cell alone groups and neutrophil coculture groups. Riluzole (2.5 μM) or glutamate (100 μM) pretreated neutrophil/tumor cells for 6 h and resuspended with complete DMEM. Subsequently, these pretreated neutrophils/tumor cells were cocultured at different ratios for 24 h. Tumor cell viability was determined by luciferase signal intensity. (G–I) Cell viability of LLC/B16/MC38 after coculturing with neutrophils. */**Differences between riluzole group and blank group. #/##Differences between glutamate group and blank group. n = 5
3.4. Expression of pSTAT3, ARF4, and RAB10 highly induced after glutamate treatment in neutrophils
We next used mass spectrometry to screen out differential proteins in neutrophils after glutamate treatment. As shown in Figure 4A–C, glutamate treatment resulted in several protein levels and related‐pathway changes from membrane to nucleus, including STAT3, calcium‐binding mitochondrial carrier protein, ARF4, and Ras‐related protein. The Kyoto Encyclopedia of Genes and Genomes pathway analysis further showed that glutamate activated several tumorigenesis‐associated pathways (Figure 4C), including the TGF‐β and hypoxia inducible factor‐1 pathways, which are important for tumor cell migration and neutrophil transformation. 37 , 38 Using the TISCH database, we analyzed the mRNA levels of glutamate‐changed proteins (STAT3, SCaMC‐1, ARF4, and RAB10) from NSCLC patients. The data revealed that all these genes were widely distributed (Figure 4D–G). For neutrophils in TME (Figure 4D–G, red circle), the SCaMC‐1 mRNA level was extremely low, while STAT3, ARF4, and RAB10 levels were considerably high. We also used immunofluorescence and western blot analyses to confirm this. As Figure 5A,B shown, protein levels of ARF4, RAB10, and pSTAT3 (functional form of STAT3) in neutrophils were significantly induced by glutamate. Also, levels of these three proteins in neutrophils from LLC tumor tissues were strikingly increased compared to blood‐derived neutrophils (Figure 5C). Quantified data (Figure 5D,E) supported that this induction was consistent with increased glutamate concentration in tumor tissues (Figure 1B,C) and decreased neutrophil cytotoxic effects (Figure 2A–C). Specifically, Vasquez‐Dunddel et al. 39 confirmed that activated STAT3 drove neutrophils into an immunosuppressive phenotype by enhancing arginase‐I activity directly. Fan et al. 40 and Gu et al. 41 also proved that RAB10 phosphorylation in neutrophils leads to activation of leucine rich repeat kinase 2, which was associated with poor prognosis in cancer patients. The detailed mechanisms that explain how STAT3/ARF4/RAB10 influenced neutrophil‐mediated cell killing effects were the subject of the following investigations.
FIGURE 4.
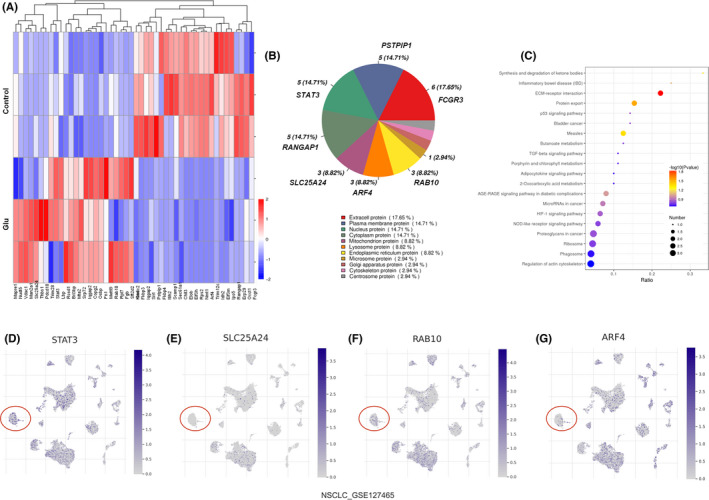
Differential protein expressions on neutrophils after glutamate treatment. (A) Mass spectrometry protein analysis for neutrophils with/without glutamate stimulation (100 μM). Differential proteins were visualized after log2 conversion. Red, upregulation; blue, downregulation. n = 3. (B) Subcellular distributions of differential proteins after glutamate treatment. Proteins are listed in each subcellular compartment. (C) Enrichment of differential protein‐related pathways based on the Kyoto Encyclopedia of Genes and Genomes database. (D–G) Tumor Immune Single‐cell Hub database analysis of STAT3/SLC25A24/RAB10/ARF4 expression comparison in human non‐small‐cell lung carcinoma (NSCLC) tumor tissue. Red circle indicates neutrophil subpopulation (cell population annotation is shown in Figure 1E). Purple bar represents log2(transcripts per million) of intended gene
FIGURE 5.
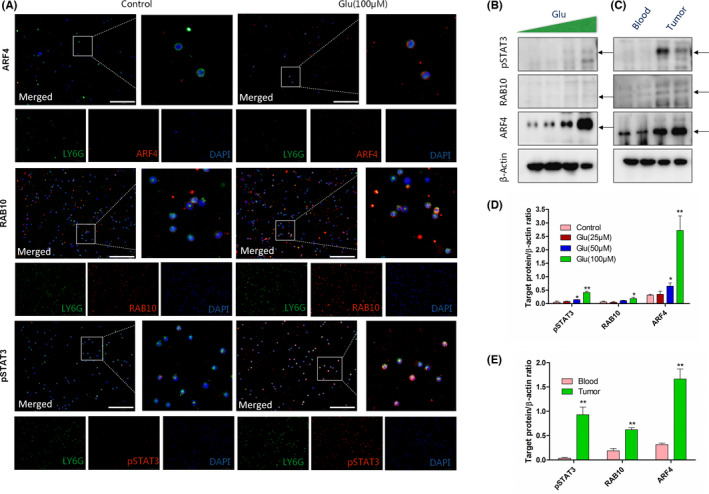
Immunofluorescence and western blot (WB) analysis of ADP‐ribosylation factor 4 (ARF4)/Ras‐related GTP‐binding protein 10 (RAB10)/signal transducer and activator of transcription 3 (pSTAT3) on neutrophils. Blood‐derived neutrophils were treated with/without glutamate (100 μM) for 24 h. (A) Triple immunostaining of neutrophils. Green, LY6G; red, specific proteins (ARF4/RAB10/pSTAT3); blue, nucleus. Scale bar, 100 μm. (B) WB analysis of ARF4/RAB10/pSTAT3 expression on blood‐derived neutrophils treated with gradient glutamate (0, 25, and 100 μM) for 24 h. In LLC‐tumor bearing mice, neutrophils were collected from blood and tumor tissues. (C) WB analysis of ARF4/RAB10/pSTAT3 expression on neutrophils isolated from blood and tumor tissues. β‐Actin was detected as loading control. (D) Quantifications of target proteins in (B) compared to loading control. */**Differences between glutamate and control group. (E) Quantifications of target proteins in (C) compared to loading control. */**Differences between blood and tumor group. n = 3
3.5. Riluzole decreases pSTAT3 level on neutrophils and increases CD8+ T cell frequency in TME
The above data suggested that glutamate abundance in TME was possible for suppressing neutrophil antitumor effects. Riluzole treatment significantly inhibited glutamate release, which was speculated to reshape immune responses in TME. To further investigate how riluzole influenced the TME and the antitumor response, we characterized neutrophils and T cells in an LLC tumor model by flow cytometry. As shown in Figure 6A,B, the number of neutrophils (CD11b+Gr‐1+) was significantly decreased in LLC tumors after riluzole treatment. The intracellular pSTAT3 levels of neutrophils in TME were also dramatically decreased after riluzole treatment (Figure 6C,D). The pSTAT3 levels of neutrophils from blood were relatively low compared to those from tumor tissues, consistent with western blot results. This indicated that decreased levels of pSTAT3 in neutrophils were accompanied with increased cytotoxicity toward tumor cells. The frequency of CD8+ T cells was increased in riluzole‐treated tumors (Figure 6E,F). We did not see a difference in CD4 T cells and regulatory T cell (CD4+FOXP3+) frequency in riluzole‐treated tumor tissue compared to control groups (Figure 6E–H). Of note, the percentages of circulating neutrophils, pSTAT3 positive neutrophils, as well as T cells from blood were quite even between riluzole treated/untreated mice (Figure S3), indicating that riluzole primarily affected those immune cells in tumor tissues. The TME reshaping after riluzole treatment, especially on neutrophils and CD8+ T cells, was possibly attributable to decreased glutamate release in tumor tissue.
FIGURE 6.
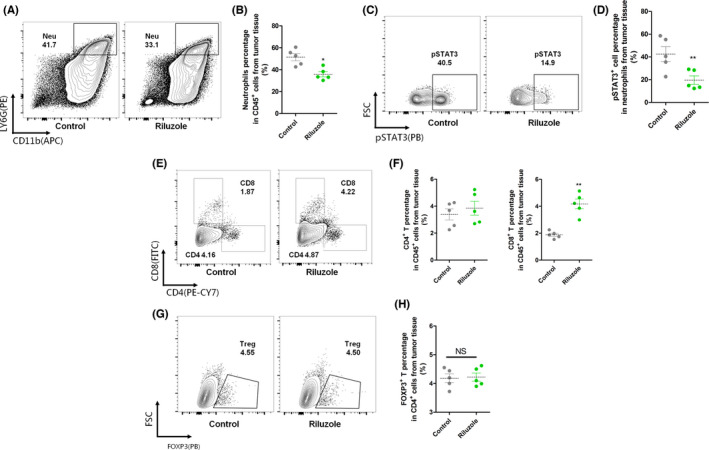
Flow cytometry analysis of immune cell subsets in tumor tissues. LLC tumor‐bearing mice were treated with/without riluzole (18 mg/kg) for five injections. FACS was used to analyze the components of specific immune cells in tumor tissues. (A) Neutrophil infiltration (CD45+CD11b+LY6G+) in tumors in LLC‐bearing mice treated with or without riluzole. n = 5. (B) These data were quantified as the percentage of MDSCs in CD45+ cells. (C) Flow cytometry analysis of pSTAT3 levels in neutrophils isolated from blood and tumors of LLC‐bearing mice treated with or without riluzole. n = 5. (D) Quantification of pSTAT3 percentage in neutrophils. (E) Flow cytometry CD4+ (CD45+CD4+) and CD8+ (CD45+CD8+) T cells in tumors in LLC‐bearing mice treated with or without riluzole. n = 5. (F) Quantitative analysis of the number of CD4+ and CD8+ T cells in tumors. (G) Flow cytometry regulatory T (Treg) (CD4+FOXP3+) cells in tumors in LLC‐bearing mice treated with or without riluzole. n = 5. (H) Quantitative analysis of the number of Tregs in tumors. Control, LLC‐bearing mice without riluzole treatment. *p < 0.05 between two groups; **p < 0.01 between two groups. NS, not significant
3.6. Antitumor effects of riluzole is partially dependent on the presence of neutrophils and CD8+ T cells in TME
Our data (Figure 7A) and other studies both showed that riluzole inhibited tumor growth in a dose‐dependent manner. 17 , 33 , 35 However, the antitumor effects in SCID mice (T and B cell deficiency) were reduced when riluzole was injected alone. 17 , 34 Given that glutamate stimulation was capable of modulating T cell activation and neutrophil migration, 42 , 43 these two populations were both tightly engaged with immune responses against cancer. 8 , 10 We explored whether riluzole‐mediated tumor growth inhibition was dependent on neutrophils and CD8+ T cells. As shown in Figure 7B, injection of either anti‐LY6G or anti‐CD8 mAb dramatically abolished the tumor growth inhibition in riluzole‐treated tumor‐bearing mice. This implied that both neutrophils and CD8+ T cells were essential to the antitumor effects of riluzole. One of the immunosuppressive potentials of neutrophils was through suppressing stimulated T cell proliferation. 44 Given the fact that riluzole alone did not influence CD8+ T cell proliferation rate directly (Figures 7C and S4), we wondered whether neutrophils from riluzole‐treated tumors could affect CD8+ T cell proliferation. We assessed the proliferation of anti‐CD3/CD28–stimulated T cells cocultured with neutrophils either from blood or tumor tissues for 24 h. When the neutrophil/CD8+ T cell coculture ratio was 1:4 and 1:2, tumor‐derived neutrophils dramatically decreased the T cell proliferation rate compared to blood‐derived neutrophils (Figure 7D). The inhibitory effects were significantly blocked after coculturing with neutrophils from riluzole‐treated tumor tissues. These results proved that riluzole treatment increased CD8+ T cell proliferation by decreasing neutrophil suppressive effects. The presence of neutrophils and CD8+ T cells were indispensable for riluzole‐mediated tumor growth inhibition.
FIGURE 7.
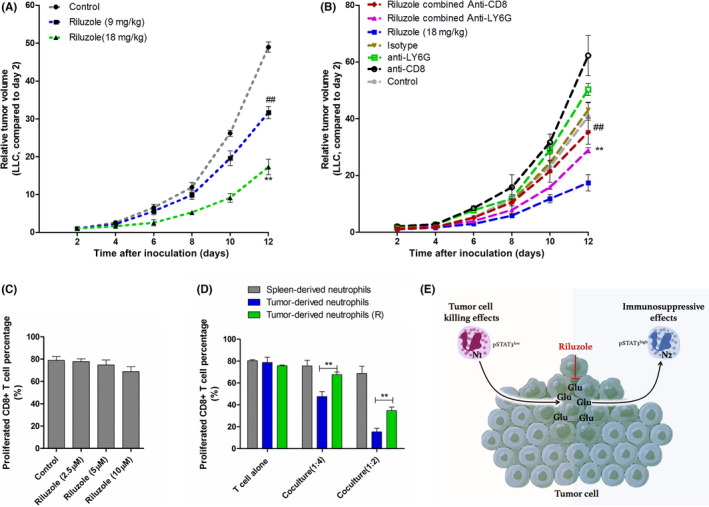
Consequences of neutrophil/CD8+ T cell depletion on riluzole antitumor effects. (A) LLC‐inoculated mice were injected with riluzole (9 mg/kg or 18 mg/kg) five times. Tumor sizes were measured every 2 days (n = 5). **/## p < 0.01 (riluzole vs. control at day 12). (B) LLC‐bearing mice treated in seven ways: control (DMSO), riluzole, riluzole combined anti‐LY6G Ab, riluzole combined anti‐CD8 Ab, isotype IgG, anti‐CD8, and anti‐LY6G. Bars represent mean ± SEM. **/## p < 0.01 (compared to riluzole alone at day 12). (C) Flow cytometry analysis of diffused CFSE‐tagged CD8+ T cells. CD8+ T cells were treated with anti‐CD3/CD28 combined with riluzole for 24 h. (D) CD8+ T cell proliferation rates after coculturing with intended neutrophils. Neutrophils from LLC‐bearing mice treated with/without riluzole (R). **p < 0.01 between intended groups. (E) Schematic presentation of the possible effects of glutamate on neutrophils in tumor microenvironment
4. DISCUSSION
In this study, we established the connection between glutamate release and neutrophil reshaping in TME. Abundant glutamate releasing from tumor cells blunted the cytotoxic effects of neutrophils in vitro and in vivo. In addition, glutamate upregulates the expression of pSTAT3, ARF4, and RAB10 in neutrophils and facilitates neutrophils forming immunosuppressive phenotypes (decreased cytotoxicity toward tumor cells and increased inhibitory effects on T cells) in TME. Moreover, riluzole (glutamate release inhibitor) effectively restores the antitumor effects of neutrophils by reducing glutamate abundance in TME. This confirms the important roles of glutamate in neutrophil transformation and TME modulation.
Glutamate, an important excitatory neurotransmitter in the brain, is also a major bioenergetic substrate for proliferating neoplastic cells. 24 A previous study confirmed that glutamate over‐release from glioma cells is associated with fast tumor growth. 27 Here, we further prove that glutamate release is prevailing in tumor cells, which can possibly be attributed to the enhanced glutamate uptake/release systems in tumor cells. 45 Tumor cell‐released glutamate promotes its proliferation in an autocrine‐dependent manner. 46 Overexpression and activation of glutamate receptors are highly involved in tumor malignancy. 47 The effectiveness and pharmacological properties of glutamate receptor antagonists are still under evaluation. 2 However, most of these investigations mainly focused on tumor cells themselves, neglecting glutamate‐mediated effects on TME. In this study, we proved that glutamate stimulation could inhibit the cell‐killing effects of neutrophils, during which STAT3 activation was possibly involved. Glutamate generally exerts biological functions by binding to GluRs or iGluRs. 2 Abnormal activations of GluRs/iGluRs are responsible for tumor development. 48 , 49 The glutamate mediated pathways, such as GluRs/β‐arrestin/STAT3 and iGluRs/Ca2+/STAT3, 50 , 51 are possible mechanisms for explaining neutrophil or other immune cell modulation in TME.
The origins and relationships between neutrophils and MDSCs in TME are still debatable. 52 Recently, it was reported that the overexpression of LOX‐1, ARG, and FATP2 are possible molecules to distinguish them. 18 , 53 , 54 However, some researchers believe that MDSCs represent polarized immature neutrophils, 55 making it possible for cell reprogramming and transformation mediated by surroundings. 54 Others argue that neutrophils are a separate cell lineage of MDSCs, in which activation and recruitment are different. 56 Even though the origins and relationships between them are controversial, the functions and phenotypes of tumor‐associated neutrophils are well‐defined. Based on the effect of neutrophils toward tumor cells, two neutrophil subtypes are introduced. 18 The N1 subset represents those neutrophils that are recruited to kill tumor cells by H2O2 secretion and myeloperoxidase granule transfer, 12 , 13 and N2 cells are capable of facilitating tumor growth in a TGF‐β or ARG‐dependent manner. 18 The tumorigenesis generally transformed N1 into the N2 cell type by reshaping TME. 10 , 11 , 57 Here we proved that tumor cell‐released glutamate was one of the factors that drove this transformation. The inductions of pSTAT3, ARF4, and RAB10 mediated by glutamate are possible molecules to distinguish N1 and N2 combined with other markers, such as LOX‐1, ARG, and FATP2.
Riluzole has been approved by the FDA for amyotrophic lateral sclerosis treatment. 32 , 58 Interestingly, riluzole also shows obvious inhibitory effects on several types of cancers. 17 , 33 , 35 Apart from the direct inhibitory effects on tumor cells, riluzole was deduced to inhibit tumor growth by modulating immune cells in TME as riluzole did not show significantly inhibitory effects in SCID‐tumor bearing mice (T and B cell deficiency). 17 , 34 Here, we confirm that riluzole restores neutrophil cell‐killing effects and CD8+ T cell proliferation in TME. This action is mainly attributed to inhibition of glutamate release in TME instead of direct effects on immune cells. Apart from tumor cells, T cells and macrophages are also the source for glutamate in TME., 59 , 60 It needs to be verified in the future whether these immune cells could influence the expansion and polarization of neutrophils by regulating glutamate secretion and reuptake.
Collectively, our data indicate that glutamate is a crucial tumor‐released AA for TME. During this process, induction of STAT3, ARF4, and RAB10 mediated by glutamate embodies the neutrophil immunosuppressive phenotype. Glutamate release inhibitor riluzole dramatically blunts neutrophil transformation by reducing glutamate abundance in TME. Therefore, glutamate is an important immunosuppressive AA for neutrophil transformation. Targeting glutamate metabolism could improve immunotherapy for cancer patients.
DISCLOSURE
The authors have no conflict of interests.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (32000670, 82071779, 81771693). We would like to thank Dr. Sun Jianbin (Department of Biochemistry and Molecular Biology, Army Medical University) for helping with us with cell culture and animal breeding during revision.
Xiong T, He P, Zhou M, et al. Glutamate blunts cell‐killing effects of neutrophils in tumor microenvironment. Cancer Sci. 2022;113:1955–1967. doi: 10.1111/cas.15355
Funding information
National Natural Science Foundation of China, Grant/Award Number: 32000670, 82071779, 81771693
Contributor Information
Zongtao Chen, Email: yjczt21@163.com.
Yang‐Wuyue Liu, Email: leo90lau@tmmu.edu.cn.
Shuang‐Shuang Dai, Email: tmmubiodss66@aliyun.com.
REFERENCES
- 1. Vettore L, Westbrook RL, Tennant DA. New aspects of amino acid metabolism in cancer. Br J Cancer. 2020;122(2):150‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stepulak A, Rola R, Polberg K, Ikonomidou C. Glutamate and its receptors in cancer. J Neural Transm. 2014;121(8):933‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sousa CM, Biancur DE, Wang X, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536(7617):479‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jakoš T, Pišlar A, Pečar FU, Švajger U, Kos J. Cysteine cathepsins L and X differentially modulate interactions between myeloid‐derived suppressor cells and tumor cells. Cancer Immunol Immunother. 2020;69(9):1869‐1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mossmann D, Park S, Hall MN. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat Rev Cancer. 2018;18(12):744‐757. [DOI] [PubMed] [Google Scholar]
- 6. Wang Q, Tiffen J, Bailey CG, et al. Targeting amino acid transport in metastatic castration‐resistant prostate cancer: effects on cell cycle, cell growth, and tumor development. J Natl Cancer Inst. 2013;105(19):1463‐1473. [DOI] [PubMed] [Google Scholar]
- 7. Karunakaran S, Ramachandran S, Coothankandaswamy V, et al. SLC6A14 (ATB0,+) protein, a highly concentrative and broad specific amino acid transporter, is a novel and effective drug target for treatment of estrogen receptor‐positive breast cancer. J Biol Chem. 2011;286(36):31830‐31838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017;27(11):863‐875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elia I, Haigis MC. Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nature Metab. 2021;3(1):21‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Powell DR, Huttenlocher A. Neutrophils in the tumor microenvironment. Trends Immunol. 2016;37(1):41‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mollinedo F. Neutrophil degranulation, plasticity, and cancer metastasis. Trends Immunol. 2019;40(3):228‐242. [DOI] [PubMed] [Google Scholar]
- 12. Yee PP, Wei Y, Kim S‐Y, et al. Neutrophil‐induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nature Commun. 2020;11(1):5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gershkovitz M, Caspi Y, Fainsod‐Levi T, et al. TRPM2 mediates neutrophil killing of disseminated tumor cells. Cancer Res. 2018;78(10):2680‐2690. [DOI] [PubMed] [Google Scholar]
- 14. Furumaya C, Martinez‐Sanz P, Bouti P, Kuijpers TW, Matlung HL. Plasticity in pro‐ and anti‐tumor activity of neutrophils: shifting the balance. Front Immunol. 2020;11:2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mühling J, Fuchs M, Fleck C, et al. Effects of arginine, L‐alanyl‐L‐glutamine or taurine on neutrophil (PMN) free amino acid profiles and immune functions in vitro. Amino Acids. 2002;22(1):39‐53. [DOI] [PubMed] [Google Scholar]
- 16. Belokrylov GA, Popova O, Molchanova IV, Sorochinskaya EI, Anokhina VV. Peptides and their constituent amino acids influence the immune response and phagocytosis in different ways. Int J Immunopharmacol. 1992;14(7):1285‐1292. [DOI] [PubMed] [Google Scholar]
- 17. Speyer CL, Bukhsh MA, Jafry WS, Sexton RE, Bandyopadhyay S, Gorski DH. Riluzole synergizes with paclitaxel to inhibit cell growth and induce apoptosis in triple‐negative breast cancer. Breast Cancer Res Treat. 2017;166(2):407‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fridlender ZG, Sun J, Kim S, et al. Polarization of tumor‐associated neutrophil phenotype by TGF‐beta: "N1" versus "N2" TAN. Cancer Cell. 2009;16(3):183‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu Y‐W, Yang T, Zhao LI, et al. Activation of Adenosine 2A receptor inhibits neutrophil apoptosis in an autophagy‐dependent manner in mice with systemic inflammatory response syndrome. Sci Rep. 2016;6:33614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mosca T, Forte WC. Comparative efficiency and impact on the activity of blood neutrophils isolated by percoll, ficoll and spontaneous sedimentation methods. Immunol Invest. 2016;45(1):29‐37. [DOI] [PubMed] [Google Scholar]
- 21. Arshid S, Tahir M, Fontes B, et al. High performance mass spectrometry based proteomics reveals enzyme and signaling pathway regulation in neutrophils during the early stage of surgical trauma. Proteomics Clin Appl. 2017;11(1–2):1600001. [DOI] [PubMed] [Google Scholar]
- 22. Sun D, Wang J, Han YA, et al. TISCH: a comprehensive web resource enabling interactive single‐cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 2021;49(D1):D1420‐D1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98‐W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eid T, Gruenbaum SE, Dhaher R, Lee TW, Zhou Y, Danbolt NC. The glutamate‐glutamine cycle in epilepsy. Adv Neurobiol. 2016;13:351‐400. [DOI] [PubMed] [Google Scholar]
- 25. Dang CV, Le A, Gao P. MYC‐induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15(21):6479‐6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seidlitz EP, Sharma MK, Saikali Z, Ghert M, Singh G. Cancer cell lines release glutamate into the extracellular environment. Clin Exp Metastasis. 2009;26(7):781‐787. [DOI] [PubMed] [Google Scholar]
- 27. de Groot J, Sontheimer H. Glutamate and the biology of gliomas. Glia. 2011;59(8):1181‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Geck RC, Toker A. Nonessential amino acid metabolism in breast cancer. Adv Biol Regul. 2016;62:11‐17. [DOI] [PubMed] [Google Scholar]
- 29. Lavender N, Yang J, Chen S‐C, et al. The Yin/Yan of CCL2: a minor role in neutrophil anti‐tumor activity in vitro but a major role on the outgrowth of metastatic breast cancer lesions in the lung in vivo. BMC Cancer. 2017;17(1):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Singel KL, Segal BH. Neutrophils in the tumor microenvironment: trying to heal the wound that cannot heal. Immunol Rev. 2016;273(1):329‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohms M, Möller S, Laskay T. An attempt to polarize human neutrophils toward N1 and N2 phenotypes in vitro. Front Immunol. 2020;11:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hunsberger HC, Weitzner DS, Rudy CC, et al. Riluzole rescues glutamate alterations, cognitive deficits, and tau pathology associated with P301L tau expression. J Neurochem. 2015;135(2):381‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Le MN, Chan J‐K, Rosenberg SA, et al. The glutamate release inhibitor Riluzole decreases migration, invasion, and proliferation of melanoma cells. J Invest Dermatol. 2010;130(9):2240‐2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Benavides‐Serrato A, Saunders JT, Holmes B, Nishimura RN, Lichtenstein A, Gera J. Repurposing potential of riluzole as an ITAF inhibitor in mTOR therapy resistant glioblastoma. Int J Mol Sci. 2020;21(1):344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lemieszek MK, Stepulak A, Sawa‐Wejksza K, Czerwonka A, Ikonomidou C, Rzeski W. Riluzole inhibits proliferation, migration and cell cycle progression and induces apoptosis in tumor cells of various origins. Anticancer Agents Med Chem. 2018;18(4):565‐572. [DOI] [PubMed] [Google Scholar]
- 36. Gross MI, Demo SD, Dennison JB, et al. Antitumor activity of the glutaminase inhibitor CB‐839 in triple‐negative breast cancer. Mol Cancer Ther. 2014;13(4):890‐901. [DOI] [PubMed] [Google Scholar]
- 37. Yang L, Pang Y, Moses HL. TGF‐beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31(6):220‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Corzo CA, Condamine T, Lu L, et al. HIF‐1α regulates function and differentiation of myeloid‐derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207(11):2439‐2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vasquez‐Dunddel D, Pan F, Zeng QI, et al. STAT3 regulates arginase‐I in myeloid‐derived suppressor cells from cancer patients. J Clin Invest. 2013;123(4):1580‐1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fan Y, Tonelli F, Padmanabhan S, et al. Human peripheral blood neutrophil isolation for interrogating the Parkinson's associated LRRK2 kinase pathway by assessing Rab10 phosphorylation. J Vis Exp. 2020(157):58956. [DOI] [PubMed] [Google Scholar]
- 41. Gu S, Chen J, Zhou Q, et al. LRRK2 is associated with recurrence‐free survival in intrahepatic cholangiocarcinoma and downregulation of LRRK2 suppresses tumor progress in vitro. Dig Dis Sci. 2020;65(2):500‐508. [DOI] [PubMed] [Google Scholar]
- 42. Gupta R, Palchaudhuri S, Chattopadhyay D. Glutamate induces neutrophil cell migration by activating class I metabotropic glutamate receptors. Amino Acids. 2013;44(2):757‐767. [DOI] [PubMed] [Google Scholar]
- 43. Pacheco R, Gallart T, Lluis C, Franco R. Role of glutamate on T‐cell mediated immunity. J Neuroimmunol. 2007;185(1–2):9‐19. [DOI] [PubMed] [Google Scholar]
- 44. Aarts CEM, Hiemstra IH, Tool ATJ, et al. Neutrophils as suppressors of T cell proliferation: does age matter? Front Immunol. 2019;10:2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sharma MK, Seidlitz EP, Singh G. Cancer cells release glutamate via the cystine/glutamate antiporter. Biochem Biophys Res Commun. 2010;391(1):91‐95. [DOI] [PubMed] [Google Scholar]
- 46. Namkoong J, Shin S‐S, Lee HJ, et al. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007;67(5):2298‐2305. [DOI] [PubMed] [Google Scholar]
- 47. Yu LJ, Wall BA, Wangari‐Talbot J, Chen S. Metabotropic glutamate receptors in cancer. Neuropharmacology. 2017;115:193‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wan Z, Sun R, Liu Y‐W, et al. Targeting metabotropic glutamate receptor 4 for cancer immunotherapy. Sci Adv. 2021;7(50):eabj4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Elnagar MR, Walls AB, Helal GK, Hamada FM, Thomsen MS, Jensen AA. Functional characterization of α7 nicotinic acetylcholine and NMDA receptor signaling in SH‐SY5Y neuroblastoma cells in an ERK phosphorylation assay. Eur J Pharmacol. 2018;826:106‐113. [DOI] [PubMed] [Google Scholar]
- 50. Li X‐R, Cheng X, Sun J, et al. Acetylation‐dependent glutamate receptor GluR signalosome formation for STAT3 activation in both transcriptional and metabolism regulation. Cell Death Discov. 2021;7(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li H, Zhang Q, Zhang G. Signal transducer and activator of transcription‐3 activation is mediated by N‐methyl‐D‐aspartate receptor and L‐type voltage‐gated Ca2+ channel during cerebral ischemia in rat hippocampus. Neurosci Lett. 2003;345(1):61‐64. [DOI] [PubMed] [Google Scholar]
- 52. Zhou J, Nefedova Y, Lei A, Gabrilovich D. Neutrophils and PMN‐MDSC: their biological role and interaction with stromal cells. Semin Immunol. 2018;35:19‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Condamine T, Dominguez GA, Youn J‐I, et al. Lectin‐type oxidized LDL receptor‐1 distinguishes population of human polymorphonuclear myeloid‐derived suppressor cells in cancer patients. Sci Immunol. 2016;1(2):8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Veglia F, Tyurin VA, Blasi M, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569(7754):73‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Veglia F, Perego M, Gabrilovich D. Myeloid‐derived suppressor cells coming of age. Nat Immunol. 2018;19(2):108‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fridlender ZG, Albelda SM. Tumor‐associated neutrophils: friend or foe? Carcinogenesis. 2012;33(5):949‐955. [DOI] [PubMed] [Google Scholar]
- 57. Shaul ME, Levy L, Sun J, et al. Tumor‐associated neutrophils display a distinct N1 profile following TGFβ modulation: a transcriptomics analysis of pro‐ vs. antitumor TANs. Oncoimmunology. 2016;5(11):e1232221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585‐591. [DOI] [PubMed] [Google Scholar]
- 59. Moidunny S, Matos M, Wesseling E, et al. Oncostatin M promotes excitotoxicity by inhibiting glutamate uptake in astrocytes: implications in HIV‐associated neurotoxicity. J Neuroinflammation. 2016;13(1):144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Coffelt SB, Kersten K, Doornebal CW, et al. IL‐17‐producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4