

Abstract
Parkinson’s disease (PD) is characterized by the formation of Lewy bodies (LBs) in the brain. LBs are mainly composed of phosphorylated and aggregated α-synuclein (α-Syn). Thus, strategies to reduce the expression of α-Syn offer promising therapeutic avenues for PD. DNA/RNA heteroduplex oligonucleotides (HDOs) are a novel technology for gene silencing. Using an α-Syn-HDO that specifically targets α-Syn, we examined whether α-Syn-HDO attenuates pathological changes in the brain of mouse models of PD. Overexpression of α-Syn induced dopaminergic neuron degeneration through inhibition of cyclic AMP-responsive-element-binding protein (CREB) and activation of methyl CpG binding protein 2 (MeCP2), resulting in brain-derived neurotrophic factor (BDNF) downregulation. α-Syn-HDO exerted a more potent silencing effect on α-Syn than α-Syn-antisense oligonucleotides (ASOs). α-Syn-HDO attenuated abnormal α-Syn expression and ameliorated dopaminergic neuron degeneration via BDNF upregulation by activation of CREB and inhibition of MeCP2. These findings demonstrated that inhibition of α-Syn by α-Syn-HDO protected against dopaminergic neuron degeneration via activation of BDNF transcription. Therefore, α-Syn-HDO may serve as a new therapeutic agent for PD.
Keywords: MT: Oligonucleotides, Therapies and Applications, alpha-synuclein, BDNF, oligonucleotide, transcription, Parkinson’s disease
Graphical abstract
CRISPR-Cas9 derived prime editor and base editors are powerful tools in precise genome editing. We demonstrate HDAC inhibition increases the efficiency of prime editor and base editors, which advances our understanding of regulating endogenous mechanisms to improve genome editing.
Introduction
Parkinson’s disease (PD), the second most prevalent age-related neurodegenerative disease, is characterized by progressive selective loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) with the concomitant loss of nigrostriatal dopaminergic termini and the resulting motor symptoms.1 Both genetic and environmental factors play a key role in the etiology of PD.2 However, most PD occurs sporadically with unknown disease etiology, and approximately 5% to 10% of PD cases are caused by genetic abnormalities.2 Both sporadic and familial PD have the same pathological hallmarks as follows: dopaminergic neuron degeneration in the SNc; and the presence of intraneuronal proteinaceous cytoplasmic inclusions, known as Lewy bodies (LBs), in the remaining dopaminergic neurons.3,4 Alpha-synuclein (α-Syn) is the main component of LBs, and its aggregation is believed to be the major step in the pathogenesis of PD.5 Mutation or multiplication of α-Syn has been identified as the pathogenesis of both sporadic and familial PD.6, 7, 8 Soluble monomers, toxic oligomers, and insoluble fibrils of α-Syn have been detected in the brains of patients with PD.9 Several mutations in the gene that encodes α-Syn (SNCA), such as A53T, A30P, E46K, H50Q, G51D, and A53E, cause autosomal-dominant PD.10 Moreover, phosphorylation of α-Syn at the Ser129 site promotes the formation of pathogenic α-Syn aggregates, which is one of the most crucial posttranslational modifications.11 Based on the above findings, downregulation of α-Syn offers a promising therapeutic avenue preventing the progression of PD.
Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin (NT) family.12, 13, 14, 15 BDNF colocalizes with dopaminergic neurons in the SNc, promoting dopaminergic neuron survival.16,17 Clinical research has revealed that BDNF levels are decreased in PD patients, suggesting that reduced levels of BDNF may be involved in the pathogenesis of PD.12,18 BDNF transcription is regulated by cyclic AMP-responsive-element-binding protein (CREB) and methyl CpG binding protein 2 (MeCP2).19,20 CREB is a transcriptional activator, and MeCP2 is a transcriptional repressor of BDNF.19,20 It has been shown that overexpression of α-Syn reduces BDNF expression;21 however, the mechanisms by which α-Syn reduces BDNF expression, resulting in PD pathology, have not been defined.
In the present study, with in vitro and in vivo systems, we provide the evidence that overexpression of α-Syn induces dopaminergic neuron degeneration via BDNF downregulation by inhibition of CREB and activation of MeCP2. DNA/RNA heteroduplex oligonucleotides (HDOs) are a newly developed technology for gene silencing.22, 23, 24 Compared with the parent single-stranded gapmer antisense oligonucleotides (ASOs), a DNA/locked nucleotide acid gapmer duplex with an α-tocopherol-conjugated complementary RNA is significantly more potent in reducing the expression of the targeted mRNA with fewer side effects.22,23 We therefore designed an α-Syn-HDO that specifically targets α-Syn. α-Syn-HDO exerted a more potent silencing effect on α-Syn than α-Syn-ASO. α-Syn-HDO attenuated abnormal α-Syn expression, activated BDNF transcription, and ameliorated dopaminergic neuron degeneration. These findings suggested that abnormal α-Syn expression induces dopaminergic neuron degeneration via inhibition of BDNF transcription, which is alleviated by attenuating abnormal α-Syn expression.
Results
Overexpression of α-Syn inhibits BDNF expression via inhibition of CREB and activation of MeCP2
BDNF plays an important role in neuronal survival in the dopaminergic neurons, and the level of BDNF is reduced in the SNc of PD patients.18 Here, we investigated whether overexpression of α-Syn inhibits BDNF expression via inhibition of CREB and activation of MeCP2. SH-SY5Y cells were transfected with GFP-α-Syn and lysed for western blot analysis. Overexpression of α-Syn decreased the ratio of p-CREB/CREB and BDNF levels; however, it increased MeCP2 expression (Figure 1A). These findings suggest that α-Syn causes inhibition of CREB and activation of MeCP2 expression, resulting in BDNF downregulation.
Figure 1.

Overexpression α-Syn inhibits BDNF expression
(A) Western blot assay for p-CREB, CREB, BDNF, and MeCP2 in SH-SY5Y cells 24 h after GFP-α-Syn transfection (mean ± SEM, n = 4 per group, Student’s t test, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). (B) Protein expression of p-CREB, CREB, BDNF, and MeCP2 in the SNc of human AAV-α-Syn-treated mice (mean ± SEM, n = 6 per group, Student’s t test, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). (C) Protein expression of p-CREB, CREB, BDNF, and MeCP2 in the striatum from DLB patients and controls (mean ± SEM, n = 10 per group, Student’s t test, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). There was a positive correlation between BDNF levels and the ratio of phosphorylated CREB/CREB in DLB patients (n = 10). Furthermore, there was a negative correlation between BDNF levels and MeCP2 levels in DLB patients (n = 10).
Next, we injected AAV9-hSyn-human SNCA virus into the SNc of wild-type (WT) mice. Subsequently, we extracted proteins from the SNc for western blot analysis. Injection of AAV9-hSyn-human SNCA decreased the ratio of p-CREB/CREB and BDNF levels; however, it increased MeCP2 expression (Figure 1B).
Dementia with Lewy bodies (DLB) is pathologically characterized by α-Syn, and phosphorylated α-Syn aggregates in the brain.25 Deposition of α-Syn has been shown in multiple brain regions of PD and DLB patients.25,26 Therefore, postmortem brain samples from patients with DLB were used. We measured the protein expression of p-CREB/CREB, BDNF, and MeCP2 in the striatum from DLB patients and age-matched control subjects. The ratio of p-CREB/CREB and the levels of BDNF were significantly lower in patients with DLB than in controls. Furthermore, the levels of MeCP2 in patients with DLB were significantly higher than those of controls (Figure 1C). Interestingly, there was a positive correlation between BDNF levels and p-CREB/CREB ratio in the striatum from DLB patients (Figure 1C). Furthermore, there was a negative correlation between BDNF levels and MeCP2 levels in the striatum from DLB patients (Figure 1C). Collectively, these findings indicated that overexpression of α-Syn causes inhibition of CREB and activation of MeCP2, resulting in BDNF downregulation.
Silencing α-Syn expression activates BDNF transcription
In the present study, we designed an α-Syn-HDO that harbors locked nucleic acids (LNAs) at each end flanking the central base of DNA and 2′-O-methyl at each end flanking the central base of cRNA with conjugated α-tocopherol. The α-Syn-HDO was also tagged with or without FAM labels for tracing (Figure 2A). FAM-α-Syn-HDO was absorbed in SH-SY5Y cells in a time-dependent manner (Figure 2B). Western blot analysis showed that α-Syn-HDO decreased α-Syn and MeCP2 expression in a dose-dependent manner (Figure 2C), while α-Syn-HDO increased the ratio of p-CREB/CREB and BDNF expression in a dose-dependent manner (Figure 2C). Moreover, compared with α-Syn-ASO (200 nM), α-Syn-HDO (200 nM) exerted a more potent silencing effect on α-Syn at both the mRNA and protein levels (Figures S1A and S1B). In addition, the half maximal inhibitory concentration (IC50) of α-Syn-HDO (64.06 nM) is more potent than α-Syn-ASO (99.81 nM) (Figures S1C and S1D). The scrambled α-Syn-HDO did not show any silencing effect for α-Syn (Figures S2A and S2B). The results suggest that α-Syn-HDO effectively silences α-Syn expression. Based on the in vitro results, we examined the silencing effects of α-Syn-HDO for α-Syn in vivo. Mice were subjected to intracerebroventricular (ICV) injection of α-Syn-HDO (200 nM/2 μL/week, total four times). Western blot analysis showed that α-Syn-HDO significantly decreased α-Syn expression in the SNc of WT mice (Figure S3A). In addition, we compared the silencing effects of α-Syn-ASO and α-Syn-HDO in vivo. ICV injection of α-Syn-ASO or α-Syn-HDO (200 nM/2 μL/week, total four times) decreased α-Syn and MeCP2 expression (Figure S3B), while α-Syn-ASO or α-Syn-HDO increased the p-CREB/CREB ratio and BDNF expression in the SNc of WT mice (Figure S3B). Importantly, α-Syn-HDO was more potent than α-Syn-ASO. These results suggest that α-Syn-HDO is associated with the activation of BDNF transcription by silencing α-Syn expression.
Figure 2.

α-Syn-HDO activates BDNF transcription
(A) Schematic illustration of the construction of α-Syn-HDO. (B) The internalization of FAM-α-Syn-HDO visualized by microscopy at 0 min, 30 min, and 1 h following transfection of FAM-α-Syn-HDO (200 nM). Scale bar, 50 μm. (C) Western blot analysis of α-Syn, p-CREB, CREB, MeCP2, and BDNF in SH-SY5Y cells treated with various dosages of α-Syn-HDO (mean ± SEM, n = 4 per group, one-way ANOVA, ∗p < 0.05 and ∗∗p < 0.01). (D) Luciferase assay for BDNF IV promoters. BDNF exon IV luciferase promoters and/or α-Syn-HDO were transfected into HEK293T cells (mean ± SEM, n = 4 per group, one-way ANOVA, ∗∗∗p < 0.001). (E) BDNF exon IV luciferase promoter and α-Syn-HDO, siRNA-CREB plasmids, or mutation (Mut) plasmids were cotransfected into HEK293T cells (mean ± SEM, n = 4 per group, one-way ANOVA, ∗∗p < 0.01 and ∗∗∗p < 0.001). (F) ChIP-PCR assays demonstrated that p-CREB specifically binds to genomic DNA of BDNF exon IV promoter binding motifs. p-CREB protein-DNA crosslinking samples were obtained from SH-SY5Y cells treated with α-Syn-HDO or vehicle via coimmunoprecipitation with an anti-p-CREB antibody. PCR was performed with primers targeting the BDNF exon IV promoter. An anti-histone H3 antibody coupled with GAPDH primers was used as the positive control (mean ± SEM, n = 4 per group, Student’s t test, ∗p < 0. 05). (G) qPCR assay for BDNF in SH-SY5Y cells treated with α-Syn-HDO (mean ± SEM, n = 5 per group, Student’s t test, ∗p < 0. 05).
To further elucidate the action of α-Syn-HDO in stimulating BDNF transcription, we performed luciferase reporter, chromatin immunoprecipitation (ChIP)-PCR, and quantitative PCR (qPCR) assays. The data showed that α-Syn-HDO activated the Bdnf exon IV promoter, which was blocked by CREB knockdown (Figures 2D and 2E). In addition, mutation in the CREB-binding motif completely abolished promoter activity (Figure 2E). In addition, ChIP-PCR analysis of genomic DNA immunoprecipitated with the p-CREB antibody demonstrated that α-Syn-HDO induced the interaction between p-CREB and the Bdnf exon IV promoter (Figure 2F). Moreover, α-Syn-HDO enhanced the Bdnf mRNA levels (Figure 2G). Collectively, these data demonstrated that α-Syn-HDO activates CREB, resulting in BDNF transcription.
α-Syn-HDO is associated with activation of CREB and inhibition of MeCP2, resulting in BDNF upregulation in α-Syn-treated SH-SY5Y cells
Overexpression of α-Syn leads to inhibition of CREB and activation of MeCP2, thereby causing BDNF downregulation. Hence, we further explored whether α-Syn-HDO associates with BDNF upregulation in α-Syn-treated SH-SY5Y cells. To address this hypothesis, SH-SY5Y cells were transfected with GFP-α-Syn or glutathione S-transferase (GST)-α-Syn. Overexpression of α-Syn significantly decreased the ratio of p-CREB/CREB and BDNF levels but it increased MeCP2 expression (Figure 3A). α-Syn-HDO reversed the effects of α-Syn overexpression in GFP-α-Syn-transfected SH-SY5Y cells (Figure 3A). Immunofluorescence staining revealed that overexpression of α-Syn caused the redistribution of p-CREB and MeCP2 in the nucleus of SH-SY5Y cells. α-Syn induced MeCP2 nuclear localization and more punctate p-CREB in the nucleus, and this redistribution was reversed by α-Syn-HDO (Figure 3B). These data demonstrated that α-Syn-HDO can attenuate BDNF downregulation in α-Syn-treated SH-SY5Y cells.
Figure 3.

α-Syn-HDO attenuates BDNF downregulation in α-Syn-treated SH-SY5Y cells
(A) Western blot assay for p-CREB, CREB, BDNF, and MeCP2 in GFP-α-Syn-transfected SH-SY5Y cells treated with α-Syn-HDO for 24 h (mean ± SEM, n = 5 per group, one-way ANOVA, ∗p < 0.05 and ∗∗p < 0.01). (B) Immunofluorescence staining for p-CREB and MeCP2 in GST-α-Syn-transfected SH-SY5Y cells treated with α-Syn-HDO for 24 h. Scale bar, 50 μm.
α-Syn-HDO attenuates dopaminergic neuron degeneration in an α-syn-induced PD mouse model
To examine potential therapeutic efficacy of α-Syn-HDO, we investigated whether α-Syn-HDO attenuates dopaminergic neuron degeneration via activation of BDNF transcription in AAV9-hSyn-human SNCA-treated mice. First, AAV9-hSyn-human SNCA was injected into the SNc of WT mice to construct a PD mouse model (Figure 4A). Subsequently, mice were subjected to ICV injection of FAM-α-Syn-HDO or α-Syn-HDO (200 nM/2 μL/wk, total four times) (Figure 4A). Following confirmation of the distribution of FAM-α-Syn-HDO in the mouse brains (Figure 4B), behavioral tests showed that α-Syn-HDO significantly prolonged the duration of AAV9-hSyn-human SNCA-treated mice on the rotarod test compared with those of the vehicle group (Figure 4C). Immunofluorescence staining demonstrated that AAV9-hSyn-human SNCA administration significantly decreased tyrosine-hydroxylase (TH) immunoreactivity but increased IBA1 and glial fibrillary acidic protein (GFAP) immunoreactivity in the SNc, and these changes were reversed by α-Syn-HDO (Figures 4D and S4). Using western blot analysis, we found that AAV9-hSyn-human SNCA significantly downregulated TH expression but increased α-Syn levels in the SNc, which was reversed by α-Syn-HDO (Figure 4E). Collectively, these data suggest that α-Syn-HDO attenuates dopaminergic neuron degeneration and ameliorates PD-like pathology in AAV9-hSyn-human SNCA-treated mice.
Figure 4.

α-Syn-HDO attenuates dopaminergic neuron degeneration in AAV9-hSyn-human SNCA-treated mice
(A) Schedule of treatment and graphical illustration of human AAV-α-Syn injection. (B) Graphical illustration of the intracerebroventricular injection site. (C) Results of the rotarod test (mean ± SEM, n = 10–12 per group, one-way ANOVA, ∗p < 0.05 and ∗∗p < 0.01). (D) Immunofluorescence staining for TH in the SNc. Quantification analysis of TH (mean ± SEM, n = 5 per group, one-way ANOVA, ∗∗p < 0.01 and ∗∗∗p < 0.001). Scale bar, 50 μm. (E) Western blot assay for TH and α-Syn in the SNc (mean ± SEM, n = 5 per group, one-way ANOVA, ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001). (F) ChIP-PCR assays for p-CREB and BDNF exon IV promoter in the SNc (mean ± SEM, n = 5 per group, one-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). (G) Western blot assay for p-CREB/CREB, BDNF, and MeCP2 in the SNc (mean ± SEM, n = 5 per group, one-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001).
To further validate the connections of therapeutic activity and the above regulation of α-Syn-HDO in vivo, we examined signaling in the SNc of AAV9-hSyn-human SNCA-treated mice. ChIP-PCR results showed that p-CREB partially dissociated from the Bdnf exon IV promoter, which was reversed by α-Syn-HDO (Figure 4F). Next, we examined the ratio of p-CREB/CREB and the protein levels of BDNF and MeCP2 in the SNc of AAV9-hSyn-human SNCA-treated mice. The data showed that the p-CREB/CREB ratio and BDNF expression were decreased and that MeCP2 expression was increased. Interestingly, α-Syn-HDO increased the ratio of p-CREB/CREB and decreased MeCP2 expression, leading to BDNF upregulation (Figure 4G). Therefore, these findings indicated that α-Syn-HDO can produce neuroprotective effects by promoting BDNF expression in AAV9-hSyn-human SNCA-treated mice via activation of CREB and inhibition of MeCP2.
α-Syn-HDO attenuates dopaminergic neuron degeneration in MTPT-treated α-Syn-A53T mice
MPTP is the best characterized toxin that causes PD pathology, and injection of MPTP accelerates PD pathology in SNCA mice in vivo.12,27 Hence, we further examined the neuroprotective effect of α-Syn-HDO in MTPT-treated α-Syn-A53T mice. ICV injection of α-Syn-HDO significantly increased the duration of MPTP-treated α-Syn-A53T mice on the rotarod test compared with the vehicle group (Figures 5A and 5B). Immunofluorescence staining indicated that α-Syn-HDO significantly increased TH immunoreactivity but decreased IBA1 and GFAP immunoreactivity in the SNc of MPTP-treated α-Syn-A53T mice compared with the vehicle group (Figures 5C and S5). Western blot analysis showed that α-Syn-HDO increased TH immunoreactivity but decreased α-Syn protein expression in the SNc of MPTP-treated α-Syn-A53T mice (Figure 5D). These data demonstrated that α-Syn-HDO ameliorates PD-like pathology in MPTP-treated α-Syn-A53T mice.
Figure 5.

α-Syn-HDO attenuates dopaminergic neuron degeneration in MPTP-treated A53T mice
(A) Schedule of treatment. (B) Results of the rotarod test (mean ± SEM, n = 10–12 per group, one-way ANOVA, ∗p < 0.05 and ∗∗p < 0.01). (C) Immunofluorescence staining for TH in the SNc. Quantification analysis of TH (mean ± SEM, n = 5 per group, one-way ANOVA, ∗∗p < 0.01 and ∗∗∗p < 0.001). Scale bar, 50 μm. (D) Western blot assay for TH and α-Syn in the SNc (mean ± SEM, n = 5 per group, one-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). (E) ChIP-PCR assays for p-CREB and BDNF exon IV promoter in the SNc (mean ± SEM, n = 5 per group, one-way ANOVA, ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001). (F) Western blot assay for p-CREB/CREB, BDNF, and MeCP2 in the SNc (mean ± SEM, n = 5 per group, one-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001).
ChIP-PCR results showed that p-CREB partially dissociated from the Bdnf exon IV promoter in MPTP-treated α-Syn-A53T mice, which was reversed by α-Syn-HDO (Figure 5E). We also found that the p-CREB/CREB ratio and BDNF expression were decreased and that MeCP2 expression was increased, and these changes in MPTP-treated α-Syn-A53T mice were reversed by α-Syn-HDO (Figure 5F). Therefore, these findings suggested that α-Syn-HDO can produce neuroprotective effects by promoting BDNF expression in MPTP-treated α-Syn-A53T mice.
α-Syn-HDO blocks α-Syn pathology in vitro and in vivo
To determine whether the reduction of α-Syn expression by α-Syn-HDO ameliorates α-Syn aggregation, we assessed the effects of α-Syn-HDO on the aggregation and phosphorylation of α-Syn in α-Syn-preformed fibrils (PFFs). We detected the aggregation of α-Syn in HEK293-α-Syn cells. After treatment with PFFs for 24 h, YFP-α-Syn started to aggregate into small fluorescence spots located in the intracellular space, and this effect was abolished by α-Syn-HDO treatment (Figures S6A and S6B). Western blot analysis showed that the PFFs induced phosphorylation of α-Syn at S129 (p-S129), which was attenuated by α-Syn-HDO (Figure S6C). PFFs were injected into the SNc of α-Syn-A53T mice in vivo, which led to the cell-to-cell transmission of pathologic α-Syn and PD-like Lewy pathology in the SNc (Figure 6A). Treatment with α-Syn-HDO significantly attenuated the PFF-induced Lewy pathology in the SNc (Figure 6A). In behavioral tests, α-Syn-HDO prolonged the duration of PFFs-treated α-Syn-A53T mice on the rotarod test compared with the vehicle group (Figure 6B). Immunofluorescence staining demonstrated that α-Syn-HDO significantly increased TH immunoreactivity but decreased IBA1 and GFAP immunoreactivity in the SNc of PFFs-treated α-Syn-A53T mice (Figures 6C, S7A, and S7B). Western blot assays showed that α-Syn-HDO treatment significantly ameliorated the decreased expression of TH and increased expression of p-S129 and α-Syn in the SNc of PFFs-treated α-Syn-A53T mice (Figure 6D). These results indicated that α-Syn-HDO could ameliorate the phosphorylation and aggregation of α-Syn, resulting in attenuation of PD pathology.
Figure 6.

α-Syn-HDO prevents α-Syn-induced PD pathology
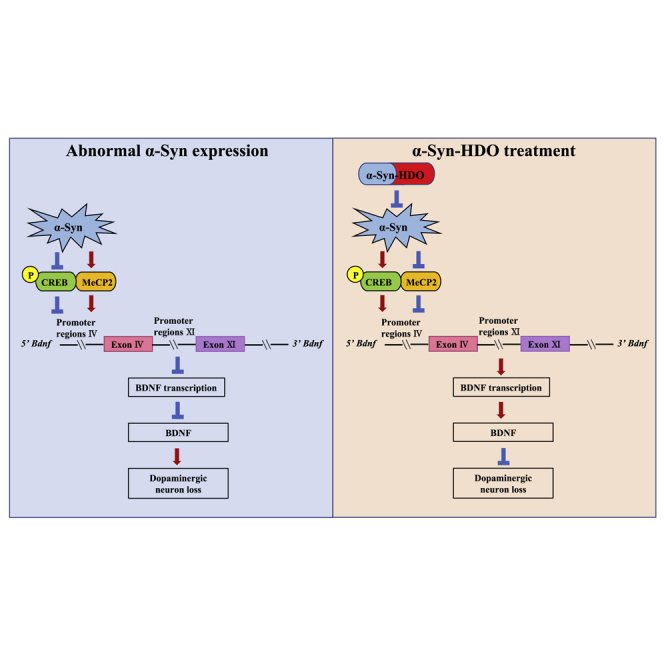
(A) Immunofluorescence staining for TH and p-α-Syn in the SNc. Scale bar, 50 μm. (B) The schedule of treatment and the rotarod test results (mean ± SEM, n = 11 or 12 per group, one-way ANOVA, ∗∗p < 0.01 and ∗∗∗p < 0.001). (C) Immunofluorescence staining for TH in the SNc. Quantification analysis of TH (mean ± SEM, n = 5 per group, one-way ANOVA, ∗p < 0.05 and ∗∗p < 0.01). Scale bar, 50 μm. (D) Western blot assay for TH, p-α-Syn, and α-Syn in the SNc (mean ± SEM, n = 5 per group, one-way ANOVA, ∗p < 0.05 and ∗∗p < 0.01). (E) ChIP-PCR assays for p-CREB and BDNF exon IV promoter in the SNc (mean ± SEM, n = 5 per group, one-way ANOVA, ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001). (F) Western blot assay for p-CREB/CREB, BDNF, and MeCP2 in the SNc (mean ± SEM, n = 5 per group, one-way ANOVA, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001). Abnormal α-Syn expression induces dopaminergic neuron degeneration via inhibition of BDNF transcription. The novel nucleic acid agent α-Syn-HDO can attenuate dopaminergic neuron degeneration in PD mouse models via activation of BDNF transcription.
In addition, ChIP-PCR results showed that p-CREB partially dissociated from the Bdnf exon IV promoter in PFFs-treated α-Syn-A53T mice, which was reversed by α-Syn-HDO (Figure 6E). Western blot results found that the p-CREB/CREB ratio and BDNF expression were decreased and that MeCP2 expression was increased, and these changes were reversed by α-Syn-HDO in PFFs-treated α-Syn-A53T mice (Figure 6F). Therefore, these findings indicated that α-Syn-HDO exerts neuroprotective effects by promoting BDNF expression in PFFs-treated α-Syn-A53T mice.
Discussion
In the present study, overexpression of α-Syn induced dopaminergic neuron degeneration via inhibition of CREB and activation of MeCP2, resulting in BDNF downregulation. Silencing abnormal α-Syn expression using α-Syn-HDO activated CREB and inhibited MeCP2, resulting in BDNF upregulation and amelioration of dopaminergic neuron degeneration in PD mouse models. Our results suggest that overexpression of α-Syn inhibits BDNF expression, resulting in PD pathology. Thus, downregulation of abnormal α-Syn expression offers a promising therapeutic avenue preventing the progression of PD.
Accumulating studies have shown that BDNF colocalizes with dopaminergic neurons in the SNc and that BDNF promotes dopaminergic neuronal survival.12,28,29 Reduced levels of BDNF have been demonstrated in the postmortem brains of PD patients.12,18,30 α-Syn-induced blockade of TrkB neurotrophic activation triggers dopaminergic neuronal death in a PD mouse model.12 In contrast, overexpression of BDNF attenuates 6-OHDA- or MPTP-induced nigrostriatal degeneration, and it improves rotational behavioral deficits by regulating dopaminergic neurotransmission.31,32 Therefore, BDNF is integral to both the pathophysiology of PD and the therapeutic mechanisms for PD. In the present study, we found that overexpression of α-Syn inhibited BDNF expression by decreasing the ratio of p-CREB/CREB and increasing MeCP2 expression. In addition, the ratio of p-CREB/CREB and the levels of BDNF were significantly lower in the postmortem brain samples from patients with DLB, whereas the levels of MeCP2 were significantly higher in these samples. Therefore, these data indicated that overexpression of α-Syn inhibits CREB and activates MeCP2, resulting in BDNF downregulation, which plays a role in the pathogenesis of PD.
Overexpression of BDNF has been shown to attenuate dopaminergic neuron degeneration.31,32 Our data showed that α-Syn-HDO attenuated dopaminergic neuronal degeneration in PD mouse models. Therefore, we examined whether α-Syn-HDO promotes BDNF transcription by inhibiting of abnormal α-Syn expression. In vitro data revealed that α-Syn-HDO promoted p-CREB binding with Bdnf exon IV promoter, resulting in Bdnf mRNA expression. The results indicated that α-Syn-HDO activates BDNF transcription. In vivo data suggested that α-Syn-HDO attenuated dopaminergic neuron degeneration in the SNc of PD mouse models, and that α-Syn-HDO attenuated the dissociated effects of the p-CREB binding with Bdnf exon IV promoter in the SNc of PD mouse models. In addition, α-Syn-HDO restored the reduction of p-CREB/CREB ratio and increased MeCP2 expression, resulting in BDNF expression through inhibition of abnormal α-Syn expression. Thus, it is likely that α-Syn-HDO might produce neuroprotective effects through inhibition of α-Syn expression in PD mouse models, leading to upregulation of CREB activity and downregulation of MeCP2 expression, which activated BDNF transcription. Besides, the abnormal α-Syn promotes the production of reactive oxygen species through interaction with complex I of the mitochondrial respiratory chain and interferes with its function.33 Accumulating evidence suggests that the toxic interaction among dopamine (DA), DA metabolites, and abnormal α-Syn might promote an oxidative environment within dopaminergic neurons. Oxidative modification of α-Syn by DA metabolites has been proposed to be responsible for the selective vulnerability to dopaminergic neurons.34,35 The oligomeric α-Syn has been suggested to represent the primary toxic species responsible for dopaminergic neurotoxicity.34 This evidence suggests that abnormal α-Syn may cause neurodegeneration in other pathways excluding the BDNF pathway. Suppression of abnormal α-Syn by α-Syn-HDO may prevent neurodegeneration beyond the CREB-BDNF signaling pathway. Therefore, it is of interest to investigate the role of other signaling pathways on the neuroprotective effects of α-Syn-HDO. In addition, altered levels of p-CREB and MeCP2 can affect the regulation of numerous genes. It is, therefore, possible that changes in widespread genes could affect MPTP-induced neurotoxicity, contributing to the effects of α-Syn-HDO in MPTP-treated α-Syn-A53T mice.
Chronic neuroinflammation, one of the key pathogenic factors responsible for neurodegenerative disorders, can lead to elevated levels of glia-derived cytokines, which exert neurotoxic effects on vulnerable dopaminergic neurons.36, 37, 38 In the animal models of PD and PD patients, reactive microglia/astrocytes (CD11b/GFAP) were found in the SNc, indicating the possible involvement of gliosis-derived inflammatory processes responsible for PD.39 Inhibition of glial activation-derived inflammatory response contributes to the protection of dopaminergic neurons in vivo and in vitro.40 In this study, we found that chronic administration of α-Syn-HDO could prevent glial activation and attenuate TH neuron degeneration in the SNc of PD mouse models. Taken together, the present data suggest that the neuroprotective effects of α-Syn-HDO might be partly mediated by inhibiting the activation of glial SNc of PD mouse models, although further study is needed.
As demonstrated by various genetic and biochemical studies, α-Syn is the major component of LBs and plays a predominant role in the pathogenesis of PD and DLB.41,42 There is extensive phosphorylation of α-Syn at S129 in LBs.43 Therefore, the most likely hypothesis is that phosphorylation of α-Syn at Ser129 accelerates the formation of insoluble α-Syn aggregates during the onset of PD.44 Moreover, exogenous PFFs have been reported to induce the aggregation of endogenous α-Syn.42,45 Using HEK293 cells stably transfected with human α-Syn, we found that α-Syn-HDO decreased the expression, phosphorylation, and aggregation of α-Syn. In addition, dopaminergic neuron degeneration in PFFs-treated α-Syn-A53T mice was attenuated by α-Syn-HDO. These data suggested that α-Syn-HDO reduces α-Syn levels, consequently alleviating α-Syn-induced pathological changes.
The present study has some limitations. The previous study has shown that the α-Syn knockout (KO) mice did exhibit abnormalities in synaptic morphology and function, along with fairly subtle behavioral changes.46,47 The α-Syn-HDO is widely distributed throughout the brain by ICV injection. Therefore, the neurotoxic effects and off-target effects of α-Syn-HDO should be further studied, especially in normal mice for long periods of time. Moreover, the striatum includes caudate, putamen, and globus pallid, which is innervated from multiple brain regions, so any changes observed cannot be exclusively attributed to the nigrostriatal pathway. In addition, the striatum is also only the terminal region of the nigrostriatal system, and changes in transcription factors may not only reflect what is happening in the soma of the neurons in the nigra. Future study using postmortem samples of nigra is needed. Finally, it has been shown that overexpression of human-α-Syn under the Thy1 regulatory element promotes expression of human-α-Syn in multiple neuronal subpopulations. Intriguingly, this did not include TH-positive dopaminergic neurons, which do not degenerate in these mice,48 inconsistent with our results. The difference may be due to different promoters for human-α-Syn. Future detailed studies are necessary to explore these differences.
In conclusion, the current study suggests that overexpression of α-Syn induces dopaminergic neuron degeneration through inhibition of BDNF transcription, and that the novel nucleic acid agent α-Syn-HDO can attenuate dopaminergic neuron degeneration in PD mouse models via activation of BDNF transcription. Therefore, α-Syn-HDO would be a potential new therapeutic agent for PD.
Materials and methods
Mice and cell lines
Male adult C57BL/6 mice (8 weeks old, 20–25 g) were obtained from Guangdong Experimental Animal Center. Male transgenic mice expressing A53T human α-Syn (12 weeks old, 25–30 g) were obtained from the Jackson Laboratory (gift from Dr. Zhentao Zhang). The animals were housed under controlled temperature and kept in a 12-h light/dark cycle with ad libitum access to food and water. The animal protocol was approved by the Jinan University Institutional Animal Care and Use Committee, and all experiments were performed following the Guide for Animal Experimentation of Jinan University. HEK293T cells, SH-SY5Y cells, and HEK293T cells stably expressing YFP-labeled human α-synuclein (HEK293-α-Syn) were cultured in DMEM or DMEM/F-12 (basal media) supplemented with 10% fetal bovine serum (Excell Bio.) and penicillin (100 units/mL)–streptomycin (100 μg/mL). Cells were cultured at 37°C in a humidified incubator containing 5% CO2. HEK293-α-Syn cells were kindly gifted by Prof. Dimond.49
Materials
MPTP (1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine) was purchased from Yuanye Bio-Technology (Shanghai, China) and dissolved in 0.9% sterile saline. MPTP (30 mg/kg) was administered intraperitoneally to mice. The doses of MPTP selected correspond to those previously reported.50 The pEGFP-α-Syn and mGST-α-Syn plasmids were kindly gifted by Dr. Zhentao Zhang (Department of Neurology, Renmin Hospital of Wuhan University).
Antisense oligonucleotides (ASOs) for α-Syn and cRNA were purchased from TsingKe Biological Technology (Wuhan, China) or Ajinomoto Bio-Pharma (Osaka, Japan) and solubilized in 0.9% sterile saline before use. For the ion of α-Syn-HDO, equimolar amounts of DNA and cRNA strands were heated in 0.9% sterile saline at 95°C for 5 min and slowly cooled to room temperature. HDO harbored LNAs at each end flanking the central base of DNA with or without an FAM (6-carboxy-fluorescein) label, and HDO harbored 2′-O-methyl at each end flanking the central base of cRNA with conjugated α-tocopherol. The sequences of ASOs and cRNA targeting α-Syn used in our experiments are as follows: ASO-α-Syn, G(L)ˆC(L)ˆtˆcˆcˆcˆtˆcˆcˆaˆcˆtˆgˆT(L)ˆC(L)ˆT(L);4 cRNA, a(M)ˆg(M)ˆa(M)ˆcaguggagggaˆg(M)ˆc(M); where L indicates the LNAs, M indicates the 2′-O-methyl modifications, and ˆ indicates the phosphorothioate bond. SH-SY5Y cells were transfected with different doses of α-Syn-HDO for 24 h using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. After transfection for 24 h, cells were collected for luciferase reporter, ChIP-PCR, immunofluorescence staining, qPCR, and western blot assays.
Full-length human α-Syn was expressed in BL21 (DE3) competent E. coli (Life Technologies) and purified as previously described.51 The purified recombinant α-Syn was stored at −80°C until use. PFFs were made by diluting recombinant α-Syn to 5 mg/mL in sterile Dulbecco’s PBS (Cellgro, Mediatech; pH adjusted to 7.0, without Ca2+ or Mg2+) followed by incubation at 37°C with constant agitation at 1,000 rpm for 7 days. PFFs were sonicated with a water-bath cup-horn sonicator (Fisher Scientific, USA) at 50% power for 5 minutes before use.
Treatment with AAV9-hSyn-human SNCA, PFFs, and α-syn-HDO
Mice were anesthetized with isoflurane and fixed to a stereotaxic apparatus. AAV9-hSyn-human SNCA (6.58 × 1013 vg/mL, Vigenebio Biosciences, Jinan, China) or PFFs were injected into the substantia nigra (1.2 mm lateral, −4.3 mm ventral, and −3.1 mm from bregma).1 Virus (2 μL) or PFFs (2.5 μL) were injected into each site using a 10 μL Hamilton syringe with a fixed needle at a rate of 0.25 μL/min using a microinjector pump (KDS, Stoelting). The needle remained in place for 5 min after the viral suspension or PFFs were completely injected followed by slow removal (over 2 min). The mice were placed on a heating pad until recovery from anesthesia.
α-Syn-HDO was injected into the right lateral ventricle using the following stereotaxic coordinates: 0.8 mm lateral, −2.1 mm ventral, and 0.74 mm from bregma following anesthetization. For multiple injections of α-Syn-HDO over 4 weeks (α-Syn-HDO: 200 nM/2 μL/week, total four times), a guiding cannula (RWD Life Science, China) was implanted using the coordinates described above. The drugs were injected by an injection cannula through a guiding cannula.
Rotarod test
For the rotarod test, mice were trained for 3 sequential days on the rotarod. Each daily practice session consisted of placing the subject on the rotarod at a slow rotational speed (5 rpm) for a maximum of 5 min. Mice were given three test trials on the test day. The rotational speed of rotarod was modulated from 0 rpm to a maximum 40 rpm. It was gradually increased during the trial at a rate of 0.1 rpm/s. Each trial was started and then sustained for 5 min. The trial was stopped when the mouse fell (activating a switch that automatically stopped the timer) or when 5 min had elapsed. The residence time on the rotarod was counted using a stopwatch. The results showed the average value of the three trials.
qPCR assay
Levels of α-Syn and Bdnf mRNA were examined by quantitative PCR (qPCR). RNA was extracted using an Eastep Super Kit (Promega) followed by reverse transcription with GoScript Reverse Transcriptase Mix, Oligo (dT) (Promega) to generate cDNA. The qPCR assays were performed with the ChamQ SYBR qPCR Master Mix Kit (Vazyme) using the 788BR05175 qPCR System. The PCR amplification protocol was as follows: 40 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension for 30 s at 72°C. The primer sequences were as follows: α-Syn forward, 5′-TGACGGGTGTGACAGCAGTAG-3′; α-Syn reverse, 5′-CAGTGGCTGCTGCAATG-3′; Bdnf forward, 5′-TTGTTTTGTGCCGTTTACCA-3′; Bdnf reverse, 5′-GGTAAGAGAGCCAGCCACTG-3′ for mouse sample; Bdnf forward, 5′-CATCCGAGGACAAGGTGGCTTGG-3′; and Bdnf reverse, 5′-GTCCTCATCCAACAGCTCTTCTATC-3′ for the human sample.4,52,53 The target genes were analyzed by the 2−ΔΔCt method.
Western blot analysis
Cell and brain homogenates were lysed in RIPA buffer. Protein concentrations were determined by a Coomassie Brilliant Blue protein assay kit (Bio-Rad). Postmortem brain samples (striatum) from DLB patients and age-matched controls were collected at Tokyo Metropolitan Geriatric Hospital and Institute of Gerontology (Tokyo, Japan). Brain samples were selected using the Brain Bank for Aging Research (BBAR) Lewy bodies rating system.54 Total protein (20 μg) was separated on 10% to 12% SDS-polyacrylamide gels and then transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% milk at room temperature for 1 h followed by incubation with primary antibodies at 4°C for 12 h. Membranes were then washed three times with TBST and incubated with the corresponding secondary antibody for 1 h at room temperature. After an additional three washes, targeted proteins were detected using the enhanced chemiluminescence method scanned by the Tanon-5200CE imaging system (Tanon, Shanghai, China). The expression levels of target proteins were normalized to β-actin as a loading control. The following primary antibodies were used: anti-phospho-CREB antibody (1:1,000, 9198S) and CREB antibody (1:1,000, 9197S) were purchased from Cell Signaling Technology; anti-MeCP2 antibody (1:1,000, M6818) was purchased from Sigma; anti-BDNF antibody (1:1,000, ab108319), anti-phospho-α-Syn antibody (1:1,000, ab51253), and anti-α-Syn antibody (1:1,000, ab1903) were purchased from Abcam; anti-TH antibody (1:1,000, GTX10372) was purchased from GTX (GeneTex); and anti-β-actin antibody was purchased from EarthOx. The horseradish peroxidase-conjugated anti-rabbit/mouse immunoglobulin G antibody was purchased from Bio-Rad.
Immunofluorescence staining
Cell or mouse brain sections were preplated on cover glasses and fixed in 4% paraformaldehyde for 10 min at room temperature. After treatment, the glasses were washed with PBS three times and blocked using 3% BSA with 0.3% Triton X-100 for 30 min followed by incubation with anti-TH (1:500, GTX10372), anti-IBA1 (1:500, GTX632426), or anti-GFAP (1:500, Affinity, DF6040) primary antibodies for 24 h at 4°C. Following washing with PBS, cells were incubated with Alexa Fluor 488/594 anti-mouse/rabbit secondary antibody (1:500) for 2 h at room temperature in the dark followed by staining with DAPI to visualize the nuclei. Cells were washed with PBS and visualized by a fluorescence microscope (Olympus BX53, Japan).
Luciferase reporter assay
HEK293T cells were cotransfected with BDNF exon IV luciferase reporter plasmid together with pRL-TK Renilla luciferase plasmid (Promega) and α-Syn-HDO, small interfering RNA (siRNA)-CREB, or CREB mutant plasmid. After transfection for 24 h, cells were collected and analyzed using the dual-luciferase reporter assay kit (Promega) according to the manufacturer’s protocol.
ChIP-PCR assay
Following treatment with α-Syn-HDO, cells or brain samples were analyzed by a ChIP-PCR assay using the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling) according to the manufacturer’s protocol. For the ChIP assay, 7.5 μg of p-CREB antibody was added to the sample homogenate, mixed, and incubated overnight at 4°C. The washing, elution, and reverse crosslinking to free DNA were performed according to the manufacturer’s protocol. BDNF exon IV-specific primers were used for amplification of the promoter region using the following primer sequences: forward 5′-GGCTTCTGTGTGCGTGAATTTGC-3′ and reverse 5′-AAAGTGGGTGGGAGTCCACGAG-3′.20 The PCR amplicon was separated on a 2% agarose gel after 35 cycles of PCR (denaturation at 95°C for 30 s, annealing at 58°C for 30 s, and extension at 72°C for 30 s).
Statistical analysis
All data results are expressed as the mean ± standard error of the mean (SEM) and were analyzed using PASW Statistics 20 software (formerly SPSS Statistics, SPSS). Potential differences between the mean values were evaluated using one-way analysis of variance followed by post hoc Fisher’s least significant difference test or two-way analysis of variance; when appropriate, post hoc comparisons were performed using the unpaired t test. Student’s t test was used to compare the differences between two groups unless otherwise specified. Asterisks were used to indicate significance: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Values >0.05 were considered not significant (ns).
Acknowledgments
We are thankful to Z.Z. (Department of Neurology, Renmin Hospital of Wuhan University) for providing the A53T mice for us. This work is supported by the National Natural Science Foundation of China (81973341 to Q.Q., 81822016 and 81771382 to Z.Z.), the Science and Technology Program of Guangzhou (202002030010 to Q.Q.), the Fundamental Research Funds for the Central Universities (11620425 to J.Z., 21620426 to Q.Q.), Huang Zhendong Research Fund for Traditional Chinese Medicine of Jinan University (201911 to J.C.), and grant-in-Aid for Scientific Research (B) of Japan Society for the Promotion of Science (21H02846 to K.H.).
Author contributions
J.Z., Q.Q., and K.H. conceived of the project, designed the experiments, analyzed the data, and wrote the manuscript. Q.C., S.L., and W.Y. designed and performed most of the experiments and analyzed the data. Y.Q. performed western blot analysis of postmortem brain samples. N.W. assisted in behavior tests. S.M. provided postmortem brain samples from control and DLB patients. Z.Z. provided pEGFP-α-Syn, mGST-α-Syn plasmid, and A53T mice. J.H. and J.C. assisted with data analysis and interpretation and critically read the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.05.037.
Contributor Information
Kenji Hashimoto, Email: hashimoto@faculty.chiba-u.jp.
Qi Qi, Email: qiqikc@jnu.edu.cn.
Ji-chun Zhang, Email: jczhang@jnu.edu.cn.
Supplemental information
References
- 1.Kang S.S., Zhang Z., Liu X., Manfredsson F.P., He L., Iuvone P.M., Cao X.B., Sun Y.E., Jin L.J., Ye K.Q. α-Synuclein binds and sequesters PIKE-L into Lewy bodies, triggering dopaminergic cell death via AMPK hyperactivation. Proc. Natl. Acad. Sci. U S A. 2017;114:1183–1188. doi: 10.1073/pnas.1618627114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ascherio A., Schwarzschild M.A. The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol. 2016;15:1257–1272. doi: 10.1016/s1474-4422(16)30230-7. [DOI] [PubMed] [Google Scholar]
- 3.Dauer W., Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 4.Yang J., Luo S., Zhang J., Yu T., Fu Z., Zheng Y., Xu X., Liu C.Y., Fan M.X., Zhang Z.T. Exosome-mediated delivery of antisense oligonucleotides targeting alpha-synuclein ameliorates the pathology in a mouse model of Parkinson’s disease. Neurobiol. Dis. 2021;148 doi: 10.1016/j.nbd.2020.105218. [DOI] [PubMed] [Google Scholar]
- 5.Volpicelli-Daley L.A., Luk K.C., Patel T.P., Tanik S.A., Riddle D.M., Stieber A., Meaney D.F., Trojanowski J.Q., Lee V.Y., Lee V.M.Y. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellis C.E., Schwartzberg P.L., Grider T.L., Fink D.W., Nussbaum R.L. α-Synuclein is phosphorylated by members of the src family of protein-tyrosine kinases. J. Biol. Chem. 2001;276:3879–3884. doi: 10.1074/jbc.m010316200. [DOI] [PubMed] [Google Scholar]
- 7.Okochi M., Walter J., Koyama A., Nakajo S., Baba M., Iwatsubo T., Meijer L., Kahle P.J., Haass C. Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J. Biol. Chem. 2000;275:390–397. doi: 10.1074/jbc.275.1.390. [DOI] [PubMed] [Google Scholar]
- 8.Jiang P., Gan M., Ebrahim A.S., Castanedes-Casey M., Dickson D.W., Yen S.H.C. Adenosine monophosphate-activated protein kinase overactivation leads to accumulation of alpha-synuclein oligomers and decrease of neurites. Neurobiol. Aging. 2013;34:1504–1515. doi: 10.1016/j.neurobiolaging.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lashuel H.A., Overk C.R., Oueslati A., Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toffoli M., Vieira S.R.L., Schapira A.H.V. Genetic causes of PD: a pathway to disease modification. Neuropharmacology. 2020;170 doi: 10.1016/j.neuropharm.2020.108022. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J., Li X., Li J.D. The roles of post-translational modifications on α-synuclein in the pathogenesis of Parkinson’s diseases. Front. Neurosci. 2019;13:381. doi: 10.3389/fnins.2019.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang S.S., Zhang Z., Liu X., Manfredsson F.P., Benskey M.J., Cao X., Xu J., Sun Y.E., Ye K.Q. TrkB neurotrophic activities are blocked by alpha-synuclein, triggering dopaminergic cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. U S A. 2017;114:10773–10778. doi: 10.1073/pnas.1713969114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang E.J., Reichardt L.F. Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- 14.Mizui T., Ishikawa Y., Kumanogoh H., Kojima M. Neurobiological actions by three distinct subtypes of brain-derived neurotrophic factor: multi-ligand model of growth factor signaling. Pharm. Res. 2016;105:93–98. doi: 10.1016/j.phrs.2015.12.019. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J.C., Yao W., Hashimoto K. Brain-derived neurotrophic factor (BDNF)-TrkB signaling in inflammation-related depression and potential therapeutic targets. Curr. Neuropharmacol. 2016;14:721–731. doi: 10.2174/1570159x14666160119094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seroogy K.B., Lundgren K.H., Tran T.M.D., Guthrie K.M., Isackson P.J., Gall C.M. Dopaminergic neurons in rat ventral midbrain express brain-derived neurotrophic factor and neurotrophin-3 mRNAs. J. Comp. Neurol. 1994;342:321–334. doi: 10.1002/cne.903420302. [DOI] [PubMed] [Google Scholar]
- 17.Baydyuk M., Xu B. BDNF signaling and survival of striatal neurons. Front. Cell. Neurosci. 2014;8:254. doi: 10.3389/fncel.2014.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howells D.W., Porritt M.J., Wong J.Y., Batchelor P.E., Kalnins R., Hughes A.J., Donnan G.A. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Exp. Neurol. 2000;166:127–135. doi: 10.1006/exnr.2000.7483. [DOI] [PubMed] [Google Scholar]
- 19.Bambah-Mukku D., Travaglia A., Chen D.Y., Pollonini G., Alberini C.M. A positive autoregulatory BDNF feedback loop via C/EBPβ mediates hippocampal memory consolidation. J. Neurosci. 2014;34:12547–12559. doi: 10.1523/jneurosci.0324-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinowich K., Hattori D., Wu H., Fouse S., He F., Hu Y., Fan G.P., Sun Y.E., Sun Y. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 21.Yuan Y., Sun J., Zhao M., Hu J., Wang X., Du G., Chen N.H. Overexpression of alpha-synuclein down-regulates BDNF expression. Cell. Mol. Neurobiol. 2010;30:939–946. doi: 10.1007/s10571-010-9523-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishina K., Piao W., Yoshida-Tanaka K., Sujino Y., Nishina T., Yamamoto T., Nitta K., Yoshioka K., Kuwahara H., Yasuhara H., et al. DNA/RNA heteroduplex oligonucleotide for highly efficient gene silencing. Nat. Commun. 2015;6:7969. doi: 10.1038/ncomms8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshioka K., Kunieda T., Asami Y., Guo H., Miyata H., Yoshida-Tanaka K., Sujino Y., Piao W.Y., Kuwahara H., Nishina K., et al. Highly efficient silencing of microRNA by heteroduplex oligonucleotides. Nucleic Acids Res. 2019;47:7321–7332. doi: 10.1093/nar/gkz492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asada K., Sakaue F., Nagata T., Zhang J.C., Yoshida-Tanaka K., Abe A., Nawa M., Nishina K., Yokota T. Short DNA/RNA heteroduplex oligonucleotide interacting proteins are key regulators of target gene silencing. Nucleic Acids Res. 2021;49:4864–4876. doi: 10.1093/nar/gkab258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren Q., Ma M., Yang J., Nonaka R., Yamaguchi A., Ishikawa K.I., Kobayashi K., Murayama S., Hwang S.H., Saiki S., et al. Soluble epoxide hydrolase plays a key role in the pathogenesis of Parkinson’s disease. Proc. Natl. Acad. Sci. U S A. 2018;115:E5815–E5823. doi: 10.1073/pnas.1802179115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickson D.W. Neuropathology of Parkinson disease. Parkinsonism Relat. Disord. 2018;46:S30–S33. doi: 10.1016/j.parkreldis.2017.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo S., Kang S.S., Wang Z.H., Liu X., Day J.X., Wu Z., Peng J., Xiang D.X., Springer W., Ye K.Q. Akt phosphorylates NQO1 and triggers its degradation, abolishing its antioxidative activities in Parkinson’s disease. J. Neurosci. 2019;39:7291–7305. doi: 10.1523/jneurosci.0625-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fumagalli F., Racagni G., Riva M.A. Shedding light into the role of BDNF in the pharmacotherapy of Parkinson’s disease. Pharmacogenomics J. 2006;6:95–104. doi: 10.1038/sj.tpj.6500360. [DOI] [PubMed] [Google Scholar]
- 29.Jin W. Regulation of BDNF-TrkB signaling and potential therapeutic strategies for Parkinson’s disease. J. Clin. Med. 2020;9:257. doi: 10.3390/jcm9010257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bathina S., Das U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015;6:1164–1178. doi: 10.5114/aoms.2015.56342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molina-Holgado F., Doherty P., Williams G., Molina-Holgado F., Doherty P., Doherty P. Tandem repeat peptide strategy for the design of neurotrophic factor mimetics. CNS Neurol. Disord. Drug Targets. 2008;7:110–119. doi: 10.2174/187152708783885200. [DOI] [PubMed] [Google Scholar]
- 32.Sun M., Kong L.X., Wang X.D., Lu X.G., Gao Q.S., Geller A.I. Comparison of the capability of GDNF, BDNF, or both, to protect nigrostriatal neurons in a rat model of Parkinson’s disease. Brain Res. 2005;1052:119–129. doi: 10.1016/j.brainres.2005.05.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Devi L., Raghavendran V., Prabhu B.M., Avadhani N.G., Anandatheerthavarada H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008;283:9089–9100. doi: 10.1074/jbc.m710012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conway K.A., Rochet J.C., Bieganski R.M., Lansbury P.T. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 35.Xu J., Kao S.Y., Lee F.J.S., Song W.H., Jin L.W., Yankner B.A. Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat. Med. 2002;8:600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
- 36.Barcia C., Barreiro A.F., Poza M., Herrero M.T. Parkinson’s disease and inflammatory changes. Neurotox. Res. 2003;5:411–417. doi: 10.1007/bf03033170. [DOI] [PubMed] [Google Scholar]
- 37.Halliday G.M., Stevens C.H. Glia: initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 2011;26:6–17. doi: 10.1002/mds.23455. [DOI] [PubMed] [Google Scholar]
- 38.Taylor J.M., Main B.S., Crack P.J. Neuroinflammation and oxidative stress: co-conspirators in the pathology of Parkinson’s disease. Neurochem. Int. 2013;62:803–819. doi: 10.1016/j.neuint.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 39.Costa G., Frau L., Wardas J., Pinna A., Plumitallo A., Morelli M. MPTP-induced dopamine neuron degeneration and glia activation is potentiated in MDMA-pretreated mice. Mov. Disord. 2013;28:1957–1965. doi: 10.1002/mds.25646. [DOI] [PubMed] [Google Scholar]
- 40.Wi R., Chung Y.C., Jin B.K., Duan L. Functional crosstalk between CB and TRPV1 receptors protects nigrostriatal dopaminergic neurons in the MPTP model of Parkinson’s disease. J. Immunol. Res. 2020;2020:1–11. doi: 10.1155/2020/5093493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shahmoradian S.H., Lewis A.J., Genoud C., Hench J., Moors T.E., Navarro P.P., Castaño-Díez D., Schweighauser G., Graff-Meyer A., Goldie K.N., et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019;22:1099–1109. doi: 10.1038/s41593-019-0423-2. [DOI] [PubMed] [Google Scholar]
- 42.Luk K.C., Kehm V., Carroll J., Zhang B., O’Brien P., Trojanowski J.Q., Lee V.M.Y. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson J.P., Walker D.E., Goldstein J.M., de Laat R., Banducci K., Caccavello R.J., Barbour R., Huang J.P., Kling K., Lee M., et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006;281:29739–29752. doi: 10.1074/jbc.m600933200. [DOI] [PubMed] [Google Scholar]
- 44.Arawaka S., Sato H., Sasaki A., Koyama S., Kato T. Mechanisms underlying extensive Ser129-phosphorylation in alpha-synuclein aggregates. Acta Neuropathol. Commun. 2017;5:48. doi: 10.1186/s40478-017-0452-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karampetsou M., Ardah M.T., Semitekolou M., Polissidis A., Samiotaki M., Kalomoiri M., Majbour N., Xanthou G., El-Agnaf O.M.A., Vekrellis K. Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci. Rep. 2017;7 doi: 10.1038/s41598-017-15813-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cabin D.E., Shimazu K., Murphy D., Cole N.B., Gottschalk W., McIlwain K.L., Orrison B., Chen A., Ellis C.E., paylor R., et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 2002;22:8797–8807. doi: 10.1523/jneurosci.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ekstrand M.I., Terzioglu M., Galter D., Zhu S., Hofstetter C., Lindqvist E., Thams S., Bergstrand A., Hansson F.S., Trifunovic A., et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. U S A. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frahm S., Melis V., Horsley D., Rickard J.E., Riedel G., Fadda P., Scherma M., Harrington C.R., Wischik C.M., Theuring F., Schwab K. Alpha-Synuclein transgenic mice, h-alpha-SynL62, display alpha-Syn aggregation and a dopaminergic phenotype reminiscent of Parkinson’s disease. Behav. Brain Res. 2018;339:153–168. doi: 10.1016/j.bbr.2017.11.025. [DOI] [PubMed] [Google Scholar]
- 49.Sanders D.W., Kaufman S.K., DeVos S.L., Sharma A.M., Mirbaha H., Li A., Barker S.J., Foley A.C., Thorpe J.R., Serpell L.C., et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–1288. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kang S.S., Ahn E.H., Zhang Z.T., Liu X., Manfredsson F.P., Sandoval I.M., Dhakal S., Iuvone P.M., Cao X.B., Ye K.Q. α-Synuclein stimulation of monoamine oxidase-B and legumain protease mediates the pathology of Parkinson’s disease. EMBO J. 2018;37 doi: 10.15252/embj.201798878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Volpicelli-Daley L.A., Luk K.C., Lee V.M.Y. Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 2014;9:2135–2146. doi: 10.1038/nprot.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim J., Lee S., Choi B.R., Yang H., Hwang Y., Park J.H.Y., LaFerla F.M., Han J.S., Lee K.W., Kim J. Sulforaphane epigenetically enhances neuronal BDNF expression and TrkB signaling pathways. Mol. Nutr. Food Res. 2017;61:1600194. doi: 10.1002/mnfr.201600194. [DOI] [PubMed] [Google Scholar]
- 53.Pruunsild P., Sepp M., Orav E., Koppel I., Timmusk T. Identification of cis-elements and transcription factors regulating neuronal activity-dependent transcription of human BDNF gene. J. Neurosci. 2011;31:3295–3308. doi: 10.1523/jneurosci.4540-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saito Y., Kawashima A., Ruberu N.N., Fujiwara H., Koyama S., Sawabe M., Arai T., Nagura H., Yamanouchi H., Hasegawa M., et al. Accumulation of phosphorylated alpha-synuclein in aging human brain. J. Neuropathol. Exp. Neurol. 2003;62:644–654. doi: 10.1093/jnen/62.6.644. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.