

Abstract
Viral immunotherapies are being recognized in cancer treatment, with several currently approved or undergoing clinical testing. While contemporary approaches have focused on oncolytic viral therapies, our efforts center on the development of plant virus-based cancer immunotherapies. In a previous work, we demonstrated the potent efficacy of the cowpea mosaic virus (CPMV), a plant virus that does not replicate in animals, applied as an in situ vaccine. CPMV is an immunostimulatory drug candidate, and intratumoral administration remodels the tumor microenvironment leading to activation of local and systemic antitumor immunity. Efficacy has been demonstrated in multiple tumor mouse models and canine cancer patients. As wild-type CPMV is infectious toward various legumes and because shedding of infectious virus from patients may be an agricultural concern, we developed UV-inactivated CPMV (termed inCPMV) which is not infectious toward plants. We report that as a monotherapy, wild-type CPMV outperforms inCPMV in mouse models of dermal melanoma or disseminated colon cancer. Efficacy of inCPMV is less than that of CPMV and similar to that of RNA-free CPMV. Immunological investigation using knockout mice shows that inCPMV does not signal through TLR7 (toll-like receptor); structure–function studies indicate that the RNA is highly cross-linked and therefore unable to activate TLR7. Wild-type CPMV signals through TLR2, -4, and -7, whereas inCPMV more closely resembles RNA-free CPMV which signals through TLR2 and -4 only. The structural features of inCPMV explain the increased potency of wild-type CPMV through the triple pronged TLR activation. Strikingly, when inCPMV is used in combination with an anti-OX40 agonist antibody (administered systemically), exceptional efficacy was demonstrated in a bilateral B16F10 dermal melanoma model. Combination therapy, with in situ vaccination applied only into the primary tumor, controlled the progression of the secondary, untreated tumors, with 10 out of 14 animals surviving for at least 100 days post tumor challenge without development of recurrence or metastatic disease. This study highlights the potential of inCPMV as an in situ vaccine candidate and demonstrates the power of combined immunotherapy approaches. Strategic immunocombination therapies are the formula for success, and the combination of in situ vaccination strategies along with therapeutic antibodies targeting the cancer immunity cycle is a particularly powerful approach.
Keywords: in situ vaccine, cowpea mosaic virus (CPMV), UV inactivation, OX40 agonist, combination therapy, cancer immunotherapy
Graphical Abstract
INTRODUCTION
The approval of the oncolytic viral immunotherapy Imlygic (Amgen) for treatment of melanoma1 and the development of recombinant poliovirus for treatment of recurrent glioblastoma2 highlight the potential of viruses in cancer immunotherapy. Imlygic is a genetically engineered oncolytic virus, a modified form of the herpes simplex virus (HSV). When administered within a lesion, it acts as an in situ vaccine by two mechanisms: (1) direct tumor cell killing as a function of the oncolytic nature of the virus; and (2) innate immune cell recruitment as a function of expression of the cytokine granulocyte-macrophage colony-stimulating factor—the gene that was inserted as part of the genetic modification of HSV.3 While mammalian viral vectors have made headway in human medicine, limitations are that these attenuated viruses can be infectious. In addition, Imlygic and other oncolytic virotherapies have a high price per dose and poor stability at room temperature, requiring storage at −80 °C that will significantly limit the market for and access to this technology. Additionally, the necessary repeated treatments of oncolytic therapies increase the risk of development of neutralizing antibodies against the biologic, thereby hindering their infectivity and potentially reducing their efficacy.4,5
To develop effective in situ vaccination approaches that address the challenges associated with oncolytic viral therapies, we have investigated plant virus-based biologics. Specifically, we have developed nanoparticles based on the cowpea mosaic virus (CPMV) for in situ vaccination. We have demonstrated that, when applied intratumorally, CPMV initiates remodeling of the tumor microenvironment and stimulates a potent antitumor immune response in mouse models of melanoma, ovarian cancer, breast cancer, colon cancer,6,7 and glioma.8 The antitumor response is systemic and accompanied by immunological memory, which can prevent recurrence. Ongoing trials in companion dogs with melanoma indicate that the potent antitumor efficacy of CPMV or virus-like particles (VLPs) of CPMV that lack RNA can be replicated in these animals.9-11
In a recent study, we delineated the mechanism of immune activation of CPMV, which provides insights into the differences in potency observed comparing native versus RNA-free formulations of CPMV, termed empty or eCPMV. Although CPMV is noninfectious toward mammals, it is immunogenic. The repetitive, multivalent protein assemblies are recognized as pathogen-associated molecular patterns (PAMPs) that act as danger signals and activate the innate immune cells, including neutrophils, dendritic cells, macrophages, and natural killer cells. The primary PAMP receptors that recognize proteins are toll-like receptors (TLRs).12 RNA-free, empty CPMV (eCPMV) particles are recognized by TLR2 and TLR4, while wild-type RNA-containing CPMV particles also signal through TLR7.13 In particular, CPMV generates a variety of proinflammatory cytokines, including interferon gamma6,7 as well as type-I interferons.13 The triple-pronged activation of RNA-laden CPMV likely underlies its increased potency.
The plant virus in situ vaccination strategy differs from oncolytic viral therapies: while the oncolytic virus relies on the tumor cell uptake to execute its cytotoxic function, the plant virus orchestrates tumor cell killing indirectly through innate immune cell activation: (1) CPMV engages with innate immune cells in the tumor microenvironment, (2) innate immune cells respond to CPMV by activating, and (3) these innate immune cells kill tumor cells and process tumor-associated antigens. These antigen-presenting cells then subsequently elicit an adaptive immune response that generates systemic immunity and immune memory.7,14 Therefore, CPMV-based immunotherapy is not subject to neutralization by antidrug antibodies. In contrast, the anti-CPMV antibodies (which are formed during the repeated treatment cycles) lead to opsonization, which stimulates innate immune cell uptake, and enhanced immune-mediated antitumor efficacy.15 Another positive attribute of CPMV and of plant viruses, in general, is that these biologics have outstanding thermal stability. CPMV withstands temperatures of 37 °C for 46 days16 and 60 °C for up to 1 h,17 and therefore it does not require freezers for storage or distribution.
While wild-type CPMV is not known to be infectious or communicated by humans or mammals, it does infect plants. In addition to the natural hosts, that is, black-eyed peas, species from several families—including legumes and Nicotiana benthamiana—are known to be susceptible to the virus, and transmission can be achieved experimentally by mechanical inoculation. In systemically infected plants CPMV typically causes mosaic or mottling symptoms.18 Therefore, agricultural safety must be considered when developing plant virus-based therapeutics. In our efforts toward environmentally safe plant virotherapy, we recently established chemical [using β-propiolactone (βPL) or formalin] and UV light inactivation of CPMV. We reported that 750 mM βPL, 1 mM formalin, or 7.5 J cm−2 UV sufficiently inactivated CPMV (inCPMV) and prevented plant infection. When used as in situ vaccine treatment, chemically inCPMV outperformed RNA-free VLPs but was less efficacious than wild-type CPMV in a murine dermal melanoma model; UV inCPMV was not tested.19
Building on these findings, we aimed to further investigate the efficacy of UV inCPMV. We assayed whether the structural changes resulting from UV exposure, such as RNA cross-linking within the nanoparticles and/or cross-linked to the protein capsid, could underlie these differences. The structural studies were complemented with immunological investigations of the relative accessibility of inCPMV RNA as a target of TLR signaling pathways. Finally, we evaluated inCPMV in combination dual therapy with OX40 agonist. OX40 is an immune stimulatory surface receptor that promotes T cell clonal expansion and activation with ligand binding. We chose OX40 therapy because we previously reported synergistic effects of combination therapy with wild-type CPMV and OX40 in multiple mouse tumor models.20 Here, we report the efficacy, structural, and immunological mechanism data of inCPMV in both solo and combination cancer immunotherapy strategies.
MATERIALS AND METHODS
CPMV Production and UV Inactivation.
Native CPMV was propagated in Vigna unguiculata (Burpee black-eyed pea No. 5) plants by mechanical inoculation; propagation and purification were as previously described.21 eCPMV was produced in N. benthamiana plants by agroinfiltration; propagation and purification were as previously described.22 Cowpea chlorotic mottle virus (CCMV) was used as a control and propagated in V. unguiculata (Burpee black-eyed pea No. 5) plants by mechanical inoculation; propagation and purification were as previously described.23 UV inactivation was carried out using previously established methods;19 in brief, native CPMV (1 mg mL−1) was exposed to UV light at a wavelength of 254 nm using a dose of 7.5 J·cm−2 using a UVP cross-linker (Analytik Jena AG).
Characterization of CPMV and inCPMV.
The concentration of CPMV and inCPMV was determined by UV–vis spectroscopy using a NanoDrop spectrophotometer (Thermo Fisher Scientific) and the Beer–Lambert law. CPMV: ε(260nm) = 8.1 mL mg−1 cm−1; molecular weight of CPMV = 5.6 × 106 g·mol−1.
The hydrodynamic diameter of native and inCPMV was determined by dynamic light scattering (DLS). Samples in 1 mg/mL in 0.01 M potassium phosphate buffer (pH 7.4) were analyzed using a Zetasizer Nano ZSP/Zen5600 instrument (Malvern Panalytical, Malvern, UK).
For transmission electron microscopy (TEM), the samples were diluted to 0.5 mg/mL in Milli-Q water; 10 μL of inCPMV was absorbed onto FCF400-CU 400-mesh copper grids (Electron Microscopy Sciences) for 2 min at room temperature, then the grid was washed twice for 30 s with Milli-Q water, and the sample was stained with 10 μL of 2% (w/v) uranyl acetate for 1 min. The grid was blotted with the Whatman filter paper to remove excess solution and examined at 80 kV using a FEI Company (Hillsboro, OR, USA) Tecnai G2 Spirit transmission electron microscope.
200 μL (1 mg mL−1) of CPMV and inCPMV were analyzed by size exclusion chromatography (SEC) using a Superose 6 Increase column and an ÄKTA Explorer chromatography system (GE Healthcare). The flow rate was set to 0.5 mL min−1 in 0.1 M potassium phosphate buffer (pH 7.0), and the absorbance at 260 and 280 nm was recorded.
Native CPMV and inCPMV (10 μg) were analyzed on agarose gels (0.8% w/v) run for 30 min at 120 V in 1× Trisacetate-ETDA running buffer in the presence of nucleic acid gel stain (GelRed, Biotium). Gels were imaged before and after staining with Coomassie brilliant blue G-250 (0.25% w/v) using the FluorChem R imaging system under UV light or white light.
CPMV and inCPMV (10 μg) were denatured in 4× LDS loading dye (Thermo Fisher Scientific) at 100 °C for 5 min, and the L and S coat proteins were separated on 4–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) precast gels in 1× morpholinepropanesulfonic acid buffer (Thermo Fisher Scientific) for 35 min at 200 V and 120 mA in the presence of SeeBlue Plus2 ladder size markers (Thermo Fisher Scientific). Gels were stained with GelRed (Biotium) and Coomassie brilliant blue G-250 (0.25% w/v) and subsequently imaged with the FluorChem R imaging system under UV light or white light. ImageJ software (https://imagej.nih.gov/ij/download.html) and band analysis tool were used for image analysis.
Immunological Assays.
Female C57BL/6J, MyD88−/−, TLR2−/−, TLR4−/−, TLR5−/−, TLR7−/−, and TLR9−/− mice (6–8 weeks old) were purchased from The Jackson Laboratory and bred at the Dartmouth College. These mouse studies were approved by the Institutional Animal Care and Use Committee of Dartmouth College. Splenocyte assays established previously13 were modified and set up from different mouse strains. Briefly, spleens from corresponding mouse strains were harvested, and 5 × 105 splenocytes were cultured with either CPMV or inCPMV (12 μg/mL) in 200 μL of complete RPMI for 24 h in 96-well plates. The conditioned medium was stored frozen (−20 °C) for cytokine quantification prior to assay by enzyme-linked immunosorbent assay (ELISA) to detect interleukin-6 (IL-6) or interferon β (IFN-β). Unless otherwise mentioned, these cells were cultured in RPMI1640 supplemented with 10% v/v fetal bovine serum and antibiotics. All cell cultures were maintained in a 5% CO2 incubator at 37 °C.
Data are presented as the mean ± standard error of the mean (SEM). The results shown here are representative responses from biological triplicates. T-test p-values <0.05 were required to assign significance and are represented as an asterisk (*) in the figures, p < 0.01 as **, and p < 0.001 as ***.
Tumor Mouse Models and CPMV Immunotherapy.
All tumor treatment experiments were conducted in accordance with the University of California, San Diego Institutional Animal Care and Use Committee and involved female C57BL6 or Balb/c mice (The Jackson Laboratory) 6–8 weeks of age.
For the dermal melanoma tumor model (one tumor), 2.5 × 105 B16F10 cells were suspended in 50 μL of PBS and were injected intradermally (i.d.) into the right flank of C57BL6 mice on day 0. CPMV, eCPMV, CCMV, and inCPMV (100 μg) were administered by intratumoral injection in 10 μL of PBS; weekly treatment was given starting ~7 days post tumor challenge when the tumors reached ~60 mm3. For the bilateral tumor model, 1.5 × 105 B16F10 cells were suspended in 20 μL of PBS and were injected (i.d.) into the left flank of C57BL6 mice on day 0; on day 3, 7.5 × 104 B16F10 cells were suspended in 10 μL of PBS and were injected (i.d.) into the right flank of C57BL6 mice to induce the secondary tumor. Only the primary tumor was treated using CPMV or inCPMV (100 μg) solo or in combination with rat anti-mouse OX40 IgG1 (clone OX86, BioXCell; 100 μg) on days 3, 8, and 13 post primary tumor challenge. OX40 was administered systemically via intraperitoneal injection. The primary tumors were surgically removed either when their volumes reach 500 mm3 or on day 15, whichever was earlier. These tumors were subsequently processed for immunofluorescence; see below. Tumor volumes were measured using a digital caliper. The tumor volume (mm3) was calculated as follows: (long diameter × short diameter2)/2. Animals were euthanized when the tumor volume exceeded 1000 mm3.
For the colon tumor model, 5 × 105 CT26-luc cells/200 μL of PBS were injected intraperitoneally (i.p.) into Balb/c mice. CPMV or inCPMV (100 μg) was administered on days 7, 14, and 21 by injection (i.p.) in 200 μL of PBS. Tumor growth was monitored by body weight and total bioluminescence imaging based on the i.p. injection of 100 mg kg−1 luciferin (Thermo Fisher Scientific), followed by analysis in an IVIS Spectrum Imaging System (PerkinElmer). Mice were euthanized when their weight reached 30 g.
Statistical analysis.
All results are expressed as the mean ± SEM, as indicated. Student’s t-test was used to compare the statistical difference between two groups, and one-way or two-way analysis of variance (ANOVA) with Sidak’s or Tukey’s multiple comparison tests were used to compare three or more groups. Survival rates were analyzed using the log-rank (Mantel–Cox). P values <0.05 were considered statistically significant: (*p < 0.05, **p < 0.05, etc.). All statistical tests were performed using GraphPad Prism v7.0 (GraphPad Software).
Immunofluorescence Staining of Tumor Tissue.
Two staining panels were used, one to target CD8+ T cells and the other to identify CD4+ T cells and Tregs. For the CD8+ T cell panel, primary antibody was rabbit anti-mouse CD8 monoclonal antibody (Abcam, ab209775, 1:500 dilution) and secondary antibody was goat anti-rabbit Alexa Fluor 488 (Abcam, ab150077, 1:500 dilution). For the CD4+ T cell and Treg panel, primary antibodies were rabbit anti-mouse CD4 monoclonal antibody (Abcam, ab183685, 1:400 dilution) and rat anti-mouse FoxP3 monoclonal antibody (ThermoFisher Scientific, FJK-16S, 1:400 dilution) and secondary antibodies were goat anti-rabbit Alexa Fluor 555 polyclonal antibody (Abcam, ab150078, 1:500 dilution) and goat anti-rat Alexa Fluor 488 polyclonal antibody (Abcam, ab150165, 1:500 dilution).
Tissue was flash frozen in OCT media with isopentane (cooled by dry ice to −78.5 °C). Tumors were cryo-sectioned into 5 μm transverse sections (orthogonal to the longest axis). Tumor sections were fixed with cooled 100% acetone (−20 °C), then washed with PBS, and blocked (1× PBS/5% (v/v) normal goat serum (Cell Signaling Technology, 5425S)/0.3% (v/v) Triton X-100) for 1 h at room temperature. Primary antibody staining was subsequently performed overnight at 4 °C. The sections were then washed in PBS and stained with secondary antibodies at room temperature for 2 h. After washing with PBS and drying, the sections were stained and mounted with Prolong Gold Antifade reagent with DAPI (Cell Signaling Technology, 8961S). Sections were visualized on a Keyence BZ-X710 all-in one microscope (Keyence Corporation) with a filter set (DAPI, TRITC, and FITC) and accompanying imaging analysis software.
RESULTS AND DISCUSSION
CPMV Outperforms inCPMV as Solo Treatment in Mouse Models of Melanoma and Colon Cancer.
We treated dermal melanoma using C57Bl6. Treatment began ~7 days post tumor challenge when palpable B16F10 tumors ~60 mm3 in volume were apparent, and animals were sacrificed when the tumors reached 1500 mm3. In the CT26 i.p. disseminated colon cancer model using Balb/C mice, tumor establishment was confirmed by bioluminescence imaging of the luc-expressing CT26 cells (not shown). Because imaging is only informative at early time points before the development of ascites, we applied imaging only to confirm successful tumor formation. We subsequently monitored the body weight and the abdominal circumference to assess tumor burden, and 30 g body weight was defined as the endpoint for the study. Weekly treatments were administered as a bolus injection either intratumorally (B16F10) or i.p. (CT26). The treatment groups were as follows: CPMV, inCPMV, PBS (negative control), eCPMV (which does not contain RNA22), or CCMV. Here, we used CCMV as a control particle: we previously reported that CCMV is not an effective in situ vaccine reagent. Based on the RAW Blue TLR reporter cell line assay,23 it is only moderately immunostimulatory. It does not engage with TLRs in HEK reporter cell lines24 and does not prime antitumor efficacy.23 Therefore, CCMV served as a negative control. Compared to PBS or any other treatment group, CPMV demonstrated the most potent efficacy in both tumor models, increasing and prolonging survival. Neither inCPMV, eCPMV, nor CCMV conferred statistically significant survival benefit under the treatment and dosing scheme used (Figure 1).
Figure 1.

CPMV outperforms inCPMV as monotherapy in mouse models of melanoma and colon cancer. (A) Treatment of B16F10 dermal melanoma bearing female C57Bl6 mice by weekly intratumoral injection of CPMV (n = 8), inCPMV (n = 7), or CCMV (n = 8) (100 μg) vs PBS (n = 7); three treatments were given. Treatment began when the tumors reached 60 mm3=, and endpoint was defined as tumors exceeding 1500 mm3. (B) Treatment of i.p. disseminated CT26 tumors in female Balb/C mice; treatment began 7 days post tumor challenge, and mice reaching 30 g was defined at the endpoint. Treatment was by weekly i.p. administration using CPMV (n = 8), inCPMV (n = 9), or eCPMV (n = 8) (100 μg) vs PBS (n = 9) (three treatments were given before endpoints were reached). Survival data were plotted, and statistical analysis was performed using Mantel–Cox test (*p < 0.05, **p < 0.05, ***p < 0.0005).
This does not necessarily indicate that none of these formulations can induce antitumor efficacy. Based on previous investigations, we can hypothesize that further modifications to the treatment dosing and administration may augment the efficacy observed in vivo. For example, while CCMV is not an effective in situ vaccine reagent, when loaded with CpG, a TLR-9 agonist, CpG-CCMV induces potent efficacy as was demonstrated in a mouse model of triple negative breast cancer.24 We have also reported that the eCPMV has antitumor efficacy in tumor mouse models and canine cancer patients,7,11 albeit at lower degree compared to CPMV.25 This relative deficit in efficacy of eCPMV was attributed to the lack of RNA TLR7 signaling as eCPMV is devoid of RNA.13 Additionally, our previous data demonstrated that chemically inCPMV was less efficacious than CPMV but more so than eCPMV.19 Furthermore, our previous report indicated that UV inCPMV—just like chemically inCPMV—maintained immunostimulatory properties as measured by TLR activation in the RAW Blue TLR reporter cell line.19 Indeed, we find that initiating treatment earlier resulted in measurable efficacy of inCPMV in a bilateral tumor model of B16F10 dermal melanoma; see data and discussion below. Therefore, it is also possible that an alternate treatment schedule, with earlier treatment, more frequent treatments, or higher dose would result in efficacy of inCPMV therapy.
Cross-linked RNA of inCPMV is Inaccessible for TLR7 Stimulation.
Since the RNA containing inCPMV behaved similarly to RNA-lacking eCPMV, we investigated the condition of the RNA in the inCPMV. First, we performed DLS, TEM, and SEC to assess whether inCPMV was intact, broken, or aggregated. Consistent with our previous reports,19 inCPMV showed some degree of aggregation. This was evidenced by an increased size and polydispersity index (PDI) in DLS measurements with inCPMV measuring ~56 nm (PDI of 0.43) versus ~37 nm (PDI of 0.19) for CPMV (Figure 2A). Also, TEM imaging confirmed the presence of intact particles. inCPMV were observed in clumps which may indicate some degree of interparticle cross-linking (Figure 2B). Aggregation was also observed by SEC and evident by a sharp peak at <10 mL elution volume from the Superose 6 Increase column. Unaggregated inCPMV eluted at ~12 mL which is consistent with the elution profile of native CPMV. The A260/280 nm ratio of 1.7 is indicative of intact CPMV with an RNA-to-protein ratio similar to that of native CPMV. Broken particles and free coat proteins or RNA, which would have higher elution volumes, were not detectable. Therefore, SEC indicates that UV inactivation results in intact inCPMV particles without loss of encapsulated RNA, albeit with some degree of interparticle aggregation (Figure 2C).
Figure 2.

Characterization of inCPMV nanoparticles by SEC and electrophoresis. (A) DLS of CPMV and inCPMV. (B) TEM of negatively stained (UAc) inCPMV. (C) SEC of CPMV (inset) and inCPMV using a Superose 6 Increase column and an ÄKTA Explorer chromatography system. (D) SDS-PAGE under white light after Coomassie staining and under UV light after GelRed staining; + and − denote inCPMV treated with UV light and native CPMV (untreated), respectively. M = SeeBlue Plus2 molecular weight marker. (E) Native agarose gel electrophoresis images of CPMV and inCPMV with and without lyophilization treatment (lyo-CPMV and lyo-inCPMV) prior to electrophoresis; gels were imaged with UV light after GelRed staining (RNA detection) or white light after Coomassie staining (protein detection). (F) Lane 4 of C (see red box), GelRed stain was analyzed using a band analysis tool and ImageJ software. The area under the curve was measured to quantify RNA associated with CPMV capsids (packaged RNA) vs free RNA (higher mobility bands).
To gain further insight into the mechanism of action of inCPMV, we performed structure–function studies. First, CPMV and inCPMV preparations were denatured, and the L and S coat protein subunits were separated and analyzed by SDS-PAGE; L and S were visualized as single bands at 42 and 24 kDa, respectively (Figure 2D). The band intensity of inCPMV coat proteins was reduced compared to the bands of CPMV coat proteins which indicates intercoat protein or coat protein–RNA cross-links resulting in reduced yields of free L and S protein. Low mobility, high-molecular-weight protein bands were not detectable by SDS-PAGE. These bands may be smeared and, therefore, are below the detection limit of the Coomassie blue staining. GelRed staining was added to SDS-PAGE gels to assess the RNA content of the particles. In contrast to native CPMV, much of the RNA from inCPMV did not travel through the gel and was detected in the well of the gel (Figure 2D). This band pattern is indicative of cross-linking of the RNA molecules and between RNA and the coat protein.
Next, we analyzed CPMV versus inCPMV by native agarose gel electrophoresis. Here, we also considered CPMV versus inCPMV stored in aqueous buffer versus lyophilized and reconstituted preparations. Lyophilization was utilized because we previously observed that freeze-drying results in loss of RNA from CPMV.26 CPMV and inCPMV exhibit comparable electrophoretic mobility under native agarose gel electrophoresis; the GelRed- and Coomassie-stained bands colocalize, indicating that intact, RNA-laden CPMV, and inCPMV migrate through the electrophoretic field and agarose gel (Figure 2E, lanes 1 + 2).
We note that the RNA staining for inCPMV is much stronger compared to that for native CPMV (the latter is hardly detectable but present), indicating that the cross-linked RNA is a better template for intercalation of the dye (GelRed). This was also consistent with previous findings.19 The band pattern observed after the samples were freeze-dried and restored in aqueous buffer prior to electrophoretic separation was remarkable. Lyophilization of native CPMV (lyo-CPMV) resulted in a loss of RNA as free RNA and free empty capsids were imaged (Figure 2E, lane 3). In contrast, RNA loss was only partial when inCPMV is lyophilized (lyo-inCPMV, Figure 2E, lane 4, and Figure 2F). Lane analysis using ImageJ software indicates that lyo-CPMV has complete loss of genomic RNA, whereas 60% of the RNA content remains associated with lyoinCPMV. Hence, only 40% of the RNA was no longer inside the protein coat.
We also considered the more extensive interparticle aggregation resulting from lyophilization of inCPMV. While aggregation of inCPMV was also apparent by SEC analysis (Figure 2C), it was not apparent by agarose gel electrophoresis for samples stored in aqueous buffer. The freeze-drying process, however, also concentrated the samples and fostered aggregation behavior as evident from the band pattern observed (Figure 2E, lane 4). Together, these data support the hypothesis that RNA is cross-linked to the protein capsid in inCPMV, leading to a higher degree of dye intercalation and resulting in more stable packaging. In part, this could be due to RNA-coat protein cross-linking or simply be steric hindrance for the cross-linked RNA to pass through the sub-nanometersized pores at CPMV’s five-fold axis.27
inCPMV is an Ineffective TLR7 Agonist and Inducer of Cytokine Production.
We set out to address whether the cross-linked RNA within inCPMV could still engage with and activate TLR7. MyD88 is a central adaptor protein for TLR recognition. In a recent work, we delineated that CPMV signals through MyD88-dependent TLR2, −4, and −7 and that the triple-pronged immune activation induces IL-6 and IFN-β.19 Using transgenic knockout (KO) mice and our established splenocyte assays,19 we compared the levels of IL-6 and IFN-β induction following stimulation with CPMV or inCPMV. Both CPMV and inCPMV induced IL-6 and IFN-β production in splenocytes from C57Bl6 mice; however, CPMV led to 2-fold higher levels of IL-6 and a 3.7-fold increase in IFN-β levels compared to inCPMV. Cytokine expression was lost when splenocytes from MyD88−/− were used, indicating that both CPMV and inCPMV are immunostimulatory and signal through MyD88-dependent pathways (Figure 3A,C).
Figure 3.

InCPMV is a poor TLR7 stimulant, leading to low cytokine induction compared to CPMV. Splenocyte assays established previously19 were set up from C57BL6, MyD88−/−, TLR7−/−, TLR2−/−, TLR4−/−, TLR5−/−, and TLR9−/− mice (n = 3/strain). Supernatant was analyzed by (A,B) IL-6 ELISA and (C,D) IFN-β ELISA. Data for bar graphs were calculated using unpaired Student’s t-test with p < 0.05 as *, p < 0.01 as **, and p < 0.001 as ***.
Next, we investigated the induction of cytokines in relevant MyD88-dependent TLR KO mice, that is, TLR2−/−, TLR4−/− TLR5−/−, TLR7−/−, and TLR9−/−.28 IL-6 and IFN-β levels were comparable when splenocytes from TLR7−/− mice were stimulated with either CPMV or inCPMV (Figure 3A,C). A direct comparison between wild-type C57BL6 and TLR7−/− mouse splenocyte assays (Figure 3 and Figure S1) showed that the superior induction ability of CPMV observed in wild-type mouse splenocyte assay was lost, when compared to that of inCPMV, in the TLR7−/− KO mouse splenocyte assay.
This indicates that the increased IL-6 and IFN-β levels observed in wild-type mouse splenocytes with CPMV stimulation, relative to the levels produced in response to inCPMV stimulation, reflected differences between CPMV and inCPMV in signaling through TLR7. TLR7 is responsible for viral ssRNA recognition and can induce type I interferon secretion.29,30 That inCPMV does not signal (or signals poorly) through TLR7 is consistent with the structural data provided in Figure 2. Data suggest that the cross-linked RNA (RNA–RNA and RNA–protein) within inCPMV is a poor TLR7 agonist compared to the intact ssRNAs that are encapsulated within CPMV. Since type I interferon is critical for the superior antitumor efficacy,31 this also explains the reduced efficacy when inCPMV was used as monotherapy for treatment of melanoma or colon cancer in mice (see Figure 1).
Last, KO models of TLR2, -4, -5, and -9 did not completely abolish the induction of IL-6 by CPMV or inCPMV (Figure 3B). This finding is aligned with our previous data demonstrating that TLR2, -4, and -7 are responsible for CPMV recognition. TLR2 and -4 are complimentary for CPMV capsid recognition (i.e., RNA-free eCPMV signals only through TLR2 and -4), and TLR7 is responsible for recognition of the packaged ssRNA.19 Although there is no statistical difference in IL-6 induction between CPMV- and inCPMV-treated splenocytes from TLR2, -4, -5, or -9 KO mice, there is a trend of increased average IL-6 induction by CPMV than inCPMV across all KO models. This is consistent with stimulation of either TLR2, -4, or -7 leading to increased proinflammatory cytokines; moreover, this is in line with our previous report that CPMV induced higher IL-6 and better antitumor efficacy compared to eCPMV, and this was attributed to additional TLR7 signaling through the packaged ssRNAs of CPMV.19 inCPMV only showed negligible-to-no IFN-β induction compared to CPMV when splenocytes from TLR2, -4, -5, or -9 KO mice were stimulated with inCPMV versus CPMV (Figure 3D). These data further strengthened our conclusion that inCPMV is a poor TLR7 agonist and that UV cross-linking likely renders the RNA inaccessible to TLR7 recognition or activation.
inCPMV in Combination with OX40 Agonist Serves as Highly Potent Cancer Immunotherapy.
While preliminary efficacy studies (Figure 1) and structure–function studies (Figures 2 and 3) clearly indicate that inCPMV is less potent compared to CPMV and exhibits cross-linked RNA inaccessible for TLR7 stimulation, we were not discouraged to further elucidate the potential of inCPMV as a cancer immunotherapy. Previous data indicate potent efficacy of eCPMV as an in situ vaccine,7,11 and the efficacy of eCPMV (which does not contain RNA) is mediated solely through TLR2 and TLR4 signaling.13 inCPMV is immunostimulatory in the RAW Blue cell assay.19 These data motivated us to investigate the therapeutic potential of inCPMV in combination with immune checkpoint therapy. The application of inCPMV may be of benefit because it is safer from an agricultural perspective, and we previously reported that inCPMV is noninfectious toward its host V. unguiculata.19 Plant viruses have been reported in human feces,32 and it is possible that “shedding” of the plant viral therapy may occur in patients. While it is unlikely to present a translational barrier, non-infectious formulations would be generally safer from an agricultural perspective.
In previous work, we demonstrated that CPMV treatment leads to various changes within the tumor microenvironment by reversing immunosuppression and launching potent and systemic antitumor immunity. Along with the observed changes, we also observed increased expression of checkpoint regulators on Foxp3− CD4+ effector T cells in the tumor microenvironment, including OX40. OX40 is an immune stimulatory surface receptor that is expressed on CD4+ and CD8+ effector T cells, as well as regulatory T cells. When OX40 is engaged with its ligand, the NF-κB signaling pathway is activated leading to T cell clonal expansion and activation.33,34 Agonistic OX40-specific antibodies can promote T cell activation.35 We observed that synergistic efficacy can be achieved through a combination of CPMV with agonistic OX40-specific antibodies. We observed reduced tumor burden, prolonged survival, and induced tumor antigen-specific immunologic memory to prevent relapse in three immunocompetent syngeneic mouse tumor models, including melanoma.20 Building on these data, we tested the efficacy of inCPMV combined with agonistic OX40-specific antibodies in a bilateral dermal melanoma model using B16F10 cells and C57Bl6 mice.
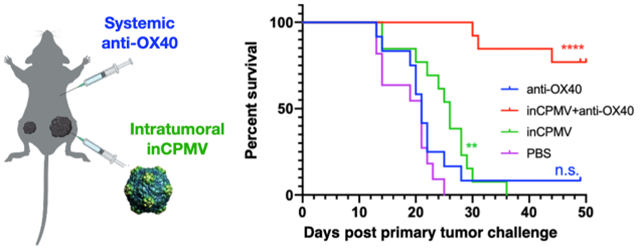
First, the mice were challenged with one tumor (left flank). Three days later, the mice were challenged with a second tumor inoculation on the contralateral flank (right flank). The primary tumor was removed when its volume reached 500 mm3 or on day 15 after the primary tumor challenge, whichever was earlier. Tumor resection kept the tumor burden below the humane endpoint threshold and to enable longitudinal monitoring of the secondary tumor’s progression. Treatment began 3 days after the left flank tumor challenge, and a total of three treatments of inCPMV were administered intratumorally treating only the primary tumor (left flank) as a monotherapy or in combination with systemically administered OX40 agonist (Figure 4A).
Figure 4.

In situ vaccination with inCPMV and OX40 agonist generates potent systemic antitumor immunity. (A) Treatment of a bilateral B16F10 dermal melanoma model using female C57Bl6 mice; secondary tumor challenge was performed 3 days after the primary tumor challenge; primary tumors were surgically removed on day 15; and treatment was given by intratumoral injection of anti-OX40 antibody (n = 9), inCPMV (n = 13), inCMV + anti-OX40 antibody (n = 14), or PBS (n = 11). (B,C) Tumor burden was measured, and the volume is reported of the primary tumor (B) and secondary tumor (C) [in panel (C), data points are shown for the groups with at least n = 3 animals remaining in the study]. (D) Survival data were plotted, and the statistical analysis was performed using Mantel–Cox test (****p < 0.00005).
Treatment with the OX40 agonist alone did not slow the growth rate of the treated tumor. While there was a moderate slowing of growth rate of some of the untreated tumors, the treatment conferred no improvement on survival compared to the survival of the PBS-treated control group. In contrast to the earlier treatment studies (Figure 1), inCPMV exhibited efficacy, and this difference may be explained by the difference in the treatment schedule: In this experiment, the primary tumor was treated 3 days after it was inoculated, when the tumors were small but palpable (Figure 4). The treatment start point in the study from Figure 1 was 7 days after inoculation of the primary tumor, when the tumors reached 60 mm3 (Figure 1A). inCPMV significantly reduced primary tumor burden and exhibited efficacy in delaying progression of the secondary tumor, evidenced by reduced tumor burden compared to the PBS-treated control group (Figure 4B,C). inCPMV also increased survival; the last animal in the inCPMV reached endpoint at 36 days post primary tumor challenge, while all PBS animals reached endpoint by 25 days post primary tumor challenge (p < 0.005) (Figure 4C).
The combination of inCPMV and OX40 agonist reached unmatched potency. The therapeutic effects were dramatic when assessing systemic efficacy by elimination of the established untreated tumor. Tumor growth was controlled over the 100 day longitudinal study with 10 out of 14 animals surviving; notably the survivors were tumor-free with no palpable tumors and no signs of metastatic disease (Figure 4C,D; p < 0.00005 vs PBS).
These data highlight the potential of inCPMV as an in situ vaccine candidate and demonstrate the power of agonistic OX40 antibody as part of immunocombination therapies. The immunostimulatory agent used for in situ vaccination leads to innate immune cell activation and recruitment, leading to tumor cell death and processing of tumor-associated antigens, thus connecting activation of the innate arm of the immune system with the adaptive arm for a more targeted response.36 Immunofluorescence staining of the sectioned tumor tissue of the treated primary tumors, 12 days following initial treatment, clearly showed infiltration of CD4+ and CD8+ T cells within the tumor microenvironment, both within the peripheral and deep tumors (because the treatment was effective, secondary tumors could not be collected). Animals treated with inCPMV and inCPMV + OX40 agonist led to increase in CD4+ and CD8+ T cells within the tumor (Figure 5); in both treatment groups, only a negligible number of FoxP3 regulatory T cells (Tregs) were observed—this is desired because Tregs are often associated with acquired resistance to immunotherapy. In stark contrast, there was no significant staining for CD4, CD8, or FoxP3 when animals received PBS or OX40 agonist alone (not shown). These data further support that the combination with an in situ vaccination strategy enables expansion of the pool of tumor-specific T cells generated by in situ vaccination to maximize the antitumor response.
Figure 5.

Immunofluorescence staining of secondary tumors indicate infiltration of CD4 and CD8 cells. Treated primary tumors were collected from animals treated with PBS, OX40 agonist, inCPMV, and inCPMV + OX40 12 days after the initial treatment. Tumor sectioned from animals treated with PBS or OX40 agonist showed no staining for CD4, CD8, or FoxP3 (not shown). Significant staining with CD4 and CD8 marker is apparent in both the peripheral and deep tumors from animals receiving inCPMV and inCPMV + OX40 agonist. Only a small number of Tregs (FoxP3) were detected. Representative images are shown; blue = DAPI staining of the cell nuclei; red and green = antibody-specific staining as indicated in the legends.
CONCLUSIONS
UV inCPMV is an immunostimulatory reagent that is noninfectious toward plants. The UV treatment cross-links the encapsulated ssRNA molecules. Immunological investigation further indicates that these structural changes render inCPMV a poor TLR7 agonist. Nevertheless, inCPMV remains immunostimulatory through the TLR2 and -4 pathways, which are activated by the protein, not the RNA cargo. We demonstrate potent, systemic antitumor immunity in a bilateral dermal melanoma mouse model when inCPMV is used in combination with an OX40 agonist. Together, these findings support inCPMV as a suitable therapeutic candidate for human or veterinary oncology applications, particularly when used in combination with other approaches to stimulate T cell effector function.
Supplementary Material
ACKNOWLEDGMENTS
We thank Angela Chen (UC San Diego) for helpful discussions. We thank Dr. Ivonne González Gamboa (UC San Diego) for assistance with TEM imaging.
Funding
This work was funded in part by the following grants from the National Institute of Health (NIH) (U01-CA218292 to N.F.S. and S.N.F. and R01-CA224605 to N.F.S.) and CDMRP (W81XWH2010742 to N.F.S.), a Pilot Research Grant from the Society of Interventional Radiology Foundation (to C.E.B.). C.E.B. and E.C.K. were supported in part by the UCSD Clinician–Scientist Radiology Residency Program grant T32EB005970 from the NIH.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.molpharmaceut.1c00681.
Cytokine data from WT and TLR7 KO mice (PDF)
The authors declare the following competing financial interest(s): Drs. Steven N. Fiering and Nicole F. Steinmetz are co-founders of and have a financial interest with Mosaic ImmunoEngineering Inc.
Contributor Information
Edward C. Koellhoffer, Department of Radiology, University of California, San Diego, La Jolla, California 92093, United States
Chenkai Mao, Department of Microbiology and Immunology, Geisel School of Medicine, Dartmouth College, Hanover, New Hampshire 03755, United States.
Veronique Beiss, Department of NanoEngineering, University of California, San Diego, La Jolla, California 92093, United States.
Lu Wang, Department of Bioengineering, University of California, San Diego, La Jolla, California 92093, United States.
Steven N. Fiering, Department of Microbiology and Immunology, Geisel School of Medicine, Dartmouth College, Hanover, New Hampshire 03755, United States; Norris Cotton Cancer Center, Geisel School of Medicine and Dartmouth Hitchcock Medical System, Lebanon, New Hampshire 03755, United States.
Christine E. Boone, Department of Radiology, University of California, San Diego, La Jolla, California 92093, United States.
Nicole F. Steinmetz, Department of Radiology, University of California, San Diego, La Jolla, California 92093, United States; Department of NanoEngineering, Department of Bioengineering, Moores Cancer Center, Center for Nano-ImmunoEngineering, and Institute for Materials Design and Discovery, University of California, San Diego, La Jolla, California 92093, United States.
REFERENCES
- (1).Andtbacka RHI; Kaufman HL; Collichio F; Amatruda T; Senzer N; Chesney J; Delman KA; Spitler LE; Puzanov I; Agarwala SS; Milhem M; Cranmer L; Curti B; Lewis K; Ross M; Guthrie T; Linette GP; Daniels GA; Harrington K; Middleton MR; Miller WH Jr.; Zager JS; Ye Y; Yao B; Li A; Doleman S; VanderWalde A; Gansert J; Coffin RS Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol 2015, 33, 2780–2788. [DOI] [PubMed] [Google Scholar]
- (2).Desjardins A; Gromeier M; Herndon JE, 2nd; Beaubier N; Bolognesi DP; Friedman AH; Friedman HS; McSherry F; Muscat AM; Nair S; Peters KB; Randazzo D; Sampson JH; Vlahovic G; Harrison WT; McLendon RE; Ashley D; Bigner DD Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med 2018, 379, 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ferrucci PF; Pala L; Conforti F; Cocorocchio E Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers 2021, 13, 1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhang Q; Liu F Advances and potential pitfalls of oncolytic viruses expressing immunomodulatory transgene therapy for malignant gliomas. Cell Death Dis. 2020, 11, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kaufman HL; Kohlhapp FJ; Zloza A Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discovery 2016, 15, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Murray AA; Wang C; Fiering S; Steinmetz NF In Situ Vaccination with Cowpea vs Tobacco Mosaic Virus against Melanoma. Mol. Pharm 2018, 15, 3700–3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lizotte PH; Wen AM; Sheen MR; Fields J; Rojanasopondist P; Steinmetz NF; Fiering S In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat. Nanotechnol 2016, 11, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kerstetter-Fogle A; Shukla S; Wang C; Beiss V; Harris PLR; Sloan AE; Steinmetz NF Plant Virus-Like Particle In Situ Vaccine for Intracranial Glioma Immunotherapy. Cancers 2019, 11, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hoopes PJ; Mazur CM; Osterberg B; Song A; Gladstone DJ; Steinmetz NF; Veliz FA; Bursey AA; Wagner RJ; Fiering SN Effect of intra-tumoral magnetic nanoparticle hyperthermia and viral nanoparticle immunogenicity on primary and metastatic cancer. Proc. SPIE-Int. Soc. Opt. Eng 2017, 10066, 100660G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hoopes PJ; Moodie KL; Petryk AA; Petryk JD; Sechrist S; Gladstone DJ; Steinmetz NF; Veliz FA; Bursey AA; Wagner RJ; Rajan A; Dugat D; Crary-Burney M; Fiering SN Hypofractionated Radiation, Magnetic Nanoparticle Hyper-thermia and a Viral Immunotherapy Treatment of Spontaneous Canine Cancer. Proc. SPIE-Int. Soc. Opt. Eng 2017, 10066, 1006605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hoopes PJ; Wagner RJ; Duval K; Kang K; Gladstone DJ; Moodie KL; Crary-Burney M; Ariaspulido H; Veliz FA; Steinmetz NF; Fiering SN Treatment of Canine Oral Melanoma with Nanotechnology-Based Immunotherapy and Radiation. Mol. Pharm 2018, 15, 3717–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Kawamura T; Ogawa Y; Aoki R; Shimada S Innate and intrinsic antiviral immunity in skin. J. Dermatol. Sci 2014, 75, 159–166. [DOI] [PubMed] [Google Scholar]
- (13).Mao C; Beiss V; Fields J; Steinmetz NF; Fiering S Cowpea mosaic virus stimulates antitumor immunity through recognition by multiple MYD88-dependent toll-like receptors. Biomaterials 2021, 275, 120914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wang C; Fiering SN; Steinmetz NF Cowpea Mosaic Virus Promotes Anti-Tumor Activity and Immune Memory in a Mouse Ovarian Tumor Model. Adv. Ther 2019, 2, 1900003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Shukla S; Wang C; Beiss V; Steinmetz NF Antibody Response against Cowpea Mosaic Viral Nanoparticles Improves In Situ Vaccine Efficacy in Ovarian Cancer. ACS Nano 2020, 14, 2994–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Madi M; Mioulet V; King DP; Lomonossoff GP; Montague NP Development of a non-infectious encapsidated positive control RNA for molecular assays to detect foot-and-mouth disease virus. J. Virol. Methods 2015, 220, 27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wang Q; Lin T; Tang L; Johnson JE; Finn MG Icosahedral virus particles as addressable nanoscale building blocks. Angew. Chem., Int. Ed 2002, 41, 459–462. [DOI] [PubMed] [Google Scholar]
- (18).Lomonossoff GP; Shanks M Comoviruses (Comoviridae). Encyclopaedia of Virology, 2nd ed.; Academic Press, 1999. [Google Scholar]
- (19).Chariou PL; Beiss V; Ma Y; Steinmetz NF In situ vaccine application of inactivated CPMV nanoparticles for cancer immunotherapy. Mater. Adv 2021, 2, 1644–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang C; Steinmetz NF A Combination of Cowpea mosaic virus and Immune Checkpoint Therapy Synergistically Improves Therapeutic Efficacy in Three Tumor Models. Adv. Funct. Mater 2020, 30, 2002299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Murray AA; Sheen MR; Veliz FA; Fiering SN; Steinmetz NF In Situ Vaccination of Tumors Using Plant Viral Nanoparticles. Methods Mol. Biol 2019, 2000, 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Saunders K; Sainsbury F; Lomonossoff GP Efficient generation of cowpea mosaicvirus empty virus-like particles by the proteolytic processing of precursors in insect cells and plants. Virology 2009, 393, 329–337. [DOI] [PubMed] [Google Scholar]
- (23).Shukla S; Wang C; Beiss V; Cai H; Washington T 2nd; Murray AA; Gong X; Zhao Z; Masarapu H; Zlotnick A; Fiering S; Steinmetz NF The unique potency of Cowpea mosaic virus (CPMV) in situ cancer vaccine. Biomater. Sci 2020, 8, 5489–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Cai H; Shukla S; Steinmetz NF The Antitumor Efficacy of CpG Oligonucleotides is Improved by Encapsulation in Plant Virus-Like Particles. Adv. Funct. Mater 2020, 30, 1908743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang C; Beiss V; Steinmetz NF Cowpea Mosaic Virus Nanoparticles and Empty Virus-Like Particles Show Distinct but Overlapping Immunostimulatory Properties. J. Virol 2019, 93, No. e00129–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zheng Y; Lee PW; Wang C; Thomas LD; Stewart PL; Steinmetz NF; Pokorski JK Freeze-Drying To Produce Efficacious CPMV Virus-like Particles. Nano Lett. 2019, 19, 2099–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lin T; Chen Z; Usha R; Stauffacher CV; Dai J-B; Schmidt T; Johnson JE The refined crystal structure of cowpea mosaic virus at 2.8 A resolution. Virology 1999, 265, 20–34. [DOI] [PubMed] [Google Scholar]
- (28).Kawasaki T; Kawai T Toll-like receptor signaling pathways. Front. Immunol 2014, 5, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Reizis B; Bunin A; Ghosh HS; Lewis KL; Sisirak V Plasmacytoid dendritic cells: recent progress and open questions. Annu. Rev. Immunol 2011, 29, 163–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gilliet M; Cao W; Liu Y-J Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol 2008, 8, 594–606. [DOI] [PubMed] [Google Scholar]
- (31).Kranz LM; Diken M; Haas H; Kreiter S; Loquai C; Reuter KC; Meng M; Fritz D; Vascotto F; Hefesha H; Grunwitz C; Vormehr M; Hüsemann Y; Selmi A; Kuhn AN; Buck J; Derhovanessian E; Rae R; Attig S; Diekmann J; Jabulowsky RA; Heesch S; Hassel J; Langguth P; Grabbe S; Huber C; Türeci Ö; Sahin U Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [DOI] [PubMed] [Google Scholar]
- (32).Rosario K; Symonds EM; Sinigalliano C; Stewart J; Breitbart M Pepper mild mottle virus as an indicator of fecal pollution. Appl. Environ. Microbiol 2009, 75, 7261–7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gramaglia I; Jember A; Pippig SD; Weinberg AD; Killeen N; Croft M The OX40 Costimulatory Receptor Determines the Development of CD4 Memory by Regulating Primary Clonal Expansion. J. Immunol 2000, 165, 3043. [DOI] [PubMed] [Google Scholar]
- (34).Rogers PR; Song J; Gramaglia I; Killeen N; Croft M OX40 Promotes Bcl-xL and Bcl-2 Expression and Is Essential for Long-Term Survival of CD4 T Cells. Immunity 2001, 15, 445–455. [DOI] [PubMed] [Google Scholar]
- (35).Guo Z; Wang X; Cheng D; Xia Z; Luan M; Zhang S PD-1 Blockade and OX40 Triggering Synergistically Protects against Tumor Growth in a Murine Model of Ovarian Cancer. PLoS One 2014, 9, No. e89350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sheen MR; Fiering S In situ vaccination: Harvesting low hanging fruit on the cancer immunotherapy tree. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol 2019, 11, No. e1524. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.